

Reprogramming of gene transcription and chromatin structure plays vital roles in the modulation of plant meiotic progression (Jiang and Zheng, 2021). However, chromatin profiling is still not fully elucidated in the plant male meiocytes, which is largely due to technical challenges for the collection of enough cells for the large‐scale chromatin immunoprecipitation (ChIP). Therefore, how chromatin modifications affect gene expression in meiosis is still unclear. To address this, we developed a low cell number (10 000 cells) ChIP‐seq, termed as LCNChIP‐seq (Figure 1a), for global profiling of H3K4me3 and H3K27me3 in the rice male meiocytes at prophase I stage. After analysing two well correlated biologically replicated data for each mark (Figure S1), we obtained 25 715 H3K4me3, 16 414 H3K27me3 reproducible peaks and 3598 genomic loci with potential co‐occurrence of both marks, termed as H3K4‐K27me3 bivalent (Figure 1b). Biological reproducibility of each mark is illustrated in Figure 1c. After associating normalized ChIP‐seq read counts of each mark with gene expression levels, we found that the abundance of H3K4me3 and H3K27me3 was correlated and anti‐correlated with gene expression levels, respectively (Figure 1d). H3K4me3/H3K27me3‐only genes had the highest and lowest expression levels (Figure 1e), corresponding to the active and repressive regulation of gene expression, respectively.
Figure 1.
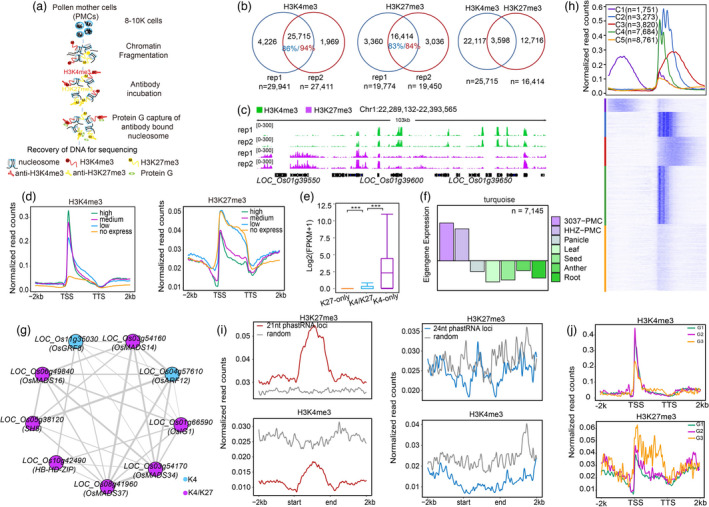
Characterization of H3K4me3 and H3K27me3 in the rice male meiocytes. (a) The flowchart illustrating the main procedures for LCNChIP‐seq. (b) Venn plots illustrating overlaps of H3K4me3 and H3K27me3 peaks between replicates and co‐presence of H3K4me3 and H3K27me3, termed as H3K4‐K27me3, respectively. (c) IGV illustrating the reproducible peaks of H3K4me3 and H3K27me3. (d) Profiling of normalized ChIP‐seq read counts across ±2 kb from the TSSs to TTSs of all genes with different expression levels (FPKM values, high, medium, low and no expression). (e) The boxplot showing the average expression levels of genes with H3K4‐K27me3 and H3K4me3/H3K27me3‐only, respectively. Significance test was determined using Wilcoxon rank sum test, ***P < 0.001. (f) Bar plots showing the consensus expression pattern of the coexpressed genes in the turquoise module. (g) TF network generated from the TF‐cluster analyses. Blue and purple colour dots indicate H3K4me3 and H3K4‐K27me3 marked TFs, respectively. The thickness of grey lines indicates the number of co‐regulatory TFs. (h) Curve plots (upper) and heatmap (bottom) showing the distributions of H3K4me3 around ±2 kb of the TSSs of genes in each subcluster. (i) Profiling of normalized ChIP‐seq read counts across ±2 kb from the start to the end point of reported 21‐nt (left) and 24‐nt (right) phasiRNA encoding loci (Jiang et al., 2020), respectively. (j) The enrichment levels of H3K4me3 or H3K27me3 across ±2 kb from the TSSs to the TTSs of genes in G1 containing genes highly expressed in all species, G2 containing genes highly expressed in rice and G3 containing genes lowly or non‐expressed in rice but highly expressed in the other two species, respectively.
We then conducted WGCNA (weighted correlation network analysis) using 19 RNA‐seq data sets (Figure S2) and identified a MEturquoise module containing 7145 genes highly expressed at the early prophase I (Figure 1f), indicative of their importance at the stage. To investigate how TFs regulate gene expression in pollen mother cells (PMCs), we constructed a hub‐TF (refers to 307 TFs identified in MEturquoise module) centred regulatory network (Figure 1g) and found that a subcluster, which was associated with the flower development, contained 9 co‐regulated TFs, including 4 MADS box TFs (OsMADS14/16/34/37), a TALE TF (SH5). 7 (ca. 78%) TFs were marked by H3K4‐K27me3, and only 2 TFs were enriched with H3K4me3 (Figure S3). These results indicate that a subset of key TFs and their target genes highly expressed at the early prophase I of PMC tend to be differentially regulated by H3K4‐K27me3 and H3K4me3, respectively.
To investigate roles of broad H3K4me3 at the early prophase I of PMCs, we conducted k‐means clustering analyses and obtained five subclusters (C1–C5, k values = 5) with distinct profiles of H3K4me3. We found that genes in C3, containing 849 genes (79 TFs), tended to be enriched with broad H3K4me3 (Figure 1h; Figure S4). GO enrichment analyses showed that they were primarily enriched in GO terms essential for normal meiosis processes in plants (Li et al., 2018), including gluco/glycol/hexo‐syltransferase activities for sugar metabolism, acetyltransferase activities, protein kinase activity, protein ubiquitination and DNA binding activities (Figure S5). These results show that broad H3K4me3 plays vital roles in the regulation of gene expression in meiosis.
21‐nt‐phased small interfering RNAs (phasiRNAs) were accumulated at rice meiocytes and gametes (Jiang et al., 2020; Li et al., 2020; Zhang et al., 2020b). To investigate their chromatin features, we plotted normalized ChIP‐seq read counts across ±2 kb of genomic loci encoding 21 and 24 nt phasiRNAs (Jiang et al., 2020). We found that H3K27me3 was more enriched in 21 nt instead of 24 nt phasiRNAs encoding loci, and we did not observe any enrichment of H3K4me3 in both types of phasiRNAs encoding loci as compared to the random control (Figure 1i). A similar trend was detected in the rice gametes (Li et al., 2020; Figure S6). Moreover, we observed differential enrichment of H3K27me3 between miR172 (Zhu et al., 2009) and OsmiR528 (Zhang et al., 2020a) encoding loci (Figure S7). These results show that H3K4me3 and H3K27me3 are differentially enriched in phasiRNAs or other non‐coding RNAs (ncRNAs) encoding loci in the rice meiotic cells.
After analysing the meiocyte‐related transcriptomic data in Arabidopsis (Walker et al., 2018), maize (Dukowic‐Schulze et al., 2014) and rice (Jiang et al., 2020). We identified 4389 homologous gene pairs with protein sequence identity >50% between rice and the other two species (Figure S8, left). We further got three subclusters, G1 (n = 530) with genes highly expressed in three species, G2 (n = 71) with genes only highly expressed in rice, and G3 (n = 60) with genes highly expressed in maize and Arabidopsis (Figure S8, right). After plotting ChIP‐seq read counts across ±2 kb from the TSSs to TTSs of the genes in each subcluster, we found that genes in G2 had the highest abundance of H3K4me3, by contrast genes in G3 had the lowest abundance of H3K4me3 but the highest abundance of H3K27me3 (Figure 1j). Furthermore, we did similar analyses in maize (Figure S9) and found that the intensity of H3K4me3 did not exhibit a direct correlation with the expression levels of genes in each subcluster (Figure S9). These results indicate that H3K4me3 and/or H3K27me3 may differentially regulate homologous genes at meiotic stages among different species.
Thus, our study provides a turning point for profiling epigenomic landscapes at the early meiotic prophase I or other key developmental stages using low cell input, thereby advancing understanding of epigenomic regulation of plant meiotic processes.
Conflict of interest
No conflict of interest was declared.
Author contributions
W.L.Z. and Z.K.C. conceived and designed the study. A.C.Z. analysed the data. Y.L.S. conducted experiments. H.L.Y., L.C., J.W.C and Y.S. collected cells. Z.K.C. supervised the experiment. W.L.Z. wrote the manuscript.
Supporting information
Appendix S1 Materials and methods.
Figure S1 Correlation analyses of biologically replicated H3K4me3 and H3K27me3 ChIP‐seq datasets.
Figure S2 Principal Component Analysis (PCA) of RNA‐seq datasets generated from different rice tissues (HHZ represents the rice variety Huanghuazhan).
Figure S3 IGV illustrating enrichment of H3K4me3 and H3K27me3 in 9 TFs in the network.
Figure S4 Barlpot showing the length distributions of H3K4me3 peaks.
Figure S5 GO term enrichment analyses of genes with broad H3K4me3.
Figure S6 Profiling of normalized H3K4me3 and H3K27me3 read counts from 2 kb upstream of the start point to 2 kb downstream of the end point of 21‐nt phasiRNA encoding loci (left) and 24‐nt phasiRNA encoding loci (right), respectively, which were reported in the rice gametes (Li et al., 2020).
Figure S7 IGV snapshots showing abundance of H3K4me3 and H3K27me3 in miR172d and OsmiR528.
Figure S8 Homologous gene pairs in three plant species.
Figure S9 Homologous gene pairs in the maize meiocytes at early prophase I.
Acknowledgements
We thank the Bioinformatics Center, Nanjing Agricultural University for providing computing facilities for data processing and analyses. This research was supported by grants from the National Natural Science Foundation of China (32070561 and U20A2030). The Fundamental Research Funds for the Central Universities (KYZZ2022003).
Contributor Information
Zhukuan Cheng, Email: zkcheng@genetics.ac.cn.
Wenli Zhang, Email: wzhang25@njau.edu.cn.
Data availability statement
The ChIP‐seq data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number: GSE203393.
References
- Dukowic‐Schulze, S. , Sundararajan, A. , Mudge, J. , Ramaraj, T. , Farmer, A.D. , Wang, M. , Sun, Q. et al. (2014) The transcriptome landscape of early maize meiosis. BMC Plant Biol. 14, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, P. , Lian, B. , Liu, C. , Fu, Z. , Shen, Y. , Cheng, Z. and Qi, Y. (2020) 21‐nt phasiRNAs direct target mRNA cleavage in rice male germ cells. Nat. Commun. 11, 5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, T. and Zheng, B. (2021) Epigenetic regulation of megaspore mother cell formation. Front. Plant Sci. 12, 826871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. , Xu, H. , Fu, F.F. , Russell, S.D. , Sundaresan, V. and Gent, J.I. (2020) Genome‐wide redistribution of 24‐nt siRNAs in rice gametes. Genome Res. 30, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Ye, J. , Ma, H. and Lu, P. (2018) Proteomic analysis of lysine acetylation provides strong evidence for involvement of acetylated proteins in plant meiosis and tapetum function. Plant J. 93, 142–154. [DOI] [PubMed] [Google Scholar]
- Walker, J. , Gao, H. , Zhang, J. , Aldridge, B. , Vickers, M. , Higgins, J.D. and Feng, X. (2018) Sexual‐lineage‐specific DNA methylation regulates meiosis in Arabidopsis . Nat. Genet. 50, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.C. , He, R.R. , Lian, J.P. , Zhou, Y.F. , Zhang, F. , Li, Q.F. , Yu, Y. et al. (2020a) OsmiR528 regulates rice‐pollen intine formation by targeting an uclacyanin to influence flavonoid metabolism. Proc. Natl. Acad. Sci. USA, 117, 727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.C. , Lei, M.Q. , Zhou, Y.F. , Yang, Y.W. , Lian, J.P. , Yu, Y. , Feng, Y.Z. et al. (2020b) Reproductive phasiRNAs regulate reprogramming of gene expression and meiotic progression in rice. Nat. Commun. 11, 6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Q.H. , Upadhyaya, N.M. , Gubler, F. and Helliwell, C.A. (2009) Over‐expression of miR172 causes loss of spikelet determinacy and floral organ abnormalities in rice (Oryza sativa). BMC Plant Biol. 9, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Materials and methods.
Figure S1 Correlation analyses of biologically replicated H3K4me3 and H3K27me3 ChIP‐seq datasets.
Figure S2 Principal Component Analysis (PCA) of RNA‐seq datasets generated from different rice tissues (HHZ represents the rice variety Huanghuazhan).
Figure S3 IGV illustrating enrichment of H3K4me3 and H3K27me3 in 9 TFs in the network.
Figure S4 Barlpot showing the length distributions of H3K4me3 peaks.
Figure S5 GO term enrichment analyses of genes with broad H3K4me3.
Figure S6 Profiling of normalized H3K4me3 and H3K27me3 read counts from 2 kb upstream of the start point to 2 kb downstream of the end point of 21‐nt phasiRNA encoding loci (left) and 24‐nt phasiRNA encoding loci (right), respectively, which were reported in the rice gametes (Li et al., 2020).
Figure S7 IGV snapshots showing abundance of H3K4me3 and H3K27me3 in miR172d and OsmiR528.
Figure S8 Homologous gene pairs in three plant species.
Figure S9 Homologous gene pairs in the maize meiocytes at early prophase I.
Data Availability Statement
The ChIP‐seq data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number: GSE203393.