

Summary
Cotton fibre is a unicellular seed trichome, and lint fibre initials per seed as a factor determines fibre yield. However, the mechanisms controlling fibre initiation from ovule epidermis are not understood well enough. Here, with single‐cell RNA sequencing (scRNA‐seq), a total of 14 535 cells were identified from cotton ovule outer integument of Xu142_LF line at four developmental stages (1.5, 1, 0.5 days before anthesis and the day of anthesis). Three major cell types, fibre, non‐fibre epidermis and outer pigment layer were identified and then verified by RNA in situ hybridization. A comparative analysis on scRNA‐seq data between Xu142 and its fibreless mutant Xu142 fl further confirmed fibre cluster definition. The developmental trajectory of fibre cell was reconstructed, and fibre cell was identified differentiated at 1 day before anthesis. Gene regulatory networks at four stages revealed the spatiotemporal pattern of core transcription factors, and MYB25‐like and HOX3 were demonstrated played key roles as commanders in fibre differentiation and tip‐biased diffuse growth respectively. A model for early development of a single fibre cell was proposed here, which sheds light on further deciphering mechanism of plant trichome and the improvement of cotton fibre yield.
Keywords: cotton fibre initiation, single cell transcriptomic atlas, Gossypium hirsutum, regulatory network, cell fate determination
With scRNA‐seq, cotton fibre cell was identified differentiated at −1 DPA. It further refines the spatiotemporal patterns of two command genes, MYB25‐like and HOX3, who determine fibre differentiation and tip‐biased diffuse growth respectively.

Introduction
Cotton is an important cash crop worldwide, and supplies the largest proportion of natural fibre to textile industry. Cotton fibres are unicellular, and initiate from ovule epidermis, making it a good model for studying mechanisms of cell fate determination. There are two types of cotton fibre according to mature fibre length: lint and fuzz. The initiation of lint (long) fibre proceeds from the day post‐anthesis (0 DPA) to 3 DPA, with the initiation of fuzz (short) fibre typically commencing afterwards. The number of lint fibre initials per seed as a factor determines fibre yield, while lint initiation is typically from 25% of all epidermal cells (Stewart, 1975). Therefore, exploring the mechanisms and analysing key factors and networks that regulate lint fibre cell fate, can provide a theoretical basis for the genetic improvement of fibre yield, so as to help improve the economic benefits of cotton planting.
Numerous studies have investigated the mechanisms of fibre development by means of expression profiling. Since early 21st century, 14 highly expressed cDNAs were identified in cotton fibre using cDNA arrays (Li et al., 2002), after that several expression profiles have been performed on various stages of fibre development (Gou et al., 2007; Shi et al., 2006), between different cotton species (Tu et al., 2007), during domestication (Hovav et al., 2008; Rapp et al., 2010), between normal cotton and fibre‐related mutants (Wu et al., 2006). A few transcription factor (TF) genes, such as R2R3‐type MYB TFs GhMYB109 (Pu et al., 2008), GhMYB25 (Machado et al., 2009), GhMYB25‐like (Walford et al., 2011; Wan et al., 2016), and HD‐ZIP family TFs GhHD‐1 (Walford et al., 2012), GhHOX3 (Shan et al., 2014) have been verified positively regulating lint fibre initiation. A model has been proposed in which no lint fibre will initiate if the combined expression levels of MYB25‐like_At and MYB25‐like_Dt are below a critical threshold level at 0 DPA (Zhu et al., 2018). In addition, suppression of sucrose synthase activity by at least 70% in the ovule epidermis (Ruan et al., 2003), or of a vacuolar invertase gene GhVIN1 (Wang et al., 2014), led to a fibreless phenotype. Furthermore, naked seeds were produced when ovules were cultured with adding no indoleacetic acid (IAA) (Zeng et al., 2019) or excess IAA transport inhibitor (Zhang et al., 2011; Zhang et al., 2017) or with high concentration of zeatin (ZT), a kind of cytokinin (Zeng et al., 2019). Recently, some review papers have summarized that fibre initiation was affected by complex cross‐talk among MYB MIXTA‐like TFs, sugar signals and plant hormones (Huang et al., 2021; Tian and Zhang, 2021; Wang et al., 2021a). Despite these studies, when did the members in the complex start work, whether they take action only in fibre cells or also in adjacent cells during the continuous fibre initiation process, were partly ambiguous and needs more details.
Now, single‐cell RNA sequencing (scRNA‐seq) has been developed and brings unprecedented opportunities to the field of plant research (Denyer and Timmermans, 2022; Mo and Jiao, 2022; Ryu et al., 2021; Seyfferth et al., 2021; Shaw et al., 2021). The first effective high‐throughput scRNA‐seq in plants exploited single‐cell transcriptome sequencing of Arabidopsis root tissue protoplasts (Ryu et al., 2019). Subsequently, a series of developmental processes in Arabidopsis had been explored at single cell resolution, such as the development of root tips (Denyer et al., 2019; Jean‐Baptiste et al., 2019; Shahan et al., 2022; Wendrich et al., 2020; Zhang et al., 2019), lateral root (Gala et al., 2021), vegetative shoot apex (Zhang et al., 2021b), stomatal cell lineage (Liu et al., 2020), developing leaf (Kim et al., 2021; Liu et al., 2022a; Lopez‐Anido et al., 2021; Tenorio Berrio et al., 2022) and vein pattern in the cotyledons (Liu et al., 2022b). At the same time, scRNA‐seq has also been widely used in other plants, including rice (Liu et al., 2021b; Wang et al., 2021b; Zhang et al., 2021a; Zong et al., 2022), corn (Li et al., 2022; Ortiz‐Ramirez et al., 2021; Satterlee et al., 2020; Sun et al., 2022; Xu et al., 2021), peanut (Liu et al., 2021a), tea plant (Wang et al., 2022), tomato (Omary et al., 2022) and poplar (Chen et al., 2021; Li et al., 2021; Xie et al., 2022). These studies provide new insights into heterogeneity of gene expression between different cell types, and molecular trajectory of cell differentiation during development.
To further decipher the detailed gene regulatory network in fibre initiation, we performed scRNA‐seq on cotton ovules. The developmental trajectory starting from early differentiated fibre cell was reconstructed. All the results can interactively be mined on the web, which is freely available at http://cotton.hzau.edu.cn/CLC/. The single cell resolution transcriptomes provide a valuable resource, and give us a deep understanding on the elaborate cotton fibre initiation process.
Results
Fibre cells begin to protrude at 0.5 days before anthesis
The phenotype of cotton Xu142_LF line (seed index 10–12 g, lint index 6–7 g and lint percentage 35%–38%) has been described in our previous work (Hu et al., 2018). A more detailed morphological observations on Xu142_LF ovules were performed at −1.5, −1, −0.5 and 0 DPA respectively (Figure 1). By SEM, no fibre cell protrusion detected on ovule epidermis at −1.5 DPA (Figure 1a) and −1 DPA (Figure 1b), while at −0.5 DPA can be detected (Figure 1c), and protrusions number increased at 0 DPA (Figure 1d,e).
Figure 1.

The phenotype of ovule epidermis of Xu142_LF line during fibre initiation. (a–d) Xu142_LF ovule epidermis observed by scanning electron microscopy (SEM) at −1.5, −1, −0.5 and 0 DPA respectively. (e) Statistics of fibre initials number on ovule epidermis per unit area (100 × 100 μm2) from (a–d). (f) Statistics of fibre initials number on ovule epidermis per unit area (100 × 100 μm2) from Figure S1. The blue solid triangles and hollow triangles marked stages in (e) and (f) mean the same developmental stage respectively. (g–j) Ovule epidermal cells observed by transmission electron microscope (TEM) at −1.5, −1, −0.5 and 0 DPA respectively. Red arrows in (i) and (j) point to fibre cells. Bars: 100 μm (a‐d); 10 μm (g–j).
To further investigate the dynamic process of fibre initiation, samples were taken every 4 h (h) before and after fibre cell protrusion (from −1 to 0 DPA, Figure S1). No epidermal cell protruded at 08 : 00, 12 : 00 and 16 : 00 of −1 DPA. At 20 : 00, fibre cells began to protrude. At 24 : 00, the number of fibre initials increased. At 4 : 00 of 0 DPA, fibre initials grew larger, similar in size to those at 8 : 00 of 0 DPA (Figure S1). From 20 : 00 (−0.5 DPA), the number of fibre initials shows an increasing trend (Figure 1f).
Ovule epidermis at those four stages were also observed by TEM. Consistent with SEM observations, epidermal cells did not protrude at −1.5 and −1 DPA (Figure 1g,h), but began to protrude at −0.5 DPA (Figure 1i) and continued to expand at 0 DPA (Figure 1j). Considering lint fibre cell start protruding at −0.5 DPA, their fate is likely to be determined as early as −1 DPA, or −1.5 DPA, or possibly earlier.
Fibre cell cluster is identified from cotton ovule outer integument by scRNA‐seq
To explore the molecular mechanism determining fibre cell fate, we performed scRNA‐seq on Xu142_LF ovules. Ovule samples collected at −1.5, −1, −0.5 and 0 DPA were used for protoplast isolating (Figure S2a,b). About 20 000 protoplasts were initially loaded onto the 10× Genomics platform. After separation, RNA from individual protoplasts were extracted for library construction followed by high‐throughput sequencing (Figure S2c). As shown in longitudinal section of ovules after enzymolysis, only cells from ovule outer integument were released (Figure S2d–g).
In Xu142_LF 0 DPA sample (LF_0d), a total of 3679 cells with 50 753 genes were detected (Table S1). After a strict gene/cell filtering process (Appendix S1), LF_0d sample obtained 35 169 gene transcripts with high reliability across 1703 cells. This filtration was performed on other samples one by one, then high‐quality gene‐cell matrices were obtained. Overall, 738–2045 filtered cells per sample were obtained (Table S1).
To examine the robustness of the scRNA‐seq results, LF_0d scRNA‐seq data were compared with 0 DPA ovule outer integument bulk RNA‐seq data (Hu et al., 2018). The correlation coefficient (R) was 0.63 with P < 2.2e‐16, showing a very significant correlation between them (Figure S3a). There were 3974 protoplasting‐induced differentially expressed genes (DEGs) identified, with 2233 DEGs up‐regulated and 1741 down‐regulated (Table S2). GO analysis suggested that the up‐regulated genes were involved in ‘response to stress and stimulus’ and ‘regulation of cell death’, among others (Figure S3b); the down‐regulated genes were mainly involved in ‘primary metabolic process’ and ‘biosynthetic process’ (Figure S3c). Next, UMAP and t‐SNE algorithm were used to visualize and explore LF_0d dataset after linear dimensional reduction. Unsupervised analyses grouped 1703 cells into nine clusters (Figure 2a,b). Similarly, nine clusters were also observed after 3974 DEGs in response to protoplasting were removed (Figure S3d), and clustered cells were almost kept in the same cell types as before (Figure S3e). This suggested that cell wall enzymolysis had only a minor effect on cell clustering, the same as reported in rice root (Liu et al., 2021b).
Figure 2.
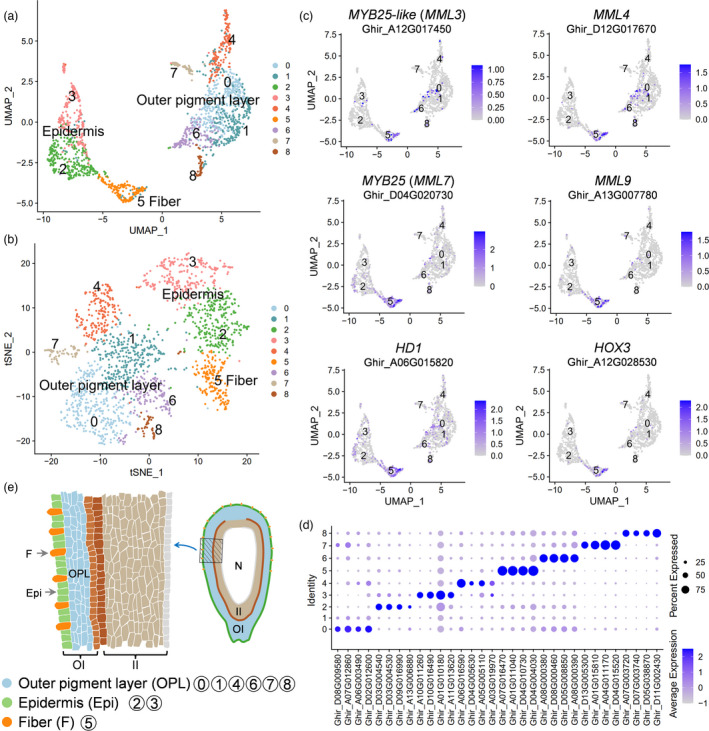
Cluster annotation of single‐cell transcriptomes from cotton ovule outer integument. (a) UMAP visualization of putative clusters from 1703 cells in cotton ovule outer integument of LF_0d sample. Each dot denotes a single cell. Colours denote corresponding cell clusters. Resolution was 0.8. (b) t‐SNE projection plot showing major clusters of the 1703 individual cell transcriptomes of LF_0d sample. (c) UMAP projection plots showing transcript accumulation for known fibre markers in individual cells. Colour intensity indicates the relative transcript level for the indicated gene in each cell. (d) Expression pattern of top four genes enriched in each cluster of LF_0d. Dot diameter, proportion of cluster cells expressing a given gene; colour, average expression across cells in that cluster. (e) Schematic diagram of longitudinal section of 0 DPA cotton ovule showing spatial distribution of cell clusters in ovule outer integument. The left part is a magnified view of the shaded part on the right. F, fibre; Epi, epidermis; OPL, outer pigment layer; OI, outer integument layer; II, inner integument layer; N, nucellus.
For cell cluster definition, accumulation of reported fibre gene transcripts in single‐cell populations were analysed (Figure 2c, Figure S4a). The fibre genes included MYB25‐like (Walford et al., 2011; Wan et al., 2016), MYB25 (Machado et al., 2009), MML4 (Wu et al., 2018), MML9 (Bedon et al., 2014), HD1 (Walford et al., 2012) and HOX3 (Shan et al., 2014). All these genes tended to be highly or preferentially expressed in cluster 5 (Figure 2c, Figure S4b), indicated that cluster 5 may be fibre cell. To further enable cell type assignment to particular clusters, a series of enriched genes for each cluster were identified (Figure 2d, Figure S4c, Table S3). With RNA in situ hybridization of the representative genes, all cell clusters from cotton ovule outer integument can be defined into three major types: fibre cell, non‐fibre epidermis and outer pigment layer (Figures 2e and 3). For example, DUF (Ghir_D07G016770, gene function unknown) was preferentially expressed in fibre cells, Erg6 (Ghir_A04G010380, a methyltransferase encoding gene) in non‐fibre epidermis and HbdA (Ghir_D01G005520, 3‐hydroxyacyl‐CoA dehydrogenase) in outer pigment layer (Figure 3, Figure S5).
Figure 3.

RNA in situ hybridization of cell‐type representative marker genes. (a) UMAP projection plots showing transcript accumulation for DUF (Ghir_D07G016770, function unknown), a novel fibre marker gene from cluster 5. (b) RNA in situ hybridization of DUF with the sense probe as a negative control. (c) Expression of non‐fibre epidermis novel marker gene Erg6 (Ghir_A04G010380, a methyltransferase encoding gene) in cluster 2 and 3. (d) RNA in situ hybridization of epidermis marker (Erg6) with the sense probe as a negative control. (e) UMAP projection plots showing transcript accumulation for outer pigment layer novel marker gene HbdA (Ghir_D01G005520, 3‐hydroxyacyl‐CoA dehydrogenase family protein) in cluster 0, 1, 4, 6, 7 and 8. (f) RNA in situ hybridization of HbdA with the sense probe as a negative control. The hybridization signals of these marker genes in whole cotton ovules were shown in Figure S5. Sections (10 μm) from Xu142_LF 0 DPA ovules were used for in situ hybridization. OI, outer integument; II, inner integument. Red arrows indicate fibre cells, and red arrowheads indicate non‐fibre epidermal cells. Yellow arrows indicate outer pigment layer. Scale bars, 50 μm.
Fibre cell definition is further proved by scRNA‐seq of Xu142 fl fibreless mutant
Single‐cell transcriptomes were generated from 0 DPA ovule protoplasts of wild type cultivar Xu142 and its fibreless mutant Xu142 fl. Clustering these two samples together generated five clusters (Figure 4a, Table S3), which were assigned to three major cell types using in situ hybridization‐verified genes: fibre (cluster 4), non‐fibre epidermis (cluster 0, 2 and 3), outer pigment layer (cluster 1, Figure 4a, Figure S6a). When comparing the clustering results between Xu142 and Xu142 fl, the fibre cell cluster (cluster 4) was found to exist only in Xu142, not in Xu142 fl (Figure 4b, Table S4). The expression distribution of three fibre marker genes (MYB25, MML9 and HOX3) also showed expressing in Xu142, not in Xu142 fl (Figure S6b). These again proved that the fibre cell definition was reliable.
Figure 4.

Clarifying of fibre cell cluster and its enriched pathways. (a) UMAP visualization of putative clusters in cotton ovule outer integument of Xu142 versus Xu142 fl sample. Each dot denotes a single cell. Colours denote corresponding cell clusters. Resolution was 0.2. (b) UMAP visualization of putative clusters which shown separately according to Xu142 and Xu142 fl. (c) Schematic diagram showing the main pathways and regulatory factors active in 0 DPA fibre cells. The red protrusions on the ovule epidermis represent protruded fibre cells.
‘Ribosome’ pathway is enriched in 0 DPA fibre cells
To compare the fibre cluster enriched genes between LF_0d and Xu142_0d, a clustering analysis on Xu142_0d sample was performed. The 738 high‐quality cells from Xu142_0d clustered into 6 clusters. Cluster 2 was identified as fibre cell with expression pattern analysis of fibre markers (MYB25 and MML9, Figure S6c,d), and 984 genes were identified enriched in fibres (Table S3). When comparing these 984 genes with the 517 genes enriched in LF_0d fibres (Table S3), it was found that 474 genes overlapped, which means a similarity of gene expression between them (Figure S6e).
To explore the pathways that are active in fibre cells, KEGG enrichment analysis was performed on the 517 genes enriched in LF_0d fibres. These genes were involved in 38 different signalling pathways (Table S5). Among them, 312 genes were involved in the most significant pathway ‘Ribosome’ (Table S5), which indicated that a major activity in 0 DPA fibres was ‘peptide biosynthetic process’. It was consistent with the characteristics of expanding fibre initial cells, that is, more abundant transcripts involved in protein synthesis to meet the high demand for new cell wall and membrane components (Wu et al., 2007). The remaining genes were enriched in several pathways such as ‘Fatty acid’, ‘Flavonoid’, ‘Oxidative phosphorylation’, ‘Sucrose’ and ‘Phagosome’ (Figure 4c, Table S5). Besides, some TFs were identified from the 517 genes, including MML9, MYB25, HD1, PDF2, HOX3, HDA3 and VFP5 (Figure 4c). These TFs should play positive roles in 0 DPA fibre development, as some of them have been experimentally verified (Machado et al., 2009; Shan et al., 2014; Walford et al., 2012).
Fibre cell was differentiated at −1 DPA
In order to clarify the dynamic changes of gene expression accompanying fibre cell initiation, the single‐cell transcriptomes in Xu142_LF across four stages (−1.5, −1, −0.5 and 0 DPA) were combined. After unsupervised clustering, totally 5137 cells were clustered into six clusters (Figure 5a), and genes enriched in each cluster were identified (Table S3). Based on the well‐defined cell types in LF_0d and cell barcode mapping (Table S6), the combined sample can be defined as fibre (cluster 5), epidermis (cluster 1, 3) and outer pigment layer (cluster 0, 2 and 4; Figure 5a). The expression pattern of fibre marker genes (MYB25, MML9 and HD1) further verified that cluster 5 was fibre cell cluster (Figure S7a).
Figure 5.

Developmental trajectory of fibre cells. (a) UMAP visualization of putative clusters of the combined sample. The scRNA‐seq data of −1.5, −1, −0.5 and 0 DPA samples were combined for clustering. Each dot denotes a single cell. Colours denote corresponding cell clusters. Resolution was 0.2. (b) Cell number of each type identified from the combined sample, which shown at −1.5, −1, −0.5 and 0 DPA respectively. (c) Re‐clustering of all fibre cells identified in the combined sample. (d) A pseudotime trajectory showing fibre cells development. The developmental branch locations of 5 sub‐clusters. Different colours represent the cells from each sub‐cluster. (e) Expression patterns of fibre marker genes (MYB25‐like, MML4, MYB25 and HD1). The colours represent expression levels of these genes in individual cells. (f) Heat map displays the 167 branch‐dependent genes with qval <0.001. Each row represents one gene. These genes were clustered into 5 modules with distinct expression patterns. Different colours represent the gene expression level. The representative genes of each module are shown in the middle panel. The gene ontology (GO) terms for each module are shown on the right panel. (g) Representative marker genes (ERF4, CINV2, ACS6 and BZIP53) expressed in branch 1 (upper panel), and expression of representative marker genes (RPL24, RPS5B, LTP1 and E6) in branch 2 (lower panel). The colour bar indicates relative expression levels. (h) RNA in situ hybridization of E6 gene (Ghir_D05G016260) in LF_0d ovules with the sense probe as a negative control. OI, outer integument; II, inner integument. Red arrows indicate fibre cells, and red arrowheads indicate non‐fibre epidermal cells. Scale bars, 50 μm.
When displaying the clustering result separately at four stages (Figure S7b–e), no fibre cell was identified at −1.5 DPA (Figure S7b), while the next three stages contained fibre cluster (Figure S7c–e). The expression pattern of MYB25 and MML9 also showed that they did not express at −1.5 DPA, but expressed at next three stages (Figure S7f,g). According to previous phenotypic observation, −0.5 DPA was the time point showing visible fibre initials (Figure 1i). Here, the transcriptome data suggested that fibre cells were differentiated at −1 DPA (Figure S7c), although they were morphologically undistinguishable to its neighbouring cells at this stage (Figure 1h). Since then, the number of fibre cell (cluster 5) increases stepwise along development (Figure 5b).
Next, the developmental trajectory of fibre cells was explored. In total, 226 fibre cells (16 cells from LF_‐1d, 30 cells from LF_‐0.5d and 180 cells from LF_0d, Figure 5b) were selected for re‐clustering. There were five sub‐clusters identified (Figure 5c). When displaying the re‐clustering result separately (Figure S7h), it was found that −1 DPA fibre cells were mainly located to sub‐cluster 5.2. Then, pseudotime analysis on these fibre cells built a developmental trajectory (Figure 5d). The starting of the trajectory was specified at the left branch, as sub‐cluster 5.2 cells (early fibre cells) were located on this branch. There was a node in the trajectory, with branching into two directions (Figure 5d). The MYB‐MIXTA‐like TFs, such as MYB25‐like, MML4 and MYB25, were prominently highly expressed at the beginning branch (Figure 5e), in agreement with their key roles in fibre cell differentiation (Machado et al., 2009; Walford et al., 2011; Wu et al., 2018). The homeodomain leucine zipper transcription factor, HD1 (Walford et al., 2012), was expressed almost at all the branches (Figure 5e), suggesting that HD1 gene has a broader temporal pattern during fibre initiation, distinct from MYB‐MIXTA‐like TFs.
It is known that once early fibre cell differentiated, they will undergo a process of diffuse growth then transformed into tip‐biased diffuse growth (Qin and Zhu, 2011; Yu et al., 2019). To further define the cell types on branch 1 and branch 2, a total of 167 genes were firstly identified as most significantly related to branching (qval < 0.001, Table S7). A heat map containing these 167 genes was produced and they were sorted into five modules (Figure 5f). Among them, 59 genes in gene module 1 (M1) are predominantly expressed in branch 1 cells. GO results show that they are mainly enriched in ‘regulation of ethylene‐activated signalling pathway’, ‘sucrose alpha‐glucosidase activity’, ‘cuticle hydrocarbon biosynthetic process’ and so on (Figure 5f, Table S8). These GO terms were reported mainly enriched in fibre elongation (Shi et al., 2006). In details, multiple genes involved in ethylene signalling, such as ethylene responsive element binding factor (ERF4, ERF9 and ERF11), and 1‐aminocyclopropane‐1‐carboxylic acid (ACC) synthase gene (ACS6) (Shi et al., 2006) were all prominently expressed in branch 1 cells (Figure 5g, Table S7). There are 79 genes in M5, which are predominantly expressed in branch 2 cells. They are mainly enriched in ‘ribosome’, ‘translation’, ‘lipid transport’ and ‘ATP transmembrane transporter activity’ (Figure 5f, Table S8), these GO terms were reported highly enriched in diffuse growing fibre cells (Wu et al., 2007). The genes encoding large and small ribosomal subunit protein (RPL24, RPS5B, RPL3A and RPS6B) and lipid transfer protein (LTP1) (Wu et al., 2006) were highly enriched in branch 2 (Figure 5g, Table S7). Moreover, an E6 protein encoding gene (Ghir_D05G016260), which plays a role in diffuse growing fibre cell (Ji et al., 2003), was identified and verified by in situ hybridization (Figure 5g,h).
As described above, fibre initiation can be subdivided into several processes. Precursor fibre cell have not differentiated at −1.5 DPA (Figure S7b), and then differentiated into fibre cell at −1 DPA (Figure S7c and Figure 5b) and start protruding by diffuse growth at −0.5 DPA (Figure 1i), then transformed into tip‐biased diffuse growth from 0 DPA (Figure 1j).
The highly interconnected gene regulatory networks coordinate fibre cell initiation
The results above have shown that these four stages (−1.5, −1, −0.5 and 0 DPA) represented distinct processes of fibre initiation. To explore gene regulatory networks in each process, clustering analysis were performed on four samples individually (Figure S8a). By analysing transcript accumulation of fibre marker gene MML4 and the in situ hybridization‐verified genes (DUF, Erg6 and HbdA, Figure S8b), the clusters in each sample can be defined into fibre, epidermis and outer pigment layer (Figure S8a). Then, genes enriched in each cluster were identified (Table S9), and WGCNA was performed on four samples respectively. Several distinct gene modules (labelled by different colours) were identified as shown in the dendrogram (Figure S9, Table S10). The modules containing genes enriched in fibre cells (−1, −0.5 and 0 DPA) or cells that may differentiate into fibre (−1.5 DPA) were selected for co‐expression network construction (Figures S9 and S10, Table S11).
Among these four networks, some core components shared between adjacent stages, while some were specific at one stage. For example, the PROTODERMAL FACTOR1 (PDF1) gene, whose silencing caused retardation of fibre initiation (Deng et al., 2012), was identified as a core network component both at −1.5, −0.5 and 0 DPA (Figure S10). Two genes coding lipid transfer protein (LTP3 and LTP6), which can transport metabolites around the cell for their membrane biosynthesis (Wu et al., 2006; Wu et al., 2007), were the core components at both −1.5 and −1 DPA network. At −0.5 DPA network, AMP‐dependent synthetase and ligase (LACS1), ABC‐2 type transporter (ABCG13), Ribosomal protein (RPS29A) and a transcription factor MYB25 were located in the core (Figure S10). At 0 DPA, the core network components including some function unknown genes, such as Ghir_D08G021100, HUTL, Era and others.
Filtering the network down by TFs, four new TF regulatory networks were obtained (Figure 6). The core network components in latter three stages (−1, −0.5 and 0 DPA) were MYB25‐like, HD1 and HOX3 respectively. These three genes are superstars in fibre cell development (Cao et al., 2020; Shan et al., 2014; Walford et al., 2011, 2012; Wan et al., 2016), but the detailed temporal pattern of how they work in individual fibre cell has not yet been answered. After comparative analysis, MYB25‐like_At (Ghir_A12G017450) and MYB25‐like_Dt (Ghir_D12G017660) were only identified at −1 and −0.5 DPA network (Figure 6), as both of them have been reported associated with lint fibre development (Zhu et al., 2018). While HD1 gene, who expressed predominantly in epidermal tissues during early fibre development (Walford et al., 2012), was identified in all these four stages. Meanwhile, the expression pattern of PROTODERMAL FACTOR 2 (PDF2) gene (Abe et al., 2003) was consistent with HD1 (Figure 6). Another HD‐ZIP TF HOX3, its silencing greatly reduces fibre length whereas overexpression leads to longer fibre (Shan et al., 2014), was only identified at 0 DPA network (Figure 6). And also, a R2R3 MYB TF MYB109, who plays a role in fibre elongation (Pu et al., 2008), showed the same pattern as HOX3.
Figure 6.

Transcription factors (TFs) regulatory network predicts key regulators in the four distinctive developmental processes during fibre initiation. A total of 15, 15, 16 and 9 TFs were identified related to fibre cell development at −1.5, −1, −0.5 and 0 DPA respectively. Each circle represents a gene, and triangle represents transcription factor. Node size is equivalent to the number of predicted connections. Node colour represents the weight abundance of predicted connections. Lines indicate edge weight (>0.01) for each pair of genes. Edge width represents the strength of the predicted connection.
As core network components, MIXTA‐like MYB genes MYB25 and MML9 were only identified at −0.5 and 0 DPA networks (Figure 6). MYB25 played a role in regulating specialized outgrowth of fibre cell (Machado et al., 2009). MML9 was reported preferentially expressed during fibre initiation (Zhang et al., 2015). At −1.5 DPA, the core gene WRKY44 (Ghir_A04G008530), also named TTG2, was reported in Arabidopsis involved in a regulatory module for regulation of seed coat mucilage synthesis (Xu et al., 2022).
MYB25‐like determines fibre differentiation at −1 DPA and HOX3 determines fibre tip‐biased diffuse growth at 0 DPA
MYB25‐like defunction was previously reported to be the determinant of cotton fibreless mutant (Walford et al., 2011; Wan et al., 2016). Histochemical localization of MYB25‐like‐GUS expression was localized in the epidermal layer of 0 DPA ovules (Walford et al., 2011). Here, compared to sense probe hybrid result, MYB25‐like expressed in ovule outer integument layer, including outer pigment layer and epidermis, and more highly in fibre cells (Figure 7a). Two independent lines with different editing types resulting both MYB25‐like_At and MYB25‐like_Dt knocked out were obtained (Figure S11a) and they showed totally fibreless phenotype (Figure S11b). Distinct from GhMYB25‐like RNA interference suppression lines, which still has few lint fibres attached on mature seeds (Walford et al., 2011), the seeds of our CRISPR mutant lines were totally glabrous. When observing 0 DPA ovules with SEM, no fibre initials was found in both MYB25‐like_CR lines as compared to WT (Jin668, the transgenic receptor material, Figure 7b,c).
Figure 7.

Function verification of cotton MYB25‐like, PDF2 and HOX3 genes. (a) MYB25‐like RNA in situ hybridization in LF_0d ovules. The sense probe was used as the negative control. OI, outer integument; II, inner integument. Red arrows indicate fibre cells, and red arrowheads indicate non‐fibre epidermal cells. Yellow arrows indicate outer pigment layer. Scale bars, 50 μm. (b) Fibre initials morphology between WT (Jin668, the transgenic receptor material) and two types of MYB25‐like CRISPR mutant (MYB25‐like _CR, both MYB25‐like homologues from At and Dt subgenomes knocked out) plants at 0 DPA. The upper panel shows the whole ovule, the lower panel is a magnified view of the ovule middle region. Scale bars, 500 μm (upper panel) and 50 μm (lower panel). (c) Statistics analysis of initial fibre densities (numbers per 0.01 mm2) on 0 DPA ovules between WT and MYB25‐like_CR plants. (d) RNA in situ hybridization of PDF2 in LF_0d ovules with the sense probe as a negative control. OI, II, red arrows and red arrowheads have the same meaning as described in (a). Scale bars, 50 μm. (e) Fibre initials morphology in two types of PDF2_CR (both PDF2 homologues from At and Dt subgenomes knocked out) lines and WT plants at 0 DPA. The lower panel is a magnified view of the middle region of whole ovule (upper panel). Scale bars, 200 μm (upper panel) and 50 μm (lower panel). (f) Statistics analysis of initial fibre densities (numbers per 0.01 mm2) on 0 DPA ovules between WT and PDF2_CR plants. All asterisks indicate significant differences when compared with the WT plants (T‐test, ** P < 0.01). (g) RNA in situ hybridization of HOX3 in LF_0d ovules with the sense probe as a negative control. OI, II, red arrows and red arrowheads have the same meaning as described in (a). Scale bars, 50 μm. (h) Fibre initials morphology in WT and HOX3_CR (both HOX3 homologues from At and Dt subgenomes knocked out) line at 0 DPA and 1 DPA respectively. The lower panel is a magnified view of the middle region of whole ovule (upper panel). Scale bars, 200 μm (upper) and 50 μm (lower).
Cotton PDF2 was a member of HD‐ZIP class IV homeodomain protein family, which shares the highest homology with AtPDF2 in Arabidopsis. AtPDF2 and its paralogue ATML1 are functionally interchangeable and act on Arabidopsis shoot epidermal cell differentiation (Abe et al., 2003; Rombola‐Caldentey et al., 2014). Cotton PDF2 was validated highly expressed in ovule epidermis and fibre cells by in situ hybridization assays (Figure 7d). Two types of PDF2 CRISPR mutant lines (PDF2_CR, both PDF2 homologues from At and Dt subgenomes knocked out) were created (Figure S12a) and their fibre initials were significantly decreased on 0 DPA ovules as compared to WT (Figure 7e,f). However, the mature fibre phenotype and fibre quality of PDF2_CR lines were nearly the same as WT, except for a decrease in fuzz fibre density (Figure S12b‐c).
Cotton HOX3 was highly expressed in 0 DPA fibre cells validated by in situ hybridization assays (Figure 7g). HOX3 transcript level was sharply decreased in homozygous transgene co‐suppression lines, and resulting in retarded fibre elongation (Shan et al., 2014). Here, through CRISPR‐Cas9 based gene editing technology, the HOX3 CRISPR mutant line (HOX3_CR, both HOX3 homologues from At and Dt subgenomes knocked out) was obtained (Figure S13a). Compared to WT, the phenotype of HOX3_CR line was seemingly naked seeds (Figure S13b), different from the HOX3 transgene co‐suppression lines which attached with very short fibres (Shan et al., 2014). When observed with SEM, normal fibre initials can be seen on 0 DPA ovules both in WT and HOX3_CR line (Figure 7h). At 1 DPA, fibre initials normally elongated on WT ovules but not on HOX3_CR line (Figure 7h).
Above all, the specific function of MYB25‐like and HOX3 were more clearly clarified using CRISPR gene editing technology. MYB25‐like determines fibre differentiation, so defunctionalization of MYB25‐like produces no fibre initials; HOX3 determines fibre tip‐biased diffuse growth, so cotton plants with HOX3 defunctionalization can perform normal fibre differentiation and diffuse growth, but cannot transform into tip‐biased diffuse growth. Besides, PDF2 play a role in fibre initiation, while it is not as critical as MYB25‐like and HOX3.
Discussion
The challenges of scRNA‐seq in non‐model crops
scRNA‐seq has flourished in plants (Ryu et al., 2021), but challenges remain, especially for non‐model crops. The biggest obstacle is definition for each cell type. In model plants Arabidopsis and poplars, a large number of known markers had been reported, which was enough to define almost all the cell types (Chen et al., 2021; Zhang et al., 2019). While in rice, corn and cotton (in this study), cell type definition still relies on RNA in situ hybridization (Liu et al., 2021b; Satterlee et al., 2020; Wang et al., 2021b; Xu et al., 2021), which is still a technologically dependent and time‐consuming challenge. Spatial transcriptome technology overcame this disadvantage and has been successfully applied in Arabidopsis leaves, which showed the bona fide single‐cell spatial transcriptome profiles (Xia et al., 2022). However, this technology still has many technical barriers in plant, for example, tissue optimization, due to the existence of cell wall.
The mutants are very effective to verify cell type definition. After scRNA‐seq on root tips of rhd6 (lack root hair) and gl2 (lack non‐hair cells) mutant, the cluster of root hair cells and non‐hair cells were respectively reduced compared to wild type (Ryu et al., 2019). The similar trend was observed in Arabidopsis root tips before and after heat stress treatment. For example, root hair cluster cell number was decreased in heat shock sample (Jean‐Baptiste et al., 2019). Here, we sequenced cotton Xu142 fl fibreless mutant ovules, and no fibre cell cluster was identified in Xu142 fl (Figure 4b). This not only proves the reliability of scRNA‐seq in cotton ovules but also confirms the accuracy of our cell type definition.
Fibre cell initiation successively experiences differentiation, diffuse growth and tip‐biased diffuse growth
The study on fibre initiation started from last century (Stewart, 1975). The morning of anthesis (0 DPA) was always selected as a representative stage for fibre initiation (Haigler et al., 2012; Lee et al., 2007). And a lot of SEM observation and comparative transcriptome profiling was performed on 0 DPA ovules (Hu et al., 2018; Qin et al., 2019; Walford et al., 2012; Zhang et al., 2011). In this study, fibre initials can be observed on Xu142_LF line ovule epidermis at −0.5 DPA (Figure 1, Figure S1). Further, Xu142_LF fibre cell was identified differentiated at −1 DPA (Figure 5b). Therefore, with Xu142_LF line growing at Wuhan city, its −1 DPA samples should be collected for fibre differentiation study and −0.5 DPA samples for fibre diffuse growth study.
scRNA‐seq has been widely used in plants (Ryu et al., 2021; Seyfferth et al., 2021; Shaw et al., 2021), but reported studies were limited to one specific developmental stage, such as primary root tips of 5 days after germination in Arabidopsis (Ryu et al., 2019), root tips of 5 days rice seedlings (Zhang et al., 2021a), leaf blades of 7 days seedlings in peanut (Liu et al., 2021a), 5–10 mm developing ears of corn (Xu et al., 2021) and stem below the third internode of 4‐month‐old poplar (Chen et al., 2021). Here, our sampling strategy included four developmental stages during fibre initiation. In single sample, cell heterogeneity can be identified (Figure 2). In multiple samples, the elaborate developmental dynamics of fibre cell differentiation, diffuse growth and tip‐biased diffuse growth could be recognized (Figure 5).
Integrating fibre cell developmental trajectory, TF regulatory networks, and core network components functional validation, a model was proposed for fibre initiation focusing on a single cell (Figure 8a). A fibre cell successively experiences the process of differentiation, diffuse growth and tip‐biased diffuse growth, with different key regulators involved in each process. HD1 and PDF2 expressed at all four stages (Figure 6 and Figure 8a). They play roles in ovule epidermis before fibre differentiation, and also in fibre cell after then. MYB25‐like was identified preferentially expressed at −1 DPA (Figure 5e and Figure 6), promoting fibre cell differentiation. After fibre cell differentiated, MYB25 and MML9 start to express and gradually increased (Figure S7f,g), accompanying with fibre cell diffuse growth (Figure 8a). MYB25 was likely regulated by MYB25‐like (Walford et al., 2011) then play a role in promoting fibre diffuse growth. At 0 DPA, a new core network component HOX3 appeared (Figure 6) and promoted fibre tip‐biased diffuse growth (Figure 8a).
Figure 8.
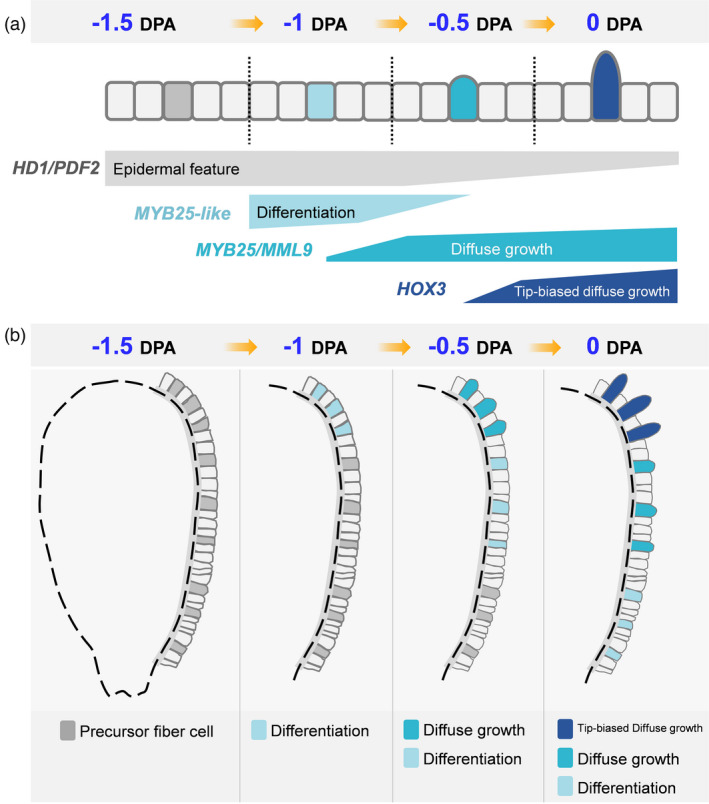
The proposed model for fibre initiation focusing on a single cell. (a) On cotton ovule epidermis, a precursor fibre cell successively experienced the process of differentiation, diffuse growth and tip‐biased diffuse growth during its initiation (above), as represented by four stages (−1.5, −1, −0.5 and 0 DPA). Different hub transcription factors that regulating each process are shown (below). (b) On the whole ovule, lint fibre differentiation firstly occurred on chalazal end at −1 DPA (marked by light sky blue). The differentiation progress gradually towards micropylar end. At −0.5 DPA, fibre initials could be seen at chalazal end (marked by cyan) due to its diffuse growth. At the same time, fibre differentiation has occurred in the middle of the ovule (light sky blue). At 0 DPA, the previously protruded fibre initials transformed into tip‐biased diffuse growth (dark blue), and the previously differentiated fibres began to protrude (cyan) due to diffuse growth. At the same time, fibre differentiation occurred at ovule micropylar end (light sky blue). To sum up, there were no fibre cell differentiated at −1.5 DPA; fibre differentiation occurred on chalazal end at −1 DPA; both fibre diffuse growth and differentiation occurred at −0.5 DPA; at 0 DPA, fibre tip‐biased diffuse growth, diffuse growth and differentiation occurred simultaneously.
On whole ovule, fibre initiation firstly occurred on chalazal end and progressed gradually towards micropylar end (Figures 7 and 8b), according to earlier morphological observations (Stewart, 1975). Once fibre cell differentiated at −1 DPA, the continuous development leads to multiple processes occurring simultaneously on ovule epidermis (Figure 8b). As shown in re‐clustering result (Figure S7h), −1 DPA cells located only at the start of trajectory. At −0.5 DPA, cell distribution expanded (Figure S7h), means these cells including both early fibre cell and diffuse growing cells (Figure 8b). The 0 DPA cells were located at all the five sub‐clusters (Figure S7h), including cells in differentiation, diffuse growth and tip‐biased diffuse growth (Figure 8b). With scRNA‐seq, gene expression pattern in cells at different developmental stages can be more finely characterized, which is an important reason why this study can subdivide fibre initiation into three processes and identify the regulatory network of each process.
MYB25‐like and HOX3 play most important roles as commanders in fibre differentiation and fibre tip‐biased diffuse growth
Through cotton transgenic verification, PDF2 gene mutation only decreased fibre initials at 0 DPA (Figure 7e). In Arabidopsis, single mutant of AtPDF2 or its paralogue ATML1 display normal shoot development, while double mutant results in severe defects in shoot epidermal cell differentiation (Abe et al., 2003; Rombola‐Caldentey et al., 2014). In cotton, PDF2 homologues GhHOX1 (Wang et al., 2004) and GhHD1 (Walford et al., 2012) had been cloned and verified promoting fibre development. PDF2 might also functionally interchangeable to GhHOX1 or GhHD1, or both of them. So, double mutants on PDF2 and its paralogue might display obvious phenotype in cotton.
There is no report to clarify the spatiotemporal pattern of MYB25‐like gene at single cell resolution. Here, expression pattern on fibre developmental trajectory showed that MYB25‐like was preferentially expressed in early fibre cells (Figure 5e). With qRT‐PCR measurements, MYB25‐like showed higher expression in −1 to 3 DPA ovules (Walford et al., 2011), in can be inferred that the expression of MYB25‐like mainly contributed by the newly differentiated fibre cells. On one hand, MYB25‐like gene starts to be highly expressed at −1 DPA (Walford et al., 2011), which is consistent with our result (Figure 6). On the other hand, its high expression lasts until 3 DPA, a stage when fuzz fibre begins to differentiate, which indicated that MYB25‐like is also participated in fuzz initiation.
Hormone response and sugar signal genes were reported involved in fibre initiation (Huang et al., 2021; Tian and Zhang, 2021; Wang et al., 2021a). Interestingly, MYB25‐like was down‐regulated in fibre‐deficient mutants of hormone‐ or sugar‐signalling genes. For example, MYB25‐like activity was suppressed in GhJAZ2 overexpressing lines (Hu et al., 2016); the transcripts of MYB25‐like was dramatically reduced in GhVIN1‐RNAi lines (Wang et al., 2014) and down‐regulated in GhPIN1a‐RNAi ovules (Zhang et al., 2017); MYB25‐like was severely down‐regulated in ovules cultured with excess ZT (trans‐zeatin, a kind of cytokinin) (Zeng et al., 2019). These suggested that hormone and sugar signalling may act on the upstream of MYB25‐like and through this gene to exert their effects on fibre development. At least among currently known fibreless mutants (such as Xu142 fl, GhVIN1‐RNAi lines and GhPIN1a‐RNAi lines) (Walford et al., 2011; Wang et al., 2014; Zhang et al., 2017), MYB25‐like gene was the direct commander controlling fibre cell differentiation.
HOX3 gene highly expressed at 0 DPA (Figure 6) and mainly in fibre cells (Figures 2c and 7g). Fibre cell can normally differentiate and diffuse grow but cannot transform into tip‐biased diffuse growth in HOX3_CR line (Figure 7h). Therefore, HOX3 seems to act as a commander controlling fibre cell development transformation. As shown in Figure 8a, two commanders (MYB25‐like and HOX3) positively regulating early fibre development are functioning like a ‘relay race’ model. The RNA‐seq, chromatin immunoprecipitation sequencing (ChIP‐seq) with these cotton mutants and identification of TF regulatory multiple complexes have been ongoing in our lab, the molecular mechanism hidden below ‘relay race’ model will be revealed in the near future.
Core regulators controlling the differentiation and tip‐biased diffuse growth of Arabidopsis trichomes and cotton fibres may be conserved
In Arabidopsis, mutant of two genes, GL1 (R2R3‐type MYB transcription factor GLABRA1) and GL2 (homeobox transcription factor GLABRA2), produced no leaf trichomes (Hulskamp et al., 1994; Szymanski et al., 1998), but they were functioning in different ways. For gl2 leaves some large cells bigger than normal epidermal cells appeared and their nuclei were also in the same size as wild‐type trichomes, which means trichome cells can differentiate and enlarge, but no local outgrowth (Hulskamp et al., 1994). However, all the epidermal cells of gl1 leaves were uniform in size and shape, and no specialized cells was found, which suggested that GL1 is required for trichome cell differentiation (Hulskamp et al., 1994). In this aspect, MYB25‐like gene functions similarly to GL1, and HOX3 to GL2. Because they were in same gene family respectively, but also the similar ways they acting on fibre cell or trichomes. In addition, other signals affecting trichome development such as phytohormones (Qi et al., 2011; Qi et al., 2014; Zhou et al., 2013), miRNAs (Xue et al., 2014; Yu et al., 2010), R3 MYBs (Gan et al., 2011; Vadde et al., 2019; Wang et al., 2007; Zhao et al., 2008) or histone demethylase (Hung et al., 2020), were always acting on the upstream of GL1. This also showed a similar pattern as cotton MYB25‐like. Therefore, the core regulators controlling differentiation and tip‐biased diffuse growth of Arabidopsis leaf trichomes and cotton fibres appear to be conserved. This also provides some inspiration for exploring other types of epidermal tissue, such as tomato trichomes, kapok fibre, tea trichomes, and okra seed coat mucilage.
Conclusion
In summary, with the help of scRNA‐seq technology and reasonable multi‐stage sampling strategy, fibre cell differentiation, diffuse growth and tip‐biased diffuse growth process during fibre initiation were more finely delineated. Through gene regulatory network analysis and CRISPR‐Cas9‐based gene function verification, the MYB25‐like gene was newly defined as a commander acting at −1 DPA on fibre cell differentiation. In addition, HOX3 was proved to be another commander controlling fibre development transformation into tip‐biased diffuse growth. It is the first report by applying scRNA‐seq technology in cotton fibre cell, and our result provides a more refined and detailed stage definition of fibre initiation. The valuable resource provided here will help to further explore the mechanism of fibre development, plant trichomes differentiation and single cell fate determination.
Materials and methods
Plant growth and sample collection
The cotton Lint‐Fuzz (Xu142_LF) line, derived from recombinant inbred lines of Xu142 × Xu142 fl (Hu et al., 2018), was planted in the experimental field at Huazhong Agriculture University, Wuhan, China. Cotton plants were grown under conventional field management. Samples were collected when cotton plants began flowering. −1 DPA flower buds, 0 DPA flowers were collected in the morning (8 : 00 a.m.), and −1.5 DPA and −0.5 DPA buds were collected in the evening (8 : 00 p.m.).
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM)
Cotton bolls of Xu142_LF were collected at −1.5 DPA and every 4 h from −1 DPA to 0 DPA (i.e. 8 : 00, 12 : 00, 16 : 00, 20 : 00, 24 : 00 at −1 DPA and 4 : 00, 8 : 00 at 0 DPA). Ovules were fixed for further SEM observation. The detailed processes were performed as reported (Hu et al., 2018).
Ovules at −1.5, −1, −0.5 and 0 DPA were collected and fixed with 2.5% glutaraldehyde, followed by fixing with 1% OsO4, acetone gradient dehydration, resin infiltration and embedment. Sections of 90 nm were cut, counterstained, then visualized using a TEM (New Bio‐TEM H‐7500, HITACHI, Japan), according to the methods reported (Liu et al., 2015; Min et al., 2013).
Tissue digestion and scRNA‐seq library preparation
Ovules collected from Xu142_LF at −1.5, −1, −0.5 and 0 DPA, and from 0 DPA of Xu142 and Xu142 fl were respectively placed into a 30‐mm‐diameter Petri dish containing 3 mL enzyme solution (1.5% [w/v] cellulose [‘ONOZUKA’ R‐10, Yakult], 1% [w/v] hemicellulose [Sigma‐Aldrich], 0.75% [w/v] Macerozyme [R‐10, Solarbio], 0.4 M Mannitol, 20 mm MES [pH 5.7], 20 mm KCl, 10 mm CaCl2, 0.1% [w/v] bovine serum albumin (BSA)). Four cotton balls were harvested and all ovules in these balls were collected together as one sample. Petri dishes were placed in a vacuum drying oven and kept at 0.1 atmospheric pressure for 5 min to promote the removal of gases in the ovule to facilitate the full submersion of the ovule in the enzyme solution. The dish was rotated at 60 rpm for 4 h at 25°C.
The enzyme solution was filtered with a 40 μm cell strainer, 1× PBS solution containing 0.04% BSA was added to the filtrate, gently inverted and mixed, centrifuged at 100 rcf for 2 min, and the supernatant was carefully discarded. Filtering, rinsing, and centrifuging steps was repeated twice to obtain a single cell suspension. The cell suspension was kept on ice to prevent cell death. Protoplast concentration was determined using a haemocytometer, and the ideal concentration is 700–1200 cells/μl. Protoplasts were stained with 0.4% trypan blue solution for detecting viability.
A commercially available droplet‐based system from 10× Genomics Inc. (Zheng et al., 2017) was used to isolate protoplasts. The protoplast suspension was loaded into Chromium microfluidic chips with 3′ v2 chemistry. RNA from the barcoded cells was subsequently reverse‐transcribed, then sequencing libraries were constructed and sequenced on NovaSeq (Illumina) platform using Hiseq PE150 strategy.
Processing of scRNA‐seq data
The detailed calculation methods and parameter settings involved in this process are described in Appendix S1. In brief, the raw data in FASTQ format were first processed to obtain clean reads. Gossypium hirsutum TM‐1 genome (Wang et al., 2019) was used as reference genome. Only reads that were uniquely mapped were used for UMI counting. The Seurat package (v. 4.0.1) implemented in R (v. 4.0.0) was used for gene‐cell matrices analysis, including doublets, no‐load cells and dead cells filtration. Principal Components Analysis (PCA), Uniform Manifold Approximation and Projection (UMAP) and t‐Stochastic Neighbour Embedding (t‐SNE) analyses were performed for visualizing data in 2‐d space. Cluster‐enriched genes were identified with Seurat function ‘FindAllMarkers’. The subset of related clusters was extracted and processed for pseudo‐time analysis. The weighted gene co‐expression network analysis (WGCNA) was performed in R software (v. 4.0.0) following the official process. The gene regulatory network displaying, personalized Gene Ontology (GO) and pathway enrichment analysis were performed using OmicShare tools, a free online platform for data analysis (www.omicshare.com/tools).
Analysis of RNA‐seq data
Bulk RNA‐seq data of Xu142_LF 0 DPA ovule outer integument from our previous study (Hu et al., 2018) was used here. Clean reads were newly mapped to the updated TM‐1 reference genome (Wang et al., 2019) using Hisat2 (v2.1.0) software (Kim et al., 2015). The mapping reads were sorted to filter those reads representing PCR duplicates. Sequencing reads with mapping quality of <25 were filtered using SAMTOOLS (v.0.1.19; Li et al., 2009), the remaining were used to calculate gene expression levels using STRINGTIE software (v.1.3.4) with default settings (Pertea et al., 2015).
RNA in situ hybridization
In situ hybridization was carried out as described in cotton research previously (Zhang et al., 2017). Briefly, 0 DPA ovaries of Xu142_LF were collected and embedded in paraffin. 10 μm paraffin sections were de‐paraffinized, rehydrated and incubated overnight with the Dig‐labelled RNA probe (Roche). Sections were then incubated with alkaline phosphatase‐conjugated anti‐digoxigenin (anti‐Dig‐AP, Roche) and the signal was detected by nitro‐blue tetrazolium/5‐bromo‐4‐chloro‐3‐inodyl‐phosphate (NBT/BCIP) colour substrate solution (Roche). Sections incubated with sense RNA probe were used as negative control. Images were captured using fully motorized upright microscope (Leica DM6B) in bright‐field mode. Primers are listed in Table S12.
Vector construction and plant transformation
CRISPR technology was employed to create the GhMYB25‐like‐CR (Ghir_A12G017450/Ghir_D12G017660), GhHOX3‐CR (Ghir_A12G028530/Ghir_D12G028680) and GhPDF2‐CR (Ghir_A10G001030/Ghir_D10G001810) mutants in cotton. For targeting MYB25‐like genes, two sgRNAs (CTCCATGTAGCGACAAGGTG; CGCCCTTCTTGGAAACAGGT) were designed for targeting MYB25‐like. The primers listed in the Table S12 were used to amplify tRNA and gRNA from the template pGTR vector. Two different fragments containing tRNA‐sgRNA1 and gRNA‐tRNA‐sgRNA2 were integrated together by an overlapping extension PCR. Finally, the PCR products were purified and inserted the Bsa I‐digested pRGEB32‐GhU6.9 vector using ClonExpress II One Step Cloning Kit (Vazyme) (Wang et al., 2018). For targeting GhHOX3 (ACCGGTACAACCTGTTCATA) and GhPDF2 (CAACTGTGTCTCCTTACTTA) genes, single sgRNA was used in the GhHOX3‐CR and GhPDF2‐CR vector. The positive vectors were transformed into Agrobacterium tumefaciens strain EHA105 for cotton transformation. JIN668 was the transgenic receptor (Li et al., 2019).
On‐target analysis of gene‐edited plants
T0 positive transgenic plants were screened by PCR analysis using Cas9 forward and reverse primers (Table S12). For identification of mutated alleles in T1 transgenic lines, high‐throughput (Hi‐Tom) sequencing was adopted (Liu et al., 2019). First, the targeted regions were amplified by PCR using site‐specific primers. Second, barcode primers were used to add barcodes to the first‐round PCR products. After the second‐round PCR amplification, the products of all samples were mixed in equal amounts and purified to perform next‐generation sequencing (NGS). Finally, NGS data was analysed by Hi‐Tom platform (http://hi‐tom.net/hi‐tom/).
Conflicts of interest
The authors declared that they have no conflict of interest.
Author contributions
L.T. and X.Z. conceived the project and designed the experiments. Y.Q. performed protoplast isolating, M.S. performed the transgenic experiments. Y.Q., M.S. and L.S. performed in situ hybridization experiment. Y.Q., M.S. and J.Y. performed bioinformatics analysis. Z.Y., W.L., J.X., M.X., L.S., Y.L. and G.Z. contributed to the vector construction. Y.Q., Z.L. and Z.X. built a website for data sharing. Y.Q., M.S. and L.T. wrote the manuscript, X.Y., M.W., K.L. and X.Z. revised the manuscript with feedback from all other authors.
Supporting information
Figure S1 Dynamic changes on Xu142_LF ovule epidermis during fibre initiation.
Figure S2 Tissue digestion and protoplast isolation.
Figure S3 Comparison between single‐cell RNA‐seq and bulk RNA‐seq of Xu142_LF.
Figure S4 Expression and identification of cluster enriched genes.
Figure S5 RNA in situ hybridization of cell‐type representative genes.
Figure S6 Single‐cell RNA‐seq and clusters identification on Xu142 and Xu142 fl.
Figure S7 Fibre cell cluster identification on the LF combined sample.
Figure S8 Clustering and annotation of four LF samples.
Figure S9 Hierarchical cluster tree showing co‐expression modules identified by WGCNA.
Figure S10 Networks related to fibre cell initiation.
Figure S11 Characterization of cotton CRISPR editing lines on MYB25‐like genes.
Figure S12 Characterization of cotton CRISPR editing lines on PDF2 genes.
Figure S13 Characterization of cotton CRISPR editing lines on HOX3 genes.
Table S1 Summary of single‐cell RNA sequencing results for each sample.
Table S2 Summary of protoplasting induced differentially expressed genes.
Table S3 List of enriched genes for each cluster.
Table S4 Cell number of each cluster identified from Xu142‐vs‐Xu142 fl.
Table S5 KEGG analysis of LF_0d fibre cell enriched genes.
Table S6 Cell barcode mapping between single sample and combined sample.
Table S7 Summary of 167 branch‐dependent genes.
Table S8 GO enrichment analysis of 167 branch‐dependent genes.
Table S9 Summary of cluster‐enriched genes.
Table S10 WGCNA on four LF samples.
Table S11 Weight coefficient of fibre development‐related genes.
Table S12 The primers used in this study.
Appendix S1 Details of bioinformatics analysis.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31830062 and 31901576), and the funds from the Interdisciplinary Sciences Research Institute of Huazhong Agricultural University (2662021JC005). We thank Novogene Corporation (http://www.novogene.com/) for assistance with scRNA‐seq library preparation and sequencing.
Data availability statement
The scRNA‐seq data have been deposited in the NCBI SRA database (https://www.ncbi.nlm.nih.gov/bioproject/) with BioProject number PRJNA600131.
References
- Abe, M. , Katsumata, H. , Komeda, Y. and Takahashi, T. (2003) Regulation of shoot epidermal cell differentiation by a pair of homeodomain proteins in Arabidopsis. Development, 130, 635–643. [DOI] [PubMed] [Google Scholar]
- Bedon, F. , Ziolkowski, L. , Walford, S.A. , Dennis, E.S. and Llewellyn, D.J. (2014) Members of the MYBMIXTA‐like transcription factors may orchestrate the initiation of fiber development in cotton seeds. Front. Plant Sci. 5, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, J.F. , Zhao, B. , Huang, C.C. , Chen, Z.W. , Zhao, T. , Liu, H.R. , Hu, G.J. et al. (2020) The miR319‐Targeted GhTCP4 promotes the transition from cell elongation to wall thickening in cotton fiber. Mol. Plant, 13, 1063–1077. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Tong, S. , Jiang, Y. , Ai, F. , Feng, Y. , Zhang, J. , Gong, J. et al. (2021) Transcriptional landscape of highly lignified poplar stems at single‐cell resolution. Genome Biol. 22, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, F. , Tu, L. , Tan, J. , Li, Y. , Nie, Y. and Zhang, X. (2012) GbPDF1 is involved in cotton fiber initiation via the core cis‐element HDZIP2ATATHB2. Plant Physiol. 158, 890–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denyer, T. , Ma, X. , Klesen, S. , Scacchi, E. , Nieselt, K. and Timmermans, M.C.P. (2019) Spatiotemporal developmental trajectories in the Arabidopsis root revealed using high‐throughput single‐Cell RNA sequencing. Dev. Cell, 48, 840–852. [DOI] [PubMed] [Google Scholar]
- Denyer, T. and Timmermans, M.C.P. (2022) Crafting a blueprint for single‐cell RNA sequencing. Trends Plant Sci. 27, 92–103. [DOI] [PubMed] [Google Scholar]
- Gala, H.P. , Lanctot, A. , Jean‐Baptiste, K. , Guiziou, S. , Chu, J.C. , Zemke, J.E. , George, W. et al. (2021) A single‐cell view of the transcriptome during lateral root initiation in Arabidopsis thaliana . Plant Cell, 33, 2197–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan, L. , Xia, K. , Chen, J.G. and Wang, S. (2011) Functional characterization of TRICHOMELESS2, a new single‐repeat R3 MYB transcription factor in the regulation of trichome patterning in Arabidopsis. BMC Plant Biol. 11, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou, J.Y. , Wang, L.J. , Chen, S.P. , Hu, W.L. and Chen, X.Y. (2007) Gene expression and metabolite profiles of cotton fiber during cell elongation and secondary cell wall synthesis. Cell Res. 17, 422–434. [DOI] [PubMed] [Google Scholar]
- Haigler, C.H. , Betancur, L. , Stiff, M.R. and Tuttle, J.R. (2012) Cotton fiber: a powerful single‐cell model for cell wall and cellulose research. Front. Plant Sci. 3, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovav, R. , Udall, J.A. , Chaudhary, B. , Hovav, E. , Flagel, L. , Hu, G. and Wendel, J.F. (2008) The evolution of spinnable cotton fiber entailed prolonged development and a novel metabolism. PLoS Genet. 4, e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, H. , He, X. , Tu, L. , Zhu, L. , Zhu, S. , Ge, Z. and Zhang, X. (2016) GhJAZ2 negatively regulates cotton fiber initiation by interacting with the R2R3‐MYB transcription factor GhMYB25‐like. Plant J. 88, 921–935. [DOI] [PubMed] [Google Scholar]
- Hu, H. , Wang, M. , Ding, Y. , Zhu, S. , Zhao, G. , Tu, L. and Zhang, X. (2018) Transcriptomic repertoires depict the initiation of lint and fuzz fibres in cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 16, 1002–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, G. , Huang, J.Q. , Chen, X.Y. and Zhu, Y.X. (2021) Recent advances and future perspectives in cotton research. Annu. Rev. Plant Biol. 72, 437–462. [DOI] [PubMed] [Google Scholar]
- Hulskamp, M. , Misra, S. and Jurgens, G. (1994) Genetic dissection of trichome cell development in Arabidopsis. Cell, 76, 555–566. [DOI] [PubMed] [Google Scholar]
- Hung, F.Y. , Chen, J.H. , Feng, Y.R. , Lai, Y.C. , Yang, S. and Wu, K. (2020) Arabidopsis JMJ29 is involved in trichome development by regulating the core trichome initiation gene GLABRA3. Plant J. 103, 1735–1743. [DOI] [PubMed] [Google Scholar]
- Jean‐Baptiste, K. , McFaline‐Figueroa, J.L. , Alexandre, C.M. , Dorrity, M.W. , Saunders, L. , Bubb, K.L. , Trapnell, C. et al. (2019) Dynamics of gene expression in single root cells of Arabidopsis thaliana . Plant Cell, 31, 993–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, S.J. , Lu, Y.C. , Feng, J.X. , Wei, G. , Li, J. , Shi, Y.H. , Fu, Q. et al. (2003) Isolation and analyses of genes preferentially expressed during early cotton fiber development by subtractive PCR and cDNA array. Nucleic Acids Res. 31, 2534–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. and Salzberg, S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nat. Methods, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J.Y. , Symeonidi, E. , Pang, T.Y. , Denyer, T. , Weidauer, D. , Bezrutczyk, M. , Miras, M. et al. (2021) Distinct identities of leaf phloem cells revealed by single cell transcriptomics. Plant Cell, 33, 511–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.J. , Woodward, A.W. and Chen, Z.J. (2007) Gene expression changes and early events in cotton fibre development. Ann. Bot. 100, 1391–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C.H. , Zhu, Y.Q. , Meng, Y.L. , Wang, J.W. , Xu, K.X. , Zhang, T.Z. and Chen, X.Y. (2002) Isolation of genes preferentially expressed in cotton fibers by cDNA filter arrays and RT‐PCR. Plant Sci. 163, 1113–1120. [Google Scholar]
- Li, H. , Dai, X. , Huang, X. , Xu, M. , Wang, Q. , Yan, X. , Sederoff, R.R. et al. (2021) Single‐cell RNA sequencing reveals a high‐resolution cell atlas of xylem in Populus. J. Integr. Plant Biol. 63, 1906–1921. [DOI] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Wang, M. , Li, Y. , Zhang, Q. , Lindsey, K. , Daniell, H. , Jin, S. et al. (2019) Multi‐omics analyses reveal epigenomics basis for cotton somatic embryogenesis through successive regeneration acclimation process. Plant Biotechnol. J. 17, 435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Zhang, X. , Gao, S. , Cui, F. , Chen, W. , Fan, L. and Qi, Y. (2022) Single‐cell RNA sequencing reveals the landscape of maize root tips and assists in identification of cell type‐specific nitrate‐response genes. Crop J. 10.1016/j.cj.2022.02.004 [DOI] [Google Scholar]
- Liu, H. , Hu, D. , Du, P. , Wang, L. , Liang, X. , Li, H. , Lu, Q. et al. (2021a) Single‐cell RNA‐seq describes the transcriptome landscape and identifies critical transcription factors in the leaf blade of the allotetraploid peanut (Arachis hypogaea L.). Plant Biotechnol. J. 19, 2261–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Pang, C. , Wei, H. , Song, M. , Meng, Y. , Ma, J. , Fan, S. et al. (2015) iTRAQ‐facilitated proteomic profiling of anthers from a photosensitive male sterile mutant and wild‐type cotton (Gossypium hirsutum L.). J. Proteomics, 126, 68–81. [DOI] [PubMed] [Google Scholar]
- Liu, Q. , Liang, Z. , Feng, D. , Jiang, S.J. , Wang, Y.F. , Du, Z.Y. , Li, R.X. et al. (2021b) Transcriptional landscape of rice roots at the single‐cell resolution. Mol. Plant, 14, 384–394. [DOI] [PubMed] [Google Scholar]
- Liu, Q. , Wang, C. , Jiao, X. , Zhang, H. , Song, L. , Li, Y. , Gao, C. et al. (2019) Hi‐TOM: a platform for high‐throughput tracking of mutations induced by CRISPR/Cas systems. Sci. China Life Sci. 62, 1–7. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Zhang, Y. , Fang, X. , Tran, S. , Zhai, N. , Yang, Z. , Guo, F. et al. (2022a) Transcriptional landscapes of de novo root regeneration from detached Arabidopsis leaves revealed by time‐lapse and single‐cell RNA sequencing analyses. Plant Commun. 3, 100306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Wang, J. , Zhou, Y. , Zhang, Y. , Qin, A. , Yu, X. , Zhao, Z. et al. (2022b) Identification of novel regulators required for early development of vein pattern in the cotyledons by single‐cell RNA‐sequencing. Plant J. 110, 7–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Zhou, Y. , Guo, J. , Li, J. , Tian, Z. , Zhu, Z. , Wang, J. et al. (2020) Global dynamic molecular profiling of stomatal lineage cell development by single‐cell RNA sequencing. Mol. Plant, 13, 1178–1193. [DOI] [PubMed] [Google Scholar]
- Lopez‐Anido, C.B. , Vaten, A. , Smoot, N.K. , Sharma, N. , Guo, V. , Gong, Y. , Anleu Gil, M.X. et al. (2021) Single‐cell resolution of lineage trajectories in the Arabidopsis stomatal lineage and developing leaf. Dev. Cell, 56, 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado, A. , Wu, Y. , Yang, Y. , Llewellyn, D.J. and Dennis, E.S. (2009) The MYB transcription factor GhMYB25 regulates early fibre and trichome development. Plant J. 59, 52–62. [DOI] [PubMed] [Google Scholar]
- Min, L. , Zhu, L. , Tu, L. , Deng, F. , Yuan, D. and Zhang, X. (2013) Cotton GhCKI disrupts normal male reproduction by delaying tapetum programmed cell death via inactivating starch synthase. Plant J. 75, 823–835. [DOI] [PubMed] [Google Scholar]
- Mo, Y. and Jiao, Y. (2022) Advances and applications of single‐cell omics technologies in plant research. Plant J. 110, 1551–1563. [DOI] [PubMed] [Google Scholar]
- Omary, M. , Gil‐Yarom, N. , Yahav, C. , Steiner, E. , Hendelman, A. and Efroni, I. (2022) A conserved superlocus regulates above‐ and belowground root initiation. Science, 375, eabf4368. [DOI] [PubMed] [Google Scholar]
- Ortiz‐Ramirez, C. , Guillotin, B. , Xu, X. , Rahni, R. , Zhang, S. , Yan, Z. , Coqueiro Dias Araujo, P. et al. (2021) Ground tissue circuitry regulates organ complexity in maize and Setaria. Science, 374, 1247–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea, M. , Pertea, G.M. , Antonescu, C.M. , Chang, T.C. , Mendell, J.T. and Salzberg, S.L. (2015) StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat. Biotechnol. 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu, L. , Li, Q. , Fan, X. , Yang, W. and Xue, Y. (2008) The R2R3 MYB transcription factor GhMYB109 is required for cotton fiber development. Genetics, 180, 811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, T. , Huang, H. , Wu, D. , Yan, J. , Qi, Y. , Song, S. and Xie, D. (2014) Arabidopsis DELLA and JAZ proteins bind the WD‐repeat/bHLH/MYB complex to modulate gibberellin and jasmonate signaling synergy. Plant Cell, 26, 1118–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, T.C. , Song, S.S. , Ren, Q.C. , Wu, D.W. , Huang, H. , Chen, Y. , Fan, M. et al. (2011) The Jasmonate‐ZIM‐Domain proteins interact with the WD‐Repeat/bHLH/MYB complexes to regulate Jasmonate‐mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana . Plant Cell, 23, 1795–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, Y. , Sun, H. , Hao, P. , Wang, H. , Wang, C. , Ma, L. , Wei, H. et al. (2019) Transcriptome analysis reveals differences in the mechanisms of fiber initiation and elongation between long‐ and short‐fiber cotton (Gossypium hirsutum L.) lines. BMC Genomics, 20, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, Y.M. and Zhu, Y.X. (2011) How cotton fibers elongate: a tale of linear cell‐growth mode. Curr. Opin. Plant Biol. 14, 106–111. [DOI] [PubMed] [Google Scholar]
- Rapp, R.A. , Haigler, C.H. , Flagel, L. , Hovav, R.H. , Udall, J.A. and Wendel, J.F. (2010) Gene expression in developing fibres of Upland cotton (Gossypium hirsutum L.) was massively altered by domestication. BMC Biol. 8, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombola‐Caldentey, B. , Rueda‐Romero, P. , Iglesias‐Fernandez, R. , Carbonero, P. and Onate‐Sanchez, L. (2014) Arabidopsis DELLA and two HD‐ZIP transcription factors regulate GA signaling in the epidermis through the L1 box cis‐element. Plant Cell, 26, 2905–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan, Y.L. , Llewellyn, D.J. and Furbank, R.T. (2003) Suppression of sucrose synthase gene expression represses cotton fiber cell initiation, elongation, and seed development. Plant Cell, 15, 952–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu, K.H. , Huang, L. , Kang, H.M. and Schiefelbein, J. (2019) Single‐cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiol. 179, 1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu, K.H. , Zhu, Y. and Schiefelbein, J. (2021) Plant cell identity in the era of single‐cell transcriptomics. Annu. Rev. Genet. 55, 479–496. [DOI] [PubMed] [Google Scholar]
- Satterlee, J.W. , Strable, J. and Scanlon, M.J. (2020) Plant stem‐cell organization and differentiation at single‐cell resolution. Proc. Natl. Acad. Sci. USA, 117, 33689–33699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfferth, C. , Renema, J. , Wendrich, J.R. , Eekhout, T. , Seurinck, R. , Vandamme, N. , Blob, B. et al. (2021) Advances and opportunities in single‐cell transcriptomics for plant research. Annu. Rev. Plant Biol. 72, 847–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahan, R. , Hsu, C.W. , Nolan, T.M. , Cole, B.J. , Taylor, I.W. , Greenstreet, L. , Zhang, S. et al. (2022) A single‐cell Arabidopsis root atlas reveals developmental trajectories in wild‐type and cell identity mutants. Dev. Cell, 57, 543–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan, C.M. , Shangguan, X.X. , Zhao, B. , Zhang, X.F. , Chao, L.M. , Yang, C.Q. , Wang, L.J. et al. (2014) Control of cotton fibre elongation by a homeodomain transcription factor GhHOX3. Nat. Commun. 5, 5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, R. , Tian, X. and Xu, J. (2021) Single‐cell transcriptome analysis in plants: advances and challenges. Mol. Plant, 14, 115–126. [DOI] [PubMed] [Google Scholar]
- Shi, Y.H. , Zhu, S.W. , Mao, X.Z. , Feng, J.X. , Qin, Y.M. , Zhang, L. , Cheng, J. et al. (2006) Transcriptome profiling, molecular biological, and physiological studies reveal a major role for ethylene in cotton fiber cell elongation. Plant Cell, 18, 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, J.M. (1975) Fiber initiation on the cotton ovule (Gossypium hirsutum). Am. J. Bot. 62, 723–730. [Google Scholar]
- Sun, G. , Xia, M. , Li, J. , Ma, W. , Li, Q. , Xie, J. , Bai, S. et al. (2022) The maize single‐nucleus transcriptome comprehensively describes signaling networks governing movement and development of grass stomata. Plant Cell, 34, 1890–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski, D.B. , Jilk, R.A. , Pollock, S.M. and Marks, M.D. (1998) Control of GL2 expression in Arabidopsis leaves and trichomes. Development, 125, 1161–1171. [DOI] [PubMed] [Google Scholar]
- Tenorio Berrio, R. , Verstaen, K. , Vandamme, N. , Pevernagie, J. , Achon, I. , Van Duyse, J. , Van Isterdael, G. et al. (2022) Single‐cell transcriptomics sheds light on the identity and metabolism of developing leaf cells. Plant Physiol. 188, 898–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, Y. and Zhang, T. (2021) MIXTAs and phytohormones orchestrate cotton fiber development. Curr. Opin. Plant Biol. 59, 101975. [DOI] [PubMed] [Google Scholar]
- Tu, L.L. , Zhang, X.L. , Liang, S.G. , Liu, D.Q. , Zhu, L.F. , Zeng, F.C. , Nie, Y.C. et al. (2007) Genes expression analyses of sea‐Island cotton (Gossypium barbadense L.) during fiber development. Plant Cell Rep. 26, 1309–1320. [DOI] [PubMed] [Google Scholar]
- Vadde, B.V.L. , Challa, K.R. , Sunkara, P. , Hegde, A.S. and Nath, U. (2019) The TCP4 transcription factor directly activates TRICHOMELESS1 and 2 and suppresses trichome initiation. Plant Physiol. 181, 1587–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walford, S.A. , Wu, Y. , Llewellyn, D.J. and Dennis, E.S. (2011) GhMYB25‐like: a key factor in early cotton fibre development. Plant J. 65, 785–797. [DOI] [PubMed] [Google Scholar]
- Walford, S.A. , Wu, Y. , Llewellyn, D.J. and Dennis, E.S. (2012) Epidermal cell differentiation in cotton mediated by the homeodomain leucine zipper gene, GhHD‐1. Plant J. 71, 464–478. [DOI] [PubMed] [Google Scholar]
- Wan, Q. , Guan, X. , Yang, N. , Wu, H. , Pan, M. , Liu, B. , Fang, L. et al. (2016) Small interfering RNAs from bidirectional transcripts of GhMML3_A12 regulate cotton fiber development. New Phytol. 210, 1298–1310. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Cook, A. , Patrick, J.W. , Chen, X.Y. and Ruan, Y.L. (2014) Silencing the vacuolar invertase gene GhVIN1 blocks cotton fiber initiation from the ovule epidermis, probably by suppressing a cohort of regulatory genes via sugar signaling. Plant J. 78, 686–696. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Kartika, D. and Ruan, Y.L. (2021a) Looking into ‘hair tonics’ for cotton fiber initiation. New Phytol. 229, 1844–1851. [DOI] [PubMed] [Google Scholar]
- Wang, M. , Tu, L. , Yuan, D. , Zhu, D. , Shen, C. , Li, J. , Liu, F. et al. (2019) Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 51, 224–229. [DOI] [PubMed] [Google Scholar]
- Wang, P.C. , Zhang, J. , Sun, L. , Ma, Y.Z. , Xu, J. , Liang, S.J. , Deng, J.W. et al. (2018) High efficient multisites genome editing in allotetraploid cotton (Gossypium hirsutum) using CRISPR/Cas9 system. Plant Biotechnol. J. 16, 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Wu, Y. , Peng, A. , Cui, J. , Zhao, M. , Pan, Y. , Zhang, M. et al. (2022) Single‐cell transcriptome atlas reveals developmental trajectories and a novel metabolic pathway of catechin esters in tea leaves. Plant Biotechnol. J. 10.1111/pbi.13891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Kwak, S.H. , Zeng, Q. , Ellis, B.E. , Chen, X.Y. , Schiefelbein, J. and Chen, J.G. (2007) TRICHOMELESS1 regulates trichome patterning by suppressing GLABRA1 in Arabidopsis. Development, 134, 3873–3882. [DOI] [PubMed] [Google Scholar]
- Wang, S. , Wang, J.W. , Yu, N. , Li, C.H. , Luo, B. , Gou, J.Y. , Wang, L.J. et al. (2004) Control of plant trichome development by a cotton fiber MYB gene. Plant Cell, 16, 2323–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Huan, Q. , Li, K. and Qian, W. (2021b) Single‐cell transcriptome atlas of the leaf and root of rice seedlings. J. Genet. Genomics, 48, 881–898. [DOI] [PubMed] [Google Scholar]
- Wendrich, J.R. , Yang, B. , Vandamme, N. , Verstaen, K. , Smet, W. , Van de Velde, C. , Minne, M. et al. (2020) Vascular transcription factors guide plant epidermal responses to limiting phosphate conditions. Science, 370, 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, H. , Tian, Y. , Wan, Q. , Fang, L. , Guan, X. , Chen, J. , Hu, Y. et al. (2018) Genetics and evolution of MIXTA genes regulating cotton lint fiber development. New Phytol. 217, 883–895. [DOI] [PubMed] [Google Scholar]
- Wu, Y. , Llewellyn, D.J. , White, R. , Ruggiero, K. , Al‐Ghazi, Y. and Dennis, E.S. (2007) Laser capture microdissection and cDNA microarrays used to generate gene expression profiles of the rapidly expanding fibre initial cells on the surface of cotton ovules. Planta, 226, 1475–1490. [DOI] [PubMed] [Google Scholar]
- Wu, Y. , Machado, A.C. , White, R.G. , Llewellyn, D.J. and Dennis, E.S. (2006) Expression profiling identifies genes expressed early during lint fibre initiation in cotton. Plant Cell Physiol. 47, 107–127. [DOI] [PubMed] [Google Scholar]
- Xia, K. , Sun, H.X. , Li, J. , Li, J. , Zhao, Y. , Chen, L. , Qin, C. et al. (2022) The single‐cell stereo‐seq reveals region‐specific cell subtypes and transcriptome profiling in Arabidopsis leaves. Dev. Cell, 57, 1299–1310. [DOI] [PubMed] [Google Scholar]
- Xie, J. , Li, M. , Zeng, J. , Li, X. and Zhang, D. (2022) Single‐cell RNA sequencing profiles of stem‐differentiating xylem in poplar. Plant Biotechnol. J. 20, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. , Crow, M. , Rice, B.R. , Li, F. , Harris, B. , Liu, L. , Demesa‐Arevalo, E. et al. (2021) Single‐cell RNA sequencing of developing maize ears facilitates functional analysis and trait candidate gene discovery. Dev. Cell, 56, 557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y. , Wang, Y. , Du, J. , Pei, S. , Guo, S. , Hao, R. , Wang, D. et al. (2022) A DE1 BINDING FACTOR 1‐GLABRA2 module regulates rhamnogalacturonan I biosynthesis in Arabidopsis seed coat mucilage. Plant Cell, 34, 1396–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, X.Y. , Zhao, B. , Chao, L.M. , Chen, D.Y. , Cui, W.R. , Mao, Y.B. , Wang, L.J. et al. (2014) Interaction between two timing MicroRNAs controls trichome distribution in arabidopsis. PLoS Genet. 10, e1004266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, N. , Cai, W.J. , Wang, S. , Shan, C.M. , Wang, L.J. and Chen, X.Y. (2010) Temporal control of trichome distribution by microRNA156‐targeted SPL genes in Arabidopsis thaliana . Plant Cell, 22, 2322–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y. , Wu, S. , Nowak, J. , Wang, G. , Han, L. , Feng, Z. , Mendrinna, A. et al. (2019) Live‐cell imaging of the cytoskeleton in elongating cotton fibres. Nat. Plants, 5, 498–504. [DOI] [PubMed] [Google Scholar]
- Zeng, J. , Zhang, M. , Hou, L. , Bai, W. , Yan, X. , Hou, N. , Wang, H. et al. (2019) Cytokinin inhibits cotton fiber initiation by disrupting PIN3a‐mediated asymmetric accumulation of auxin in the ovule epidermis. J. Exp. Bot. 70, 3139–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M. , Zeng, J.Y. , Long, H. , Xiao, Y.H. , Yan, X.Y. and Pei, Y. (2017) Auxin regulates cotton fiber initiation via GhPIN‐mediated auxin transport. Plant Cell Physiol. 58, 385–397. [DOI] [PubMed] [Google Scholar]
- Zhang, M. , Zheng, X. , Song, S. , Zeng, Q. , Hou, L. , Li, D. , Zhao, J. et al. (2011) Spatiotemporal manipulation of auxin biosynthesis in cotton ovule epidermal cells enhances fiber yield and quality. Nat. Biotechnol. 29, 453–458. [DOI] [PubMed] [Google Scholar]
- Zhang, T. , Hu, Y. , Jiang, W. , Fang, L. , Guan, X. , Chen, J. , Zhang, J. et al. (2015) Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM‐1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537. [DOI] [PubMed] [Google Scholar]
- Zhang, T.Q. , Chen, Y. , Liu, Y. , Lin, W.H. and Wang, J.W. (2021a) Single‐cell transcriptome atlas and chromatin accessibility landscape reveal differentiation trajectories in the rice root. Nat. Commun. 12, 2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T.Q. , Chen, Y. and Wang, J.W. (2021b) A single‐cell analysis of the Arabidopsis vegetative shoot apex. Dev. Cell, 56, 1056–1074. [DOI] [PubMed] [Google Scholar]
- Zhang, T.Q. , Xu, Z.G. , Shang, G.D. and Wang, J.W. (2019) A single‐cell RNA sequencing profiles the developmental landscape of Arabidopsis root. Mol. Plant, 12, 648–660. [DOI] [PubMed] [Google Scholar]
- Zhao, M. , Morohashi, K. , Hatlestad, G. , Grotewold, E. and Lloyd, A. (2008) The TTG1‐bHLH‐MYB complex controls trichome cell fate and patterning through direct targeting of regulatory loci. Development, 135, 1991–1999. [DOI] [PubMed] [Google Scholar]
- Zheng, G.X. , Terry, J.M. , Belgrader, P. , Ryvkin, P. , Bent, Z.W. , Wilson, R. , Ziraldo, S.B. et al. (2017) Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z. , Sun, L. , Zhao, Y. , An, L. , Yan, A. , Meng, X. and Gan, Y. (2013) Zinc Finger Protein 6 (ZFP6) regulates trichome initiation by integrating gibberellin and cytokinin signaling in Arabidopsis thaliana . New Phytol. 198, 699–708. [DOI] [PubMed] [Google Scholar]
- Zhu, Q.H. , Yuan, Y. , Stiller, W. , Jia, Y. , Wang, P. , Pan, Z. , Du, X. et al. (2018) Genetic dissection of the fuzzless seed trait in Gossypium barbadense. J. Exp. Bot. 69, 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong, J. , Wang, L. , Zhu, L. , Bian, L. , Zhang, B. , Chen, X. , Huang, G. et al. (2022) A rice single cell transcriptomic atlas defines the developmental trajectories of rice floret and inflorescence meristems. New Phytol. 234, 494–512. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Dynamic changes on Xu142_LF ovule epidermis during fibre initiation.
Figure S2 Tissue digestion and protoplast isolation.
Figure S3 Comparison between single‐cell RNA‐seq and bulk RNA‐seq of Xu142_LF.
Figure S4 Expression and identification of cluster enriched genes.
Figure S5 RNA in situ hybridization of cell‐type representative genes.
Figure S6 Single‐cell RNA‐seq and clusters identification on Xu142 and Xu142 fl.
Figure S7 Fibre cell cluster identification on the LF combined sample.
Figure S8 Clustering and annotation of four LF samples.
Figure S9 Hierarchical cluster tree showing co‐expression modules identified by WGCNA.
Figure S10 Networks related to fibre cell initiation.
Figure S11 Characterization of cotton CRISPR editing lines on MYB25‐like genes.
Figure S12 Characterization of cotton CRISPR editing lines on PDF2 genes.
Figure S13 Characterization of cotton CRISPR editing lines on HOX3 genes.
Table S1 Summary of single‐cell RNA sequencing results for each sample.
Table S2 Summary of protoplasting induced differentially expressed genes.
Table S3 List of enriched genes for each cluster.
Table S4 Cell number of each cluster identified from Xu142‐vs‐Xu142 fl.
Table S5 KEGG analysis of LF_0d fibre cell enriched genes.
Table S6 Cell barcode mapping between single sample and combined sample.
Table S7 Summary of 167 branch‐dependent genes.
Table S8 GO enrichment analysis of 167 branch‐dependent genes.
Table S9 Summary of cluster‐enriched genes.
Table S10 WGCNA on four LF samples.
Table S11 Weight coefficient of fibre development‐related genes.
Table S12 The primers used in this study.
Appendix S1 Details of bioinformatics analysis.
Data Availability Statement
The scRNA‐seq data have been deposited in the NCBI SRA database (https://www.ncbi.nlm.nih.gov/bioproject/) with BioProject number PRJNA600131.