

ABSTRACT
Accumulating evidence suggests that macroautophagy/autophagy dysfunction plays a critical role in myocardial ischemia-reperfusion (I/R) injury. However, the underlying mechanisms responsible for malfunctional autophagy in cardiomyocytes subjected to I/R are poorly understood. As a result, there are no effective therapeutic options that target autophagy to prevent myocardial I/R injury. We recently revealed that MCOLN1/TRPML1, a lysosomal cationic channel, directly contributes to the inhibition of autophagic flux in cardiomyocytes post I/R. We found that MCOLN1 is activated secondary to reactive oxygen species (ROS) elevation following I/R, which in turn induces the release of lysosomal zinc into the cytosol. This ultimately blocks autophagic flux in cardiomyocytes by disrupting the fusion between autophagosomes containing engulfed mitochondria and lysosomes. Furthermore, we discovered that the MCOLN1-mediated inhibition of autophagy induced by I/R impairs mitochondrial function, which results in further detrimental ROS release that directly contributes to cardiomyocyte death. More importantly, restoration of blocked autophagic flux in cardiomyocytes subjected to I/R achieved by blocking MCOLN1 channels significantly rescues cardiomyocyte death in vitro and greatly improves cardiac function of mice subjected to I/R in vivo. Therefore, targeting MCOLN1 represents a novel therapeutic strategy to protect against myocardial I/R injury.
Abbreviations: I/R: ischemia-reperfusion; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MCOLN1/TRPML1: mucolipin TRP cation channel 1; ROS: reactive oxygen species; SQSTM1/p62: sequestosome 1.
KEYWORDS: Autophagy inhibition, cardiomyocyte death, ischemia-reperfusion injury, MCOLN1, mitochondria turnover
Acute myocardial infarction is a life-threatening condition that occurs when the heart muscle is severely damaged resulting from a lack of blood flow supply that leads to the formation of an infarct. The primary treatment strategy is myocardial reperfusion and can reduce the size of a myocardial infarct. However, although beneficial for myocardial salvage, the process of reperfusion induces injury called myocardial ischemia-reperfusion (I/R) injury, which contributes to cardiomyocyte cell death. Experimental studies have identified several mediators of myocardial reperfusion injury, including mitochondria dysfunction, oxidative stress, intracellular calcium overload, pH paradox, and inflammation. However, the reduction of the myocardial infarction size by targeting any of these aforementioned mediators proves to be a significant challenge in the clinical setting.
It has been well accepted that autophagy, an evolutionarily conserved degradation process, is one of the most important ways for damaged mitochondria to be eliminated. Conversely, mitochondrial dysfunction has been considered as the most significant molecular mechanism underling the pathogenesis of myocardial I/R injury. Therefore, targeting autophagy could be a therapeutic strategy to protect against myocardial I/R injury. However, there is a debate over autophagy being beneficial or detrimental for such injury. As such, meticulous investigation into the role of myocardial autophagic flux following I/R is demanded.
To address this question, we used both an in vitro and in vivo I/R model to meticulously track myocardial autophagic flux during the I/R process [1]. We found that autophagy is induced following ischemia, as reflected by an increase in LC3-II protein levels and a decrease in SQSTM1 protein levels in cardiomyocytes. In contrast, autophagic flux is blocked during reperfusion, as determined by an increase in both LC3-II and SQSTM1 protein levels post I/R, a reduction in the fusion process between autophagosomes and lysosomes in vitro, and an increase in the punctate structures of LC3 and SQSTM1 in vivo. Thus, our observations demonstrate that in contrast to the ischemia process, myocardial autophagic flux is blocked during reperfusion (Figure 1).
Figure 1.
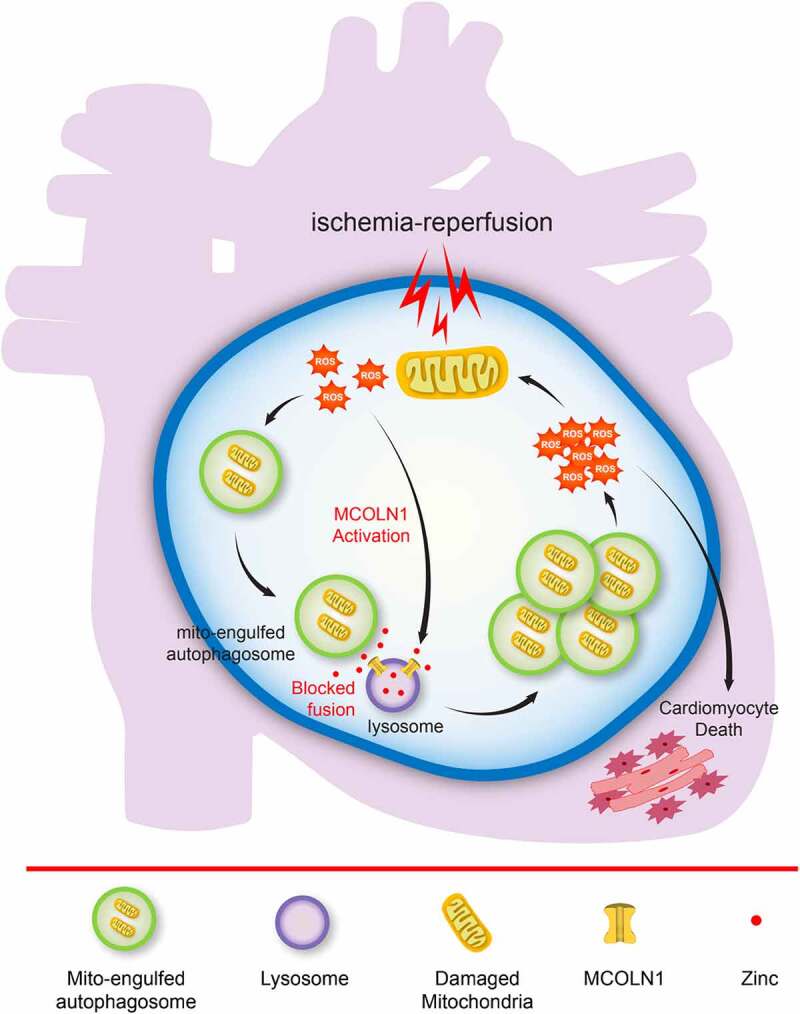
A working model illustrates a network of myocardial autophagic flux blocked by the activation of MCOLN1/TRPML1 channels during the process of myocardial ischemia-reperfusion injury. The blocked autophagic flux disrupts the fusion between autophagosomes containing engulfed mitochondria and lysosomes, leading to accumulation of damaged mitochondria and detrimental ROS release, which directly causes cardiomyocyte death.
As supported by our previous studies, the activation of the lysosomal cationic channel MCOLN1/TRPML1 (mucolipin TRP cation channel 1) results in the release of lysosomal zinc into the cytosol. This in turn disrupts fusion between autophagosomes and lysosomes and ultimately inhibits autophagic flux. In parallel, as a potent endogenous activator of MCOLN1 channels, the activity of ROS theoretically inhibits autophagic flux. In addition to this, a substantial quantity of ROS is generated following the restoration of oxygen flow during the myocardial I/R process. Given this, we wanted to provide critical new mechanistic insight into the potential role of MCOLN1 in the inhibition of autophagic flux in cardiomyocytes when subjected to I/R, and where excessive ROS may function as a mediator integrating MCOLN1 activity with the inhibition of autophagic flux in response to myocardial I/R injury.
We found that MCOLN1 is activated secondary to ROS generation during the reperfusion process following ischemia, which in turn inhibits myocardial autophagic flux by mediating the release of lysosomal zinc into the cytosol. Consequently, the inhibition of autophagic flux downstream of MCOLN1 activation disrupts the fusion process between autophagosomes containing engulfed mitochondria and lysosomes, leading to the accumulation of damaged mitochondria and detrimental ROS release, which directly causes cardiomyocyte death following I/R (Figure 1).
Importantly, blocking MCOLN1 channels using pharmacological or genetic approaches exhibits significant effects on protecting cardiac function following I/R injury in mice. Accordingly, our findings identify a promising strategy of targeting MCOLN1 to protect cardiac function against myocardial I/R injury (Figure 1).
In summary, activation of MCOLN1 underlies the pathogenesis of impaired autophagic flux in cardiomyocytes subjected to I/R. Blunting MCOLN1 channels greatly improves both short-term and long-term heart function in vivo in I/R mouse models. Therefore, targeting MCOLN1 represents a promising new strategy to effectively protect against I/R injury in the clinical setting (Figure 1).
Acknowledgments
We appreciate the encouragement and helpful comments from other members of the Wang laboratory.
Funding Statement
This work was supported by National Natural Science Foundation of China (NSFC) grants (81772559 to W.W; 82101314 to Y. X), Key University Science Research Project of Jiangsu Province (20KJA310001 to W.W), Jiangsu Specially Appointed Professor award to W.W (2017), and Jiangsu Province Innovative and Entrepreneurial Talent program to W.W (2018) and Jiangsu Province Innovative and Entrepreneurial Team program to W.W (2020). Natural Science Foundation of Liaoning Province (2021-MS-161 to M.M. W).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- [1].Xing Y, Sui Z, Liu Y, et al. Blunting TRPML1 channels protects myocardial ischemia/reperfusion injury by restoring impaired cardiomyocyte autophagy. Basic Res Cardiol. 2022;117:20. PMID: 35389129. [DOI] [PubMed] [Google Scholar]