

Abstract
Trinucleotide repeat instability is a driver of human disease. Large expansions of (GAA)n repeats in the first intron of the FXN gene are the cause Friedreich’s ataxia (FRDA), a progressive degenerative disorder which cannot yet be prevented or treated. (GAA)n repeat instability arises during both replication-dependent processes, such as cell division and intergenerational transmission, as well as in terminally differentiated somatic tissues. Here, we provide a brief historical overview on the discovery of (GAA)n repeat expansions and their association to FRDA, followed by recent advances in the identification of triplex H-DNA formation and replication fork stalling. The main body of this review focuses on the last decade of progress in understanding the mechanism of (GAA)n repeat instability during DNA replication and/or DNA repair. We propose that the discovery of additional mechanisms of (GAA)n repeat instability can be achieved via both comparative approaches to other repeat expansion diseases and genome-wide association studies. Finally, we discuss the advances towards FRDA prevention or amelioration that specifically target (GAA)n repeat expansions.
Keywords: GAA repeat instability, trinucleotide repeats, DNA triplex, H-DNA, replication fork stalling, Friedreich’s ataxia
1. Introduction
DNA microsatellites, 1-to-9 base-pair long tandemly duplicated sequences, comprise up to 3% of the human genome [1–3]. Expansions of a subset of microsatellites are associated with over 50 repeat expansion diseases (REDs), and the number is ever-growing [4]. Disease-associated repeats are characterized by length variability (expansions and contractions) and are known to induce fragility and repeat-induced mutagenesis, resulting in genomic instability (for an extensive review, see [5]). In the first years after their discovery in 1991 [6], most repeat expansion diseases were identified as autosomal dominant disorders associated with expansions of (CNG)n repeats, resulting in a toxic gain of function at the protein level. (CNG)n expansion diseases include Huntington’s disease (HD), spinocerebellar ataxias (SCAs), myotonic dystrophy (MD) and Fragile X syndrome (FXS).
All expandable (CNG)n repeats can form imperfect hairpins stabilized by CG base pairs or slipped strand DNA structures that result from the formation of two such hairpins in complementary DNA strands [7]. Thus, strand slippage and hairpin formation during DNA replication was initially proposed to be at the center of trinucleotide repeat (TNR) instability [8]. Therefore, the discovery of the genetic basis of Friedreich’s ataxia (FRDA) in 1996 came as a surprise [9]. FRDA is an autosomal recessive disease caused by the expansion of a (GAA)n repeat, which, unlike (CGN)n repeats, cannot form a hairpin structure [9]. Overall, (GAA)n runs are amongst the most expansion-prone trinucleotide repeats in the human genome, the majority of which originated from 3′ poly(A) tracts of various Alu elements upon AAA to GAA transition [10], [11]. In the case of FRDA, however, the (GAA)n repeat originated from the An(TAC)An sequence at the center of the Alu Sq element located in the first intron of the FXN gene [10].
FRDA is the most common form of hereditary ataxia in humans [12]. (GAA)1-33 repeats are in the normal range, (GAA)34-65 repeats are pre-mutational, and longer repeats are pathogenic. The length of the (GAA)n repeat positively correlates with the age of disease onset, its severity and progression [13–17]. (GAA)n expansions ultimately lead to chromatin changes and FXN gene silencing, resulting in a drastic reduction in the levels of the mitochondrial protein frataxin [18–22]. Reduced frataxin levels lead to increased oxidative stress, accumulation of iron species in the mitochondria and subsequent cell death, primarily affecting neuronal tissues [18], [23–25]. The loss of gene function upon repeat expansions accounts for the recessive mode of inheritance. FRDA is associated with cerebellar and sensory ataxia, diabetes mellitus, and cardiomyopathies, leading to early death (for a clinical review, see [26]). There is currently no effective cure or treatment for FRDA [27].
2. (GAA)n repeats form triplex DNA structures during transcription and replication
Being a homopurine-homopyrimidine (hPu/hPy) mirror repeat, (GAA)n runs can assume the non-B DNA structure termed DNA triplex or H-DNA. H-DNA is formed when a DNA strand corresponding to one half of the repeat folds back forming a triplex with the duplex half of the repeat via Hoogsteen base pairing, while its complementary strand remains single-stranded (Fig. 1A–D) [28], [29]. Like other alternative DNA structures, H-DNA is thermodynamically unfavorable in linear double-stranded DNA, but becomes favorable in negatively supercoiled DNA, which is topologically equivalent to unwound DNA, as it relieves torsional stress [30], [31]. Consequently, H-DNA is not a steady-state presence in genomic DNA but can rather form at different stages of the cell-cycle during specific genetic processes such as replication and transcription, hence it is a dynamic DNA structure.
Figure 1:
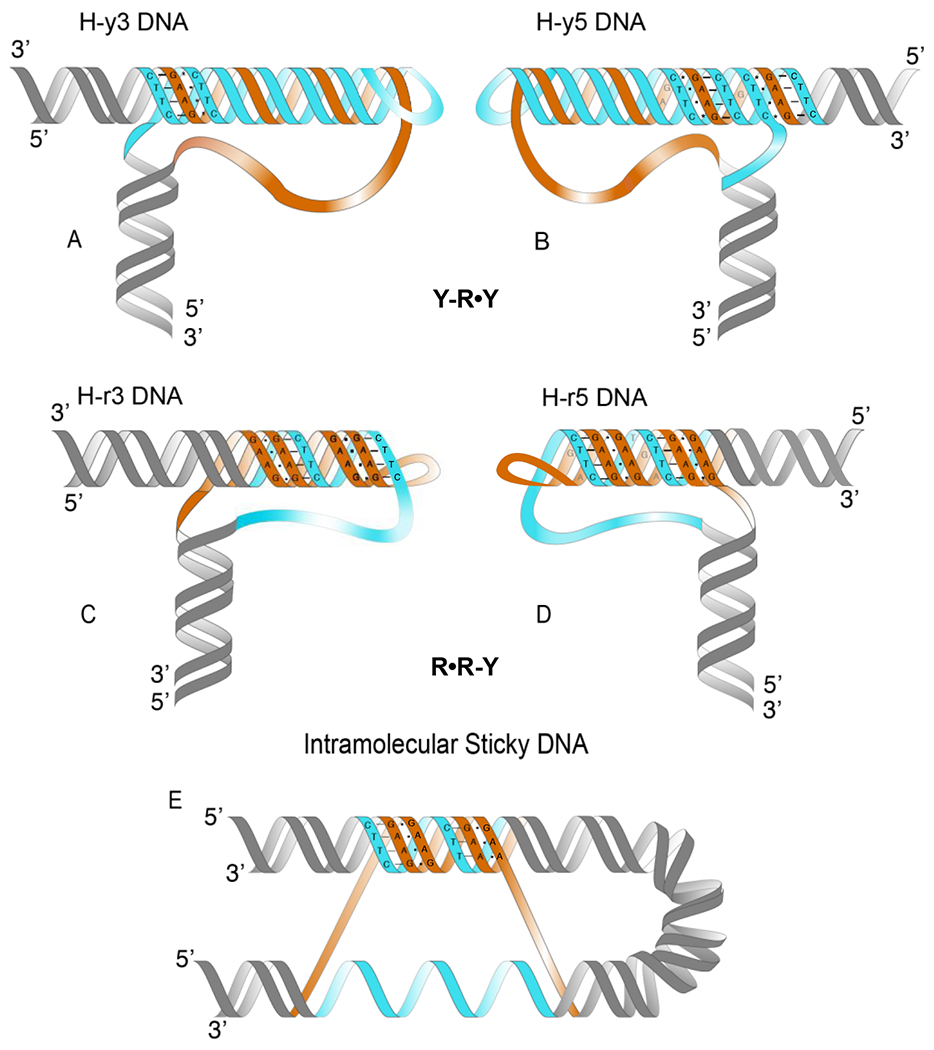
Various types of triplex DNA structures formed by long (GAA)n repeats. Each H-DNA conformation, YRY or RRY, can exists in two isoforms, depending on whether the 3’ or the 5’ of a strand is donated to the triplex. Homopurine strands are in orange, homopyrimidine strands are in blue. Dashes indicate Watson-Crick base-pairing, circles indicate Hoogsteen or reverse Hoogsteen base-pairing, asterisks indicate protonated cytosines. See Section 2 for more detail.
H-DNA can exist in several conformations [32]. In an H-y triplex (YRY), the pyrimidine strand contributes to triplex formation by Hoogsteen base pairing (Fig. 1A–B). Normally, H-y formation is favored at low pH since it requires cytosine protonation, but because of their high AT-content, (GAA)n repeats can readily form H-y triplexes under physiological conditions [31], [33]. In an H-r triplex (RRY), the purine strand contributes to triplex formation through reverse Hoogsteen base pairing (Fig. 1C–D). H-r triplexes, including those formed by the (GAA)n repeat, were observed at neutral pH in the presence of divalent cations [34], [35]. Finally, two distant (GAA)n repeats within the same supercoiled DNA molecule can form a structure termed sticky DNA (Fig. 1E) [34], [36]. In this case, a homopurine strand from one of the repeats forms an H-r triplex with another repeat, also in the presence of divalent cations [34], [37].
While all these structures can be formed under specific conditions in vitro, it remains unclear which one is the most common at the FRDA locus and how each contributes to repeat instability and disease. It is generally challenging to detect dynamic DNA structures at endogenous genomic loci in vivo, given that they may only be formed transiently. It is particularly difficult in mammalian cells, in which chemical probing has proven to be highly cumbersome, partially due to the extreme genome size (reviewed in [38]). Triplex-specific antibodies were shown to bind in situ to multiple sites in human chromosomes, some of which contained (GAA)n repeats [39–41]. Note however, that the resolution power of this technique is not at the nucleotide level.
As discussed above, negative DNA supercoiling is the main driver for H-DNA formation in vitro. In mammalian nuclei, transcription is the main source of negative DNA supercoiling ([42] and references therein). Kouzine et al. used permanganate treatment, which oxidizes thymine residues in single-stranded DNA (ssDNA), in combination with S1 nuclease cleavage and high-throughput sequencing to detect non-B DNA structures in the genome of mouse B-cells [43]. This study identified approximately 17,000 sites of H-DNA out of ~728,000 predicted H-DNA motifs, the prevalence of which positively correlated with transcription levels (Fig. 2A) [43]. More recently, an S1-seq based method, which relies on mapping of S1-cleavage sites in permeabilized cells [44], identified about 144,000 H-DNA forming structures, preferably H-y5 triplexes (Fig. 2B) [45]. Notably, most H-DNA motifs were observed at relatively short homopurine-homopyrimidine repeats, and the authors believe that they could have formed ex vivo during sample preparation owing to the acidic pH of the S1-nuclease reaction buffer. In FRDA patient cells, a similar S1-END-seq approach revealed H-DNA at the expanded (GAA)n repeats specifically when the FXN locus was transcribed (Fig. 2C) ([46] preprint). It is concluded, therefore, that in this case, the triplex is formed in vivo during transcription of the (GAA)n repeat.
Figure 2:
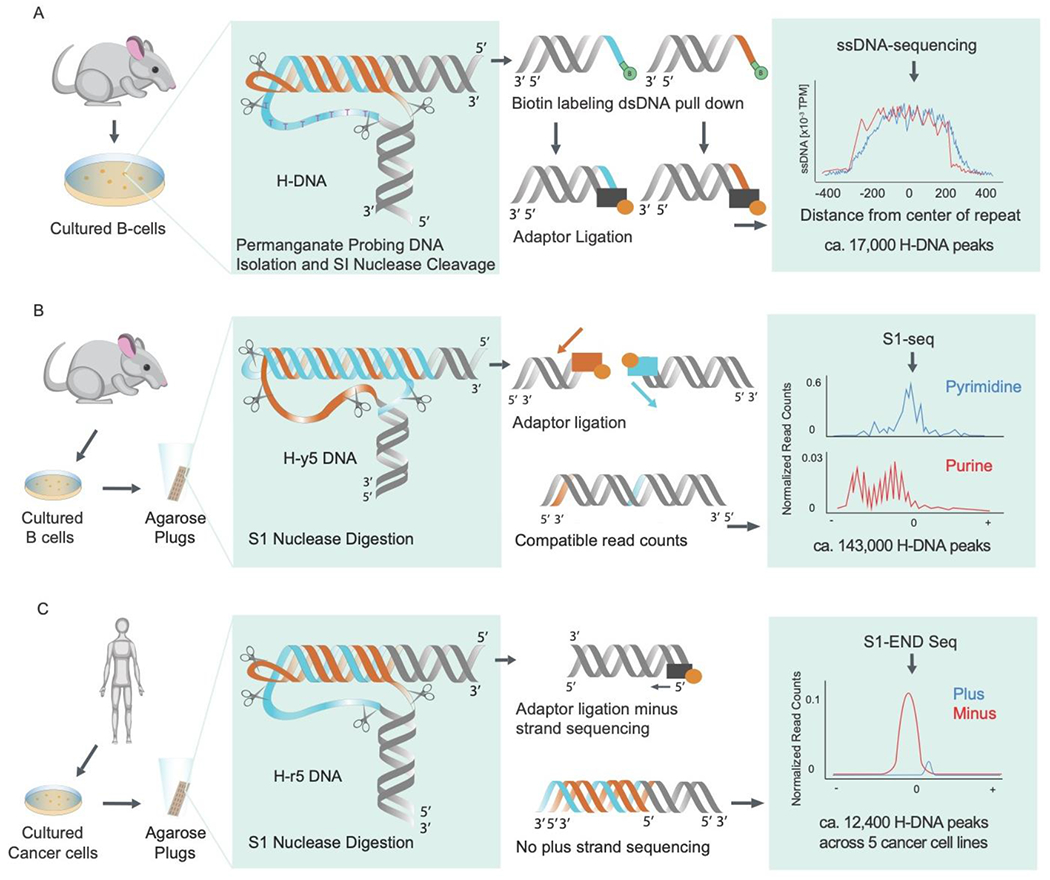
Methods to detect in vivo formation of H-DNA in higher eukaryotes (see section 2). (A) Genome-wide H-DNA formation in cultured mouse B-cells (adapted from [43]). (B) Genome-wide H-DNA formation in primary mouse cells via S1-seq (adapted from [45]). (C) Genome-wide H-DNA formation in human cancer cell lines via S1-END Seq (adapted from [46] preprint).
Besides transcriptional supercoiling, formation of H-DNA can be promoted by DNA strand unwinding during DNA replication. During DNA polymerization in vitro, a DNA strand from a partially unwound repeat can fold into a triplex structure, effectively blocking further DNA polymerase progression. Consequently, triplex forming motifs were called “suicidal sequences” for DNA polymerization [47]. A similar mechanism accounts for the blockage of DNA polymerization by expanded (GAA)n repeats [31], [48]. Recently, analysis of DNA synthesis through a reconstituted eukaryotic replication fork revealed a weak but reproducible stalling only when a long (GAA)n repeat was located on the leading strand template [49].
Long (GAA)n repeats were also shown to stall replication in vivo both in yeast and mammalian cells [50–54]. In yeast, (GAA)n-mediated fork stalling is orientation-dependent, as it was only observed when the (GAA)n run was on the lagging strand template [53], [55]. This was interpreted as the formation of an H-r triplex in front of the fork, followed by strand unwinding by the replicative CMG (Cdc45/Mcm2-7/GINS) helicase [53]. In human cells, (GAA)n repeats cause fork stalling in SV40-based episomes in an orientation-independent manner [50], [51], [56] (preprint). This inconsistency between the two systems could be explained by the fact that in an SV40 replisome, the T-antigen helicase is used instead of the endogenous CMG helicase, and synthesis is performed by Pol δ on both the leading and the lagging strand [57]. Fork stalling at (GAA)n repeats result in fork reversal [51], [56] (preprint),which can lead to subsequent fragility and instability [55], [56], [58].
It is important, therefore, to understand whether triplexes can cause the stalling of regular replication forks at endogenous chromosomal locations. The use of the S1-END-seq approached described above [44] identified two types of non-B DNA structures in human cells: DNA cruciforms formed by (AT)n repeats and H-DNA formed by long hPu/hPy mirror repeats ([46] preprint). H-DNA was detected in multiple cancer cell types, and a considerable portion of sequences was shared between genomes, indicating the presence of highly conserved H-DNA regions (Fig. 2C). Most of the detected triplexes were formed by (GAAA)n, (GAA)n and (GGAA)n repeats and corresponded to the H-r5 conformation. To address the concern that the acidic pH of S1-nuclease treatment could promote triplex formation ex vivo, the authors replaced S1-nuclease with P1-nuclease, which cleaves ssDNA at neutral pH. This new approach, called P1-END-seq, revealed a similar number and distribution of triplex peaks in cancer cell lines. Another important argument supporting H-DNA formation in vivo is that the triplex peaks spike in S-phase, which correlates with orientation-dependent replication fork stalling, similar to that observed in yeast.
Replication fork stalling at expanded (GAA)n repeats at the FXN locus was directly shown using single-molecule analysis of replicated DNA (SMARD) isolated from FRDA patient cells [52]. Stalling was observed in both orientations, being particularly pronounced when the (TTC)n run is on the lagging strand template. This polarity is opposite to that observed in yeast and in human cells via S1-END-seq. While the reason for this difference remains unclear, one possibility is that head-on collision of FXN transcription with replication going in the opposite direction adds to the strength of the stall. Importantly, GAA-specific polyamides that disrupt triplex DNA rescue replication fork stalling, implying that H-DNA causes fork stalling at the repeat (preprint) [46], [52].
Further characterization of non-B DNA structures at the single nucleotide level has the potential to provide insight into not only location and frequency of H-DNA, but also requirements for its formation, mutagenic potential and association with cell or tissue type. This information will be important to determine whether H-DNA is associated with genetic diseases such as cancer besides FRDA, as proposed by recent computational analyses [59], [60]. So far, individual sequencing methods show detection biases toward specific H-DNA isomers, and different types of sample processing might introduce artificial H-DNA formation to a certain extent. Newer methods based on the use of small molecules as modifiers are being developed to allow more sensitive and accurate detection of alternative DNA structures [61], [62]. Where, then, are the commonalities? Firstly, all studies agree that even though both replication and transcription can promote H-DNA formation, the major contributor varies based on the specific repeat and its genetic environment, such as chromatin context and specific location. In addition, interruptions in the repeat decrease its ability to form H-DNA and stall the replication fork [45], [46] (preprint), in accordance with previous in vitro studies for long interrupted (GAA)n repeats [36]. Importantly, interrupted (GAA)n repeats are rarely found in FRDA patients, and when present lead to delayed disease onset and milder phenotypes, strongly suggesting that the ability of the repeat to form a triplex is essential for disease pathogenesis [13], [63]. Finally, the prevalence of H-DNA forming sequence warrants extensive studies of their potential biological functions, which likely remain widely underestimated.
3. Genome instability mediated by (GAA)n repeats
As is true for other repeat expansion diseases, the longer the (GAA)n repeat is, the more prone to length instability it becomes [64], [65]. Instability is observed both during intergenerational transmission and in post-mitotic somatic cells [66–70]. Expansions predominantly happen during intergenerational transmission and cell division. In somatic cells, contractions are the most prevalent form of (GAA)n instability, although expansions were observed in affected tissues, including the heart, pancreas and neuronal tissues [66], [71]. Somatic mosaicism – the presence of a variety of (GAA)n lengths in the same patient – was observed in multiple patient tissues in an age-dependent manner, with the length of the largest allele determining the scale of the observed mosaicism [66], [71–74].
Since (GAA)n repeat expansions cause a human hereditary disease, research has focused on the study of their expansion mechanisms. Somewhat less appreciated is the fact that (GAA)n repeats can cause additional local and global genome rearrangements. The original examples came from studies in yeast. First, it was directly demonstrated that expanded (GAA)n repeats are fragile, resulting in double-strand breaks [55]. Second, (GAA)n repeats appear to cause mutagenesis at a distance, in a process that we called repeat-induced mutagenesis (RIM) [75]. Expanded (GAA)n repeats at the FXN locus also increase mutagenesis in the area surrounding the repeat, likely through double-strand break (DSB) repair processes [75–79]. Note that other triplex-forming sequences were also associated with increased break-induced mutagenesis in mammalian cells [77], [80], [81].
Altogether, various types of (GAA)n repeat-mediated instability contribute to the accumulation of mutations at and around the expanded locus, as well as rearrangements in other genomic regions. These events can modulate the age and onset of FRDA and/or lead to the emergence of other pathogenic mutations, which can in turn modulate (GAA)n length stability. We will cover the mechanisms of (GAA)n mediated genome instability during replication and in non-dividing cells in the next sections.
4. (GAA)n repeat instability during DNA replication
4.1. Fragility
As is common for other disease-related repeats, (GAA)n repeats were shown to be fragile in multiple model systems, causing DSBs and genome rearrangements. In yeast, (GAA)n fragility was shown to be dependent on its orientation relative to the replication origin, being higher when the (GAA)n is on the lagging-strand template – the same orientation that causes replication stalling [55]. In this case, fragility was dependent on the mismatch repair (MMR) machinery. We hypothesize that yeast MMR cleaves the single-stranded loops of H-DNA, erroneously perceiving them as mismatched loop-outs. It would be of great interest to substantiate this idea in biochemical studies. Indirect evidence also indicates that there is increased fragility at the endogenous (GAA)n repeat in the FXN locus in FRDA patient cells [58].
4.2. Expansions
Expanded (GAA)n repeats were shown to affect replication fork progression in every experimental system studied to date, likely owing to their triplex-forming potential. In yeast, (GAA)n repeats start expanding at the carrier length of (GAA)52, and the rate of expansion increases exponentially with the repeat’s length [65]. This begs the question, is there a link between replication through the repeat and its instability? An unambiguous affirmative answer came from a yeast experimental system. A genome-wide screen identified genes that modulate (GAA)n instability, including its fragility and propensity for expansions [82]. The screen had hits in three main categories: replication-associated genes, transcription initiation genes, and two components of the CST (Cdc13-Stn1-Ten1) complex, which regulates telomere maintenance. Further studies expanded on the role of each category in (GAA)n repeat instability.
First, an intact and processive core replisome was shown to counteract instability [82], [83], as mutations in Pol ε and δ, as well as in the fork stabilization complex (Tof1-Csm3-Mrc1), promote large-scale (GAA)n repeat expansions [83]. Mutations in subunits of the CMG helicase also increase mid-scale expansions of short (GAA)25 repeats, through a mechanism consistent with template switching (TS) and break-induced replication (BIR) [84], [85]. During lagging strand synthesis, each Okazaki fragment needs to be processed by 5’ flap endonucleases prior to ligation. The major flap endonucleases in yeast are Rad27 and Dna2, which cleave short and long flaps, respectively. Mutation of either flap endonuclease dramatically increases (GAA)n repeat expansions, possibly due to an imbalance in the total amount of ssDNA in the cells which can result in increased formation of triplexes on the flaps, ultimately resulting in (GAA)n repeat instability (Fig. 3A) [65], [83].
Figure 3:
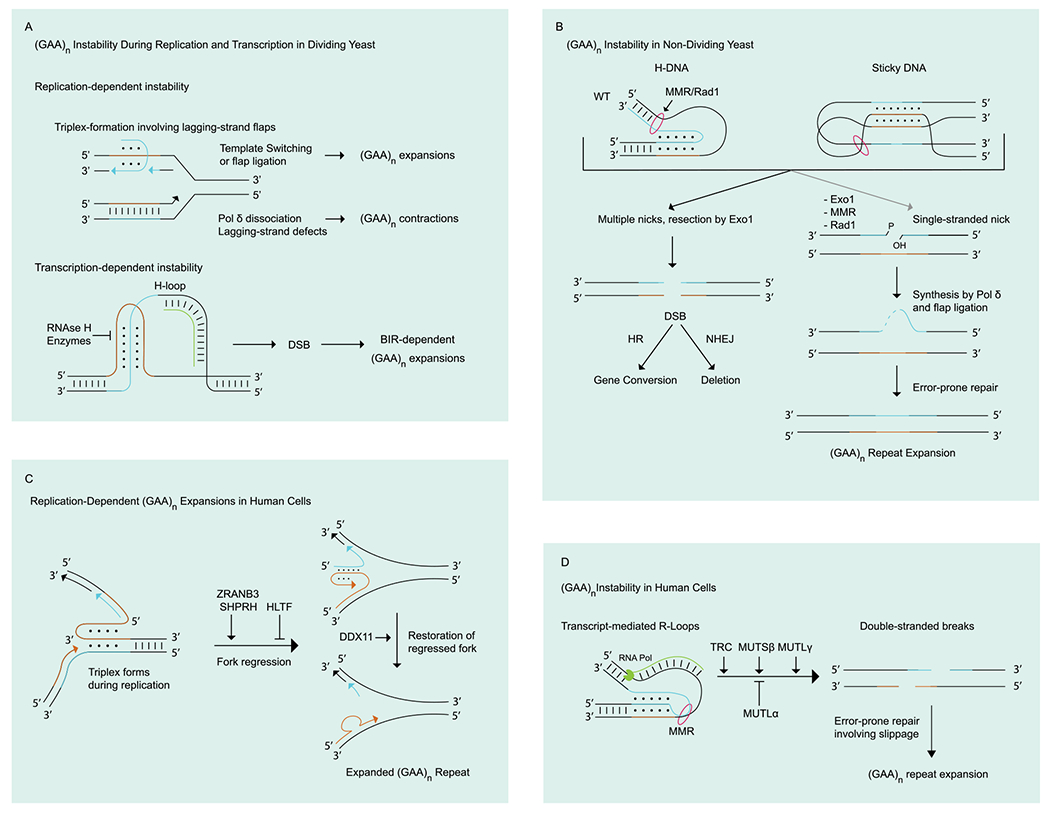
Models of (GAA)n repeat instability. (A) (GAA)n repeat instability in dividing yeast mainly occurs via replication-dependent mechanisms, such as template switching (TS) or flap ligation, leading to expansions (see 4.2) [65], [83]. Pol δ dissociation during lagging strand synthesis causes contractions (see 4.3) [64]. During transcription, formation of a triplex-stabilizing R-loop (H-loop) can trigger break-induced recombination leading to repeat expansions (see 5) [113]. (B) (GAA)n repeat instability in non-dividing yeast is characterized by two different types of events, depending on whether the MMR machinery is functional. MMR drives incisions in H-DNA and sticky DNA structures, which are then converted into DSBs. DSB repair by HR leads to gene conversions events, while repair by NHEJ results in deletions. In MMR-deficient strains, nick repair leads to expansions (see 6.1). (C) Replication dependent (GAA)n repeat expansions in human cells is initiated by triplex formation ahead of the fork leading to its regression. Repeats can expand upon strand slippage during the restoration of the regressed fork [56] (preprint) (see 4.2). (D) (GAA)n repeat instability in non-dividing and somatic human cells is promoted by the formation of R-loops during transcription and/or upon transcription-replication collisions (TRCs), which are then recognized by MMR and converted into DSBs. Other sources of DSBs can initiate this process as well. Error-prone repair results in (GAA)n repeat expansions (see 6.1 and 6.2) [52], [114–116].
A strong replication stall could lead to dramatic consequences such as template switching or replication fork reversal [51]. The goal of both of these processes is to bypass a template “lesion”, but can result in (GAA)n repeat instability [86]. Nevertheless, whereas fork stalling is orientation-dependent [52], [53], [65], [69], large-scale (GAA)n repeat instability seems largely orientation-independent, even though there is a slight bias for increased instability when the (GAA)n run is on the lagging strand template in all of the studied systems [64], [65]. This bias has been related to the asymmetrical nature of DNA replication and led to the proposal of the “ori-switch” model, in which origin activity and orientation relative to the position of the repeat influences predisposition for either expansions or contractions [87]. What other processes, in addition to fork stalling, are then causing instability during replication? Template-switching during DNA synthesis could also occur independently of stalling, especially at the site of long repetitive templates in which TS could happen at a higher rate (Fig. 3A).
In addition, factors other than the ones involved directly in the replication fork can contribute to (GAA)n expansions during replication, as illustrated by the identification of the CST complex in the genetic screen described above [82]. Recently, mutations in the CST complex were shown to lead to large-scale (GAA)n repeat expansions through a mechanism involving the Rad9-dependent G2/M DNA damage checkpoint activation and post-replicative repair (PPR) of gaps and nicks [88]. Even though (GAA)n repeats, unlike expanded (CAG)n repeats, do not activate the checkpoint by themselves, the checkpoint response likely becomes relevant in the context of other sources of replicative stress [89].
Very recently, an episomal experimental system was set up to study the link between replication and expansions of (GAA)100 repeats in human cells [56] (preprint). Using siRNA to deplete specific proteins, this study shows that large-scale repeat expansions are promoted by proteins involved in replication fork reversal (SHPRH/SMARCAL1/HLTF) and counteracted by proteins that are involved in the restoration of reversed forks (RAD52/RECQL1/WRN). Further, the DDX11 helicase, which was shown to untangle triplex DNA in vitro [90], also counteracts repeat expansions. Altogether, this data indicates that expansions occur while the replication fork attempts to bypass a triplex formed by the (GAA)n repeat (Fig. 3C).
4.3. Contractions
Repeat contractions are another type of (GAA)n repeat instability which has been intimately tied with ongoing DNA replication. (GAA)n repeats become more prone to contractions the longer they are, and contractions occur progressively in somatic cells of FRDA patients, as well as during intergenerational transmission [66], [67], [70]. The question of whether contractions and expansions occur through a shared mechanism remained a mystery for a long time. A recent study in yeast set up a genetic assay to study large-scale contractions (> 20 repeats) of long (GAA)124 tracts [64]. First, contractions were associated with the ability of the repeat to form H-DNA, directly tying contraction events to triplex formation during DNA synthesis. Second, mutations of lagging strand synthesis polymerases (Pol α and Pol δ) and flap-processing nucleases (Rad27 and Dna2), which result in the accumulation of long ssDNA tracts, promoted large contractions in an orientation-independent manner. Third, the ssDNA-binding replication protein A (RPA) strongly counteracted triplex formation between the nascent lagging strand and its template, preventing contractions (Fig. 3A) [64]. At the same time, all major DSB-repair pathways did not influence (GAA)n repeat contractions. This led us to conclude that large-scale contractions occur during triplex bypass during lagging strand synthesis.
Importantly, the average contraction size of the starting (GAA)124 was of ca. 60 repeats in this system, and likely to be the result of a one-step process. This corresponds to a contraction of a (GAA)124 repeat in the mutational range down to pre-mutational or normal repeat sizes, which is very encouraging from a potential therapeutic standpoint. The challenge remains to identify proteins which exclusively promote contractions or prevent expansions, without affecting the other side of instability or have genome-wide mutagenic effects.
Altogether, the expansion and contraction data point to a link between replication through (GAA)n repeats and their instability. Both fork stalling at the repeat and instability become apparent at (GAA)n repeat lengths corresponding to carrier sizes in patients and become more pronounced at disease length. That being said, in the experimental systems studied so far, there is no obvious direct correlation between the strength of fork stalling and repeat instability causing disease. This ambiguity warrants further studies of the mechanisms of replication-dependent repeat instability.
4.4. Complex genome rearrangements
Two classes of repeat-mediated genome rearrangements were revealed in a yeast experimental system. First, repair of DSBs within expanded (GAA)n repeats occasionally leads to the formation of large deletions which encompass the repeat and its adjacent regions [65]. Second, genetic assays combined with Nanopore sequencing found that (GAA)n repeats cause complex genome rearrangements (CGRs) of the yeast genome that are yet another byproduct of DSB repair [91]. These CGRs resulted from a mixture of reciprocal and non-reciprocal gene-conversion events, which involve both intra- and inter-chromosomal interactions [91].
We also want to emphasize that with the advent of long-read sequencing and dedicated computational tools, the sequencing of expanded repeats appears more practical, and provides the opportunity to widely survey both the general and affected populations to assemble a comprehensive picture of (GAA)n repeat sizes and distributions [91–98]. Recently, Nanopore sequencing was used to detect replication fork stalling associated with structure formation during sequencing [99], [100]. The combination of long-read sequencing with GWAS, such as the ones conducted on (CAG)n repeats and Huntington’s disease [101–103], could provide invaluable information on genetic modifiers of (GAA)n repeat stability and FRDA onset and severity, revealing new mechanisms of instability and guiding the development of novel therapeutic avenues.
4.5. Repeat-induced mutagenesis
As mentioned above, expanded (GAA)n repeats in the FXN locus increase mutagenesis in surrounding genomic regions in FRDA patients. The mechanisms of this mutagenesis were thus far studied only in a yeast experimental system. Repeat-induced mutagenesis (RIM) was detected up to 10 kb upstream and downstream of long (GAA)n repeats [79]. Conditional mutations in Pol Ɛ dramatically elevated the rate of RIM, implicating DNA replication in the process [83]. It is not yet clear what causes the mutagenesis. In two studies, RIM depended on translesion synthesis (TLS) by DNA Polymerase ζ [79], [104]. In another study, translesion synthesis was only involved in RIM if Pol δ activity was compromised [83]. Two mechanisms are being considered: repair of post-replication gaps that involves Pol ζ [79], [104], and a BIR-like pathway [83].
5. Role of transcription and R-loops in (GAA)n repeat instability
Expanded (GAA)n repeats pose an obstacle to transcription in an orientation-dependent manner [54], [105], [106]. During transcription, a sense r(GAA)n strand or an antisense r(UUC)n strand can participate in the formation of stable DNA:RNA duplexes, or R-loops, consisting of two DNA and one RNA strands [107], [108]. Interestingly, recent analyses have shown that R-loop formation might be promoted at repetitive elements across eukaryotic genomes [109], [110]. Enrichment of unscheduled R-loops can result in genomic instability and has been shown to modulate stability of other TNRs (reviewed in [111], [112]).
R-loop formation during transcription of expanded (GAA)n repeats has been proposed as a pathogenic mechanism contributing to transcriptional silencing at the FXN locus [117]. The formation of DNA:RNA hybrids at expanded (GAA)n repeats is thermodynamically favorable since the sense RNA is homopurine [118], [119]. Interestingly, transcription has been shown to increase (GAA)n repeat instability in both human and yeast experimental systems [82], [120–122]. In yeast, the transcription-dependent increase in instability was further exacerbated in the absence of the RNase H enzymes, which counteract the accumulation of R-loops [113]. Furthermore, this increased instability was caused by BIR. Altogether, these data point to the role of R-loops and transcription-replication collisions (TRC) in (GAA)n repeat instability (Fig.3A).
It is generally believed that R-loops can cause genome instability when replication and transcription collide head-on. It was surprising, therefore, that R-loop-dependent instability of (GAA)n repeats did not depend on relative orientation of replication and transcription [113]. To explain this difference, it was hypothesized that H-DNA transiently formed upstream of elongating RNA polymerase interacts with the repetitive RNA transcript forming the so-called H-loop [5]. This H-loop is much more stable than either H-DNA or R-loop alone. Therefore, DNA replication could be blocked notwithstanding its directionality. It is yet to be determined whether transcription can promote (GAA)n repeat instability independently of DNA replication. The answer this question will allow us to determine whether transcription-mediated instability might be a universally shared mechanism in both dividing and non-dividing cells. It will be important to study whether other factors which contribute to R-loop balance and/or transcription-coupled repair (TCR) affect (GAA)n repeat stability. One promising candidate is the Senataxin helicase (Sen1 in S. cerevisiae), which associates with the replication fork to resolve R-loops and prevents their accumulation during DNA replication and DNA damage repair [123–127].
6. Replication-independent pathways of (GAA)n repeat instability
The tissues which are most severely affected in repeat expansion diseases are usually terminally differentiated and do not undergo cellular division. In such tissues, DNA replication-dependent processes cannot be a major source of repeat instability. Nevertheless, (GAA)n repeats progressively expand in neuronal tissues, such as the cerebellum and dorsal root ganglia (DRG), as well as in cardiac muscles, in both human and mouse models [66], [128–132]. A longitudinal study in FRDA patients confirmed lifetime-long addition of (GAA)n repeats in multiple non-dividing tissues and shows that longer starting repeat sizes lead to a greater magnitude in expansions over time [71]. DNA damage and subsequent repair, involving tracts of DNA synthesis through the repetitive tract, occur in post-mitotic tissues such as neuronal tissues at every stage of the cell cycle [133], [134]. This is relevant for (GAA)n repeat instability since DNA damage repair can be affected by the presence of the triplex H-DNA structure, which may be perceived as a lesion by DNA repair machineries. It is also foreseeable that repeat instability could arise during cell-cycle reactivation in postmitotic neurons, which occasionally involves aberrant DNA synthesis [135–138]. In the next section, we focus on the DNA repair mechanism that has received most of the recent attention regarding (GAA)n repeat instability – mismatch repair.
6.1. Mismatch repair in (GAA)n repeat instability
Mismatch repair (MMR) is responsible for the correction of base mismatches and small insertions and deletions (indels), which can arise during DNA replication, recombination, and repair. In MMR, mismatches and small loops (1-3 bp) are recognized by MutSα (Msh2-Msh6), and larger loop-outs by MutSβ (Msh2-Msh3). Subsequently, the MutL complexes, MutLα (Mlh1-Pms2) and MutLγ (Mlh1-Mlh3), excise the mismatch, followed by DNA strand resection and fill-in synthesis. In this capacity, MMR is essential for the maintenance of genome stability. Among other things, defects in MMR lead to microsatellite instability – a characteristic feature of various cancers (reviewed in [139]). But not all microsatellites are equal in the eyes of the MMR machinery. Counterintuitively, functional MMR overall promotes instability of the microsatellites responsible for repeat expansion diseases, including (CAG)n, (CGG)n and (GAA)n [140], [141]. The individual effects of the MMR components on repeat instability depend on both the model organism and whether somatic or intergenerational instability is under investigation (Table 1).
Table 1:
Role of MMR proteins during length instability of (GAA)n and (CAG)n repeats
| Complex | MMR protein | GAA somatic and non-dividing | GAA intergenerational and replication-models | CAG somatic and non-dividing | CAG intergenerational and replication-models |
|---|---|---|---|---|---|
| MutLα MutLγ |
MLH1 | Promotes expansions Mouse model [143] No effect on expansions Promotes deletions Non-dividing yeast cells [149] |
Promotes expansions Mouse model and human cells [116], [143] |
Promotes expansions HD mouse model [156] Human HD population [157] |
Prevents expansions S. cerevisiae [158] |
| MutLα | PMS2 (Pms1 in S. cerevisiae) | Prevents expansions Mouse model Non-dividing yeast cells [114], [149] Promotes deletions Non-dividing yeast cells [149] |
Prevents expansions Promotes contractions Mouse model [142] Prevents expansions Human cells [116] |
Promotes expansions Prevents deletions Mouse model [159] |
Prevents expansions S. cerevisiae [158] |
| MutLγ | MLH3 | Prevents expansions Human cells [116] |
Prevents expansions Human cells [116] |
Promotes expansions HD mouse models and patient derived cells [156], [160] |
No effect S. cerevisiae [158] |
| MutSβ MutSα |
MSH2 | Promotes expansions Mouse model Human cells [114], [115] |
Prevents contractions Mouse model [142] Promotes expansions iPSC GAA expansion model [144], [161] |
Promotes expansions Mouse models Human cell model [126], [162], [163] |
Promotes expansions S. cerevisiae Mouse model [164], [165] Prevents contractions S. cerevisiae [158] |
| MutSβ | MSH3 | Prevents expansions Promotes deletions Non-dividing yeast cells [149] Promotes expansions Human cells and FRDA fibroblasts [115] |
No effect on expansions G0 yeast cells [149] Prevents contractions Mouse model [142] |
Promotes expansions Human cell model [126] |
Promotes expansions HD mouse model DM1 mouse Human cell lines S. cerevisiae [164], [166–170] |
| MutSα | MSH6 | Promotes expansions Mouse model [114] No effect on expansions and promotes deletions Non-dividing yeast cells [149] |
Prevents expansions and contractions Mouse model iPSC model [142], [161] No effect on expansions Human cells [115] |
Prevents expansions Human cell model [126] |
Prevents contractions HD mouse model [166] Prevents expansions S. cerevisiae [164] |
In humanized FRDA mice, MMR affects repeat instability, although fine molecular mechanisms are somewhat controversial. That is, MUTSβ prevents intergenerational contractions, MUTSα precludes both expansions and contractions, while MUTLα counteracts expansions but promotes contractions [142], [143]. In somatic cells, specifically in neurological tissues, MUTLα suppresses expansions, but MUTSα promotes expansions [114].
In contrast to mice, MUTSβ promotes small-scale expansions in cultured human cells [115]. This depends on the activity of MUTLγ, and MUTLα protected against expansions, as observed in mouse models (Fig. 3D) [116]. In FRDA patient-derived induced-pluripotent stem cells (iPSCs), MUTSβ promotes large-scale (GAA)n expansions [144]. Whether allele variants of the individual proteins are actually modifiers of repeat stability in humans remains to be ascertained [145–148]. Notably, genetic analysis of (GAA)n repeat instability in non-dividing somatic cells cannot be easily done in patient-derived tissues, as they present a static picture or are passaged derived cells, which underwent further replication cycles.
To address this problem, Neil et al. developed a novel experimental system to study instability of a long (GAA)100 repeat in chronologically aging quiescent (G0) cells in S. cerevisiae [149]. Three categories of mutagenic events were observed: repeat expansions, NHEJ-dependent deletions, and HR-mediated gene conversions. Whereas the main mutational event in dividing yeast cells is repeat expansions, the balance rapidly shifted to large deletions frequently including the whole (GAA)n repeat as the cells entered quiescence (Fig. 3B). Deletions were triggered by DSBs at the repeat generated during MMR as discussed in 4.1. These DSBs are then repaired by Exo1-mediated resection and NHEJ. Consequently, a functional MMR machinery suppresses expansions of (GAA)n repeats specifically in non-dividing yeast cells (Table 1) [55], [65], [149]. Inactivation of MutSβ and MutLα and, above all, Exo1 increased both the frequency and the size of (GAA)n repeat expansions in quiescent cells. Finally, expansions involved processive DNA synthesis by Pol δ, likely occurring during error-prone repair of nicks accumulated in the damage-susceptible single-stranded parts of H-DNA [150]. Whereas large-scale expansions occur in one step during replication [83], expansions in quiescent cells could result from multiple smaller-scale expansions [149].
In sum, MMR has been established as a major pathway modulating (GAA)n repeat stability, albeit with dramatic differences when it comes to the role of individual MMR components between different model systems. Note, also, that the effects of the MMR machinery on (GAA)n repeat instability are strikingly different from their role in (CAG)n repeat instability, as is comprehensively displayed in Table 1. In short, the MMR machinery binds but cannot excise long stable hairpins, further stabilizing those hairpins. Consequently, MMR promotes (CAG)n expansions in mice and humans [151–155]. The association between MMR and (GAA)n repeat instability clearly does not fit the same scheme. Furthermore, the effects of MMR on (GAA)n stability are clearly different in dividing and non-dividing cells, as evidenced by different effects of MutLα and MutSβ described above and in Table 1. The latter difference could result from different expression levels of MMR proteins as well as a different relationship between replication, transcription, and DNA repair.
6.2. Base excision repair in (GAA)n repeat instability
Base excision repair (BER) is a DNA damage repair pathway which processes lesions initiated by oxidative damage, alkylation and base deamination. In short, the modified base is removed by a DNA glycosylase and the resulting abasic site is cleaved by the AP endonuclease creating a nick in a DNA strand. DNA polymerase β carries out repair DNA synthesis, which is followed by flap removal by flap-endonucleases and ligation. BER has been proposed to be a major pathway leading to small-scale, age-dependent somatic expansions of (CAG)n repeats [171], [172]. Trinucleotide repeat stability is influenced by BER based on the site of DNA modifications within the repeat, the presence of additional proteins, and its balance with MMR processes [173].
Lai et al. showed that alkylation of (GAA)n repeats by the chemotherapeutic temozolomide results in BER-mediated (GAA)n repeat contractions both in vitro and in lymphoblasts of FRDA patients [174]. A structure-prone repetitive flap may compromise coordination between Pol β and flap cleavage by the flap endonuclease FEN1. BER was also proposed as a mechanism of repair of abasic sites within R-loops formed at (CAG)n repeat [158]. Laverde et al. conducted a biochemical study of BER activity on R-loop substrates containing a (GAA)20 repeat. The BER enzyme AP endonuclease 1 (APE1) was found to incise the abasic site in the (GAA)n R-loop, creating a double flap intermediate that hinders synthesis by Pol β while stimulating 5’-flap cleavage by FEN1. Cleavage by FEN1 promotes R-loop resolution and contractions of about half the repeat length [175]. Therefore, the data so far indicate that processing of lesions in (GAA)n repeats by BER primarily results in repeat contractions.
What remains to be determined is the direct contribution of individual oxidizing agents and lesions on (GAA)n repeat stability. The position of the lesion relative to the repeat could also be involved in determining whether repair will result in expansions or contractions, as is the case for (CAG)n repeats [176]. Oxidative damage is particularly relevant in the context of FRDA, as it is a prominent form of DNA damage in aging neurons and the frataxin protein itself is involved in the processing of oxidative damage [177], [178]. Mitochondrial dysfunction, accumulation of reactive oxygen species (ROS) and subsequent cell death has been proposed as a driving cause of FRDA [179]. ROS accumulation promotes elevated levels of oxidative damage in the cell. If oxidative damage repair modulates (GAA)n repeat instability in somatic cells, it could be one of the main drivers of age-dependent somatic instability.
6.3. (GAA)n repeats are hotspots of homologous recombination
(GAA)n repeats were shown to promote homologous recombination in bacterial and yeast experimental systems [55], [180], [181], a feature they share with other trinucleotide repeats [182]. However, there are sensitive differences between the two systems. In bacteria, the recombinogenic potential of (GAA)n repeats decreased with their length, which was attributed to the formation of sticky DNA by longer repeats (see Section 2). In yeast, in contrast, the repeat’s recombinogenic potential increased with its length. Furthermore, repeat-mediated recombination in bacteria, but not in yeast, occasionally led to length instability of the repeat itself. Finally, the data from the tetrad analysis in yeast indicated that repeat-mediated recombination occurred during the G1 phase of the cell cycle – it was replication-independent [180]. We want to emphasize that unlike (CAG)n repeats, (GAA)n repeat instability is not modulated by homologous recombination factors in yeast experimental systems, except for when the RNase H enzymes had been deleted, eliciting a BIR response [64], [82], [113], [182] (see Section 5).
7. Does FAN1 play a role in (GAA)n repeat instability?
Cells contain a variety of structure-specific nucleases, which process flaps and other structures during DNA replication, repair, and recombination. Whereas each nuclease optimally processes a specific substrate, it is possible they can mis-recognize unusual DNA structures, influencing their stability. Two such nucleases are FEN1 (Rad27 in S. cerevisiae) and the Fanconi-associated nuclease FAN1. FEN1 processes 5’ flaps of Okazaki fragments during lagging strand synthesis and DNA damage repair [183]. FAN1 is an interstrand cross-link (ICL) repair protein which exhibits 5’-to-3’ exonuclease activity as well as endonuclease activity, participates in HR and processing of stalled forks [184]. FEN1 and FAN1 belong to the same class of enzymes and have been shown to have overlapping substrates, suggesting they might have similar or redundant roles in the regulation of repeat instability.
In yeast, Rad27 (FEN1) prevents instability of both (GAA)n and (CAG)n repeats [64], [185–188], and multiple models propose that it does so through its flap equilibration abilities, which likely counteracts the formation of non-B DNA structures. In contrast, mammalian FEN1 does not seem to fully share this important role, as its depletion does not affect (GAA)n instability and has contrasting effects on (CAG)n stability, and has not been identified as a disease regulator [189–193].
What could explain this striking difference between organisms? It is possible that flap processing during Okazaki fragment synthesis might not be a major contributor to somatic instability overall. Alternatively, flap processing performed by other nucleases might be more important in human cells. Recently, FAN1 has emerged as a prominent candidate. Genome-wide association studies (GWAS) of Huntington’s disease have identified FAN1 as a strong genetic modifier of disease onset, with FAN1 mutations being associated with earlier onset [103], [194]. In addition, FAN1 prevents expansions of (CGG)n repeats in Fragile X syndrome mouse models and rare missense variants in FAN1 were associated with expanded (CGG)n repeats present in individuals with autism spectrum disorders [195–197].
Goold et al. propose that FAN1 acts through a nuclease-independent pathway to stabilize (CAG)n repeats and prevent expansions, possibly by recruiting DNA damage repair proteins to the repeat, promoting conservative repair [198]. Candidates for key FAN1 interactors are its physical interactors in the MutL family [199], [200]. FAN1 and MLH1 have been recently shown to have opposite but interdependent effects on (CAG)n repeat instability. FAN1 sequesters MLH1 through a *SPYF* motif, leaving it unable to promote (CAG)n expansions through its canonical MMR function [201–204] (Table 1). In these studies, the nuclease activity of FAN1 was needed to prevent expansions. Variants of FAN1 with reduced nuclease activity have been found in patients with particularly early HD onset and highlight the endo- and exonuclease activity of FAN1 in protecting against expansions [195], [205]. Therefore, it has become clear that FAN1 is a major regulator of (CAG)n repeat stability [206].
Since MLH1 activity has been shown to regulate (GAA)n repeat instability in yeast and mouse models as well as in cultured human cells, it is foreseeable that the FAN1-MLH1 interaction might influence the balance of (GAA)n repeat stability possibly by the processing of 5’ flaps generated by strand displacement during DNA synthesis. The latter role would more closely mirror the effects of Rad27 on (GAA)n repeat instability, as Rad27 does not interact with the MMR machinery in S. cerevisiae in otherwise unperturbed conditions [207]. Thus, we believe it is of great interest to study the role of FAN1 nuclease in (GAA)n repeat instability.
8. Therapeutic avenues targeting (GAA)n repeat stability
8.1. Gene editing and replacement therapies to modulate (GAA)n repeat size and stability
Friedreich’s ataxia is a predominantly monogenic disease. Therefore, removal of expanded (GAA)n repeats at the FXN locus via gene editing is a potentially promising approach to modulate and even prevent disease. Excision of the (GAA)n repeat and some of the flanking sequence with zinc-finger nucleases in FRDA derived cells can partially rescue defects in FXN expression and ameliorates pathological phenotypes in iPSC-derived neuronal cells [208]. More recently, CRISPR technology has been applied to the study of repeat expansion diseases [209–211]. Removal of expanded (GAA)n repeats in an FRDA mouse cell line containing one expanded (GAA)190 allele promoted partial transcriptional rescue of FXN gene expression, with an associated increase in protein levels [211]. The same result, though, was not observed in a cell line with two expanded alleles [211]. We envision that both length of the expanded allele and the relative size of the other allele can influence the success of CRISPR targeting.
Genome editing techniques to restore frataxin levels can be combined with cell replacement therapies to overcome an additional hurdle in therapy. Before the advent of CRISPR, same-species (allogeneic) transplant of healthy cells had been explored as a method to treat FRDA symptoms in mice [212]. The major hurdles of allogeneic transplantation are immunosuppression and graft rejection. The possibility of using the patient’s own cells, modifying their genome, and reintroducing them into the patient (autologous graft) is therefore much more attractive, as it circumvents the mentioned issues. Rocca et al. removed the expanded repeat from human FRDA fibroblasts and hematopoietic stem and progenitor cells (HPSC), reaching a substantial increase in frataxin protein levels and rescue of mitochondrial defects. HPSCs then underwent hematopoiesis but displayed reduced cell proliferation rates [210].
Can CRISPR succeed also in the context of a functional brain? FRDA-derived iPSCs and embryonic stem cells differentiated into neuronal derivatives are able to withstand grafting into rodent brains, survive and even mature into dorsal root ganglia (DRG) – the primary tissue affected by neurodegeneration in FRDA [213], [214]. Consequently, they constitute a model in which the utility of gene editing can be more reliably tested. FRDA-derived iPSCs have also been used to develop an in vitro 3D DRG organoid (DRGO) model [215]. Removal of the expanded (GAA)n repeat by CRISPR partially restored the frataxin protein levels and rescued FRDA-associated phenotypes, and the level of rescue was greater when shorter (GAA)n repeats were deleted. On the other hand, an almost complete deletion of the first FXN intron restored frataxin expression levels to approximately wildtype levels. The repressing chromatin markers associated with FXN transcription silencing in FRDA were permanently removed when the whole intron was removed, indicating that long (GAA)n repeats propagate chromatin silencing through its upstream and downstream regions [215]. Thus, removal of the expanded (GAA)n repeat alone might not be sufficient and will only work in association with the concomitant loss of repressive chromatin marks in its surroundings.
What are some of the caveats? First, it seems that the extent of the FXN region to be removed will have to be tailored to the starting (GAA)n repeat length of the individual patient, and different guides might be needed in each specific case. Secondly, off-target effects need to be carefully studied and minimized, and this will have to be tested for each target site. Third, whether the same approach can be applied to intergenerational instability remains to be determined. Finally, long-term studies are needed to test whether once the repeat has been shortened it remains stable over a long period of time, or whether it eventually becomes unstable again.
An alternative approach is gene addition, in which transgenic wild-type FXN is reintroduced into cells using viral vectors to rescue frataxin expression (reviewed in [216]). Since gene addition was successfully used for treating recessive genetic diseases in humans [217], [218], this approach has been broadly investigated for FRDA, using various mouse models, patient-derived fibroblasts and in non-human primates ([216] and references therein). The main caveats in the FRDA case, however, was toxicity upon delivery and failure to rescue frataxin expression in neurological tissues.
8.2. Oligonucleotide-based approaches
The availability of repeat-stabilizing agents, such as small molecules that stabilize H-DNA or promote contractions, is being investigated as a complementary approach for treating repeat expansion diseases [52], [219]. Since the protein coding sequence of frataxin remains unaltered in FRDA, upregulation of transcription and protein levels is a viable therapeutic avenue. Recently, the Napierala group has pioneered an oligonucleotide-based approach in which frataxin mRNA levels were stabilized resulting in increased frataxin protein levels in both FRDA fibroblasts and iPSC-derived neuronal progenitor cell lines [220]. Targeting the 5’ and 3’ untranslated regions of the FXN in combination led to a modest increase in mRNA half-life protein levels without altering the chromatin status of the FXN gene [220]. Since FRDA patients only have 5% to 35% of the control frataxin levels, it remains to be determined whether such an increase has the potential to alleviate frataxin-deficiency associated phenotypes [221], [222].
Oligonucleotides can also be used to directly try and prevent (GAA)n repeat expansions. The first support for this idea comes from the use of locked nucleic acids (LNA), which have been shown to be able to interfere with triplex DNA formation [223]. LNA-DNA mixmers, which are not toxic for human cells, nearly completely prevented large-scale expansions of (GAA)n repeats in human cells [224]. This approach directly inhibits repeat expansions and is therefore very promising, albeit their effectiveness in FRDA patients remains to be determined. One of the major challenges regarding patient treatment with these oligonucleotides, as well as other promising small molecules [225–227], is the inefficiency of drug delivery to the central nervous system. While delivery to other affected tissues such as heart or pancreas can be achieved with viral vectors, the current ways of delivering drugs to the central nervous system are very invasive [228]. We hope that a better understanding of the mechanism of (GAA)n repeat instability and FXN expression during human development would lead to defining the most effective spatiotemporal windows for long-lasting treatment.
Acknowledgements:
We thank Victoria Brown, Tyler Maclay and Julia Hisey for helpful discussion and proofreading the manuscript. We are also grateful to the anonymous reviewers for their insightful comments and suggestions.
Funding:
This work was supported by National Institute of General Medical Sciences grant R35GM130322 to S.M.M.
List of Abbreviations:
- CGR
Complex Genome Rearrangement
- FRDA
Friedreich’s ataxia
- hPu/hPy
Homopurine/homopyrimidine DNA run
- LNA
locked nucleic acid
- RED
Repeat Expansion Disease
- RIM
Repeat-induced Mutagenesis
- TNR
Trinucleotide Repeat
- TRC
transcription-replication collisions
Footnotes
Conflict of Interest: The authors declare that there are no conflicts of interest.
References
- [1].Gymrek M, “A genomic view of short tandem repeats,” Current Opinion in Genetics & Development, vol. 44, pp. 9–16, 2017, doi: 10.1016/j.gde.2017.01.012. [DOI] [PubMed] [Google Scholar]
- [2].Lander ES et al. , “Initial sequencing and analysis of the human genome,” Nature, vol. 409, no. 6822, Art. no. 6822, Feb. 2001, doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- [3].Subramanian S, Mishra RK, and Singh L, “Genome-wide analysis of microsatellite repeats in humans: their abundance and density in specific genomic regions,” Genome Biol., vol. 4, no. 2, p. R13, 2003, doi: 10.1186/gb-2003-4-2-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Depienne C and Mandel J-L, “30 years of repeat expansion disorders: What have we learned and what are the remaining challenges?,” The American Journal of Human Genetics, vol. 108, no. 5, pp. 764–785, Maggio 2021, doi: 10.1016/j.ajhg.2021.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Khristich AN and Mirkin SM, “On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability,” J. Biol. Chem, p. jbc.REV119.007678, Feb. 2020, doi: 10.1074/jbc.REV119.007678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Spada ARL, Wilson EM, Lubahn DB, Harding AE, and Fischbeck KH, “Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy,” Nature, vol. 352, no. 6330, Art. no. 6330, Jul. 1991, doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- [7].Kiliszek A and Rypniewski W, “Structural studies of CNG repeats,” Nucleic Acids Research, vol. 42, no. 13, pp. 8189–8199, Jul. 2014, doi: 10.1093/nar/gku536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Petruska J, Hartenstine MJ, and Goodman MF, “Analysis of strand slippage in DNA polymerase expansions of CAG/CTG triplet repeats associated with neurodegenerative disease,” J Biol Chem, vol. 273, no. 9, pp. 5204–5210, Feb. 1998, doi: 10.1074/jbc.273.9.5204. [DOI] [PubMed] [Google Scholar]
- [9].Campuzano V et al. , “Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion,” Science, vol. 271, no. 5254, pp. 1423–1427, Mar. 1996, doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- [10].Clark RM, Dalgliesh GL, Endres D, Gomez M, Taylor J, and Bidichandani SI, “Expansion of GAA triplet repeats in the human genome: unique origin of the FRDA mutation at the center of an Alu,” Genomics, vol. 83, no. 3, pp. 373–383, Mar. 2004, doi: 10.1016/j.ygeno.2003.09.001. [DOI] [PubMed] [Google Scholar]
- [11].Chauhan C, Dash D, Grover D, Rajamani J, and Mukerji M, “Origin and Instability of GAA Repeats: Insights from Alu Elements,” Journal of Biomolecular Structure and Dynamics, vol. 20, no. 2, pp. 253–263, Oct. 2002, doi: 10.1080/07391102.2002.10506841. [DOI] [PubMed] [Google Scholar]
- [12].Vankan P, “Prevalence gradients of Friedreich’s Ataxia and R1b haplotype in Europe co-localize, suggesting a common Palaeolithic origin in the Franco-Cantabrian ice age refuge,” Journal of Neurochemistry, vol. 126, no. s1, pp. 11–20, 2013, doi: 10.1111/jnc.12215. [DOI] [PubMed] [Google Scholar]
- [13].Al-Mahdawi S et al. , “Large Interruptions of GAA Repeat Expansion Mutations in Friedreich Ataxia Are Very Rare,” Frontiers in Cellular Neuroscience, vol. 12, p. 443, 2018, doi: 10.3389/fncel.2018.00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Filla A et al. , “The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia.,” Am J Hum Genet, vol. 59, no. 3, pp. 554–560, Sep. 1996. [PMC free article] [PubMed] [Google Scholar]
- [15].Montermini L et al. , “The Friedreich ataxia GAA triplet repeat: premutation and normal alleles,” Hum Mol Genet, vol. 6, no. 8, pp. 1261–1266, Aug. 1997, doi: 10.1093/hmg/6.8.1261. [DOI] [PubMed] [Google Scholar]
- [16].La Pean A, Jeffries N, Grow C, Ravina B, and Di Prospero NA, “Predictors of progression in patients with Friedreich ataxia,” Mov Disord, vol. 23, no. 14, pp. 2026–2032, Oct. 2008, doi: 10.1002/mds.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sharma R, De Biase I, Gómez M, Delatycki MB, Ashizawa T, and Bidichandani SI, “Friedreich ataxia in carriers of unstable borderline GAA triplet-repeat alleles: FRDA Unstable Borderline Alleles,” Ann Neurol., vol. 56, no. 6, pp. 898–901, Dec. 2004, doi: 10.1002/ana.20333. [DOI] [PubMed] [Google Scholar]
- [18].Campuzano V et al. , “Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes,” Hum Mol Genet, vol. 6, no. 11, pp. 1771–1780, Oct. 1997, doi: 10.1093/hmg/6.11.1771. [DOI] [PubMed] [Google Scholar]
- [19].Punga T and Bühler M, “Long intronic GAA repeats causing Friedreich ataxia impede transcription elongation,” EMBO Mol Med, vol. 2, no. 4, pp. 120–129, Apr. 2010, doi: 10.1002/emmm.201000064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rodden LN et al. , “Methylated and unmethylated epialleles support variegated epigenetic silencing in Friedreich ataxia,” Hum Mol Genet, vol. 29, no. 23, pp. 3818–3829, Feb. 2021, doi: 10.1093/hmg/ddaa267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Soragni E, Herman D, Dent SYR, Gottesfeld JM, Wells RD, and Napierala M, “Long intronic GAA•TTC repeats induce epigenetic changes and reporter gene silencing in a molecular model of Friedreich ataxia,” Nucleic Acids Res, vol. 36, no. 19, pp. 6056–6065, Nov. 2008, doi: 10.1093/nar/gkn604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li Y et al. , “Expanded GAA repeats impede transcription elongation through the FXN gene and induce transcriptional silencing that is restricted to the FXN locus,” Hum Mol Genet, vol. 24, no. 24, pp. 6932–6943, Dec. 2015, doi: 10.1093/hmg/ddv397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].González-Cabo P and Palau F, “Mitochondrial pathophysiology in Friedreich’s ataxia,” Journal of Neurochemistry, vol. 126, no. s1, pp. 53–64, 2013, doi: 10.1111/jnc.12303. [DOI] [PubMed] [Google Scholar]
- [24].Koeppen AH et al. , “The dorsal root ganglion in Friedreich’s ataxia,” Acta Neuropathol, vol. 118, no. 6, pp. 763–776, Dec. 2009, doi: 10.1007/s00401-009-0589-x. [DOI] [PubMed] [Google Scholar]
- [25].Koeppen AH, Davis AN, and Morral JA, “The cerebellar component of Friedreich’s ataxia,” Acta Neuropathol, vol. 122, no. 3, pp. 323–330, Sep. 2011, doi: 10.1007/s00401-011-0844-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cook A and Giunti P, “Friedreich’s ataxia: clinical features, pathogenesis and management,” Br Med Bull, vol. 124, no. 1, pp. 19–30, Dec. 2017, doi: 10.1093/bmb/ldx034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zesiewicz TA, Hancock J, Ghanekar SD, Kuo S-H, Dohse CA, and Vega J, “Emerging therapies in Friedreich’s Ataxia,” Expert Review of Neurotherapeutics, vol. 20, no. 12, pp. 1215–1228, Dicembre 2020, doi: 10.1080/14737175.2020.1821654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mirkin SM, Lyamichev VI, Drushlyak KN, Dobrynin VN, Filippov SA, and Frank-Kamenetskii MD, “DNA H form requires a homopurine–homopyrimidine mirror repeat,” Nature, vol. 330, no. 6147, Art. no. 6147, Dec. 1987, doi: 10.1038/330495a0. [DOI] [PubMed] [Google Scholar]
- [29].Mirkin SM and Frank-Kamenetskii MD, “H-DNA and Related Structures,” Annu. Rev. Biophys. Biomol. Struct, vol. 23, no. 1, pp. 541–576, Giugno 1994, doi: 10.1146/annurev.bb.23.060194.002545. [DOI] [PubMed] [Google Scholar]
- [30].Paleček E, “Local Supercoil-Stabilized DNA Structure,” Critical Reviews in Biochemistry and Molecular Biology, vol. 26, no. 2, pp. 151–226, Jan. 1991, doi: 10.3109/10409239109081126. [DOI] [PubMed] [Google Scholar]
- [31].Potaman VN et al. , “Length-dependent structure formation in Friedreich ataxia (GAA)n·(TTC)n repeats at neutral pH,” Nucleic Acids Res, vol. 32, no. 3, pp. 1224–1231, 2004, doi: 10.1093/nar/gkh274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Frank-Kamenetskii MD and Mirkin SM, “Triplex Dna Structures,” Annual Review of Biochemistry, vol. 64, no. 1, pp. 65–95, 1995, doi: 10.1146/annurev.bi.64.070195.000433. [DOI] [PubMed] [Google Scholar]
- [33].Bergquist H et al. , “Structure-specific recognition of Friedreich’s ataxia (GAA)n repeats by benzoquinoquinoxaline derivatives,” Chembiochem, vol. 10, no. 16, pp. 2629–2637, Nov. 2009, doi: 10.1002/cbic.200900263. [DOI] [PubMed] [Google Scholar]
- [34].Sakamoto N et al. , “Sticky DNA: Self-Association Properties of Long GAA·TTC Repeats in R·R·Y Triplex Structures from Friedreich’s Ataxia,” Molecular Cell, vol. 3, no. 4, pp. 465–475, Apr. 1999, doi: 10.1016/S1097-2765(00)80474-8. [DOI] [PubMed] [Google Scholar]
- [35].Faucon B, Mergny JL, and Héléne C, “Effect of third strand composition on the triple helix formation: purine versus pyrimidine oligodeoxynucleotides,” Nucleic Acids Res, vol. 24, no. 16, pp. 3181–3188, Aug. 1996, doi: 10.1093/nar/24.16.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sakamoto N, Larson JE, Iyer RR, Montermini L, Pandolfo M, and Wells RD, “GGA·TCC-interrupted Triplets in Long GAA·TTC Repeats Inhibit the Formation of Triplex and Sticky DNA Structures, Alleviate Transcription Inhibition, and Reduce Genetic Instabilities,” Journal of Biological Chemistry, vol. 276, no. 29, pp. 27178–27187, Jul. 2001, doi: 10.1074/jbc.M101852200. [DOI] [PubMed] [Google Scholar]
- [37].Son LS, Bacolla A, and Wells RD, “Sticky DNA: in vivo formation in E. coli and in vitro association of long GAA*TTC tracts to generate two independent supercoiled domains,” J Mol Biol, vol. 360, no. 2, pp. 267–284, Jul. 2006, doi: 10.1016/j.jmb.2006.05.025. [DOI] [PubMed] [Google Scholar]
- [38].Poggi L and Richard G-F, “Alternative DNA Structures In Vivo: Molecular Evidence and Remaining Questions,” Microbiol. Mol. Biol. Rev, vol. 85, no. 1, Feb. 2021, doi: 10.1128/MMBR.00110-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Agazie YM, Lee JS, and Burkholder GD, “Characterization of a new monoclonal antibody to triplex DNA and immunofluorescent staining of mammalian chromosomes.,” Journal of Biological Chemistry, vol. 269, no. 9, pp. 7019–7023, Mar. 1994, doi: 10.1016/S0021-9258(17)37476-8. [DOI] [PubMed] [Google Scholar]
- [40].Agazie YM, Burkholder GD, and Lee JS, “Triplex DNA in the nucleus: direct binding of triplex-specific antibodies and their effect on transcription, replication and cell growth.,” Biochem J, vol. 316, no. Pt 2, pp. 461–466, Jun. 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ohno M, Fukagawa T, Lee JS, and Ikemura T, “Triplex-forming DNAs in the human interphase nucleus visualized in situ by polypurine/polypyrimidine DNA probes and antitriplex antibodies,” Chromosoma, vol. 111, no. 3, pp. 201–213, Sep. 2002, doi: 10.1007/s00412-002-0198-0. [DOI] [PubMed] [Google Scholar]
- [42].Ma J and Wang MD, “DNA supercoiling during transcription,” Biophys Rev, vol. 8, no. Suppl 1, pp. 75–87, Jul. 2016, doi: 10.1007/s12551-016-0215-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kouzine F et al. , “Permanganate/S1 Nuclease Footprinting Reveals Non-B DNA Structures with Regulatory Potential across a Mammalian Genome,” Cell Syst, vol. 4, no. 3, pp. 344–356.e7, Mar. 2017, doi: 10.1016/j.cels.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wu W et al. , “Neuronal enhancers are hotspots for DNA single-strand break repair,” Nature, vol. 593, no. 7859, pp. 440–444, May 2021, doi: 10.1038/s41586-021-03468-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Maekawa K, Yamada S, Sharma R, Chaudhuri J, and Keeney S, “Triple-helix potential of the mouse genome,” Proceedings of the National Academy of Sciences, vol. 119, no. 19, p. e2203967119, May 2022, doi: 10.1073/pnas.2203967119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Matos-Rodrigues G et al. , “Linking Dynamic DNA Secondary Structures to Genome Instability,” p. 43. [Google Scholar]
- [47].Samadashwily GM, Raca G, and Mirkin SM, “Trinucleotide repeats affect DNA replication in vivo,” Nat Genet, vol. 17, no. 3, pp. 298–304, Nov. 1997, doi: 10.1038/ng1197-298. [DOI] [PubMed] [Google Scholar]
- [48].Gacy AM et al. , “GAA instability in Friedreich’s Ataxia shares a common, DNA-directed and intraallelic mechanism with other trinucleotide diseases,” Mol Cell, vol. 1, no. 4, pp. 583–593, Mar. 1998, doi: 10.1016/s1097-2765(00)80058-1. [DOI] [PubMed] [Google Scholar]
- [49].Casas-Delucchi CS, Daza-Martin M, Williams SL, and Coster G, “The mechanism of replication stalling and recovery within repetitive DNA,” Nat Commun, vol. 13, no. 1, Art. no. 1, Jul. 2022, doi: 10.1038/s41467-022-31657-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chandok GS, Patel MP, Mirkin SM, and Krasilnikova MM, “Effects of Friedreich’s ataxia GAA repeats on DNA replication in mammalian cells,” Nucleic Acids Res, vol. 40, no. 9, pp. 3964–3974, May 2012, doi: 10.1093/nar/gks021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Follonier C, Oehler J, Herrador R, and Lopes M, “Friedreich’s ataxia–associated GAA repeats induce replication-fork reversal and unusual molecular junctions,” Nat Struct Mol Biol, vol. 20, no. 4, pp. 486–494, Apr. 2013, doi: 10.1038/nsmb.2520. [DOI] [PubMed] [Google Scholar]
- [52].Gerhardt J et al. , “Stalled DNA Replication Forks at the Endogenous GAA Repeats Drive Repeat Expansion in Friedreich’s Ataxia Cells,” Cell Rep, vol. 16, no. 5, pp. 1218–1227, Aug. 2016, doi: 10.1016/j.celrep.2016.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Krasilnikova MM and Mirkin SM, “Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo,” Mol Cell Biol, vol. 24, no. 6, pp. 2286–2295, Mar. 2004, doi: 10.1128/mcb.24.6.2286-2295.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ohshima K, Montermini L, Wells RD, and Pandolfo M, “Inhibitory effects of expanded GAA.TTC triplet repeats from intron I of the Friedreich ataxia gene on transcription and replication in vivo,” J Biol Chem, vol. 273, no. 23, pp. 14588–14595, Jun. 1998, doi: 10.1074/jbc.273.23.14588. [DOI] [PubMed] [Google Scholar]
- [55].Kim H-M et al. , “Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair,” EMBO J, vol. 27, no. 21, pp. 2896–2906, Nov. 2008, doi: 10.1038/emboj.2008.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rastokina A, Cebrián J, Mandel N, Zain R, Lopes M, and Mirkin SM, “Large-scale expansions and replication stalling of Friedreich’s ataxia GAA repeats in an experimental mammalian system.” bioRxiv, p. 2022.07.04.498737, Jul. 05, 2022. doi: 10.1101/2022.07.04.498737. [DOI] [Google Scholar]
- [57].Fanning E and Zhao K, “SV40 DNA replication: From the A gene to a nanomachine,” Virology, vol. 384, no. 2, pp. 352–359, Feb. 2009, doi: 10.1016/j.virol.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kumari D, Hayward B, Nakamura AJ, Bonner WM, and Usdin K, “Evidence for chromosome fragility at the frataxin locus in Friedreich ataxia,” Mutat Res, vol. 781, pp. 14–21, Nov. 2015, doi: 10.1016/j.mrfmmm.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Cheloshkina K and Poptsova M, “Comprehensive analysis of cancer breakpoints reveals signatures of genetic and epigenetic contribution to cancer genome rearrangements,” PLoS Comput Biol, vol. 17, no. 3, p. e1008749, Mar. 2021, doi: 10.1371/journal.pcbi.1008749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Georgakopoulos-Soares I, Morganella S, Jain N, Hemberg M, and Nik-Zainal S, “Noncanonical secondary structures arising from non-B DNA motifs are determinants of mutagenesis,” Genome Res., vol. 28, no. 9, pp. 1264–1271, Sep. 2018, doi: 10.1101/gr.231688.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lyu R et al. , “KAS-seq: genome-wide sequencing of single-stranded DNA by N3-kethoxal–assisted labeling,” Nat Protoc, vol. 17, no. 2, Art. no. 2, Feb. 2022, doi: 10.1038/s41596-021-00647-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wu T, Lyu R, You Q, and He C, “Kethoxal-assisted single-stranded DNA sequencing captures global transcription dynamics and enhancer activity in situ,” Nat Methods, vol. 17, no. 5, pp. 515–523, May 2020, doi: 10.1038/s41592-020-0797-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Nethisinghe S et al. , “Interruptions of the FXN GAA Repeat Tract Delay the Age at Onset of Friedreich’s Ataxia in a Location Dependent Manner,” Int J Mol Sci, vol. 22, no. 14, p. 7507, Jul. 2021, doi: 10.3390/ijms22147507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Khristich AN, Armenia JF, Matera RM, Kolchinski AA, and Mirkin SM, “Large-scale contractions of Friedreich’s ataxia GAA repeats in yeast occur during DNA replication due to their triplex-forming ability,” Proc Natl Acad Sci USA, vol. 117, no. 3, pp. 1628–1637, Jan. 2020, doi: 10.1073/pnas.1913416117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Shishkin AA et al. , “Large-Scale Expansions of Friedreich’s Ataxia GAA Repeats in Yeast,” Molecular Cell, vol. 35, no. 1, pp. 82–92, Jul. 2009, doi: 10.1016/j.molcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].De Biase I et al. , “Somatic instability of the expanded GAA triplet-repeat sequence in Friedreich ataxia progresses throughout life,” Genomics, vol. 90, no. 1, pp. 1–5, Jul. 2007, doi: 10.1016/j.ygeno.2007.04.001. [DOI] [PubMed] [Google Scholar]
- [67].De Michele G et al. , “Parental gender, age at birth and expansion length influence GAA repeat intergenerational instability in the X25 gene: pedigree studies and analysis of sperm from patients with Friedreich’s Ataxia,” Human Molecular Genetics, vol. 7, no. 12, pp. 1901–1906, Nov. 1998, doi: 10.1093/hmg/7.12.1901. [DOI] [PubMed] [Google Scholar]
- [68].McMurray CT, “Mechanisms of trinucleotide repeat instability during human development,” Nat Rev Genet, vol. 11, no. 11, pp. 786–799, Nov. 2010, doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Pollard LM et al. , “Replication-mediated instability of the GAA triplet repeat mutation in Friedreich ataxia,” Nucleic Acids Research, vol. 32, no. 19, pp. 5962–5971, Ottobre 2004, doi: 10.1093/nar/gkh933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sharma R et al. , “The GAA triplet-repeat sequence in Friedreich ataxia shows a high level of somatic instability in vivo, with a significant predilection for large contractions,” Hum Mol Genet, vol. 11, no. 18, pp. 2175–2187, Sep. 2002, doi: 10.1093/hmg/11.18.2175. [DOI] [PubMed] [Google Scholar]
- [71].Long A et al. , “Somatic instability of the expanded GAA repeats in Friedreich’s ataxia,” PLoS One, vol. 12, no. 12, p. e0189990, 2017, doi: 10.1371/journal.pone.0189990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Montermini L, Kish SJ, Jiralerspong S, Lamarche JB, and Pandolfo M, “Somatic mosaicism for Friedreich’s ataxia GAA triplet repeat expansions in the central nervous system,” Neurology, vol. 49, no. 2, pp. 606–610, Aug. 1997, doi: 10.1212/wnl.49.2.606. [DOI] [PubMed] [Google Scholar]
- [73].Hellenbroich Y, Schwinger E, and Zühlke C, “Limited somatic mosaicism for Friedreich’s ataxia GAA triplet repeat expansions identified by small pool PCR in blood leukocytes,” Acta Neurol Scand, vol. 103, no. 3, pp. 188–192, Mar. 2001, doi: 10.1034/j.1600-0404.2001.103003188.x. [DOI] [PubMed] [Google Scholar]
- [74].Machkhas H, Bidichandani SI, Patel PI, and Harati Y, “A mild case of Friedreich ataxia: lymphocyte and sural nerve analysis for GAA repeat length reveals somatic mosaicism,” Muscle Nerve, vol. 21, no. 3, pp. 390–393, Mar. 1998, doi: 10.1002/(sici)1097-4598(199803)21:3<390::aid-mus13>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- [75].Shah KA and Mirkin SM, “The hidden side of unstable DNA repeats: Mutagenesis at a distance,” DNA Repair, vol. 32, pp. 106–112, Aug. 2015, doi: 10.1016/j.dnarep.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bidichandani SI et al. , “Somatic Sequence Variation at the Friedreich Ataxia Locus Includes Complete Contraction of the Expanded GAA Triplet Repeat, Significant Length Variation in Serially Passaged Lymphoblasts and Enhanced Mutagenesis in the Flanking Sequence,” Human Molecular Genetics, vol. 8, no. 13, pp. 2425–2436, Dicembre 1999, doi: 10.1093/hmg/8.13.2425. [DOI] [PubMed] [Google Scholar]
- [77].Wang G and Vasquez KM, “Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells,” Proc Natl Acad Sci U S A, vol. 101, no. 37, pp. 13448–13453, Sep. 2004, doi: 10.1073/pnas.0405116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wojciechowska M, Napierala M, Larson JE, and Wells RD, “Non-B DNA Conformations Formed by Long Repeating Tracts of Myotonic Dystrophy Type 1, Myotonic Dystrophy Type 2, and Friedreich’s Ataxia Genes, Not the Sequences per se, Promote Mutagenesis in Flanking Regions *,” Journal of Biological Chemistry, vol. 281, no. 34, pp. 24531–24543, Aug. 2006, doi: 10.1074/jbc.M603888200. [DOI] [PubMed] [Google Scholar]
- [79].Tang W, Dominska M, Gawel M, Greenwell PW, and Petes TD, “Genomic deletions and point mutations induced in Saccharomyces cerevisiae by the trinucleotide repeats (GAA·TTC) associated with Friedreich’s ataxia,” DNA Repair, vol. 12, no. 1, pp. 10–17, Jan. 2013, doi: 10.1016/j.dnarep.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhao J et al. , “Distinct Mechanisms of Nuclease-Directed DNA-Structure-Induced Genetic Instability in Cancer Genomes,” Cell Rep, vol. 22, no. 5, pp. 1200–1210, Jan. 2018, doi: 10.1016/j.celrep.2018.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wang G and Vasquez KM, “Non-B DNA structure-induced genetic instability,” Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, vol. 598, no. 1, pp. 103–119, Jun. 2006, doi: 10.1016/j.mrfmmm.2006.01.019. [DOI] [PubMed] [Google Scholar]
- [82].Zhang Y et al. , “Genome-wide Screen Identifies Pathways that Govern GAA/TTC Repeat Fragility and Expansions in Dividing and Nondividing Yeast Cells,” Molecular Cell, vol. 48, no. 2, pp. 254–265, Oct. 2012, doi: 10.1016/j.molcel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Shah KA, Shishkin AA, Voineagu I, Pavlov YI, Shcherbakova PV, and Mirkin SM, “Role of DNA Polymerases in Repeat-Mediated Genome Instability,” Cell Reports, vol. 2, no. 5, pp. 1088–1095, Nov. 2012, doi: 10.1016/j.celrep.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Grabowska E et al. , “Proper functioning of the GINS complex is important for the fidelity of DNA replication in yeast,” Mol Microbiol, vol. 92, no. 4, pp. 659–680, May 2014, doi: 10.1111/mmi.12580. [DOI] [PubMed] [Google Scholar]
- [85].Denkiewicz-Kruk M et al. , “Recombination and Pol ζ Rescue Defective DNA Replication upon Impaired CMG Helicase—Pol ε Interaction,” International Journal of Molecular Sciences, vol. 21, no. 24, Art. no. 24, Jan. 2020, doi: 10.3390/ijms21249484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Neil AJ, Kim JC, and Mirkin SM, “Precarious maintenance of simple DNA repeats in eukaryotes,” Bioessays, vol. 39, no. 9, Sep. 2017, doi: 10.1002/bies.201700077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Mirkin SM and Smirnova EV, “Positioned to expand,” Nat Genet, vol. 31, no. 1, Art. no. 1, May 2002, doi: 10.1038/ng0502-5. [DOI] [PubMed] [Google Scholar]
- [88].Spivakovsky-Gonzalez E et al. , “Rad9-mediated checkpoint activation is responsible for elevated expansions of GAA repeats in CST-deficient yeast,” Genetics, vol. 219, no. 2, Oct. 2021, doi: 10.1093/genetics/iyab125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lahiri M, Gustafson TL, Majors ER, and Freudenreich CH, “Expanded CAG Repeats Activate the DNA Damage Checkpoint Pathway,” Molecular Cell, vol. 15, no. 2, pp. 287–293, Jul. 2004, doi: 10.1016/j.molcel.2004.06.034. [DOI] [PubMed] [Google Scholar]
- [90].Guo M et al. , “A distinct triplex DNA unwinding activity of ChlR1 helicase,” J Biol Chem, vol. 290, no. 8, pp. 5174–5189, Feb. 2015, doi: 10.1074/jbc.M114.634923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].McGinty RJ et al. , “Nanopore sequencing of complex genomic rearrangements in yeast reveals mechanisms of repeat-mediated double-strand break repair,” Genome Res., vol. 27, no. 12, pp. 2072–2082, Dec. 2017, doi: 10.1101/gr.228148.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Chiu R, Rajan-Babu I-S, Friedman JM, and Birol I, “Straglr: discovering and genotyping tandem repeat expansions using whole genome long-read sequences,” Genome Biology, vol. 22, no. 1, p. 224, Agosto 2021, doi: 10.1186/s13059-021-02447-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].De Roeck A et al. , “NanoSatellite: accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION,” Genome Biol, vol. 20, no. 1, p. 239, Nov. 2019, doi: 10.1186/s13059-019-1856-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Dolzhenko E et al. , “Detection of long repeat expansions from PCR-free whole-genome sequence data,” Genome Res., vol. 27, no. 11, pp. 1895–1903, Nov. 2017, doi: 10.1101/gr.225672.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Dolzhenko E et al. , “ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions,” Bioinformatics, vol. 35, no. 22, pp. 4754–4756, Nov. 2019, doi: 10.1093/bioinformatics/btz431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Liu Q, Zhang P, Wang D, Gu W, and Wang K, “Interrogating the ‘unsequenceable’ genomic trinucleotide repeat disorders by long-read sequencing,” Genome Medicine, vol. 9, no. 1, p. 65, Luglio 2017, doi: 10.1186/s13073-017-0456-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Liu Q, Tong Y, and Wang K, “Genome-wide detection of short tandem repeat expansions by long-read sequencing,” BMC Bioinformatics, vol. 21, no. 21, p. 542, Dicembre 2020, doi: 10.1186/s12859-020-03876-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Mantere T, Kersten S, and Hoischen A, “Long-Read Sequencing Emerging in Medical Genetics,” Front. Genet, vol. 0, 2019, doi: 10.3389/fgene.2019.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Boemo MA, “DNAscent v2: detecting replication forks in nanopore sequencing data with deep learning,” BMC Genomics, vol. 22, no. 1, p. 430, Giugno 2021, doi: 10.1186/s12864-021-07736-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Müller CA et al. , “Capturing the dynamics of genome replication on individual ultra-long nanopore sequence reads,” Nature Methods, vol. 16, no. 5, Art. no. 5, May 2019, doi: 10.1038/s41592-019-0394-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Ciosi M et al. , “A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes,” EBioMedicine, vol. 48, pp. 568–580, Oct. 2019, doi: 10.1016/j.ebiom.2019.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium, “Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease,” Cell, vol. 162, no. 3, pp. 516–526, Jul. 2015, doi: 10.1016/j.cell.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. and Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium, “CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset,” Cell, vol. 178, no. 4, pp. 887–900.e14, Aug. 2019, doi: 10.1016/j.cell.2019.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Saini N et al. , “Fragile DNA Motifs Trigger Mutagenesis at Distant Chromosomal Loci in Saccharomyces cerevisiae,” PLOS Genetics, vol. 9, no. 6, p. e1003551, giu 2013, doi: 10.1371/journal.pgen.1003551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Krasilnikova MM, Kireeva ML, Petrovic V, Knijnikova N, Kashlev M, and Mirkin SM, “Effects of Friedreich’s ataxia (GAA)n*(TTC)n repeats on RNA synthesis and stability,” Nucleic Acids Res, vol. 35, no. 4, pp. 1075–1084, 2007, doi: 10.1093/nar/gkl1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Bidichandani SI, Ashizawa T, and Patel PI, “The GAA Triplet-Repeat Expansion in Friedreich Ataxia Interferes with Transcription and May Be Associated with an Unusual DNA Structure,” The American Journal of Human Genetics, vol. 62, no. 1, pp. 111–121, Gennaio 1998, doi: 10.1086/301680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Reddy K et al. , “Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats,” Nucleic Acids Res, vol. 39, no. 5, pp. 1749–1762, Mar. 2011, doi: 10.1093/nar/gkq935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Zhang J, Fakharzadeh A, Pan F, Roland C, and Sagui C, “Atypical structures of GAA/TTC trinucleotide repeats underlying Friedreich’s ataxia: DNA triplexes and RNA/DNA hybrids,” Nucleic Acids Res, vol. 48, no. 17, pp. 9899–9917, Sep. 2020, doi: 10.1093/nar/gkaa665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Zeng C, Onoguchi M, and Hamada M, “Association analysis of repetitive elements and R-loop formation across species,” Mobile DNA, vol. 12, no. 1, p. 3, Gennaio 2021, doi: 10.1186/s13100-021-00231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Li H, Long C, Hong Y, Chao L, Peng Y, and Zuo Y, “The Cumulative Formation of R-loop Interacts with Histone Modifications to Shape Cell Reprogramming,” International Journal of Molecular Sciences, vol. 23, no. 3, Art. no. 3, Jan. 2022, doi: 10.3390/ijms23031567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Freudenreich CH, “R-loops: targets for nuclease cleavage and repeat instability,” Curr Genet, vol. 64, no. 4, pp. 789–794, Aug. 2018, doi: 10.1007/s00294-018-0806-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].García-Muse T and Aguilera A, “R Loops: From Physiological to Pathological Roles,” Cell, vol. 179, no. 3, pp. 604–618, Oct. 2019, doi: 10.1016/j.cell.2019.08.055. [DOI] [PubMed] [Google Scholar]
- [113].Neil AJ, Liang MU, Khristich AN, Shah KA, and Mirkin SM, “RNA-DNA hybrids promote the expansion of Friedreich’s ataxia (GAA)n repeats via break-induced replication,” Nucleic Acids Res, vol. 46, no. 7, pp. 3487–3497, Apr. 2018, doi: 10.1093/nar/gky099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Bourn RL et al. , “Pms2 suppresses large expansions of the (GAA·TTC)n sequence in neuronal tissues,” PLoS One, vol. 7, no. 10, p. e47085, 2012, doi: 10.1371/journal.pone.0047085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Halabi A, Ditch S, Wang J, and Grabczyk E, “DNA Mismatch Repair Complex MutSβ Promotes GAA·TTC Repeat Expansion in Human Cells,” J Biol Chem, vol. 287, no. 35, pp. 29958–29967, Aug. 2012, doi: 10.1074/jbc.M112.356758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Halabi A, Fuselier KTB, and Grabczyk E, “GAA•TTC repeat expansion in human cells is mediated by mismatch repair complex MutLγ and depends upon the endonuclease domain in MLH3 isoform one,” Nucleic Acids Res, vol. 46, no. 8, pp. 4022–4032, May 2018, doi: 10.1093/nar/gky143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Groh M, Lufino MMP, Wade-Martins R, and Gromak N, “R-loops Associated with Triplet Repeat Expansions Promote Gene Silencing in Friedreich Ataxia and Fragile X Syndrome,” PLOS Genetics, vol. 10, no. 5, p. e1004318, mag 2014, doi: 10.1371/journal.pgen.1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Belotserkovskii BP, Neil AJ, Saleh SS, Shin JHS, Mirkin SM, and Hanawalt PC, “Transcription blockage by homopurine DNA sequences: role of sequence composition and single-strand breaks,” Nucleic Acids Research, vol. 41, no. 3, pp. 1817–1828, Feb. 2013, doi: 10.1093/nar/gks1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Belotserkovskii BP, Liu R, Tornaletti S, Krasilnikova MM, Mirkin SM, and Hanawalt PC, “Mechanisms and implications of transcription blockage by guanine-rich DNA sequences,” Proc Natl Acad Sci U S A, vol. 107, no. 29, pp. 12816–12821, Jul. 2010, doi: 10.1073/pnas.1007580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Ditch S, Sammarco MC, Banerjee A, and Grabczyk E, “Progressive GAA.TTC repeat expansion in human cell lines,” PLoS Genet, vol. 5, no. 10, p. e1000704, Oct. 2009, doi: 10.1371/journal.pgen.1000704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Shah KA, McGinty RJ, Egorova VI, and Mirkin SM, “Coupling transcriptional state to large-scale repeat expansions in yeast,” Cell Rep, vol. 9, no. 5, pp. 1594–1602, Dec. 2014, doi: 10.1016/j.celrep.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].McGinty RJ et al. , “A Defective mRNA Cleavage and Polyadenylation Complex Facilitates Expansions of Transcribed (GAA)n Repeats Associated with Friedreich’s Ataxia,” Cell Rep, vol. 20, no. 10, pp. 2490–2500, Sep. 2017, doi: 10.1016/j.celrep.2017.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Zardoni L et al. , “Elongating RNA polymerase II and RNA:DNA hybrids hinder fork progression and gene expression at sites of head-on replication-transcription collisions,” Nucleic Acids Res, vol. 49, no. 22, pp. 12769–12784, Dec. 2021, doi: 10.1093/nar/gkab1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Alzu A et al. , “Senataxin Associates with Replication Forks to Protect Fork Integrity across RNA-Polymerase-II-Transcribed Genes,” Cell, vol. 151, no. 4, pp. 835–846, Nov. 2012, doi: 10.1016/j.cell.2012.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Cohen S et al. , “Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations,” Nat Commun, vol. 9, no. 1, Art. no. 1, Feb. 2018, doi: 10.1038/s41467-018-02894-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Nakatani R, Nakamori M, Fujimura H, Mochizuki H, and Takahashi MP, “Large expansion of CTG•CAG repeats is exacerbated by MutSβ in human cells,” Sci Rep, vol. 5, no. 1, Art. no. 1, Jun. 2015, doi: 10.1038/srep11020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Appanah R, Lones EC, Aiello U, Libri D, and De Piccoli G, “Sen1 Is Recruited to Replication Forks via Ctf4 and Mrc1 and Promotes Genome Stability,” Cell Rep, vol. 30, no. 7, pp. 2094–2105.e9, Feb. 2020, doi: 10.1016/j.celrep.2020.01.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Al-Mahdawi S, Pinto RM, Ruddle P, Carroll C, Webster Z, and Pook M, “GAA repeat instability in Friedreich ataxia YAC transgenic mice,” Genomics, vol. 84, no. 2, pp. 301–310, Aug. 2004, doi: 10.1016/j.ygeno.2004.04.003. [DOI] [PubMed] [Google Scholar]
- [129].Anjomani Virmouni S, Sandi C, Al-Mahdawi S, and Pook MA, “Cellular, molecular and functional characterisation of YAC transgenic mouse models of Friedreich ataxia,” PLoS One, vol. 9, no. 9, p. e107416, 2014, doi: 10.1371/journal.pone.0107416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Anjomani Virmouni S et al. , “Identification of telomere dysfunction in Friedreich ataxia,” Mol Neurodegener, vol. 10, p. 22, Jun. 2015, doi: 10.1186/s13024-015-0019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Clark RM, De Biase I, Malykhina AP, Al-Mahdawi S, Pook M, and Bidichandani SI, “The GAA triplet-repeat is unstable in the context of the human FXN locus and displays age-dependent expansions in cerebellum and DRG in a transgenic mouse model,” Hum Genet, vol. 120, no. 5, pp. 633–640, Jan. 2007, doi: 10.1007/s00439-006-0249-3. [DOI] [PubMed] [Google Scholar]
- [132].De Biase I et al. , “Progressive GAA expansions in dorsal root ganglia of Friedreich’s ataxia patients,” Ann Neurol, vol. 61, no. 1, pp. 55–60, Jan. 2007, doi: 10.1002/ana.21052. [DOI] [PubMed] [Google Scholar]
- [133].Konopka A and Atkin JD, “The Role of DNA Damage in Neural Plasticity in Physiology and Neurodegeneration,” Frontiers in Cellular Neuroscience, vol. 16, 2022, Accessed: Jul. 27, 2022. [Online]. Available: 10.3389/fncel.2022.836885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Li X, Cao G, Liu X, Tang T-S, Guo C, and Liu H, “Polymerases and DNA Repair in Neurons: Implications in Neuronal Survival and Neurodegenerative Diseases,” Frontiers in Cellular Neuroscience, vol. 16, 2022, Accessed: Jul. 27, 2022. [Online]. Available: 10.3389/fncel.2022.852002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Merlo D et al. , “DNA repair in post-mitotic neurons: a gene-trapping strategy,” Cell Death Differ, vol. 12, no. 3, Art. no. 3, Mar. 2005, doi: 10.1038/sj.cdd.4401572. [DOI] [PubMed] [Google Scholar]
- [136].Schwartz EI et al. , “Cell cycle activation in postmitotic neurons is essential for DNA repair,” Cell Cycle, vol. 6, no. 3, pp. 318–329, Feb. 2007, doi: 10.4161/cc.6.3.3752. [DOI] [PubMed] [Google Scholar]
- [137].Shanbhag NM et al. , “Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease,” Acta Neuropathol Commun, vol. 7, no. 1, p. 77, May 2019, doi: 10.1186/s40478-019-0723-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Reid DA et al. , “Incorporation of a nucleoside analog maps genome repair sites in postmitotic human neurons,” Science, vol. 372, no. 6537, pp. 91–94, Apr. 2021, doi: 10.1126/science.abb9032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Pećina-Šlaus N, Kafka A, Salamon I, and Bukovac A, “Mismatch Repair Pathway, Genome Stability and Cancer,” Frontiers in Molecular Biosciences, vol. 7, p. 122, 2020, doi: 10.3389/fmolb.2020.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Richard G-F, “The Startling Role of Mismatch Repair in Trinucleotide Repeat Expansions,” Cells, vol. 10, no. 5, p. 1019, Apr. 2021, doi: 10.3390/cells10051019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Schmidt MHM and Pearson CE, “Disease-associated repeat instability and mismatch repair,” DNA Repair, vol. 38, pp. 117–126, Feb. 2016, doi: 10.1016/j.dnarep.2015.11.008. [DOI] [PubMed] [Google Scholar]
- [142].Ezzatizadeh V et al. , “The mismatch repair system protects against intergenerational GAA repeat instability in a Friedreich ataxia mouse model,” Neurobiol Dis, vol. 46, no. 1, pp. 165–171, Apr. 2012, doi: 10.1016/j.nbd.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Ezzatizadeh V, Sandi C, Sandi M, Anjomani-Virmouni S, Al-Mahdawi S, and Pook MA, “MutLα heterodimers modify the molecular phenotype of Friedreich ataxia,” PLoS One, vol. 9, no. 6, p. e100523, 2014, doi: 10.1371/journal.pone.0100523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Ku S et al. , “Friedreich’s ataxia induced pluripotent stem cells model intergenerational GAA⋅TTC triplet repeat instability,” Cell Stem Cell, vol. 7, no. 5, pp. 631–637, Nov. 2010, doi: 10.1016/j.stem.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Flower M et al. , “MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1.,” Brain, vol. 142, no. 7, pp. 1876–1886, Jul. 2019, doi: 10.1093/brain/awz115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Flower M, Lomeikaite V, Holmans P, Jones L, Tabrizi SJ, and Monckton DG, “Reply: The repeat variant in MSH3 is not a genetic modifier for spinocerebellar ataxia type 3 and Friedreich’s ataxia,” Brain, vol. 143, no. 4, p. e26, Apr. 2020, doi: 10.1093/brain/awaa044. [DOI] [PubMed] [Google Scholar]
- [147].Mirkin SM, “Getting to the Core of Repeat Expansions by Cell Reprogramming,” Cell Stem Cell, vol. 7, no. 5, pp. 545–546, Nov. 2010, doi: 10.1016/j.stem.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Yau WY et al. , “The repeat variant in MSH3 is not a genetic modifier for spinocerebellar ataxia type 3 and Friedreich’s ataxia,” Brain, vol. 143, no. 4, p. e25, Apr. 2020, doi: 10.1093/brain/awaa043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Neil AJ et al. , “Replication-independent instability of Friedreich’s ataxia GAA repeats during chronological aging,” PNAS, vol. 118, no. 5, Feb. 2021, doi: 10.1073/pnas.2013080118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Panigrahi GB, Lau R, Montgomery SE, Leonard MR, and Pearson CE, “Slipped (CTG)•(CAG) repeats can be correctly repaired, escape repair or undergo error-prone repair,” Nat Struct Mol Biol, vol. 12, no. 8, Art. no. 8, Aug. 2005, doi: 10.1038/nsmb959. [DOI] [PubMed] [Google Scholar]
- [151].Guo J, Gu L, Leffak M, and Li G-M, “MutSβ promotes trinucleotide repeat expansion by recruiting DNA polymerase β to nascent (CAG)n or (CTG)n hairpins for error-prone DNA synthesis,” Cell Res, vol. 26, no. 7, Art. no. 7, Jul. 2016, doi: 10.1038/cr.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Iyer RR and Pluciennik A, “DNA Mismatch Repair and its Role in Huntington’s Disease,” J Huntingtons Dis, vol. 10, no. 1, pp. 75–94, 2021, doi: 10.3233/JHD-200438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Kadyrova LY, Gujar V, Burdett V, Modrich PL, and Kadyrov FA, “Human MutLγ, the MLH1–MLH3 heterodimer, is an endonuclease that promotes DNA expansion,” Proc Natl Acad Sci USA, vol. 117, no. 7, pp. 3535–3542, Feb. 2020, doi: 10.1073/pnas.1914718117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Owen BAL et al. , “(CAG)(n)-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition,” Nat Struct Mol Biol, vol. 12, no. 8, pp. 663–670, Aug. 2005, doi: 10.1038/nsmb965. [DOI] [PubMed] [Google Scholar]
- [155].Zhang T, Huang J, Gu L, and Li G-M, “In vitro repair of DNA hairpins containing various numbers of CAG/CTG trinucleotide repeats,” DNA Repair (Amst), vol. 11, no. 2, pp. 201–209, Feb. 2012, doi: 10.1016/j.dnarep.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Pinto RM et al. , “Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: genome-wide and candidate approaches,” PLoS Genet, vol. 9, no. 10, p. e1003930, Oct. 2013, doi: 10.1371/journal.pgen.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Lee J-M et al. , “A modifier of Huntington’s disease onset at the MLH1 locus,” Human Molecular Genetics, vol. 26, no. 19, pp. 3859–3867, Ottobre 2017, doi: 10.1093/hmg/ddx286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Su XA and Freudenreich CH, “Cytosine deamination and base excision repair cause R-loop-induced CAG repeat fragility and instability in Saccharomyces cerevisiae,” Proc Natl Acad Sci U S A, vol. 114, no. 40, pp. E8392–E8401, Oct. 2017, doi: 10.1073/pnas.1711283114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Gomes-Pereira M, Fortune MT, Ingram L, McAbney JP, and Monckton DG, “Pms2 is a genetic enhancer of trinucleotide CAG·CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion,” Human Molecular Genetics, vol. 13, no. 16, pp. 1815–1825, Agosto 2004, doi: 10.1093/hmg/ddh186. [DOI] [PubMed] [Google Scholar]
- [160].Roy JCL et al. , “Somatic CAG expansion in Huntington’s disease is dependent on the MLH3 endonuclease domain, which can be excluded via splice redirection,” Nucleic Acids Research, vol. 49, no. 7, pp. 3907–3918, Apr. 2021, doi: 10.1093/nar/gkab152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Du J et al. , “Role of mismatch repair enzymes in GAA·TTC triplet-repeat expansion in Friedreich ataxia induced pluripotent stem cells,” J Biol Chem, vol. 287, no. 35, pp. 29861–29872, Aug. 2012, doi: 10.1074/jbc.M112.391961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Kovalenko M et al. , “Msh2 Acts in Medium-Spiny Striatal Neurons as an Enhancer of CAG Instability and Mutant Huntingtin Phenotypes in Huntington’s Disease Knock-In Mice,” PLOS ONE, vol. 7, no. 9, p. e44273, set 2012, doi: 10.1371/journal.pone.0044273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Manley K, Shirley TL, Flaherty L, and Messer A, “Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice.,” Nature Genetics, vol. 23, no. 4, pp. 471–473, Dec. 1999, doi: 10.1038/70598. [DOI] [PubMed] [Google Scholar]
- [164].Kantartzis A, Williams GM, Balakrishnan L, Roberts RL, Surtees JA, and Bambara RA, “Msh2-Msh3 interferes with Okazaki fragment processing to promote trinucleotide repeat expansions,” Cell Rep, vol. 2, no. 2, pp. 216–222, Aug. 2012, doi: 10.1016/j.celrep.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Tomé S et al. , “MSH2 ATPase domain mutation affects CTG*CAG repeat instability in transgenic mice,” PLoS Genet, vol. 5, no. 5, p. e1000482, May 2009, doi: 10.1371/journal.pgen.1000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Dragileva E et al. , “Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes,” Neurobiol Dis, vol. 33, no. 1, pp. 37–47, Jan. 2009, doi: 10.1016/j.nbd.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Foiry L et al. , “Msh3 is a limiting factor in the formation of intergenerational CTG expansions in DM1 transgenic mice,” Hum Genet, vol. 119, no. 5, pp. 520–526, Jun. 2006, doi: 10.1007/s00439-006-0164-7. [DOI] [PubMed] [Google Scholar]
- [168].Keogh N, Chan KY, Li G-M, and Lahue RS, “MutSβ abundance and Msh3 ATP hydrolysis activity are important drivers of CTG•CAG repeat expansions,” Nucleic Acids Res, vol. 45, no. 17, pp. 10068–10078, Sep. 2017, doi: 10.1093/nar/gkx650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Tomé S et al. , “MSH3 Polymorphisms and Protein Levels Affect CAG Repeat Instability in Huntington’s Disease Mice,” PLOS Genetics, vol. 9, no. 2, p. e1003280, Feb. 2013, doi: 10.1371/journal.pgen.1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Williams GM and Surtees JA, “MSH3 Promotes Dynamic Behavior of Trinucleotide Repeat Tracts In Vivo,” Genetics, vol. 200, no. 3, pp. 737–754, Jul. 2015, doi: 10.1534/genetics.115.177303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Cilli P et al. , “Oxidized dNTPs and the OGG1 and MUTYH DNA glycosylases combine to induce CAG/CTG repeat instability,” Nucleic Acids Research, vol. 44, no. 11, pp. 5190–5203, Giugno 2016, doi: 10.1093/nar/gkw170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, and McMurray CT, “OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells,” Nature, vol. 447, no. 7143, pp. 447–452, May 2007, doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Lai Y, Beaver JM, Laverde E, and Liu Y, “Trinucleotide repeat instability via DNA base excision repair,” DNA Repair (Amst), vol. 93, p. 102912, Sep. 2020, doi: 10.1016/j.dnarep.2020.102912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Lai Y et al. , “Base excision repair of chemotherapeutically-induced alkylated DNA damage predominantly causes contractions of expanded GAA repeats associated with Friedreich’s ataxia,” PLoS One, vol. 9, no. 4, p. e93464, 2014, doi: 10.1371/journal.pone.0093464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Laverde EE, Lai Y, Leng F, Balakrishnan L, Freudenreich CH, and Liu Y, “R-loops promote trinucleotide repeat deletion through DNA base excision repair enzymatic activities,” Journal of Biological Chemistry, vol. 295, no. 40, pp. 13902–13913, Oct. 2020, doi: 10.1074/jbc.RA120.014161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Lai Y, Xu M, Zhang Z, and Liu Y, “Instability of CTG Repeats is Governed by the Position of a DNA Base Lesion through Base Excision Repair,” PLOS ONE, vol. 8, no. 2, p. e56960, Feb. 2013, doi: 10.1371/journal.pone.0056960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].La Rosa P, Petrillo S, Bertini ES, and Piemonte F, “Oxidative Stress in DNA Repeat Expansion Disorders: A Focus on NRF2 Signaling Involvement,” Biomolecules, vol. 10, no. 5, Art. no. 5, May 2020, doi: 10.3390/biom10050702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Cobley JN, Fiorello ML, and Bailey DM, “13 reasons why the brain is susceptible to oxidative stress,” Redox Biology, vol. 15, pp. 490–503, Maggio 2018, doi: 10.1016/j.redox.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Lupoli F, Vannocci T, Longo G, Niccolai N, and Pastore A, “The role of oxidative stress in Friedreich’s ataxia,” FEBS Letters, vol. 592, no. 5, pp. 718–727, 2018, doi: 10.1002/1873-3468.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Tang W et al. , “Friedreich’s ataxia (GAA)n•(TTC)n repeats strongly stimulate mitotic crossovers in Saccharomyces cerevisae,” PLoS Genet, vol. 7, no. 1, p. e1001270, Jan. 2011, doi: 10.1371/journal.pgen.1001270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Napierala M, Dere R, Vetcher A, and Wells RD, “Structure-dependent recombination hot spot activity of GAA.TTC sequences from intron 1 of the Friedreich’s ataxia gene,” J Biol Chem, vol. 279, no. 8, pp. 6444–6454, Feb. 2004, doi: 10.1074/jbc.M309596200. [DOI] [PubMed] [Google Scholar]
- [182].Polleys EJ and Freudenreich CH, “Homologous recombination within repetitive DNA,” Curr Opin Genet Dev, vol. 71, pp. 143–153, Dec. 2021, doi: 10.1016/j.gde.2021.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Balakrishnan L and Bambara RA, “Flap Endonuclease 1,” Annual Review of Biochemistry, vol. 82, no. 1, pp. 119–138, 2013, doi: 10.1146/annurev-biochem-072511-122603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Michl J, Zimmer J, and Tarsounas M, “Interplay between Fanconi anemia and homologous recombination pathways in genome integrity,” EMBO J, vol. 35, no. 9, pp. 909–923, May 2016, doi: 10.15252/embj.201693860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Yang J and Freudenreich CH, “Haploinsufficiency of Yeast FEN1 Causes Instability of Expanded CAG/CTG Tracts in a Length-Dependent Manner,” Gene, vol. 393, no. 1–2, pp. 110–115, May 2007, doi: 10.1016/j.gene.2007.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Freudenreich CH, Stavenhagen JB, and Zakian VA, “Stability of a CTG/CAG trinucleotide repeat in yeast is dependent on its orientation in the genome,” Mol Cell Biol, vol. 17, no. 4, pp. 2090–2098, Apr. 1997, doi: 10.1128/mcb.17.4.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Tsutakawa SE et al. , “Phosphate steering by Flap Endonuclease 1 promotes 5’-flap specificity and incision to prevent genome instability,” Nat Commun, vol. 8, p. 15855, Jun. 2017, doi: 10.1038/ncomms15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Liu Y, Zhang H, Veeraraghavan J, Bambara RA, and Freudenreich CH, “Saccharomyces cerevisiae Flap Endonuclease 1 Uses Flap Equilibration to Maintain Triplet Repeat Stability,” Molecular and Cellular Biology, vol. 24, no. 9, pp. 4049–4064, 2004, doi: 10.1128/MCB.24.9.4049-4064.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].van den Broek WJAA, Nelen MR, van der Heijden GW, Wansink DG, and Wieringa B, “Fen1 does not control somatic hypermutability of the (CTG)(n)*(CAG)(n) repeat in a knock-in mouse model for DM1,” FEBS Lett, vol. 580, no. 22, pp. 5208–5214, Oct. 2006, doi: 10.1016/j.febslet.2006.08.059. [DOI] [PubMed] [Google Scholar]
- [190].Spiro C and McMurray CT, “Nuclease-deficient FEN-1 blocks Rad51/BRCA1-mediated repair and causes trinucleotide repeat instability,” Mol Cell Biol, vol. 23, no. 17, pp. 6063–6074, Sep. 2003, doi: 10.1128/MCB.23.17.6063-6074.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Moe SE, Sorbo JG, and Holen T, “Huntingtin triplet-repeat locus is stable under long-term Fen1 knockdown in human cells,” J Neurosci Methods, vol. 171, no. 2, pp. 233–238, Jun. 2008, doi: 10.1016/j.jneumeth.2008.03.012. [DOI] [PubMed] [Google Scholar]
- [192].Otto CJ, Almqvist E, Hayden MR, and Andrew SE, “The ‘flap’ endonuclease gene FEN1 is excluded as a candidate gene implicated in the CAG repeat expansion underlying Huntington disease,” Clin Genet, vol. 59, no. 2, pp. 122–127, Feb. 2001, doi: 10.1034/j.1399-0004.2001.590210.x. [DOI] [PubMed] [Google Scholar]
- [193].Liu G, Chen X, Bissler JJ, Sinden RR, and Leffak M, “Replication-dependent instability at (CTG)•(CAG) repeat hairpins in human cells,” Nat Chem Biol, vol. 6, no. 9, Art. no. 9, Sep. 2010, doi: 10.1038/nchembio.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Kim K-H et al. , “Genetic and Functional Analyses Point to FAN1 as the Source of Multiple Huntington Disease Modifier Effects,” Am J Hum Genet, vol. 107, no. 1, pp. 96–110, Jul. 2020, doi: 10.1016/j.ajhg.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Deshmukh AL et al. , “FAN1 exo- not endo-nuclease pausing on disease-associated slipped-DNA repeats: A mechanism of repeat instability,” Cell Reports, vol. 37, no. 10, p. 110078, Dec. 2021, doi: 10.1016/j.celrep.2021.110078. [DOI] [PubMed] [Google Scholar]
- [196].Trost B et al. , “Genome-wide detection of tandem DNA repeats that are expanded in autism,” Nature, vol. 586, no. 7827, pp. 80–86, Oct. 2020, doi: 10.1038/s41586-020-2579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Zhao X-N and Usdin K, “FAN1 protects against repeat expansions in a Fragile X mouse model,” DNA Repair (Amst), vol. 69, pp. 1–5, Sep. 2018, doi: 10.1016/j.dnarep.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Goold R et al. , “FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat,” Hum Mol Genet, vol. 28, no. 4, pp. 650–661, Feb. 2019, doi: 10.1093/hmg/ddy375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Cannavo E, Gerrits B, Marra G, Schlapbach R, and Jiricny J, “Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2,” J Biol Chem, vol. 282, no. 5, pp. 2976–2986, Feb. 2007, doi: 10.1074/jbc.M609989200. [DOI] [PubMed] [Google Scholar]
- [200].Smogorzewska A et al. , “A genetic screen identifies FAN1, a Fanconi anemia associated nuclease necessary for DNA interstrand crosslink repair,” Mol Cell, vol. 39, no. 1, pp. 36–47, Jul. 2010, doi: 10.1016/j.molcel.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Goold R et al. , “FAN1 controls mismatch repair complex assembly via MLH1 retention to stabilize CAG repeat expansion in Huntington’s disease,” Cell Rep, vol. 36, no. 9, p. 109649, Aug. 2021, doi: 10.1016/j.celrep.2021.109649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Lahue RS, “SPYing on triplet repeat expansions: Insights into FAN1-MLH1 interaction and regulation,” Cell Reports, vol. 36, no. 11, p. 109736, Sep. 2021, doi: 10.1016/j.celrep.2021.109736. [DOI] [PubMed] [Google Scholar]
- [203].Loupe JM et al. , “Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington’s disease knock-in mice is blocked by Mlh1 knock-out,” Hum Mol Genet, vol. 29, no. 18, pp. 3044–3053, Nov. 2020, doi: 10.1093/hmg/ddaa196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Porro A et al. , “FAN1-MLH1 interaction affects repair of DNA interstrand cross-links and slipped-CAG/CTG repeats,” Sci Adv, vol. 7, no. 31, p. eabf7906, Jul. 2021, doi: 10.1126/sciadv.abf7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].McAllister B et al. , “Exome sequencing of individuals with Huntington’s disease implicates FAN1 nuclease activity in slowing CAG expansion and disease onset,” Nat Neurosci, vol. 25, no. 4, Art. no. 4, Apr. 2022, doi: 10.1038/s41593-022-01033-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Deshmukh AL et al. , “FAN1, a DNA Repair Nuclease, as a Modifier of Repeat Expansion Disorders,” J Huntingtons Dis, vol. 10, no. 1, pp. 95–122, 2021, doi: 10.3233/JHD-200448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Calil FA et al. , “Rad27 and Exo1 function in different excision pathways for mismatch repair in Saccharomyces cerevisiae,” Nat Commun, vol. 12, no. 1, Art. no. 1, Sep. 2021, doi: 10.1038/s41467-021-25866-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Li Y et al. , “Excision of Expanded GAA Repeats Alleviates the Molecular Phenotype of Friedreich’s Ataxia,” Molecular Therapy, vol. 23, no. 6, pp. 1055–1065, Jun. 2015, doi: 10.1038/mt.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Oura S et al. , “Precise CAG repeat contraction in a Huntington’s Disease mouse model is enabled by gene editing with SpCas9-NG,” Commun Biol, vol. 4, no. 1, pp. 1–13, Jun. 2021, doi: 10.1038/s42003-021-02304-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Rocca CJ et al. , “CRISPR-Cas9 Gene Editing of Hematopoietic Stem Cells from Patients with Friedreich’s Ataxia,” Mol Ther Methods Clin Dev, vol. 17, pp. 1026–1036, Jun. 2020, doi: 10.1016/j.omtm.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Ouellet DL, Cherif K, Rousseau J, and Tremblay JP, “Deletion of the GAA repeats from the human frataxin gene using the CRISPR-Cas9 system in YG8R-derived cells and mouse models of Friedreich ataxia,” Gene Ther, vol. 24, no. 5, pp. 265–274, May 2017, doi: 10.1038/gt.2016.89. [DOI] [PubMed] [Google Scholar]
- [212].Anjomani Virmouni S et al. , “A novel GAA-repeat-expansion-based mouse model of Friedreich’s ataxia,” Dis Model Mech, vol. 8, no. 3, pp. 225–235, Mar. 2015, doi: 10.1242/dmm.018952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Bird MJ et al. , “Functional characterization of Friedreich ataxia iPS-derived neuronal progenitors and their integration in the adult brain,” PLoS One, vol. 9, no. 7, p. e101718, 2014, doi: 10.1371/journal.pone.0101718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Viventi S et al. , “In vivo survival and differentiation of Friedreich ataxia iPSC-derived sensory neurons transplanted in the adult dorsal root ganglia,” Stem Cells Transl Med, vol. 10, no. 8, pp. 1157–1169, Aug. 2021, doi: 10.1002/sctm.20-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Mazzara PG et al. , “Frataxin gene editing rescues Friedreich’s ataxia pathology in dorsal root ganglia organoid-derived sensory neurons,” Nat Commun, vol. 11, no. 1, p. 4178, Aug. 2020, doi: 10.1038/s41467-020-17954-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Sivakumar A and Cherqui S, “Advantages and Limitations of Gene Therapy and Gene Editing for Friedreich’s Ataxia,” Frontiers in Genome Editing, vol. 4, 2022, Accessed: May 31, 2022. [Online]. Available: 10.3389/fgeed.2022.903139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Ribeil J-A et al. , “Gene Therapy in a Patient with Sickle Cell Disease,” N Engl J Med, vol. 376, no. 9, pp. 848–855, Mar. 2017, doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- [218].Jacobson SG et al. , “Safety and improved efficacy signals following gene therapy in childhood blindness caused by GUCY2D mutations,” iScience, vol. 24, no. 5, p. 102409, May 2021, doi: 10.1016/j.isci.2021.102409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Nakamori M et al. , “A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo,” Nat Genet, vol. 52, no. 2, pp. 146–159, Feb. 2020, doi: 10.1038/s41588-019-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Li Y et al. , “Targeting 3′ and 5′ untranslated regions with antisense oligonucleotides to stabilize frataxin mRNA and increase protein expression,” Nucleic Acids Research, vol. 49, no. 20, pp. 11560–11574, Nov. 2021, doi: 10.1093/nar/gkab954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Lazaropoulos M et al. , “Frataxin levels in peripheral tissue in Friedreich ataxia,” Ann Clin Transl Neurol, vol. 2, no. 8, pp. 831–842, Aug. 2015, doi: 10.1002/acn3.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Pianese L et al. , “Real time PCR quantification of frataxin mRNA in the peripheral blood leucocytes of Friedreich ataxia patients and carriers,” J Neurol Neurosurg Psychiatry, vol. 75, no. 7, pp. 1061–1063, Jul. 2004, doi: 10.1136/jnnp.2003.028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Bergquist H et al. , “Disruption of Higher Order DNA Structures in Friedreich’s Ataxia (GAA)n Repeats by PNA or LNA Targeting,” PLOS ONE, vol. 11, no. 11, p. e0165788, Nov. 2016, doi: 10.1371/journal.pone.0165788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Rastokina A, Mozafari N, Cebrián J, Smith CIE, Mirkin SM, and Zain R, “Large-scale expansions of Friedreich’s ataxia GAA•TTC repeats in human cells are prevented by LNA-DNA oligonucleotides and PNA oligomers.” bioRxiv, p. 2022.07.04.498742, Jul. 05, 2022. doi: 10.1101/2022.07.04.498742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Grant L, Sun J, Xu H, Subramony SH, Chaires JB, and Hebert MD, “Rational selection of small molecules that increase transcription through the GAA repeats found in Friedreich’s ataxia,” FEBS Lett, vol. 580, no. 22, pp. 5399–5405, Oct. 2006, doi: 10.1016/j.febslet.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Lufino MMP, Silva AM, Németh AH, Alegre-Abarrategui J, Russell AJ, and Wade-Martins R, “A GAA repeat expansion reporter model of Friedreich’s ataxia recapitulates the genomic context and allows rapid screening of therapeutic compounds,” Hum Mol Genet, vol. 22, no. 25, pp. 5173–5187, Dec. 2013, doi: 10.1093/hmg/ddt370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Burnett R et al. , “DNA sequence-specific polyamides alleviate transcription inhibition associated with long GAA·TTC repeats in Friedreich’s ataxia,” Proceedings of the National Academy of Sciences, vol. 103, no. 31, pp. 11497–11502, Aug. 2006, doi: 10.1073/pnas.0604939103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Nance E, Pun SH, Saigal R, and Sellers DL, “Drug delivery to the central nervous system,” Nat Rev Mater, vol. 7, no. 4, Art. no. 4, Apr. 2022, doi: 10.1038/s41578-021-00394-w. [DOI] [PMC free article] [PubMed] [Google Scholar]