

ABSTRACT
Escherichia coli strains are responsible for a majority of human extra-intestinal infections, resulting in huge direct medical and social costs. We had previously shown that HlyF encoded by a large virulence plasmid harbored by pathogenic E. coli is not a hemolysin but a cytoplasmic enzyme leading to the overproduction of outer membrane vesicles (OMVs). Here, we showed that these specific OMVs inhibit the macroautophagic/autophagic flux by impairing the autophagosome-lysosome fusion, thus preventing the formation of acidic autolysosomes and autophagosome clearance. Furthermore, HlyF-associated OMVs were more prone to activate the non-canonical inflammasome pathway. Because autophagy and inflammation are crucial in the host’s response to infection especially during sepsis, our findings revealed an unsuspected role of OMVs in the crosstalk between bacteria and their host, highlighting the fact that these extracellular vesicles have exacerbated pathogenic properties.
Abbreviations: AIEC: adherent-invasive E. coliBDI: bright detail intensityBMDM: bone marrow‐derived macrophagesCASP: caspaseE. coli: Escherichia coliEHEC: enterohemorrhagic E. coliExPEC: extra-intestinal pathogenic E. coliGSDMD: gasdermin DGFP: green fluorescent proteinHBSS: Hanks’ balanced salt solutionHlyF: hemolysin FIL1B/IL‐1B: interleukin 1 betaISX: ImageStreamX systemLPS: lipopolysaccharideMut: mutatedOMV: outer membrane vesicleRFP: red fluorescent proteinTEM: transmission electron microscopyWT: wild-type
KEYWORDS: Autophagy, Escherichia coli, HlyF, inflammasome, outer membrane vesicle, pathogenesis
Introduction
Although the majority of E. coli strains are commensal bacteria colonizing the digestive tract, some are pathogenic because they have acquired an arsenal of virulence factors that allow them to overcome innate or acquired defense mechanisms of their host [1]. HlyF is a bacterial protein encoded by a gene present on a virulence plasmid found in strains of E. coli responsible for extraintestinal infection in both humans and animals [2]. More specifically, it is found in strains responsible for neonatal meningitis, avian colibacillosis [3–5] and more recently in an emerging serovar of enterohemorrhagic E. coli (EHEC) responsible for hemolytic and uremic syndrome in human [6]. The hlyF gene does not code for hemolysin as its name would suggest, but for a bacterial cytoplasmic protein that contributes to the bacterial virulence [7]. The HlyF protein displays a predicted catalytic domain of the short-chain dehydrogenase/reductase superfamily. This conserved domain is involved in the ability of HlyF to promote the production of bacterial OMVs [7]. Noteworthy, HlyF protein is not detected in OMVs. OMVs are nanoparticles consisting of a lipid bilayer envelope including membranous proteins and LPS originating from the parental strain and containing lumenal cargos [8,9]. Similarly to eukaryotic exosomes and microvesicles, OMVs are a mean of communication between bacteria and eukaryotic host cells [10,11]. Interestingly, we have observed that eukaryotic cell treatment with culture supernatants of E. coli strains expressing HlyF induced MAP1LC3/LC3-positive vesicles accumulation in host cells. At that time, we thought we were observing an exacerbation of autophagy and we interpreted this phenotype as related to the greater quantity of OMVs present in the supernatant [7].
Here, we showed that extracellular vesicles from E. coli strains producing HlyF had exacerbated pathogenic properties compared to OMVs produced by isogenic strains unable to produce a functional HlyF. Our results revealed that these OMVs inhibit the autophagic flux and thus are more prone to promote the activation of the non-canonical inflammasome pathway, the main host defense mechanism against infections that must be tightly regulated to ensure a correct antimicrobial response [12–15]. Thus, these OMVs, produced by bacteria responsible of extraintestinal infections, could destabilize the host response to the infection, thus facilitating bacterial pathogenesis.
Results
OMVs from HlyF-expressing E. coli specifically induced an increase of LC3-positive vesicle number
In agreement with our previous results [7], bacterial expression of wild-type (WT) HlyF was associated with an overproduction of OMVs (Figure 1A). Moreover, HeLa cells treatment with culture supernatant of these HlyF WT strains induced an increase of LC3-positive structure number in host cells, as denoted by vesicle-associated LC3 foci (Figure 1B). To take into account a dose effect due to overproduction of OMVs from bacteria producing HlyF, eukaryotic cells were then treated with the same number of purified OMVs from isogenic E. coli strains expressing or not HlyF. To do so, we analyzed OMVs by transmission electron microscopy (TEM) and checked that the concentration of OMVs was correlated to protein concentration (data not shown). At the same concentration, only OMVs from the wild-type strain expressing HlyF specifically induced an increase of LC3-positive structure number in host cells (Figure 1C). We repeated these experiments using the same amount of OMVs purified from the laboratory E. coli strain BL21(DE3) devoid of virulence factors and flagellin, expressing wild-type or inactivated HlyF (Fig. S1A). OMVs were analyzed by TEM (Figure 1D), but also by dynamic light scattering (Fig. S1B) and we performed protein and LPS dosages (Fig. S1C) to refine their characterization. These results showed that OMVs produced from both strains expressing active and inactive HlyF had a similar diameter (25.95 nm ± 4.91 and 24.5 nm ± 4.86 respectively) and comparable concentration of protein and LPS per vesicle. The increase of GFP-LC3positive foci (Figure 1E) as well as the significant increase of LC3-II level (Figure 1F-G) confirmed the specific HlyF-dependent increase of LC3-positive structure number following cell treatment with OMVs from BL21-producing wild-type HlyF.
Figure 1.
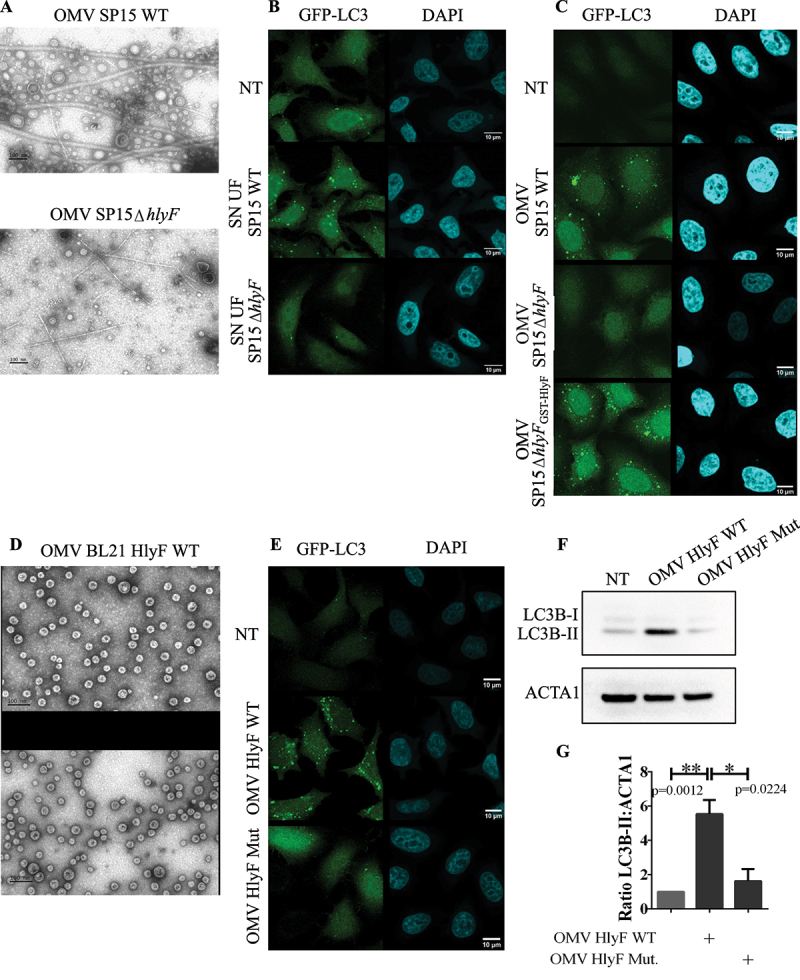
Increased LC3-positive structure number in epithelial cells treated by OMVs from E. coli producing HlyF. (A) Images of purified OMVs with equivalent volumes from wild-type SP15 (SP15 WT) and from SP15ΔhlyF visualized by negative staining transmission electron microscopy (TEM). Scale bar: 100 nm. Images representative of 3 independent experiments. (B-C) Images of confocal microscopy of GFP-LC3 (green) and DAPI (blue) in GFP-LC3-HeLa cells treated 3 h with supernatant from wild-type SP15 (SP15 WT) and SP15ΔhlyF 250 µL/mL after ultrafiltration (B) or with purified OMVs with equivalent protein dosage from wild-type SP15 (SP15 WT) 10 µg/mL, SP15ΔhlyF 10 µg/mL and SP15ΔhlyFGST-HlyF 1 µg/mL (C). Scale bar: 10 µm. Images representative of 3 independent experiments. (D) Images of purified OMVs with equivalent protein dosage from BL21 expressing HlyF WT or inactivated (Mut) visualized by negative staining TEM. Scale bar: 100 nm. Images representative of 3 independent experiments. (E) Images of confocal microscopy of GFP-LC3 (green) and DAPI (blue) in GFP-LC3-HeLa cells treated 3 h with OMV BL21 HlyF WT or OMV BL21 HlyF Mut 5 µg/mL. Scale bar: 10 µm. Images representative of 3 independent experiments. (F) Western blot analysis of LC3 and ACTA1 in cell extracts of HeLa cells treated 3 h with OMVs from BL21 HlyF WT and BL21 HlyF Mut 5 µg/mL. Experiment reproduced 3 times independently. (G) Quantification of the LC3B-II:ACTA1 ratios of the Figure 1F obtained by densitometric analysis of 3 independent experiments. The graph shows the mean and the standard deviation for each condition. ** p < 0,01 and *p < 0,05t test.
Increased LC3-positive vesicle number induced by OMVs from HlyF-expressing E. coli was dose-dependent.
To quantify the number of LC3-positive foci per cell, we counted the fluorescently labeled GFP-LC3 puncta per cell using the imaging flow cytometry technology (ImageStreamX system – ISX) [16]. The quantification of dots of GFP-LC3 per cell showed that OMVs HlyF WT treatment induced a 5-fold increase of LC3-positive structure in eukaryotic cells compared to non-treated cells (Figure 2A–B). We also showed that the accumulation of LC3-positive structure increased substantially with the concentration of OMVs HlyF WT until it reached a plateau from a concentration of 4 µg/mL of OMVs (Figure 2C–D). A dose effect showed that no matter how much OMVs of the non-HlyF producing strain were put on the HeLa cells, no LC3-positive structure accumulation was ever observed (Figure 2C–D). Finally, measurement of the level of LC3-II at different time points in HeLa cells showed that autophagosome increase in HeLa cells lasted at least 72 h after the beginning of OMV HlyF WT treatment (Fig. S2).
Figure 2.
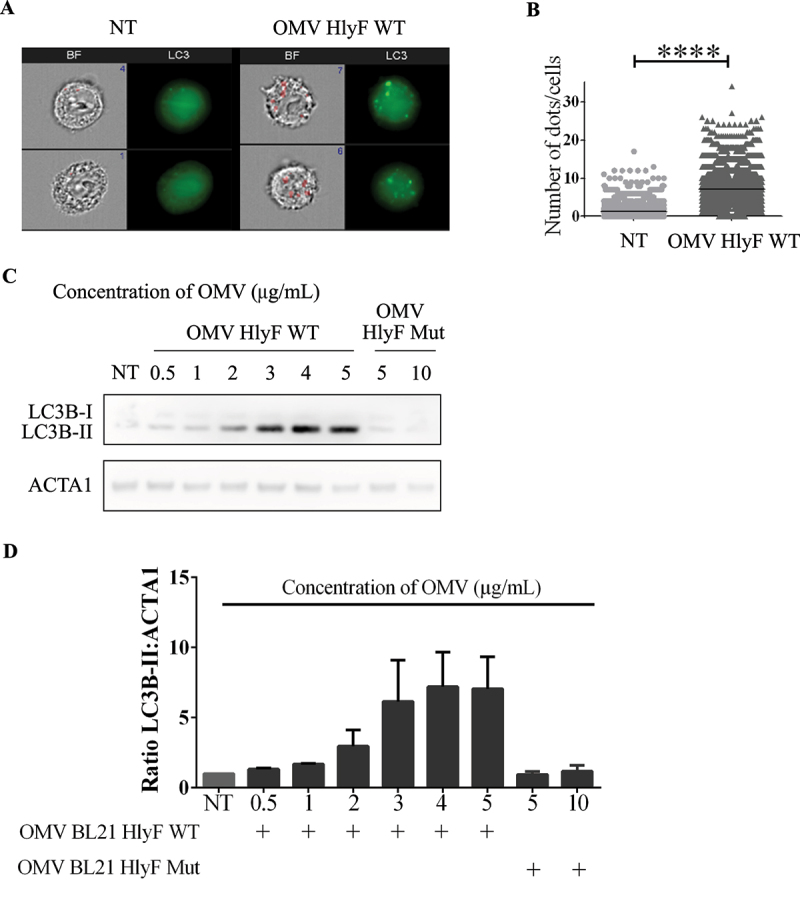
OMVs-induced increase of LC3-positive structure number wasdose-dependent. (A) Images acquired by ISX of GFP-LC3 HeLa cells treated 3 h with OMVs from BL21 HlyF WT 5 µg/mL. On the left panel, brightfield channel (BF) images the cells according to their size and morphology; on the right panel, LC3 channel images cells according to their fluorescence in GFP-LC3 in green. Number of LC3 green dots of GFP-LC3 per cell is indicated in blue in the dial at the top right of the BF channel. The mask is shown in red on the BF. Scale bars: 7 µm. Images representative of 4 independent experiments. (B) Quantification of (A). Number of LC3 green dots of GFP-LC3 per cell counted with the Spot count function is indicated on the vertical axis on a minimum of 3000 cells per condition. Each square represents a cell image acquired by ISX. The graphs show the mean for each condition. ****p < 0.0001 t test. Graph representative of 4 independent experiments. (C) HeLa cells were treated with increasing concentrations of OMVs from BL21 HlyF WT for 3 h then LC3 and ACTA1 were analyzed by western blot. Experiment reproduced 3 times independently. (D) Quantification of the LC3B-II:ACTA1 ratios of the Figure 2C obtained by densitometric analysis of 3 independent experiments. The graph shows the mean and the standard deviation for each condition.
OMVs from HlyF-expressing E. coli triggered the specific accumulation of autophagosomes in host cells.
Accumulation of LC3-positive structures in eukaryotic cells following OMVs treatment could result from an accumulation of autophagosomes or from LC3-associated phagocytosis vesicles (LAP). To distinguish between these two types of vesicles, we first observed that OMVs were not sequestered inside the LC3-positive vesicles, as there was no colocalization between GFP-LC3 structures and OMVs stained with DiI (Figure 3A), suggesting that the accumulation of LC3-positive structures did not result from LAP and the phagocytosis of OMVs prior to their degradation but from autophagosomes. We confirmed this result by transmission electron microscopy analysis of HeLa cells treated by OMVs, in which we observed the accumulation of vesicles (Figure 3B), and checked that these vesicles consisted of a double-membrane indicative of autophagosomes, rather than the single-membrane vesicles of LAP (Figure 3C). Additionally, we treated cells with a potent phagocytosis inhibitor, cytochalasin D [17,18] that also blocks LAP [19]. Pharmacological blockage of phagocytosis by cytochalasin did not block LC3-II accumulation in cells treated with OMV HlyF WT while it did not increase LC3-II levels in the absence of OMV or in the presence of OMV HlyF Mut after 3 h (Figure 3D). This result suggested that the increase of LC3-II level induced by OMV HlyF WT was not a result of LAP but rather depended on autophagosome accumulation.
Figure 3.
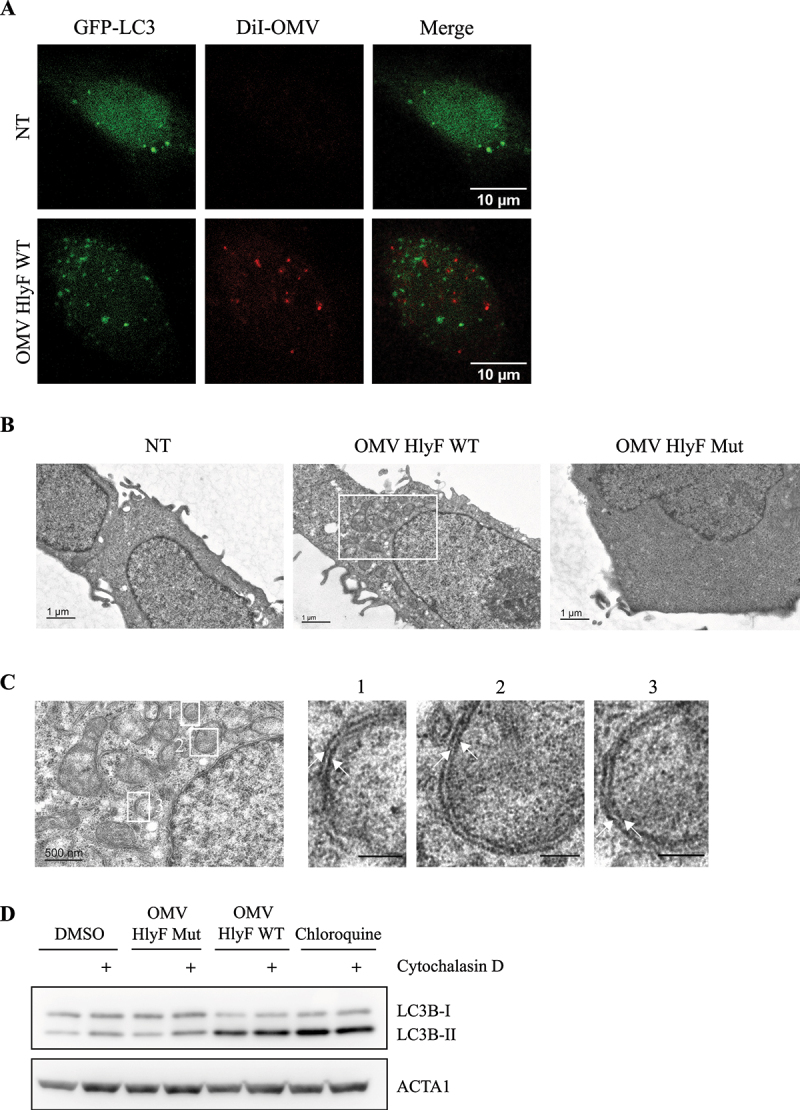
OMVs HlyF WT triggered the specific accumulation of autophagosomes in host cells. (A) Confocal images of GFP-LC3 (green) and fluorescently labeled OMVs with DiI (Red) fluorescence in GFP-LC3 HeLa cells. The cells were incubated with OMVs from BL21 HlyF WT 5 µg/mL for 3 h. 100 cells were analyzed per condition. Scale bar: 10 µm. Images representative of 3 independent experiments. (B-C) Ultrastructure of HeLa cells visualized by TEM. The cells were incubated with OMVs from BL21 HlyF WT or from BL21 HlyF Mut 5 µg/mL for 3 h. 15 cells were analyzed per condition. (B) Scale bar: 1 µm. (C) The image is a magnification of the part of the image (B) within the rectangle. Scale bar: 500 nm. The images on the right panel are a magnification of the part of the image of the left panel within the rectangles 1, 2 and 3. Arrows indicate double-membrane of autophagosomes. (D) HeLa cells incubated with DMSO or 5 µM cytochalasin D were left untreated or treated with 5 µg/mL of OMVs from BL21 HlyF WT, from BL21 HlyF Mut or 50 μM chloroquine for 3 h before LC3 and ACTA1 western-blot analysis.
Altogether, these data indicated that OMV HlyF WT promoted the accumulation of autophagosomes in host cells.
OMVs from HlyF-expressing E. coli induced an autophagic blockade.
Autophagy is a dynamic process in which the number of autophagosomes depends on their formation and further degradation. This is referred as autophagic flux. Thus, OMV-dependent accumulation of autophagosomes could result either from an increased synthesis of autophagosomes or from a decreased degradation following their fusion with lysosomes (Figure 4A) [20,21]. To decipher the cellular basis of increase of autophagosomes number after OMV treatment, we monitored the level of autophagosomes-associated LC3-II in HeLa cells. As expected, LC3-II level increased when lysosomal-dependent LC3-II degradation was blocked by the lysosomal inhibitor chloroquine. However, these levels were comparable in cells treated with OMVs in presence of chloroquine (Figure 4B, lines 2 and 4, quantified in Figure 4C). Absence of cumulative effect of OMVs and chloroquine on LC3-II level suggested that OMVs HlyF WT induced an autophagy blockade in HeLa cells similarly to chloroquine treatment. These results were confirmed by quantification of GFP-LC3 cells treated by OMVs HlyF WT in presence or absence of chloroquine by imaging flow cytometry. We observed that the number of GFP-LC3 fluorescent dots in OMV-treated cells was not further increased by chloroquine which would have been the case if the autophagic flux had been stimulated (Figure 4D–E). Altogether, these results showed that OMVs HlyF WT treatment induced an autophagic blockade in eukaryotic cells rather than an induction of the flux.
Figure 4.
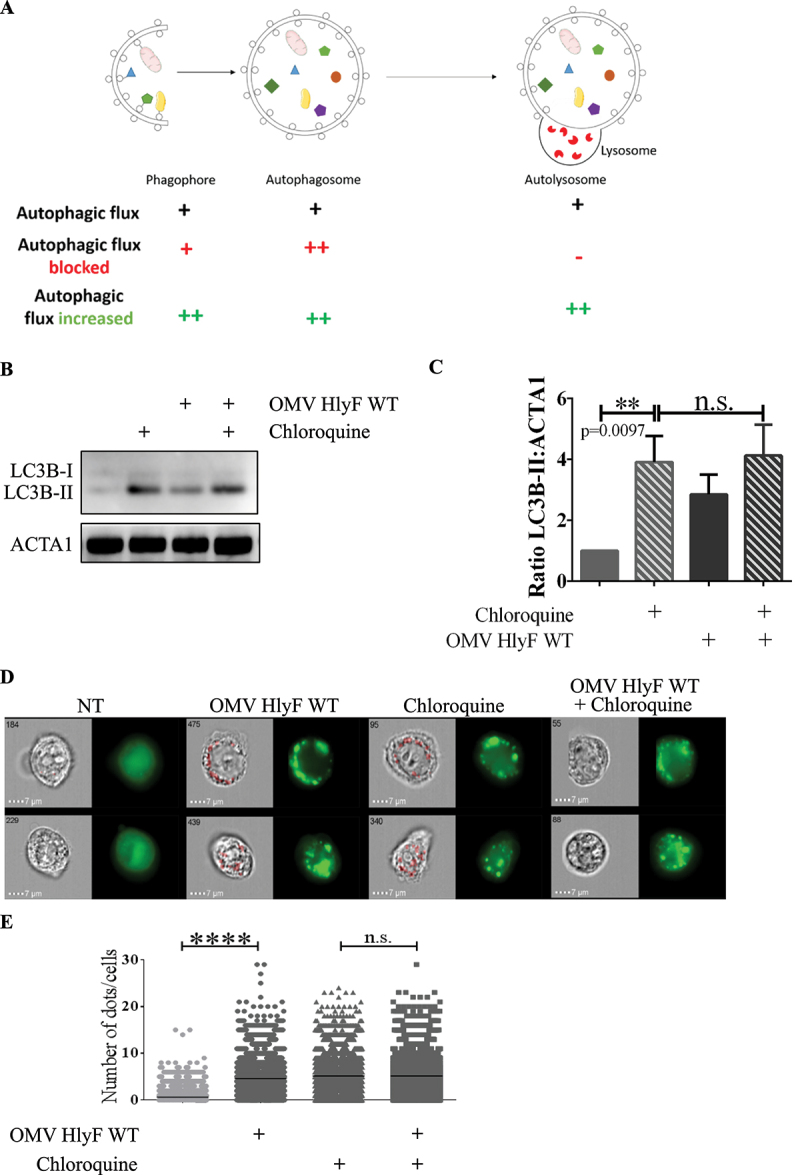
Autophagy flux was blocked by OMVs HlyF WT. (A) Schema of the autophagic flux and of the accumulation of different autophagic structures depending on the autophagic dynamic regulation (adapted from Mizushima et al., 2010). (B) HeLa cells were treated with OMVs from BL21 HlyF WT 5 µg/mL and/or 50 μM chloroquine for 3 h before LC3 and ACTA1 western-blot analysis. Experiment reproduced 3 times independently. (C) Quantification of the LC3B-II:ACTA1 ratios of the Figure 4B obtained by densitometric analysis of 3 independent experiments. The graph shows the mean and the standard deviation for each condition. **p: 0.0097 t test, n.s.: non-significant. (D) GFP-LC3 HeLa cells were treated with OMVs from BL21 HlyF WT 5 µg/mL and/or 50 μM chloroquine for 3 h before ISX analysis. On the left panel, brightfield channel (BF) images; on the right panel, LC3 channel image cells according to their fluorescence in GFP-LC3 in green. Scale bars: 7 µm. Images representative of 3 independent experiments. (E) Quantification of (D). Number of LC3 green dots of GFP-LC3 per cell counted with the Spot count function is indicated on the vertical axis, on a minimum of 3000 cells per condition. Each square represents a cell image acquired by ISX. The graphs show the mean for each condition. Experiment reproduced 3 times independently, ****p < 0.0001 t test.
OMVs from HlyF-expressing E. coli prevented the autophago-lysosome fusion and autophagosomes clearance.
To characterize which step of the autophagy pathway was inhibited, we used HeLa-Difluo™ hLC3 reporter cells. These cells express a fusion protein RFP-GFP-LC3, in which the N terminus of LC3 is fused to two fluorescent reporter proteins: an RFP (acid stable) and a GFP (acid sensitive). Autophagosomes that contain intact RFP-GFP-LC3 proteins emit both RFP and GFP signals resulting in a colocalization of GFP- and RFP-positive puncta. After fusion of the autophagosomes with the lysosomes, the GFP fluorescence diminishes due to the acidification of the autolysosome, while the red fluorescence corresponding to the remaining acidic-resistant RFP-LC3 signal is maintained [22,23]. We performed confocal imaging of HeLa-Difluo™ hLC3 reporter cells. As expected, in the control condition treated with lysosomal inhibitor chloroquine, we observed an accumulation of colocalization of GFP- and RFP-positive puncta corresponding to autophagosomes harboring RFP-GFP-LC3. When cells were starved with HBSS, known to induce the autophagic flux, a proportion of GFP fluorescence was decreased by the acidic pH environment while acid-resistant red fluorescence persisted, denoting that fusion of autophagosome with lysosome occurred in these cells. In contrast, in cells treated with OMVs HlyF WT, we observed an increased number of colocalization of GFP- and RFP-positive puncta and absence of red-fluorescent-associated autolysosome, indicating that the autophagic blockade occurred before the acidification of the autophagosomal compartment (Figure 5A).
Figure 5.
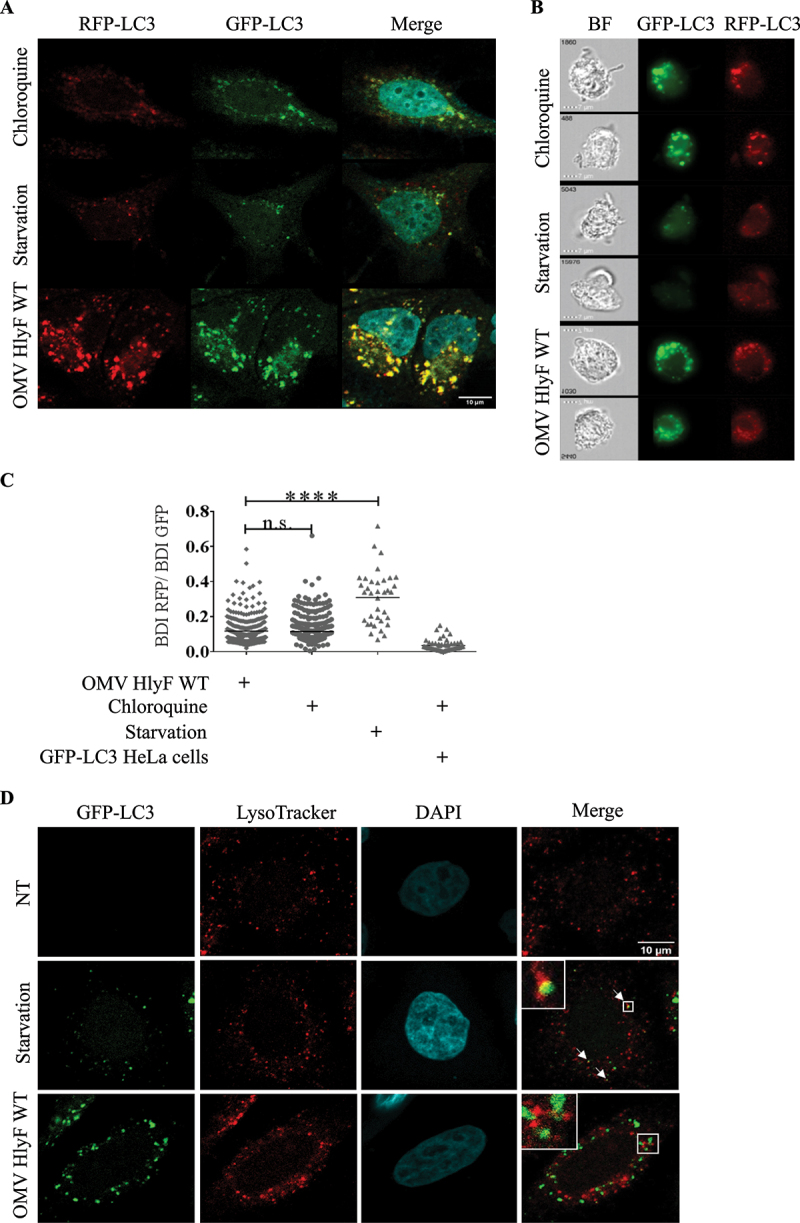
Autophagy flux blocked by OMVs HlyF WT at lysosomal fusion step. (A) Confocal images of HeLa-Difluo™ hLC3 reporter cells deprived with HBSS or treated for 3 h with 50 μM chloroquine or with OMVs from BL21 HlyF WT 5 µg/mL. Images of GFP-LC3 fluorescence (green), RFP-LC3 fluorescence (red), and the overlay with DAPI (blue) which shows colocalization of GFP- and RFP-positive puncta. Scale bar: 10 µm. 50 cells were analyzed per condition. Images representative of 3 independent experiments. (B) Images acquired by ISX of HeLa-Difluo™ hLC3 reporter cells deprived with HBSS or treated for 3 h with 50 μM chloroquine or with OMVs from BL21 HlyF WT 5 µg/mL. On the left panel, brightfield channel (BF), on the medium panel, GFP-LC3 channel images cells according to their fluorescence in GFP in green and on the right panel RFP-LC3 channel images cells according to their fluorescence in RFP in red. Scale bars: 7 µm. Images representative of 3 independent experiments. (C) Quantification of BDI (GFP and RFP) by ISX of HeLa-Difluo™ hLC3 reporter cells deprived with HBSS or treated for 3 h with chloroquine 50 µM or with OMVs from BL21 HlyF WT 5 µg/mL. Each square represents the ratio of the intensity of RFP and GFP within a cell. The graph shows the mean of each condition. Experiment reproduced 3 times independently. ****p < 0.0001 t test, n.s.: non-significant. (D) Confocal images of GFP-LC3 (green), LysoTracker Red (red) and DAPI (blue) fluorescence in GFP-LC3 HeLa cells. The cells were incubated with OMVs from BL21 HlyF WT 5 µg/mL for 3 h and LysoTracker Red for 1 h. 100 cells were analyzed per condition. Scale bar: 10 µm. Arrows indicates colocalization foci. Images representative of 3 independent experiments.
To confirm this result, we quantified the level of fluorescently labeled RFP and GFP LC3 in HeLa-Difluo™ hLC3 reporter cells by imaging flow cytometry. In contrast to starvation but similarly to chloroquine treatment, we observed that in cells treated by OMVs the ratio RFP:GFP was low, indicating that the GFP is preserved from autophagosome acidification (Figure 5B–C).
Finally, we stained acidic organelles in GFP-LC3 cells with LysoTracker Red labeling. We checked that upon starvation, GFP-LC3 HeLa cells harbored colocalization of GFP- and RFP-positive puncta (highlighted by white arrows) resulting from the colocalization of autophagosomes and lysosomes merged signals. In contrast, absence of colocalization of GFP- and RFP-positive puncta indicated that OMVs HlyF WT treatment of GFP-LC3 HeLa cells prevented lysosomal acidification of autophagosomes (Figure 5D). These results showed that autophagy flux was blocked by OMVs at lysosomal fusion step. Moreover, persistance of signal corresponding LC3-II up to 72 h after OMV addition (Fig. S2) supported the fact that autophagosomal clearance was inhibited.
OMVs from HlyF-expressing E. coli induced a stronger activation of the non-canonical inflammasome pathway compared to OMVs from E. coli expressing a nonfunctional HlyF protein.
Numerous studies show that autophagy is a negative regulator that prevents excessive activation of inflammasomes [12,24–30]. We checked that OMVs HlyF WT caused a substantial autophagosome accumulation in mouse bone marrow-derived macrophages (BMDMs) (Fig. S3A-B) and in the THP1 cell line (Fig. S3C-D). We therefore speculated that OMV-dependent inhibition of autophagic flux could exacerbate inflammasome activation. OMV-treatment of unprimed BMDMs from wild-type mouse led to cell death (measured by LDH [lactase dehydrogenase] release) and to the excretion of mature IL1B in the supernatant of cell culture (Figure 6A). These phenotypes were dependent of the activation of the non-canonical inflammasome pathway as they were abolished in inflammasome-deficient BMDM invalidated for CASP11, CASP1 CASP11 or GSDMD (gasdermin D) (Figure 6A), and as the cleaved forms of GSDMD and CASP1 were detected only in the supernatants of OMV-treated wild-type BMDM but not in the supernatants of OMV-treated gsdmd KO and casp11 KO BMDM (Fig. S3 E-F). Importantly, these phenotypes were significantly exacerbated when BMDM were treated with OMVs HlyF WT compared to OMVs produced in bacteria expressing inactive HlyF (Figure 6A and Fig S3E-F).
Figure 6.
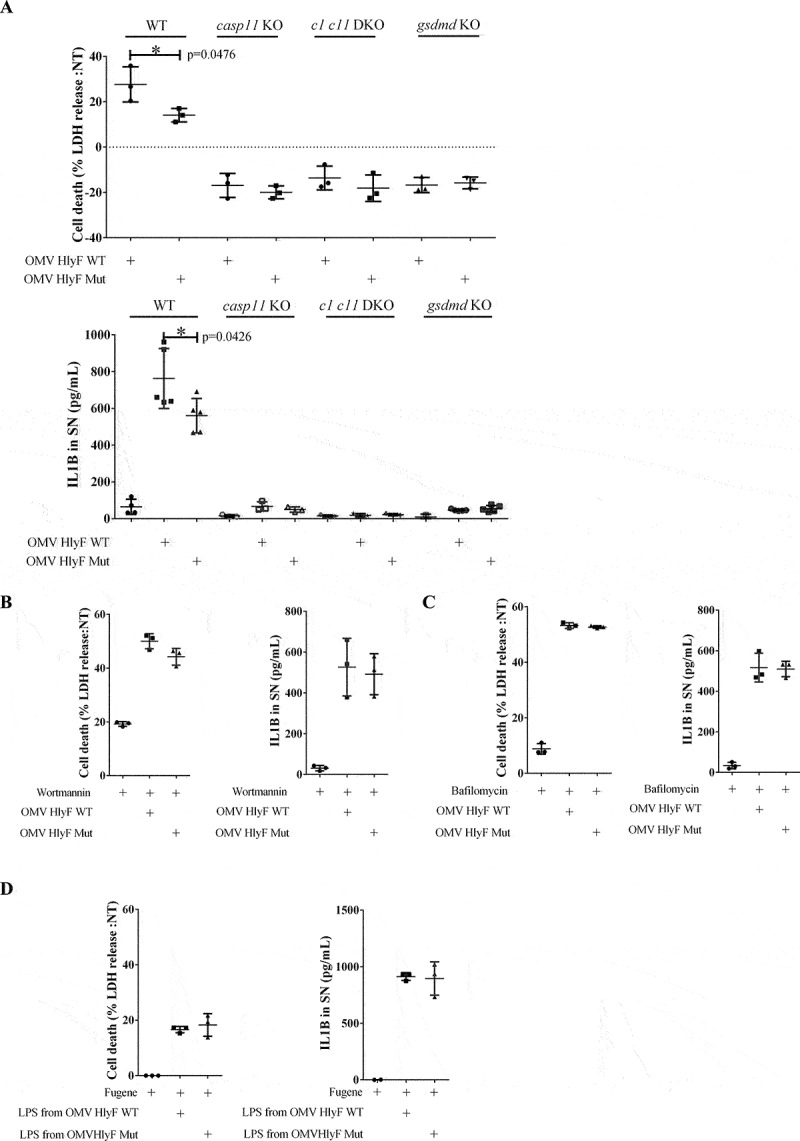
OMVs HlyF WT induced a stronger activation of the non-canonical inflammasome pathway compared to OMVs HlyF Mut. (A) Release of LDH and IL1B from unprimed WT, casp11−/−, casp1−/−casp11−/− and gsdmd−/− mouse BMDM treated for 24 h with OMV HlyF WT or OMV HlyF Mut 5 µg/mL. The graphs show the mean and the standard deviation of at least 3 independent experiments for each condition. Each dot represents the value obtained in one experiment. *p < 0.05 t test. (B) Release of LDH and IL1B from unprimed WT mouse BMDM treated for 24 h with OMV HlyF WT or OMV HlyF Mut 5 µg/mL with 10 µM wortmaninn. The graphs show the mean and the standard deviation of 3 independent experiments for each condition. Each dot represents the value obtained in one experiment. (C) Release of LDH and IL1B from unprimed WT mouse BMDM treated for 24 h with OMV HlyF WT or OMV HlyF Mut 5 µg/mL with 10 nM bafilomycin. The graphs show the mean and the standard deviation of 3 independent experiments for each condition. Each dot represents the value obtained in one experiment. (D) Release of LDH and IL1B from unprimed WT mouse BMDM transfected with LPS purified from OMVs BL21 HlyF Mut or from OMVs BL21 HlyF WT (1 µg/mL using FuGENE HD, Promega) for 24 h. The graphs show the mean and the standard deviation of 3 independent experiments for each condition. Each dot represents the value obtained in one experiment.
Furthermore, we deciphered the impact of autophagic blockage on the inflammasome activation following OMV treatment, using 2 different autophagic inhibitors (wortmannin and bafilomycin A1). As expected, autophagic inhibition alone did not activate the inflammasome machinery. Interestingly, we observed that autophagic inhibition by bafilomycin and wortmannin abolished the autophagic negative feedback loop and thus restored the overactivation of the inflammasome in OMV HlyF Mut treated cells to the level of OMV HlyF WT treated BMDM (Figure 6B–C). In accordance with the literature, these results suggested that the specific exacerbation of the non-canonical inflammasome pathway activation induced by OMVs HlyF WT treatment was due to their ability to block the autophagic flux [12,24–30]. Finally, since activation of the non-canonical inflammasome pathway is related to the sensing of LPS though host guanylate-binding proteins in the cytosol [31], we transfected purified LPS from OMVs HlyF WT and from OMVs HlyF Mut directly into the cytosol of BMDM. We observed that the activation of the non-canonical inflammasome pathway, measured by cell death and IL1B release, was similar regardless of the LPS transfected (Figure 6D). This result suggested that the overactivation of the non-canonical inflammasome pathway did not depend on LPS difference but rather resulted from OMV HlyF WT ability to block autophagy.
Discussion
In this study, we confirmed that pathogenic E. coli strains harboring a virulence plasmid coding for hlyF overproduce OMVs that modulate autophagy [7]. However, we showed that this modulation was due to an autophagic blockage instead of an induction of the flux as initially thought [7]. Moreover, we demonstrated that OMVs HlyF WT blocked autophagosome maturation and fusion with lysosomes and inhibited their clearance from host cytosol. Autophagy is recognized as an innate antibacterial mechanism by direct lysosomal degradation of intracellular pathogen. After engulfment of the cytosolic pathogen, autophagosomes fuse with lysosomes, leading to degradation of the sequestered content [27]. Thus, autophagy, that is crucial to maintain cellular homeostasis, acts also as the first line of host defense against bacterial pathogen as it fights against the intruder very early during infection [32]. It has been shown recently that some ExPEC strains hijack the host autophagy machinery to promote their intracellular survival and replication in macrophages [33]. This ability is due to the inhibition of the fusion of ExPEC-containing phagosomes with lysosomes and their subsequent degradation [33]. This observation is consistent with the properties we demonstrated here, i.e. OMVs preventing the autophagosome-lysosome fusion and acidification in macrophages. In line with this idea, the fact that the adherent-invasive E. coli (AIEC) reference strain NRG 857Cc harbors the hlyF gene suggests that OMVs actively participate in the virulence of these pathogenic bacteria by favoring their survival and their replication into eukaryotic cells [34]. Pathogenic bacteria could exploit HlyF OMV property to block their autophagosomal clearance into epithelial cells and macrophages.
Thus, the contribution of OMVs produced by HlyF-expressing E. coli to bacterial virulence includes autophagic blockage but also the dissemination and delivery of their virulence factors potentially far from the source of infection, directly into the cytoplasm of target cells. In line with this idea, OMVs play a very important role in mediating the cytosolic localization of lipopolysaccharide (LPS), an endotoxin that is present at their surface. Once in the host cytosol, LPS is recognized by the non-canonical inflammasome machinery and thereby activates an inflammatory immune response [31,35,36]. However, excessive inflammation activation by LPS can be deleterious for the host. The inflammatory burst can also lead to organ dysfunction which can worsen up to sepsis in severe cases [37,38]. At the early stage of sepsis, macrophages release large amounts of pro-inflammatory molecules that exacerbate inflammatory response. It is then followed by an immunosuppression due to the massive apoptosis of macrophages, which makes host susceptible to recurrent infections [39,40]. The role of OMVs in sepsis is highlighted by the fact that injection of OMVs alone in mice recapitulates sepsis-associated exacerbated inflammation and mortality [41]. Thus, this inflammation machinery initiated by OMV recognition must be tightly regulated. Here, we showed that OMV HlyF WT provoked an exacerbated activation of the non-canonical inflammasome activation and IL1B release, one of the main mechanisms though which innate and adaptive immune responses are induced by an infection [38,41,42]. We showed that not only OMVs HlyF WT induced inflammation, they also blocked autophagy, a major negative regular of excessive inflammation activation and IL1B release [29,43]. In particular, autophagy induction prevents the onset of sepsis [12]. This negative regulation mainly relies through effects on the secretion of immune mediators and on the removal of endogenous inflammasome agonists such as PAMPs [24–28]. Therefore, not only OMVs favor intracellular delivery of LPS and PAMPs into cells but inhibition of autophagosomal clearance of these inflammasome activators by OMV HlyF WT impairs inflammasome negative feedback control, leading to an excessive and uncontrolled inflammatory condition detrimental for the host.
Our study unraveled the mechanisms by which HlyF expression by E. coli confers hyper-virulent properties to OMVs, as they gain the property to manipulate the host cell autophagic and inflammatory responses to bacterial benefit both at the site and at great distance from the site of infection. An additional contribution of OMVs HlyF to bacterial virulence could be associated to the dissemination of toxins [7,44]. Overall, OMVs HlyF contributed significantly to the virulence of pathogenic bacteria.
Materials and methods
Bacterial strains and growth conditions
SP15 is an extra-intestinal pathogenic E. coli (ExPEC) strain isolated from a neonatal meningitis case [45]. SP15, SP15∆hlyf and SP15∆hlyfGST-hlyF were previously described in [7]. pGEX6P-1 plasmids (Cytiva, 28,954,651) expressing GST-HlyF protein, wild-type or inactivated by a mutation on its putative catalytic site (Fig. S1A), were transformed into E. coli BL21(DE3) (Sigma, CMC0014) strain. SP15 and BL21(DE3) strains and derivatives were inoculated and grown in Terrific Broth (Sigma, T5574) with appropriate antibiotics for 8 h at 37°C with agitation.
Purification of bacterial OMVs
After 8 h of culture the bacterial suspension was centrifuged at 6500 g for 10 min at 4°C. The supernatant was filtered (pore size 0.45 μm; Merck-Millipore, S2GPU10RE) to obtain bacterial-free supernatant. The filtered supernatant was ultrafiltered and concentrated using tangential concentrator and PES membrane cassette (pore size 100 kDa; Sartorius, VF20H4) and ultracentrifuged at 150,000 g for 1.5 h at 4°C. After removing the supernatant, pelleted OMV were resuspended in sterile PBS with Ca2+ and Mg2+ (Eurobio scientific, CS1PBS00K-BP) and stored at 4°C. To visualize the concentration of OMVs and the purity of the preparation, negative staining transmission electron microscopy (TEM) was performed according to standard procedures. The concentration of OMVs in the suspension correlates with protein concentration evaluated by BCA protein assay (Bio-Rad, RC DCTM Protein Assay kit II 5000122) (Fig. S1C). LPS concentration in OMV was evaluated by Purpald assay as described in [46]. Values were normalized to glycerol concentration. OMV diameter was calculated by dynamic light scattering using Zetasizer Nano instrument (Malvern) (Fig. S1B).
OMVs in PBS were fluorescently labeled using 1% (v:v) DiI (Invitrogen, V22885) for 30 min at 37°C.
Purification and transfection of LPS from OMVs
LPS was extracted from purified OMV using LPS extraction kit (iNtRON Biotechnology, 17,141) according to manufacturer’s instruction. LPS concentration was determined by Purpald assay. Transfection of cells with purified LPS was done at a concentration of 500 ng/50,000 cells, using FuGeneHD (Promega, E2311) transfection reagent in Opti‐MEM (Gibco, 11,058,021), as previously described [31].
Eukaryotic cell culture and treatments
HeLa cells (ATCC, HeLa CCL2), HeLa-GFP-LC3 cells (gift from Patrice Codogno, Insitut Necker, Paris) and HeLa-Difluo™ hLC3 reporter cells (Invivogen, heldf-hlc3) were cultured in DMEM with Glutamax (Life Technology, 10,566,016) supplemented with 10% fetal bovine serum (FBS) (Eurobio scientific, CVFSVF06-01). HeLa-Difluo™ hLC3 reporter cells were maintained in growth medium supplemented with the selection antibiotic Zeocin 200 µg/mL (Invivogen, ant-zn-05). One day before the experiment, HeLa cells were seeded at a density of 5 × 104 cells per mL in DMEM supplemented with 10% FBS. OMV treatment were done by adding directly in the medium of cell culture the volume of OMV corresponding to the amount of proteins indicated. The cells were collected 3 h after OMV addition. When indicated, cells were pre-treated for 40 min and treated with cytochalasin D (Sigma, C2618) at a final concentration of 5 µM.
THP1 cells were cultured in RPMI with Glutamax (Life Technology, 12,027,599) supplemented with 10% FBS. One day before the experiment, THP1 cells were seeded at a density of 3 × 105 cells per mL. OMV treatment were done by adding directly in the medium of cell culture the volume of OMV corresponding to the amount of proteins indicated. The cells were collected 3 h after OMV addition.
Bone marrow‐derived macrophages (BMDM) were prepared from fresh bone marrow isolated from WT, casp11−/−, casp1−/− casp11−/− or gsdmd−/− mutant mice. Bone marrow cells were allowed to differentiate in macrophages by incubation for 7 days in DMEM supplemented with 10% FBS, 25 ng/ml CSF1/M-CSF (Immunotools, 12,343,113), 10 mM HEPES, pH 7.2, and nonessential amino acids (Life Technology, 11,140,050). One day before treatment, BMDM were seeded into 24-well plates at a density of 2.5 × 105 cells per well. OMV treatment, were done by adding the volume of OMV corresponding to the amount of proteins indicated in Opti-MEM medium. The cells and supernatant were collected 24 h after OMV addition.
Immunoblotting
Cells were treated with the OMV or left untreated for the indicated times. Then cells were washed twice with cold PBS, lysed in NuPAGE LDS sample buffer with reducing agent (Invitrogen, NP0007) and sonicated. Cell extracts were boiled for 5 min at 95°C, separated by SDS-PAGE (Invitrogen, NP0323) and transferred to PVDF membranes (Invitrogen, LC2000). The membranes were blocked in 5% BSA (Dominique Dutscher, P6154,) in 0.1% Tween® 20 (Sigma, P1379) in TBS (Euoromedex, ET220) and probed with primary antibodies. The primary antibodies used were rabbit anti-LC3 (Cell Signaling Technology, S 3868), mouse anti-ACTA1 (Abcam, Ab3280), mouse anti-CASP1/caspase 1 (Addipogen, AG20B-0042), rabbit anti-GSDMD/Gasdermin D (Abcam, ab209845), rabbit anti-GAPDH (GeneTex, GTX100118), rat anti-CASP11 (Novus, NB120-10,454). After incubation with goat anti-rabbit (Promega, W4011), goat anti-rat (Advansta, R-05075-500) and goat anti-mouse HRP-conjugated (Promega, W4021) secondary antibodies, proteins were visualized using chemiluminescent peroxidase substrat detection reagents (Sigma, CPS3100-1KT), and acquired by the ChemiDoc XRS+ System (Bio-Rad). Densitometric analyses of immunoblots were performed using the ImageJ software.
Fluorescence microscopy
GFP-LC3 HeLa cells or HeLa-Difluo™ hLC3 reporter cells were seeded onto Nunc Lab-Tek Chamber Slide system 8 wells (Sigma, C7182). The day of the experiment, cells were incubated with the indicated treatment or left untreated for the indicated times. LysoTracker Red (Invitrogen, L7528) staining was achieved by adding the LysoTracker probe at a final concentration of 50 nM directly in the medium of cell culture for 1 h. Then the cells were washed and fixed with 4% paraformaldehyde for 15 min on ice. DNA was visualized using Fluoroshield with 4,6-diamidino-2-phenylindole (DAPI; Sigma, F6057). Images were acquired using a Zeiss LSM 710 confocal microscope and subsequently processed using the ImageJ software package.
Transmission electron microscopy
Negative staining TEM for OMV was performed according to standard procedures. Briefly, 5 µL of OMV samples were added to formvar carbon coated copper mesh grids (EMS, EMS400-Cu) and stained with 1% uranyl acetate for 1 min and imaged with Jeol JEM-1400 transmission electron microscope. For HeLa cell ultrastructure visualization, cells were cultured on 6-well plates at a density of 1.5 × 106 cells/well one day prior to the experiment. After OMV treatment, cells were fixed with final concentrations of 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1 M Sorensen buffer pH 7,2 (EMS, EMS11600-05) volume:volume in the culture medium for 15 min at room temperature. After this primary fixation, the cells were secondarily fixed with 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1 M Sorensen buffer (pH 7.2) for 1.15 h at room temperature. Then cells were rinsed three times in Sorensen buffer for 10 min each time and let in Sorensen buffer with 1% paraformaldehyde at 4°C until they were then scraped off, pelleted, concentrated in agarose, and treated for 1 h with 2% aqueous uranyl acetate. The samples were then dehydrated in a graded ethanol series and embedded in Epon (EMS, EMbed-812). After 48 h of polymerization at 60°C, ultrathin sections (80-nm thick) were mounted on 200-mesh formvar-carbon-coated copper grids (EMS, EMS200-Cu). Finally, sections were stained with uranyless 3% Reynolds lead citrate (Delta Microscopies, 11,300). Grids were examined with a TEM (Jeol JEM-1400, JEOL Inc, Peabody, MA, USA) at 80 kV. Images were acquired using a digital camera (Gatan Orius, Gatan Inc, Pleasanton, CA, USA) and subsequently processed using the ImageJ software package.
Measurement of LDH release
Cell death was quantified by measuring LDH release, using the CyQUANTTM LDH Cytotoxicity Assay (Invitrogen, C20300). To normalize for spontaneous cell lysis, the percentage of cell death was calculated as follows: ([LDH sample] – [LDH non-treated control])/([LDH positive control] – [LDH non-treated control]) X 100.
Measurement of IL1B release
Quantification of IL1B secretion in cell supernatant was measured by ELISA (IL1 beta Mouse Uncoated ELISA Kit; Invitrogen, 88–7013-88), according to the manufacturer’s instructions.
Imaging flow cytometry
GFP-LC3 HeLa cells or HeLa-Difluo™ hLC3 reporter cells were incubated with the indicated treatment or left untreated for the indicated times. Then the cells were washed, trypsinized, fixed with 4% paraformaldehyde for 15 min on ice. Image of each cell was acquired while they were run on the ImageStreamX Mark II (Amnis-EMD Millipore) at 5 × 107 cells per mL in PBS. In each experiment 5,000 (GFP+) events per sample were acquired (ensuring a sufficient number of events remaining for statistically robust analysis) at 60X magnification. Single stain samples were also collected using the same settings and used as compensation controls to generate compensation matrix.
Image analysis was completed using image-based algorithms in the ImageStream Data Exploration and Analysis Software (IDEAS_6.2.64.0, EMD Millipore). The number of GFP-LC3 puncta per cell was assessed with the feature “Spot Counting”. A mask based on GFP-LC3 staining was employed (SpotCount (M02, LC3, Bright, 16, 2, 1)) to visualize the GFP-LC3 puncta. A feature in the IDEAS software called Bright Detail Intensity R7 (BDI) was used to assess the intensity of bright spots in the cell image that have radii smaller than a threshold size (7 pixels in this study), while neglecting background staining. The ratio of BDI RFP:BDI GFP brightness was thus assessed. The same mask and feature were automatically applied for each condition.
Statistical analyses
Statistical analyses were performed on at least three independent experiments with the unpaired t-test using the Prism software package (GraphPad Software). Differences were considered significant for the following P-values: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Supplementary Material
Acknowledgments
We gratefully acknowledge the imaging platform of INFINITy institute. This work also benefited from the assistance of Stephanie Balor and Vanessa Soldan from the Multiscale Electron Imaging platform (METi) of the Centre de Biologie Intégrative (CBI). We also thank Emmanuel Ravet (Invivogen, Toulouse) for providing us HeLa-Difluo™ hLC3 reporter cells, Dr Patrice Codogno for providing the HeLa-GFP-LC3 cell line, Marie-Charline Blatche (LAAS institute, Toulouse) for assistance in DLS analysis and Miriam Pinilla (IPBS institute, Toulouse) for her valuable help in the preparation of BMDM cells and the LPS transfection experiments. Finally, we are indebted to Dr MS, Dr J-PM and Dr J-PN for their insightful comments on the manuscript.
Funding Statement
This project was funded by ERC StG grant (INFLAME 804249) to EM and RP. This project was supported by INCa-Cancéropôle GSO
Disclosure statement
The authors declare that they have no conflict of interest.
Supplemental material
Supplemental data for this article can be accessed here
References
- [1].Sarowska J, Futoma-Koloch B, Jama-Kmiecik A, et al. Virulence factors, prevalence and potential transmission of extraintestinal pathogenic Escherichia coli isolated from different sources: recent reports. Gut Pathog. 2019;11:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Peigne C, Bidet P, Mahjoub-Messai F, et al. The plasmid of Escherichia coli strain S88 (O45:K1:H7) that causes neonatal meningitis is closely related to avian pathogenic E. coli plasmids and is associated with high-level bacteremia in a neonatal rat meningitis model. Infect Immun. 2009;77:2272–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Johnson TJ, Wannemuehler Y, Doetkott C, et al. Identification of minimal predictors of avian pathogenic Escherichia coli virulence for use as a rapid diagnostic tool. J Clin Microbiol. 2008;46:3987–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kim YB, Yoon MY, Ha JS, et al. Molecular characterization of avian pathogenic Escherichia coli from broiler chickens with colibacillosis. Poult Sci. 2020;99:1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].de Oliveira AL, Rocha DA, Finkler F, et al. Prevalence of ColV plasmid-linked genes and in vivo pathogenicity of avian strains of Escherichia coli. Foodborne Pathog Dis. 2015;12:679–685. [DOI] [PubMed] [Google Scholar]
- [6].Soysal N, Mariani-Kurkdjian P, Smail Y, et al. Enterohemorrhagic Escherichia coli hybrid pathotype O80:H2 as a new therapeutic challenge. Emerg Infect Dis. 2016;22:1604–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Murase K, Martin P, Porcheron G, et al. HlyF produced by extraintestinal pathogenic Escherichia coli is a virulence factor that regulates outer membrane vesicle biogenesis. J Infect Dis. 2016;213:856–865. [DOI] [PubMed] [Google Scholar]
- [8].Jan AT. Outer membrane vesicles (OMVs) of gram-negative bacteria: a perspective update. Front Microbiol. 2017. [cited 2021 Jan 15];8. [Internet]. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5465292/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schwechheimer C, Kuehn MJ. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat Rev Microbiol. 2015;13:605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kuehn MJ, Kesty NC. Bacterial outer membrane vesicles and the host–pathogen interaction. Genes Dev. 2005;19:2645–2655. [DOI] [PubMed] [Google Scholar]
- [11].Kulp A, Kuehn MJ. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol. 2010;64:163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Biasizzo M, Kopitar-Jerala N. Interplay between NLRP3 inflammasome and autophagy. Front Immunol. 2020. [cited 2021 Jan 14];11. [Internet]. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7583715/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Feng Y, Liu B, Zheng X, et al. The protective role of autophagy in sepsis. Microb Pathog. 2019;131:106–111. [DOI] [PubMed] [Google Scholar]
- [14].Ho J, Yu J, Wong SH, et al. Autophagy in sepsis: degradation into exhaustion? Autophagy. 2016;12:1073–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Qiu P, Liu Y, Zhang J. Review: the role and mechanisms of macrophage autophagy in sepsis. Inflammation. 2019;42:6–19. [DOI] [PubMed] [Google Scholar]
- [16].Pugsley H. Quantifying autophagy: measuring LC3 puncta and autolysosome formation in cells using multispectral imaging flow cytometry. Methods San Diego Calif. 2017;112:147–156. [DOI] [PubMed] [Google Scholar]
- [17].Simone LC, Caplan S, Naslavsky N. Role of phosphatidylinositol 4,5-bisphosphate in regulating ehd2 plasma membrane localization. PLOS ONE. 2013;8:e74519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Soldati T, Schliwa M. Powering membrane traffic in endocytosis and recycling. Nat Rev Mol Cell Biol. 2006;7:897–908. [DOI] [PubMed] [Google Scholar]
- [19].Huang J-H, Liu C-Y, S-y W, et al. NLRX1 facilitates histoplasma capsulatum-induced LC3-associated phagocytosis for cytokine production in macrophages. Front Immunol. 2018. [cited 2022 Jan 30];9. [Internet]. Available from: https://www.frontiersin.org/article/10.3389/fimmu.2018.02761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Muñoz-Braceras S, Escalante R. Analysis of relevant parameters for autophagic flux using hela cells expressing EGFP-LC3. In: Matthiesen R,editor. Proteostasis methods protoc [Internet]. New York NY:Springer.2016[cited 2020 Mar 30]; 313–329.Available from: 10.1007/978-1-4939-3756-1_20. [DOI] [PubMed] [Google Scholar]
- [22].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. [DOI] [PubMed] [Google Scholar]
- [23].Loos B, du Toit A, Hofmeyr J-HS. Defining and measuring autophagosome flux—concept and reality. Autophagy. 2014;10:2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Harris J, Lang T, Thomas JPW, et al. Autophagy and inflammasomes. Mol Immunol. 2017;86:10–15. [DOI] [PubMed] [Google Scholar]
- [26].Krakauer T. Inflammasomes, autophagy, and cell death: the trinity of innate host defense against intracellular bacteria. Mediators Inflamm. 2019;2019:2471215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Netea-Maier RT, Plantinga TS, van de Veerdonk FL, et al. Modulation of inflammation by autophagy: consequences for human disease. Autophagy. 2015;12:245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Qian S, Fan J, Billiar TR, et al. Inflammasome and autophagy regulation: a two-way street. Mol Med. 2017;23:188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL1beta production. Nature. 2008;456:264–268. [DOI] [PubMed] [Google Scholar]
- [30].Shi C-S, Shenderov K, Huang -N-N, et al. Activation of autophagy by inflammatory signals limits IL1B production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Santos JC, Dick MS, Lagrange B, et al. LPS targets host guanylate‐binding proteins to the bacterial outer membrane for non‐canonical inflammasome activation. EMBO J. 2018;37:e98089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Losier TT, Akuma M, McKee-Muir OC, et al. AMPK promotes xenophagy though priming of autophagic kinases upon detection of bacterial outer membrane vesicles. Cell Rep. 2019;26:2150–2165.e5. [DOI] [PubMed] [Google Scholar]
- [33].Zhuge X, Sun Y, Xue F, et al. A Novel PhoP/PhoQ regulation pathway modulates the survival of extraintestinal pathogenic Escherichia coli in macrophages. Front Immunol. 2018;9:788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].McPhee JB, Small CL, Reid-Yu SA, et al. Host Defense Peptide Resistance Contributes to Colonization and Maximal Intestinal Pathology by Crohn’s Disease-Associated Adherent-Invasive Escherichia coli. Infect Immun. 2014;82:3383–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kaparakis-Liaskos M, Ferrero RL. Immune modulation by bacterial outer membrane vesicles. Nat Rev Immunol. 2015;15:375–387. [DOI] [PubMed] [Google Scholar]
- [36].Vanaja SK, Russo AJ, Behl B, et al. Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell. 2016;165:1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, von Pein JB, Broz P, Sweet MJ, Schroder K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol. 2018;3:26. [DOI] [PubMed] [Google Scholar]
- [38].Schoder K, Kanneganti T-D, Shao F, et al. Mechanisms and consequences of inflammasome activation. J Mol Biol. 2018;430:131–132. [DOI] [PubMed] [Google Scholar]
- [39].Cecconi M, Evans L, Levy M, et al. Sepsis and septic shock. Lancet Lond Engl. 2018;392:75–87. [DOI] [PubMed] [Google Scholar]
- [40].van der Poll T, van de Veerdonk FL, Scicluna BP, et al. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. 2017;17:407–420. [DOI] [PubMed] [Google Scholar]
- [41].Eren E, Planès R, Bagayoko S, Bordignon PJ, Chaoui K, Hessel A, Santoni K, Pinilla M, Lagrange B, Burlet-Schiltz O, Howard JC, Henry T, Yamamoto M, Meunier E. Irgm2 and Gate-16 cooperatively dampen Gram-negative bacteria-induced caspase-11 response. EMBO Rep. 2020;21:11:e50829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell. 2014;54:224–233. [DOI] [PubMed] [Google Scholar]
- [43].Crişan TO, Plantinga TS, van de Veerdonk FL, et al. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PloS One. 2011;6:e18666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bielaszewska M, Rüter C, Bauwens A, et al. Host cell interactions of outer membrane vesicle-associated virulence factors of enterohemorrhagic Escherichia coli O157: intracellular delivery, trafficking and mechanisms of cell injury. PLOS Pathog. 2017;13(2):e1006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Johnson JR, Oswald E, O’Bryan TT, et al. Phylogenetic distribution of virulence-associated genes among Escherichia coli isolates associated with neonatal bacterial meningitis in the Netherlands. J Infect Dis. 2002;185:774–784. [DOI] [PubMed] [Google Scholar]
- [46].Lee CH, Tsai CM. Quantification of bacterial lipopolysaccharides by the purpald assay: measuring formaldehyde generated from 2-keto-3-deoxyoctonate and heptose at the inner core by periodate oxidation. Anal Biochem. 1999;267:161–168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.