

Abstract
LIMKs are important regulators of actin and microtubule dynamics, and they play essential roles in many cellular processes. Deregulation of LIMKs has been linked to the development of diverse diseases, including cancers and cognitive disabilities, but well-characterized inhibitors known as chemical probes are still lacking. Here, we report the characterization of three highly selective LIMK1/2 inhibitors covering all canonical binding modes (type I/II/III) and the structure-based design of the type II/III inhibitors. Characterization of these chemical probes revealed a low nanomolar affinity for LIMK1/2, and all inhibitors 1 (LIMKi3; type I), 48 (TH470; type II), and 15 (TH257; type III) showed excellent selectivity in a comprehensive scanMAX kinase selectivity panel. Phosphoproteomics revealed remarkable differences between type I and type II inhibitors compared with the allosteric inhibitor 15. In phenotypic assays such as neurite outgrowth models of fragile X-chromosome, 15 showed promising activity, suggesting the potential application of allosteric LIMK inhibitors treating this orphan disease.
Graphical Abstract

INTRODUCTION
LIM kinases (LIMKs) 1 and 2 are two structurally conserved protein kinases that belong to the tyrosine kinase-like family (TKL). The LIMKs are dual specificity kinases recognizing serine/threonine as well as tyrosine-containing substrates and are represented in humans by two paralogues, LIMK1 and LIMK2. Both LIMKs share the same domain organization, which comprises two N-terminal LIM domains, a PDZ domain and a proline/serine-rich region followed by the C-terminal kinase domain. Human LIMK1 and LIMK2 share a high structural similarity, with >50% overall sequence conservation and >70% sequence similarity within the kinase domain. LIMKs show a distinct cell-type-specific expression pattern and subcellular localization.1 LIMKs have been reported to be activated by at least three different signaling pathways.2 The best-known pathway resulting in LIMK activation is via the small GTPases of the Rho family, such as RhoA, Rac, or CDC42, which activate Rho kinases (ROCK) or p21-activated kinases (PAKs). These kinases activate LIMKs by phosphorylating Thr508 in LIMK1 or Thr505 in LIMK2, located in the activation loop. In addition, Takemura et al. demonstrated that phosphorylation of Thr508 in LIMK1 by Ca2+/calmodulin-dependent protein kinase IV (CaMKIV) is critical for calcium-induced neurite outgrowth.3 LIMK1 has also been reported to be phosphorylated and activated by the MKK6/p38/MK-2 signaling cascade on Ser323, which is located between the PDZ and PK domains.4
Fragile X syndrome (FXS), a common inherited form of intellectual disability and autism, is characterized by abnormal synapse and dendritic spine development. FXS occurs because of genetic alterations, leading to an increased number of CGG trinucleotide repeats in the 5’ untranslated region of the RNA binding protein FMR1 (fragile X messenger ribonucleoprotein 1), resulting in methylation of the FMR1 locus and silencing of FMR1 transcripts. However, in some cases inactivating mutations of the FMR1 gene have been observed. FMR1 binds to and thereby represses the translation of a number of mRNAs including the mRNA encoding for the long isoform of bone morphogenetic protein type 2 receptor (BMPR2), resulting in the derepression of the full-length isoform of this kinase. This isoform interacts and strongly activates LIMK1, resulting in the stimulation of the noncanonical BMP signal transduction pathway, actin reorganization, and promotion of neurite outgrowth and synapse formation.5,6 Recently, it has been demonstrated that the pharmacological inhibition of LIMK1 ameliorates the aberrant spine development in FMR1-KO mice, suggesting LIMK1 as a potential target for the treatment of FXS (Figure 1).7
Figure 1.

(A) Activation of LIMK by different pathways (according to ref 2). (B) Downstream effects of LIM kinases. LIMK influences actin dynamics via phosphorylation and inactivation of cofilin (left) and microtubule disassembly (middle). In addition, other interaction partners that can be phosphorylated by LIMKs have been reported (right), but the phenotypic effects of these interaction partners are less well described.
Well-known substrates for the LIMKs are actin-binding proteins, namely cofilin 1, cofilin 2, and destrin, also called actin-depolymerizing factors (ADFs). Cofilin/ADF is phosphorylated by LIMKs at Ser3, which results in its inactivation, and causes polymerization and stabilization of actin filaments and suppression of actin turnover. Thus, phosphorylation of cofilin by LIMKs plays a pivotal role in the actin cytoskeletal reorganization and many cellular activities such as cell migration or morphogenesis.2,8 Besides their central role in actin filament dynamics, it has been demonstrated that LIMKs are regulators of the microtubule disassembly, which is independent of the regulation of actin microfilament. It is believed that the LIMK1 PDZ domain interacts with tubulin, and overexpression of LIMK1 has been associated with microtubule destabilization9,10 as pharmacological inhibition of LIMKs leads to a microtubule stabilizing effect.11–14 Many additional macromolecular interaction partners of LIMKs have been identified; however, only a few small molecules have been identified inhibiting LIMK activity and their mode of action has been poorly characterized1,15–17 (Figure 2). One of the most studied LIMK inhibitors is the dual LIMK1/2 inhibitor compound 1 (BMS-5/LIMKi3), which has originally been developed by Bristol–Meyer–Squibbs. BMS-5 is known as an ATP competitive LIMK1/2 inhibitor with IC50 values in the low nanomolar range.18 Other inhibitors such as compound 2 (Damnacanthal)19 or 3 (Pyr1)12 have also been disclosed; however, no selectivity data have been reported. These compounds seem to be reactive or have a limited solubility, which makes them less attractive as starting compounds for the development of a chemical probe for LIMK1/2. More recently, compound 4 has been published as an allosteric type III inhibitor with preference for LIMK2 versus LIMK1, although no cellular data are available and only a limited selectivity screen has been reported.20 Some additional LIMK1/2 inhibitors have been published based on a pyrrolopyrimidine scaffold, which include SR7826 (5) or LX7101 (6). However, a significant number of off-targets have been reported for both of these inhibitors. LX7101, which has been investigated in phase 1/2a clinical trials for primary open-angle glaucoma or ocular hypertension,21 is known as a dual LIMK/ROCK inhibitor with off-target PKA activity.22–24 Thus, a more comprehensive evaluation of inhibitor selectivity and the development of selective LIMK inhibitors is needed for a mechanistic evaluation of LIMK1/2 roles in human pathology and physiology. Here, we describe the development of an allosteric dual LIMK1/2 inhibitor as well as a type II inhibitor together with a detailed characterization of the type I inhibitor LIMKi3. All inhibitors exhibited excellent selectivity for LIMK1/2, with no additional off-targets identified for the allosteric type III inhibitor (15) and only one and four additional off-targets identified for the type I (1) and type II (33) inhibitors, respectively. All three inhibitors showed submicromolar cellular on-target activity, making them suitable as chemical probes for LIMK1/2.
Figure 2.

Reported LIMK inhibitors in the literature.
RESULTS AND DISCUSSION
For our chemical probe development, we chose compound 4 as the starting point, as allosteric inhibitors have usually favorable properties in terms of selectivity profile.20,25 Therefore, we resynthesized 4 and modified this lead structure at three different positions. To understand the structure–activity relationship, modifications were introduced at the sulfonamide residue (R1) and at the amide nitrogen as well as the pending ring system (R2 and R3). Compounds 15–27 were synthesized in a two- or three-step synthesis using the previously reported synthetic strategy.20 The first step of the synthetic route consisted of the installation of the sulfonamide using aniline or benzylamine, and the sulfonamide intermediates were subsequently coupled by secondary amines to the corresponding amides (Scheme 1). The binding affinity of this first set of compounds to LIMK1 and LIMK2 was determined by differential scanning fluorimetry (DSF). Interestingly, only small modifications were tolerated on the sulfonamide moiety (R1; see Table 1). Introduction of chlorines, especially in para-position (23), led to strong reduction in Tm shifts in comparison to 4, as well as the introduction of an additional methylene group from the phenyl moiety in 4 to a benzyl moiety in 22 reduced binding. Surprisingly, the introduction of a chlorine in meta-position (19) led to a decrease in ΔTM in the DSF assay for LIMK1 but this modification had a lesser effect on LIMK2 Tm shifts compared to the lead structure 4. Likewise, it was impossible to replace the benzyl moiety on the tertiary amide in position R3 either by shortening (25) or by elongation (21) without losing activity. The largest impact of this SAR series with respect to ΔTM values was achieved in position R2 by elongation of the methyl group to ethyl (16), n-propyl (17), or an n-butyl (15) derivative. In turn, the reduction of the methyl group in this position to a secondary amide (24) nearly completely abrogated binding to both LIMK1 and LIMK2. To understand the binding affinity and to correlate ΔTM in the DSF assay into affinities, isothermal titration calorimetry (ITC) experiments were performed using recombinant LIMK1 kinase domain with compounds 15–18. These experiments revealed that KD values of compounds 15–17 were 64, 53, and 89 nM, respectively (Table 1). In addition, the combination of the propyl-residue in R2 with chlorine in meta-position on R1 (18) resulted in a significant decrease in ΔTM for LIMK1 and LIMK2; however, ITC revealed that 18 still had a KD of 386 nM (Table 1).
Scheme 1. Synthesis of Sulfamoylbenzamides (15–27)a.
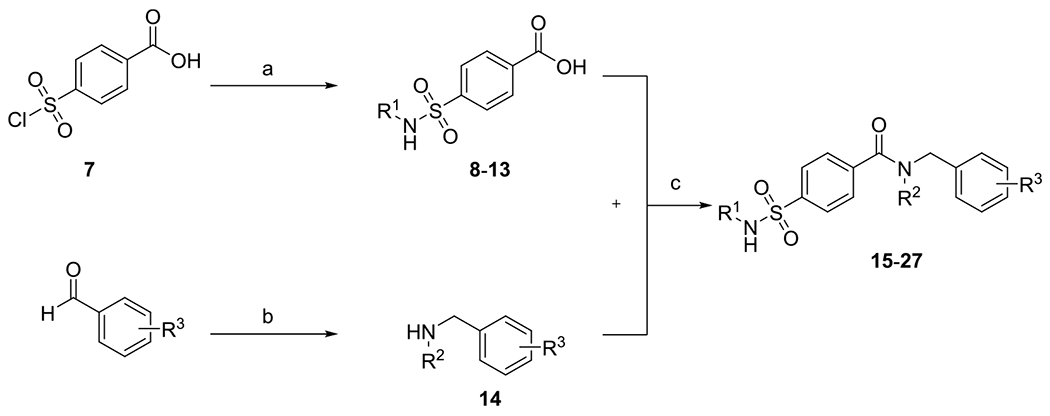
aReagents and conditions: (a) substituted anilines or benzylamine (10 equiv), 4-(chlorosulfonyl)benzoic acid (1.00 equiv), THF, RT, 18 h; (b) primary amines (1.00 equiv), benzaldehyde (1.00 equiv), sodium triacetoxyborohydride (1.40 equiv), glacial acetic acid (2.00 equiv), DCE, RT, 20 h, 56%; and (c) EDC*HCl (1.20 equiv), HOBT (1.00 equiv), secondary amine (1.50 equiv), DCM, RT, 20 h, 45–86%.
Table 1.
Structure–Activity Relationship (SAR) on the 4-Sulfamoylbenzamide Moiety for the Allosteric Type III Inhibitorsa

| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| compound | R1 | R2 | R3 | LIMK1 ΔTm [°C] | LIMK2 ΔTm [°C] | LIMK1 KD [nM] |
| 15 | Ph | n-butyl | Bn | 5.6 ± 2.0 | 12.8 ± 0.1 | 64 ± 21 |
| 16 | Ph | ethyl | Bn | 6.7 ± 0.9 | 12.4 ± 0.1 | 53 ± 9 |
| 17 | Ph | n-propyl | Bn | 6.2 ± 0.9 | 12.2 ± 0.3 | 89 ± 18 |
| 4 | Ph | methyl | Bn | 4.5 ± 0.7 | 9.3 ± 0.1 | |
| 18 | 3-Cl-Ph | n-propyl | Bn | 2.9 ± 0.4 | 7.3 ± 2.4 | 386 ± 55 |
| 19 | 3-Cl-Ph | methyl | Bn | 0.9 ± 1.6 | 7.1 ± 0.2 | |
| 20 | 2-Cl-Ph | methyl | Bn | 1.5 ± 1.2 | 5.1 ± 0.0 | |
| 21 | Ph | methyl | (CH2)2-Ph | 1.3 ± 0.3 | 3.2 ± 1.3 | |
| 22 | Bn | methyl | Bn | 1.6 ± 0.7 | 2.6 ± 0.3 | |
| 23 | 4-Cl-Ph | methyl | Bn | −1.0 ± 2.3 | 2.2 ± 0.4 | |
| 24 | Ph | H | Bn | 0.4 ± 1.7 | 1.7 ± 0.1 | |
| 25 | Ph | methyl | Ph | 0.2 ± 0.1 | 1.7 ± 0.1 | |
| 26 | Bn | H | Bn | 1.2 ± 1.0 | 0.7 ± 0.1 | |
| 27 | (CH2)2-Ph | methyl | Bn | 0.9 ± 0.6 | 0.1 ± 0.2 | |
Binding of the substituted sulfamoylbenzamide derivatives (compounds 4 and 15–27) to LIMK1/2, as determined by DSF. ΔTm shifts are reported as means ± SD of three independent experiments. KD values for LIMK1 were determined by isothermal titration calorimetry (ITC). Errors were determined based on the nonlinear least-squares fit of the isotherms using a single binding site model.
Cocrystallization of 18 with LIMK2 Guided the SAR-Driven Back-to-Front-Pocket Optimization.
After our initial TM screen, we chose 18 for cocrystallization experiment because the ΔTm for LIMK1 was lower compared to the ΔTm for LIMK2, indicating possible isoform selectivity. However, ITC experiments reveal that 18 is still a submicromolar binder to LIMK1 (Table 1). The structure of the complex revealed the anticipated type III binding mode of 18 inducing an “αC-out” and “DFG-out” conformations (Figure 3A). This allosteric binding mode induced a strong rearrangement of the αC-helix resulting in a displacement of about 11.8 Å compared to the position of the αC-helix in the AMP–PNP complex (PDB-ID: 5HVJ).26 In addition, a large shift in the position of the P-loop and the DFG motif was observed, indicating that these three structural elements are highly flexible in unphosphorylated LIMK2 (Figure 3B). These findings were in agreement with the binding mode of the closely related derivative of the lead structure 4 (PDB-ID: 4TPT;20 Figure 3C). The most striking feature of the structure of 18 with LIMK2 was a strong distortion of the P-loop compared to the binding mode of 4, as well as a flip of F470 of the DFG motif. While in the parental compound 4, the access to the hinge was blocked by three phenylalanine residues (F341, F342, F470), the elongation of the alkyl chain causes a rearrangement of these flexible regions and opened access to the ATP-binding pocket (Figure 3D).
Figure 3.

(A) Cocrystal structure of the allosteric inhibitor 18 with LIMK2 (PDB-ID: 5NXD). (B) Superimposition of AMP–PNP bound to LIMK1 (PDB-ID: 5HVJ26) and 18 bound to LIMK2 (PDB-ID: 5NXD) revealed three flexible regions that led to the formation of the allosteric binding site. (C). Superimposition of the lead structure bound to LIMK2 (PDB-ID: 4TPT20) and 18 bound to LIMK2 (PDB-ID: 5NXD) revealed the distortion of the P-loop. (D) Superimposition of PDB-ID: 4TPT and PDB-ID: 5NXD revealed that access to the ATP-binding pocket was blocked by three phenylalanine residues. Structural rearrangement upon type III inhibitor binding opens access to the ATP pocket allowing the development of type II inhibitors.
These findings encouraged us to continue the SAR mainly in position R2 to increase the potency of the sulfamoylbenzamide moiety in a back-to-front-pocket optimization. In a first attempt, we tried to replace the n-butyl chain in 15 by terminal alkynes with different carbon chains (29, 31, 32) with a nitrile (28) or by a phenethyl moiety (30) to explore the space in this position (Table 2). In particular, 30–32 showed comparable TM shifts on LIMK2 and slightly superior TM shifts on LIMK1. All of these agents were still effective in a cellular system showing two- to three-digit nanomolar IC50 values in a NanoBRET assay for LIMK1 and LIMK2. The functional groups employed should be used as a bridging unit for the implementation of various potential hinge-binders to develop a highly potent and selective type II inhibitor. The terminal alkynes in 31 and 32 were used for click chemistry with benzyl azide to grow the inhibitor toward the ATP pocket, resulting in 33 and 34, with 33 showing a slightly better stabilization in TM shifts as well as better inhibition in the cellular assay system. For this reason, we kept the same linker in 33 and used different azides to study the ATP-binding pocket. Introduction of one chlorine in the ortho-position in 35 showed little lower IC50 values for both LIMK1/2 compared with 33, whereas the introduction of two chlorines in the meta- and para-positions in 37 displayed the same IC50 value for LIMK2 but was slightly worse for LIMK1. In the next step, we introduced more heterocycles in this position, aiming to achieve a possible polar interaction within the ATP-binding pocket. Introduction of a 4-pyridinyl residue in 36 still showed a good inhibition of LIMK2 (IC50: 118 nM) but was worse in LIMK1 (IC50: 540 nM). The imidazopyrimidine residue in 38 and the 2-methylthiazole in 39 were even slightly worse for both LIMK1/2, while the oxazolidinone in 40 completely abolished the activity for both (Table 3). The synthesis of these compounds (28–40) is described in Schemes 2 and 3, starting with the 4-chlorosulfonylbenzoic acid, which was coupled to various anilines. The corresponding carboxylic acids were then coupled to various secondary amines using EDC*HCl and HOBT to obtain the corresponding benzenesulfonamide derivatives. Alkynes 31 and 32 were then used for a CuAAC (“click-reaction”) to give triazoles 33–40 (Scheme 3).
Table 2.
Structure–Activity Relationship (SAR) on the 4-Sulfamoylbenzamide Moiety with the Main Focus on Position R2 to Enter the ATP-Binding Pocket. Binding of the substituted sulfamoylbenzamide derivatives (compounds 28–32) to LIMK1/2 as determined by DSF and NanoBRETa

|
ΔTm shifts are reported as means of three independent experiments. IC50 values for LIMK1 and LIMK2 were determined in a cellular assay system using NanoBRET. IC50 values are reported as means ± SD of three independent experiments.
Table 3.
Structure–Activity Relationship (SAR) on the 4-Sulfamoylbenzamide Moiety with the Implementation of Various Potential Hinge-Binders for the Development of Type II Inhibitorsa

|
Binding of the substituted sulfamoylbenzamide derivatives (compounds 33–40 and 48) to LIMK1/2 as determined by DSF. ΔTm shifts are reported as means of three independent experiments. IC50 values for LIMK1 and LIMK2 were determined in a cellular assay system using NanoBRET. IC50 values are reported as means ± SD of three independent experiments.
Scheme 2. Synthesis of Sulfamoylbenzamides (28, 30)a.
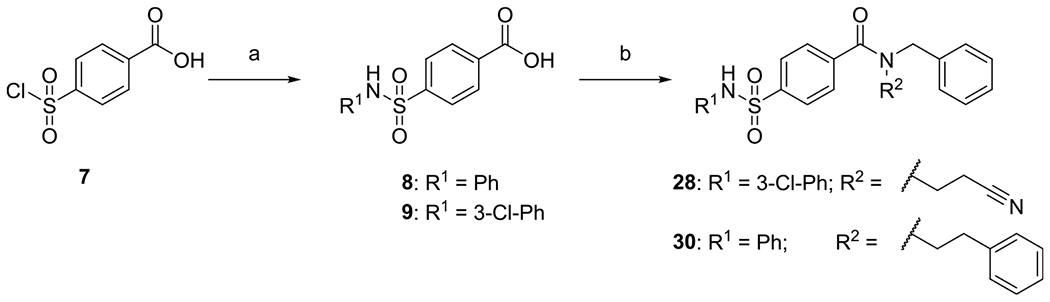
aReagents and conditions: (a) substituted anilines (10 equiv), 4-(chlorosulfonyl)benzoic acid (1.00 equiv), THF, RT, 18 h; and (b) EDC*HCl (1.20 equiv), HOBT (1.00 equiv), secondary amine (1.50 equiv), CH2Cl2, RT, 20 h.
Scheme 3. Synthesis of Sulfamoylbenzamides (29, 31, 32) and Potential Type II Inhibitors (33–40)a.
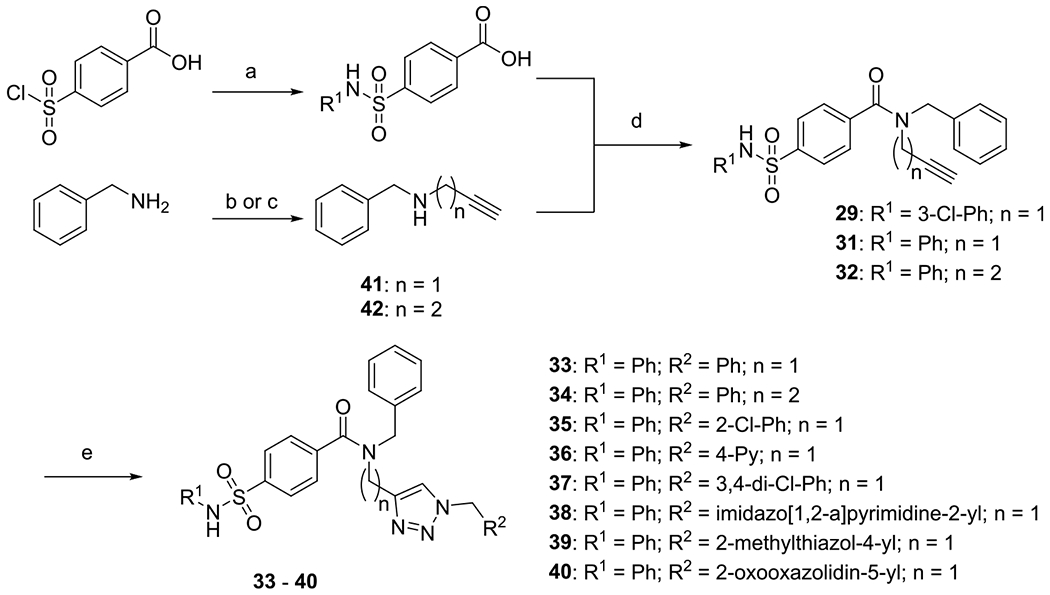
aReagents and conditions: (a) substituted anilines (10 equiv), 4-(chlorosulfonyl)benzoic acid (1.00 equiv), THF, RT, 18 h; (b) 3-bromoprop-1-yne (1 equiv), benzylamine (6 equiv), RT, 72 h; (c) benzylamine (1.00 equiv), but-3-yn-1-yl 4-methylbenzene-1-sulfonate (1 equiv), K2CO3 (2 equiv), MeCN, reflux, 16 h; (d) EDC*HCl (1.20 equiv), HOBT (1.00 equiv), secondary amine (1.50 equiv), CH2Cl2, RT, 20 h; and (e) alkyne (1 equiv), azide (1 equiv), NaAsc (0.1 equiv), CuSO4*5H2O (0.01 equiv), H2O/t-BuOH, RT, 24–120 h.
Structural Insights Facilitated the Rational Design of a Highly Potent Type II Inhibitor.
Simultaneously, cocrystallization experiments of compound 1 with LIMK1 were performed. LIMKi3 has been reported as an ATP competitive inhibitor although no crystal structure of this compound in complex with LIMK1 or 2 has been published so far. The structure of the LIMKi3 complex revealed a canonical type I binding mode with “DFG-in” conformation (Figures 4 and S3). The aminothiazole moiety acted as an ATP mimetic hinge-binding moiety, forming two hydrogen bonds with Ile416 of the hinge backbone (Figure 4). In addition, the 2,6-dichlorophenyl residue was perpendicular to the pyrazole ring of 1, efficiently occupying the binding pocket (Figure 4). The combined insights of these two crystal structures were the basis for the rationale design of a type II inhibitor by maintaining the hinge-binder moiety of 1 and combining it with the N-phenylsulfonamide, which targeted the DFG- and αC-out-pocket, and the N-benzylamide of 18, which was oriented toward the P-loop pocket (Figure 4).
Figure 4.

(A) Cocrystal of 1 (salmon) bound to LIMK1 (marine blue), PDB-ID: 8AAU; 18 (slate blue) bound to LIMK2 (yellow orange), PDB-ID: 5NXD; alignment of 1 and 18 reveals an intersection point; cocrystal of 48 (orange) bound to LIMK2 (lime), PDB-ID: 7QHG. (B) Rational design of a type II inhibitor by combining types I (1) and III (18).
The synthesis of this compound is described in Scheme 4. The “allosteric” binding motif was synthesized as described above; the sulfonamide 8 was linked to the BOC-protected secondary amine to obtain the tertiary amide 46, and finally, the BOC-protecting group was cleaved using TFA in dichloromethane to obtain the primary amine 47. For the synthesis of the “hinge” binding motif, ethyl 2-amino-1,3-thiazole-5-carboxylate (43) was coupled to isobutyryl chloride to obtain the corresponding secondary amide (44) and afterward, the ester was cleaved to obtain the carboxylic acid 45. In the last step, the primary amine 47 and the carboxylic acid 45 were coupled using EDC*HCl and HOBT to obtain 48. The potency of this potential type II inhibitor was evaluated using DSF assay, and the ΔTM was about a magnitude higher both for LIMK1 (ΔTM = 17.8 °C) and for LIMK2 (ΔTM = 20.6 °C) (Table 3).
Scheme 4. Synthesis of Type II Binder (33)a.

aReagents and conditions: (a) amine (1.00 equiv), 2-methylpropanoyl chloride (1.20 equiv), pyridine (1.50 equiv), CH2Cl2, RT, 20 h, 87%; (b) LiOH*H2O (5.00 equiv), THF/H2O, 30 °C, 72 h, quant.; (c) EDC*HCl (1.20 equiv), HOBT (1.00 equiv), secondary amine (1.50 equiv), CH2Cl2, RT, 20 h, 83%; (d) TFA, CH2Cl2, RT, 2 h, quant.; and (e) EDC*HCl (1.20 equiv), HOBT (1.00 equiv), secondary amine (1.50 equiv), CH2Cl2, RT, 20 h, 66%.
In agreement with the significant increase in temperature shift data, the KD value for 48 for LIMK1 was determined using ITC and was found to be lower (KD = 7 nM) compared to the allosteric inhibitors (15–8). We also obtained the cocrystal structure of 48 with LIMK2. The structure confirmed that 48 bound as a type II inhibitor with the 2-aminothiazole moiety as a hinge-binder and the phenylsulfamoyl moiety in the DFG-out pocket, as expected (Figures 4 and S4). An additional hydrogen bond to the gatekeeper T405 was observed, engaging the nitrogen of the secondary amide. To determine the activity of the most interesting inhibitors, enzyme kinetic data (IC50) were measured using a RapidFire mass spectrometry (RF-MS) assay with the endogenous substrate cofilin. Both the type I inhibitor 1 (LIMKi3) and compound 15 (TH257), the developed allosteric inhibitor (type III), revealed low nanomolar activity in the RF-MS assay. For the type II inhibitor 48, potency was increased to 7 and 20 nM for LIMK1 and LIMK2, respectively. Compound 26 (TH263) had IC50 values >50 μM for LIMK1 and LIMK2, which was in agreement with the ΔTM in the DSF assay (Table 4), identifying 26 as a negative control compound. To evaluate the potential in vivo use of these new type II (48) and type III inhibitors (15), we evaluated the pharmacokinetic properties of 15, 18, and 48. It has been shown that attachment of the hinge-binding moiety to the allosteric part has not only an impact on the potency of these compounds but also an increased solubility and metabolic stability (Table S12).
Table 4.
Summary and Comparative Analysis of the Biological Activity of Type I (1), Type II (48), and Type III (15) Inhibitors and the Negative Control Compound 26 on LIMK1 and LIMK2 in Thermal Shift Assay (DSF), In Vitro Activity by RapidFire MS, and Cellular Activity by NanoBRET
| DSF ΔTm [°C] |
RapidFire MS IC50 [nM] |
NanoBRET IC50 [nM] |
||||
|---|---|---|---|---|---|---|
| compound | LIMK1 | LIMK2 | LIMK1 | LIMK2 | LIMK1 | LIMK2 |
| Type I 1 (LIMKi3) |
6.7 ± 0.3 | 3.7 ± 0.3 | 16.6 ± 4 | 11.6 ± 3 | 62 ± 19 | 17 ± 3.9 |
| Type II 48 (TH470) |
17.8 ± 0.4 | 20.6 ± 4 | 7 ± 2 | 20 ± 3 | 9.8 ± 1.7 | 13 ± 4.3 |
| Type III 15 (TH257) |
5.6 ± 2.0 | 12.8 ± 0.1 | 83.8 ± 14 | 15.5 ± 2 | 238 ± 97 | 91 ± 34 |
| neg. control 26 (TH263) |
1.2 ± 1.0 | 0.7 ± 0.1 | >50,000 | >50,000 | >25,000 | >25,000 |
KINOMEscan and Kinobead PD Assay Revealed Excellent Selectivity for All Types (I–III).
To determine the selectivity of these different types of inhibitors, we chose compound 1 as type I inhibitor, 48 as our newly designed type II inhibitor, and compound 15 as the allosteric type III inhibitor, since 15 has the highest TM shift on LIMK2 for the allosteric compounds (Table 1). Compounds 1, 48, and 15 were evaluated in a comprehensive selectivity panel KINOMEscan (scanMAX, DiscoverX). The allosteric inhibitor 15 showed excellent selectivity when screened at 1 μM with KD values of 120 nM for LIMK1 and 64 nM for LIMK2, respectively (determined by DiscoverX; Tables S6 and S8). No significant interaction with other kinases of the 468 kinases was detected. Also the inactive control, 26, was evaluated in the KINOMEscan selectivity panel and revealed no interacting kinase (Tables S7 and S9). The type I 1 and the type II inhibitor 48 were less selective in the KINOMEscan panel when screened at 1 μM (Tables S2 and S4). However, as 1 and 48 had low nM potencies, we concluded that the concentration used for the selectivity assessment was too high. Both inhibitors were re-evaluated in the KINOMEscan panel at a concentration of 100 nM (Tables S3 and S5). The selectivity screen revealed an S(35)-score ranging from 0.01 to 0.02 (Figure 5). Recently, Klaeger et al. have shown that some compounds, which had been designated as chemical probes, were not selective in kinobead pulldown assays.27 Thus, all four compounds (1, 15, 48, and 26) were evaluated in a dose–response (30 μM to 1 nM) in the kinobead assay. In this assay, typically more than 200 full-length kinases were detected in cellular lysate. No other hits except LIMK1 and LIMK2 were identified, highlighting the specificity of these compounds. However, the IC50 values determined by the kinobead assay were slightly higher than those determined by RapidFire MS, which is likely due to the different readout, as the RapidFire MS is performed in an in vitro activity assay, whereas the kinobead PD assay determined the binding affinity in lysates (Figure 6C, D). Other potential reasons leading to differences in these two binding assays have been discussed elsewhere.27
Figure 5.

Comparison of types I, II, and III and the negative control compound. (A) Chemical structure of 1 (LIMKi3 = type I inhibitor), 48 (TH470 = type II inhibitor), 15 (TH257 = type III inhibitor), and 26 (TH263 = neg. control compound for TH257). (B) Selectivity profile using the scanMAX kinome wide selectivity assay (Eurofins) of 1, 48, 15, and 26. Data are illustrated using the TREEspot analysis (DiscoverX).
Figure 6.

Cellular target engagement for type I (LIMKi3, 1), type II (TH470, 48), type III (TH257, 15), and negative control compound (TH263, 26). (A) Results from NanoBRET assay on LIMK1. (B) Results from NanoBRET assay on LIMK2. (C) Results from kinobead pulldown (PD) assays. The kinobead pulldown assay covered around 300 kinases. Radar plots of each inhibitor (LIMKi3 (1), TH257 (15), TH263 (26), and TH470 (48)) showing all identified targets. The bars represent the pKD app for all identified targets. (D) A table that summarizes all IC50s and KD apps for the four ligands (LIMKi3 (1), TH257 (15), TH263 (26), and TH470 (48)) on LIMK1 and LIMK2 determined in cellular lysates by the kinobead pulldown assay.
To determine the cellular activity of these compounds, NanoBRET assays28–31 were performed, demonstrating one to two-digit nanomolar IC50 values for the type I (1) and type II (48) inhibitor: also, the allosteric inhibitor (15) had an IC50 of 238 nM for LIMK1 and 91 nM for LIMK2. Again, compound 26 had IC50 values higher than 25 μM on both LIMK1 and LIMK2, confirming its usefulness as a negative control (Table 4 and Figure 6). To assess all possible off-targets detected in kinome-wide screening, we performed nanoBRET assays on the off-targets detected for 1 and 48, which showed binding of less than 50% in scanMAX KINOMEscan assays (Eurofins). Dose–response titrations confirmed excellent selectivity in cells. Titration curves of all LIMK chemical probes are shown in Figure 6A,B and off-target activities are shown in Figure S1. In addition, temperature shift data measured on more than 100 human kinase domains as well as common off-targets of kinase inhibitors showed no significant temperature shifts (Table S11). Thus, the set of developed LIMK inhibitors (1, 15, and 48) together with the negative control 26 represent an excellent toolbox of three chemical probes with diverse binding modes covering all canonical binding modes.
Phosphoproteomic Approach Displays Remarkable Differences between Types I/II and III.
LIMKs are known as macromolecules with many different interaction partners.1 The best-known substrate of LIMKs to date is cofilin, although other phosphorylation substrates have been described (Figure 1), and therefore, LIMKs are fundamental for various cellular functions through regulating the actin and microtubule cytoskeleton. It is important to use highly selective LIMK inhibitors to understand the signaling pathway that might be upregulated or downregulated. In addition, differences in inhibitors on cellular signaling based on diverse binding modes have been well documented.25,32–34 To evaluate functional differences of the three chemical probes based on their diverse binding modes, we evaluated the developed LIMK inhibitors by analyzing differences in phosphorylation response using phosphoproteomics. All in all, over 4000 proteins were detected using HeLa cells, and over 10,000 phosphorylation sites were identified. Interestingly, phosphorylation sites that were significantly altered after inhibitor treatment differed between the allosteric inhibitors 15 and the negative control 26 and the type I inhibitor 1 and the type II inhibitor 48. The type III inhibitor (15) showed modest changes in phosphorylation compared to ATP competitive inhibitors (Figure 7).
Figure 7.

Results of quantitative phosphoproteomic data analysis. (A) At the top: volcano plot for LIMKi3 (1) revealing 4140 proteins with 10387 phosphosites. 362 sites were significantly regulated, of which 189 were upregulated and 173 were downregulated. In the middle: volcano plot for TH470 (48) revealing 4139 proteins with 10344 phosphosites. 339 sites were significantly regulated, of which 146 were upregulated and 193 were downregulated. At the bottom: volcano plot for TH257 (15) revealing 4076 proteins with 10158 phosphosites. 11 sites were significantly regulated, of which 4 were upregulated and 7 were downregulated. Most significant hits were marked in red. All kinases were highlighted in yellow. Remarkably, the known LIMK substrate cofilin (CFL1/2) was not significantly affected by any of the used chemical probes. (B) On the left: GO-term analysis revealed up- and downregulated genes that are involved in actin dynamics for LIMKi3 (1), TH470 (48), and TH257 (15). On the right: GO-term analysis revealed up- and downregulated genes that are involved in microtubule dynamics for LIMKi3 (1), TH470 (48), and TH257 (15).
We speculate that these differences are due to the diverse binding modes and the preferential inhibition of the unphosphorylated inactive state of LIMK1/2, causing minimal effects in unstimulated cells. Intriguingly, the ATP competitive type I inhibitor (1) and type II inhibitor (48) showed similar behavior with respect to up- and downregulated phosphorylation sites (Figure 7). As expected, GO-term analysis revealed a strong link to processes regulating the actin cytoskeleton or microtubules (Figure 8B). Surprisingly, we also found a strong link to phosphorylation changes in proteins involved in RNA processing and RNA binding function as well as mRNA processing (Figure 8). Thus, it is tempting to speculate that due to the role of FMRP, a regulator of the BMPR2-LIMK1 signaling axis, as an RNA binding protein that regulates mRNA metabolism,45 LIMK1/2 may modulate FMRP-dependent regulation of mRNA metabolism.
Figure 8.

Gene ontology (GO) term enrichment analysis of both ATP competitive inhibitors (1) and (48). Significant phosphosites were chosen for both compounds and analyzed using the analysis tool from the PANTHER classification system.35 Results were sorted by the gene ontology category biological process, molecular function, and cellular component. (A) Results for the type I inhibitor (1). (B) Results for the type II inhibitor (48).
Phenotypic Effects of LIMK Inhibitors in a Neurite Outgrowth Model.
To confirm the reported roles of LIMK1/2 in an FXS model, compounds 1, 15, 48, and 26 were evaluated in a neurite outgrowth model. Previously, it has been shown that both LIMK1 and LIMK2 activities are required for NGF-induced neurite extension.2,36 Therefore, N1E-115 cells were transfected with siRNAs against FMR1 (siFMR1) or control siRNAs (siCtr). LIMK inhibitors at different concentrations were added for 12 h followed by stimulation with BMP7 (10 ng/mL). The experiment showed that all inhibitors, compounds 1 (LIMKi3), 15 (TH257), and 48 (TH470) inhibited neurite outgrowth in a dose-dependent manner, whereas 26 (TH263; negative control) showed no inhibition in comparison to the control (siFMR1+BMP7), suggesting that the observed effect is due to the inhibition of LIMK1/2 activity (Figure 9). Interestingly, the type I inhibitor showed a small effect, whereas 15 and 48 revealed more robust dose-dependent effects on the neurite outgrowth, suggesting that allosteric inhibitors might be more effective in modulating neuronal phenotypes mediated by the deregulation of LIMKs.
Figure 9.

Inhibition of neurite outgrowth by the treatment with LIMK inhibitors. (A) N1E-115 cells were transfected with siFMR1 or siCtr. Cell culture media were replaced with 0.2% FBS-containing medium containing 0.05, 0.5, or 5 μM LIMK inhibitors or DMSO (mock) and incubated for 12 h, followed by 10 ng/mL of BMP7 treatment for 24 h. Representative pictures of each group of cells are shown. (B) The number of cells bearing neurite-like structure with longer than a cell body (neurite-positive, gray) was counted, and the fraction (%) of neurite-positive cells was plotted. A minimum of 100 cells were counted in each condition. Mean + SD was plotted (N = 4). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, no significance by ANOVA with post hoc Dunnett test.
CONCLUSIONS
In this manuscript, we reported the characterization of a LIMK1/2 chemical probe set covering all canonical binding modes (type I (1/LIMKi3), II (48/TH470), and III (15/TH257)), and the structure-based design for the type II and type III. All chemical probes had low nanomolar potency, robust cellular activity on target in NanoBRET assays, and excellent selectivity determined by a comprehensive selectivity panel (KINOMEscan). In addition, high selectivity was also observed in cellular lysates using the kinobead assay. Our SAR study also identified a suitable inactive negative control compound (26). The chemical probes were active in a phenotypic assay of neurite outgrowth but showed remarkable differences in signaling evaluated by phosphoproteomics. We have identified a number of significantly up- and downregulated phosphosites altered by the pharmacological inhibition of LIMKs, which give a basis for insights into the signaling pathways mediated by LIMKs. Interestingly, the allosteric inhibition of LIMKs appears to have little to no effect on the upstream and downstream phospho signaling pathways. However, functional and time course studies are needed to elucidate the details of mechanistic differences among the three types of LIMK inhibitors. The characterized chemical tool compounds will facilitate the elucidation of the roles of LIMKs and their mechanisms of action in pathogenesis and in normal physiology.
EXPERIMENTAL SECTION
Differential Scanning Fluorimetry Assay.
The assay was performed according to a previously established protocol.37 A 2 μM solution of the respective LIMK in assay buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 0.5 mM TCEP, 5% glycerol) was mixed 1:1000 with SYPRO Orange (Sigma-Aldrich). The compounds to be tested were added to a final concentration of 10 μM. Twenty microliters of each sample were placed in a 96-well plate and heated from 25 to 95 °C. Fluorescence was monitored using an Mx3005P real-time PCR instrument (Stratagene) with excitation and emission filters set to 465 and 590 nm, respectively. Data were analyzed with the MxPro software.
Isothermal Titration Calorimetry (ITC).
Measurements were performed at 20 °C on a MicroCal VP-ITC (GE Healthcare). LIMK1330-637 was dialyzed overnight into assay buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 0.5 mM TCEP, 5% glycerol). The syringe was loaded with 105 μM LIMK1330-637, and the cell was filled with assay buffer containing 10 μM of the respective inhibitor. Every 5 minutes, 10 μL of the protein solution was injected into the cell for a total of 28 injections. The heat flow data were analyzed with the MicroCal ORIGIN software package employing a single binding site model.
Protein Expression and Purification.
The recombinant LIMK kinase domains LIMK1330-637 and LIMK2330-632 were expressed in insect cells and purified as previously described.16 In brief, exponentially growing TriEx cells (Novagen) at 2 × 106 cells/mL were infected 1:64 with baculovirus stock, incubated for 66 h at 27 °C under constant shaking, and harvested by centrifugation. Cells were then resuspended in lysis buffer (50 mM HEPES pH 7.4, 500 mM NaCl, 20 mM imidazole, 0.5 mM TCEP, 5% glycerol) and lysed by sonication. The lysate was cleared by centrifugation and loaded onto a Ni NTA column. After vigorous rinsing with lysis buffer, the His6-tagged proteins were eluted in lysis buffer containing 300 mM imidazole. While the proteins were subjected to dialysis to remove the imidazole, the N-terminal tags were cleaved by TEV protease. Contaminating proteins, the cleaved tags and TEV protease itself were removed with another Ni NTA step. Finally, the LIMK kinase domains were concentrated and subjected to gel filtration using an AKTA Xpress system combined with an S200 16/600 gel filtration column (GE Healthcare). The elution volumes of 91.8 mL (LIMK1330-637) and 91.6 mL (LIMK2330-632) indicated the proteins to be monomeric in solution. The final yields were 2.0 mg/L of insect cell medium for LIMK1330-637 and 0.2 mg/L of insect cell medium for LIMK2330-632.
Crystallization.
One hundred nanoliters of drops of the protein solution with the respective ligand were transferred to a 3-well crystallization plate (Swissci), mixed with 50 nL of the precipitant solution, and incubated at 4 °C (details in Table S1). Crystals appeared overnight and did not change appearance after 7 days. They were mounted with additional 25% ethylene glycol for cryoprotection. Data were collected at Diamond Light Source beamline I03, analyzed, scaled, and merged with Xia2.38 The structures were solved by molecular replacement with Phaser39 using a LIMK2 model as a template (PDB-ID 4TPT) and refined with Refmac5.40 The models were validated using MolProbity.41 A summary of data collection and refinement statistics is given in Table S1. The model and the structure factors have been deposited with the PDB IDs 5NXD,7QHG and 8AAU (crystallographic parameters are included in Table S1).
LIMK1/2 RapidFire Mass Spectrometry Assay.
RapidFire Mass Spectrometry Assay was performed as previously described.16 In brief, RapidFire mass spectrometry offers a high-throughput, label-free, and direct measurement of substrate modifications, including protein phosphorylation. This technology couples desalting by solid-phase extraction (SPE) with rapid automated sample injection and processing to achieve high sampling rates. For IC50 determination, the respective inhibitors in DMSO (11-point concentration series, all points in technical duplicates) were dispensed to a 384-well polypropylene plate using an ECHO 550 acoustic dispenser (Labcyte). Then, the LIMK1 kinase domain was added (final concentration 40 nM) to allow for a 10 min preincubation at room temperature. The phosphorylation reaction was initiated by adding a mixture of CFL1 (final concentration 2 mM) and ATP (final concentration 800 mM). After 60 min of incubation at room temperature, the reaction was stopped by adding formic acid to a final concentration of 1%. The reaction volume was 50 ml, and the assay buffer composition was 50 mM tris pH 7.5, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM MgCl2. The plate was transferred to a RapidFire RF360 high-throughput sampling robot (Agilent). Samples were aspirated under vacuum and loaded onto a C4 SPE cartridge and washed for 5.5 s with 0.1% (v/v) formic acid in LC-MS grade water to remove nonvolatile buffer components. After the aqueous wash, analytes of interest were eluted from the C4 SPE onto a 6530 Q-TOF LC/MS (Agilent) in an organic elution step (85% acetonitrile in LC-MS grade water containing 0.1% formic acid). Ion data for the CFL1 substrate and the Phospho-CFL1 product were extracted, and peak area data were integrated using RapidFire integrator software (Agilent). Finally, IC50 curves were generated using GraphPad Prism 7.
NanoBRET.
The assay was performed as described previously.29–31 In brief, full-length LIMK1 and LIMK2 cloned in frame in a NanoLuc-vector (Promega) were transfected into HEK293T cells (ATCC CRL-1573) using FuGENE HD (Promega, E2312), and proteins were allowed to express for 20 h. The serially diluted inhibitor and NanoBRET Kinase Tracer (Promega) were pipetted into white 384-well plates (Greiner 781 207) using an ECHO 550 acoustic dispenser (Labcyte). All constructs and tracers used are summarized in Supporting Table S10. The corresponding transfected cells were added and reseeded at a density of 2 × 105 cells/mL after trypsinization and resuspension in Opti-MEM without phenol red (Life Technologies). The system was allowed to equilibrate for 2 h at 37 °C and 5% CO2 prior to BRET measurements. To measure BRET, NanoBRET NanoGlo Substrate + Extracellular NanoLuc Inhibitor (Promega, N2160) were added as per the manufacturer’s protocol, and filtered luminescence was measured on a PHERAstar plate reader (BMG Labtech) equipped with a luminescence filter pair (450 nm BP filter (donor) and 610 nm LP filter (acceptor)). Competitive displacement data were then plotted using GraphPad Prism 9 software using a normalized 3-parameter curve fit with the following equation: Y = 100/(1 + 10∧((X-Log IC50))).
Chemistry.
All starting materials, reagents, and solvents were purchased from commercial suppliers like Alfa-Aesar, Sigma-Aldrich, TCI, and Apollo Scientific Ltd., and used without further purification unless otherwise stated. Reactions were monitored by thin-layer chromatography (TLC) using precoated TLC sheets ALUGRAM Xtra SIL G/UV254 (thickness of layer: 0.20 mm; mean pore size: 60 Å) purchased from Machery-Nagel (Düren, Germany).
The structures of the presented compounds were verified by 1H-, 13C-NMR, mass spectrometry (ESI), and high-resolution mass spectrometry (HRMS); the purity of the final compounds (>95%) was determined by HPLC. Compound 1 (BMS-5/LIMKi3) was purchased from Tocris Bioscience. Compounds 8–48 were synthesized as described in Figure 2B and Schemes 1–3. All commercial chemicals and solvents are of reagent grade and were used without further purification. 1H- and 13C-NMR spectra were measured in DMSO-d6 or CDCl3 on a Bruker DPX 250 or AV 500 spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) in the scale relative to the solvent signals: 2.50 ppm (1H-NMR) and 39.52 ppm (13C-NMR) for DMSO-d6, 7.26 ppm (1H-NMR) and 77.16 ppm (13C-NMR) for CDCl3. Coupling constants (J) were reported in hertz (Hz), and multiplicities were designated as follows: s (singlet), bs (broad singlet), d (doublet), dd (double doublet), ddd (doublet of doublet of doublets), t(triplet), dt (doublet of triplets), q (quartet), m (multiplet). Mass spectra were obtained on a Fisons Instruments VG Platform II spectrometer (ESI-MS system) or on a PerSeptive Biosystems Mariner biospectrometry workstation (nanospray ESI-MS system) measuring in the positive- and/or negative-ion mode. High-resolution mass spectra were recorded on a MALDI LTQ Orbitrap XL system from Thermo Scientific (Waltham, MA). Purity of the synthesized compounds was determined by HPLC with either method-A: on a Shimadzu (Duisburg, Germany) LC-20AD HPLC set at 254 and 280 nm equipped with a Shimadzu LC-MS-2020 detector using a Phenomenex LTD (Aschaffenburg, Germany) Luna 10 μm 21.2 × 250 mm reversed-phase (C18) column. As mobile phase, Milli-Q water (A) and acetonitrile (B) + 0.1% formic acid were used with a flow rate of 1 mL/min. The gradient was running over 25 min starting with 95% A and 5% B, going down on 10% A and 90% B, and finishing at 95% A and 5% B. Or method-B: on a Varian ProStar HPLC equipped with a MultoHigh100 RP18-5μ 240 mm + 4 mm column (CS-Chromatographie Service GmbH, Langerwehe, Germany) using a gradient (H2O + 0.1% formic acid/MeOH 80:20 isocratic for 5 min to MeOH after an additional 45 min and MeOH for an additional 10 min) at a flow rate of 1 mL/min and UV detection at 254 and 280 nm.
Scheme 1; Step a: general procedure for the formation of sulfonamides (compounds 8–13).
4-(Chlorosulfonyl)benzoic acid (7) (1.00 equiv) was dissolved in 11 mL of THF/mmol 7. Amine (10 equiv) was added dropwise to the stirred solution at 20 °C. The mixture was stirred at room temperature for 18 h. The reaction was diluted with 100 mL of EtOAc. The organic layer was washed with 1 N HCl (3 × 50 mL). Afterward, the water layer was extracted once more with 50 mL of EtOAc and the combined organic layers were dried over MgSO4. The organic layer was concentrated under reduced pressure. The residue was recrystallized with hot MeOH/H2O.
Synthesis of 4-(phenylsulfamoyl)benzoic acid (8): synthesis Scheme 1; step a: 4-(chlorosulfonyl)benzoic acid (7; 1.00 g, 4.53 mmol), aniline (4.14 mL, 45.3 mmol); product: pale crystalline solid; yield: 1.12 g ≡ 89%. 1H-NMR (500 MHz, (CD3)2SO): δ = 13.44 (s, 1H), 10.43 (s, 1H), 8.06 (d, J = 8.5 Hz, 2H), 7.85 (d, J = 8.5 Hz, 2H), 7.23 (t, J = 7.9 Hz, 2H), 7.08 (d, J = 7.6 Hz, 2H), 7.04 (t, J = 7.4 Hz, 1H). 13C-NMR (126 MHz, (CD3)2SO): δ = 166.09, 143.08, 137.27, 134.55, 130.14, 129.26, 127.01, 124.46, 120.42. MS (ESI−): m/z = 276.07 [M – H]−.
Synthesis of 4-[(3-chlorophenyl)sulfamoyl]benzoic acid (9): synthesis Scheme 1; step a: 4-(chlorosulfonyl)benzoic acid (7; 500 mg, 2.27 mmol), 3-chloroaniline (2.89 g, 22.66 mmol); product: pale crystalline solid; yield: 0.54 g ≡ 76%. 1H-NMR (500 MHz, (CD3)2SO): δ = 13.48 (s, 1H), 10.74 (s, 1H), 8.09 (d, J = 8.6, 2H), 7.88 (d, J = 8.5, 2H), 7.27 (t, J = 8.4, 1H), 7.14–7.09 (m, 2H), 7.09–7.04 (m, 1H). 13C-NMR (126 MHz, (CD3)2SO): δ = 166.03, 142.67, 138.90, 134.84, 133.45, 131.08, 130.31, 127.01, 124.15, 119.41, 118.32. MS (ESI−): m/z = 309.94 [M – H]−.
Synthesis of 4-[(2-chlorophenyl)sulfamoyl]benzoic acid (10): synthesis Scheme 1; step a: 4-(chlorosulfonyl)benzoic acid (7; 500 mg, 2.27 mmol), 2-chloroaniline (2.89 g, 22.67 mmol); product: pale crystalline solid; yield: 0.35 g ≡ 50%. 1H-NMR (500 MHz, (CD3)2SO): δ = 13.47 (s, 1H), 10.23 (s, 1H), 8.08 (d, J = 8.6 Hz, 2H), 7.81 (d, J = 8.6 Hz, 2H), 7.41 (dd, J = 7.7, 1.1 Hz, 1H), 7.30 (ddd, J = 8.2, 7.0, 1.5 Hz, 1H), 7.26–7.19 (m, 2H). 13C-NMR (126 MHz, (CD3)2SO): δ = 166.21, 144.03, 134.52, 133.13, 130.14, 130.00, 129.60, 128.13, 128.10, 127.89, 126.98. MS (ESI−): m/z = 309.89 [M – H]−.
Synthesis of 4-[(4-chlorophenyl)sulfamoyl]benzoic acid (11): synthesis Scheme 1; step a: 4-(chlorosulfonyl)benzoic acid (7; 500 mg, 2.27 mmol); 4-chloroaniline (2.89 g, 22.67 mmol); product: pale crystalline solid; yield: 0.36 g ≡ 51%. 1H-NMR (500 MHz, (CD3)2SO): δ = 13.44 (s, 1H), 10.56 (s, 1H), 8.07 (d, J = 8.7 Hz, 2H), 7.85 (d, J = 8.7 Hz, 2H), 7.31 (d, J = 9.0 Hz, 2H), 7.09 (d, J = 9.0 Hz, 2H).
Synthesis of 4-(benzylsulfamoyl)benzoiinsights facilitated the rational design of a highly potentc acid (12): synthesis Scheme 1; step a: 4-(chlorosulfonyl)benzoic acid (7; 500 mg, 2.27 mmol); benzylamine (2.47 mL, 22.67 mmol); product: pale crystalline solid; yield: 0.55 g ≡ 83%. 1H-NMR (500 MHz, (CD3)2SO): δ = 13.43 (s, 1H), 8.35 (t, J = 6.3 Hz, 1H), 8.08 (d, J = 8.6 Hz, 2H), 7.89 (d, J = 8.6 Hz, 2H), 7.29–7.24 (m, 2H), 7.21 (dd, J = 9.9, 4.4 Hz, 3H), 4.02 (d, J = 6.3 Hz, 2H). 13C-NMR (126 MHz, (CD3)2SO): δ = 166.27, 144.49, 137.42, 134.01, 130.08, 128.25, 127.61, 127.21, 126.77, 46.13. MS (ESI−): m/z = 292.08 [M + H]+.
Synthesis of 4-[(2-phenylethyl)sulfamoyl]benzoic acid (13): synthesis Scheme 1; step a: 4-(chlorosulfonyl)benzoic acid (7; 500 mg, 2.27 mmol), 2-phenylethan-1-amine (2.85 mL, 22.67 mmol); product: pale crystalline solid; yield: 0.59 g ≡ 85%. 1H-NMR (500 MHz, (CD3)2SO): δ = 13.42 (s, 1H), 8.10 (d, J = 8.3 Hz, 2H), 7.91 (t, J = 5.8 Hz, 1H), 7.88 (d, J = 8.3 Hz, 2H), 7.25 (t, J = 7.3 Hz, 2H), 7.19 (d, J = 7.3 Hz, 1H), 7.15 (dd, J = 7.3, 5.9 Hz, 2H), 3.03–2.96 (m, 2H), 2.67 (t, J = 7.5 Hz, 2H). 13C-NMR (126 MHz, (CD3)2SO): δ = 166.27, 144.12, 138.57, 134.07, 130.15, 128.69, 128.33, 126.77, 126.28, 44.05, 35.28. MS (ESI+): m/z = 306.06 [M + H]+.
Synthesis of benzyl(propyl)amine (14): synthesis Scheme 1; step b: to a solution of benzaldehyde (711 mg, 6.70 mmol) and n-propylamine (396 mg, 6.70 mmol) in DCE was added glacial acetic acid (0.77 mL, 13.40 mmol) and the solution was stirred for 1 h at RT. After 1 h, NaHB(OAc)3 (1.99 g, 9.38 mmol) was added and the reaction was stirred for 20 h. Afterward, the reaction was quenched using 1 M NaOH and the product was extracted with Et2O. The organic layer was washed with brine, dried over MgSO4, and evaporated under a reduced pressure. The crude product was purified by column chromatography using n-hexane/EtOAc to obtain benzyl(propyl)amine as a yellow oil. Yield: 0.56 g ≡ 56%. 1H-NMR (500 MHz, (CD3)2SO): δ = 7.36–7.25 (m, 4H), 7.24–7.16 (m, 1H), 3.67 (s, 2H), 2.43 (t, J = 7.1, 2H), 1.48–1.37 (m, 2H), 0.86 (t, J = 7.4, 3H). 13C-NMR (126 MHz, (CD3)2SO): δ = 141.20, 128.02, 127.84, 126.37, 53.05, 50.70, 22.68, 11.85. MS (ESI+): m/z = 150.12 [M + H]+.
Scheme 1; step c: general procedure for the formation of amides (compounds 15–32).
Benzoic acid (1.00 equiv), EDC*HCl (1.20 equiv), and HOBT (1.00 equiv) were dissolved in 10 mL of CH2Cl2 and stirred at room temperature. After 1 h, amine (1.5 equiv) was added and the mixture was stirred at room temperature for 20 h. The reaction was quenched with 40 mL of H2O. The product was extracted with 50 mL of EtOAc. The organic layer was washed with 1 N HCl and brine and dried over MgSO4. The organic layer was concentrated under a reduced pressure. The residue was further purified by column chromatography, using n-hexane/ethyl acetate.
Synthesis of N-benzyl-4-(benzylsulfamoyl)benzamide (15): synthesis Scheme 1; step c: 4-(phenylsulfamoyl)benzoic acid (8; 150 mg, 0.54 mmol), EDC*HCl (124 mg, 0.65 mmol), HOBT (73 mg, 0.54 mmol), benzyl(butyl)amine (132 mg, 0.81 mmol); product: resinlike solid; yield: 0.17 g ≡ 74%. 1H-NMR (500 MHz, CDCl3, rotamers observed) δ: 0.71, 0.93 (t, 3H, J = 7.20 Hz), 1.00–1.07, 1.31–1.37 (m, 2H), 1.39–1.45, 1.59–1.62 (m, 2H), 3.01, 3.47 (t, 2H, J = 7.50 Hz), 4.37, 4.74 (s, 2H), 7.02–7.08 (m, 4H), 7.12 (t, 1H, J = 6.79 Hz), 7.18–7.23 (m, 2H), 7.28–7.37 (m, 4H), 7.41–7.45 (m, 2H), 7.69–7.78 (m, 2H). 13C-NMR (126 MHz, CDCl3, rotamers observed) δ: 170.43, 141.23, 141.10, 140.17, 140.01, 136.97, 136.33, 136.20, 129.54, 129.08, 128.91, 128.22, 127.96, 127.78, 127.62, 127.57, 127.26, 126.71, 125.97, 125.90, 122.31, 122.18, 52.49, 48.01, 47.56, 45.03, 30.32, 29.23, 20.30, 19.76, 13.99, 13.65. MS (ESI+): m/z = 423.13 [M + H]+. HRMS: m/z calcd for C24H27N2O3S1 [M + H]+ = 423.17369. Found [M + H]+ = 423.17329. HPLC (method-B): tR = 35.473, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-N-ethyl-4-(phenylsulfamoyl)benzamide (16): synthesis Scheme 1; step c: 4-(phenylsulfamoyl)benzoic acid (8; 100 mg, 0.36 mmol), EDC*HCl (83 mg, 0.43 mmol), HOBT (49 mg, 0.36 mmol), benzyl(ethyl)amine (80 μL, 0.54 mmol); product: white solid; yield: 0.09 g ≡ 63%. 1H-NMR (500 MHz, CDCl3, rotamers observed) δ: 1.03, 1.21 (t, 3H, J = 6.70 Hz), 3.09, 3.54 (q, 2H, J = 6.82 Hz), 4.38, 4.75 (s, 2H), 6.56, 6.60 (s, 1H), 7.02–7.16 (m, 4H), 7.21–7.25 (m, 2H), 7.30–7.35 (m, 4H), 7.44–7.48 (m, 2H), 7.70–7.79 (m, 2H). 13C-NMR (126 MHz, CDCl3, rotamers observed) δ: 170.30, 170.21, 141.27, 141.04, 140.18, 140.12, 137.00, 136.32, 136.14, 129.58, 129.10, 128.92, 128.23, 127.97, 127.80, 127.67, 127.59, 127.34, 127.12, 126.72, 126.02, 122.33, 122.22, 52.10, 47.22, 42.92, 40.26, 13.75, 12.36. MS (ESI+): m/z = 395.15 [M + H]+. HRMS: m/z calcd for C22H23N2O3S1 [M + H]+ = 395.14239. Found [M + H]+ = 395.14218. HPLC (method-B): tR = 33.552, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-4-(phenylsulfamoyl)-N-propylbenzamide (17): synthesis Scheme 1; step c: 4-(phenylsulfamoyl)benzoic acid (8; 150 mg, 0.54 mmol), EDC*HCl (124 mg, 0.65 mmol), HOBT (73 mg, 0.54 mmol), benzyl(propyl)amine (12; 121 mg, 0.81 mmol); product: resinoid pale yellow solid; yield: 0.12 g ≡ 52%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ: 0.54, 0.85 (t, 3H, J = 7.20 Hz), 1.34–1.42, 1.52–1.59 (m, 2H), 2.93, 3.29 (t, 2H, J = 7.50 Hz), 4.34, 4.67 (s, 2H), 7.02–7.09 (m, 4H), 7.19–7.23 (m, 2H), 7.26–7.38 (m, 4H), 7.51, 7.57 (d, 2H, J = 7.80 Hz), 7.73, 7.79 (d, 2H, J = 7.80 Hz), 10.32 (s, 1H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ: 169.48, 169.42, 140.82, 139.82, 139.69, 137.55, 137.43, 129.17, 128.67, 128.56, 127.50, 127.15, 127.05, 126.95, 126.87, 124.45, 124.36, 120.60, 120.42, 51.69, 49.84, 46.92, 46.00, 21.00, 19.89, 11.18, 10.76. MS (ESI+): m/z = 409.12 [M + H]+. HRMS: m/z calcd for C23H24N2O3S1 [M] = 408.15076. Found [M] = 408.15235. HPLC (method-A): tR = 15.447, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-N-methyl-4-(phenylsulfamoyl)benzamide (4): synthesis Scheme 1; step c: 4-(phenylsulfamoyl)benzoic acid (8; 120 mg, 0.43 mmol), EDC*HCl (100 mg, 0.52 mmol), HOBT (58 mg, 0.43 mmol), benzyl(methyl)amine (79 mg, 0.65 mmol); product: white solid; yield: 0.13 g ≡ 76%. 1H-NMR (500 MHz, CDCl3, rotamers observed) δ: 2.78, 3.05 (s, 3H), 4.39, 4.73 (s, 2H), 6.84, 6.87 (s, 1H), 7.03–7.14 (m, 4H), 7.20–7.25 (m, 2H), 7.32–7.36 (m, 4H), 7.47–7.49 (m, 2H), 7.72–7.79 (m, 2H). 13C-NMR (126 MHz, CDCl3, rotamers observed) δ: 170.66, 169.95, 140.78, 140.67, 140.38, 140.32, 136.53, 136.12, 135.94, 129.60, 129.17, 128.98, 128.38, 128.07, 127.93, 127.72, 127.60, 127.53, 126.68, 126.02, 122.28, 122.22, 55.13, 51.02, 36.96, 33.61. MS (ESI+): m/z = 381.04 [M + H]+. HRMS: m/z calcd for C21H21N2O3S1 [M + H]+ = 381.12674. Found [M + H]+ = 381.12608. HPLC (method-B): tR = 32.986, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-4-[(3-chlorophenyl)sulfamoyl]-N-propylbenzamide (18): synthesis Scheme 1; step c: 4-[(3-chlorophenyl)-sulfamoyl]benzoic acid (9; 150 mg, 0.48 mmol), EDC*HCl (111 mg, 0.58 mmol), HOBT (65 mg, 0.48 mmol), benzyl(propyl)amine (12, 108 mg, 0.72 mmol); product: resinoid pale solid; yield: 0.18 g ≡ 85%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ: 0.54, 0.86 (t, 3H, J = 7.26 Hz), 1.34–1.42, 1.53–1.60 (m, 2H), 2.93, 3.30 (t, 2H, J = 7.30 Hz), 4.35, 4.67 (s, 2H), 7.03–7.10 (m, 4H), 7.23–7.38 (m, 5H), 7.54, 7.61 (d, 2H, J = 8.10 Hz), 7.77, 7.83 (d, 2H, J = 8.00 Hz), 10.62 (s, 1H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ: 169.39, 169.34, 141.11, 139.40, 139.26, 139.02, 137.52, 136.83, 133.39, 130.97, 129.18, 128.65, 128.56, 127.50, 127.44, 127.33, 127.15, 127.07, 126.96, 126.86, 124.14, 124.08, 119.58, 119.44, 118.58, 118.39, 51.71, 49.83, 46.93, 46.03, 21.01, 19.88, 11.18, 10.74. MS (ESI+): m/z = 443.15 [M + H]+. HRMS: m/z calcd for C23H24ClN2O3S1 [M + H]+ = 443.11907. Found [M + H]+ = 443.11867. HPLC (method-A): tR = 16.164, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-4-[(3-chlorophenyl)sulfamoyl]-N-methylbenzamide (19): synthesis Scheme 1; step c: 4-[(3-chlorophenyl)-sulfamoyl]benzoic acid (9; 150 mg, 0.48 mmol), EDC*HCl (111 mg, 0.58 mmol), HOBT (65 mg, 0.48 mmol), benzyl(methyl)amine (87 mg, 0.72 mmol); product: pale yellow solid; yield: 0.14 g ≡ 70%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ: 2.74, 2.90 (s, 3H), 4.35, 4.66 (s, 2H), 7.04–7.11 (m, 4H), 7.23–7.39 (m, 5H), 7.58, 7.65 (d, 2H, J = 8.20 Hz), 7.78, 7.84 (d, 2H, J = 8.10 Hz), 10.55 (s, 1H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ: 169.38, 168.84, 140.81, 140.64, 139.70, 139.57, 139.03, 136.99, 136.51, 133.41, 131.04, 128.74, 128.65, 127.86, 127.64, 127.53, 127.29, 126.92, 126.80, 124.09, 119.43, 118.38, 53.96, 49.79, 36.67, 32.78. MS (ESI+): m/z = 415.07 [M + H]+. HRMS: m/z calcd for C21H20ClN2O3S1 [M + H]+ = 415.08777. Found [M + H]+ = 415.08763. HPLC (method-A): tR = 15.184, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-4-[(2-chlorophenyl)sulfamoyl]-N-methylbenzamide (20): synthesis Scheme 1; step c: 4-[(2-chlorophenyl)-sulfamoyl]benzoic acid (10; 150 mg, 0.48 mmol), EDC*HCl (111 mg, 0.58 mmol), HOBT (65 mg, 0.48 mmol), benzyl(methyl)amine (87 mg, 0.72 mmol); product: white solid; yield: 0.09 g ≡ 45%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ: 2.77, 2.91 (s, 3H), 4.39, 4.68 (s, 2H), 7.13–7.41 (m, 9H), 7.57, 7.63 (d, 2H, J = 8.20 Hz), 7.69, 7.75 (d, 2H, J = 8.10 Hz), 10.13 (s, 1H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ: 169.56, 169.02, 141.00, 140.83, 140.59, 140.41, 137.04, 136.54, 133.28, 129.96, 129.88, 129.46, 128.76, 128.66, 128.18, 128.08, 127.97, 127.85, 127.67, 127.61, 127.49, 127.29, 126.88, 126.80, 53.93, 49.79, 36.68, 32.69. MS (ESI+): m/z = 415.09 [M + H]+. HRMS: m/z calcd for C21H20ClN2O3S1 [M + H]+ = 415.08777. Found [M + H]+ = 415.08748. HPLC: tR = 14.837, purity ≥95% (UV: 254/280 nM).
Synthesis of N-methyl-N-(2-phenylethyl)-4-(phenylsulfamoyl)-benzamide (21): synthesis Scheme 1; step c: 4-(phenylsulfamoyl)-benzoic acid (8; 250 mg, 0.90 mmol), EDC*HCl (207 mg, 0.11 mmol), HOBT (122 mg, 0.90 mmol), methyl(2-phenylethyl)amine (183 mg, 1.35 mmol); product: pale yellow solid; yield: 0.18 g ≡ 49%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ: 10.34, 10.30 (s, 1H), 7.78–7.64 (m, 2H), 7.46–7.03 (m, 11H), 6.87 (d, J = 6.9 Hz, 1H), 3.65, 3.26 (t, J = 7.1 Hz, 2H), 3.01, 2.73 (s, 3H), 2.88, 2.71 (t, J = 7.3 Hz, 2H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ: 169.08, 168.45, 140.78, 140.67, 139.94, 139.43, 138.98, 138.21, 137.42, 129.21, 129.15, 128.77, 128.73, 128.39, 127.42, 126.95, 126.83, 126.61, 126.35, 126.25, 124.39, 124.35, 120.44, 120.32, 52.05, 48.23, 37.13, 33.47, 32.56, 32.23. MS (ESI+): m/z = 395.16 [M + H]+. HRMS: m/z calcd for C22H23N2O3S1 [M + H]+ = 395.14239. Found [M + H]+ = 395.14200. HPLC (method-A): tR = 14.435, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-4-(benzylsulfamoyl)-N-methylbenzamide (22): synthesis Scheme 1; step c: 4-(benzylsulfamoyl)benzoic acid (12; 200 mg, 0.69 mmol), EDC*HCl (158 mg, 0.82 mmol), HOBT (93 mg, 0.69 mmol), benzyl(methyl)amine (125 mg, 1.03 mmol); product: colorless crystals; yield: 0.22 g ≡ 81%. 1H-NMR (500 MHz, CDCl3, rotamers observed) δ: 2.83, 3.07 (s, 3H), 4.15 (t, 2H, J = 7.00 Hz), 4.45, 4.76 (s, 2H), 4.83–4.87 (m, 1H), 7.14–7.25 (m, 5H), 7.28–7.40 (m, 5H), 7.56 (d, 2H, J = 7.50 Hz), 7.85, 7.91 (d, 2H, J = 8.05 Hz), 10.32 (s, 1H). 13C-NMR (126 MHz, CDCl3, rotamers observed) δ: 170.69, 170.01, 141.23, 141.14, 140.69, 140.54, 136.60, 136.04, 129.20, 128.99, 128.91, 128.87, 128.41, 128.22, 128.10, 128.03, 127.93, 127.83, 127.63, 127.53, 126.71, 55.16, 51.01, 47.47, 36.98, 33.48. MS (ESI+): m/z = 395.07 [M + H]+. HRMS: m/z calcd for C22H23N2O3S1 [M + H]+ = 395.14239. Found [M + H]+ = 395.14212. HPLC (method-A): tR = 14.541, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-4-[(4-chlorophenyl)sulfamoyl]-N-methylbenzamide (23): synthesis Scheme 1; step c: 4-[(4-chlorophenyl)-sulfamoyl]benzoic acid (11; 150 mg, 0.48 mmol), EDC*HCl (111 mg, 0.58 mmol), HOBT (65 mg, 0.48 mmol), benzyl(methyl)amine (87 mg, 0.72 mmol); product: white solid; yield: 0.10 g ≡ 50%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ: 2.74, 2.90 (s, 3H), 4.35, 4.66 (s, 2H), 7.07–7.11 (m, 3H), 7.27–7.39 (m, 6H), 7.57, 7.64 (d, 2H, J = 8.00 Hz), 7.75, 7.81 (d, 2H, J = 8.00 Hz), 10.51 (s, 1H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ: 169.42, 168.86, 140.68, 140.52, 139.81, 139.67, 137.00, 136.52, 136.43, 129.24, 128.75, 128.65, 128.50, 127.81, 127.65, 127.47, 127.29, 126.90, 126.78, 121.94, 53.96, 49.80, 36.70, 32.79. MS (ESI +): m/z = 415.07 [M + H]+. HRMS: m/z calcd for C21H20ClN2O3S1 [M + H]+ = 415.08777. Found [M + H]+ = 415.08755. HPLC: tR = 15.207, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-4-(phenylsulfamoyl)benzamide (24): synthesis Scheme 1; step c: 4-(phenylsulfamoyl)benzoic acid (8; 141 mg, 0.51 mmol), EDC*HCl (117 mg, 0.61 mmol), HOBT (69 mg, 0.51 mmol), benzylamine (83 μL, 0.76 mmol); product: white solid; yield: 0.16 g ≡ 83%. 1H-NMR (500 MHz, (CD3)2SO) δ: 4.46 (d, 2H, J = 5.95 Hz), 7.03 (t, 1H, J = 7.30 Hz), 7.09 (d, 2H, J = 7.50 Hz), 7.21–7.25 (m, 3H), 7.29–7.33 (m, 4H), 7.83 (d, 2H, J = 8.50 Hz), 7.99 (d, 2H, J = 8.50 Hz), 9.21 (t, 1H, J = 5.95 Hz), 10.38 (s, 1H). 13C-NMR (126 MHz, (CD3)2SO) δ: 165.05, 141.65, 139.22, 138.21, 137.38, 129.24, 128.32, 128.19, 127.28, 126.85, 126.79, 124.36, 120.30, 42.75. MS (ESI+): m/z = 367.05 [M + H]+. HRMS: m/z calcd for C20H19N2O3S1 [M + H]+ = 367.11109. Found [M + H]+ = 367.11121. HPLC (method-A): tR = 14.112, purity ≥95% (UV: 254/280 nM).
Synthesis of N-methyl-N-phenyl-4-(phenylsulfamoyl)benzamide (25): synthesis Scheme 1; step c: 4-(phenylsulfamoyl)benzoic acid (8; 200 mg, 0.72 mmol), EDC*HCl (166 mg, 0.87 mmol), HOBT (97 mg, 0.72 mmol), N-methylaniline (117 μL, 1.08 mmol); product: white solid; yield: 0.23 g ≡ 86%. 1H-NMR (500 MHz, (CD3)2SO) δ: 10.19 (s, 1H), 7.52 (d, J = 8.1, 2H), 7.36 (d, J = 7.9, 2H), 7.24–7.08 (m, 7H), 7.05 (t, J = 7.4, 1H), 6.94 (d, J = 7.6, 2H), 3.35 (s, 3H). 13C-NMR (126 MHz, (CD3)2SO) δ: 168.18, 143.66, 140.61, 139.53, 137.22, 129.13, 129.10, 128.66, 127.14, 126.80, 126.19, 124.50, 120.75, 37.52. MS (ESI+): m/z = 367.14 [M + H]+. HRMS: m/z calcd for C20H19N2O3S1 [M + H]+ = 367.11109. Found [M + H]+ = 367.11132. HPLC (method-A): tR = 14.116, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-4-(benzylsulfamoyl)benzamide (26): synthesis Scheme 1; step c: 4-(benzylsulfamoyl)benzoic acid (12; 114 mg, 0.39 mmol), EDC*HCl (90 mg, 0.47 mmol), HOBT (53 mg, 0.39 mmol), benzylamine (63 mg, 0.59 mmol); product: white solid; yield: 0.08 g ≡ 55%. 1H-NMR (500 MHz, (CD3)2SO) δ: 9.25 (t, J = 6.0 Hz, 1H), 8.28 (s, 1H), 8.04 (d, J = 8.5 Hz, 2H), 7.88 (d, J = 8.5 Hz, 2H), 7.37–7.31 (m, 4H), 7.31–7.19 (m, 6H), 4.50 (d, J = 6.0 Hz, 2H), 4.00 (s, 2H). 13C-NMR (126 MHz, (CD3)2SO) δ: 165.10, 142.99, 139.34, 137.62, 137.50, 128.35, 128.27, 128.13, 127.60, 127.27, 127.21, 126.87, 126.57, 46.13, 42.76. MS (ESI+): m/z = 381.07 [M + H]+. HRMS: m/z calcd for C21H21N2O3S1 [M + H]+ = 381.12674. Found [M + H]+ = 381.12615. HPLC (method-A): tR = 14.245, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-N-methyl-4-[(2-phenylethyl)sulfamoyl]-benzamide (27): synthesis Scheme 1; step c: 4-[(2-phenylethyl)-sulfamoyl]benzoic acid (13; 150 mg, 0.49 mmol), EDC*HCl (113 mg, 0.59 mmol), HOBT (66 mg, 0.49 mmol), benzyl(methyl)amine (89 mg, 0.74 mmol); product: brownish solid; yield: 0.15 g ≡ 75%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ: 2.63–2.70 (m, 2H), 2.80, 2.91 (s, 3H), 2.98–3.02 (m, 2H), 4.43, 4.69 (s, 2H), 7.11–7.41 (m, 10H), 7.61, 7.66 (d, 2H, J = 8.00 Hz), 7.78–7.97 (m, 3H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ: 169.67, 169.11, 141.08, 140.06, 139.93, 138.61, 137.08, 136.59, 128.77, 128.66, 128.33, 127.67, 127.49, 127.37, 127.30, 126.78, 126.71, 126.26, 54.01, 49.82, 44.04, 36.76, 35.27, 32.70. MS (ESI+): m/z = 409.15 [M + H]+. HRMS: m/z calcd for C23H25N2O3S1 [M + H]+ = 409.15804. Found [M + H]+ = 409.15779. HPLC (method-A): tR = 14.926, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-4-(N-(3-chlorophenyl)sulfamoyl)-N-(2-cyanoethyl)benzamide (28): synthesis Scheme 2; step b: 4-(N-(3-chlorophenyl)sulfamoyl)benzoic acid (9; 250 mg, 0.80 mmol), EDC*HCl (184 mg, 0.96 mmol), HOBT (108 mg, 0.80 mmol), 3-(benzylamino)propanenitrile (128 mg, 1.20 mmol); product: white solid; yield: 0.25 g ≡ 70%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ 10.64 (s, 1H), 7.83 (d, J = 6.3 Hz, 2H), 7.61 (d, J = 7.2 Hz, 2H), 7.43–7.22 (m, 5H), 7.17–7.01 (m, 4H), 4.74, 4.44 (s, 2H), 3.62, 3.35 (s, 2H), 2.86, 2.76 (s, 2H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ 169.78, 140.22, 139.79, 138.98, 136.87, 136.31, 133.41, 130.99, 128.73, 127.81, 127.61, 127.46, 127.05, 126.92, 124.11, 119.50, 118.90, 118.42, 51.91, 46.30, 43.72, 40.37, 16.30, 15.36. MS (ESI+): m/z = 454.12 [M + H]+. HPLC (method-A): tR = 14.877, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-4-(N-(3-chlorophenyl)sulfamoyl)-N-(prop-2-yn-1-yl)benzamide (29): synthesis Scheme 3; step d: 4-(N-(3-chlorophenyl)sulfamoyl)benzoic acid (9; 200 mg, 0.64 mmol), EDC*HCl (148 mg, 0.77 mmol), HOBT (87 mg, 0.64 mmol), N-benzylprop-2-yn-1-amine (41; 140 mg, 0.96 mmol); product: white solid; yield: 0.21 g ≡ 75%. 1H-NMR (500 MHz, CDCl3, rotamers observed) δ 7.82–7.79 (m, 2H), 7.64–7.55 (m, 2H), 7.44–7.28 (m, 4H), 7.24–6.97 (m, 5H), 6.93 (d, J = 6.5 Hz, 1H), 4.86, 4.54 (s, 2H), 4.30, 3.76 (s, 2H), 2.37, 2.29 (s, 2H). 13C-NMR (126 MHz, CDCl3, rotamers observed) δ 170.09, 169.67, 140.57, 140.09, 137.48, 135.86, 135.21, 130.60, 129.72, 129.21, 129.04, 128.80, 128.20, 127.85, 127.67, 127.11, 125.94, 121.69, 119.58, 77.92, 73.98, 72.92, 51.86, 47.90, 38.23, 34.04. MS (ESI−): m/z = 437.04 [M-H]−. HPLC (method-A): tR = 15.573, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-N-phenethyl-4-(N-phenylsulfamoyl)-benzamide (30): synthesis Scheme 2; step b: 4-(N-phenylsulfamoyl)-benzoic acid (8; 150 mg, 0.54 mmol), EDC*HCl (124 mg, 0.65 mmol), HOBT (73 mg, 0.54 mmol), benzyl(2-phenylethyl)amine (171 mg, 0.81 mmol); product: white solid; yield: 0.22 g ≡ 84%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ 10.33 (s, 1H), 7.77–7.68 (m, 2H), 7.48–6.98 (m, 16H), 6.80 (d, J = 6.9 Hz, 1H), 4.76, 4.24 (s, 2H), 3.54, 3.19 (t, J = 7.1 Hz, 2H), 2.86, 2.68 (t, J = 7.1 Hz, 2H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ 169.50, 169.31, 140.65, 140.46, 139.94, 139.67, 138.95, 138.05, 137.46, 136.66, 129.18, 128.72, 128.68, 128.63, 128.45, 127.69, 127.54, 127.25, 127.16, 126.98, 126.81, 126.40, 126.34, 124.40, 120.49, 120.41, 52.01, 49.79, 46.70, 46.08, 33.65, 32.71. MS (ESI+): m/z = 471.09 [M + H]+. HRMS: m/z calcd for C28H27N2O3S1 [M + H]+ = 471.17369. Found [M + H]+ = 471.17302. HPLC (method-A): tR = 16.167, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-4-(N-phenylsulfamoyl)-N-(prop-2-yn-1-yl)-benzamide (31): synthesis Scheme 3; step d: 4-(N-phenylsulfamoyl)-benzoic acid (8; 250 mg, 0.90 mmol), EDC*HCl (207 mg, 1.08 mmol), HOBT (122 mg, 0.90 mmol), N-benzylprop-2-yn-1-amine (41, 196 mg, 1.35 mmol); product: white solid; yield: 0.33 g ≡ 91%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ 10.37 (s, 1H), 7.83–7.76 (m, 2H), 7.64–7.55 (m, 2H), 7.44–6.98 (m, 10H), 4.71, 4.44 (s, 2H), 4.19, 3.85 (s, 2H), 3.28 (s, 1H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ 169.18, 140.60, 140.36, 139.50, 137.40, 136.48, 135.86, 129.23, 128.65, 127.78, 127.45, 127.02, 124.42, 120.46, 78.78, 78.67, 76.04, 74.97, 51.59, 47.79, 38.66, 34.03. MS (ESI+): m/z = 405.02 [M + H]+. HRMS: m/z calcd for C23H21N2O3S1 [M + H]+ = 405.12674. Found [M + H]+ = 405.12639. HPLC (method-A): tR = 14.926, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-N-(but-3-yn-1-yl)-4-(phenylsulfamoyl)-benzamide (32): synthesis Scheme 3; step d: 4-(N-phenylsulfamoyl)-benzoic acid (8; 130 mg, 0.47 mmol), EDC*HCl (108 mg, 0.56 mmol), HOBT (63 mg, 0.47 mmol), benzyl(but-3-yn-1-yl)amine (42, 112 mg, 0.70 mmol); product: white solid; yield: 0.15 g ≡ 74%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ 10.33 (s, 1H), 7.83–7.72 (m, 2H), 7.65–7.49 (m, 2H), 7.39–7.17 (m, 6H), 7.16–6.96 (m, 4H), 4.71, 4.41 (s, 2H), 3.46, 3.16 (t, J = 6.7 Hz, 2H), 2.88 (d, J = 14.2 Hz, 1H), 2.36 (s, 1H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ 169.69, 169.53, 140.37, 140.01, 139.94, 137.39, 137.17, 136.54, 129.21, 128.73, 128.60, 127.60, 127.26, 126.98, 126.94, 124.50, 124.38, 120.64, 120.44, 81.64, 81.00, 73.47, 72.61, 52.08, 46.38, 43.25, 17.40, 16.41. MS (ESI+): m/z = 419.08 [M + H]+. HRMS: m/z calcd for C24H23N2O3S1 [M + H]+ = 419.14239. Found [M + H]+ = 419.14198. HPLC (method-A): tR = 14.967, purity ≥95% (UV: 254/280 nM).
Scheme 3; step e: general procedure for CuAAc reaction (compounds 33–40).
Alkyne (1 equiv) and azide (1 equiv) were dissolved in 6 mL of H2O/t-BuOH (1:1). Copper (II) sulfate pentahydrate (0.01 equiv, 0.1 M in H2O) and sodium ascorbate (0.1 equiv, 0.1 M in H2O) were added to this solution, and the reaction was stirred at room temperature until all starting material was consumed (identified per TLC, about 24–72 h). The reaction was quenched with saturated NH4Cl solution, and the product was extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated under a reduced pressure. The residue was further purified by column chromatography using n-hexane/ethyl acetate as an eluent.
Synthesis of N-benzyl-N-[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]-4-(phenylsulfamoyl)benzamide (33): synthesis Scheme 3; step e: N-benzyl-4-(phenylsulfamoyl)-N-(prop-2-yn-1-yl)benzamide (31; 50 mg, 0.12 mmol), (azidomethyl)benzene (247 μL, 0.5 M in DCM, 0.12 mmol), copper (II) sulfate pentahydrate (12.36 μL, 0.1 M in H2O, 1.24 μmol), sodium ascorbate (124 μL, 0.1 M in H2O, 12.4 μmol); product: white solid; yield: 0.06 g ≡ 97%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ 10.35, 10.32 (s, 1H), 8.12, 8.02 (s, 1H), 7.82–7.67 (m, 3H), 7.53 (d, J = 7.4 Hz, 1H), 7.40–7.18 (m, 11H), 7.11–6.97 (m, 4H), 5.58, 5.55 (s, 2H), 4.58 (s, 2H), 4.36, 4.29 (s, 2H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ 169.42, 142.37, 140.13, 137.38, 136.82, 129.20, 128.76, 128.68, 128.53, 128.11, 127.87, 127.76, 127.65, 127.30, 126.93, 124.40, 123.98, 120.47, 52.80, 51.79, 47.13, 43.47. MS (ESI+): m/z = 538.09 [M + H]+. HRMS: m/z calcd for C30H28N5O3S1 [M + H]+ = 538.19074. Found [M + H]+ = 538.19018. HPLC (method-A): tR = 15.004, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-N-[2-(1-benzyl-1H-1,2,3-triazol-4-yl)ethyl]-4-(phenylsulfamoyl)benzamide (34): synthesis Scheme 3; step e: N-benzyl-N-(but-3-yn-1-yl)-4-(phenylsulfamoyl)benzamide (32; 50 mg, 0.12 mmol), (azidomethyl)benzene (287 μL, 0.5 M in DCM, 0.14 mmol), copper (II) sulfate pentahydrate (23.89 μL, 0.1 M in H2O, 2.38 μmol), sodium ascorbate (239 μL, 0.1 M in H2O, 23.89 μmol); product: white solid; yield: 0.06 g ≡ 83%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ 10.33, 10.30 (s, 1H), 8.03–7.63 (m, 3H), 7.47–7.15 (m, 13H), 7.11–6.97 (m, 4H), 5.55, 5.51 (s, 2H), 4.71, 4.22 (s, 2H), 3.58, 3.29 (t, J = 6.9 Hz, 2H), 2.94, 2.79 (t, J = 7.0 Hz, 2H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ 169.48, 144.23, 143.31, 140.58, 140.42, 139.87, 139.77, 137.33, 136.59, 136.28, 136.08, 129.18, 128.75, 128.69, 128.61, 128.15, 128.02, 127.97, 127.79, 127.55, 127.21, 127.13, 127.03, 126.86, 124.48, 124.38, 122.74, 122.65, 120.74, 120.38, 52.70, 51.88, 47.70, 46.55, 44.11, 23.80, 22.83. MS (ESI+): m/z = 552.11 [M + H]+. HRMS: m/z calcd for C31H30N5O3S1 [M + H]+ = 552.20639. Found [M + H]+ = 552.20668. HPLC (method-A): tR = 14.860, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-N-((1-(2-chlorobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-4-(N-phenyl-sulfamoyl)benzamide (35): synthesis Scheme 3; step e: N-benzyl-N-(but-3-yn-1-yl)-4-(phenylsulfamoyl)-benzamide (31; 100 mg, 0.25 mmol), 1-(azidomethyl)-2-chlorobenzene (494 μL, 0.5 M in MTBE, 0.25 mmol), copper (II) sulfate pentahydrate (0.6 mg, 3.0 μmol), sodium ascorbate (9.8 mg, 49 μmol); product: white solid; yield: 0.09 g ≡ 41%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ 10.34 (s, 1H), 8.09, 7.99 (s, 1H), 7.85–7.68 (m, 3H), 7.53 (d, J = 7.8 Hz, 2H), 7.45–7.24 (m, 6H), 7.20 (t, J = 7.8 Hz, 2H), 7.17–6.98 (m, 5H), 5.69, 5.66 (s, 2H), 4.62, 4.59 (s, 2H), 4.37, 4.30 (s, 2H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ 169.48, 142.88, 142.28, 140.12, 137.40, 136.82, 133.20, 132.50, 130.25, 130.20, 129.62, 129.19, 128.68, 128.53, 127.74, 127.66, 127.29, 126.94, 124.38, 120.47, 51.81, 50.61, 47.15, 43.44. MS (ESI+): m/z = 572.20 [M + H]+. HRMS: m/z calcd for C30H27ClN5O3S1 [M + H]+ = 572.15176. Found [M + H]+ = 572.15251. HPLC (method-A): tR = 15.411, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-4-(N-phenylsulfamoyl)-N-((1-(pyridin-4-ylmethyl)-1H-1,2,3-triazol-4-yl)methyl)benzamide (36): synthesis Scheme 3; step e: N-benzyl-N-(but-3-yn-1-yl)-4-(phenylsulfamoyl)-benzamide (31; 100 mg, 0.25 mmol), 4-(azidomethyl)pyridine (494 μL, 0.5 M in MTBE, 0.25 mmol), copper (II) sulfate pentahydrate (0.6 mg, 3.0 μmol), sodium ascorbate (9.8 mg, 49 μmol); product: white solid; yield: 0.10 g ≡ 74%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ 10.34 (s, 1H), 8.56 (s, 2H), 8.19, 8.09 (s, 1H), 7.85–7.69 (m, 3H), 7.54 (d, J = 7.4 Hz, 1H), 7.39–7.24 (m, 4H), 7.22–7.17 (m, 3H), 7.13–6.97 (m, 5H), 5.67, 5.64 (s, 2H), 4.63, 4.62 (s, 2H), 4.38, 4.33 (s, 2H). 13C-NMR (126 MHz, (CD3)2SO) δ 169.43, 150.03, 144.79, 142.54, 140.12, 137.41, 136.84, 129.19, 128.67, 128.52, 127.74, 127.61, 127.32, 126.95, 124.56, 124.37, 122.14, 120.46, 51.49, 47.21, 43.54. MS (ESI+): m/z = 539.20 [M + H]+. HRMS: m/z calcd for C29H27N6O3S1 [M + H]+ = 539.18599. Found [M + H]+ = 539.18606. HPLC (method-A): tR = 11.654, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-N-((1-(3,4-dichlorobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-4-(N-phenylsulfamoyl)benzamide (37): synthesis Scheme 3; step e: N-benzyl-N-(but-3-yn-1-yl)-4-(phenylsulfamoyl)-benzamide (31; 100 mg, 0.25 mmol), 4-(azidomethyl)-1,2-dichlorobenzene (494 μL, 0.5 M in MTBE, 0.25 mmol), copper (II) sulfate pentahydrate (0.6 mg, 3.0 μmol), sodium ascorbate (9.8 mg, 49 μmol); product: white solid; yield: 0.09 g ≡ 63%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ 10.35 (s, 1H), 8.17, 8.07 (s, 1H), 7.84–7.68 (m, 3H), 7.65–7.59 (m, 2H), 7.53 (d, J = 7.7 Hz, 1H), 7.35–7.17 (m, 7H), 7.11–6.97 (m, 4H), 5.60, 5.58 (s, 2H), 4.60, 4.59 (s, 2H), 4.36, 4.30 (s, 2H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ 169.45, 143.17, 142.53, 140.33, 140.06, 137.57, 137.06, 136.89, 136.81, 136.21, 131.28, 131.01, 130.91, 130.09, 129.17, 128.65, 128.49, 128.37, 128.30, 127.70, 127.64, 127.51, 127.28, 127.18, 126.93, 124.27, 124.15, 124.04, 120.47, 51.84, 51.42, 47.14, 43.49. MS (ESI+): m/z = 606.25 [M + H]+. HRMS: m/z calcd for C30H26Cl2N5O3S1 [M + H]+ = 606.11279. Found [M + H]+ = 606.11353. HPLC (method-A): tR = 16.068, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-N-((1-(imidazo[1,2-a]pyrimidin-2-ylmethyl)-1H-1,2,3-triazol-4-yl)methyl)-4-(N-phenylsulfamoyl)benzamide (38): synthesis Scheme 3; step e: N-benzyl-N-(but-3-yn-1-yl)-4-(phenylsulfamoyl)benzamide (31; 100 mg, 0.25 mmol), 2-(azidomethyl)imidazo[1,2-a]pyrimidine (494 μL, 0.5 M in MTBE, 0.25 mmol), copper (II) sulfate pentahydrate (0.6 mg, 3.0 μmol), sodium ascorbate (9.8 mg, 49 μmol); product: white solid; yield: 0.08 g ≡ 53%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ 10.32 (s, 1H), 8.97 (dd, J = 6.8, 1.9 Hz, 1H), 8.55 (dd, J = 4.1, 2.0 Hz, 1H), 8.14, 8.04 (s, 1H), 7.88–7.72 (m, 4H), 7.53 (d, J = 7.8 Hz, 1H), 7.33–7.18 (m, 6H), 7.12–6.96 (m, 5H), 5.74, 5.72 (s, 2H), 4.60, 4.58 (s, 2H), 4.36, 4.29 (s, 2H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ 169.44, 150.88, 147.65, 144.46, 142.78, 142.14, 141.77, 141.67, 140.23, 140.11, 137.45, 136.80, 135.47, 129.18, 128.69, 128.52, 127.70, 127.30, 127.16, 126.93, 124.35, 124.23, 120.47, 109.91, 109.01, 51.68, 47.72, 46.99, 43.36. MS (ESI+): m/z = 579.24 [M + H]+. HRMS: m/z calcd for C30H27N8O3S1 [M + H]+ = 579.19213. Found [M + H]+ = 579.19159. HPLC (method-A): tR = 12.533, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-N-((1-((2-methylthiazol-4-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)-4-(N-phenylsulfamoyl)benzamide (39): synthesis Scheme 3; step e: N-benzyl-N-(but-3-yn-1-yl)-4-(phenylsulfamoyl)benzamide (31; 100 mg, 0.25 mmol), 4-(azidomethyl)-2-methylthiazole (494 μL, 0.5 M in MTBE, 0.25 mmol), copper (II) sulfate pentahydrate (0.6 mg, 3.0 μmol), sodium ascorbate (9.8 mg, 49 μmol); product: white solid; yield: 0.10 g ≡ 71%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ 10.34 (s, 1H), 8.07, 7.97 (s, 1H), 7.84–7.68 (m, 3H), 7.57–7.16 (m, 8H), 7.13–6.98 (m, 4H), 5.61, 5.59 (s, 2H), 4.58 (s, 2H), 4.36, 4.28 (s, 2H), 2.61 (s, 3H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ 169.43, 166.47, 149.60, 149.53, 142.19, 140.20, 140.14, 137.41, 136.82, 136.23, 129.21, 128.70, 128.54, 127.73, 127.67, 127.54, 127.30, 127.23, 126.94, 124.40, 124.12, 123.93, 120.47, 118.01, 117.85, 51.74, 48.92, 47.03, 43.36, 18.73. MS (ESI+): m/z = 559.21 [M + H]+. HRMS: m/z calcd for C28H27N6O3S2 [M + H]+ = 559.15806. Found [M + H]+ = 559.15812. HPLC (method-A): tR = 14.084, purity ≥95% (UV: 254/280 nM).
Synthesis of N-benzyl-N-((1-((2-oxooxazolidin-5-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)-4-(N-phenylsulfamoyl)benzamide (40): synthesis Scheme 3; step e: N-benzyl-N-(but-3-yn-1-yl)-4-(phenylsulfamoyl)benzamide (31; 100 mg, 0.25 mmol), 5-(azidomethyl)oxazolidin-2-one (494 μL, 0.5 M in MTBE, 0.25 mmol), copper (II) sulfate pentahydrate (0.6 mg, 3.0 μmol), sodium ascorbate (9.8 mg, 49 μmol); product: white solid; yield: 0.06 g ≡ 44%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ 10.34 (s, 1H), 8.07, 7.98 (s, 1H), 7.89–7.70 (m, 3H), 7.62–7.49 (m, 2H), 7.41–7.26 (m, 4H), 7.09–7.03 (m, 2H), 7.13–6.97 (m, 4H), 4.95 (s, 1H), 4.73–4.48 (m, 4H), 4.32, 4.28 (s, 2H), 3.61, 3.27 (t, J = 9.0 Hz, 2H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ 169.43, 158.01, 142.76, 142.13, 140.23, 140.11, 137.41, 136.74, 136.15, 129.22, 128.73, 128.60, 127.79, 127.71, 127.57, 127.30, 126.97, 124.77, 124.61, 124.40, 120.46, 73.35, 73.25, 52.12, 51.54, 48.61, 46.74, 43.12, 42.12. MS (ESI+): m/z = 569.20 [M + Na]+. HRMS: m/z calcd for C27H27N6O5S2 [M + H]+ = 547.17582. Found [M + H]+ = 547.17560. HPLC (method-A): tR = 12.640, purity ≥95% (UV: 254/280 nM).
Synthesis of benzyl(prop-2-yn-1-yl)amine (41): synthesis Scheme 3; step b: 3-bromoprop-1-yne (3.0 mL, 26.9 mmol) was added slowly to benzylamine (17.65 mL, 162 mmol) over 30 minutes via addition funnel. The reaction was stirred at room temperature for 72 h. The reaction was diluted in Et2O and extracted with saturated aq. NaHCO3. The organic layer was dried over MgSO4 and concentrated under a reduced pressure. The residue was further purified by column chromatography using n-hexane/EtOAc 9:1 as an eluent. Yield: 2.35 g ≡ 60%. 1H-NMR (250 MHz, (CD3)2SO) δ 7.43–7.07 (m, 5H), 3.74 (s, 2H), 3.27 (d, J = 2.4 Hz, 2H), 3.07 (t, J = 2.4 Hz, 1H), 2.44 (s, 1H). MS (ESI+): m/z = 146.12 [M + H]+.
Synthesis of benzyl(but-3-yn-1-yl)amine (42): synthesis Scheme 3; step c: potassium carbonate (1.29 g; 9.33 mmol) and benzylamine (0.50 g; 4.66 mmol) were suspended in acetonitrile (10 mL). But-3-yn-1-yl 4-methylbenzene-1-sulfonate (1.05 g, 4.66 mmol) was added, and the reaction was refluxed overnight. The resulting solution was quenched with H2O (50 mL), and the product was extracted using EtOAc (3 × 50 mL). The organic layer was washed with brine and dried over MgSO4. The organic layer was concentrated under a reduced pressure. The residue was further purified by column chromatography as an eluent. Yield: 0.40 g ≡ 54%. 1H-NMR (500 MHz, (CD3)2SO) δ 7.34–7.27 (m, 4H), 7.22 (dt, J = 8.6, 2.3 Hz, 1H), 3.69 (s, 2H), 2.77 (t, J = 2.7 Hz, 1H), 2.62 (t, J = 7.2 Hz, 2H), 2.29 (td, J = 7.2, 2.6 Hz, 2H). 13C-NMR (126 MHz, (CD3)2SO) δ 140.76, 128.12, 127.88, 126.55, 83.18, 71.75, 52.40, 47.36, 19.03.
Synthesis of ethyl 2-(2-methylpropanamido)-1,3-thiazole-5-carboxylate (44): synthesis Scheme 4; step a: ethyl 2-amino-1,3-thiazole-5-carboxylate (43; 500 mg, 2.90 mmol) and pyridine (0.35 mL, 4.36 mmol) were dissolved in 10 mL of CH2Cl2. 2-Methylpropanoyl chloride (371 mg, 3.50 mmol) was added slowly, and the mixture was stirred at room temperature for 20 h. The reaction was washed with 1 N HCl and brine and dried over MgSO4. The organic layer was concentrated under reduced pressure. The residue was recrystallized with n-hexane/methylene chloride yielding ethyl 2-(2-methylpropanamido)-1,3-thiazole-5-carboxylate (31) as a brownish solid. Yield: 0.45 g ≡ 63%. 1H-NMR (250 MHz, DMSO-d6): δ 12.54 (s, 1H), 8.13 (s, 1H), 4.27 (q, J = 7.1 Hz, 2H), 2.77 (hept, J = 6.9 Hz, 1H), 1.28 (t, J = 7.1 Hz, 3H), 1.13 (d, J = 6.9 Hz, 6H). MS (ESI+): m/z = 243.14 [M + H]+.
Synthesis of 2-(2-methylpropanamido)-1,3-thiazole-5-carboxylic acid (45): synthesis Scheme 4; step b: to a solution of ethyl 2-(2-methylpropanamido)-1,3-thiazole-5-carboxylate (44; 300 mg, 1.24 mmol) in THF (10 ml) was added LiOH*H2O (260 mg, 6.19 mmol) dissolved in water (2 ml). The reaction was stirred for 72 h at 30 °C. The solvent was evaporated under a reduced pressure, and the residue was solved in water. The water layer was acidified by 2 N HCl, and the resulting precipitate was extracted with ethyl acetate. The organic layer was dried over anhydrous MgSO4, and the solvent was evaporated under a reduced pressure. The crude product was finally recrystallized with n-hexane/ethyl acetate to obtain 2-(2-methylpropanamido)-1,3-thiazole-5-carboxylic acid (32) as a brownish yellow solid. Yield: 0.08 g ≡ 35%. 1H-NMR (250 MHz, DMSO-d6) δ 13.06 (s, 1H), 12.45 (s, 1H), 8.04 (s, 1H), 2.76 (hept, J = 6.8 Hz, 1H), 1.12 (d, J = 6.9 Hz, 6H). MS (ESI+): m/z = 215.00 [M + H]+.
Synthesis of tert-butyl N-(2-{N-benzyl-1-[4-(phenylsulfamoyl)-phenyl]formamido}ethyl) carbamate (46): synthesis Scheme 4; step c: 4-(phenylsulfamoyl)benzoic acid (8; 370 mg, 1.33 mmol), EDC*HCl (306 mg, 1.60 mmol), and HOBT (180 mg, 1.33 mmol) were dissolved in 10 mL of CH2Cl2 and stirred at room temperature. After 1 h, tert-butyl N-[2-(benzylamino)ethyl]carbamate (500 mg, 2.00 mmol) was added and the mixture was stirred at room temperature for 20 h. The reaction was quenched with 40 mL of H2O. The product was extracted with 50 mL of EtOAc. The organic layer was washed with 1 N HCl and brine and dried over MgSO4. The organic layer was concentrated under a reduced pressure. The residue was further purified by column chromatography using n-hexane/ethyl acetate as an eluent to obtain tert-butyl N-(2-{N-benzyl-1-[4-(phenylsulfamoyl)phenyl]formamido}ethyl) carbamate (46) as a white solid. Yield: 0.57 g ≡ 83%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ: 10.33 (s, 1H), 7.79–7.72 (m, 2H), 7.54 (d, J = 8.3 Hz, 2H), 7.38–7.18 (m, 6H), 7.09–6.89 (m, 5H), 4.68, 4.35 (s, 2H), 3.37–3.35 (m, 1H), 3.19 (d, J = 5.6 Hz, 1H), 3.08–2.98 (m, 2H), 1.38, 1.34 (s, 9H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ: 169.64, 169.57, 155.85, 155.44, 140.67, 140.56, 139.89, 137.45, 137.25, 136.69, 129.21, 129.16, 128.71, 128.63, 127.52, 127.37, 127.24, 127.21, 126.93, 126.84, 124.38, 124.32, 120.51, 120.41, 77.89, 77.79, 52.64, 47.51, 46.72, 44.49, 37.69, 37.55, 28.26, 28.22. MS (ESI+): m/z = 510.15 [M + H]+. HPLC (method-A): tR = 15.265, purity ≥95% (UV: 254/280 nM).
Synthesis of N-(2-aminoethyl)-N-benzyl-4-(phenylsulfamoyl)-benzamide (47): synthesis Scheme 4; step d: tert-butyl N-(2-{N-benzyl-1-[4-(phenylsulfamoyl)phenyl]formamido}ethyl) carbamate (46; 472 mg, 0.93 mmol) was dissolved in 15 mL of methylene chloride. Trifluoroacetic acid (3 mL; 39.18 mmol) was added slowly under stirring and cooling with ice. The reaction was stirred at room temperature for 2 h. The resulting mixture was diluted with ethyl acetate (50 mL) and neutralized with a saturated NaHCO3 solution. The organic layer was washed with brine, dried over MgSO4, and concentrated under a reduced pressure. The residue was taken as such without further purification for the next step.
Synthesis of N-benzyl-N-(2-{[2-(2-methylpropanamido)-1,3-thiazol-5-yl]formamido}ethyl)-4-(phenylsulfamoyl)benzamide (48): synthesis Scheme 4; step e: 2-(2-methylpropanamido)-1,3-thiazole-5-carboxylic acid (45; 75 mg, 0.35 mmol), EDC*HCl (81 mg, 0.42 mmol), and HOBT (47 mg, 0.35 mmol) were dissolved in 10 mL of CH2Cl2 and stirred at room temperature. After 1 h, N-(2-aminoethyl)-N-benzyl-4-(phenylsulfamoyl)benzamide (47; 215 mg, 0.53 mmol) was added and the mixture was stirred at room temperature for 20 h. The reaction was quenched with 40 mL of H2O. The product was extracted with 50 mL of EtOAc. The organic layer was washed with 1 N HCl and brine and dried over MgSO4. The organic layer was concentrated under a reduced pressure. The residue was further purified by column chromatography using n-hexane/ethyl acetate as an eluent to obtain N-benzyl-N-(2-{[2-(2-methylpropanamido)-1,3-thiazol-5-yl]formamido}ethyl)-4-(phenylsulfamoyl)-benzamide (48) as a white solid. Yield: 0.14 g ≡ 66%. 1H-NMR (500 MHz, (CD3)2SO, rotamers observed) δ: 1.12 (d, 6H, J = 6.88 Hz), 2.76 (m, 1H), 3.21–3.31 (m, 2H), 3.50 (s, 2H), 4.37, 4.74 (s, 2H), 6.99–7.08 (m, 4H), 7.18–7.30 (m, 4H), 7.34–7.39 (m, 2H), 7.51 (d, 2H, J = 8.15 Hz), 7.73 (d, 2H, J = 7.59 Hz), 7.93, 8.01 (s, 1H), 8.46, 8.63 (s, 1H), 10.30, 10.33 (s, 1H), 12.29 (s, 1H). 13C-NMR (126 MHz, (CD3)2SO, rotamers observed) δ: 175.62, 169.71, 169.64, 161.17, 160.86, 160.69, 160.60, 140.62, 140.39, 139.94, 139.88, 139.62, 139.41, 137.37, 137.22, 136.65, 129.22, 129.17, 128.70, 128.66, 127.55, 127.32, 127.19, 126.89, 124.40, 120.55, 120.49, 52.33, 47.19, 46.92, 44.16, 36.95, 36.43, 33.82, 19.05. MS (ESI−): m/z = 604.27 [M-H]−. HRMS: m/z calcd for C30H32N5O5S2 [M + H]+ = 606.18394. Found [M + H]+ = 606.18367. HPLC (method-A): tR = 13.925, purity ≥95% (UV: 254/280 nM).
KINOBEAD PULL DOWNS
Hela cells were grown in IBM medium (Biochrom Ltd.) supplemented with 10% FBS (Biochrom Ltd.). For lysis, cells were washed twice with PBS and then lysed by scraping in the presence of lysis buffer (0.8% NP40, 50 mM tris–HCl pH 7.5, 5% glycerol, 1.5 mM MgCl2, 150 mM NaCl, 1 mM Na3VO4, 25 mM NaF, 1 mM DTT, SigmaFast protease inhibitors). The lysate was cleared with centrifugation for 1 h at 52,000g. Protein concentration was determined with a Bradford Assay and adjusted to 5 mg/ml. Kinobead pulldown assays were performed as previously described.27 Inhibitors of interest were spiked into 1 mL of lysate at increasing concentrations (DMSO, 1, 3, 10, 30, 100, 300 nM, 1, 3, and 30 mM) and incubated for 45 min at 4 °C. This was followed by incubation with kinobeads (35 μL settled beads) for 30 min at 4 °C. To assess the degree of protein depletion from the lysates by the kinobeads, a second kinobead pulldown (with fresh beads) was performed on the unbound fraction of the vehicle control (so-called pulldown of pulldown). This enabled the determination of a correction factor for each protein that was used to calculate apparent dissociation constants for a drug–target complex. Proteins bound to kinobeads were eluted with LDS sample buffer (NuPAGE, Invitrogen) containing 50 mM DTT. Kinobead eluates were alkylated with 55 mM CAA and run into a 4–12% NuPAGE gel (Invitrogen; approximately 1 cm). In-gel digestion was performed according to standard procedures.42
Generated peptides were analyzed by LC-MS/MS on a Dionex Ultimate 3000 UHPLC system coupled to a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific). After reconstitution in 0.1% formic acid (FA), an amount corresponding to 1 μg peptides was injected. Peptides were delivered to a trap column (75 μm × 2 cm, packed in-house with 5 μm C18 resin; Reprosil PUR AQ, Dr. Maisch, Ammerbruch-Entringen, Germany) and washed using 0.1% FA at a flow rate of 5 μL/min for 10 min Subsequently, peptides were transferred to an analytical column (75 μm × 45 cm, packed in-house with 3 μm C18 resin; Reprosil Gold, Dr. Maisch) applying a flow rate of 300 nL/min and separated using a 60 min linear gradient from 2 to 32% LC solvent B (0.1% FA, 5% DMSO in ACN) in LC solvent A (0.1% FA in 5% DMSO). The HF-X was operated in data-dependent acquisition (DDA) and positive ionization mode. Full scan MS1 spectra were recorded in the orbitrap from 360 to 1300 m/z at a resolution of 60 K (automatic gain control (AGC) target value of 3e6 charges, maximum injection time (maxIT) of 45 ms). MS2 spectra for peptide identification were recorded in the orbitrap at 30 K resolution after sequential isolation of up to 12 precursors (isolation window 1.7 m/z, AGC target value of 2e5, maxIT of 75 ms, dynamic exclusion of 30 s) and fragmentation via HCD (NCE of 26%).
Peptide/protein identification and quantification were performed with MaxQuant (v.1.5.7.4) by searching against the Swissprot database (human, 42,145 entries, including splice variants, downloaded on 01.02.2016) using the embedded search engine Andromeda.43 Trypsin/P was specified as the proteolytic enzyme, with up to two missed cleavage sites allowed, and quantification was done with the LFQ option. Results were adjusted to 1% peptide spectrum match (PSM) and 1% protein false discovery rate (FDR) employing a target–decoy approach using reversed protein sequences. IC50 and EC50 values were deduced by a four-parameter log-logistic regression using an internal pipeline that utilizes the “drc” package in R. A Kd,app was calculated by multiplying the estimated EC50 with a protein-dependent correction factor (depletion factor) that was limited to a maximum value of 1. The correction factor (cf) for a protein is defined as the ratio of the amount of protein captured from the consecutive pulldown of the same DMSO control lysate. Each compound was processed separately.
PHOSPHOPROTEOMIC ANALYSIS
Hela cells were grown in IBM medium (Biochrom) supplemented with 10% FBS (Biochrom). For phosphoproteomic analysis, cells were treated for 30 min with 1 mM (TH263, TH257) or 100 μM (LIMKi3, TH470) of each compound in triplicate. After treatment, cells were washed twice with PBS and then lysed by scraping in the presence of lysis buffer (40 mM tris–HCl pH 7.8, 8 M urea, EDTA-free protease inhibitor complete mini (Roche) and phosphatase inhibitor cocktail at a 1× final concentration). The lysate was cleared with centrifugation for 1 h at 21,000g, and the protein concentration was determined with a Bradford assay. Two hundred micrograms of protein per sample were further processed for the phosphoproteomic workflow. After reduction (10 mM DTT, 30 °C, 30 min) and alkylation (50 mM chloroacetamide, room temperature, 30 min), lysates were diluted to 1.6 M urea using 40 mM tris–HCl (pH 7.6). Digestion was performed by adding trypsin (Promega, 1:50 enzyme-to-substrate ratio) and incubating overnight at 37 °C at 700 rpm. Digests were acidified to 1% FA and desalted using 50 mg C18, reversed-phase (RP) solid-phase extraction cartridges (Waters Corp., Eschborn, Germany; wash solvent: 0.1% FA; elution solvent: 0.1% FA in 50% ACN). The cleaned peptides were frozen at −80 °C and dried in a SpeedVac. Digests were reconstituted in 20 μL of 50 mM HEPES (pH 8.5), and 5 μL of an 11.6 mM TMT stock (Thermo Fisher) in 100% anhydrous ACN was added to each sample. After incubation for 1 h at 25 °C and 500 rpm, the labeling reaction was stopped by adding 2 μL of 5% hydroxylamine. Peptide solutions were pooled and acidified using 20 μL of 10% FA. Reaction vessels in which the labeling took place were rinsed with 20 μL of 10% FA in 10% ACN, and the solvent was added to the pooled sample. In total, two TMT10plex sets were used. Triplicate of two drugs was combined with triplicate of the DMSO control to one TMT set, leaving one channel empty. DMSO triplicate in each TMT set allowed comparison across TMT batches. The pools were frozen at −80 °C and dried down in a SpeedVac. Subsequently, pooled samples were desalted using 50 mg C18, RP solid-phase extraction cartridges (Waters Corp.; wash solvent: 0.07% TFA; elution solvent: 0.07% TFA in 60% ACN). The eluted peptides were diluted to a volume of 500 μL with IMAC loading buffer (0.07% TFA, 30% ACN) and loaded onto an analytical Fe-IMAC column (950 mm ProPac IMAC-10, Thermo Fisher Scientific) connected to an HPLC system (ÄKTA Explorer FPLC system, Amersham Biosciences Pharmacia) with a 1 mL sample loop. Phosphopeptide enrichment was performed as described previously.44 Flow-through and phosphopeptide fractions were collected according to the UV signal (280 nm). Peptide solutions were frozen at −80 °C and dried in a SpeedVac. For desalting and high pH RP tip fractionation, self-packed StageTips (five disks, Ø 1.5 mm, C18 material, 3 M EmporeTM via Sigma-Aldrich) were activated and equilibrated using 250 μL of 100% ACN, followed by 250 μL of 0.1% FA in 50% ACN and twice 250 μL of 0.1% FA. The dried phosphopeptides were reconstituted in 250 μL of 0.1% FA and loaded onto the C18 material by gentle centrifugation. Thereafter, peptides are washed with 250 μL of 0.1% FA and rebuffered to pH 10 with 50 μL of 25 mM ammonium formate. Flow-through was collected and reapplied. Peptides were sequentially eluted using 40 μL of 25 mM ammonium formate containing increasing concentrations of ACN (5, 7.5, 10, 12.5, 15, 17.5, and 50% ACN). The sample flow-through was combined with the 17.5% ACN eluate and the 5% ACN fraction with the 50% ACN fraction, resulting in a total of six fractions, which were dried down in a SpeedVac and stored at −20 °C until LC-MS measurement.
The generated peptides were analyzed by LC-MS/MS on a Dionex Ultimate 3000 UHPLC system coupled to a Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific). After reconstitution in 0.1% FA, an amount corresponding to 1 μg peptides was twice injected. Peptides were delivered to a trap column (75 μm × 2 cm, packed in-house with 5 μm C18 resin; Reprosil PUR AQ, Dr. Maisch, Ammerbruch-Entringen, Germany) and washed using 0.1% FA at a flow rate of 5 μL/min for 10 min. Subsequently, peptides were transferred to an analytical column (75 μm × 45 cm, packed in-house with 3 μm C18 resin; Reprosil Gold, Dr. Maisch) applying a flow rate of 300 nL/min and separated using a 100 min linear gradient from 4 to 32% LC solvent B (0.1% FA, 5% DMSO in ACN) in LC solvent A (0.1% FA in 5% DMSO). The Fusion Lumos was operated in data-dependent acquisition (DDA) and positive ionization mode. Full scan MS1 spectra were recorded in the orbitrap from 360 to 1300 m/z at a resolution of 60 K, an automatic gain control (AGC) target value of 4e5 charges, and a maximum injection time (maxIT) of 50 ms. MS2 spectra for peptide identification were recorded in the orbitrap at a 15 K resolution via sequential isolation of up to 10 precursors (isolation window 0.7 m/z, AGC target value of 5e4, maxIT of 22 ms, dynamic exclusion of 90 s) and fragmentation via CID (NCE of 35%, activation Q of 0.25). For each peptide precursor, an additional MS3 spectrum for TMT quantification was obtained in the orbitrap at a 50 K resolution (scan range 100–1000 m/z, charge-dependent isolation window from 1.3 (2+) to 0.7 (5–6+) m/z, AGC of 1.2e5 charges, maxIT of 120 ms). For this, the precursor was again fragmented for MS2 analysis, followed by the synchronous selection of the 10 most intense peptide fragments in the ion trap and further fragmentation via HCD using an NCE of 55%.
Peptide/protein identification and MS3-based TMT quantification were performed using MaxQuant (v.1.5.7.4) with its built-in search engine Andromeda.43 Tandem mass spectra were searched against the Swissprot database (human, 42,145 entries, including splice variants, downloaded on 01.02.2016) supplemented with common contaminants. Carbamidomethylated cysteine was set as fixed modification and oxidation of methionine, N-terminal protein acetylation, and phosphorylation on S, T, and Y as variable modifications. Isotope impurities of the TMT batch were specified in the configuration of TMT modifications to allow MaxQuant the automated correction of TMT intensities. Trypsin/P was specified as the proteolytic enzyme, with up to two missed cleavage sites allowed. The precursor tolerance was set to 5 ppm, and the fragment ion tolerance was set to 20 ppm. Results were adjusted to 1% peptide spectrum match (PSM) and 1% protein false discovery rate (FDR), employing a target–decoy approach using reversed protein sequences. Fold changes for each inhibitor against the vehicle control were calculated per peptide and tested for significance using a t-test with the Benjamini–Hochberg adjustment. Only phosphorylation sites with a p-value of <0.01 and fold change > |1| in all replicates were considered as significantly regulated.
NEURITE OUTGROWTH
Cell Culture and siRNA Transfection.
HEK293 and N1E-115 cells were purchased from American Type Culture Correction and maintained in Dulbecco’s modified Eagle’s media (DMEM) containing 10% fetal bovine serum (FBS). Cells were cultured at 37 °C in the presence of 5% CO2. Transfection of siRNA was performed using Lipofectamine RNAiMAX (Life Technologies).
Chemicals and siRNAs for Cell Culture.
LIMKi3 was purchased from TOCRIS Bioscience. LIMK inhibitors were dissolved in DMSO at a final concentration of 10 mM, followed by dilution to 0.05–10 μM in cell culture medium. siRNAs targeting both human and mouse FMR1 (5′-GGAUGAUAAAGGGUGAGUUdTdT-3′)7 were purchased from Dharmacon. Control siRNA was purchased from Qiagen (#1027281).
Quantitation of Neurite Outgrowth.
Twenty-five thousand N1E-115 cells were seeded on a 3.5 cm plate and transfected with siRNA for FMR1 (siFMR1) or nonspecific control (siCon).7 Sixty hours after transfection, cell culture media were replaced with low serum media with 0.2% FBS with or without LIMK1 inhibitors, and the cells were incubated for 12 h, followed by 10 ng/mL BMP7 treatments for 24 h. Cells were observed under an optical microscope (Nikon), and the number of cells bearing neurite-like structures with lengths corresponding to more than one time cell body (neurite-positive) was counted and the fraction (%) of neurite-positive cells out of total cells was calculated.
Statistical Analysis.
Statistical analysis was performed using the Prism 7 GraphPad package. Statistical significance is denoted in the figure legend.
Supplementary Material
ACKNOWLEDGMENTS
T.H., S.M., B.-T.B., B.SK, and S.K. are grateful for the support from the Structural Genomics Consortium (SGC), a registered charity (no: 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN grant 875510], Janssen, Merck KGaA (aka EMD in Canada and USA), Pfizer, and Takeda. B.-T.B. would like to acknowledge funding by the SFB1399, and S.K. is grateful for the support of the DKTK translational cancer network and the Frankfurt Cancer Institute (FCI).
ABBREVIATIONS USED
- ACN
acetonitrile
- BMPR2
bone morphogenetic protein receptor type-2
- CDC42
cell division control protein 42 homolog
- CuAAC
copper-catalyzed azide–alkyne cycloadditions
- DCE
1,2-dichloroethane
- DCM
dichloromethane
- DFG motif
Asp–Phe–Gly motif
- DSF
differential scanning fluorimetry
- EDC
1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide
- EtOAc
ethyl acetate
- FMR1
fragile X mental retardation 1
- FMRP
fragile X mental retardation protein
- FXS
fragile X syndrome
- GO
gene ontology
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- HOBT
N-hydroxybenzotriazole
- ITC
isothermal titration calorimetry
- NaAsc
sodium ascorbate
- PK domain
protein kinase domain
- Rac
Ras-related C3 botulinum toxin substrate
- RhoA
transforming protein RhoA
- TCEP
tris(2-carboxyethyl)-phosphine
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.2c01106
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01106.
Selectivity data of 1 (LIMKi3), 15 (TH257), 26 (TH263), and 48 (TH470); BRET data, as well as differential scanning calorimetry data measured on more than 100 human kinases and analytical data for compounds 15–40 and 48. Molecular formula strings are provided as a supplementary file (PDF)
SMILES codes of final compounds (CSV)
The authors declare no competing financial interest.
Contributor Information
Thomas Hanke, Institute for Pharmaceutical Chemistry, Johann Wolfgang Goethe-University, D-60438 Frankfurt am Main, Germany; Structural Genomics Consortium (SGC), Buchmann Institute for Molecular Life Sciences, Johann Wolfgang Goethe-University, D-60438 Frankfurt am Main, Germany.
Sebastian Mathea, Institute for Pharmaceutical Chemistry, Johann Wolfgang Goethe-University, D-60438 Frankfurt am Main, Germany; Structural Genomics Consortium (SGC), Buchmann Institute for Molecular Life Sciences, Johann Wolfgang Goethe-University, D-60438 Frankfurt am Main, Germany.
Julia Woortman, Chair of Proteomics and Bioanalytics, Technical University of Munich (TUM), D-85354 Freising, Germany.
Eidarus Salah, Chemistry Research Laboratory, University of Oxford, Oxford OX1 3TA, United Kingdom.
Benedict-Tilman Berger, Institute for Pharmaceutical Chemistry, Johann Wolfgang Goethe-University, D-60438 Frankfurt am Main, Germany; Structural Genomics Consortium (SGC), Buchmann Institute for Molecular Life Sciences, Johann Wolfgang Goethe-University, D-60438 Frankfurt am Main, Germany.
Anthony Tumber, Chemistry Research Laboratory, University of Oxford, Oxford OX1 3TA, United Kingdom.
Risa Kashima, Cardiovascular Research Institute, University of California, San Francisco, San Francisco, California 94143, United States.
Akiko Hata, Cardiovascular Research Institute, University of California, San Francisco, San Francisco, California 94143, United States.
Bernhard Kuster, Chair of Proteomics and Bioanalytics, Technical University of Munich (TUM), D-85354 Freising, Germany; German Translational Cancer Network (DKTK), Site Frankfurt/Mainz, German Cancer Research Center (DKFZ), D-69120 Heidelberg, Germany.
Susanne Müller, Institute for Pharmaceutical Chemistry, Johann Wolfgang Goethe-University, D-60438 Frankfurt am Main, Germany; Structural Genomics Consortium (SGC), Buchmann Institute for Molecular Life Sciences, Johann Wolfgang Goethe-University, D-60438 Frankfurt am Main, Germany.
Stefan Knapp, Institute for Pharmaceutical Chemistry, Johann Wolfgang Goethe-University, D-60438 Frankfurt am Main, Germany; Structural Genomics Consortium (SGC), Buchmann Institute for Molecular Life Sciences, Johann Wolfgang Goethe-University, D-60438 Frankfurt am Main, Germany; German Translational Cancer Network (DKTK), Site Frankfurt/Mainz, German Cancer Research Center (DKFZ), D-69120 Heidelberg, Germany.
REFERENCES
- (1).Manetti F LIM kinases are attractive targets with many macromolecular partners and only a few small molecule regulators. Med. Res. Rev 2012, 32, 968–998. [DOI] [PubMed] [Google Scholar]
- (2).Mizuno K Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013, 25, 457–469. [DOI] [PubMed] [Google Scholar]
- (3).Takemura M; Mishima T; Wang Y; Kasahara J; Fukunaga K; Ohashi K; Mizuno K Ca2+/calmodulin-dependent protein kinase IV-mediated LIM kinase activation is critical for calcium signal-induced neurite outgrowth. J. Biol. Chem 2009, 284, 28554–28562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kobayashi M; Nishita M; Mishima T; Ohashi K; Mizuno K MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO J. 2006, 25, 713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lee-Hoeflich ST; Causing CG; Podkowa M; Zhao X; Wrana JL; Attisano L Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J. 2004, 23, 4792–4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Foletta VC; Lim MA; Soosairajah J; Kelly AP; Stanley EG; Shannon M; He W; Das S; Massague J; Bernard O Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J. Cell Biol 2003, 162, 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kashima R; Roy S; Ascano M; Martinez-Cerdeno V; Ariza-Torres J; Kim S; Louie J; Lu Y; Leyton P; Bloch KD; Kornberg TB; Hagerman PJ; Hagerman R; Lagna G; Hata A Augmented noncanonical BMP type II receptor signaling mediates the synaptic abnormality of fragile X syndrome. Sci. Signal 2016, 9, ra58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bernard O Lim kinases, regulators of actin dynamics. Int. J. Biochem. Cell Biol 2007, 39, 1071–1076. [DOI] [PubMed] [Google Scholar]
- (9).Gorovoy M; Niu J; Bernard O; Profirovic J; Minshall R; Neamu R; Voyno-Yasenetskaya T LIM kinase 1 coordinates microtubule stability and actin polymerization in human endothelial cells. J. Biol. Chem 2005, 280, 26533–26542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Prunier C; Prudent R; Kapur R; Sadoul K; Lafanechére L LIM kinases: cofilin and beyond. Oncotarget. 2017, 8, 41749–41763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mardilovich K; Gabrielsen M; McGarry L; Orange C; Patel R; Shanks E; Edwards J; Olson MF Elevated LIM kinase 1 in nonmetastatic prostate cancer reflects its role in facilitating androgen receptor nuclear translocation. Mol. Cancer Ther 2015, 14, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Prudent R; Vassal-Stermann E; Nguyen CH; Pillet C; Martinez A; Prunier C; Barette C; Soleilhac E; Filhol O; Beghin A; Valdameri G; Honoré S; Aci-Séche S; Grierson D; Antonipillai J; Li R; Di Pietro A; Dumontet C; Braguer D; Florent JC; Knapp S; Bernard O; Lafanechére L Pharmacological inhibition of LIM kinase stabilizes microtubules and inhibits neoplastic growth. Cancer Res. 2012, 72, 4429–4439. [DOI] [PubMed] [Google Scholar]
- (13).Oku Y; Tareyanagi C; Takaya S; Osaka S; Ujiie H; Yoshida K; Nishiya N; Uehara Y Multimodal effects of small molecule ROCK and LIMK inhibitors on mitosis, and their implication as anti-leukemia agents. PLoS One. 2014, 9, No. e92402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bhardwaj A; Srivastava SK; Singh S; Arora S; Tyagi N; Andrews J; McClellan S; Carter JE; Singh AP CXCL12/CXCR4 signaling counteracts docetaxel-induced microtubule stabilization via p21-activated kinase 4-dependent activation of LIM domain kinase 1. Oncotarget 2014, 5, 11490–11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chatterjee D; Preuss F; Dederer V; Knapp S; Mathea S Structural Aspects of LIMK Regulation and Pharmacology. Cells 2022, 11, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Salah E; Chatterjee D; Beltrami A; Tumber A; Preuss F; Canning P; Chaikuad A; Knaus P; Knapp S; Bullock AN; Mathea S Lessons from LIMK1 enzymology and their impact on inhibitor design. Biochem. J 2019, 476, 3197–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Manetti F Recent advances in the rational design and development of LIM kinase inhibitors are not enough to enter clinical trials. Eur. J. Med. Chem 2018, 155, 445–458. [DOI] [PubMed] [Google Scholar]
- (18).Ross-Macdonald P; de Silva H; Guo Q; Xiao H; Hung CY; Penhallow B; Markwalder J; He L; Attar RM; Lin TA; Seitz S; Tilford C; Wardwell-Swanson J; Jackson D Identification of a nonkinase target mediating cytotoxicity of novel kinase inhibitors. Mol. Cancer Ther 2008, 7, 3490–3498. [DOI] [PubMed] [Google Scholar]
- (19).Ohashi K; Sampei K; Nakagawa M; Uchiumi N; Amanuma T; Aiba S; Oikawa M; Mizuno K Damnacanthal, an effective inhibitor of LIM-kinase, inhibits cell migration and invasion. Mol. Biol. Cell 2014, 25, 828–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Goodwin NC; Cianchetta G; Burgoon HA; Healy J; Mabon R; Strobel ED; Allen J; Wang S; Hamman BD; Rawlins DB Discovery of a type III inhibitor of LIM kinase 2 that binds in a DFG-out conformation. ACS Med. Chem. Lett 2015, 6, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21). https://clinicaltrials.gov/ct2/show/NCT01528111.
- (22).Yin Y; Zheng K; Eid N; Howard S; Jeong JH; Yi F; Guo J; Park CM; Bibian M; Wu W; Hernandez P; Park H; Wu Y; Luo JL; LoGrasso PV; Feng Y Bis-aryl urea derivatives as potent and selective LIM kinase (Limk) inhibitors. J. Med. Chem 2015, 58, 1846–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Boland S; Bourin A; Alen J; Geraets J; Schroeders P; Castermans K; Kindt N; Boumans N; Panitti L; Vanormelingen J; Fransen S; Van de Velde S; Defert O Design, synthesis and biological characterization of selective LIMK inhibitors. Bioorg. Med. Chem. Lett 2015, 25, 4005–4010. [DOI] [PubMed] [Google Scholar]
- (24).Harrison BA; Almstead ZY; Burgoon H; Gardyan M; Goodwin NC; Healy J; Liu Y; Mabon R; Marinelli B; Samala L; Zhang Y; Stouch TR; Whitlock NA; Gopinathan S; McKnight B; Wang S; Patel N; Wilson AG; Hamman BD; Rice DS; Rawlins DB Discovery and development of LX7101, a dual LIM-Kinase and ROCK inhibitor for the treatment of glaucoma. ACS Med. Chem. Lett 2015, 6, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cowan-Jacob SW; Jahnke W; Knapp S Novel approaches for targeting kinases: allosteric inhibition, allosteric activation and pseudokinases. Future Med. Chem 2014, 6, 541–561. [DOI] [PubMed] [Google Scholar]
- (26).Hamill S; Lou HJ; Turk BE; Boggon TJ Structural basis for noncanonical substrate recognition of Cofilin/ADF proteins by LIM kinases. Mol. Cell 2016, 62, 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Klaeger S; Heinzlmeir S; Wilhelm M; Polzer H; Vick B; Koenig PA; Reinecke M; Ruprecht B; Petzoldt S; Meng C; Zecha J; Reiter K; Qiao H; Helm D; Koch H; Schoof M; Canevari G; Casale E; Depaolini SR; Feuchtinger A; Wu Z; Schmidt T; Rueckert L; Becker W; Huenges J; Garz AK; Gohlke BO; Zolg DP; Kayser G; Vooder T; Preissner R; Hahne H; Tõnisson N; Kramer K; Götze K; Bassermann F; Schlegl J; Ehrlich HC; Aiche S; Walch A; Greif PA; Schneider S; Felder ER; Ruland J; Médard G; Jeremias I; Spiekermann K; Kuster B The target landscape of clinical kinase drugs. Science 2017, 358, No. eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Machleidt T; Woodroofe CC; Schwinn MK; Méndez J; Robers MB; Zimmerman K; Otto P; Daniels DL; Kirkland TA; Wood KV NanoBRET–a novel BRET platform for the analysis of protein-protein interactions. ACS Chem. Biol 2015, 10, 1797–1804. [DOI] [PubMed] [Google Scholar]
- (29).Vasta JD; Corona CR; Wilkinson J; Zimprich CA; Hartnett JR; Ingold MR; Zimmerman K; Machleidt T; Kirkland TA; Huwiler KG; Ohana RF; Slater M; Otto P; Cong M; Wells CI; Berger BT; Hanke T; Glas C; Ding K; Drewry DH; Huber KVM; Willson TM; Knapp S; Müller S; Meisenheimer PL; Fan F; Wood KV; Robers MB Quantitative, wide-spectrum kinase profiling in live cells for assessing the effect of cellular ATP on target engagement. Cell Chem. Biol 2018, 25, 206–214 DOI: 10.1016/j.chembiol.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Robers MB; Wilkinson JM; Vasta JD; Berger LM; Berger BT; Knapp S Single tracer-based protocol for broad-spectrum kinase profiling in live cells with NanoBRET. STAR Protoc. 2021, 2, No. 100822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Robers MB; Friedman-Ohana R; Huber KVM; Kilpatrick L; Vasta JD; Berger BT; Chaudhry C; Hill S; Müller S; Knapp S; Wood KV Quantifying target occupancy of small molecules within living cells. Annu. Rev. Biochem 2020, 89, 557–581. [DOI] [PubMed] [Google Scholar]
- (32).Chaikuad A; Tacconi EM; Zimmer J; Liang Y; Gray NS; Tarsounas M; Knapp S A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol 2014, 10, 853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Deniston CK; Salogiannis J; Mathea S; Snead DM; Lahiri I; Matyszewski M; Donosa O; Watanabe R; Böhning J; Shiau AK; Knapp S; Villa E; Reck-Peterson SL; Leschziner AE Structure of LRRK2 in parkinson’s disease and model for microtubule interaction. Nature 2020, 588, 344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Attwood MM; Fabbro D; Sokolov AV; Knapp S; Schiöth HB Trends in kinase drug discovery: targets, indications and inhibitor design. Nat. Rev. Drug Discovery 2021, 20, 839–861. [DOI] [PubMed] [Google Scholar]
- (35).Mi H; Muruganujan A; Casagrande JT; Thomas PD Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc 2013, 8, 1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Endo M; Ohashi K; Mizuno K LIM kinase and slingshot are critical for neurite extension. J. Biol. Chem 2007, 282, 13692–13702. [DOI] [PubMed] [Google Scholar]
- (37).Niesen FH; Berglund H; Vedadi M The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc 2007, 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- (38).Winter G; Lobley CM; Prince SM Decision making in xia2. Acta Crystallogr., Sect. D: Biol. Crystallogr 2013, 69, 1260–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).McCoy AJ; Grosse-Kunstleve RW; Storoni LC; Read RJ Likelihood-enhanced fast translation functions. Acta Crystallogr., Sect. D: Biol. Crystallogr 2005, 61, 458–464. [DOI] [PubMed] [Google Scholar]
- (40).Murshudov GN; Vagin AA; Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (41).Chen VB; Arendall WB III; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Shevchenko A; Tomas H; Havlis J; Olsen JV; Mann M In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc 2006, 1, 2856–2860. [DOI] [PubMed] [Google Scholar]
- (43).Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- (44).Ruprecht B; Koch H; Domasinska P; Frejno M; Kuster B; Lemeer S Optimized enrichment of phosphoproteomes by Fe-IMAC column chromatography. Methods Mol. Biol 2017, 1550, 47–60. [DOI] [PubMed] [Google Scholar]
- (45).Ule J; Darnell RB RNA binding proteins and the regulation of neuronal synaptic plasticity. Curr. Opin. Neurobiol 2006, 16, 102–110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.