

Summary
Biologically derived redox-active motifs have great potential in energy storage due to their inherent functionality and availability from natural resources. In this protocol, we describe the synthesis and characterization of a class of bio-derived 4-electron-accepting carbonyl-N-methylpyridinium species for lithium-organic batteries. This protocol enables the synthesis of three small molecules and two conjugated polymers with carbonyl-N-methylpyridinium units in good quantities. We also detail the fabrication process and performance evaluation of the coin-cell-type lithium-organic batteries.
For complete details on the use and execution of this protocol, please refer to Wang et al. (2022).1
Subject areas: Energy, Chemistry, Material sciences
Graphical abstract
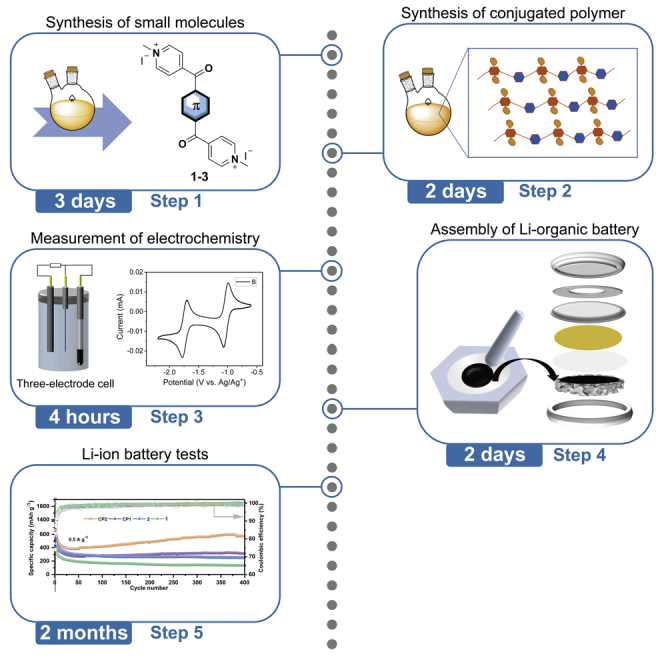
Highlights
-
•
Detailed synthesis of 4-electron-accepting carbonyl-N-methylpyridinium molecules
-
•
Preparation of conjugated polymers from brominated precursor through Sonogashira coupling
-
•
Detailed fabrication and characterization of lithium-organic batteries
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Biologically derived redox-active motifs have great potential in energy storage due to their inherent functionality and availability from natural resources. In this protocol, we describe the synthesis and characterization of a class of bio-derived 4-electron-accepting carbonyl-N-methylpyridinium species for lithium-organic batteries. This protocol enables the synthesis of three small molecules and two conjugated polymers with carbonyl-N-methylpyridinium units in good quantities. We also detail the fabrication process and performance evaluation of the coin-cell-type lithium-organic batteries.
Before you begin
Compared with the limited resources of inorganic materials, organic compounds have become an attractive alternative electrode for the next generation of lithium organic battery (LOB) because of their unique advantages such as lightweight, low-cost, flexible, environmentally friendly and structure diversity. Among them, new organic molecules that exhibit multiple and stable redox reactions, limited solubility and improved conductivity can compete with high-performance inorganic electrode materials.2,3,4
As the key motif in NAD+/NADH couple involved in electron-transfer reaction in natural system, carbonylpyridinium redox-active unit has attracted our attention. The early basic work of Leventis et al. has proved that the benzoyl-N-methylpyridinium (BMP) part can accept two electrons per unit, and has two reversible redox events and a high theoretical capacity.5 Sanford et al. have certificated that BMP derivatives are very promising for redox flow batteries. However, the exploration of the application of this potential biologically derived redox active structure in lithium-ion batteries is still at an early stage, mainly due to its high solubility in common electrolytes.6 Recently, we reported a series of novel carbonylpyridinium-based small molecules and polymers with extended conjugation to solve the above issue. These materials show promising performance as organic anodes for LOB applications, and both cycling stability and rate performance are significantly enhanced through polymerization.1
Therefore, the current protocol describes the step-by-step synthesis of the class of 4-electron-accepting carbonyl-N-methylpyridinium species (Scheme 1), with a specific focus on S3, 3 and CP2. The protocol can also be extended to the synthesis of other derivatives (S1, S2, 1, 2 and CP1). Moreover, detailed fabrication and characterization of coin-cell LOB has been described. For the same experiment performed in batch, please refer to Wang et al.1
Scheme 1.

Synthetic routes
(A) Synthesis of molecular compounds 1–3.
(B) Synthesis of conjugated polymers CP1 and CP2.
Preparation of raw materials and reaction bottles (for the synthesis of small molecules)
Timing: 2 h
-
1.Prepare a 500 mL Schlenk flask, magnetic stirrer, a 16 μm syringe (for solvent) and a 12 μm syringe (for butyllithium),
-
a.Place them in an air-blast drying oven and dry at 110°C for 1 h.
-
b.Quickly transfer to an exchange chamber in the glove box for vacuum cooling to 25°C.
-
a.
-
2.
Take out n-BuLi from the refrigerator 30 min in advance, allowing it warm to 25°C before use.
Preparation of solutions and reagent (for the synthesis of small molecules)
-
3.
Slowly add 20 mL concentrated HCl (12 M) to 100 mL of deionized water under vigorous stirring to obtain 2 M HCl solution.
CRITICAL: HCl is a corrosive chemical; thus, the preparation should be handled in the fume hood and the researcher should also wear protective gloves, masks and goggles.
-
4.
Weigh 8 g NaOH into a 250 mL beaker, and add 100 mL deionized water while stirring to obtain 2 M NaOH solution.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| 5,5′-Dibromo-2,2′-bithiophene (98%) | Adamas | CAS: 4805-22-5 |
| 4,4′-dibromobiphenyl | Adamas | CAS: 92-86-4 |
| 3,3′,5,5′-tetrabromo-2,2′-bithiophene | Adamas | CAS: 125143-53-5 |
| 4-cyanopyridine (99%+) | Adamas | CAS: 100-48-1 |
| n-Butyllithium (2.5 M solution of Hexanes, Safeseal) | Adamas | CAS: 109-72-8 |
| Iodomethane (MeI) | Energy Chemical | CAS: 74-88-4 |
| Bis(triphenylphosphine)palladium(II) chloride (Pd(PPh3)2Cl2) | AMATEK | CAS: 13965-03-2 |
| Copper iodide | Adamas | CAS: 7681-65-4 |
| Triethylamine (AR) | SCR | CAS: 121-44-8 |
| Tetrahydrofuran (Dry THF, 99.9%, Safe Dry, with molecular sieves, Water≤ 30 ppm) | Adamas | CAS: 109-99-9 |
| N,N′-dimethylformamide (Dry DMF, 99.8%, Safe Dry, with molecular sieves) | Adamas | CAS: 68-12-2 |
| N,N′-dimethylformamide (AR, 99.5%) | SCR | CAS: 68-12-2 |
| Tetrahydrofuran (AR) | SCR | CAS: 109-99-9 |
| Hydrochloric acid (HCl) (AR, 36.0–38.0%) | SCR | CAS: 7647-01-0 |
| Sodium hydrate (NaOH) (AR) | SCR | CAS: 1310-73-2 |
| Dichloromethane (CH2Cl2) (AR) | SCR | CAS: 75-09-2 |
| Diethyl ether (Et2O) (AR) | SCR | CAS: 60-29-7 |
| Methanol (MeOH) (AR, 99.5%) | SCR | CAS: 67-56-1 |
| Polyvinylidene fluoride (PVDF) | Shenzhen Kejing Zhida Technology Co., Ltd | CAS: 24937-79-9 |
| N-methyl pyrrolidone (NMP) | Energy Chemical | CAS: 872-50-4 |
| Other | ||
| Schlenk line | Synthware Co., Ltd | N/A |
| Analytical balance | METTLER TOLEDO | ME204 |
| Stir plate | IKA | Model RCT B S025 |
| Vacuum drying oven | Shanghai Yiheng Scientific Instrument Co., Ltd | DZF-6030A |
| Blast drying oven | Shanghai Yiheng Scientific Instrument Co., Ltd | DHG-9070A |
| Centrifuge | Sichuan Shuke Instrument Co., Ltd | TG16-WS |
| Low-constant-temperature reaction bath | Zhengzhou Greatwall Scientific Industrial and Trade Co., Ltd | DHJF-8002 |
| Rotary evaporator | IKA | RV10 |
| Glassy carbon working electrode | Shanghai Chenhua Instrument Co., Ltd | CHI102 |
| Pt wire counter electrode | Shanghai Chenhua Instrument Co., Ltd | CHI115 |
| Ag/AgNO3 reference electrode | Shanghai Chenhua Instrument Co., Ltd | CHI112 |
| Nickel wafer | Shenzhen Kejing Zhida Technology Co., Ltd | 350 |
| Conductive carbon black (ECP-600JD) | Shenzhen Kejing Zhida Technology Co., Ltd | ECP-600JD |
| Electrochemical workstation | Shanghai Chenhua Instrument Co., Ltd | CHI760E |
| Glove box | Vigor | SG1200/750TS-F |
| Battery packaging machine | HF-Kejing | MSK-160E |
| Constant temperature incubator | DHP-420BS | Beijing Yongguangming Medical Instrument Co., Ltd |
| LANHE CT3001A | Wuhan Lantian Electronics Co., Ltd | BT2016A |
| 400 MHz/ 600 MHz Bruker NMR spectrometers | Bruker | Ascend TM 400 |
| JEOL RESONANCE ECZ 400R NMR spectrometer | JEOL | JNM-ECZ400R/S1 |
| Bruker maxis UHR-TOF mass spectrometer | Bruker Daltonics | Maxis Ultimate 300hplc |
| Fourier-transform infrared spectrometer (FTIR) | Bruker | TENSOR27 |
| X-ray Photoelectron Spectrometer | Shimadzu | Axis UltraDLD |
| Scanning electron microscope (SEM) | HITACHI | SU8220 |
| X-ray powder diffractometer | Bruker | Bruker D8 Discover |
| Physical adsorption instrument | Micromeritics | ASAP2460 |
| Thermogravimetric analyzer | TA Instruments | Q-600/Q1000 |
| UV-vis spectrometer | Agilent | Cary 3500 |
Alternatives: In this protocol, the dry THF could be prepared by distillation over CaH2.
Materials and equipment
Stock solution of 2 M HCl solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Concentrated HCl | 12 M | 20 mL |
| Deionized water | N/A | 100 mL |
| Total | 2 M | 120 mL |
Note: The HCl solution can be stored at 25°C for 3 months.
Stock solution of 2 M NaOH solution
| Reagent | Final concentration | Amount |
|---|---|---|
| NaOH | N/A | 8 g |
| Deionized water | N/A | 100 mL |
| Total | 2 M | 100 mL |
Note: The NaOH solution can be stored at 25°C for one month.
Step-by-step method details
Part 1: Synthesis of small molecules
In this part, we take 4,4'-(3,3′-dibromo-[2,2′-bithiophene]-5,5′-dicarbonyl)bis(1-methylpyridinium) iodide (3) as an example to describe the synthesis in detail. This protocol has also been extended to the synthesis of 4,4'-([1,1′-biphenyl]-4,4′-dicarbonyl)bis(1-methylpyridinium) iodide (1) and 4,4'-([2,2′-bithiophene]-5,5′-dicarbonyl)bis(1-methylpyridinium) iodide (2). The overall synthetic route is presented in Scheme 1.
-
1.Synthesis of (3,3′-dibromo-[2,2′-bithiophene]-5,5′-diyl)bis(pyridin-4-ylmethanone) (S3, step i in Scheme 1).
-
a.Weigh 3,3′,5,5′-tetrabromo-2,2′-bithiophene (5 g, 10.4 mmol) on the analytical balance and add it into the 500 mL flame-dried Schlenk flask.
-
b.Place the glass stopper in the neck of the flask, and take the reaction flask to fume hood setup, fit it to Schlenk line.
-
c.The flask was vacuum treated and back filled with nitrogen for 3 times.
-
d.Replace the glass stopper with septum, and add 150 mL of dry THF to the reaction bottle using syringe under N2.
-
e.Stir the suspension for 15 min at 25°C.
-
f.Place the reaction bottle into −78°C cooling bath and stir for 30 min.
-
g.Add n-butyl lithium (2.5 M, 8.7 mL, 22 mmol) slowly to the reaction bottle using syringe for 30 min. The mixture was stirred at −78°C for 1 h.CRITICAL: A dry needle should be used to access the butyllithium and a sealing film should be wrapped around the connection between the syringe and the needle. In detail, first extend the nitrogen-passed needle above the liquid level; then aspirate the butyllithium and transfer it quickly to the reaction flask, pushing it in gradually and without venting. The whole process should preferably be completed within three minutes. An ethanol wash bottle should be prepared to clean the used needles.
-
h.Add 4-cyanopyridine (2.2 g, 22 mmol) dissolved in 15 mL dry THF to the reaction flask using syringe in 5 min. Allow the mixture stir at −78°C for 2 h and then warm up to 25°C slowly for a further 2 h.Note: The reaction mixture changes from off-white to yellow, then to brown and finally to black.
-
i.Add 2 M HCl (80 mL) to quench the reaction, and the mixture was stirred for 2 h at 25°C.
-
j.Basicify the mixture to pH 9–10 by adding 2 M NaOH solution (80 mL).
-
k.Collect the solid by vacuum filtration using a Buchner funnel, wash it with H2O (3 × 20 mL), CH3OH (3 × 20 mL) and CH2Cl2 (3 × 20 mL).
-
l.Purify the product by washing with hot THF.Note: The crude product was suspended in 100 mL THF, heated to 70°C for 4 h. After cooling down, the pure product was collected by vacuum filtration and dried in vacuo. The desired product was obtained yellow solids in 55%.
-
m.Analyze S3 by 1H NMR (600 MHz, CDCl3): δ = 8.89 (d, J = 5.9 Hz, 4H), 7.68 (d, J = 6.0 Hz, 4H), 7.63 (s, 2H) ppm; HRMS (APCI): m/z calculated for C20H11Br2N2O2S2+ [M+H]+ 534.8603, found 534.8600.Note: No well-resolved 13C NMR spectrum of S3 could be obtained due to its extremely limited solubility.Note: The synthesis of [1,1′-biphenyl]-4,4′-diylbis(pyridin-4-ylmethanone) (S1) and [2,2′-bithiophene]-5,5′-diylbis(pyridin-4-ylmethanone) (S2) was similar to that used to prepare S3, except 4,4′-dibromobiphenyl (3.2 g, 10.4 mmol) or 5,5′-tetrabromo-2,2′-bithiophene (3.4 g, 10.4 mmol) was used in place of 3,3′,5,5′-tetrabromo-2,2′-bithiophene (5 g, 10.4 mmol). Analytical data for S1: Yield: 86%; white solid. 1H NMR (600 MHz, CDCl3): δ (ppm) = 8.85 (d, J = 5.9 Hz, 4H), 7.96 (d, J = 8.2 Hz, 4H), 7.80 (d, J = 8.3 Hz, 4H), 7.63 (d, J = 6.1 Hz, 4H); 13C NMR (100 MHz, CDCl3): δ (ppm) = 194.53, 150.47, 144.56, 144.24, 135.56, 130.90, 127.58, 122.80; HRMS: m/z calculated for C24H17N2O2+ [M+H]+ 365.1285, found 365.1284. Analytical data for S2: Yield: 65%; yellow solid. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.85 (d, J = 5.4 Hz, 4H), 7.66 (d, J = 5.4 Hz, 4H), 7.60 (d, J = 4.1 Hz, 2H), 7.42 (d, J = 4.1 Hz, 2H); No well-resolved 13C NMR spectrum could be obtained due to its extremely limited solubility; HRMS (APCI): m/z calculated for C20H13N2O2S2+ [M+H]+ 377.0413, found 377.0408.
-
a.
-
2.Synthesis of 4,4'-(3,3′-dibromo-[2,2′-bithiophene]-5,5′-dicarbonyl)bis(1-methylpyridinium) iodide (3, step ii in Scheme 1).
-
a.Assemble the 100 mL two-neck round bottom flask, the condenser, the gas inlet adaptor and magnetic stirrer onto the oil bath.
-
b.Purge the flask with nitrogen for 10 min.
-
c.Load the reaction flask with S3 (1.5 g, 2.8 mmol) and dry DMF (15 mL). The mixture was stirred for 10 min at room temperature.
-
d.Add iodomethane (4.0 g, 28 mmol) to the flask, and heat the reaction mixture to 90°C for 16 h.
-
e.Allow the solution to cool down to room temperature and add diethyl ether (40 mL) to precipitate out the product.
-
f.Filter the obtained solids using Buchner funnel, and wash with diethyl ether (3 × 20 mL) and CH2Cl2 (20 mL).
-
g.Dry the solid under vacuum (-0.1 MPa) at 40°C for 2 h in the vacuum drying oven. The desired product was obtained as brown solids in 98%.
-
h.Analyze 3 by 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 9.23 (d, J = 6.7 Hz, 4H), 8.50 (d, J = 6.7 Hz, 4H), 8.00 (s, 2H), 4.46 (s, 6H); 13C NMR (100 MHz, DMSO-d6): δ (ppm) = 188.48, 154.44, 152.00, 147.20, 145.38, 142.08, 131.94, 119.67, 53.51. HRMS (ESI): m/z calculated for C22H16Br2N2O2S22+ [M-2I]2+ 281.9494, found 281.9492.
-
a.
Note: The two-step synthesis of 4,4'-([1,1′-biphenyl]-4,4′-dicarbonyl)bis(1-methylpyridinium) iodide (1) and 4,4'-([2,2′-bithiophene]-5,5′-dicarbonyl)bis(1-methylpyridinium) iodide (2) was similar to that used to prepare 3, except S1 (500 mg, 1.4 mmol) or S2 (500 mg, 1.3 mmol) was used in place of S3 (1.5 g, 2.8 mmol). Analytical data for 1: Yield: 95%; yellow solid. 1H NMR (400 MHz, DMSO-d6): δ = 9.21 (d, J = 6.8 Hz, 4H), 8.39 (d, J = 6.7 Hz, 4H), 8.07 (d, J = 8.5 Hz, 4H), 7.97 (d, J = 8.5 Hz, 4H), 4.45 (s, 6H); 13C NMR (100 MHz, DMSO-d6): δ = 192.17, 151.59, 147.04, 144.56, 134.58, 131.55, 128.26, 127.02, 48.71; HRMS: m/z calculated for C24H17N2O2+ [M-I]+ 521.0720, found 521.0723. Analytical data for 2: Yield: 98%; brown solid. 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 9.22 (d, J = 6.8 Hz, 4H), 8.47 (d, J = 6.7 Hz, 4H), 7.93 (d, J = 4.1 Hz, 2H), 7.81 (d, J = 4.2 Hz, 2H), 4.45 (s, 6H); 13C NMR (100 MHz, DMSO-d6): δ (ppm) = 183.95, 150.51, 147.18, 145.55, 141.87, 139.68, 129.37, 126.89, 48.74; HRMS: m/z calculated for C22H18IN2O2S2+ [M-I]+ 532.9849, found 532.9843.
Part 2: Synthesis of conjugated polymers
Polymerization of 3 via Sonogashira coupling by reacting it with the corresponding arylethynylenes (4,7-diethynyl-2,1,3-benzothiadiazole or 1,4-diethynylbenzene) in the presence of Pd(PPh3)2Cl2, copper iodide (CuI) in DMF/Et3N (1:1, v/v) afforded poly{[(2,2′-bithiophene)-5,5′-dicarbonyl)bis(1-methylpyridinium)-3,3′-diynyl]-alt-phenyl}iodide} (CP1) and poly{[(2,2′-bithiophene)-5,5′-dicarbonyl)bis(1-methylpyridinium)-3,3′-diynyl]-alt-benzothiadiazole}iodide} (CP2), respectively (Scheme 1). Due to the similar procedure, the synthesis of CP2 is described in detail in this part.
-
3.Synthesis of CP2 (step iii in Scheme 1).
-
a.Assemble the 100 mL two neck round bottom flask, the condenser, the gas inlet adaptor and magnetic stirrer on to the oil bath.
-
b.Charge the reaction flask with 3 (409 mg, 0.5 mmol), 4,7-diethynyl-2,1,3-benzothiadiazole (92 mg, 0.5 mmol), Pd2(PPh3)2Cl2 (18 mg, 0.025 mmol), CuI (10 mg, 0.05 mmol).
-
c.The flask was vacuum treated and back filled with nitrogen for 3 times under Schlenk line.
-
d.Add degassed solvent mixture of DMF/Et3N (15 mL, 2:1, v/v) by syringe bubbling method. The reaction mixture was stirred at 90°C for 48 h in the atmosphere of nitrogen.
-
e.After being cooled to room temperature, the polymer was collected by centrifugation with 4000 rpm and washed the solids in turn with excess DMF, methanol, THF and finally diethyl ether. After drying under vacuum, the resultant product appears as tawny solids with a yield of 78%.
-
a.
Note: The synthesis of CP1 was similar to that used to prepare CP2, except 1,4-bis(ethynyl)benzene (63 mg, 0.5 mmol) was used in place of 4,7-diethynyl-2,1,3-benzothiadiazole. The resulting two polymers are not soluble in water, organic solvents, or organic carbonates (electrolyte), indirectly affirming their polymeric structure. The chemical structures of the polymers were verified by Fourier transform infrared spectroscopy (FTIR) and solid-state 13C NMR spectroscopy.
Part 3: Measurement of electrochemistry
To confirm the four-electron accepting property, Cyclic voltammograms (CV) and differential pulse voltammograms (DPV) were conducted on a 1 mM 1 or 2 solution in degassed DMF containing 0.1 M TBAPF6 as a supporting electrolyte (Figure 1).
-
4.Prepare three electrodes (Figure 1A).
-
a.Working electrode: glassy carbon disk electrode with 4 mm diameter.
-
b.Counter electrode: Pt wire.
-
c.Reference electrode: Ag/AgNO3 (0.01 M) pesudo-reference electrode in fritted compartment.
-
a.
-
5.
Clean working electrode (Figure 1B).
-
6.
Assemble the electrochemical cell, and add 5 mL DMF solution of small molecules (1 mM) containing 0.1 M TBAPF6 as a supporting electrolyte (Figure 1C).
-
7.
Record the CV and DPN (Figures 1C and 1D).
Figure 1.

Electrochemical measurement
(A) Photographs of working, counter and reference electrodes.
(B) Schematic illustration of polishing glass carbon electrodes.
(C) Instruments for electrochemical testing.
(D) CVs of 1 and 2 in DMF solution (c = 1 mM) at a scan rate of 0.1 mV s-1, with 0.1 M TBAPF6 as the electrolyte.
(E) DPV of 1 and 2 in DMF solution (c = 1 mM), with 0.1 M TBAPF6 as the electrolyte. Parameters: Increase E = 4 mV, amplitude = 50 mV, pulse width = 60.0 ms, sampling width = 20.0 ms, pulse period = 500.0 ms.
Part 4: Assembly of Li-organic batteries
The overall preparation of the working electrodes is illustrated in Figure 2.
-
8.Fabrication of working electrode.
-
a.Material Preparation.
-
i.The materials: active material (CP2), conductive carbon black (ECP-600JD), the binder (polyvinylidene fluoride, PVDF) and the solvent (N-methyl pyrrolidone, NMP).
-
ii.The utensils: the foam nickel wafer with a diameter of 12 mm (weigh and record the mass of each disk), a weighing spoon, a scraping spoon and an agate mortar.
-
i.
-
b.Weighing and Mixing.
-
i.Weigh 18 mg CP2 into the agate mortar and grind it into delicate powder (5 min);
-
ii.Weigh 9 mg ECP-600JD and continue grinding (30 min);
-
iii.weigh 3 mg PVDF and continue grinding (10 min).CRITICAL: Grind the slurry in one direction scraping solids off of the agate mortar walls and push them back to the center every five minutes before continuing to grind.
-
i.
-
c.Adding NMP.CRITICAL: NMP should be added drop by drop during the grinding process, avoiding adding a lot of NMP at one time. Slowly add about 200 μL NMP with continuous grinding to form a well-dispersed slurry in 15–30 min.
-
d.Electrode coating.CRITICAL: Evenly coat the slurry on the foam nickel wafer using a brush one by one.
-
e.Dry the electrodes.CRITICAL: Make sure to fully dry the prepared working electrodes in a vacuum drying oven at 80°C for 12 h to remove any solvent residues.
-
f.Flatten the electrodes.CRITICAL: Use a Manual infrared tablet press to tablet the electrodes at a pressure of 4 MPa to prevent the foam nickel wafer from penetrating the separator.
-
g.Calculate the mass loading.Note: Weigh the mass of the dried electrode sheet and calculate the mass of the coated active substance. The mass loading of active material in this protocol is 0.8–1.0 mg cm-2.
-
a.
-
9.Assemble CR2032 coin cells in the argon-filled glove box (Figure 3).
-
a.Transfer the prepared electrodes into a glove box under an Ar atmosphere (<0.01 ppm of oxygen and water).Note: Foam nickel wafer pasted active materials was used as the working electrode, 1 mol/L LiPF6+EC/EMC/DMC (1:1:1, v/v, 1% vinyl carbonate as additive) as the electrolyte, celgard2400 as the separator, and lithium foil of the corresponding size as the counter electrode. These materials are commercially available and stored in glove box.
-
b.Assembly the coin cells.Note: Put the positive plate, working electrolyte, separator, lithium plate, spacer (0.8 mm) and gasket into the positive shell in order, and cover the negative shell of the battery.
-
c.Use the battery sealing machine to pressurize the battery to ∼8.5 MPa for sealing. Place the assembled battery in a box and place it in a 30°C incubator for 8 h to be tested.
-
a.
Figure 2.

Preparation of the working electrodes
Figure 3.

Assembly of coin-cell-type lithium-organic batteries (LOB)
(A) Schematic illustration of the fabrication of LOB.
(B) Pictures for each components.
(C) Picture showing the assembly details for coin cell LOB.
Part 5: Li-ion battery tests
For the characterization of the battery performance, the coin cells were connected to the LANHE CT3001A battery test system to record the galvanostatic charge/discharge behaviors, cycling performance at constant current and rate performance at different current densities. In addition, the CV and electrochemical impedance spectra were tested by an electrochemical workstation (CHI760E). The key parameter setting and results of the battery test are presented in Figures 4 and 5.
-
10.Test lithium battery performance by the electrochemical workstation and the LANHE CT3001A battery test system.
- a.
-
b.Record the galvanostatic charge-discharge (GCD) curves at a current density of 0.5 A g-1 (Figure 5B).Note: Firstly discharge to 0.05 V and then charge to 3.00 V at a constant current of 0.5 A g-1. It can also be used to test the GCD curves at other current densities.
- c.
- d.
-
e.Analyze the electrochemical impedance spectroscopy (EIS).Note: It was carried out using an electrochemical workstation (CHI760E) at a frequency range between 100 kHz to 0.1 MHz. The detailed program settings are displayed in Figure 4E.
Figure 4.

Battery testing
(A) LANHE CT3001A battery test system.
(B) The detailed program of Cycling performance test.
(C) Rate performance test.
(D) Cyclic voltammograms measurement.
(E) Electrochemical impedance spectroscopy (EIS) measurement.
Figure 5.

Electrochemical and battery performance
(A) CV curves of 1, 2, CP1, and CP2 electrodes in a battery configuration at 0.2 mV s-1.
(B) Galvanostatic charge-discharge behaviors of 1, 2, CP1, and CP2 at 0.5 A g-1.
(C and D) Cycling performance of 1, 2, CP1, and CP2 at 0.5 A g-1 and 0.2 A g-1.
(E) Rate performance of 1, 2, CP1, and CP2 at different current densities. Figure reprinted with permission from Wang et al.1
Expected outcomes
This protocol provides the synthesis of a class of 4-electron-accepting carbonyl-N-methylpyridinium species, with a focus on the procedure of 3 and CP2. Other materials (1, 2 and CP1) can be obtained following the similar protocol. It also describes the detailed procedure for battery assembly and analysis of the battery performance. The designed molecular compounds 1–3 can reversibly store up to four electrons, which is highly beneficial to increase the charge-storage capacity in battery. Both cycling stability and rate performance are further enhanced through polymerization, as a result of high electrochemical activity and effective suppression of dissolution. Impressively, polymer CP2 delivers not only the highest capacity but also the best cycling stability, reaching up to 574 mAh g-1 after 400 cycles at 0.5 A g-1, and even as high as 807 mAh g-1 after 300 cycles at 0.2 A g-1.
Limitations
There are several limitations to this protocol. First, n-butyllithium is extremely sensitive to water. When using it for synthesis, it is necessary to ensure that the reaction solvents and glassware used are dried; otherwise the yield of the reaction will be low. Secondly, the quality of polymers may slightly vary from different batches, and therefore strictly keep the reaction conditions as same is important. If available, we would recommend to synthesize polymers in sufficient quantities at one time to ensure that all characterizations use the same batch of polymer. Thirdly, the slurry must be ground evenly when fabricating the working electrodes; otherwise the mass distribution of the active material will be uneven, resulting in a large difference in battery performance.
Troubleshooting
Problem 1
Low yield of S3 arising from air and water sensitivity of n-BuLi (step 1g in part 1: synthesis of small molecules).
Potential solution
Butyllithium is highly sensitive to water and air. Therefore, the reaction bottle, solvents and the needle for solvent transfer much be dried in advance before the experiment. We recommend using the fresh distilled tetrahydrofuran.
Problem 2
Polymerization reactions may not occur or polymer yields are low (step 3c in part 2: synthesis of conjugated polymers).
Potential solution
Remove oxygen in the solvent during the polymerization, which plays a crucial role in the occurrence and yield of the polymerization reaction. During the synthesis of conjugated polymers CP1 and CP2, the solvents are measured with help of syringes and must contain no air. It is necessary to remove the air inside by bubbling the solvent-filled syringe for 2 min before injection.
Problem 3
Different batches of polymers may lead to differences in cell performance due to differences in polymerization (part 2: synthesis of conjugated polymers).
Potential solution
Because of the variation in compounds synthesized from batch to batch, we need to synthesize enough polymers for all characterizations at once to ensure that the same batch is used for all characterizations.
Problem 4
Spurious peaks may appear during the CV test (part 3: measurement of electrochemistry).
Potential solution
Oxygen is electroactive and can be reduced quite easily, interfering with the CV testing. Before measuring the reduction processes for 1–3 by CV, it is better to bubbling an inter gas (e.g., N2) through the solution for about 2 min.
Problem 5
There are differences in the electrochemical properties of cells made from the same batch (part 4: assembly of Li-organic batteries and part 5: Li-ion battery tests).
Potential solution
In the process of making battery cathode sheets, the unevenness of the ground slurry may lead to differences in battery performance. Care should be taken to always grind in one direction throughout the process of grinding the slurry. Also, scrape it to the bottom of the mortar after every five minutes of grinding before continuing to grind. In the process of grinding the active material and carbon black, it should be ground for at least 30 min to ensure full contact between the active material and carbon black. In addition, differences mass loading of active material pasted onto the foam nickel wafer can lead to differences in battery performance. The best approach is to apply a uniform coating and select working electrodes with a similar mass of mass loading of active material (difference of no more than 0.05 mg) for the fabrication of cell.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xiaoming He (xmhe@snnu.edu.cn).
Materials availability
All reagents generated in this study are available from the lead contact.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (no. 51703166 and no. 22105121) and the Innovation Capability Support Program of Shaanxi (no. 2020TD024). X.W. acknowledges the financial support from China Postdoctoral Science Foundation (no. 2020M683419). X.H. thanks Shaanxi Normal University for the funding support.
Author contributions
Q.L. and L.C. repeated the experiments, made the figures, and wrote the manuscript. X.W. performed the experiments and data analysis. X.H. conceived and supervised the project.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Xiujuan Wang, Email: xjwang@snnu.edu.cn.
Xiaoming He, Email: xmhe@snnu.edu.cn.
Data and code availability
No unique datasets or codes were generated in this study.
References
- 1.Wang X., Xue W., Gao G., Chen L., Baumgartner T., He X. Bio-derived 4-electron-accepting carbonyl-N-methylpyridinium species for high-performance lithium/organic batteries. Cell Rep. Phys. Sci. 2022;3:100951. doi: 10.1016/j.xcrp.2022.100951. [DOI] [Google Scholar]
- 2.Lu Y., Chen J. Prospects of organic electrode materials for practical lithium batteries. Nat. Rev. Chem. 2020;4:127–142. doi: 10.1038/s41570-020-0160-9. [DOI] [PubMed] [Google Scholar]
- 3.Kim J., Kim J.H., Ariga K. Redox-active polymers for energy storage nanoarchitectonics. Joule. 2017;1:739–768. doi: 10.1016/j.joule.2017.08.018. [DOI] [Google Scholar]
- 4.Lee S., Kwon G., Ku K., Yoon K., Jung S.K., Lim H.-D., Kang K. Recent progress in organic electrodes for Li and Na rechargeable batteries. Adv. Mater. 2018;30:1704682. doi: 10.1002/adma.201704682. [DOI] [PubMed] [Google Scholar]
- 5.Leventis N., Rawaswdeh A.M.M., Zhang G., Elder I.A., Sotiriou-Leventis C. Tuning the redox chemistry of 4-benzoyl-N-methylpyridinium cations through para substitution. Hammett linear free energy relationships and the relative aptitude of the two-electron reduced forms for H-bonding. J. Org. Chem. 2002;67:7501–7510. doi: 10.1021/jo020489+. [DOI] [PubMed] [Google Scholar]
- 6.Sevov C.S., Hickey D.P., Cook M.E., Robinson S.G., Barnett S., Minteer S.D., Sigman M.S., Sanford M.S. Physical organic approach to persistent, cyclable, low-potential electrolytes for flow battery applications. J. Am. Chem. Soc. 2017;139:2924–2927. doi: 10.1021/jacs.7b00147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No unique datasets or codes were generated in this study.