

Summary
The regulation of gene expression plays an essential role in both the phenotype and adaptation of plants. Transcriptome sequencing enables simultaneous identification of exonic variants and quantification of gene expression. Here, we sequenced the leaf transcriptomes of 287 rice accessions from around the world and obtained a total of 177 853 high‐quality single nucleotide polymorphisms after filtering. Genome‐wide association study identified 44 354 expression quantitative trait loci (eQTLs), which regulate the expression of 13 201 genes, as well as 17 local eQTL hotspots and 96 distant eQTL hotspots. Furthermore, a transcriptome‐wide association study screened 21 candidate genes for starch content in the flag leaves at the heading stage. HS002 was identified as a significant distant eQTL hotspot with five downstream genes enriched for diterpene antitoxin synthesis. Co‐expression analysis, eQTL analysis, and linkage mapping together demonstrated that bHLH026 acts as a key regulator to activate the expression of downstream genes. The transgenic assay revealed that bHLH026 is an important regulator of diterpenoid antitoxin synthesis and enhances the disease resistance of rice. These findings improve our knowledge of the regulatory mechanisms of gene expression variation and complex regulatory networks of the rice genome and will facilitate genetic improvement of cultivated rice varieties.
Keywords: population RNA‐seq, expression quantitative trait loci (eQTLs), diterpenoid antitoxin, key regulators, transcriptome‐wide association study (TWAS)
Introduction
The phenotypic polymorphism of a species is generally defined by genetic variations, and the associations between them are established through genetic methods such as genome‐wide association studies (GWAS). (Huang et al., 2012; Wang et al., 2015). Gene expression is an essential molecular mechanism linking genomic polymorphisms and the phenotype of the organism, and its regulation has long been studied and plays important role in the phenotypic variations of various organisms (Fu et al., 2021; Wang et al., 2018). Identification of expression quantitative trait loci (eQTLs) that affect gene expression levels is critical to understanding how genomic variations regulate gene expression levels, and how information on the genome is transmitted to morphological phenotypes through the genetic central dogma. With the advancement of technology and decrease in the cost of next‐generation sequencing, many natural populations have been frequently used for eQTL studies (Albert and Kruglyak, 2015; Fu et al., 2013; Li et al., 2020; Zhang et al., 2017), providing novel and important insights into the genetic basis of natural variations at the transcriptome level and the influence of gene expression in phenotypic variations.
Variations in gene expression usually arise from cis‐ and/or trans‐regulation (Wittkopp et al., 2004). Cis‐regulation tends to occur in the vicinity of the target gene and affects gene expression levels with variations in various cis‐acting elements, and cis‐regulatory variations are usually detected as local eQTLs in natural populations. In contrast, trans‐regulation acts at a certain distance, usually in the form of transcription factors (Zhang et al., 2017) or certain metabolites (Wang et al., 2018), which affect the expression of downstream genes; besides, trans‐regulatory variants are usually detected as distant eQTLs in natural populations. Previous studies have suggested that local eQTLs tend to explain more variations than distant eQTLs and play a major role in determining the variations in gene expression (Cubillos et al., 2012; Kliebenstein, 2009). However, many other studies have demonstrated that distant regulation at the transcriptional level is also important for normal plant development (Narula and Igoshin, 2010; van Heyningen and Bickmore, 2013; Xiang et al., 2014). In addition, hotspots of distant eQTLs (trans‐regulation) are thought to comprise key regulators, which regulate the expression of a wide range of development‐ and/or metabolism‐related downstream genes (Li et al., 2020; Wang et al., 2018; Zhang et al., 2017).
Rice (Oryza sativa L.) is a major cereal crop in Asia, as well as a major model crop for genetic improvement. Large amounts of high‐density genotype data and related high‐throughput phenotypic data of rice have been accumulated and associated with many important candidate genes through GWAS approaches (Huang et al., 2012; Wang et al., 2015). GWAS can identify candidate associations by detecting variations across genotypes and phenotypes, but cannot accurately identify candidate genes and gene functions due to the decay of linkage disequilibrium and limited gene annotation. In recent studies, some researchers have used methods such as transcriptome‐wide association study (TWAS) or Camoco that can combine genomic and transcriptomic data (Gusev et al., 2016; Schaefer et al., 2018) to more accurately predict the candidate genes for phenotypic variations (Tang et al., 2021; Walker et al., 2019). eQTLs and co‐localized QTLs are equally important for dissecting the genetic architecture of complex traits (Giambartolomei et al., 2014). For example, by combining GWAS with eQTL data, some studies of the growth and phenotypic variations in poplar (Drost et al., 2010) identified multiple candidate genes for lettuce leaf color (Zhang et al., 2017), and a combined approach using eQTLs and metabolic QTLs revealed the history of metabolic breeding in tomato (Zhu et al., 2018). Such studies are expected to enhance the comprehension of regulatory strategies of plants and facilitate a more accurate explanation of related mechanisms.
Here, we analysed the transcriptomes of flag leaves at the heading stage in 287 cultivated rice accessions. Subsequently, we identified 44 354 eQTLs regulating the expression of 13 201 genes, as well as 17 local and 96 distant eQTL hotspots. A key transcription factor, bHLH026, was identified in a distant eQTL hotspot (HS002), which activates the expression of downstream genes related to the synthesis of diterpenoid antitoxins. A transgenic assay revealed that bHLH026 affects the metabolic level of diterpenoid antitoxins and disease resistance in rice. The findings will enhance our comprehension of the regulatory mechanisms of transcriptomic variations and the complex regulatory network of the rice genome, and facilitate future genetic improvement of cultivated rice varieties.
Results
Transcriptome sequencing and exonic SNP identification
A total of 287 accessions of O. sativa, which represent both landraces and elite germplasms from all over the world, were selected from 533 minicore germplasms (Xie et al., 2015) for genome‐wide analysis (Table S1). RNA was extracted from the top fully expanded leaves at the heading stage from each accession. Transcriptome sequencing generated 11 billion paired‐end reads with an average of 38 million reads for each accession after the removal of low‐quality reads. The obtained reads from each accession were mapped to the Oryza genome (MSU 7.0) (Kawahara et al., 2013) to quantify the gene expression levels, and the average mapping rate of unique reads was 70.07% (Table S2).
Based on the mapping results, 177 853 high‐quality single nucleotide polymorphisms (SNPs) were detected using a range of filtering approaches. As expected, these SNPs were mostly located within genes since they were derived from RNA‐seq data. Then, the SNPs were used to analyse the population structure of the 287 accessions with Bayesian clustering software. By progressively increasing the number of clusters (K), the 287 accessions were divided into different subpopulations (Figure 1a), and the lowest CV error was observed at K = 9 (Figure S1). Indica, japonica, and Aus subpopulations were clearly observed at K = 3. Indica was further divided into indica I and indica II subpopulations. When K = 6, japonica was further divided into tropical japonica and temperate japonica subpopulations.
Figure 1.
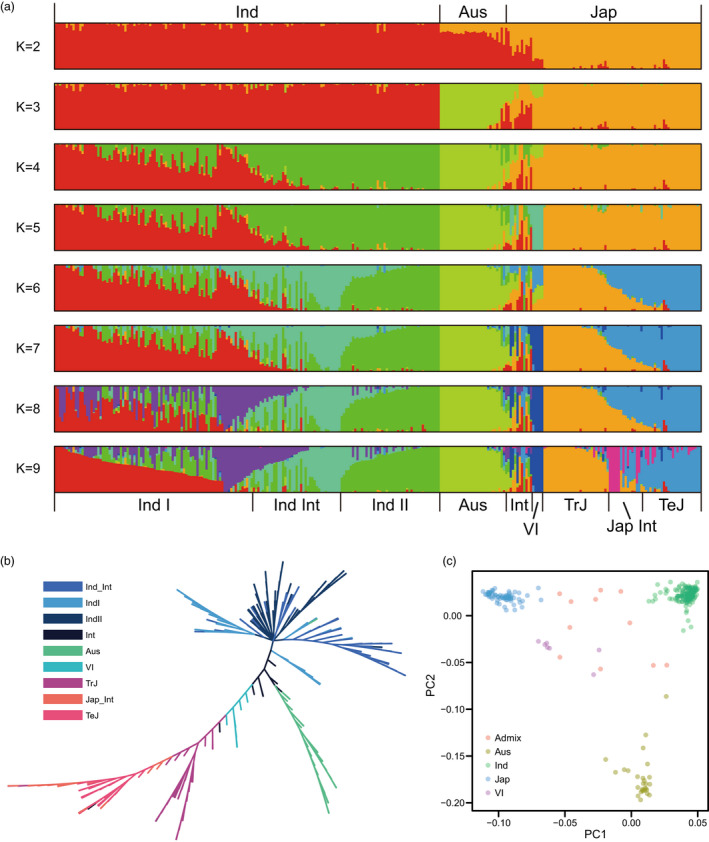
Population structure of 287 rice accessions from all over the world. (a) NJ (Neighbour‐Joining) tree of 287 rice accessions constructed from simple matching distances of genome‐wide SNPs. (b) Principal component analysis revealed that the first two principal components could explain approximately 57.6% of the genetic variations within the 287 rice accessions. (c) Genetic structure of the 287 rice accessions analysed with the ADMIXTURE program.
The maximum‐likelihood phylogenetic tree was established and the phylogenetic relationships among 287 accessions were analysed (Figure 1b). As a result, indica, japonica, and Aus subpopulations were located in different branches. Principal component analysis (PCA) also supported the phylogenetic relationships among different accessions and confirmed the clustering of indica, japonica, and Aus subpopulations (Figure 1c). These results obtained using RNA‐seq calling SNPs were consistent with the results in previous studies which quantified the population structure of O. sativa, and confirmed that our panel could capture abundant genetic variations of rice germplasm (Xie et al., 2015; Zhou et al., 2017).
Genome‐wide mapping of eQTLs
Expression quantitative trait loci mapping is a powerful approach to identifying the expression variation of each gene as well as revealing the regulatory network of genes for corresponding traits (Fu et al., 2013; Zhang et al., 2017). Here, a quantitative analysis of transcriptome data identified 23 325 genes expressed in leaves at the heading stage, accounting for about 41.8% of the total annotated genes (55 801) in the MSU 7.0 genome. Using the Fast‐LMM software (Lippert et al., 2011), with genomic SNP data from 287 accessions, the transformed expression levels of each gene were used for association analysis with the SNPs in the genome. As a result, the expression of 14 562 genes was significantly associated with at least one SNP over the Bonferroni‐corrected threshold (P = 5.43 × 10−8; α = 0.05). SNPs associated with the same gene were clustered into one unique eQTL block including at least three SNPs, and the SNP with the lowest P‐value was used to represent this block. A total of 44 354 eQTLs were identified from 13 201 genes (Table S3).
According to the relative positions of genes and their corresponding eQTLs in the genome, a strong diagonal enrichment could be observed (Figure 2a). In addition, based on the relative distance between eQTLs and genes, all eQTLs could be divided into 19 549 local eQTLs (<100 kb) and 24 805 distant eQTLs (>100 kb or on different chromosomes), and 74.6% of the genes had local eQTLs (Figure 2a, d; Table S3). A comparison of the P‐values and explanation rate (r 2) of SNPs in the association analysis of local and distant eQTLs revealed that local eQTLs have a greater effect on gene expression variations than distant eQTLs (two‐sided Wilcoxon rank sum test, P‐value <2.2 × 10−16; Figure 2b, c). Therefore, local‐regulatory effects may play a leading role in determining the expression variations of most genes, which is consistent with previous findings in other organisms (Wang et al., 2018; Zhang et al., 2017). In terms of eGenes (genes regulated by eQTL), 9853 eGenes were regulated by local eQTLs; 8427 eGenes were regulated by distant eQTLs; and 5079 eGenes (38.5%) were regulated by both local eQTLs and distant eQTLs (Figure 2d). On average, each eGene corresponded to 3.4 eQTLs, and only 4446 eGene (33.7%) were regulated by one single eQTL, while the majority of eGenes were regulated by multiple eQTLs, suggesting that the expression of most genes in rice is under complex genetic regulation (Figure 2e). As for the location of local eQTLs relative to their eGenes, most of the lead SNPs were located in or around the gene body of the eGenes; interestingly, local eQTLs had two peaks in the 5′‐ and 3′‐regions, and the peak in the 5′‐region was more prominent (Figure S2), indicating that the 5′‐sequences may play a more important role in regulating gene expression or stabilizing mRNA. The distribution of local eQTLs in the 5′ promoter region was observed. It was found that the distribution of local eQTLs gradually decreased as they moved away from the transcription start site (TSS). About one‐third of the local eQTLs fell in the first 10 kb of the TSS, and 60.5% of them fell in the first 30 kb of the TSS (Figure 2f).
Figure 2.
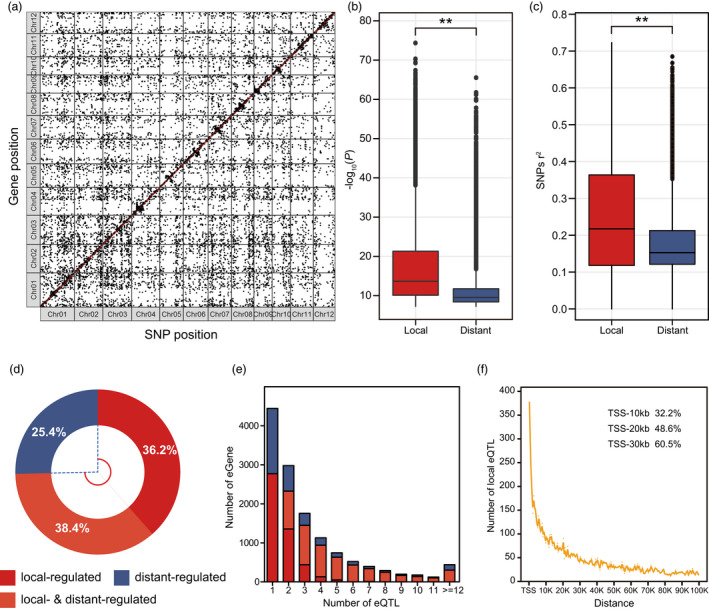
Identification of eQTLs using RNA‐Seq data from rice flag leaves at the heading stage. (a) Distribution of eQTLs and their regulatory genes on 12 chromosomes. The x‐axis is the single nucleotide polymorphism (SNP) position (bp) for each chromosome and the y‐axis is the gene position (bp) on each chromosome. Each black dot indicates a distant eQTL, and a red dot on the diagonal line indicates a local eQTL. (b) Comparison of the −log10(P) values of local and distant eQTLs. Box plots show the distribution quantiles. Two‐sided Wilcoxon rank sum test, **P‐value <2.2 × 10−16. (c) Comparison of explanation rate (r2) of SNPs for expression variation between local eQTL and distant eQTL. Two‐sided Wilcoxon rank sum test, **P‐value <2.2 × 10−16. (d) Distribution of genes regulated by local and/or distant eQTLs. (e) Distribution of the number of eGenes. Blue bars indicate genes regulated by distant eQTLs, red bars indicate genes regulated by local eQTLs, and orange bars indicate genes regulated by both distant and local eQTLs. (f) Distribution of local eQTLs in the promoter region.
Identification of local eQTLs and co‐regulated gene clusters in rice.
As described above, local eQTLs usually have a greater impact on the expression variation of eGenes. To further explore the genes regulated by local eQTLs and their biological functions, a gene enrichment analysis was performed on the identified genes with local eQTLs. In the KEGG database, these genes showed the most significant enrichment in secondary metabolite synthesis, followed by the transcriptional and translational pathways of mRNA (Figure S3). An analysis of their metabolic pathways revealed that these genes were also enriched in the pathways of cytokinin and brassinosteroid synthesis (Figure S3). Moreover, gene ontology (GO) enrichment analysis revealed that these genes with local eQTLs were enriched in GO terms of protein modification, cell death, and stress response (Figure S4). These results suggested that these genes with local eQTLs regulation probably play certain roles in secondary metabolite synthesis, protein modification, and hormone synthesis, which are processes highly responsive to the environment. Similarly, more variations in sequence and expression were found in genes involved in secondary metabolism in previous studies (Gan et al., 2011; Moore et al., 2014; Wang et al., 2018).
As reported in earlier research, a genomic region may contain a large number of eQTLs and affect the expression of multiple genes, that is, this region harbors an eQTL hotspot (Albert and Kruglyak, 2015; Fu et al., 2013; Li et al., 2020; Zhang et al., 2017). An examination of the distribution of local eQTLs showed that they were unevenly distributed across the genome. We identified 17 local eQTL hotspots by the hot_scan program (Silva et al., 2014), most of which were located at the ends of chromosomes (Table S4). Interestingly, the heat map of local eQTL distribution demonstrated that there were very few local eQTLs in the centromere region of each chromosome (Figure 3a), possibly due to the suppression of gene expression (Wu et al., 2011) or gene escape from the centromere region (Liao et al., 2018).
Figure 3.
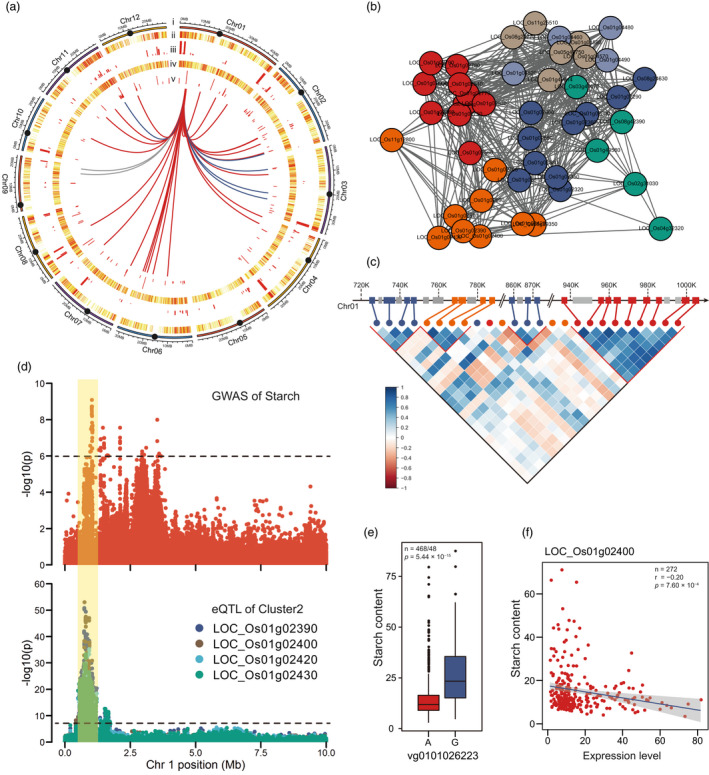
Regulatory hotspots of local eQTLs and their associations with phenotypic variations. (a) Distribution of local eQTLs and distant eQTLs on the whole genome. (i) 12 chromosomes in rice, solid black circles indicate centromeres; (ii) heatmap showing the number of local eQTLs in a 200‐kb window along the chromosome; (iii) histogram showing the number of eQTLs in each local eQTL hotspot; (iv) heatmap showing the number of distant eQTLs in a 200‐kb window along the chromosome; (v) histogram showing the number of eQTLs in each distant eQTL hotspot. (vi) Curves showing the association of the distant eQTL hotspot HS002 with downstream genes. The red and blue lines indicate different expression patterns (Figure 4b). (b) Co‐expression network of 50 genes related to sn‐glycerol 3‐phosphate synthesis. Different colors indicate different expression patterns. (c) The local eQTL hotspot L01 contains four co‐regulated gene clusters. The upper part of the image shows the location of genes in L01, with different colored rectangles indicating genes with different expression patterns. The lower part of the image shows the correlation of genes in the hotspot in inverted triangles, with the red triangles highlighting the correlation of gene expression within the four co‐regulated gene clusters. (d) Manhattan plots of GWAS for starch content and eQTLs for co‐regulated gene clusters (Cluster 2) on chromosome 1. The horizontal dashed line indicates the significance threshold (1/Me; 6.0). (e) Divergence of starch content between different alleles of the lead SNP. (f) Levels of gene expression (in the case of LOC_Os01g02400) were negatively correlated with starch content.
Enrichment analysis of the 17 local eQTL hotspots and overlap analysis with metabolic gene clusters demonstrated that several hotspots were associated with metabolic pathways (Table S4). Among them, the hotspot L01 comprised 26 genes related to sn‐glycerol 3‐phosphate synthesis. Genes in close proximity to each other and under the regulation of local eQTLs were found to form co‐regulated gene clusters, and the genes in the same cluster were found to have similar expression patterns and functions in maize (Wang et al., 2018). Subsequently, we examined the co‐expression of all annotated genes in the sn‐glycerol 3‐phosphate synthesis, which showed six different expression patterns (Figure 3b; Table S5) and four co‐regulated gene clusters in the hotspot L01 (Figure 3C; Table S6). Similarly, six brassinosteroid synthesis‐associated genes were detected in the hotspot L08, and co‐expression analysis of 43 brassinosteroid synthesis‐associated genes revealed three distinct expression patterns (Figure S5; Table S7), and one co‐regulated gene cluster was identified in the hotspot L08 (Figure S5). These results suggested that such clusters of genes co‐regulated by the same local eQTLs are also widespread in rice.
Explanation of phenotypic changes by combining genomic and transcriptomic variations
For the 287 accessions, we also determined the starch content in the flag leaves at the heading stage. As a result, 25 key loci associated with starch content in flag leaves were identified using the GWAS approach based on the same SNP datasets as the eQTL identification (Figure S6; Table S8). Subsequent TWAS analysis combined with the transcriptome expression data and phenotypic data detected 21 candidate genes significantly associated with starch content in rice flag leaves at the heading stage (Table 1; Figure S7) by strict thresholds (FDR‐corrected P‐value ≤ 0.05).
Table 1.
Results of transcriptome‐wide association study (TWAS) for starch content in the flag leaves at the heading stage
| Gene_ID | CHR | bp | P_value | FDR | Symbols | Annotation |
|---|---|---|---|---|---|---|
| LOC_Os02g02670 | Chr02 | 991 974 | 1.10 × 10−8 | 0.00014514 | NBS‐LRR disease resistance protein | |
| LOC_Os05g38950 | Chr05 | 22 838 503 | 1.24 × 10−8 | 0.00014514 | TBC domain‐containing protein | |
| LOC_Os09g27650 | Chr09 | 16 823 960 | 7.31 × 10−8 | 0.0005681 | OsIDD13 | ZOS9‐14‐C2H2 zinc finger protein |
| LOC_Os02g02690 | Chr02 | 1 010 137 | 2.77 × 10−7 | 0.00161314 | Expressed protein | |
| LOC_Os01g04920 | Chr01 | 2 276 969 | 1.24 × 10−6 | 0.00482646 | OsSQD2 | Glycosyl transferase, group 1 domain‐containing protein |
| LOC_Os05g45770 | Chr05 | 26 508 840 | 1.22 × 10−6 | 0.00482646 | Divergent PAP2 family domain‐containing protein | |
| LOC_Os02g56120 | Chr02 | 34 349 219 | 1.66 × 10−6 | 0.00483869 | OsIAA9 | OsIAA9‐Auxin‐responsive Aux/IAA gene family member |
| LOC_Os07g14700 | Chr07 | 8 382 541 | 1.57 × 10−6 | 0.00483869 | Harpin‐induced protein 1 domain‐containing protein | |
| LOC_Os02g27190 | Chr02 | 16 001 144 | 2.02 × 10−6 | 0.00522673 | Expressed protein | |
| LOC_Os02g38040 | Chr02 | 22 980 200 | 3.92 × 10−6 | 0.0091447 | OsIRL2 | Leucine‐rich repeat family protein, putative |
| LOC_Os03g29170 | Chr03 | 16 574 889 | 1.43 × 10−5 | 0.02874069 | Sterol‐4‐alpha‐carboxylate 3‐dehydrogenase | |
| LOC_Os08g06480 | Chr08 | 3 671 452 | 1.48 × 10−5 | 0.02874069 | OsTPL | Lissencephaly type‐1‐like homology motif, putative |
| LOC_Os04g32340 | Chr04 | 19 387 504 | 1.98 × 10−5 | 0.03046124 | C3H27 | RNA‐binding motif protein, putative |
| LOC_Os07g14160 | Chr07 | 8 080 858 | 2.09 × 10−5 | 0.03046124 | Polygalacturonase | |
| LOC_Os08g07970 | Chr08 | 4 510 288 | 2.06 × 10−5 | 0.03046124 | OsbZIP64 | Transcription factor |
| LOC_Os08g39370 | Chr08 | 24 884 637 | 1.87 × 10−5 | 0.03046124 | Citrate transporter | |
| LOC_Os06g08440 | Chr06 | 4 139 955 | 2.70 × 10−5 | 0.03703781 | OsRR22 | Two‐component response regulator |
| LOC_Os01g02700 | Chr01 | 922 311 | 3.89 × 10−5 | 0.04322466 | Protein kinase domain‐containing protein | |
| LOC_Os02g45850 | Chr02 | 27 933 760 | 3.57 × 10−5 | 0.04322466 | RAV6 | B3 DNA binding domain‐containing protein |
| LOC_Os07g14350 | Chr07 | 8 196 955 | 3.88 × 10−5 | 0.04322466 | OsLLB | Methyltransferase |
| LOC_Os07g37920 | Chr07 | 22 756 369 | 3.56 × 10−5 | 0.04322466 | ONAC010 | No apical meristem protein |
Generally, it is difficult to identify genes from QTL intervals as a result of the large LD intervals in rice and some other influencing factors such as artificial selection. The range of candidate genes can be narrowed through genomic annotation, the correlation between gene expression and phenotype, and co‐localization of the eQTLs of priori genes and GWAS results (Tang et al., 2021; Walker et al., 2019). Therefore, we used the fusion (Gusev et al., 2016) software to detect the correlation between the gene expression and phenotype, and employed the coloc software (Giambartolomei et al., 2014) to determine the co‐localization between eQTLs and GWAS results. A total of 338 candidate genes significantly associated with the phenotype were identified (Fusion TWAS P ≤ 0.01) by the fusion software, among which 120 were also detected (COLOC.PP3 > 0.7) by coloc software (Table S9).
In particular, the local eQTL hotspot L01 was very close to the GWAS QTL LS01 for starch content (Table S4; Table S8), and the eQTLs for four genes related to sn‐glycerol 3‐phosphate synthesis in Cluster 2 of co‐regulated genes in the eQTL hotspot L01 were overlapped with the GWAS results for starch content (Figure 3d). In addition, the lead SNP vg0101026223 was significant in the GWAS results of starch content (Figure 3e) as well as in the eQTLs for the four genes (LOC_Os01g02390: P = 4.17 × 10−9; LOC_Os01g02400: P = 2.00 × 10−8; LOC_Os01g02420: P = 7.10 × 10−7; LOC_Os01g02430: P = 5.20 × 10−6) in the co‐regulated gene Cluster 2. Therefore, it could be speculated that QTL LS01 affects rice starch content by influencing the expression of the four sn‐glycerol 3‐phosphate synthesis‐related genes. Finally, we examined the correlation between gene expression and phenotype, finding that the expression levels of all four genes were negatively correlated with starch content (Figure 3f; Figure S8).
Identification of distant eQTL hotspots and a key regulator of diterpene antitoxin synthesis
An analysis of the distant eQTLs identified in the whole genome resulted in the identification of 96 distant eQTL hotspots, which involved the regulation of 1726 genes (Figure 3a; Table S10). For each hotspot, the number of eQTLs varied from 12 to 51. Hotspot analysis results could improve the understanding of complex regulatory networks and reveal that there are key regulators of multiple downstream eGenes in these hotspots.
A distant eQTL hotspot, HS002, was identified on chromosome 1, which included a total of 32 downstream genes (Figure 3a). GO enrichment analysis revealed that these genes were enriched in the lipid metabolism pathway (Figure 4a). Further analysis of these enriched genes revealed the presence of some important genes for the synthesis of diterpenoid antitoxins, such as CPS2, KSL5, and KSL6. Previous studies have demonstrated that the key regulators in distant eQTL hotspots tend to influence their own expression through a cis‐acting mechanism, which in turn affects the expression of their downstream genes. For this reason, the master regulators tend to have the same expression patterns as their downstream genes (Li et al., 2020; Wang et al., 2018). Therefore, to find reliable master regulators in the H002 hotspot region, we performed a co‐expression analysis of the genes in this hotspot and the 32 downstream genes. As a result, all the genes related to the synthesis of diterpenoid antitoxins were co‐expressed and clustered in the red module. In addition, a bHLH‐like transcription factor bHLH026 (LOC_Os01g09930) in the HS002 hotspot region also fell into the co‐expressed red module (Figure 4b). Therefore, bHLH026 was considered a potential master regulator in this hotspot to regulate the expression of multiple downstream genes related to the synthesis of diterpenoid antitoxins.
Figure 4.
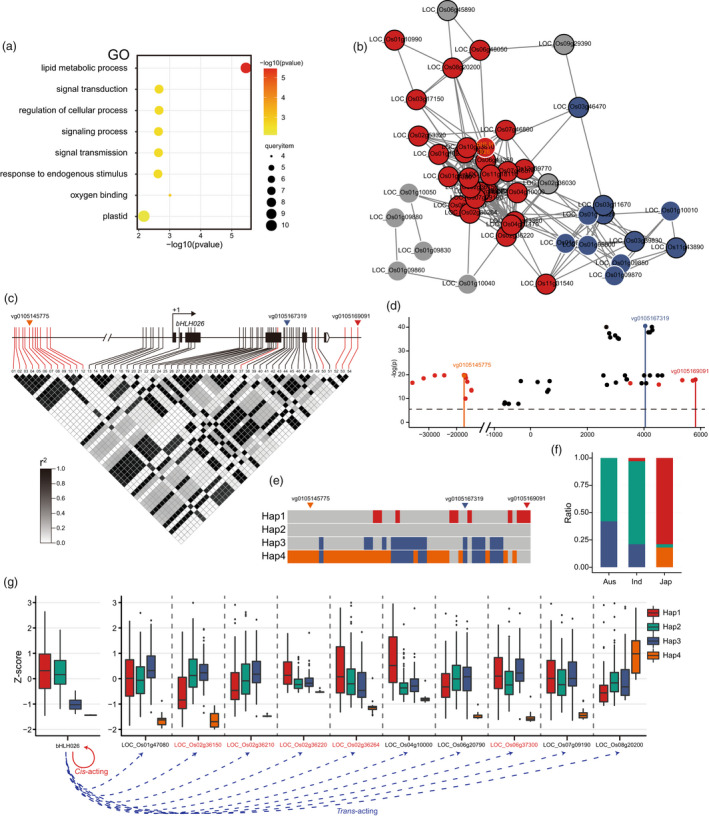
Characterization of the distant eQTL hotspot 002 (HS002) on chromosome 1. (a) GO (biological process) enrichment analysis of downstream genes regulated by HS002 on chromosome 1. (b) Co‐expression analysis of downstream genes regulated by HS002 and genes within the hotspot. The red and blue circles represent genes with different expression patterns. The black outer circle indicates downstream genes regulated by HS002. The white outer circle indicates genes within HS002. (c) Linkage disequilibrium of important SNPs in the vicinity of the master regulator bHLH026. Three small inverted triangles show the lead SNPs of the local eQTLs for bHLH026. The SNPs marked in red are the lead SNPs of the eQTLs for the downstream genes. (d) Significance levels of important SNPs near the master regulator bHLH026 in the local eQTL of bHLH026. The SNPs marked in red are the lead SNPs of the eQTLs for the downstream genes. The horizontal dashed line indicates the significance threshold (1/Me; 6). The zero point on the x‐axis indicates the translation start site of bHLH026. (e) Haplotype analysis of bHLH026. (f) Distribution of bHLH026 haplotypes in three major subpopulations. (g) Normalized expression (Z‐score) of bHLH026 and representative downstream genes in four different bHLH026 haplotypes. The genes marked in red are genes related to the synthesis of diterpenoid antitoxins.
To verify whether bHLH026 regulates its own expression and thus affects the expression of downstream genes by affecting its own local eQTLs, we further performed an eQTL analysis on bHLH026. As a result, bHLH026 had three mutually unlinked local eQTLs (Figure S9), and one lead SNP vg1015145775, which was about 20 kb upstream of bHLH026, was tightly linked with the lead SNP of the distant eQTL for downstream lipid metabolism‐related genes (Figure 4c). The lead SNPs of the distant eQTL for downstream genes also reached significant levels in the local eQTL for bHLH026 and were tightly linked to vg1015145775 (Figure 4d). Therefore, it could be speculated that the variation in this linkage region of vg1015145775 may affect the expression of master regulators in the HS002 hotspot and thus the expression of its downstream genes (Figure 4c, d). Moreover, the linkage disequilibrium map showed that vg1015145775 was tightly linked to nonsynonymous mutations in the third exon of bHLH026. Therefore, nonsynonymous mutations within these genes may also affect the expression of their downstream genes. Subsequently, we performed a haplotype analysis of bHLH026, and finally, bHLH026 was classified into four major haplotypes (Figure 4e). Two haplotypes, Hap2 and Hap3, were mainly distributed in indica and Aus, while the other two haplotypes, Hap1 and Hap4, were only distributed in japonica (Figure 4f). Based on the haplotype typing of bHLH026, the expression of downstream genes in each haplotype was also examined. The expression of bHLH026 in the Hap1 haplotype was significantly higher than that in the Hap4 haplotype in the japonica subpopulation, and all the downstream genes related to lipid metabolism exhibited the same expression pattern as bHLH026, except for LOC_08g20200, which showed an opposite expression pattern (Figure 4g). However, no significant difference in expression of downstream genes was found between the Hap2 and Hap3 haplotypes of indica and Aus (Figure 4g), possibly because of a nonsynonymous mutation in bHLH026 linked to vg1015145775, which affects the function of bHLH026 and thus has a greater impact on downstream genes.
bHLH026 activated the expression of downstream genes
To further confirm the regulatory effect of bHLH026 on downstream genes, we cloned the 2‐kb region before the transcription start site of CPS2 and KSL6 genes as their promoter regions and performed a yeast one‐hybrid assay. The results showed that bHLH026 could bind to the promoter region of CPS2 and KSL6 (Figure 5a). Then, a dual luciferase activity assay of the bHLH026 protein was performed using GAL4 binding specific sequences and the binding domain of fused GAL4. The results showed that bHLH026 has transcriptional activation activity (Figure 5b). Dual luciferase activity assay using the promoter sequences of CPS2 and KSL6 and bHLH026 protein also demonstrated that bHLH026 protein could bind to the promoter region of downstream genes and activate their expression (Figure 5b).
Figure 5.
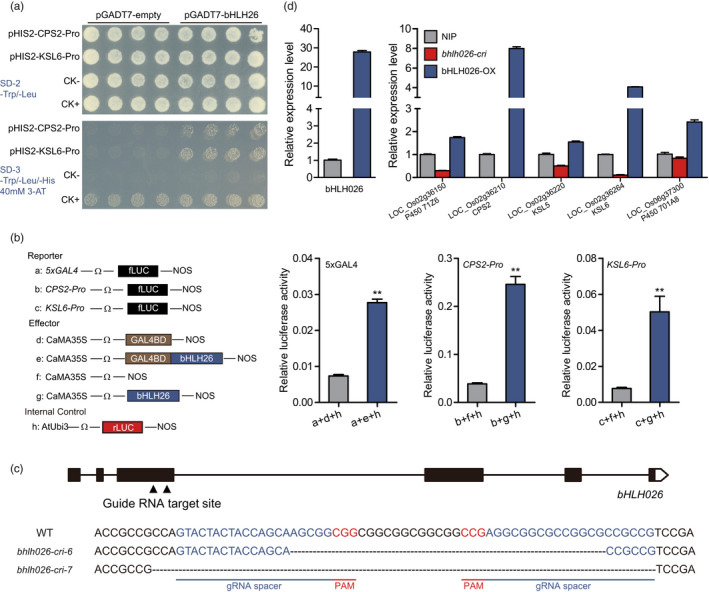
Regulation of bHLH026 on downstream gene promoters and identification of bHLH026 transgenic material. (a) Yeast‐one‐hybrid assay revealed that bHLH026 can bind to the promoter regions of CPS2 and KSL6. pGADT7‐53 and pHIS2‐53 were used as a positive control (CK+), while pGADT7 and pHIS2‐53 were used as a negative control (CK−). (b) Dual luciferase activation assay of bHLH026 shows that bHLH026 can activate the expression of downstream genes. The left is the scheme of the constructs used in the rice protoplast co‐transfection assay. The value of relative luciferase activity in each column represents the mean of three independent replicates, and the error bars indicate the standard deviation. **, P < 0.01 (Student's t‐test). (c) Schematic diagram of the gene structure of bHLH026 and the DNA sequence of the cas9‐edited bHLH026 gene. Triangles and underline‐highlighted regions indicate target sites for Cas9 guide RNA. (d) Transcript levels of bHLH026 and its downstream genes related to diterpenoid antitoxins in leaves of wild‐type and transgenic material. Error bars represent standard deviation (n ≥ 6 each).
bHLH026 affected the synthesis of diterpenoid antitoxins and disease resistance in rice
CPS2 is a key gene for the production of more specialized ent‐CPP in diterpene metabolism in rice (Krishnan et al., 2009; Lu et al., 2018), and kaurene synthase‐like (KSL) is a class of diterpenes that synthesize different families of diterpenes derived from ent‐ or syn‐CPP (Lu et al., 2018). The hotspot analysis revealed that CPS2, KSL5, KSL6, cytochrome P450 701A8, and cytochrome P450 71Z6 were downstream genes of the bHLH026 transcription factor, and thus it can be speculated that bHLH026 may be related to the synthesis of diterpenoid antitoxins and disease resistance of rice.
To further investigate the biological function of bHLH026, we constructed a loss‐of‐function mutant of bHLH026 using the CRISPR‐Cas9 system and designed two gRNA target sites in the third exon of bHLH026. The constructed vectors were transformed into Nipponbare, and a total of 20 CRISPR‐Cas9 editing events were identified. Two transgenic lines, bhlh026‐cri‐6 and bhlh026‐cri‐7, showed 37 and 61‐bp deletions at the third exon of bHLH026, respectively, which resulted in the loss of function of bHLH026 owing to a shift mutation (Figure 5c). We also constructed overexpression lines of bHLH026 using the ORF with functional bHLH026. RT‐qPCR results revealed that the expression of diterpene synthesis‐related downstream genes was significantly reduced in the bhlh026‐cri line, while significantly elevated in the bHLH026‐OX line compared with that in the wild‐type (Figure 5d). Therefore, bHLH026 is a key transcription factor regulating the expression of genes related to diterpene synthesis.
To investigate the metabolism of diterpene antitoxins in each transgenic line of bHLH026, liquid chromatography‐tandem mass spectrometry was performed to analyse the chemicals in these transgenic materials. We detected the diterpenoid antitoxins Oryzalexin S and Momilactones A&B in the syn‐CPP pathway and their intermediates syn‐stemar‐13‐ene and syn‐pimara‐7,15‐diene. The results showed that the levels of all the above metabolites were significantly higher in the bHLH026‐OX line than in the wild‐type; however, they showed almost no change in the bhlh026‐cri line except for Oryzalexin S, which was significantly reduced compared with that in the wild‐type (Figure 6a, b). In addition, we also detected the diterpenoid antitoxins phytocassanes C, D, and E and oryzalexins C and F in part of the ent‐CPP pathway, and similarly, the levels of these metabolites were significantly higher in bHLH026‐OX lines than in the wild‐type, and there were significant decreases in phytocassanes D and E in the bhlh026‐cri lines (Figure 6a, c). Recently, the casbane‐type phytoalexin ent‐10‐oxodepressin was identified in rice, which exhibited evident broad‐spectrum disease resistance, but its biosynthesis has not been elucidated (Liang et al., 2021; Zhan et al., 2020). Ent‐10‐oxodepressin was also detected in our transgenic lines by metabolome assay, and its level was significantly higher in the bHLH026‐OX line while significantly lower in the bhlh026‐cri line relative to that in the wild‐type (Figure S10). Overall, the diterpene antitoxin‐related metabolites detected in the bHLH026‐OX line were all significantly elevated, while those detected in the bhlh026‐cri lines mostly showed no significant change compared with the wild‐type, possibly due to the low level of diterpene antitoxin synthesis in both bhlh026‐cri line and wild‐type.
Figure 6.
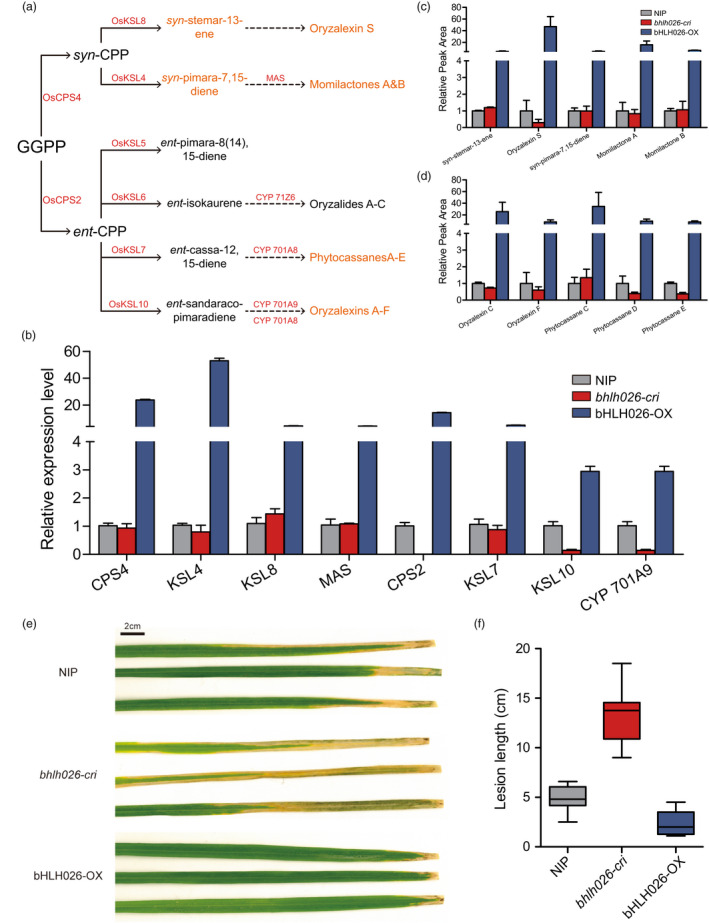
Content of diterpenoid antitoxins and disease resistance of bHLH026 transgenic material. (a) Rice Labdane‐Related Diterpenoid Biosynthetic Network. GGDP, geranylgeranyl diphosphate; CDP, copalyl diphosphate. (b) Transcript levels of key genes related to diterpenoid antitoxins in leaves of wild‐type and bHLH026 transgenic materials. Relative expression levels normalized against Ubiquitin are presented. Error bars represent standard deviation (n ≥ 6 each). (c and d) Contents of intermediate and final metabolites of diterpenoid antitoxins in the syn‐CPP pathway c and ent‐CPP pathway d in leaves of wild‐type and bHLH026 transgenic materials. Error bars represent standard deviation (n ≥ 4 each). (e) Representative leaves after Xoo infection of bHLH026 transgenic plants vs. wild‐type plants. (f) Lesion lengths of bHLH026 transgenic material compared with their wild‐type plants.
In addition, we examined the expression of genes related to diterpene antitoxin synthesis in rice using RT‐PCR. Similar to the results of the metabolome analysis, the expression of these genes was all significantly higher in bHLH026‐OX lines, while partially significantly lower in bhlh026‐cri line than in the wild‐type (Figure 6a, b). Based on all the above results, it can be concluded that bHLH026 is indeed a key transcription factor affecting diterpene antitoxin synthesis in rice.
A number of studies have reported the broad‐spectrum resistance of diterpenoid antitoxins to Xanthomonas oryzae (Xoo) in rice. We then inoculated the bHLH026 transgenic material with a native Chinese isolate of the Xoo strain Zhe134. The susceptibility of each transgenic material was determined and the bhlh026‐cri line was found to have a longer lesion length than the wild‐type, indicating that it was more susceptible. Conversely, the bHLH026‐OX line was more resistant to the Xoo (Figure 6e, f). The results revealed that bHLH026 can influence rice resistance to Xoo by affecting the metabolism of diterpenoid antitoxins.
Discussion
The resequencing of a large number of phenotypically distinct rice varieties has demonstrated the rich variations in the genome and revealed the population structure and genetic information of rice of different origins. In this study, we identified a large number of high‐quality exonic variants and their corresponding transcript levels by sequencing the transcriptome of 287 rice accessions. The widespread exon variations among subpopulations could clearly distinguish indica, japonica, and Aus. In addition, based on the expression data of each rice accession, we found that accessions within the same subpopulation had more similar expression patterns, possibly due to the presence of specifically expressed genes in each subpopulation (Figure S11).
With the recent accumulation of high‐density genotype data and associated high‐throughput transcriptome data of many species, eQTLs have been used to determine the inter‐regulation of genes on the genome. In this study, only 33.7% of the eGenes in rice had only one unique eQTL, and the remaining eGenes had more than one eQTL (Figure 2b, c), while most eGenes have one unique eQTL in other crops such as maize (about 69%) and cotton (about 67%) (Li et al., 2020; Wang et al., 2018). Moreover, about 38.4% of eGenes in rice were co‐regulated by both local eQTLs and distant eQTLs (Figure 2d). All these results suggest that there may be a relatively more complex regulatory network of gene expression in rice.
Similar to the usual phenotypic GWAS, eQTL analysis is also constrained by the large linkage disequilibrium in rice (Figure S12), making it difficult to identify candidate genes from eQTL intervals with varying sizes. In this study, bHLH026 was precisely identified as a key regulator by combining co‐expression information of downstream genes and linkage disequilibrium between the lead SNPs of downstream gene eQTLs. Three unlinked local eQTLs and one distant eQTL were also found for bHLH026. The linkage analysis revealed that the lead SNP vg1015145775 of the local eQTL is an important variant that is tightly linked to the lead SNPs of the downstream gene eQTLs and affects the expression of bHLH026 (Figure 4). The fact that the key regulator bHLH026 is both a regulator of several downstream genes and also regulated by local eQTLs and distant eQTLs indicates that the regulatory network of genes in rice is complex.
Although candidate genes in eQTL regions can be discovered by targeting co‐expression and linkage disequilibrium of individual genes, it remains challenging to build genome‐wide “gene‐gene” regulatory networks. In maize, the genome was divided into separate bins to explore their inter‐regulatory relationships (Liu et al., 2017), while in studies of cotton, the genes closest to the lead SNP were considered regulatory genes by default (Li et al., 2020). These may be efficient but fairly rough approaches to provide a macroscopic view of the gene expression regulatory network in each crop, but can hardly facilitate the understanding of more specific intergenic regulation. By contrast, for phenotypic GWAS with a limited number of QTLs, considerable progress has been achieved in using transcriptome expression data and eQTL results to assist in the identification of candidate genes, such as scoring of candidate genes within QTL regions using transcriptome information (Tang et al., 2021), or using TWAS and eQTL co‐localization to identify candidate genes (Li et al., 2020; Tang et al., 2021; Zhang et al., 2017). Here, we also employed the GWAS results for starch content in rice flag leaves at the heading stage and combined them with transcriptomic data to screen candidate genes (Figure S6; Table S8). Both TWAS and eQTL co‐localization could effectively identify some key genes that were missed by GWAS (Figure 3d; Figure S7; Table 1; Table S9). Due to the temporal (growth period) and spatial (sampling site) specificity of the transcriptome, the fitting degree between the transcriptome data and phenotype data is often critical for precise localization.
In the genome‐wide eQTL identification, we identified a total of 17 local eQTL hotspots and 96 distant eQTL hotspots (Table S4; Table S10). Only those hotspots with more significant functional clustering and clearer annotation were selected for subsequent analysis. The same practice was also adopted in many previous studies of eQTL identification, where many significant or effective loci are often ignored because they are not enriched for a specific biological pathway due to the lack of gene annotation information (Li et al., 2020; Wang et al., 2018; Zhang et al., 2017). Unlike traditional forward or reverse genetics in which candidate genes are selected based on phenotype, the use of eQTLs alone to construct regulatory networks and find key transcription factors is more dependent on gene annotation and can only be continued using the annotation information to infer the biological processes or phenotypes affected by the key genes. As for the distant eQTL hotspot HS002, GO enrichment analysis showed that it was enriched in lipid metabolism (Figure 4a), but subsequent KEGG and pathway analyses indicated that it contains key genes for diterpene antitoxin synthesis. As a result, bHLH026 was identified as a key regulator for diterpene antitoxin synthesis and disease resistance in rice, whose effect was subsequently verified using a transgenic assay (Figure 6). While eQTLs are often used as a bridge to connect the genomic and phenotypic information, more comprehensive and accurate gene function annotation will also make eQTL studies an effective tool to identify key transcription factors. The regulatory hotspots' lack of annotation information may include some key transcription factors that we have not identified.
In summary, we used the eQTL approach to explore the complex regulatory network of rice by combining transcriptomic, genomic, and phenotypic data, and identified a key regulator of diterpenoid antitoxin synthesis in rice by analysing the regulatory hotspots, which will provide more insights into the complex regulatory network of rice and a more effective method for identifying key regulators in rice.
Methods
Plant materials
Most Oryza materials were obtained from the RiceVarMap website (http://ricevarmap.ncpgr.cn/v2/). All materials (a total of 533 accessions) were sown in May, 2016 in the experimental field of Huazhong Agricultural University, Wuhan, China (30.47°N, 114.35°E). In total, 287 accessions were selected to perform the genetic analysis based on phenotypic variations. Samples of rice arriving at the heading stage were taken from 5 to 6 p.m. each day, and observations were made to mark the accessions to be collected the next day. Twenty plants were planted for each accession, and three flag leaves of the same growth trend were sampled in a mixture at the heading stage and then immediately frozen in liquid nitrogen.
RNA extraction, sequencing, and analysis
Extraction of total RNA was performed on the top fully expanded leaves at the heading stage with the TRIzol reagent (Invitrogen). The strand‐specific paired‐end RNA‐Seq library for each accession was constructed using the Illumina TruSeq RNA sample preparation kit (Version 2). The 150‐bp paired‐end reads were obtained by sequencing the libraries on the Illumina HiSeq 2500 platform.
Raw transcriptome sequencing data were screened with Trimmomatic (version 0.33) software to remove sequencing adapters and low‐quality bases. The processed fastq files were mapped to the reference genome sequence of Oryza (MSU 7.0) (Kawahara et al., 2013) using Tophat2 (Kim et al., 2013) software, and subsequently, the expected number of reads and fragments per kilobase (FPKM) were calculated for each gene using StringTie (Pertea et al., 2015).
Screening for transcriptomic SNPs and genomic SNPs
After filtering to obtain 287 transcriptomes of clean data, the transcriptomic data were mapped on the reference genome using STAR (2.7.0c) (Dobin et al., 2013) software and subsequently identified to 2 631 987 original SNPs using the sentieon toolkit. SNPs were filtered using the ‐‐minDP 4 ‐‐minQ 30 ‐‐max‐missing 0.1 ‐‐maf 0.05 parameters of VCFtools (v0.1.13) (Danecek et al., 2011), and 177 853 high‐quality SNPs were finally retained.
Based on the genomic SNPs identified by resequencing 533 accessions (http://ricevarmap.ncpgr.cn/v2/), the SNPs of 287 accessions were extracted using plink (v1.90b5.3) (Purcell et al., 2007) software and SNPs with missing <0.05 and maf >0.05 were excluded, resulting in 6 608 819 SNPs retained.
Genetic analysis of the population
A maximum likelihood tree was constructed using the RAxML software (Stamatakis, 2014) based on the SNPs called by the transcriptome. A nonparametric bootstrap analysis was performed with 100 bootstrap replicates. The final tree was visualized using iTOL software (Letunic and Bork, 2016).
The EIGENSOFT (Price et al., 2006) software was used to perform PCA analysis based on SNPs called by the transcriptome. Finally, the first two principal components of the PCA analysis were visualized using the R package ggplot2.
The ADMIXTURE (Alexander et al., 2009) program was used to infer population structure. Set progressively increasing K values and calculate the cross‐validation error at each K value. The cross‐validation error was minimized when K = 9, indicating that the 287 accessions divided into 9 subpopulations were optimal.
Identification of expression QTL (eQTL)
To identify eQTL for genes of interest in the flag leaf of rice at the heading stage, we performed gene expression level analysis and excluded genes with a median of expression (FPKM) equal to zero, and a total of 23 325 genes were filtered from the 55 801 in the reference genome genes for subsequent analysis. Using the qqnorm function in R, the expression levels of the retained genes were performed normal quantile transformation. Subsequently, GWAS was performed for each gene based on the genomic SNPs of 287 materials using the FAST‐LMM (Lippert et al., 2011) program. The effective number of SNPs (M e = 920 371.54) was calculated by GEC software (Li et al., 2012). The horizontal dashed line shows the significance threshold of GWAS (0.05/M e; 7.3). The region that had at least three significant SNPs was regarded as one eQTL block. To obtain independent association signals, multiple SNPs with values higher than the threshold in a 5‐Mb region were clustered based on r 2 of LD ≥0.1. The SNPs that had the lowest P‐value in one cluster were identified as lead SNPs.
The hot_scan software (Silva et al., 2014) was explored for the identification of distant‐eQTL hotspots. The window size was set as 20 kb and the Benjamini and Yekutieli adjusted P‐value was set to 0.01.
Enrichment analyses
Many different kinds of gene sets were obtained in the identification of local eQTL and analysis of local eQTL and distant eQTL hotspots. GO enrichment analysis of expressed genes in individual gene sets was performed using the AgriGO webserver (http://systemsbiology.cau.edu.cn/agriGOv2/index.php) (Tian et al., 2017). The enrichment analysis of KEGG and metabolic pathway PlantCyc is implemented through the PlantGSEA web server (http://systemsbiology.cau.edu.cn/PlantGSEAv2/index.php; Yi et al., 2013). When the FDR of each enrichment item is less than the threshold value of 0.05, it is considered an important item.
Co‐expression analysis
After gene counting, genes that median expression equal to 0 were removed by quality control, expression was conditional quantile normalized, and then co‐expression analysis was performed on genes in the gene expression dataset using the WGCNA program (Langfelder and Horvath, 2008) in R. The appropriate soft threshold processing capability is selected and subsequently, the topological overlap dendrogram was used to define modules using the correlation type of “pearson,” minimum module size of 5, and a merge threshold of 0.25.
Measurement of starch content
Three representative plants were taken from each material at the heading stage, and after killing in an oven at 100 °C for 30 min, the leaves were dried at 80 °C for 72 h. Soluble carbohydrates were removed with 80% ethanol, followed by starch extraction with 35% perchloric acid and finally, the anthrone‐sulfuric acid colorimetric assay (Laurentin and Edwards, 2003) was used to determine the starch content in the leaves.
Genome‐wide association analyses for starch content
The starch content of rice flag leaf at the heading stage was measured in 287 accessions, with three replicates for each accession. Based on 6 608 819 genomic SNPs, we performed GWAS using the linear mixed model (LMM) of the FaST‐LMM (Lippert et al., 2011) program. Population structure was modelled as a random effect in LMM with the kinship matrix, and it was found to be sufficient to control the spurious association. A modified Bonferroni correction was performed to determine the genomewide significance threshold of GWAS, in which the total number of SNPs (M) for threshold calculation was substituted by the effective number of SNPs (M e). The M e of SNPs (Me = 920371.54) was calculated by GEC software (Li et al., 2012). The threshold was uniformly set as P = 1.0 × 10−6 (1/M e) to obtain the suggestive significant association signals by LMM (Chen et al., 2014; Wang et al., 2015). To obtain the independent association signals, multiple SNPs with values higher than the threshold in a 5‐Mb region were clustered based on r 2 of LD ≥0.25. The SNPs with the lowest P‐value in one cluster were identified to be lead SNPs.
Transcriptome‐wide association analysis of starch content
For TWAS analysis, those genes with a median expression equal to 0 were removed in the subsequent analysis. Association analysis was carried out using the EMMAX software (Kang et al., 2010) with LMM. An IBS kinship matrix was calculated based on the genomic SNPs, and the significance threshold for TWAS analysis was taken as an FDR‐corrected P value ≤0.05.
Yeast one‐hybrid assay
Yeast one‐hybrid assay was carried out by the Matchmaker Library Construction & Screening Kits (Clontech). The promoter sequence of CPS2 snd KSL6 was cloned into the pHis2 vector harbouring the HIS3 gene, which conferred resistance to 3‐Aminotriazole (3‐AT). The full‐length cDNA of bHLH026 was amplified and cloned into the pGADT7 vector. The pHis2‐pro and pGADT7‐bHLH026 constructs were then co‐transformed into the yeast strain Y187, and selection of the transformed cells was conducted on SD‐2 (−Trp/−Leu) and SD‐3 (−Trp/−Leu/‐His/40 mM 3AT) plates.
Dual‐luciferase transcriptional activity assay in rice protoplasts
Rice protoplasts were obtained from 13‐day‐old Zhonghua 11 seedlings as previously described (Xie and Yang, 2013), and then transformation was conducted as following previous descriptions (Zong et al., 2016). The effector and reporter constructs were cotransfected together with the construct that contained the Renilla luciferase (rLUC) gene as an internal control into rice protoplasts at a ratio of 6:6:1 (effector:reporter:reference). Co‐transfected protoplast cells were cultured for 12 h at 24 °C under dark conditions, and then the Dual‐Luciferase Reporter Assay System (Promega, Madison, WI) was used to measure the luciferase activities following the manufacturer's instructions.
Transgenic lines
Specific gRNA target sites were designed to obtain the bhlh026‐cri mutants and then assembled into the expression vector pYLCRISPR/CAS9‐MH. For the construction of the bHLH026 overexpression line, the full‐length cDNA of bHLH026 was amplified and cloned into the pCAMBIA1301S vector. The constructs were then transformed into Nipponbare through Agrobacterium tumefaciens‐mediated transformation.
RT‐qPCR
Total RNA from rice leaves was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and chloroform in accordance with the manufacturer's instructions. About 3 μg of total RNA was subsequently reverse transcribed to cDNA using MMLV reverse transcriptase (Invitrogen). RT‐qPCR was carried out using the QuantStudio 7 Flex System (Applied Biosystems, Foster City, CA) with the SYBR Premix Ex Taq (TaKaRa, Tokyo, Japan). The ubiquitin gene was adopted as the reference, and the assessment of each sample was conducted in three technical replicates. All the used primers are presented in Table S11.
Diterpenoid metabolite profiling
Transgenic lines of bHLH026 and wild‐type lines were grown in sterile rooting tubes in a lighted incubator for 12 days. Above‐ground tissues of transgenic/wild‐type lines were collected using liquid nitrogen, with four biological replicate sets. Crushing of the freeze‐dried samples was conducted using a mixer mill with zirconia beads at 60 Hz for 1 min. A 100 mg of dry powder was weighed and extracted with 1.0 mL of 70% aqueous methanol containing 0.1 mg of Acy (internal standard), sonicated for 30 min. After centrifugation, the supernatant was taken over a 0.22 μm filter membrane into an injection vial and subsequently analysed for diterpenoid metabolites using an LC–ESI–MS/MS system (Chen et al., 2013; Peng et al., 2017). Qualification of metabolites was carried out using a scheduled multiple reaction monitoring methods (Chen et al., 2013).
Assay of disease resistance
Xoo infection was conducted with the leaf‐clipping method (Kauffman et al., 1973), using fully expanded leaves from 8‐week‐old rice plants and inoculation with the Xoo strain Zhe134. Xoo was first grown for 2–3 days on a solid PSA medium at 28 °C, which was then scraped off and resuspended in sterilized MgCl2 solution (10 mM). The suspension was then adjusted to an optical density of 0.5 at 600 nm before infection. After clipping, it was observed that greyish chlorotic coloration moved on the leaf along the main vein. The length of these lesions at 15 days after inoculation was analysed to measure the disease progression. The infection involved ten plants and the disease assay was repeated three times.
Conflict of interest
The authors declare no competing financial interests.
Author contributions
G.W. and D.Z. conceived and supervised the study. C.L. and G.W. designed the experiment. C.L. performed the data analysis. C.L., X.Z., J.Z., X.F., K.C, M.S., L.M., X.L., and C.Z. planted the population, scored plant phenotype, and extracted RNAs. C.L. and G.W. wrote the manuscript, and D.Z. provided help in the revision of the manuscript.
Supporting information
Figure S1 Cross‐validation error at different K values for the analysis of 287 accessions by ADMIXTURE software.
Figure S2 The distribution of the lead SNPs in local eQTLs.
Figure S3 Enrichment analysis with all genes regulated by local eQTL.
Figure S4 GO (biological process) enrichment analysis for all genes regulated by local eQTL.
Figure S5 A co‐regulated gene cluster formed by 43 brassinosteroid biosynthesis II related genes.
Figure S6 Manhattan plot of GWAS for starch content of flag leaf at the heading stage.
Figure S7 Manhattan plot of TWAS results for starch content of flag leaves at the tassel heading (FDR <0.05).
Figure S8 In the co‐regulated gene cluster (Cluster2), the expression levels of all four genes were negatively correlated with starch content.
Figure S9 Three mutually unlinked Local eQTLs for bHLH026.
Figure S10 Contents of intermediate and final metabolites of ent‐10‐oxodepressin biosynthesis in leaves of wild‐type and bHLH026 transgenic materials.
Figure S11 Expression correlation analysis of 287 accessions.
Figure S12 Linkage disequilibrium decay in different populations.
Table S1 Information on the germplasm used in the present study.
Table S2 Table transcriptome mapping information for 287 accessions.
Table S3 Detailed eQTL information identified in the present study.
Table S4 Hotspots of the local‐regulation gene.
Table S5 Co‐expression analysis of 50 sn‐glycerol 3‐phosphate synthesis associated genes.
Table S6 Co‐regulatory gene clusters in local regulatory hotspots L01.
Table S7 Co‐expression analysis of 43 brassinosteroid synthesis‐associated genes.
Table S8 GWAS results for starch content of flag leaf at the heading stage.
Table S9 Results of screening candidate genes for starch content in rice using fusion and color.
Table S10 Hotspots of distant eQTL.
Table S11 Primers used in this study.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31871224), the National Special Key Project for Transgenic Breeding (2016ZX08009001), and the earmarked fund for the China Agriculture Research System (CARS‐01‐03) of China. We thank Dr Yueming Tian for providing computing support. We are grateful to Dr Weibo Xie for his help in data analysis and Dr Meng Yuan for providing Xoo strain Zhe134. The computations in this paper were run on the bioinformatics computing platform of the National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University.
Data availability statement
The sequencing data for this project have been deposited at the NCBI Sequence Read Archive (SRA) under project PRJNA858547.
References
- Albert, F.W. and Kruglyak, L. (2015) The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 16, 197–212. [DOI] [PubMed] [Google Scholar]
- Alexander, D.H. , Novembre, J. and Lange, K. (2009) Fast model‐based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Gao, Y.Q. , Xie, W.B. , Gong, L. , Lu, K. , Wang, W.S. , Li, Y. et al. (2014) Genome‐wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 46, 714–721. [DOI] [PubMed] [Google Scholar]
- Chen, W. , Gong, L. , Guo, Z.L. , Wang, W.S. , Zhang, H.Y. , Liu, X.Q. , Yu, S.B. et al. (2013) A novel integrated method for large‐scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant, 6, 1769–1780. [DOI] [PubMed] [Google Scholar]
- Cubillos, F.A. , Coustham, V. and Loudet, O. (2012) Lessons from eQTL mapping studies: non‐coding regions and their role behind natural phenotypic variation in plants. Curr. Opin. Plant Biol. 15, 192–198. [DOI] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.A. , Banks, E. , DePristo, M.A. , Handsaker, R.E. et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C.A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. , Batut, P. et al. (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost, D.R. , Benedict, C.I. , Berg, A. , Novaes, E. , Novaes, C.R.D.B. , Yu, Q.B. , Dervinis, C. et al. (2010) Diversification in the genetic architecture of gene expression and transcriptional networks in organ differentiation of Populus. Proc. Natl. Acad. Sci. U. S. A. 107, 8492–8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, J.J. , Cheng, Y.B. , Linghu, J.J. , Yang, X.H. , Kang, L. , Zhang, Z.X. , Zhang, J. et al. (2013) RNA sequencing reveals the complex regulatory network in the maize kernel. Nat. Commun. 4, 2832. [DOI] [PubMed] [Google Scholar]
- Fu, X.K. , Liu, C. , Li, Y.Z. , Liao, S.Y. , Cheng, H.Y. , Tu, Y. , Zhu, X.Y. et al. (2021) The coordination of OsbZIP72 and OsMYBS2 with reverse roles regulates the transcription ofOsPsbS1in rice. New Phytol. 229, 370–387. [DOI] [PubMed] [Google Scholar]
- Gan, X.C. , Stegle, O. , Behr, J. , Steffen, J.G. , Drewe, P. , Hildebrand, K.L. , Lyngsoe, R. et al. (2011) Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature, 477, 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giambartolomei, C. , Vukcevic, D. , Schadt, E.E. , Franke, L. , Hingorani, A.D. , Wallace, C. and Plagnol, V. (2014) Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLoS Genet. 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev, A. , Ko, A. , Shi, H. , Bhatia, G. , Chung, W. , Penninx, B.W. , Jansen, R. et al. (2016) Integrative approaches for large‐scale transcriptome‐wide association studies. Nat. Genet. 48, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X.H. , Zhao, Y. , Wei, X.H. , Li, C.Y. , Wang, A. , Zhao, Q. , Li, W.J. et al. (2012) Genome‐wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 44, 32–U53. [DOI] [PubMed] [Google Scholar]
- Kang, H.M. , Sul, J.H. , Service, S.K. , Zaitlen, N.A. , Kong, S.Y. , Freimer, N.B. , Sabatti, C. et al. (2010) Variance component model to account for sample structure in genome‐wide association studies. Nat. Genet. 42, 348–U110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman, H.E. , Reddy, A.P.K. , Hsieh, S.P.Y. and Merca, S.D. (1973) Improved Technique for Evaluating Resistance Of Rice Varieties To Xanthomonas‐Oryzae. Plant Dis. Rep. 57, 537–541. [Google Scholar]
- Kawahara, Y. , de la Bastide, M. , Hamilton, J.P. , Kanamori, H. , McCombie, W.R. , Ouyang, S. , Schwartz, D.C. et al. (2013) Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice, 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. and Salzberg, S.L. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein, D. (2009) Quantitative genomics: analyzing intraspecific variation using global gene expression polymorphisms or eQTLs. Annu. Rev. Plant Biol. 60, 93–114. [DOI] [PubMed] [Google Scholar]
- Krishnan, A. , Guiderdoni, E. , An, G. , Hsing, Y.I.C. , Han, C.D. , Lee, M.C. , Yu, S.M. et al. (2009) Mutant resources in rice for functional genomics of the grasses. Plant Physiol. 149, 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder, P. and Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. Bmc Bioinformatics, 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurentin, A. and Edwards, C.A. (2003) A microtiter modification of the anthrone‐sulfuric acid colorimetric assay for glucose‐based carbohydrates. Anal. Biochem. 315, 143–145. [DOI] [PubMed] [Google Scholar]
- Letunic, I. and Bork, P. (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M.X. , Yeung, J.M.Y. , Cherny, S.S. and Sham, P.C. (2012) Evaluating the effective numbers of independent tests and significant p‐value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 131, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Wang, P. , You, C. , Yu, J. , Zhang, X. , Yan, F. , Ye, Z. et al. (2020) Combined GWAS and eQTL analysis uncovers a genetic regulatory network orchestrating the initiation of secondary cell wall development in cotton. New Phytol. 226, 1738–1752. [DOI] [PubMed] [Google Scholar]
- Liang, J. , Shen, Q.Q. , Wang, L.P. , Liu, J. , Fu, J.Y. , Zhao, L. , Xu, M.M. et al. (2021) Rice contains a biosynthetic gene cluster associated with production of the casbane‐type diterpenoid phytoalexin ent‐10‐oxodepressin. New Phytol. 231, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y. , Zhang, X.M. , Li, B. , Liu, T.Y. , Chen, J.F. , Bai, Z.T. , Wang, M.J. et al. (2018) Comparison of Oryza sativa and Oryza brachyantha genomes reveals selection‐driven gene escape from the centromeric regions. Plant Cell, 30, 1729–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert, C. , Listgarten, J. , Liu, Y. , Kadie, C.M. , Davidson, R.I. and Heckerman, D. (2011) FaST linear mixed models for genome‐wide association studies. Nat. Methods, 8, 833–835. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Luo, X. , Niu, L. , Xiao, Y. , Chen, L. , Liu, J. , Wang, X. et al. (2017) Distant eQTLs and non‐coding sequences play critical roles in regulating gene expression and quantitative trait variation in maize. Mol. Plant, 10, 414–426. [DOI] [PubMed] [Google Scholar]
- Lu, X. , Zhang, J. , Brown, B. , Li, R.Q. , Rodriguez‐Romero, J. , Berasategui, A. , Liu, B. et al. (2018) Inferring Roles in Defense from Metabolic Allocation of Rice Diterpenoids([OPEN]). Plant Cell, 30, 1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, B.D. , Andrew, R.L. , Kulheim, C. and Foley, W.J. (2014) Explaining intraspecific diversity in plant secondary metabolites in an ecological context. New Phytol. 201, 733–750. [DOI] [PubMed] [Google Scholar]
- Narula, J. and Igoshin, O.A. (2010) Thermodynamic models of combinatorial gene regulation by distant enhancers. IET Syst. Biol. 4, 393–U166. [DOI] [PubMed] [Google Scholar]
- Peng, M. , Shahzad, R. , Gul, A. , Subthain, H. , Shen, S. , Lei, L. , Zheng, Z. , et al. (2017) Differentially evolved glucosyltransferases determine natural variation of rice flavone accumulation and UV‐tolerance. Nat Commun. 8, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea, M. , Pertea, G.M. , Antonescu, C.M. , Chang, T.C. , Mendell, J.T. and Salzberg, S.L. (2015) StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat. Biotechnol. 33, 290‐+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, A.L. , Patterson, N.J. , Plenge, R.M. , Weinblatt, M.E. , Shadick, N.A. and Reich, D. (2006) Principal components analysis corrects for stratification in genome‐wide association studies. Nat. Genet. 38, 904–909. [DOI] [PubMed] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M.A.R. , Bender, D. , Maller, J. et al. (2007) PLINK: A tool set for whole‐genome association and population‐based linkage analyses. Am. J. Hum. Genet. 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, R.J. , Michno, J.M. , Jeffers, J. , Hoekenga, O. , Dilkes, B. , Baxter, I. and Myers, C.L. (2018) Integrating coexpression networks with GWAS to prioritize causal genes in maize. Plant Cell. 30, 2922–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, I.T. , Rosales, R.A. , Holanda, A.J. , Nussenzweig, M.C. and Jankovic, M. (2014) Identification of chromosomal translocation hotspots via scan statistics. Bioinformatics, 30, 2551–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, S. , Zhao, H. , Lu, S.P. , Yu, L.Q. , Zhang, G.F. , Zhang, Y.T. , Yang, Q.Y. et al. (2021) Genome‐ and transcriptome‐wide association studies provide insights into the genetic basis of natural variation of seed oil content in Brassica napus. Mol. Plant, 14, 470–487. [DOI] [PubMed] [Google Scholar]
- Tian, T. , Liu, Y. , Yan, H.Y. , You, Q. , Yi, X. , Du, Z. , Xu, W.Y. et al. (2017) agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 45, W122–W129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heyningen, V. and Bickmore, W. (2013) Regulation from a distance: long‐range control of gene expression in development and disease. Philos. T R Soc. B. 368, 20120372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, R.L. , Ramaswami, G. , Hartl, C. , Mancuso, N. , Gandal, M.J. , de la Torre‐Ubieta, L. , Pasaniuc, B. et al. (2019) Genetic control of expression and splicing in developing human brain informs disease mechanisms. Cell, 179, 750–771 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Xie, W. , Xing, H. , Yan, J. , Meng, X. , Li, X. , Fu, X. et al. (2015) Genetic architecture of natural variation in rice chlorophyll content revealed by a genome‐wide association study. Mol. Plant, 8, 946–957. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Chen, Q. , Wu, Y. , Lemmon, Z.H. , Xu, G. , Huang, C. , Liang, Y. et al. (2018) Genome‐wide analysis of transcriptional variability in a large maize‐teosinte population. Mol. Plant, 11, 443–459. [DOI] [PubMed] [Google Scholar]
- Wittkopp, P.J. , Haerum, B.K. and Clark, A.G. (2004) Evolutionary changes in cis and trans gene regulation. Nature, 430, 85–88. [DOI] [PubMed] [Google Scholar]
- Wu, Y.F. , Kikuchi, S. , Yan, H.H. , Zhang, W.L. , Rosenbaum, H. , Iniguez, A.L. and Jiang, J.M. (2011) Euchromatic subdomains in rice centromeres are associated with genes and transcription. Plant Cell, 23, 4054–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, J.F. , Yin, Q.F. , Chen, T. , Zhang, Y. , Zhang, X.O. , Wu, Z. , Zhang, S.F. et al. (2014) Human colorectal cancer‐specific CCAT1‐L lncRNA regulates long‐range chromatin interactions at the MYC locus. Cell Res. 24, 513–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, K. and Yang, Y. (2013) RNA‐guided genome editing in plants using a CRISPR‐Cas system. Mol. Plant, 6, 1975–1983. [DOI] [PubMed] [Google Scholar]
- Xie, W. , Wang, G. , Yuan, M. , Yao, W. , Lyu, K. , Zhao, H. , Yang, M. et al. (2015) Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection. Proc. Natl. Acad. Sci. U. S. A. 112, E5411–E5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, X. , Du, Z. and Su, Z. (2013) PlantGSEA: a gene set enrichment analysis toolkit for plant community. Nucleic Acids Res. 41, W98–W103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, C.S. , Lei, L. , Liu, Z.X. , Zhou, S. , Yang, C.K. , Zhu, X.T. , Guo, H. et al. (2020) Selection of a subspecies‐specific diterpene gene cluster implicated in rice disease resistance. Nat. Plants, 6, 1447–1454. [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Su, W. , Tao, R. , Zhang, W. , Chen, J. , Wu, P. , Yan, C. et al. (2017) RNA sequencing provides insights into the evolution of lettuce and the regulation of flavonoid biosynthesis. Nat. Commun. 8, 2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H. , Li, P. , Xie, W. , Hussain, S. , Li, Y. , Xia, D. , Zhao, H. et al. (2017) Genome‐wide association analyses reveal the genetic basis of stigma exsertion in rice. Mol. Plant, 10, 634–644. [DOI] [PubMed] [Google Scholar]
- Zhu, G.T. , Wang, S.C. , Huang, Z.J. , Zhang, S.B. , Liao, Q.G. , Zhang, C.Z. , Lin, T. et al. (2018) Rewiring of the fruit metabolome in tomato breeding. Cell, 172, 249–261. [DOI] [PubMed] [Google Scholar]
- Zong, W. , Tang, N. , Yang, J. , Peng, L. , Ma, S.Q. , Xu, Y. , Li, G.L. et al. (2016) Feedback regulation of ABA signaling and biosynthesis by a bZIP transcription factor targets drought‐resistance‐related genes. Plant Physiol. 171, 2810–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Cross‐validation error at different K values for the analysis of 287 accessions by ADMIXTURE software.
Figure S2 The distribution of the lead SNPs in local eQTLs.
Figure S3 Enrichment analysis with all genes regulated by local eQTL.
Figure S4 GO (biological process) enrichment analysis for all genes regulated by local eQTL.
Figure S5 A co‐regulated gene cluster formed by 43 brassinosteroid biosynthesis II related genes.
Figure S6 Manhattan plot of GWAS for starch content of flag leaf at the heading stage.
Figure S7 Manhattan plot of TWAS results for starch content of flag leaves at the tassel heading (FDR <0.05).
Figure S8 In the co‐regulated gene cluster (Cluster2), the expression levels of all four genes were negatively correlated with starch content.
Figure S9 Three mutually unlinked Local eQTLs for bHLH026.
Figure S10 Contents of intermediate and final metabolites of ent‐10‐oxodepressin biosynthesis in leaves of wild‐type and bHLH026 transgenic materials.
Figure S11 Expression correlation analysis of 287 accessions.
Figure S12 Linkage disequilibrium decay in different populations.
Table S1 Information on the germplasm used in the present study.
Table S2 Table transcriptome mapping information for 287 accessions.
Table S3 Detailed eQTL information identified in the present study.
Table S4 Hotspots of the local‐regulation gene.
Table S5 Co‐expression analysis of 50 sn‐glycerol 3‐phosphate synthesis associated genes.
Table S6 Co‐regulatory gene clusters in local regulatory hotspots L01.
Table S7 Co‐expression analysis of 43 brassinosteroid synthesis‐associated genes.
Table S8 GWAS results for starch content of flag leaf at the heading stage.
Table S9 Results of screening candidate genes for starch content in rice using fusion and color.
Table S10 Hotspots of distant eQTL.
Table S11 Primers used in this study.
Data Availability Statement
The sequencing data for this project have been deposited at the NCBI Sequence Read Archive (SRA) under project PRJNA858547.