

ABSTRACT
Externalization of the phospholipid cardiolipin (CL) to the outer mitochondrial membrane has been proposed to act as a mitophagy trigger. CL would act as a signal for binding the LC3 macroautophagy/autophagy proteins. As yet, the behavior of the LC3-subfamily members has not been directly compared in a detailed way. In the present contribution, an analysis of LC3A, LC3B and LC3C interaction with CL-containing model membranes, and of their ability to translocate to mitochondria, is described. Binding of LC3A to CL was stronger than that of LC3B; both proteins showed a similar ability to colocalize with mitochondria upon induction of CL externalization in SH-SY5Y cells. Besides, the double silencing of LC3A and LC3B proteins was seen to decrease CCCP-induced mitophagy. Residues 14 and 18 located in the N-terminal region of LC3A were shown to be important for its recognition of damaged mitochondria during rotenone- or CCCP-induced mitophagy. Moreover, the in vitro results suggested a possible role of LC3A, but not of LC3B, in oxidized-CL recognition as a counterweight to excessive apoptosis activation. In the case of LC3C, even if this protein showed a stronger CL binding than LC3B or LC3A, the interaction was less specific, and colocalization of LC3C with mitochondria was not rotenone dependent. These results suggest that, at variance with LC3A, LC3C does not participate in cargo recognition during CL-mediated-mitophagy. The data support the notion that the various LC3-subfamily members might play different roles during autophagy initiation, identifying LC3A as a novel stakeholder in CL-mediated mitophagy. Abbreviations: ACTB/β-actin: actin beta; Atg8: autophagy-related 8; CL: cardiolipin; CCCP: carbonyl cyanide m-chlorophenyl hydrazone; DMSO: dimethyl sulfoxide; DOPE: 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine; DTT: DL-dithiothreitol; FKBP8: FKBP prolyl isomerase 8; GABARAP: GABA type A receptor associated protein; GABARAPL1: GABA type A receptor associated protein like 1; GABARAPL2: GABA type A receptor associated protein like 2; GFP: green fluorescent protein; IMM: inner mitochondrial membrane; LUV/LUVs: large unilamellar vesicle/s; MAP1LC3A/LC3A: microtubule associated protein 1 light chain 3 alpha; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MAP1LC3C/LC3C: microtubule associated protein 1 light chain 3 gamma; NME4/NDPK-D/Nm23-H4: NME/NM23 nucleoside diphosphate kinase 4; O/A: oligomycin A + antimycin A; OMM: outer mitochondrial membrane; PA: phosphatidic acid; PC: phosphatidylcholine; PG: phosphatidylglycerol; PINK1: PTEN induced putative kinase 1; PtdIns4P: phosphatidylinositol-4-phosphate; Rho-PE: lissamine rhodamine phosphatidylethanolamine; SUV/SUVs: small unilamellar vesicle/s
KEYWORDS: Atg8, autophagosome, autophagy cargo recognition, LC3/GABARAP-protein family, lipid oxidation, lipid-protein interaction, membrane curvature, mitochondria, negatively charged phospholipids
Introduction
Mitochondria are essential organelles for energy transduction in the eukaryotic cell. Mitochondrial dysfunctions have been associated with a wide number of pathological conditions, including neurodegenerative diseases, myopathies, and cancer [1]. Cells have developed different quality control systems in order to maintain an optimal mitochondrial network, including processes that allow the removal of damaged or superfluous mitochondria without causing cell death [2]. Among them, the selective degradation of mitochondria via macroautophagy is termed mitophagy.
Macroautophagy is characterized by the formation of a double-membrane structure around the cellular components to be degraded, the autophagosome (AP). The AP then fuses with lysosomes resulting in contents degradation by the lysosomal hydrolases. In this highly conserved mechanism, a plethora of signals and proteins are involved, making it exceedingly complex [3]. The human autophagy-related LC3/GABARAP-protein family [4] is composed of at least six orthologs of the yeast protein Atg8, divided into two subfamilies, LC3 and GABARAP [5]. As in the case of Atg8, all LC3/GABARAP-family members are found in both bilayers of the nascent autophagosomes [6], bound to phosphatidylethanolamine (PE). PE binding occurs via a conserved C-terminal glycine, through the coordinated action of two ubiquitin-like conjugation systems [7]. This protein family is involved not only in membrane expansion, closure and fusion with lysosomes [8,9] but also in cargo recruitment [10].
Recognition of damaged or superfluous mitochondria is a key event in mitophagy. Many different indicators of mitochondrial damage are known. The PINK1-PRKN/Parkin axis, recognized as the main regulator of mitophagy [11], is characterized by the tagging of damaged mitochondria with ubiquitin chains that trigger their selective autophagy. This process requires the participation of different proteins such as OPTN (optineurin) and CALCOCO2/NDP52, which act as receptors. They are able not only to recognize the ubiquitin chain on the mitochondrial surface but also to bind LC3B in the phagophore membrane [12]. However, a growing body of evidence has shown that this pathway is not the only relevant one. Many mitophagy receptors, such as FUNDC1, BNIP3L/NIX, BNIP3, BCL2L13, AMBRA1 or FKBP8, that are found in the outer mitochondrial membrane (OMM), have shown the ability to recruit directly LC3/GABARAP proteins to mediate mitochondrial degradation, without ubiquitin signaling [13]. Most of these receptors have an LC3-interacting motif (LIR) that allows their association with LC3/GABARAP proteins [14]. Lipids, such as ceramide [15] or cardiolipin [16], can also serve as damaged mitochondrial signals/receptors.
The mitophagy mechanism mediated by the phospholipid cardiolipin (CL) is particularly intriguing. CL is essential for mitochondrial function and it is localized in the inner mitochondrial membrane (IMM) in healthy mitochondria [17]. In 2013, Chu et al. [16] found out that rotenone and other pro-mitophagic stimuli cause CL externalization to OMM, in turn inducing mitophagy. Furthermore, inhibition of CL synthesis or of CL externalization decreased the delivery of mitochondria to AP. Therefore, those authors proposed that CL would play a role in mitophagy; its externalization to the OMM upon mitochondrial injury would act as a signal for LC3 proteins to remove damaged mitochondria in neuronal cells, preventing CL oxidation and accumulation of proapoptotic signals [16]. Recent studies have demonstrated that CL-externalization takes place also in vivo. Traumatic brain injury (TBI) model animals externalize CL to the OMM to a similar extent than rotenone-exposed mitochondria, thus underlining the importance of CL for mitophagy induction during the early response to TBI in human and rat brain [18]. Moreover, it is known that TAFAZZIN-deficiency-related perturbations in CL remodeling, found in the Barth syndrome, cause defective mitophagosome biogenesis [19]. In addition, it seems that externalized CL could interact with SNCA/alpha-synuclein [20], a protein associated with Parkinson disease. For these reasons, studying this mechanism of CL-mediated mitophagy could lead to interpreting some neurodegenerative diseases as mitophagy-related diseases [21,22].
Chu et al. [16] demonstrated the importance of two conserved residues (R10, R11) located in the N-terminal region of LC3 proteins for its interaction with CL. They proposed a mode of interaction between LC3 proteins and CL based on the recognition of damaged mitochondria by the protein N-terminal region, while their C-terminal region would target the autophagosomal membrane. These results were extended by studies in our laboratory [23] that explored the molecular basis of LC3-CL interaction. It was shown that LC3B was able to interact preferentially with CL over other di-anionic lipids, and that after protein binding to CL-enriched membranes the C terminus of LC3B remained exposed to the hydrophilic environment allowing its interaction with the phagophore. Thus, CL appeared to serve as a specific mitophagy receptor for LC3 proteins, ultimately leading to the removal of damaged mitochondria. However, although Chu et al. [16] showed that LC3A and LC3B interacted with CL-containing liposomes, no comparative study was available on the CL- and mitochondria-binding abilities of the LC3 subfamily members LC3A, LC3B, and LC3C. The data in this paper show the different behavior of the LC3-subfamily members in the selection of damaged mitochondria as autophagosomal cargo, and describe LC3A as an LC3/GABARAP-family member capable of recognizing externalized CL, and of mediating mitophagy.
Results
The similarity of predicted CL-interacting residues in LC3-subfamily members suggests similar functions.
In mammals a variety of well conserved Atg8 orthologs can be found (Figure 1), suggesting some degree of specialization of the various family members. LC3/GABARAP proteins share a similar structure, consisting of two amino-terminal α-helices and an ubiquitin-like (UBL) core (Figure 1A). However, despite the extensive structural similarities, it has been shown that, while the first α-helix of LC3 subfamily is strongly basic, in the GABARAP subfamily this region is acidic [24] (Figure 1A, B). These differences are relevant because the N-terminal region of these proteins might be important for specific functions during autophagy, playing an important role in protein-protein interactions, lipid-protein interactions, or via post-translational modifications [25].
Figure 1.
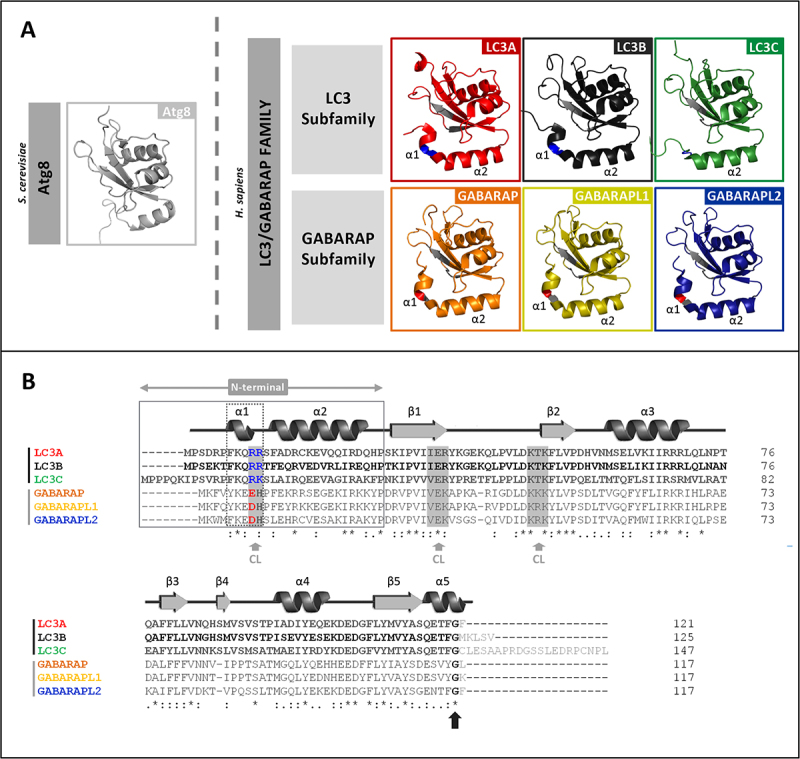
LC3/GABARAP-family members and their interaction with CL. (A) Schematic representation of the 3D structures of yeast Atg8 and of each LC3/GABARAP protein in solution, displayed with PyMOL. PDB: Atg8 (2KQ7), LC3A (5CX3), LC3B (2ZJD), LC3C (2NCN), GABARAP (1GNU), GABARAPL1 (5LXI) and GABARAPL2 (4CO7). The thermodynamically preferred residues for cardiolipin binding on LC3B, identified by docking analysis [16], and their equivalents in the other LC3/GABARAP-family members are highlighted in gray (See Figure 1B). Among them, the two residues in the α1 N-terminal region proposed to be essential for the interaction, and their equivalents in the LC3 subfamily, are colored in blue (positively charged). The amino acid corresponding to R10 of LC3B is a negatively charged residue in the GABARAP subfamily, colored in red (See Figure 1B). (B) Sequence alignment of the human orthologs of Atg8 obtained with Clustal W. UniProt: LC3A (Q9GZQ8), LC3B (Q9GZQ8), LC3C (Q9BXW4), GABARAP (O95166), GABARAPL1 (Q9H0R8) and GABARAPL2 (P60520). The secondary structure elements of LC3B (PDBsum, 2ZJD) are indicated above the alignment as an example. The N-terminal and α-helix 1 (α1) of the two subfamilies are boxed. As in Figure 1A, the thermodynamically preferred residues for cardiolipin binding of LC3B, and their equivalents in the other members of the LC3/GABARAP family are highlighted in gray (gray arrow). The two positively charged α1 N-terminal residues proposed to be essential for the interaction of LC3B with CL and their equivalents are colored; blue: positively charged, red: negatively charged. The C-terminal glycine (black arrow) is conserved in all the LC3/GABARAP-family members.
Previous studies from this laboratory have focused on the potential different roles of these two subfamilies, both in cargo recognition [23] and during autophagosome expansion [26]. Regarding cargo recognition, several aspects of the mitophagic mechanism initiated by CL externalization remain obscure. Previous work from this laboratory [23] compared the behavior of LC3B with that of GABARAP subfamily members. The study explored the differences in residues putatively involved in LC3B interaction with CL (Figure 1B). In vitro, the GABARAP-subfamily members showed less binding to CL than LC3B and this corresponded with the absence of translocation to mitochondria in cells.
Taking into account the similarity between the predicted CL-interacting residues in LC3B and those in LC3A and LC3C (Figure 1B), it could be assumed that all three LC3-subfamily members would be involved in CL recognition. This hypothesis was experimentally tested as follows.
LC3A and LC3C, as well as LC3B, interact with CL-containing model membranes
To determine whether members of the LC3 subfamily other than LC3B were also able to bind CL-enriched model membranes, the binding of LC3A and LC3C to large unilamellar vesicles (LUV) containing CL was measured. For this purpose, a vesicle flotation assay was performed [23], in which protein association with membranes was assessed by the protein ability to float together with the vesicles after equilibrium in sucrose-gradient centrifugation (Figure 2A). A highly unsaturated (mainly tetralinoleoyl) CL was chosen based on the results by Antón et al. [23], who compared the LC3B binding affinities toward CL containing acyl chains with different degrees of unsaturation. CL concentration in these experiments intended to mimic the one of the interaction zones formed in the OMM after CL exposure, and it was based on the CL levels used in similar studies with other proteins [20,27,28].
Figure 2.
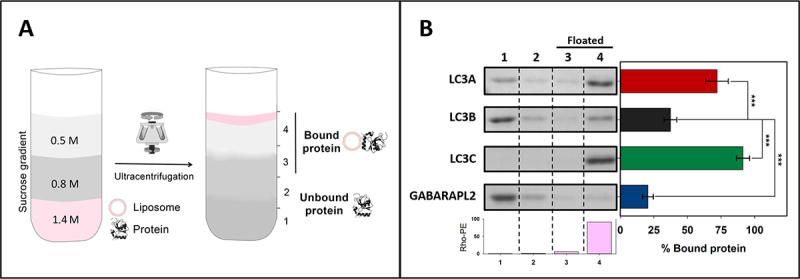
LC3A and LC3C, as well as LC3B, interact with CL-containing model membranes. Interaction of LC3/GABARAP proteins with CL-containing membranes measured by a vesicle flotation assay. (A) Representative scheme of a flotation assay in a sucrose-density gradient. Proteins (10 μM) were incubated with rhodamine-PE (Rho-PE)-labeled LUVs (3 mM) and subsequently mixed within the 1.4 M (final concentration) layer of a discontinuous sucrose gradient. After ultracentrifugation, four equal-volume fractions were collected, 1–4, starting from the bottom; protein found in fractions 3 + 4 was taken as bound protein. (B) Interaction of LC3A, LC3B, LC3C and GABARAPL2 with LUVs containing CL. LUVs were composed of PC:DOPE:CL (33:33:33 mol ratio) + 0.05% Rho-PE. The presence of vesicles and proteins in the different fractions was probed by Rho-PE fluorescence emission and by SDS-PAGE/Coomassie Brillant Blue staining respectively. Bars at the bottom: Rho-PE emission was detected only in fractions 3–4 (i.e., floating fractions). The bars at the right-hand side correspond to the percentage of bound protein, taken as protein co-floating with vesicles and calculated by gel densitometry. Data are means ± SD (n ≥ 9). ***p < 0.001.
In fact, the in vitro interaction of LC3A and LC3C with CL was higher than that of LC3B. Almost all of the LC3C was bound to membranes when CL was present, and, while not all LC3A interacted with CL, it nearly doubled the proportion of bound LC3B (Figure 2B). Binding of the three proteins was dependent on both vesicle curvature (the proteins bound more highly curved SUV better than LUV) and CL concentration in the bilayers (Fig. S1). Consistent with previous results [23] GABARAPL2 did not reach the binding level of the LC3 proteins to CL-containing membranes (Figure 2B). The results indicated that LC3B was not the only member of the LC3 subfamily able to bind CL in lipid bilayers, thus perhaps LC3B might not be the only one capable of recognizing externalized CL in damaged mitochondria.
LC3A and LC3B interact preferentially with CL over other negatively charged phospholipids
To determine whether LC3 protein interaction with membranes was specific for CL, the binding of these proteins to LUVs of different lipid compositions was investigated. None of the LC3-subfamily members was able to float in the absence of vesicles, or in the presence of liposomes composed of electrically neutral phospholipids (egg phosphatidylcholine [PC] or dioleoyl phosphatidylethanolamine [DOPE]) (Figure 3A). Moreover, the interaction of LC3 proteins with membranes containing egg phosphatidic acid (PA) or egg phosphatidylglycerol (PG), phospholipids that are structurally and metabolically related to CL, was assayed. PA and PG are also negatively charged, but they contain ≈1 charge per molecule under physiological conditions, vs. ≈2 in the case of CL [29]. Neither LC3A nor LC3B did interact with PA- or PG-containing membranes. LC3C was able to bind PA-containing bilayers, albeit the interaction was lower than with CL-vesicles. PG did not increase LC3C binding to liposomes (Figure 3).
Figure 3.
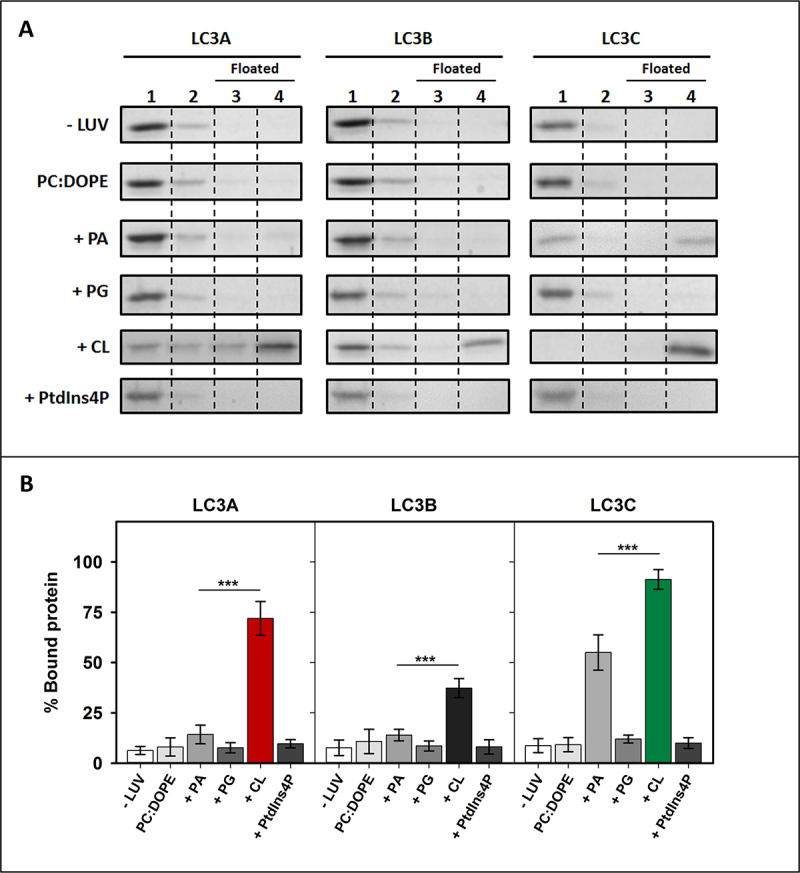
LC3A and LC3B exhibit a marked specificity for CL. Interaction of LC3 proteins with membranes of different compositions, measured by a vesicle flotation assay. (A) Representative SDS-PAGE Coomassie Brilliant Blue-stained gels of the fractions obtained from LC3A, LC3B or LC3C vesicle flotation assays performed without liposomes (-LUVs) or with liposomes of different compositions, either PC:DOPE (50:50) or PC:DOPE:X (33:33:33 mol ratio) where X was PA, PG, CL or PtdIns4P. Bound protein was computed as the proportion retrieved in fractions 3 + 4 (See Figure 2A). (B) Binding of LC3A, LC3B, and LC3C to liposomes quantified by gel densitometry. Data are means ± SD (n ≥ 3). ***p < 0.001.
In addition, the interaction with brain phosphatidylinositol-4-phosphate (PtdIns4P) was measured. PtdIns4P contains two negative charges per molecule, as in the case of CL, but two fatty acyl chains per molecule, vs. four in the case of CL. None of the LC3-subfamily members showed the ability to interact with PtdIns4P-containing membranes, suggesting that the interaction could not be explained by purely electrostatic forces (Figure 3). Note that PtdIns4P generation in autophagosomes was shown to be critically important for GABARAP-mediated fusion with lysosomes [30], another example of differentiation in the LC3/GABARAP-protein family. In summary, the above results suggest that all three LC3-subfamily members interact preferentially with CL over other negatively charged lipids, although LC3C displays also a rather high affinity toward PA, which suggests that this protein is not as specific for CL as the other LC3 homologs.
The higher capacity of LC3C to interact with CL resides in its N- terminal region
To determine the characteristics that made LC3C interact with CL with a higher affinity than LC3B, a sequence alignment of LC3C and LC3B was performed. LC3C exhibited large differences with LC3B in its amino acid sequence (46/72% identity/similarity) (Figure 4A). The main differences were found in the N terminal region. This observation was consistent with the previously mentioned role of the N-terminal region in the interaction with CL [16,23]. Therefore, the possibility that this region could participate in the interaction with CL was tested. For this purpose, a chimera protein was constructed, containing the LC3C N-terminal region (1–33) and the LC3B 29–120 residues (Figure 4B). The proportion of this chimera bound to CL-containing liposomes was larger than that of LC3B, reaching the level of LC3C (Figure 4C, D), thus implying that the N-terminal region of LC3C alone was enough to explain the higher degree of interaction with CL observed with LC3C.
Figure 4.
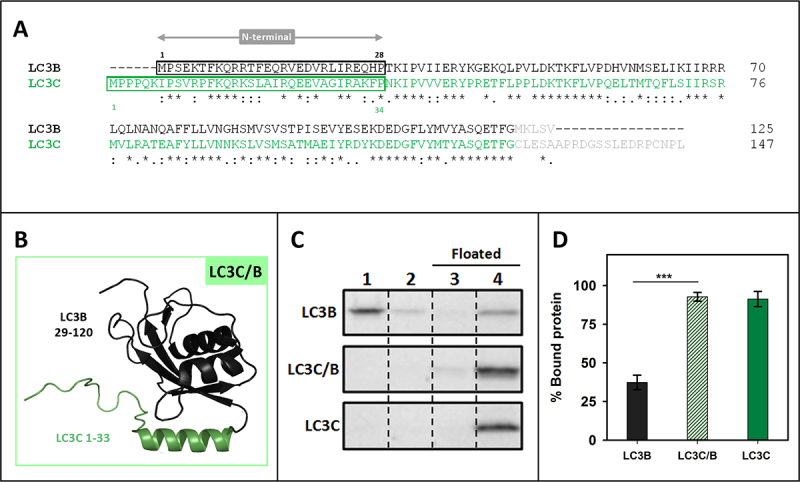
The higher capacity of LC3C to interact with CL resides in its N-terminal region. (A) Clustal W alignment of LC3B and LC3C amino acid sequences. The N-terminal region of both proteins is boxed. Residues processed by ATG4 and not present in our recombinant proteins are in light gray. (B) 3D outline of the LC3C/B chimera composed of the structures of the LC3C N-terminal region (1–33 amino acids) and 29–120 residues of LC3B. (C) Representative SDS-PAGE/Coomassie Brilliant Blue-stained gels of the fractions obtained from LC3B, LC3C/B or LC3C vesicle flotation assays performed with CL-containing liposomes (PC:DOPE:CL (33:33:33 mol ratio)). Bound protein was computed as the proportion retrieved in fractions 3 + 4 (See Figure 2A) (D) Binding of LC3B, LC3C/B, and LC3C to CL-containing liposomes quantified by gel densitometry. Data are means ± SD (n ≥ 7). ***p < 0.001.
The N-terminal regions of LC3A and LC3B are important for their differential interaction with CL
The reason why LC3A interacted with CL with a higher affinity than LC3B was also explored. These two proteins exhibited a 79/92% identity/similarity when their amino acid sequences were compared (Figure 5A) and the main differences were also found in the N-terminal region. Therefore, the implication of this region in LC3A interaction with CL was tested. In this case, a chimera composed of the LC3A N-terminal region (1–28) and the LC3B protein 29–120 residues was designed (Figure 5B). The chimera showed a higher interaction with cardiolipin than LC3B, even if it did not reach LC3A binding levels (Figure 5C, D). These results suggested a mode of interaction with CL involving other regions of the protein apart from its N terminus, similar to the mechanism proposed for LC3B, but not to the one observed with LC3C. The increased CL interaction with the LC3A/B chimera, as compared to that of LC3B, suggests that LC3A might contain some residues in its N-terminal region allowing a stronger interaction with that phospholipid.
Figure 5.
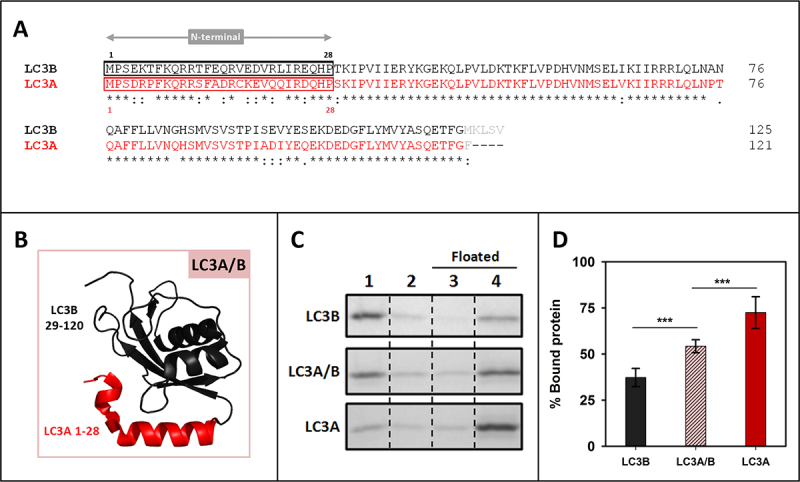
The N-terminal regions of LC3A and LC3B are important for their differential interaction with CL. (A) Clustal W alignment of LC3B and LC3A amino acid sequences. The N-terminal region of both proteins is boxed. Residues processed by ATG4 and not present in our recombinant proteins are in light gray (B) 3D outline of the LC3A/B chimera composed of the structures of the LC3A N-terminal region (1–28 amino acids) and 29–120 residues of LC3B. (C) Representative SDS-PAGE/Coomassie Brilliant Blue-stained gels of the fractions obtained from LC3B, LC3A/B or LC3A vesicle flotation assays performed with CL-containing liposomes (PC:DOPE:CL (33:33:33 mol ratio). Bound protein was computed as the proportion retrieved in 3 + 4 (See Figure 2A) (D) Binding of LC3B, LC3A/B, and LC3B to CL-containing liposomes quantified by gel densitometry. Data are means ± SD (n ≥ 8) ***p < 0.001.
A14 and K18 residues in LC3A N-terminal region are key for its higher interaction with CL
To further analyze the importance of specific residues in the N terminus of LC3A, a sequence alignment focusing on this region of LC3B and LC3A proteins (Figure 6A) was performed. This comparison revealed that residues shown by Chu et al. [16] to be involved in the interaction with CL were the same in both proteins (R10, R11), suggesting that the cause for the different behavior had to be found elsewhere. Taking into account that the object of the study was an interaction with a negatively charged phospholipid an analysis of the isoelectric points (pI) of the proteins (Fig. S2A) was performed. The effect of ionic strength was assessed using either the standard buffer (150 mM NaCl) or a buffer containing 300 mM NaCl. A decreased LC3 binding in the presence of high-ionic-strength buffer was observed in all cases (Fig. S2B, C), pointing to an implication of electrostatic interactions in LC3 proteins binding to CL-containing bilayers. Moreover, the fact that this decrease was more marked for LC3A could indicate the implication of charged amino acids in the differential interaction of LC3A and LC3B.
Figure 6.
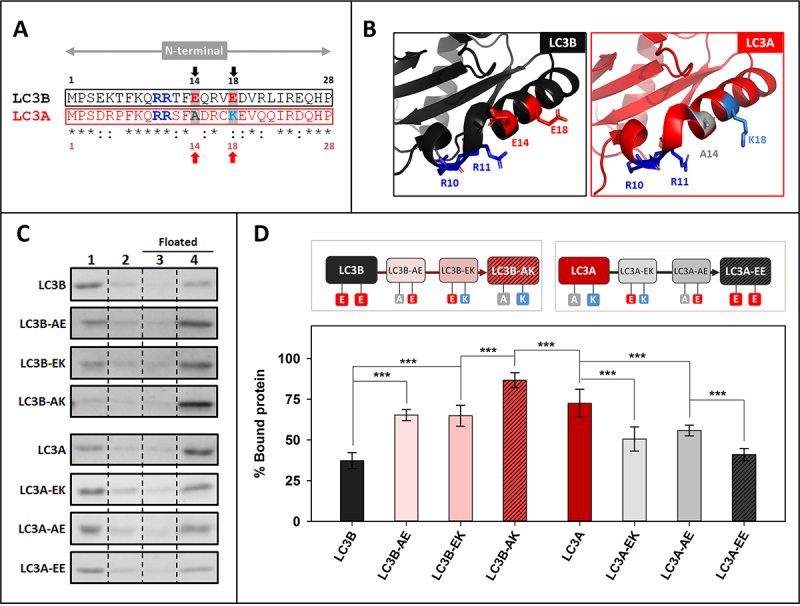
A14 and K18 residues in LC3A N-terminal region are key for its higher interaction with CL. (A) Comparative analysis of the N-terminal regions of LC3B and LC3A obtained using Clustal W. Amino acids previously proposed to be important in LC3B-CL interaction (R10, R11) and the ones chosen in this study to be mutated (positions 14 and 18) are written in bold and colored; red: negatively charged, blue: positively charged, gray: no charge (B) 3D structures of the N-terminal regions of LC3B and LC3A showing the amino acids chosen for this study and the residues already proposed to be involved in the interaction with CL. (C) Representative SDS-PAGE/Coomassie Brilliant Blue-stained gels of the fractions obtained from LC3B, LC3B-AE (LC3BE14A), LC3B-EK (LC3BE18K), LC3B-AK (LC3BE14A,E18K), LC3A, LC3A-EK (LC3AA14E), LC3A-AE (LC3AK18E), LC3A-EE (LC3AA14E,K18E) vesicle flotation assays performed with CL-containing liposomes (PC:DOPE:CL (33:33:33 mol ratio)). Bound protein was computed as the proportion retrieved in fractions 3 + 4 (see Figure 2A). (D) Binding percentage of LC3B, LC3B-AE (LC3BE14A), LC3B-EK (LC3BE18K), LC3B-AK (LC3BE14A,E18K), LC3A, LC3A-EK (LC3AA14E), LC3A-AE (LC3AK18E), LC3A-EE (LC3AA14E,K18E) to CL-containing liposomes quantified by gel densitometry. Data shown as mean ± SD (n ≥ 5) ANOVA statistical analysis, ***p < 0.001.
Two negatively charged amino acids were observed in LC3B (E14, E18) that were not present in LC3A, the latter protein containing instead one neutral and one positively charged residue (A14, K18) (Figure 6A, B). To test the involvement of residues 14 and 18 in the differential LC3A and LC3B binding to CL, a series of mutants was designed, expressed and purified. The objective was to “transform” LC3B into LC3A and vice versa LC3A into LC3B by mutating the mentioned residues, to elucidate whether the CL-binding affinity was influenced or not. As shown in Figure 6 C and D, after changing E14 of LC3B into the corresponding A14 of LC3A, i.e. obtaining the mutant LC3BE14A (LC3B-AE), the percentage of bound protein was higher than with native LC3B. Mutating residue 18, to obtain LC3BE18K (LC3B-EK), also increased binding, to the same extent than for LC3B-AE. The highest proportion of bound protein was found with LC3BE14A,E18K (LC3B-AK), above the value observed for LC3A (Figure 6C, D).
In the case of LC3A, both single-residue mutants (LC3AA14E [LC3A-EK], LC3AK18E [LC3A-AE]) exhibited a decreased interaction. Furthermore, the LC3AA14E,K18E (LC3A-EE) showed a degree of binding to CL-containing vesicles similar to that of LC3B (Figure 6C, D). Thus, while mutating the two residues of LC3B almost doubled its interaction, changing the two amino acids of LC3A significantly decreased its interaction with CL, demonstrating the importance of the nature of residues 14 and 18 in the higher CL binding activity of LC3A. In addition, when the equivalent of the LC3B E14 residue (A20) in the N-terminal region of LC3C was mutated, to obtain LC3CA20E (Fig. S3 A, B), LC3C binding was not affected (Fig. S3C, D). Thus, the very effective binding of LC3C to CL-containing vesicles should not be interpreted in the same terms described for LC3A and LC3B above, rather indicating that the whole N-terminal region of LC3C was important for its interaction with CL.
LC3A and LC3B puncta per cell and their colocalization with mitochondria increase with rotenone treatment
CL externalization to the OMM had been shown using treatments with non-lethal doses of rotenone [16]. However, the mechanism controlling CL externalization is not completely understood. CL is a phospholipid mainly found in the inner monolayer of IMM; therefore, three steps are required for it to be fully translocated to the external monolayer of the OMM. Translocation between the monolayers of the same membrane is believed to be catalyzed by PLSCR3 (phospholipid scramblase 3) [16,31]. The transfer between the IMM outer monolayer and the inner leaflet of OMM is thought to be carried out by the kinase NME4/NDPK-D/Nm23-H4 [32,33].
To assess whether the above in vitro results were indicative of the behavior of LC3 proteins in cells, the involvement of each LC3 member in CL-mediated mitophagy was tested (Figure 7). Experiments were performed using LC3A, LC3B, LC3C, and GABARAPL2 to test whether this treatment induced an increase in LC3/GABARAP- protein puncta inside the cell (as a signal of autophagy) and whether it increased the number of puncta that colocalized with mitochondria (mitophagy). The experimental conditions were as described in previous studies in which CL externalization had been shown [16]. SH-SY5Y neuroblastoma cells were transfected with GFP-tagged LC3 proteins and treated with 1 µM rotenone (6 h). GFP-GABARAPL2 was used as a control as this protein was unable to translocate to mitochondria upon rotenone treatment in U87MG cells [23].
Figure 7.
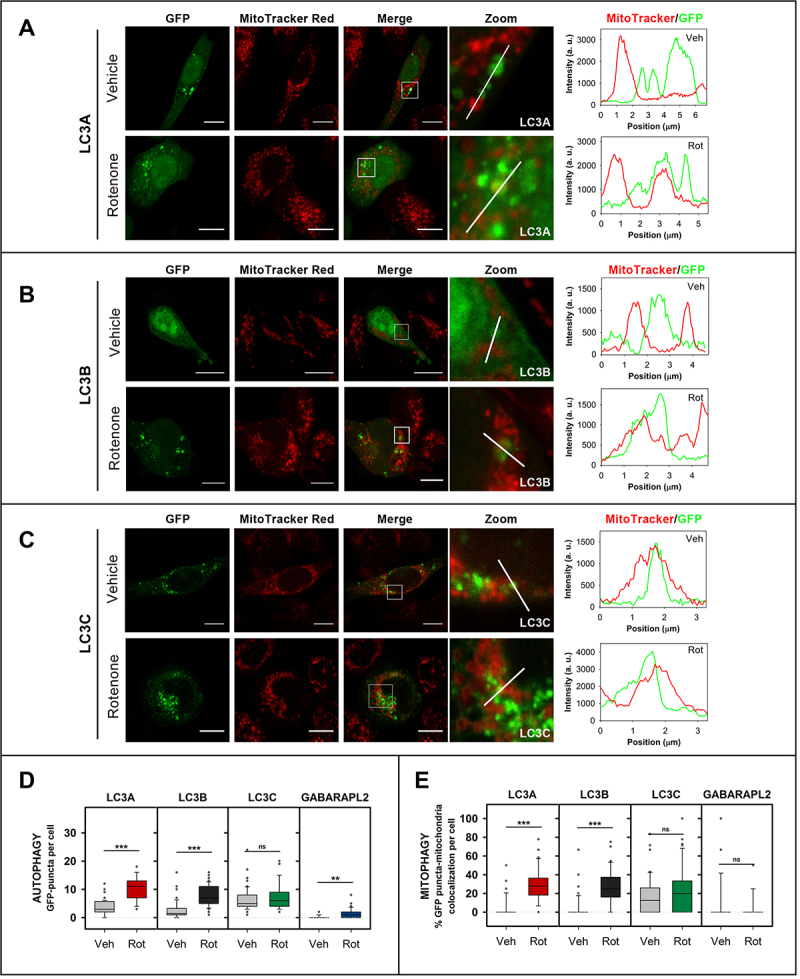
LC3A and LC3B puncta and their colocalization with mitochondria increase with rotenone treatment. SH-SY5Y cells were transfected with different members of the LC3/GABARAP family tagged with GFP. Mitochondria were labeled using MitoTracker Red, prior to treatment with 1 μM rotenone for 6 h. Vehicle (Veh) controls were treated with DMSO. Images were retrieved using a Nikon Eclipse C1 confocal microscope. Scale bar: 10 μm. At the right-hand side of each condition, MitoTracker (red) and GFP (green) line-profiles show examples of colocalization and non-colocalization events. (A) Representative images of GFP-LC3A SH-SY5Y transfected cells untreated (vehicle) or treated with rotenone. (B) Representative images of GFP-LC3B SH-SY5Y transfected cells untreated (vehicle) or treated with rotenone. (C) Representative images of GFP-LC3C SH-SY5Y transfected cells untreated (vehicle) or treated with rotenone. (Representative images of GFP-GABARAPL2 can be found in Fig. S4C). (D) Number of GFP-LC3A, GFP-LC3B, GFP-LC3C and GFP-GABARAPL2 puncta per cell, an indication of autophagy, in SH-SY5Y cells untreated (Veh) and treated with rotenone (Rot). (E) Percent GFP-LC3A, GFP-LC3B, GFP-LC3C and GFP-GABARAPL2 puncta that colocalize with mitochondria, a signal of mitophagy, in SH-SY5Y cells untreated (Veh) or treated with rotenone (Rot). To compute the percent colocalization, images were analyzed with JACop plugging of ImageJ. At least 30 images were analyzed per condition. ***p < 0.001, **p < 0.01, ns: non-significant.
The formation of puncta, as detected by conventional fluorescence microscopy, has been interpreted in the experiments below as an indicator of soluble LC3 (LC3-I) conversion to the autophagosome-associated form (LC3-II). To ascertain whether the apparent autophagy observed was indeed due to autophagy vesicles and not to protein aggregates, non-conjugatable mutants of the proteins were used. In these mutants the C-terminal Gly had been mutated into an Ala residue, resulting in a nonfunctional form of the proteins that could not be conjugated to PE, thus they could not be anchored to the phagophore or autophagosomal membrane. The mutant proteins consistently failed to give rise to puncta, indicating that the detected GFP signals did not correspond to nonspecific aggregates (Fig. S4A, B).
Six h-rotenone treatment induced an enhanced number of GFP-LC3A and GFP-LC3B puncta, and their colocalization with mitochondria was equally increased (Figure 7A, B, D, E). These results, using a CL-externalizing treatment, were consistent with the ability of LC3A and LC3B to interact with CL-containing membranes in vitro (Figure 2B). In agreement with previous results [23] and with our own in vitro studies (Figure 2B), rotenone did not increase GFP-GABARAPL2 colocalization with mitochondria (Figure 7 D, E and Fig. S4C). However, at variance with the other proteins, a high amount of GFP-LC3C puncta was observed under basal conditions and, despite the remarkable level of interaction displayed by LC3C with CL-containing membranes (Figure 2B), the amount of GFP-LC3C puncta did not increase after rotenone treatment (Figure 7C, D, E). These results suggest that LC3A and LC3B are able to recognize CL not only in model membranes but also in damaged mitochondria, while LC3A and LC3B, but not LC3C or GABARAPL2, are involved in cargo recognition during CL-mediated mitophagy.
In order to confirm the participation of LC3A in CL-mediated mitophagy we treated our cells with a different CL-externalization inductor [32], carbonyl cyanide m-chlorophenyl hydrazone (CCCP). We decided to assess the localization of LC3A after a 1 h treatment in order to study the early stages of the process. We observed an increase in the number of puncta and their colocalization with mitochondria after CCCP treatment (Fig. S5 A, B). These results support the conclusion that LC3A, the homolog recognizing CL in vitro with a higher specificity and efficiency, can also detect damaged mitochondria during rotenone- or CCCP-induced mitophagy.
The double mutation that hampers LC3A binding to CL in vitro also decreases its location to mitochondria in rotenone- and CCCP-induced mitophagy
To determine whether the residues identified as key for the differential in vitro interaction of LC3A and LC3B with CL were also important in cells, the ability of the double mutants to recognize damaged mitochondria was tested. SH-SY5Y cells were transfected with the GFP-tagged LC3 WT or mutant proteins and treated with different mitophagy inductors. LC3B-AK, which exerted a significant positive action in vitro, did not have a parallel effect in rotenone-treated cells (Fig. S6A, C), suggesting a peculiar mode of LC3B-CL interaction that would not involve residues 14 and 18 of the protein.
However, results obtained in cells after substitution of those two amino acids in LC3A pointed to an important role of those residues. The LC3A-EE mutant, which exhibited a much lower binding to CL-containing vesicles than its native counterpart (Figure 6D), also showed lower colocalization levels with mitochondria in cells treated with rotenone or CCCP, as compared to those transfected with GFP-LC3A (Figure 8A, B, C, and Fig. S6B, D). Moreover, in support of the LC3A N-terminal region implication, we used the LC3AR10,11A mutant in which the two Arg, shown by Chu et al. [16] to be important for CL recognition in LC3B, were mutated to Ala. The double mutation caused a lower binding to CL-containing vesicles (Fig. S5C) and lower colocalization levels with mitochondria after rotenone treatment (Fig. S5D, E), as compared with the native protein. These results indicate that mutations that reduce the binding of LC3A to CL in vitro, also decrease the ability of LC3A to recognize damaged mitochondria in cells, decreasing or delaying the involvement of this homolog in the mitophagic process.
Figure 8.
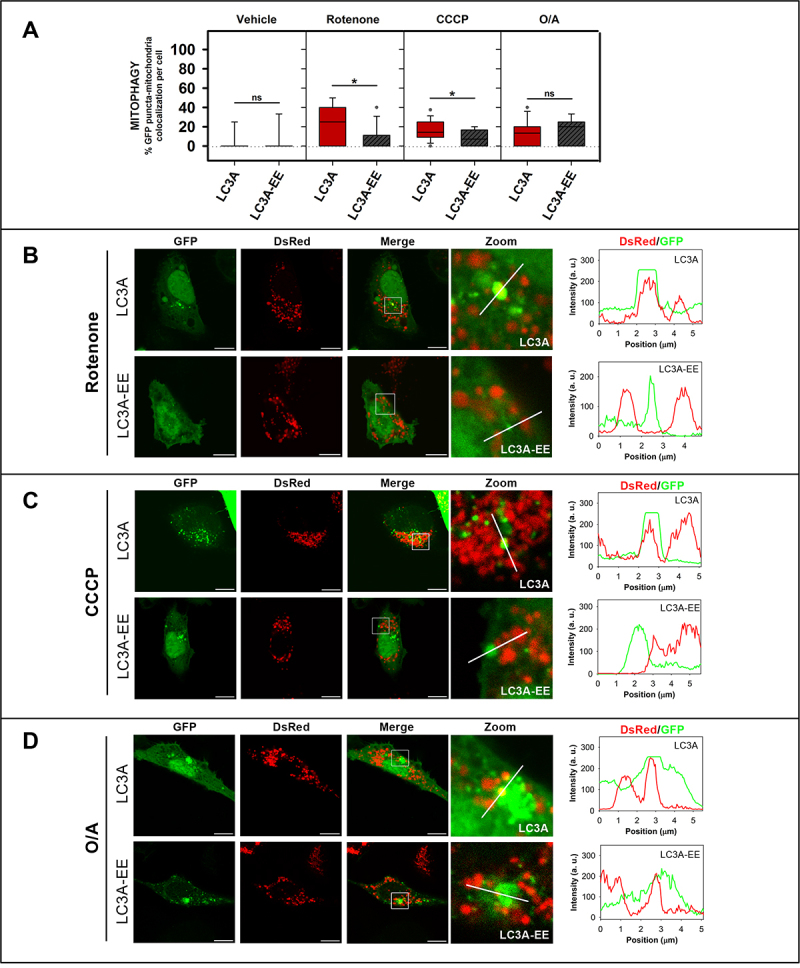
The LC3A-EE double mutation that hampers LC3A binding to CL in vitro also decreases its location to mitochondria in rotenone- and CCCP-induced mitophagy. SH-SY5Y cells were co-transfected with DsRed2-Mito7 (DsRed) and GFP-tagged WT or mutant LC3A. Vehicle (Veh) controls were treated with DMSO. (A) Percent GFP-LC3A or GFP-LC3A-EE puncta that colocalize with mitochondria, a signal of mitophagy, in SH-SY5Y cells untreated (vehicle), or treated with rotenone (1 μM, 6 h), or CCCP (20 μM, 1 h) or O/A (10/4 μM, 6 h). To compute the percent colocalization, images were analyzed with the JACop plugging of ImageJ. At least 30 images were analyzed per condition. *P < 0.05, ns: non-significant. (B) Representative images of GFP-LC3A and GFP-LC3A-EE SH-SY5Y transfected cells treated with 1 μM rotenone for 6 h. (C) Representative images of GFP-LC3A and GFP-LC3A-EE SH-SY5Y transfected cells treated with 20 μM CCCP for 1 h. (D) Representative images of GFP-LC3A and GFP-LC3A-EE SH-SY5Y transfected cells treated with O/A (10/4 μM for 6 h). Images were acquired using a Nikon Eclipse C1 confocal microscope. Scale bar: 10 μm. At the right-hand side of each condition, DsRed (red) and GFP (green) line profiles show examples of co-localization and non-colocalization events.
The above results were complemented with the use of an additional mitophagy inducer, namely oligomycin A + antimycin A (O/A), shown by Lazarou and coworkers [12] to induce colocalization of all LC3/GABARAP-family members with mitochondria. As expected, after O/A treatment LC3A gave rise to an increased number of puncta and was found to colocalize with mitochondria (Figure 8A, D). However, at variance with rotenone or CCCP treatments, neither LC3A-EE (Figure 8A, D) nor LC3AR10,11A mutants (Fig. S5D, E) caused a decreased colocalization with mitochondria. These results suggest that in O/A-induced mitophagy, the N terminus is not as important as in rotenone or CCCP treatments.
Endogenous LC3A and LC3B are involved in CCCP-induced mitophagy
Several studies have been carried out comparing the role(s) of LC3 proteins in mitophagy [12,34–36], but most of them use transfected proteins, which might have an expression level that is not comparable to the endogenous protein levels, overestimating their implication in this process. To determine if SH-SY5Y cells were suitable for the study of endogenous LC3A effects, the mRNA and protein expression levels of LC3-subfamily members were quantified (Fig. S7). Substantial levels of both mRNA and LC3A protein were detected, indicating that these cells were an adequate cellular model for the present study. Therefore, in order to confirm the results obtained with the overexpressed protein a direct evaluation of endogenous LC3A activation upon CCCP treatment was performed.
CCCP (20 µM) treatment for 4 h or 6 h caused a decrease in the inner mitochondrial membrane marker COX4I in SH-SY5Y cells (Figure 9A) indicating completion of the mitophagy flux. Moreover, an increase in LC3A-II and LC3B-II was already observed within the first hour of treatment (Figure 9B). This activation also occurred at lower CCCP concentrations, such as 5 µM or 10 µM (Figure 9C). Consistent with these results, mitochondrial fractionation experiments clearly show that in cells treated with CCCP, LC3A and LC3B were found to a larger extent in the mitochondrial fraction than in the cytosol (Figure 9D). In order to address the importance of the involvement of these proteins in this type of mitophagy each homolog was independently knocked down. For this purpose, specific siRNAs for each homolog were used [37,38]. Knocking down LC3A or LC3B had no effect on mitochondrial marker reduction, but the double silencing of both homologs was able to reduce mitophagy flux (Figure 9E). These data support the results obtained by confocal microscopy with GFP-tagged proteins, demonstrating the involvement of LC3A and LC3B in cardiolipin-mediated mitophagy induced by CCCP.
Figure 9.
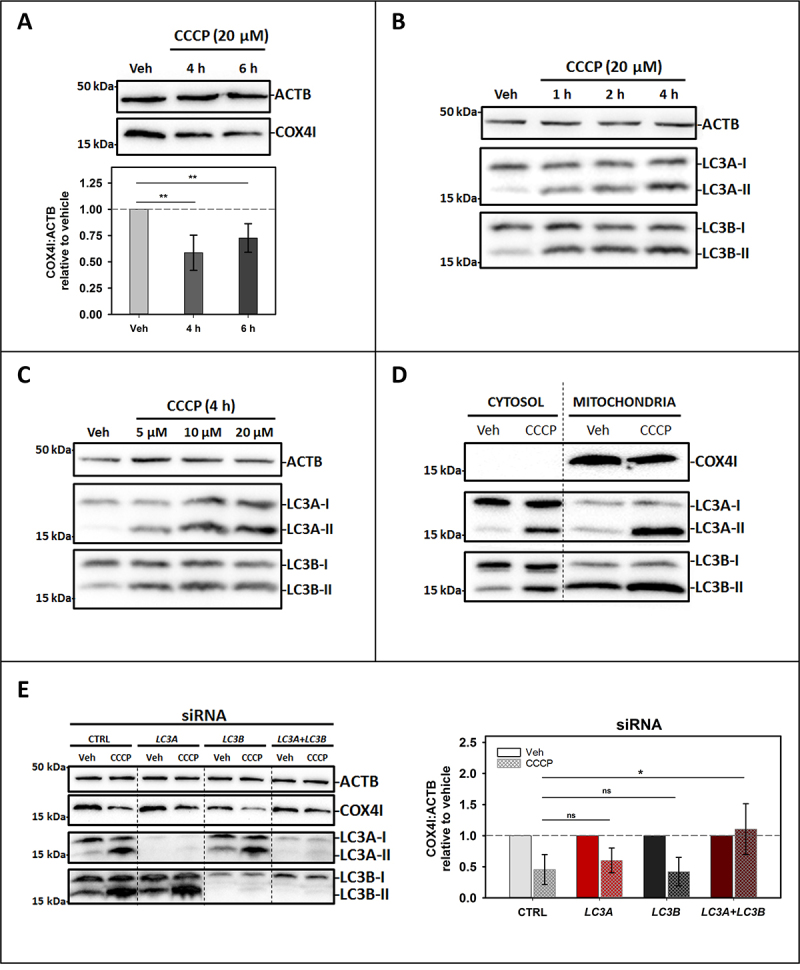
Endogenous LC3A and LC3B are involved in CCCP-induced mitophagy in SH-SY5Y cells. SH-SY5Y cells were used to study the activation of endogenous LC3A and LC3B by western blot after CCCP treatment. Vehicle (Veh) controls were treated with DMSO. (A) Top: Representative western blot of the degradation of the mitochondrial IMM marker COX4I in cells treated with 20 μM CCCP for 4 h or 6 h. Bottom: COX4I:ACTB ratio in treated cells relative to vehicle quantified by gel densitometry. Data shown are means ± SD **P < 0.01. (B) Activation of LC3A and LC3B, shown as an increase of the LC3A-II or LC3B-II corresponding band intensity, in cells treated with 20 μM CCCP for different times (1, 2 and 4 h). (C) Activation of LC3A and LC3B, shown as an increase of the LC3A-II or LC3B-II corresponding band intensity, in cells treated for 4 h with different concentrations of CCCP (5, 10 or 20 μM). (D) Detection of LC3A and LC3B in the mitochondrial fraction in cells treated with 20 μM CCCP for 4 h, COX4I is used as a marker of efficient mitochondrial fractionation. (E) Left: Representative western blot of the degradation of COX4I and the activation of LC3A and LC3B in cells treated with 10 μM CCCP for 4 h where LC3A and/or LC3B had been silenced, compared to control siRNA-transfected cells. Right: Comparison of COX4I:ACTB ratio relative to vehicle in siRNA-transfected cells treated with CCCP, quantified by gel densitometry. Data shown are means ± SD *p < 0.05; ns: non-significant.
LC3A but not LC3B is able to bind oxidized CL
It has been proposed that, when CL participates in apoptosis, it is previously oxidized due to the peroxidase activity of cytochrome C [39], while the CL fraction that is recognized by the autophagy machinery is not [16]. Taking into account that the interplay between autophagy and apoptosis implies a high level of complexity [40], understanding how the role of CL is affected by its oxidation state could help in understanding how these two important processes are regulated.
In order to assess the ability of LC3A and LC3B to recognize oxidized CL, LUVs were treated with CuCl2 for 5 h, and the increase in absorbance at 245 nm (A245) was measured to quantify lipid oxidation (Figure 10A). The highly unsaturated CL is the main lipid oxidized under those conditions. Then, protein bound to either control or oxidized vesicles was measured. The results obtained with LC3B and LC3A in these experiments showed that binding of LC3B (Figure 10C), but not of LC3A (Figure 10B), decreased with CL oxidation. At variance with the results observed with wild-type LC3A, the ability of LC3A mutants to bind CL decreased with lipid oxidation (Figure 10D), showing the importance of residues 14 and 18 for the recognition of oxidized CL. Moreover, LC3B double mutant enhanced the originally low capacity of wild-type LC3B to recognize oxidized CL (Figure 10E). Therefore, residues 14 and 18, identified as key for the differential interaction of CL with LC3A and LC3B, were shown to be also important for the LC3A-mediated recognition of oxidized CL. These results suggested that LC3A could bind oxidized CL in the OMM, perhaps preventing its recognition by the apoptotic machinery.
Figure 10.
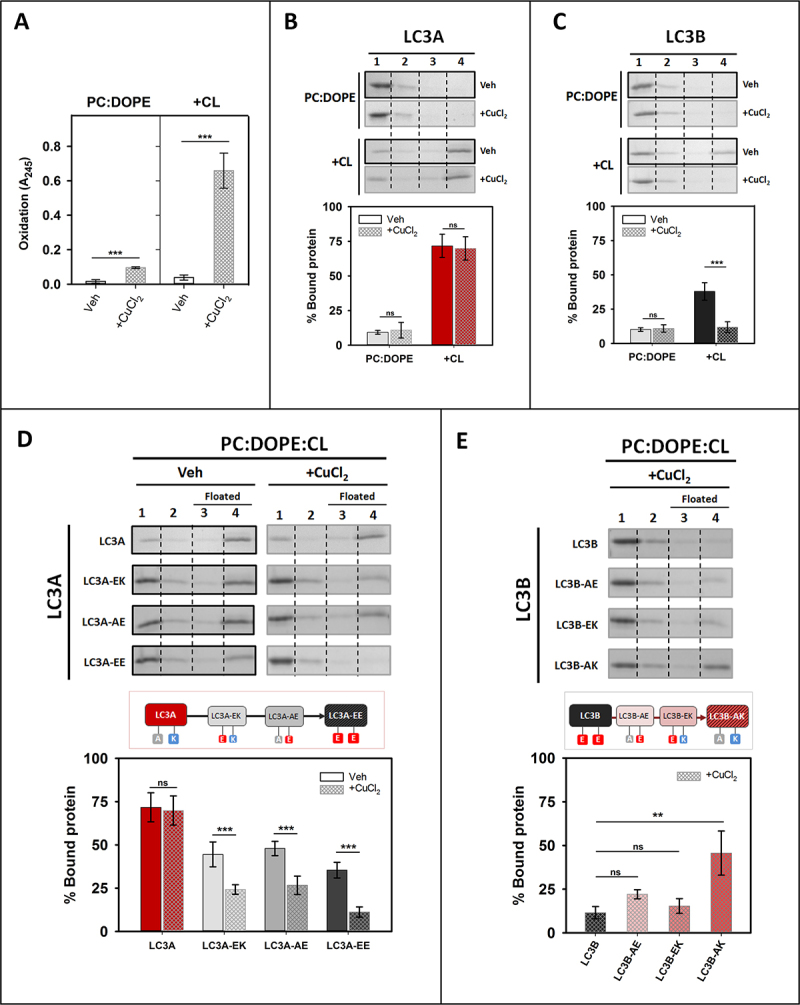
LC3A, but not LC3B, binds oxidized CL. Interaction of LC3A or LC3B proteins with oxidized membranes measured by a vesicle flotation assay. (A) Lipid oxidation was assessed measuring absorbance at 245 nm (A245) of the PC:DOPE (50:50) or PC:DOPE:CL (33:33:33) liposomes (+CL), treated without (Veh) or with CuCl2 (+CuCl2). Data shown as means ± SD (n ≥ 3). ***p < 0.001. (B and C) Top: Representative SDS-PAGE/Coomassie Brilliant Blue-stained gels of the fractions obtained from LC3A (B) or LC3B (C) vesicle flotation assays performed with liposomes composed of PC:DOPE (50:50) or PC:DOPE:CL (33:33:33) which had been previously treated without (Veh) or with CuCl2. Bound protein is computed as the proportion retrieved in fractions 3 + 4 (See Figure 2A). Bottom: Binding of LC3A (B) or LC3B (C) to non-oxidized and oxidized liposomes, quantified by gel densitometry. Data shown are means ± SD (n ≥ 3). ***p < 0.001; ns: non-significant. (D) Top: Representative SDS-PAGE/Coomassie Brilliant Blue-stained gels of the fractions obtained from LC3A, LC3A-EK (LC3AA14E), LC3A-AE (LC3AK18E)and LC3A-EE (LC3AA14E,K18E) vesicle flotation assays performed with liposomes composed of PC:DOPE:CL (33:33:33) that had been previously treated without (Veh) or with CuCl2. Bottom: Binding of LC3A and LC3A mutants to non-oxidized and oxidized liposomes, quantified by gel densitometry. Data shown are means ± SD (n ≥ 3). ***p < 0.001; ns: non-significant. (E) Top: Representative SDS-PAGE/Coomassie Brilliant Blue-stained gels of the fractions obtained from LC3B, LC3B-AE (LC3BE14A), LC3B-EK (LC3BE18K)and LC3B-AK (LC3BE14A,E18K) vesicle flotation assays performed with liposomes composed of PC:DOPE:CL (33:33:33) which had been previously treated with CuCl2. Bottom: Binding of LC3B and LC3B mutants to oxidized liposomes, quantified by gel densitometry. Data shown are means ± SD (n ≥ 3). **p < 0.01; ns: non-significant.
Discussion
Deciphering the different modes of damaged mitochondria signaling, and the role that each of the LC3/GABARAP-family members plays in their recognition is extremely important for understanding mitophagy. Many types of receptors participate in this process, and different studies reinforce the idea that the LC3/GABARAP-family members could play specific roles depending on the particular mechanism involved [14]. There are examples of proteins that act as mitophagy receptors by interacting preferentially with one or several of the LC3/GABARAP-family members, for example, BNIP3L recruits GABARAP [41] and GABARAPL1 [42], while FUNDC1, BNIP3 and BCL2L13 are found bound to LC3B [43–45], and FKBP8 is recognized by LC3A [35]. In the case of the phospholipid CL acting as a mitophagy receptor, LC3B was shown to be a mediator [16], and our previous work comparing both Atg8 subfamilies suggested that neither GABARAP nor GABARAPL2 were involved [23]. However, a comparative study of the different LC3-subfamily members function(s) does not appear to be available.
The specific interaction of LC3A with CL allows its participation in the mechanism of CL-mediated mitophagy
The interaction of LC3A with membranes was specific for the presence of CL (Figure 3). LC3A did not show any measurable interaction with other lipids containing the same negative charge of CL, such as PtdIns4P (Figure 3B), therefore, this interaction did not seem to be exclusively dependent on the lipid charge. The amino acid sequence of LC3A and LC3B is very similar, and even some residues, demonstrated to be key for the interaction with CL, R10 and R11 [16], located in the alpha-helix 1, are identical in both proteins (Figure 1B, 6A, B). However, in the above studies with model membranes a clear difference in CL binding was observed between LC3A and LC3B, the former being more active (Figure 2B). Experiments involving the LC3A/B chimera (Figure 5) and high-salt concentrations (Fig. S2), suggested that the higher binding detected with LC3A could be in part due to the existence of positively charged residues in its N-terminal region, that are not present in LC3B. Comparative binding assays using LC3A and LC3B single and double mutants showed that the key amino acids were located in positions 14 and 18 of helix 2 (Figure 6). Therefore, the CL binding site of LC3A appears to be composed of two alpha-helices containing positively charged residues, in agreement with previously described binding sites for other soluble proteins that can also bind CL in membranes [46]. However, the fact that LC3A/B chimera did not reach the same binding levels as LC3A (Figure 5) suggests the implication of other protein regions apart from the N terminus, as previously proposed for LC3B, in the interaction of LC3A with CL.
To further dissect the differences among these subfamily members, the participation of each LC3-subfamily member in CL-mediated mitophagy was studied by assessing the colocalization of transfected GFP-tagged proteins with mitochondria in SH-SY5Y cells. Even if the effect of the GFP tag in the LC3/GABARAP-protein function has been criticized [47], we decided to use this method due to the similar results obtained using anti-LC3 antibodies [20,23] and GFP-tagged LC3B [16] to study CL-mediated mitophagy in cells. This supports the notion that the GFP-tag does not disturb the recognition of externalized CL in damaged mitochondria. In addition, the results obtained with the non-conjugatable mutants of each homolog (Fig. S4A, B), in which the number of puncta was residual, supported the idea that the GFP puncta were, in fact, autophagic vesicles, and not aggregates. The above observations indicate that the results obtained are reliable.
The experiments performed with cells showed that, apart from LC3B, LC3A was also able to participate in rotenone- or CCCP-induced mitophagy (Figure 7 and Fig. S5A, B), implying that LC3A could recognize externalized CL in damaged mitochondria in cells. The participation of LC3A in mitophagy has been described previously, LC3A being able to specifically recognize the mitophagy receptor FKBP8 located in the OMM of damaged mitochondria [35]. LC3A recognition of CL and of FKBP8 are not by necessity mutually excluding events, in fact these two mechanisms might act together in cooperation. It is e.g. possible that CL externalization brings LC3A closer to mitochondria to facilitate binding to FKBP8.
Ala 14 and Lys 18 residues, identified in vitro as important for LC3A-CL interaction (Figure 6D), were also shown to be key for CL recognition during rotenone- and CCCP- induced mitophagy in cells, as demonstrated by the behavior of LC3A-EE mutant (Figure 8A). Similar results obtained with the LC3AR10,11A mutant support the notion that the two N-terminal alpha helices of LC3A are also important in CL recognition in cells (Fig. S5D). Furthermore, the fact that LC3A mutants are able to participate in O/A-induced mitophagy to the same extent as LC3A WT (Figure 8A and Fig. S5D) indicates that those mutants are functional, although the importance of CL-recognition is decreased in the case of the O/A treatment. This could be explained taking into account that O/A can activate numerous mitophagy pathways and receptors [12]. It could be possible that, at variance with rotenone and CCCP, in O/A treatment the LDS (LIR docking site) of LC3A becomes more relevant, while the importance of the N terminus residues is diminished.
The differences found in vitro between LC3A and LC3B binding to CL-containing vesicles (Figure 2B) were not observed when colocalization with mitochondria was considered (Figure 7D, E). Moreover, while the exchange of residues 14 and 18 of LC3B for the corresponding ones of LC3A in the LC3B-AK mutant increased significantly its interaction with CL-containing membranes (Figure 6D), this change did not affect its function in mitophagy: the double mutant did not show a higher level of colocalization with mitochondria when compared to native LC3B (Fig. S6). These results could suggest that the mitophagy level observed with LC3B represents a colocalization threshold for the mitophagy treatment. If that were the case, even if mitochondrial recognition by LC3A or LC3B-AK were better, they would not lead to an observable higher colocalization. This could also indicate that, at variance with the equivalent residues in LC3A, the negatively charged residues present in the LC3B N-terminal region (E14, E18) do not play a role in the interaction with externalized CL in cells (Figure 6B and Fig. S6). In turn, this would imply that the molecular mechanisms through which LC3A and LC3B interact with CL in the cell might be different, or else that their interaction might be regulated differently.
Analysis of protein levels in SH-SY5Y cells revealed marked differences in the amounts of each form of LC3 (Fig. S7C), without any clear relationship between the mRNA expression levels and the amount of protein detected for each LC3-subfamily member (Fig. S7A, C). Explaining these observations is beyond the scope of the present work, nevertheless they may be worth mentioning. The possibility that modified forms of LC3 proteins, by e.g. acetylation [48] or phosphorylation [49], could go undetected by the antibodies used in this study, leading to underestimation of the amounts of protein measured, cannot be discarded. The study of endogenous LC3 proteins in SH-SY5Y cells (Figure 9) supports the conclusions obtained with the transfected GFP-tagged LC3 proteins, demonstrating the participation of LC3A and LC3B in CCCP-induced mitophagy. The results obtained after silencing LC3A and LC3B show the important role of these proteins in CCCP-mediated mitophagy. Moreover, the need to silence both proteins to detect a reduction in mitophagy suggests some redundancy of function between the two homologs. Perhaps the main conclusion of these experiments is that although the LC3B homolog has been most extensively studied (and most frequently cited) in this context, LC3A does play a role in mitophagy and, in certain cell types such as neuroblastoma cell lines, it may be the most abundant form of LC3.
LC3C does not participate in cargo recognition during CL-mediated mitophagy
The interaction of LC3C with membranes was not specific for the presence of CL, as this protein could also interact with PA (Figure 3B). Moreover, despite the higher interaction displayed with CL-containing model membranes when compared with LC3A or LC3B (Figure 2B), LC3C did not appear to play a role in rotenone-mediated mitophagy. These results led to the conclusion that LC3C was less effective than LC3A or LC3B in recognizing damaged mitochondria (Figure 7). This LC3C behavior is not surprising, since it has been shown that LC3C has different functions and targets as compared to the other subfamily members [4], such as its specific function in antibacterial autophagy [37].
While LC3A and LC3B show a high similarity in their sequence (92%), LC3C is the most differentiated homolog (71–72% similarity). Unlike in other LC3/GABARAP-family members, the N-terminal region of LC3C does not form a stable α1-helix but a “sticky arm” (Figure 1A) consisting of a polyproline motif [50]. It has also been shown that the interactome of LC3C differs clearly from that of other members of the LC3 subfamily [51]. Another important singularity is that although LC3A and LC3B are ubiquitously expressed in almost all tissues [52] LC3C expression is extremely low and/or tissue-specific (lungs and placenta). In the current study, although mRNA expression was measurable in SH-SY5Y cells, the amounts of cellular protein were not sufficient to be detected by the antibodies, thus studying endogenous LC3C in this cell type was not feasible (Fig. S7). The results in this paper show CL-mediated mitophagy as yet another mechanism in which LC3C behaves differently from the other subfamily members.
The assays using SH-SY5Y cells showed that in the basal state GFP-LC3C was already forming puncta (Figure 7C, D) and some of these puncta colocalized with mitochondria (Figure 7D, E). Moreover, CL externalization by rotenone did not have any further effect on puncta formation/colocalization (Figure 7D, E). These unchanged levels of autophagy and mitophagy after rotenone treatment suggest that CL externalization had little or no effect on the participation of LC3C in autophagy or on its mitochondrial localization. These results are in accordance with a previous study that used Saos-2 cells and GFP-LC3C [52]. In the basal state, they also observed more puncta per cell than with LC3A or LC3B, but these values did not increase after starvation-induced autophagy.
The observed GFP-LC3C colocalization with mitochondria could be explained by the recently discovered involvement of LC3C in the maintenance of basal mitochondrial network homeostasis [36]. In their study, Le Guerroue et al. [36] described an LC3C-driven mitophagy mechanism that mediates autophagosome degradation of mitochondrial parts in a piecemeal way. Moreover, the affinity that LC3C shows toward PA-containing membranes could also allow its interaction with healthy mitochondria, which contain PA in their OMM [53]. In addition, previous results indicated that LC3C does not have a relevant function in PRKN-mediated mitophagy [47]. Those studies, together with the results in this paper suggest that the main function of LC3C in mitophagy could be related to the maintenance of a healthy mitochondrial network, by regulating degradation of the damaged mitochondrial parts [36].
LC3A could prevent excessive apoptosis activation
It has been shown that CL could interact with various proteins involved in the regulation of crucial cellular and mitochondrial processes [46]. Besides LC3A and LC3B interaction with CL studied here, this phospholipid can also interact with BECN1 [54], a key component in the autophagic core machinery, with DNM1L/Drp1 (dynamin 1 like), one of the proteins in charge of mitochondrial fission [28], or with OPA1, involved in IMM fusion [55], among others. Interestingly, CL externalization also occurs during apoptosis [39]. Externalized CL can interact with many proteins involved in this process, such as BAK1, BAX, or CASP8 [27,56,57], and it also participates in other programmed cellular death pathways such as NLRP3 inflammasome activation [58].
All these studies show that both CL and its location are key for the correct functioning of mitochondria. Among its different roles, it is surprising that CL externalization could promote both a generally “pro-survival” pathway, such as autophagy, and a “pro-death” pathway, such as apoptosis [59]. However, it has been proposed that CL participating in apoptosis is previously oxidized due to the peroxidase activity of cytochrome C [39], while CL recognized by the autophagy machinery is not [16]. Interestingly, the results obtained with LC3B and LC3A in the experiments with oxidized CL show that while LC3B binding decreases when CL is oxidized (Figure 10C), CL oxidation does not affect LC3A binding (Figure 10B). These results together with the ones of LC3A and LC3B single and double mutants (Figure 10D, E) highlight the importance of the LC3A 14 and 18 residues in oxidized CL-binding. Thus, LC3A would recognize oxidized CL in the OMM, shielding it from the apoptotic machinery, and preventing excessive activation of apoptosis. According to our in vitro data, LC3B would not be able to achieve the latter function, as its ability to interact with CL decreases with oxidation.
Concluding remarks
The importance of CL externalization as a signal to degrade damaged mitochondria via autophagy has been demonstrated in vitro and in vivo [16,18,23,32]. In this work, LC3A has been identified as an additional LC3-subfamily member involved in mitophagy, and key residues for its interaction with CL have been singled out. This contribution could facilitate the design of precise modulators for this mitophagy mechanism. However, the possibility of this process being cell-, tissue- or organ-specific, or even the possibility of a crosstalk between different types of autophagy cannot be dismissed. Further investigations would be required to improve our understanding of the mechanisms triggering mitophagy, which could in turn be involved in the appearance of important neurodegenerative diseases such as Parkinson or Alzheimer.
Materials and methods
Materials
L-α-phosphatidylcholine from hen egg yolk (PC; 840,051), bovine heart cardiolipin (CL; 90% tetralinoleoylcardiolipin, 5% tetraoleoylcardiolipin, 5% unknown; 840,012), brain phosphatidylinositol-4-phosphate (PtdIns4P; 840,045), egg phosphatidic acid (PA; 840,101), hen egg L-α-phosphatidylglycerol (PG; 841,138), 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine-N-lissamine rhodamine B sulfonyl (Rho-PE; 810,150), and 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine (DOPE; 850,725) were purchased from Avanti Polar Lipids, Inc. E. coli BL21(λDE3) cells (C600003), RPMI 1640 medium (61,870–10), Opti-MEM medium (11,058–011), Lipofectamine™ 2000 (11,668,027), Lipofectamine™ 3000 (L3000008), MitoTracker™ Red CMXRos (M7512), fetal bovine serum (10,270,106) were purchased from Thermo Fisher Scientific. Anti-LC3A (ab26628), anti-LC3B (ab51520) anti-LC3C (ab150367) anti-COX4I/COX-IV (ab14744) antibodies and rotenone (ab143145) were purchased from Abcam. Anti-ACTB/actin (sc-8432), HRP-conjugated anti-mouse (sc-516,102), HRP-conjugated anti-rabbit (sc-2357) antibodies, carbonyl cyanide m-chlorophenylhydrazone (CCCP; sc-202,984) and oligomycin A (sc-201,551) were purchased from Santa Cruz Biotechnology. DMEM medium (D5796), isopropyl-β-D-thiogalactoside (IPTG; I6758), DL-dithiothreitol (DTT; D0632), paraformaldehyde (PFA; 158,127), non-essential amino acids (M7145), 1% penicillin-streptomycin (P4333), RIPA lysis buffer (R0278), antimycin A (A8674), Immobilon®Forte Western HRP Substrate (WBLUF0100), Thrombin Protease (27–0846-01), cOmplete™, EDTA-free Protease Inhibitor Cocktail (5,056,489,001) and sucrose (84,097) were purchased from Merck.
DNA constructs and site-directed mutagenesis
The pGEX4T-1 plasmids for expression of several of the various Atg8 orthologs tagged with GST (glutathione S-transferase) (human LC3A, human LC3B, human LC3C and human GABARAPL2) were kindly provided by Dr. Ivanna Novak (School of Medicine, University of Split, Croatia). Note that each of these Atg8 orthologs was a truncated form lacking the C-terminal Gly. The Gly-exposed forms, such that no ATG4-mediated pre-processing was necessary, that were used in this work, were constructed using a QuikChange site-directed mutagenesis kit (Agilent, 200,514). The green fluorescent protein (GFP) plasmids (pHAGE-N-eGFP) were kindly provided by Dr. Christian Behrends (Munich Cluster for Systems Neurology, Munich, Germany.) Each of these GFP-tagged Atg8 orthologs contains the full-length sequence of the proteins. For the preparation of the mutant proteins LC3AA14E (LC3A-EK), LC3AK18E (LC3A-AE), LC3AA14E,K18E (LC3A-EE), LC3BE14A (LC3B-AE), LC3BE18K (LC3B-EK), LC3BE14A,E18K (LC3B-AK), LC3CA20E, GFP-LC3AG120A, GFP-LC3BG120A, GFP-LC3CG126A and GFP-GABARAPL2G116C, an inverse PCR-based site-directed mutagenesis using the KOD-Plus mutagenesis kit (Toyobo, SMK-101) was employed, following the instructions of the manufacturer. The sequences in all mutant constructs were confirmed by DNA sequencing analysis (Secugen S.L). The primers used for making the different mutants are listed in Table S1. The chimeras (LC3A/B, LC3C/B), the GFP-tagged double mutants (GFP-LC3A-EE, GFP-LC3B-AK) and the LC3AR10,11A mutant (GST- and GFP-tagged versions) were obtained by subcloning (synthesized by GenScript). Circular dichroism analysis of native and mutant proteins was performed. The results indicated that the mutations did not appreciably affect the protein secondary structure (data not shown).
Recombinant protein expression and purification
All proteins were purified from soluble fractions of bacterial extracts obtained in the absence of detergents, and they were >90% pure as evaluated by Coomassie Brilliant Blue-stained SDS-PAGE. E. coli BL21(λDE3) cells were transformed with appropriate plasmids, they were grown to OD600 = 0.8 and induced with 0.5 mM IPTG for 16 h at 20°C. Following centrifugation at 4,500 x g for 15 min, the pellet was resuspended and sonicated in Breaking Buffer (phosphate-buffered saline [PBS; 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4, pH 7] with protease inhibitors mixture and 1 mM DTT). After removal of cellular debris by centrifugation at 30,000 x g for 30 min at 4°C, the sample supernatant fraction was incubated with 1 ml Glutathione Sepharose 4B (GE Healthcare, 17–0756-01) for 3 h at 4°C to bind GST-tagged proteins. Bound proteins were cleaved with Thrombin Protease overnight at room temperature in Thrombin Buffer (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.3 with freshly added 1 mM DTT). After cleavage, they were eluted in Assay Buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA with freshly added 1 mM DTT), then concentrated to 500 µl using Amicon Ultra-4 (4 mL, 3 kDa cutoff; Millipore, UFC800324), and loaded onto a Superdex 75 10/300 GL size exclusion column (GE Healthcare, GE17-5174-01) equilibrated in Assay Buffer supplemented with freshly added 1 mM DTT. Proteins were distributed in aliquots, flash-frozen and stored in 20% glycerol at −80°C until further use.
Liposome preparation
The appropriate lipids were mixed in organic solution and the solvent was evaporated to dryness under a N2 stream. Then the sample was kept under vacuum for 1 h to remove solvent traces. The lipids were swollen in Assay Buffer in order to obtain multilamellar vesicles (MLVs). Large unilamellar vesicles (LUV) were produced from MLV according to the extrusion method described by Mayer et al. (1986) [60]. They were subjected to 10 freeze/thaw cycles, and then extruded using a LIPEX Liposome Extrusion System (Evonik Health Care, Essen, Germany) with a 0.1-μm pore size Nuclepore filters (Whatman, 110,605). Small unilamellar vesicles (SUV) were obtained by sonicating MLV with a probe tip sonicator (MSE Soniprep 150, MSE, UK) for 20 min (10 sec on, 10 sec off) on ice. Vesicle size was checked by quasi-elastic light scattering using a Malvern Zeta-Sizer 4 spectrometer (Malvern Instruments, Malvern, UK). LUV had an average diameter of ≈100 nm and SUV average diameter was ≈50 nm. Phospholipid concentration was determined by phosphate analysis [61].
Vesicle flotation assay
Protein interaction with membranes was assessed using flotation in sucrose gradients (see Figure 2A). Liposomes (3 mM), containing 0.05 mol% Rho-PE for detection, were incubated with 10 μM of the different purified proteins, for 1 h at 37°C in Assay Buffer under continuous stirring (1100 rpm). The protein/lipid mix was adjusted to 1.4 M sucrose concentration in 300 μl and transferred to a centrifuge tube. This first (bottom) layer was overlaid with successive solutions containing 0.8 M (400 μl) and 0.5 M (300 μl) sucrose. The three-layer gradients were centrifuged in a TLA-120.2 rotor (Beckman Coulter, Brea, CA, US) at 355,040 x g for 50 min at 4°C. After centrifugation, four 250-µl fractions were collected, starting from the bottom. Proteins were detected in SDS-PAGE gels by using Coomassie Brilliant Blue staining, and the presence of liposomes was monitored by measuring rhodamine (Rho-PE) fluorescence in a microplate reader Synergy HT (Bio-Tek, Winooski, VT, USA). Densitometry of the protein bands was performed using ImageJ software, and the percent liposome-bound protein was calculated from the band intensities measured in the third + fourth fractions (floating vesicle fractions), relative to the total sum of intensities measured in all fractions.
Cell culture
SH-SY5Y neuroblastoma cells (ATCC®, CRL-2266™) obtained from Innoprot S.L. (Derio, Spain) were cultured in RPMI 1640 medium containing 10% fetal bovine serum, non-essential amino acids and 1% penicillin-streptomycin at 37°C in a humidified atmosphere with 5% CO2. Cells were transfected with Lipofectamine™ 3000 following the manufacturer’s instructions
Fluorescence confocal microscopy
SH-SY5Y cells were grown on µ-Slide 8-well chambered coverslips (Ibidi, 80,826). 24 h post-transfection, cells were stained with MitoTracker Red CMXRos (100 nM 1 h 37°C) or co-transfected with DsRed2-Mito-7 plasmid (Addgene, 55,838; deposited by Michael Davidson). Cells were treated with rotenone (1 µM 6 h), CCCP (20 µM 1 h) or O/A (oligomycin A 10 µM + antimycin A 4 µM, 6 h) in serum-free medium and then fixed with 4% paraformaldehyde (PFA) for 10 min. Coverslips were mounted on glass slides and samples were visualized in an inverted confocal fluorescence microscope (Nikon Eclipse C1, Nikon Inc., Melville, NY, USA) with a x60 oil immersion objective. The excitation wavelengths used were 488 nm for GFP and 561 nm for MitroTracker Red, and emitted fluorescence was recorded using band pass filters BP515 and BP593 respectively. Autophagy vesicles and their colocalization with mitochondria were analyzed using Just Another Colocalization Plugin Software (JaCoP) in ImageJ [62]. At least 30 cells were analyzed per condition.
Cell lysis and western blot
Cells were scraped in PBS and collected after centrifugation at 1500 x g for 10 min at 4°C. They were lysed using the commercial RIPA lysis buffer supplemented with protease inhibition cocktail at 1X for 45 min. Afterward, cell lysates were centrifuged at 12,000 x g for 10 min at 4°C and supernatants were collected. Samples were separated by SDS-PAGE, using 15% polyacrylamide gels, and then transferred onto PVDF blotting membranes with a 0.2-μm pore size (AmershamTM HybondTM, 10,600,021). Membranes were blocked with 5% nonfat milk in TBST buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween 20 [Sigma-Aldrich, P7949]). Western blots were performed incubating the membranes with primary antibodies in TBST buffer containing 1% of nonfat milk overnight at 4°C. The primary antibodies employed were: anti-LC3A (rabbit) 1:1000 (Abcam, ab26628); anti-LC3B (rabbit polyclonal) 1:10,000 (Abcam, ab51520); anti-LC3C (rabbit monoclonal) 1:1000 (Abcam, ab150367); anti-COX4I/COX-IV (mouse monoclonal) 1:1000 (Abcam, ab14744); anti-ACTB/actin (mouse monoclonal) 1:1000 (Santa Cruz Biotechnology, sc-8432). After washing, blots were incubated with secondary antibodies in TBST containing 1% nonfat milk for 1 h at room temperature, using HRP-conjugated anti-mouse 1:5000 (Santa Cruz Biotechnology, sc-516,102) or HRP-conjugated anti-rabbit 1:5000 (Santa Cruz Biotechnology, sc-2357) as required. Blots were then washed in TBST and bands were visualized using Immobilon®Forte Western HRP Substrate in an iBright 1500 equipment (Invitrogen, California, CA, USA).
RT-qPCR
Total RNA was extracted from cells using NZYol (NZYtech, MB18501) according to the manufacturer’s instructions. Two µg RNA was used to perform reverse transcription and quantitative real time PCR with One-step NZY RT-qPCR Green kit (MB34302, NZYtech) on CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). ACTB mRNA levels were used as housekeeping gene. Primer information is detailed in Table S2.
siRNA
SH-SY5Y cells were transfected with 100 nM siRNA using Lipofectamine™ 2000 in Opti-MEM medium according to manufacturer instructions. Opti-MEM was replaced with complete RPMI 1640 medium after 6-h incubation. This incubation was repeated after 24 h, for a period of 72 h. siRNAs were designed to target the sequences used in other works [37,38] for the specific silencing of LC3A and LC3B proteins and are indicated in Table S3.
Mitochondrial isolation
Cells were collected and resuspended in mitochondrial insolation buffer (MIB; 10 mM HEPES, 70 mM sucrose, 210 mM mannitol, 1 mM EDTA, pH 7.5) supplied with protease inhibitors and homogenized (50 strokes) with a Potter-Elvehjem homogenizer (Fischer Scientific, 10,373,143). The homogenate was centrifuged at 1500 x g for 10 min at 4°C and the post-nuclear supernatant was collected. Supernatants were mixed and centrifuged at 10,500 x g for 5 min at 4°C. The pellet containing purified mitochondria was washed once with MB, and finally resuspended in RIPA lysis buffer supplemented with protease inhibition cocktail at 1X. Samples were examined by western blot. Immunodetection of COX4I only in mitochondrial fractions was considered as a control of mitochondrial isolation.
Statistical analysis
Statistical analyses were performed using the Student’s t-test. Results in Figure 6 were analyzed with ANOVA.
Supplementary Material
Acknowledgments
The authors are indebted to Dr. C. Behrends (Munich, Germany), Dr. I. Novak (Split, Croatia), and Dr. I. Ramos (Innoprot, Derio, Spain) for providing essential plasmids and cells. They are also grateful to Ms. Araceli Marcos for skillful technical help. Ms. Maider Garnica helped in obtaining some of the mutants and performed some preliminary flotation experiments.
Funding Statement
This work was supported in part by the Spanish Ministerio de Ciencia e Innovación (MCI), Agencia Estatal de Investigación (AEI) and Fondo Europeo de Desarrollo Regional (FEDER) (grant No. PGC2018-099857-B-I00), by the Basque Government (grants No. IT1625-22 and IT1270-19), by Fundación Ramón Areces (CIVP20A6619), by Fundación Biofísica Bizkaia and by the Basque Excellence Research Centre (BERC) program of the Basque Government. MI and YV were recipients of predoctoral FPU fellowships from the Spanish Ministry of Science Innovation and Universities (FPU16/05873, FPU18/00799), UB thanks the University of the Basque Country for a predoctoral contract, JHH was supported by a Postdoctoral Fellowship from the Basque Government.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].J-h U, Yun J.. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017;50:299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28:R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014;24:24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schaaf MBE, Keulers TG, Vooijs MA, et al. LC3/GABARAP family proteins: autophagy‐(un)related functions. FASEB J. 2016;30:3961–3978. [DOI] [PubMed] [Google Scholar]
- [5].Jatana N, Ascher DB, Pires DEV, et al. Human LC3 and GABARAP subfamily members achieve functional specificity via specific structural modulations. Autophagy. 2020;16:239–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Martens S. No ATG8s, no problem? How LC3/GABARAP proteins contribute to autophagy. J Cell Biol. 2016;215:761–763. 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nakatogawa H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. Vol. 55. 2013. pp. 39–50. [DOI] [PubMed] [Google Scholar]
- [8].Weidberg H, Shvets E, Shpilka T, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29:1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tsuboyama K, Koyama-Honda I, Sakamaki Y, et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354:1036–1041. [DOI] [PubMed] [Google Scholar]
- [10].Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495–501. [DOI] [PubMed] [Google Scholar]
- [11].Nguyen TN, Padman BS, Lazarou M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016;26:733–744. [DOI] [PubMed] [Google Scholar]
- [12].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Villa E, Marchetti S, Ricci J-E. No Parkin Zone: mitophagy without Parkin. Trends Cell Biol. 2018;28:882–895. [DOI] [PubMed] [Google Scholar]
- [14].Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol. 2020;432:80–103. [DOI] [PubMed] [Google Scholar]
- [15].Sentelle RD, Senkal CE, Jiang W, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8:831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dudek J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front Cell Dev Biol. 2017;5:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chao H, Lin C, Zuo Q, et al. Cardiolipin-Dependent mitophagy guides outcome after traumatic brain injury. J Neurosci. 2019;39:1930–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hsu P, Liu X, Zhang J, et al. Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy. 2015;11:643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ryan T, V BV, Stykel MG, et al. Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat Commun. 2018;9:817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chu CT. Mechanisms of selective autophagy and mitophagy: implications for neurodegenerative diseases. Neurobiol Dis. 2019;122:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lizama BN, Chu CT. Molecular aspects of medicine neuronal autophagy and mitophagy in Parkinson’s disease. Molecular Aspects of Medicine. 2021;82:100972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Antón Z, Landajuela A, Hervás JH, et al. Human Atg8-cardiolipin interactions in mitophagy: specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy. 2016;12:2386–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sugawara K, Suzuki NN, Fujioka Y, et al. The crystal structure of microtubule-associated protein light chain 3, a mammalian homologue of Saccharomyces cerevisiae Atg8. Genes Cells. 2004;9:611–618. [DOI] [PubMed] [Google Scholar]
- [25].Lee Y-K, Lee J-A. Role of the mammalian ATG8/LC3 family in autophagy: differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016;49:424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Landajuela A, Hervás JH, Antón Z, et al. Lipid geometry and bilayer curvature modulate LC3/GABARAP-mediated model autophagosomal elongation. Biophys J. 2016;110:411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Landeta O, Landajuela A, Gil D, et al. Reconstitution of proapoptotic bak function in liposomes reveals a dual role for mitochondrial Lipids in the BAK-driven membrane permeabilization process. J Biol Chem. 2011;286:8213–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Stepanyants N, Macdonald PJ, Francy CA, et al. Cardiolipin’s propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol Biol Cell. Vol. 26. 2015. pp. 3104–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kates M, Syz J-Y, Gosser D, et al. pH-dissociation characteristics of cardiolipin and its 2′-deoxy analogue. Lipids. 1993;28:877–882. [DOI] [PubMed] [Google Scholar]
- [30].Wang H, Sun H-Q, Zhu X, et al. GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion. Proc Natl Acad Sci. 2015;112:7015–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu J, Epand RF, Durrant D, et al. Role of phospholipid scramblase 3 in the regulation of tumor necrosis factor-α-induced apoptosis. Biochemistry. 2008;47:4518–4529. [DOI] [PubMed] [Google Scholar]
- [32].Kagan VE, Jiang J, Huang Z, et al. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ. 2016;23:1140–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schlattner U, Tokarska-Schlattner M, Ramirez S, et al. Dual function of mitochondrial nm23-H4 protein in phosphotransfer and Intermembrane lipid transfer. J Biol Chem. 2013;288:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Onishi M, Yamano K, Sato M, et al. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bhujabal Z, Birgisdottir ÅB, Sjøttem E, et al. FKBP8 recruits LC3A to mediate Parkin‐independent mitophagy. EMBO Rep. 2017;18:947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Guerroué F L, Eck F, Jung J, et al. Autophagosomal content profiling reveals an LC3C-dependent piecemeal mitophagy pathway. Mol Cell. 2017;68:786–796. [DOI] [PubMed] [Google Scholar]
- [37].von Muhlinen N, Akutsu M, Ravenhill BJ, et al. LC3C, bound selectively by a noncanonical LIR Motif in NDP52, is required for antibacterial autophagy. Mol Cell. 2012;48:329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liu X, Li Y, Wang X, et al. The BEACH-containing protein WDR81 coordinates p62 and LC3C to promote aggrephagy. J Cell Biol. 2017;216:1301–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kagan VE, Tyurin VA, Jiang J, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. [DOI] [PubMed] [Google Scholar]
- [40].Kang R, Zeh HJ, Lotze MT, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Schwarten M, Mohrlüder J, Ma P, et al. Nix directly binds to GABARAP : a possible crosstalk between apoptosis and autophagy Nix directly binds to GABARAP A possible crosstalk between apoptosis and autophagy. Autophagy. 2009;5:690–698. [DOI] [PubMed] [Google Scholar]
- [42].Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. [DOI] [PubMed] [Google Scholar]
- [44].Hanna RA, Quinsay MN, Orogo AM, et al. Microtubule-associated protein 1 light chain 3 (LC3) interacts with bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Murakawa T, Yamaguchi O, Hashimoto A, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015;6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Planas-Iglesias J, Dwarakanath H, Mohammadyani D, et al. Cardiolipin Interactions with Proteins. Biophys J. 2015;109:1282–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nguyen TN, Padman BS, Usher J, et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome–lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol. 2016;215(6):857–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Huang R, Xu Y, Wan W, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57:456–466. [DOI] [PubMed] [Google Scholar]
- [49].Cherra SJ, Kulich SM, Uechi G, et al. Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol. 2010;190:533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Krichel C, Möckel C, Schillinger O, et al. Solution structure of the autophagy-related protein LC3C reveals a polyproline II motif on a mobile tether with phosphorylation site. Sci Rep. 2019;9:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Behrends C, Sowa ME, Gygi SP, et al. Network organization of the human autophagy system. Nature. 2010;466:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bai H, Inoue J, Kawano T, et al. A transcriptional variant of the LC3A gene is involved in autophagy and frequently inactivated in human cancers. Oncogene. 2012;31:4397–4408. [DOI] [PubMed] [Google Scholar]
- [53].Basu Ball W, Neff JK, Gohil VM. The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Lett. 2018;592:1273–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Huang W, Choi W, Hu W, et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012;22:473–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ban T, Ishihara T, Kohno H, et al. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol. 2017;19:856–863. [DOI] [PubMed] [Google Scholar]
- [56].Sani M-A, Dufourc EJ, Gröbner G. How does the Bax-α1 targeting sequence interact with mitochondrial membranes? The role of cardiolipin. Biochim Biophys Acta - Biomembr. 2009;1788:623–631. [DOI] [PubMed] [Google Scholar]
- [57].Gonzalvez F, Schug ZT, Houtkooper RH, et al. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol. 2008;183:681–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Iyer SS, He Q, Janczy JR, et al. Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity. 2013;39:311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].X-x L, Tsoi B, Y-f L, et al. Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J Histochem Cytochem. 2015;63:301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mayer LD, Hope MJ, Cullis PR. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim Biophys Acta - Biomembr. 1986;858:161–168. [DOI] [PubMed] [Google Scholar]
- [61].Böttcher CJF, Van Gent CM, Pries C. A rapid and sensitive sub-micro phosphorus determination. Anal Chim Acta. 1961;24:203–204. [Google Scholar]
- [62].Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.