

Abstract
Exposure to sunlight is both beneficial, as it heats the planet to a comfortable temperature, and potentially harmful, since sunlight contains ultraviolet radiation (UVR), which is deemed detrimental for living organisms. Earth’s ozone layer plays a vital role in blocking most of the extremely dangerous UVC; however, low frequency/energy UVR (i.e., UVB and UVA) seeps through in minute amount and reach the Earth’s surface. Both UVB and UVA are physiologically most responsible for a plethora of skin ailments, including skin cancers. The UVR is readily absorbed by the genomic DNA of skin cells, causing DNA bond distortion and UV-induced DNA damage. As a defense mechanism, the DNA damage response (DDR) signaling in skin cells activates nucleotide excision repair (NER), which is responsible for the removal of UVR-induced DNA photolesions and helps maintain the genomic integrity of the cells. Failure of proper NER function leads to mutagenesis and development of skin cancers. These cancers include melanoma, widely considered the deadliest form of skin cancers, which originates upon the genetic transformation of melanocytes. NER is well studied in the skin, as a tissue, but not much is known about it in melanocytes. Therefore, this review encapsulates NER in melanocytes, with a specific focus on its functional regulators and their cross-talks due to skin heterogeneity and divulging the potential knowledge gap in the field.
Keywords: Melanocytes, Solar UVB, DDR signaling, NER, αMSH, End1, Circadian clock
Introduction:
All living organisms, including humans, receive solar radiation in the form of light and heat energy. Along with its life-giving properties, the sunlight contains various types of radiation, including ultraviolet radiation (UVR), which is harmful to living organisms. Earth’s ozone layer plays an essential role in blocking extremely harmful UVR, which is UVC, allowing only low energy UVB and UVA to seep through and reach the Earth’s surface. Nevertheless, this small fraction of UVR reaching Earth’s surface is responsible for a plethora of skin ailments, including sunburn erythema, photoaging, and melanoma and non-melanoma skin cancers in humans, a majority of which are caused by UV-mediated DNA damage in the skin cells (Schuch et al. 2017; Sample and He 2018). UVR affects all types of epidermal skin cells equally, and since keratinocytes are the most abundant cell type in the skin, the most common UV-induced non-melanoma skin cancers originate from keratinocytes, which far exceeds the number of melanoma (cancer arising from melanocytes) cases. However, melanoma is considered the most dangerous type of skin cancers due to its frequent metastasizing to the lymph nodes, lungs, and brain (Fuchs and Raghavan 2002; Guy et al. 2015). Only 4–5% of all skin cancer cases reported in the US are melanomas, but melanomas account for more than 75% of skin cancer-related deaths in the US (Neville et al. 2007; Shenenberger 2012; Society 2019; U.S. Department of health and human services 2019). The latest report estimates that the number of melanoma cases will drastically increase in the coming years, which will have a substantial economic burden, and therefore makes melanoma a significant area of research in understanding its biology and developing better therapies to tackle this surge (Arondekar et al. 2015; Guy et al. 2015). One novel therapeutic approach could be to find potential druggable targets, which can be harnessed to augment the endogenous protective mechanisms against UVR in melanocytes.
Upon UVR exposure, the genomic DNA of the skin melanocytes incur damage; in reply, cellular DNA damage response (DDR) signaling mechanisms such as checkpoint signing, nucleotide excision repair (NER), and programmed cell death gets activated in order to minimize DNA damage to protect the genomic integrity of the cells (Latonen and Laiho 2005). In addition to the above mentioned DDR signaling events, other skin protective mechanisms, including melanogenesis (synthesis of melanin) and hyperkeratosis, protects melanocytes from further UVR damage (Slominski et al. 2004; Scott et al. 2012). Failure of these skin protective mechanisms, in combination with predisposing genetic factors including skin coloration, family history, and conditions such as xeroderma pigmentosum, lead to the development of UV-induced melanomagenesis (Gloster and Neal 2006; Lehmann et al. 2011). Multiple studies have shown the presence of robust DDR signaling pathways in melanocytes; however, the various regulators of DDR in melanocytes remain unexplored. In this review, we focus on DDR signaling pathways in melanocytes, specifically nucleotide excision repair (NER), and highlight other mechanisms involved, which could provide a link to understanding the biology of melanoma progression and developing improved therapeutics.
The Skin: a focus on melanocytes
Skin is the largest organ in our body, which is part of the integumentary system. It protects the underlying tissues and maintains the homeostasis of our body. It is known as the first line of defense against physical, chemical, bacterial, and environmental challenges faced by humans on a daily basis. Broadly, the skin is divided into dermis and epidermis, originating respectively from the mesoderm and ectoderm (Figure 1). The outermost layer of the skin is the epidermis, which is primarily composed of keratinized, stratified squamous epithelium cells, generally known as keratinocytes and few langerhans cells which are macrophages of the skin. Keratinocytes constitute around 90–95% of the epidermis and are characterized by the expression and storage of keratin and formation of desmosomes. Keratin is a fibrous protein found in cells that gives hair, nails, and the skin their hardness and water-resistant property. Dead keratinocytes are regularly sloughed off and replaced by cells from the deeper layers generated through cell division and programmed differentiation of the keratinocyte stem cells. Under the epidermis lies the dermis, which is made up of fibroblasts, macrophages and mast cells and host cutaneous structures of the skin, including the hair follicles, nerves, sebaceous glands, sweat glands, and connective tissues like blood vessels and lymph nodes (Brown and Krishnamurthy 2020). The basement membrane resides between the epidermis and dermis, above which the basal layer of the epidermis resides. The basal epidermal layer houses not only the keratinocyte stem cells, but also a small fraction (5–10%) of melanocytes, the melanin pigment-producing cells. Together, both melanocytes and keratinocytes form the skin pigmentary system and are responsible for determining the complexion of our skin, as well as protecting us from adverse outcomes of the environment including genomic instability of UVR (Fuchs and Raghavan 2002; Madison 2003; Fuchs 2007; Proksch et al. 2008; Gaddameedhi and Sancar 2011).
Figure 1. Integumentary system.

The skin is the largest organ of our body, accounting for about 16% of body mass. It contains two primary layers: the epidermis and dermis. The epidermis is the functional part of the skin, which protects our body from the outside world. It comprises keratinocytes, melanocytes, and markel and langerhans cells. Dermis contains connective tissues like the nerve ending and blood vessels, and functional skin organs like the sweat glands and hair follicles. In addition, the dermis houses fibroblast cells, mast cells, and adipose tissue. The junction between epidermis and dermis is known as the basement membrane (BM) which makes a tight leak-proof junction in between the cells.
Melanocytes are derived from the neural crest cells and are found at the density of around 12.2 to 12.8 melanocytes/mm or ~1500 epidermal melanocytes/mm2 (excluding the hair follicle) in adult human epidermis, which is consistent regardless of the race (Szabo 1967; Whiteman et al. 1999; Tadokoro et al. 2005). Melanocytes are a slow-dividing cell type with a proliferation rate of less than twice per year, allowing them to stay at the basal layer of the epidermis and rarely shed (Fitzpatrick and Breathnach 1963; Downing and Roth 1974; Jimbow et al. 1975). However, to culture melanocytes in an in vitro condition, activation of cAMP, PKC, and endothelin-1 by stimulators like TPA, IBMX, Cholera toxin, or αMSH are used, which allows them to proliferate faster and live significantly longer. (Eisinger and Marko 1982; Halaban et al. 1986; Yaar and Gilchrest 1991; Medrano et al. 1993). In the epidermis, although keratinocytes house the melanin pigment, it is produced in the melanocytes. Upon exposure to UVR, the DDR signaling mechanism in epidermal keratinocytes activates p53 mediated expression of proopiomelanocortin (POMC), which upon post-translational cleavage generates a paracrine factor, α Melanocortin Stimulating Hormone” (αMSH), which specifically binds to the Melanocortin1 receptor (MC1R) found on the surface of melanocytes (Cui et al. 2007). Melanocytes then initiate cAMP-mediated melanin synthesis in specialized cellular organelles called melanosomes, which then carry the melanin to the epidermal keratinocytes and further protects the genomic DNA from the damage by UVR (Suzuki et al. 1999; Tsatmali et al. 2002; Joshi et al. 2007; Nordlund 2007; Cardinali et al. 2008; Yamaguchi and Hearing 2009). A differentiated melanocyte forms dendritic projections, which allow it to be in contact with about 30 to 40 keratinocytes and thus forming the “epidermal melanin unit” (Fitzpatrick and Breathnach 1963).
Apart from melanin synthesis, melanocytes play a vital role in producing a plethora of signaling molecules such as cytokines, eicosanoids, catecholamine, melanocortin peptides, serotonin, and nitric oxide (Iyengar and Misra 1987; Armstrong et al. 1992; Okano-Mitani et al. 1997; Wakamatsu et al. 1997; Johansson et al. 1998; Tsatmali et al. 2000). Although the primary role of melanocytes is producing the melanin pigment, melanocytes play other physiological roles, such as a stress sensor or a potential contributor to skin immune response against UVR, or mimicking neuroendocrine cells (Tsatmali et al. 2002).
Melanoma: a rogue melanocyte
As discussed above, melanocytes are multifaceted and play a vital role in the skin’s natural defense against solar UVR, but like all other cell types in the skin, melanocytes are also negatively affected by UVR. According to various epidemiological studies, it has been estimated that around 65% of melanomas, the most dangerous form of skin cancers, are caused by solar UVR (Armstrong and Kricker 1993; Pleasance et al. 2010). The Centers for Disease Control and Prevention (CDC) classifies skin cancer as one of the most common types of cancer; of these, non-melanoma skin cancers typically have better prognosis and are highly curable if detected early and treated adequately compared to cases reported as melanoma, which accounts for more than 75% of skin cancer-related deaths (Neville et al. 2007; Shenenberger 2012; Society 2019; U.S. Department of health and human services 2019). The rise in skin cancer incidents impacts the economy as well: while the average annual cost of treating all non-melanoma cancer cases combined is estimated to be ~4.8 billion, due to the unavailability of effective therapy it costs ~3.3 billion dollars to the American economy to treat melanoma-related cases (Guy et al. 2015). According to the latest report by the Skin Cancer Foundation, there could be an increase in new melanoma cases by 2% in 2020, making the total estimated number of cases 196,060, out of which 6,850 people are estimated to die (Society 2020).
Melanoma originates from melanocytes in the skin epidermis which have undergone various sequential genetic and epigenetic changes/mutations to progress to melanoma (Miller and Mihm 2006). Cutaneous melanomas are commonly classified as either chronically-sun damaged (CSD) or non-CSD melanoma. In the Caucasian population, ~90% of melanomas are CSD, while in non-Caucasian population, 60 to 75% of melanomas are non-CSD (Gloster and Neal 2006; Shain and Bastian 2016). The significant difference between CSD and non-CSD is the site of origin on the skin, the frequency of UVR exposure, age, mutation burden, and types of oncogenic alteration (Maldonado et al. 2003; Curtin et al. 2005; Viros et al. 2008; Long et al. 2011). A fully developed melanoma contains a multitude of mutations, some of which are frequently occurring somatic mutations found in both CSD and non-CSD melanomas.
If some of the most recurrent mutations found in a developed melanoma are classified according to the well-established 10 hallmarks of cancer, gene mutations associated with 6 of those hallmarks come up: 1) sustaining proliferation (BRAF, NRAS, MITF, KIT, GNAQ and NF1), 2) deregulation of cellular energetics (PTEN and KIT), 3) resisting cell death (BCL2, TP53 and PTEN), 4) enabling replicative immortality (telomerase reverse transcriptase - TERT), 5) avoiding immune destruction (AT-rich interaction domain 2 – ARID2), and 6) evading growth suppression (cyclin-dependent kinase inhibitor 2A - CDKN2A, which encodes for p16INK4A and p14ARF). Furthermore, there are a plethora of gene mutations that have been detected in melanoma cases which are very well summarized in the cited articles (Hanahan and Weinberg 2011; Hodis et al. 2012; Krauthammer et al. 2012; Horn et al. 2013; Huang et al. 2013; Shtivelman et al. 2014). The above gene mutation list indicates a complete disarray of multiple signaling pathways, which may cause total disruption of cellular homeostasis.
These mutations compile throughout the melanocyte life span and finally lead to melanoma formation; however, the order in which these pathways are disrupted is still a matter of investigation and can vary among different melanoma cases. For instance, in non-CSD melanoma, signature mutation in BRAFV600E gene, where a valine (V) at position 600 is substituted by glutamic acid (E), is prevalent and is deemed the melanoma initiation mutation. This mutation converts melanocyte to banal naevus form; over time, the accumulation of driver mutations in genes like TERT and CDKN2A due to UVR exposure progresses naevi into melanomas (Shain and Bastian 2016). In contrast, CSD melanomas have mutations in NRAS, NF1, and BRAFnonV600E genes which push the melanocytes to form melanoma in situ lesions (benign form of melanoma) and upon accumulation of UV-induced mutations in progression-associated genes, including TERT, PTEN, and TP53, it advances to a matured stage melanoma (Shain and Bastian 2016). CSD melanoma cases have most genes with UV manifested signature mutations, and in rare cases, the BRAFV600E mutation is also associated with UVR exposure (Thomas et al. 2006; Brash 2015). UVR is considered the primary causative agent for melanoma initiation and progression. Therefore, understanding the effect of UVR on the skin, specifically on melanocytes, is essential and will be discussed in detail in the next section.
Solar UV radiation: a foe in disguise
The Sun is the closest star to Earth; the heat and light energy produced by it is vital for the existence of life on Earth. Sun’s light energy is absorbed by plants, which convert it to chemical energy, i.e., glucose through photosynthesis, which then enters the food chain and helps sustain living organisms on our planet. The heat energy helps warm the planet to a comfortable temperature, which is suitable for sustaining a wide diversity of flora and fauna. In contrast, the Sun also emits harmful radiations like gamma, X, and UVR (NASA 2008; EPA 2014). As shown in figure 2, UVR is further classified into 3 distinct groups, from least to most harmful to living organisms, as UVA (320–400 nm), UVB (280–320 nm), and UVC (100–280 nm). The stratospheric ozone layer plays a vital role in completely blocking the UVC and most of the UVB radiation. However, small amounts of UVB (5–10%) and almost 90–95% of UVA seeps through the ozone layer and is known to have deleterious effects on all living organisms, including humans (Schuch et al. 2013). According to extensive studies across the globe, the Earth’s ozone layer is depleting at an alarming rate due to various human-inflicted reasons (de Gruijl and van der Leun 2000). The loss of the ozone layer increases the chances of various UVR-mediated diseases, including skin cancers, which come with a substantial economic burden as well (Lucas et al. 2015).
Figure 2. Effect of ultraviolet radiation (UVR) on skin cells.

The UVR from the Sun can be broadly classified as UVC (100–280 nm), UVB (280–320 nm) and UVA (320–400 nm). The Earth’s ozone layer helps reflect back UVC and most of UVB, allowing the majority of UVA and a minute amount of UVB to reach Earth’s surface. Therefore, UVB and UVA are more physiologically relevant when it comes to UV-induced human diseases. Genomic DNA absorbs both UVB and UVA radiation. UVB is deemed more harmful than UVA as it accounts for the majority of the UV-induced photolesion formation. There are two major types of photo products formed upon UVB or UVA exposure: cyclobutane pyrimidine dimers (CPDs) and (6–4) photoproducts [(6–4) PPs]. Around 80% of photoproduct formed are CPDs, whereas only around 20% are (6–4) PPs. Apart from these major types of DNA damage from UVA and UVB, UVA is also known to produce 7,8-dihydro-8-oxoguanine (8-OxoG) photolesions on the DNA. The primary source of such DNA lesions is reactive oxygen species (ROS) which can be produced due to the excitation of chromophores like melanin upon UVR exposure.
Tanned skin in the Caucasian population has long been a contemporary fashion statement, and the most comfortable and least expensive way to tan is through sunbathing. Increased recreational exposure to sunlight has significantly contributed to the surge of skin cancer cases because of increased exposure to UVB, which is deemed more carcinogenic, causing direct genomic DNA damage in epidermal skin cells (Khan et al. 2018). Prolonged exposure to solar UVB causes the majority of skin-related ailments, including sunburn erythema, skin thickening, photoaging, and skin cancers (Bayerl et al. 1995; Wenk et al. 2001; Scott et al. 2012). UVB affects the epidermal part of the skin and causes inflammation and formation of direct DNA photolesions known as cyclobutane pyrimidine dimers (CPDs) and (6–4) photoproducts, which leads to formation of characteristic C→T cancerous UV signature mutations if inefficiently removed by NER (Sato et al. 1993; Sarasin 1999; Sancar et al. 2004; Hoeijmakers 2009; Hodis et al. 2012; Brash 2015; Singh et al. 2019). In response, our skin defense mechanism initiates melanin synthesis in melanocytes, which are distributed to the keratinocytes and absorb UVB radiation to reduce further DNA lesion formation. In parallel, the NER mechanism works to repair UVB-induced DNA photoproducts and help maintain the genomic integrity of the skin melanocytes (Figure 3). Apart from its harmful effects, UVB exposure ensures synthesis of vitamin D in a dose-dependent manner in the skin, which is directly proportional to melanin level in the skin (Sample and He 2018). Vitamin D is an essential hormone, which aids in maintaining calcium homeostasis and essential physiological processes of the body (Bogh et al. 2011).
Figure 3. First responders against solar UVR exposure.
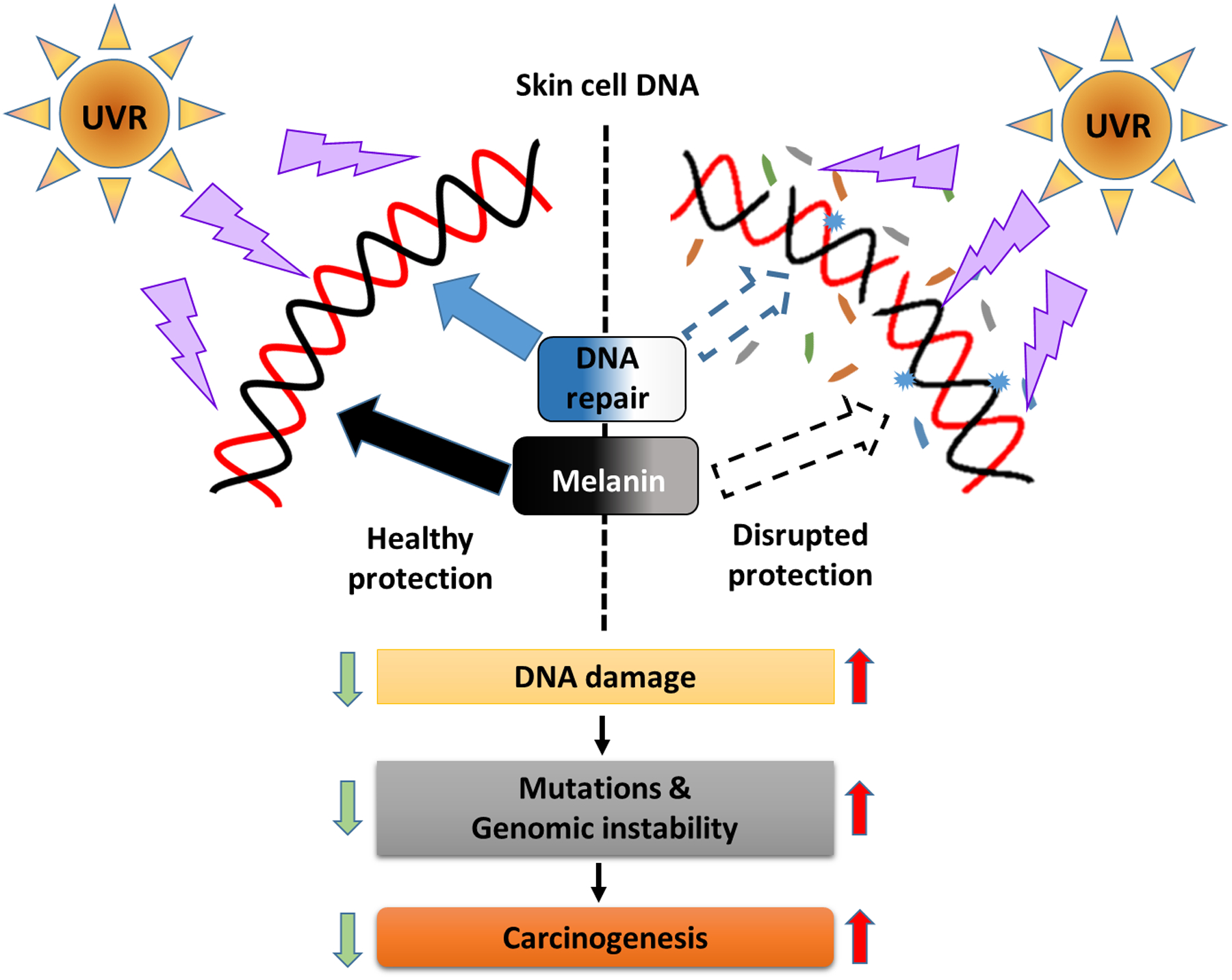
Solar UVR reaching the Earth’s surface has been associated with a plethora of skin ailments inflicted mainly by UV-induced DNA damage. In response to this environmental exposure, our skin has developed various defense mechanisms. Of these mechanisms, tanning caused upon the synthesis of melanin and DNA repair, especially nucleotide excision repair (NER), es regarded as the first responder to UVR exposure. Melanin in the skin is regarded as a natural UV protectant that is produced primarily upon UVR exposure, whereas NER is responsible for the removal of UV-induced DNA lesions including CPDs and (6–4) PPs; NER also protects genomic DNA from UV signature mutations. At a basal level, NER is always active in the skin, such that it scans the genome to find DNA damage; however, during UV exposure, DNA damage sensors like ATR are activated which may further enhance its activity. Both the mechanisms are vital for maintaining skin homeostasis, while failure of either or both processes, as with Caucasian populations or xeroderma pigmentosum patients, can increase the basal level of DNA damage caused upon UVR exposure. If this damage is not appropriately repaired, it can lead to an increase in mutation load; ultimately, the accumulation of such mutation leads to genomic instability and skin cancers including melanomas.
Conversely, UVA constitutes the majority of the UVR reaching the Earth’s surface. Due to UVA’s long wavelength, it penetrates deeper into the dermis of the skin with minimal resistance and causes direct or indirect DNA damage mostly through lipid peroxidation and oxidative stress (Jiang et al. 2009; Mouret et al. 2010; Brem and Karran 2012; Brem et al. 2017). For a long time, it was a common belief that UVA was harmless to skin and was a safe way to tan our skin, which led to the increasing use of UVA in the tanning bed industry (Choi et al. 2010). However, CPDs have been observed upon UVA exposure, and while relatively less dangerous compared to UVB exposure, claims about the harmlessness of UVA have come under doubt (Freeman et al. 1989; Mouret et al. 2006).
Recently, the reports of delayed cutaneous effects of UVA have also gained attention. UVA has been associated with formation of reactive oxygen species (ROS) including reactive singlet oxygen, superoxide anion, hydrogen peroxide, and hydroxyl radicals, upon excitation of non-DNA chromophores of the cells, which in turn could damage genomic DNA by formation of 7,8-dihydro-8-oxoguanine (8-OxoG) and may contribute to deleterious cancerous mutations including the G→T transversion or T→G mutation (Drobetsky et al. 1995; Cooke et al. 2003). Researchers estimate that melanocytes develop more 8-OxoG compared to keratinocytes upon UVA exposure, possibly due to ROS formed upon biosynthesis of melanin in melanocytes (Mouret et al. 2012; Denat et al. 2014). In light of these findings, the International Agency for Research on Cancer (IARC) reclassified UVA-emitting tanning devices from group 2A to group 1 carcinogen as of 2009 (El Ghissassi et al. 2009). In addition, both UVB and UVA have been associated with the formation of dark CPDs in melanocytes through melanin both in vitro and in vivo (Premi et al. 2015). These delayed dark CPDs formed on genomic DNA cause mutations as seen by UV exposure, but the only difference is that they are formed in the complete absence of UVR. This paradigm-changing observation that melanin, which is regarded as a broad UV protectant of the skin, a potential contributor of genomic DNA damage as well. Overall, the UV-induced effect on the skin is complicated, as the extent of UV damage depends not only on the type of UVR but also upon the cellular composition and requires further investigation at the mechanistic level.
DNA damage response (DDR) signaling: a dominion over melanocyte fate
As discussed above, UVR exposure causes direct DNA damage through CPDs and (6–4) PPs formation. UVR exposure also causes indirect DNA damage, such as 8-OxoG formation, which if not repaired correctly may lead to bulky lesions and single strand breaks (SSBs) (Dedon 2008; Kaufmann 2010; Iyer and Rhind 2017). These DNA breaks initiate two different types of DDR signaling, i.e. (ATR/CHK1) and (ATM/CHK2) signaling. Generally, UVR exposure causes the formation of long stretches of single strand DNA (ssDNA) either due to DNA polymerase stalling or transcriptional stalling, or during intermediate steps of DNA repair (Byun et al. 2005; Kemp and Sancar 2016; Kemp 2017). This ssDNA is then readily bound by RPA, which recruits ATR-ATRIP complex and further initiates CHK1-mediated cellular responses, including cell cycle arrest, replication fork stabilization, and apoptosis (Khan et al. 2018). Single strand breaks are relatively easy to fix, mostly error-free, and are mainly repaired by homologous recombination.
In contrast, ATM/CHK2 DDR signaling is activated upon DSBs, which is not a typical UV-induced DNA damage. However, recent studies have shown that pyrimidine dimers, oxidized bases, and unrepaired single strand DNA caused by UVR exposure may lead to DSBs (Guo et al. 2010; Kaufmann 2010; Kemp et al. 2011; Dungrawala et al. 2015; Iyer and Rhind 2017). Even in non-replicating cells, activation of ATM through NER dependent or independent mechanisms are observed upon UVR exposure (Wakasugi et al. 2014; Kemp and Sancar 2016). Activated ATM/CHK2 signaling initiates cell cycle arrest, replication origin firing, replication fork stabilization, DNA repair, and apoptosis, which are like ATR/CHK1 signaling targets. Therefore, it may be challenging to differentiate between the two types of DDR signaling events, as there seems to be a considerable amount of cross-talk between them (Sancar et al. 2004; Ciccia and Elledge 2010). In melanocytes, studies have shown robust DNA repair mechanisms which can be broadly classified as nucleotide excision repair (NER), base excision repair (BER) and DNA strand break repair (Non-homologous end-joining and homologous recombination) (Gaddameedhi et al. 2010; Mouret et al. 2012; Denat et al. 2014). Our review mainly focuses on understanding the NER mechanism and its regulators in melanocytes, whereas other repair systems lie beyond the scope of this review.
NER is a primary DNA repair mechanism that detects and repairs UV-induced photolesions, CPDs, and (6–4) PPs formed in the genomic DNA (Reardon and Sancar 2005; Scharer 2013). NER is subclassified as either global genomic NER (GG-NER) or transcriptional coupled NER (TC-NER). These two subclassifications of NER differ concerning how and where (non-transcribed or actively transcribed regions of the DNA, respectively) the UV-induced DNA lesions are detected in the genome (de Laat et al. 1999). In GG-NER, the pyrimidine lesions are detected by a recognition complex made up of xeroderma pigmentosum (XP) group C (XPC), RAD23B, and DNA damage-binding protein (DDB2), which bind to the opposite DNA strand of the photolesions (Sugasawa 2010; Ruthemann et al. 2016). In contrast, TC-NER is activated upon RNA-polymerase II stalling at the UV-induced photolesions, which recruits other cofactors like Cockayne syndrome (CS) proteins (CSA and CSB)(de Laat et al. 1999). Apart from the differences in the initial DNA lesion recognition step, GG-NER and TC-NER follow similar steps in removing the photolesions from the genomic DNA (Sancar et al. 2004; Wood 2010; Marteijn et al. 2014).
The complete NER process requires more than 30 proteins to act sequentially to process the DNA photoproducts and protect the genomic DNA from UV-induced mutations (Figure 4). The significance of NER in the protection of melanocytes is evident of up to 2000-fold increase of melanoma risk in xeroderma pigmentosum (XP) patients, who have compromised NER genes (Cleaver 1968; Kraemer et al. 1994; Wang et al. 2009; Bradford et al. 2011). Further, studies have shown UV signature mutations in the XP patients with melanoma indicate a correlation between UV exposure and the potential role of NER in impeding melanomagenesis (Daya-Grosjean 2008). As NER is one of the first responders in the skin against UVR exposure, it is in our best interest to identify the various regulators of this mechanism in melanocytes, which directly or indirectly augment NER to maintain genomic integrity.
Figure 4. Nucleotide excision repair.

UV-induced DNA damage lesions, particularly CPDs and (6–4) PPs, are detected by NER damage sensors, i.e., XPC-RAD23-DDB2 for global genome NER (GG-NER) and RNApol-CSA-CSB-XAB2 for transcription-coupled NER (TC-NER). Upon detection, NER damage sensors initiates recruitment of DNA structural unwinding and stabilizing proteins like the replication protein A (RPA), Transcription factor II-H (TFIIH), and XPA. The helicase enzymes XPB and XPD, which are part of TFIIH, are responsible for the unwinding of DNA, while XPA plays a vital role in stabilizing the whole complex. This is followed by binding the specific 5’ and 3’ strand endonucleases XPF-ERCC1 and XPG, respectively, to the repair fork which cleaves a 25–30 nucleotide long DNA strand (associated with TFIIH) containing the UV-induced DNA lesion, followed by DNA synthesis involving proteins like RPA, PCNA, replication factor C (RF-C) and DNA polymerase (pol) δ/ε. Briefly, RF-C binds to the 3’ open terminal of the DNA, allowing loading of PCNA, a homotrimeric ring-shaped clamp used to track duplex DNA. DNA Pol δ/ε uses this platform to synthesize single strand DNA in the presence of haloenzymes. In this step, PCNA plays a vital role as a mediator between cell cycle and DNA repair, where it participates in replication arrest and allows DNA repair to prevent generation of faulty mutagenic DNA. Finally, the new synthesized DNA is ligated at the 5’ end by DNA ligases.
αMelanocortin Stimulating Hormone (αMSH) and Endothelin-1 (ET-1): Facilitators of melanocyte-NER
At a cellular level, the microenvironment of skin is diverse and hosts multiple signaling cross-talks between numerous cell types, including keratinocytes, fibroblasts, immune cells, and melanocytes (Nordlund 2007). This interconnected nature of the skin helps it efficiently protect the body from various environmental stressors like UVR. One of the well-known examples of the heterogeneity of the skin is melanin biosynthesis upon UVR exposure (Tsatmali et al. 2002; Lin and Fisher 2007). This process initiates due to UV-induced DNA damage in keratinocytes, which triggers p53-mediated αMSH (a paracrine hormone) synthesis. The αMSH, thus produced from keratinocytes, binds to the Melanocortin 1 receptor (MC1R) found on melanocytes and further initiates cAMP-mediated melanogenesis pathway, which is responsible for absorption of excess UVR and causes tanning. In parallel, growth hormones from basic fibroblast growth factor (bFGF), hepatocyte growth factor (HGF), and soluble stem cell factor (SCF) have also been shown to influence pigment regulation in melanocytes, indicating its strong interaction with its neighboring cell types which directly or indirectly influences melanin synthesis (Imokawa et al. 1986; Halaban et al. 1988; Matsumoto et al. 1991; Wu et al. 2000; Imokawa 2004). In the past two decades, complex paracrine networks have been identified These paracrine networks affect major cellular functions of melanocytes, other than melanogenesis, like the melanocyte DDR signaling mechanisms, including NER. In the context of enhanced UV protection, it important to discuss these signaling pathways in detail.
αMSH is one of the well-studied regulatory paracrine factors of NER regulation in melanocytes. As noted earlier, αMSH is secreted by keratinocytes upon post-transcriptional cleavage of proopiomelanocortin (POMC) following UVR exposure. It specifically binds to the G protein-coupled receptor known as MC1R found on the surface of melanocytes and transcends its effect (Rees 2000; Garcia-Borron et al. 2014; Wolf Horrell et al. 2016). Concerning DDR signaling, endogenous stimulation of MC1R has been shown to enhance NER efficacy and genomic stability (Swope et al. 2014; Jarrett et al. 2017). Enhanced CPD removal in αMSH treated melanocytes post-UV exposure was first shown simultaneously by two groups, indicating the effect of MSH/MC1R signaling cascade on melanocyte DNA repair (Bohm et al. 2005; Kadekaro et al. 2005). Further, Malek’s group confirmed their finding in a follow-up study by using the loss of function MC1R expressing melanocytes, which showed that MC1R genotype influenced DNA damage response in human melanocytes (Kadekaro et al. 2010). Later, it was reported that an increase in DNA repair upon MC1R activation was due to elevated levels of XPC and γH2AX (Swope et al. 2014). In another study from D’Orazio’s group, it was elegantly demonstrated that stimulation of MC1R augments NER through PKA-mediated ATR protein phosphorylation at S435, which in turn stabilizes XPA and allows its colocalization at the UV-induced DNA photolesions in the nucleus (Jarrett et al. 2014). Researchers in this study also found MC1R-ATR-XPA signaling cascade increases XPA interaction with XPF, ERCC1, and RPA, enhancing 5’ strand incision at the site of photodamage and ultimately increasing NER activity. This concept was confirmed in a recent study by Malek’s group, where they demonstrated αMSH and endothelin-1 influenced localization of XPA in melanocyte nuclear and chromatin fraction upon UVB exposure (Swope et al. 2019).
As ATR dependent stabilization of XPA and its localization to UV-damaged DNA sites has been reported earlier, the above studies strongly indicate a possible role of αMSH and endothelin1 signaling in regulating the DNA repair mechanism through ATR-XPA signaling (Wu et al. 2007; Kang et al. 2011; Li et al. 2013; Lee et al. 2014). Though most of the essential details of the pathway are known, some questions still linger with respect to the sequential order or the cellular location of the ATR-mediated XPA phosphorylation event prior to the translocation of XPA to the UV-induced DNA damage site in chromatin. Answering these questions will potentially help us determine the hierarchical order of the MC1R-ATR-XPA signaling axis in melanocytes following UVR exposure and may provide insight to another unknown mechanism that might play a protective role as well. Also, MC1R is known to have multiple single nucleotide polymorphisms and is known to affect the color of our skin and hair along with our susceptibility to UVR; therefore, such mutations could potentially affect the regulation of NER by αMSH as well, especially in people with loss of MC1R function mutation(s) (Rana et al. 1999; Scott et al. 2002; Kadekaro et al. 2010; Morgan et al. 2018).
In addition to the MC1R-ATR-XPA pathway, microphthalmia-associated transcription factor (MITF), a primary target of the MC1R/cAMP/CREB pathway, plays a significant role in influencing UV-induced DDR signaling, especially melanin pigmentation (Strub et al. 2011). Previously, MITF has been shown to regulate vital melanocyte functions like metabolism, differentiation, survival, and cell cycle, and is known as the master regulator of melanocyte biology (Yasumoto et al. 1994; Bertolotto et al. 1998; McGill et al. 2002; Du et al. 2004; Haq et al. 2013; Kawakami and Fisher 2017). Understanding the regulation of DDR signaling by MITF stemmed from a study published by Davidson’s group, which, through ChIP Seq and RNA seq experiments, showed MITF binds to the promoter region of DNA repair proteins including BRACA1, FANCA, and DNA ligases1, which strongly indicates the influence of MITF on the DNA repair pathway; however, the nature of this regulation, i.e., positive or negative, is not well understood (Strub et al. 2011). A recent study by Wolf Horrell et al. showed that cAMP-mediated NER regulation and cAMP-induced melanogenesis are two separate events, where cAMP-mediated MITF activation does not affect the NER pathway (Wolf Horrell et al. 2017). In contrast, a study published in the same year by Xia et al. reported that MED23 acts as a mediator between pigmentation and DNA repair by regulating MITF expression (Xia et al. 2017). The researchers demonstrated that the loss of MED23 enhances NER activity and impairs pigmentation potentially through MITF and vice versa. They also showed that loss of MITF upregulates ATR, XPA, and Rad51, indicating a negative regulation of NER by MITF. Though the study makes a convincing correlation between MED23, MITF and NER, however, the molecular mechanism was not clearly addressed by the authors. As MITF regulates CDK2 expression and inhibition of CDK2 by p21 is essential for DDR progression, this could possibly explain the observation by Xia et al., however, this need further exploration (Du et al. 2004; Satyanarayana and Kaldis 2009). Another recent study by Seoane et al. brings forth a direct transcriptional regulation of GTF2H1/p62, a core element in the TFIIH complex by MITF, which positively influences NER activity (Seoane et al. 2019). To this end, some caution is recommended in interpreting the data, as the authors used unscheduled DNA synthesis assay to measure NER activity, which is an indirect assay for measuring DNA repair since it mainly accounts for DNA synthesis in proliferating cells and the effect measured through this assay could be a combination of both. Nevertheless, the study makes a case for further investigation of MITF’s role in regulating NER with a systematic experimental approach using highly sensitive DNA repair assays. Taken together, the studies mentioned above indicate a potential role of MITF in regulating NER can be justified. Though much remains uncertain about this regulatory pathway, further studies can help clear up these conflicting observations.
Like αMSH, UV-induced keratinocyte-derived endothelin-1 (ET-1) helps maintain melanocyte homeostasis by regulating its proliferation, development, and pigmentation (Yada et al. 1991; Imokawa et al. 1992; Yohn et al. 1993). ET-1 is synthesized upon activation of p53 due to UV-induced DNA damage in the keratinocytes, which binds to endothelin B receptors (ETBR) (Hyter et al. 2013). ETBR is a Gq-coupled receptor (a subtype of G protein-coupled receptor) found on the surface of melanocytes, which is responsible for mobilizing intracellular Ca2+ by activating PKC-calmodulin pathway (Kang et al. 1998). The effect of ET-1 on NER and apoptosis in UV-irradiated melanocytes was first shown by Abdel-Malek’s group, and over the years through multiple studies, they have demonstrated an increase in NER upon ETBR stimulation was due to activation of JNK and p38 signaling pathways (Kadekaro et al. 2005; von Koschembahr et al. 2015). Further studies by Abdel-Malek’s group suggested that an increase in NER with ET-1 stimulation was due to an enhanced UV-induced DNA damage recognition, which was indicated by increased clearance of XPC. Very recently, another elegant study from the same group has compared the effect of αMSH and ET-1 on DNA damage sensor protein ATR and found that both paracrine factors act as a backup for each other in phosphorylating ATR, which further enhances DNA damage recognition and NER activity (Swope et al. 2019). Therefore, distinct signaling pathways like ET-1 and αMSH pathways regulate common targets in the DDR signaling, ensuring a effective clearance of mutation-causing DNA damage by upregulating NER activity, which eventually helps maintain genomic stability of melanocytes. Such redundant signaling pathways could be one of the reasons for lower melanoma incidents in humans.
Circadian clock: Keeper of the NER function
Apart from paracrine regulators, melanocytes have an endogenous timekeeping mechanism called the circadian clock, which plays a vital role in maintaining the melanocyte homeostasis efficiently and aids in cellular protection from various environmental stressors, including UVR (Zanello et al. 2000; Sandu et al. 2012). The circadian clock can be broadly classified as a master clock, present in the suprachiasmatic nucleus of the brain, and as a peripheral clock present in almost every cell of our body, including the skin. The master clock gets entrained by external cues like the 24-hour light/dark cycles, upon which it to send out hormonal and neuronal signals to entrain the peripheral clocks present in various organs including skin (Lin et al. 2009; Matsui et al. 2016). This synchronization of master and peripheral clocks generates diverse biological rhythms including biochemical, physiological, and behavioral rhythms (Reppert and Weaver 2001; Hastings et al. 2003). Though the circadian clock gets entrained to maintain a 24-hour rhythm, at a molecular level it is self-sustained, and functions via a transcriptional translational feedback loop made up of core clock proteins called Brain and Muscle ARNT-Like 1 (BMAL1), Circadian Locomotor Output Cycles Kaput (CLOCK), Cryptochrome (CRY 1,2), and Period (Per 1, 2, 3) (Takahashi 2017). Further, through RNA seq studies, the circadian clock is known to regulate around 43% mouse and 50% human protein-coding genes somewhere in the body, but mostly in a tissue-specific manner (Zhang et al. 2014; Ruben et al. 2018).
The circadian clock has previously been shown to regulate DDR signaling either by transcriptional regulation or by direct interaction with crucial genes or proteins of the DDR pathway, respectively. Sancar’s group first showed the potential effect of the circadian clock in regulating DDR signaling in mammals. They reported a protein: protein interaction of circadian clock protein CRY2 and cell cycle protein timeless, which is known to be an integral part of the DNA damage sensing complex (Unsal-Kacmaz et al. 2005). In a succeeding series of studies, Sancar’s group has systematically demonstrated a circadian regulation of XPA, such that XPA mRNA, protein, and NER activity show circadian oscillation with 24 hours in various mouse organs including skin (Kang et al. 2009; Kang et al. 2010; Gaddameedhi et al. 2011; Kang et al. 2011). Recently, our group has reported that this effect could be due to transcriptional regulation of XPA by BMAL1, a core clock protein (Dakup et al. 2018). The Sancar group has previously performed detailed studies to tease out the effect of time-dependent changes in NER capacity toward the ability of the mouse skin to protect against UVR-induced sunburn erythema and skin cancer. They found the likelihood of developing sunburn or skin cancer in the morning UV-treated mice, when the NER activity is low, was higher compared to the evening when NER activity is high (Gaddameedhi et al. 2011; Gaddameedhi et al. 2015). Also, these studies reported that low XPA levels correlated with elevated ATR and ATR downstream effectors like CHK1 and p53. To this end, as most of these studies were done in mouse models, we hypothesized that in humans, it would be opposite because humans are diurnal, while mice are nocturnal animals. This hypothesis resonates with a well-designed human study by Nikkola et al. which recently showed significantly higher sunburn erythema index scores in human skin exposed to UVB in the evening compared to morning time (Nikkola et al. 2018; Sarkar and Gaddameedhi 2018). Apart from controlling NER, Clock has been shown to influence OGG1 expression, an essential protein in the BER pathway, in a time of the day dependent manner in human blood samples. It was reported that the genomic DNA had less 8-oxo-G products when the OGG levels were high compared to the time when it was low (Manzella et al. 2015). As 8-oxo-G DNA lesions are readily formed in melanocytes due to oxidative degradation of melanin upon UV exposure, similar regulation of BER could be hypothesized in melanocytes as well (Cooke et al. 2003). Therefore, the above studies indicate a potential regulation of DDR signaling by the circadian clock in melanocytes; however, it seems that we have just scratched the surface of this complex regulation and therefore requires further investigations.
Conclusions and future directions:
The conversion of melanocyte to melanoma is a rare incident, which is evident by the low number of melanoma cases reported in the US. Also, melanocytes are slow-dividing cells compared to keratinocytes; therefore, melanocytes have more chances of developing UVR induced DNA damage over their life span (Jimbow et al. 1975). A prompt and robust NER process keeps the DNA damage in check and maintains genomic integrity, but the failure of NER allows melanocytes to accumulate cancerous mutations, which ultimately pushes them onto the melanomagenesis path. Once transformed into melanoma there are curative therapies that can help remove the transformed cells; however, no therapies currently exist which can reverse melanoma back to normal functioning melanocytes (Lo and Fisher 2014; Johnson and Sosman 2015). Therefore, the prevention of melanocytes from transforming to melanoma could potentially be a practical approach to halt melanoma in its stride. Therefore, an in-depth understanding of the biology of innate protective mechanisms of melanocytes against UVR will be helpful. In this review, we discussed the presence of efficient NER in melanocytes and its cellular regulators; however, not much is known regarding melanocyte-specific DNA repair efficacy compared to other types of cells present in the skin. A study by Tang’s group compared melanocytes to fibroblast cells and identified melanocytes with impaired DNA damage repair (Wang et al. 2010). As the authors used cell free extracts from melanocytes and fibroblasts to measure DNA repair, they concluded that the presence of melanin negatively influences DNA repair. This observation is in accordance with our previously published data, where UV photoproduct removal was inhibited by synthetic melanin in vitro. However, physiologically melanin in melanocytes is packaged and stored in lysosome like cellular organelles and melanosomes. Therefore, the interference of melanin in DNA repair activity is physiologically irrelevant (Lin and Fisher 2007; Gaddameedhi et al. 2010). Another reason for finding a reduced repair capacity of melanocytes in this study could be due to not considering the effect of paracrine signaling on melanocytes, which can further augment the repair capacity. Therefore, unraveling the mechanistic differences in repair capacity between melanocytes and other skin cell types might help develop targeted therapies to specifically enhance the innate protective mechanism of melanocytes against UVR.
As the effect of paracrine factors on melanocyte-NER activity is spearheaded by Malek’s and D’Orazio’s group through systematic approaches, the influence of other micro-environmental factors such as circadian clock is still not well understood, which can potentially influence paracrine/DDR signaling axis in melanocytes. In line with this, a recent study by D’Orazio’s group showed cAMP-mediated SIRT1 activation regulates the deacetylation of XPA and increases ATR-mediated XPA phosphorylation, which further influences NER capacity (Jarrett et al. 2018). As SIRT1 has been known to interact with core clock proteins, a potential circadian clock/SIRT1/XPA post-translational signaling axis may impact paracrine-influenced NER capacity in melanocytes (Asher et al. 2008). In conclusion, we attempt to encompass the complexity of DDR signaling cascades, its regulators and multiple cross talks in melanocytes, which ultimately adds into the protective arsenal of the melanocytes against UVR (Figure 5).
Figure 5. Regulators of nucleotide excision repair (NER) in melanocytes.

NER is the major DNA repair pathway in mammals which removes UV-induced CPDs and (6–4) PPs from the genomic DNA. It is broadly classified as global genomic NER (GG-NER) and transcription-coupled NER (TC-NER). Of the two, GG-NER is the major type of NER which is initiated upon detection of photolesions by XPC (sensor proteins), which then recruits other NER proteins. NER in its entirety is a self-driven process which helps maintain the genomic integrity of the cell; however, it takes input from various cellular process to function effectively. In melanocytes, apart from the standard endogenous regulator of NER, it is influenced by exogenous factors like the melanin stimulating hormone (MSH) and endothelin 1 (ET1), classified as paracrine factors. These factors influence the DNA damage sensor protein ATR, which in turn influences NER either directly through a p53-mediated pathway or by stabilization of XPA, an essential rate-limiting protein of NER. In parallel, the circadian clock influences NER through transcriptional regulation of XPA. Overall, the clock regulation of NER makes the process efficient, as it anticipates the requirement of NER according to time of the day.
ACKNOWLEDGMENTS/FUNDING
We thank Dr. Jack M. Downs (Washington State University) for his valued suggestions. This work was supported by grants from the National Institutes of Health ES030113, CA227381 (S.G.), in part by the Congressionally Directed Medical Research Program Award CA171123 (S.G.) and Melanoma Research Alliance Team Science Award (S.G.).
Footnotes
CONFLICT OF INTERESTS
The authors declare that they have no conflicts of interest with the contents of this article.
REFERENCES
- Armstrong BK, Kricker A. 1993. How much melanoma is caused by sun exposure? Melanoma Res 3(6):395–401. [DOI] [PubMed] [Google Scholar]
- Armstrong CA, Tara DC, Hart CE, Kock A, Luger TA, Ansel JC. 1992. Heterogeneity of cytokine production by human malignant melanoma cells. Exp Dermatol 1(1):37–45. [DOI] [PubMed] [Google Scholar]
- Arondekar B, Curkendall S, Monberg M, Mirakhur B, Oglesby AK, Lenhart GM, Meyer N. 2015. Economic burden associated with adverse events in patients with metastatic melanoma. J Manag Care Spec Pharm 21(2):158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U. 2008. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134(2):317–328. [DOI] [PubMed] [Google Scholar]
- Bayerl C, Taake S, Moll I, Jung EG. 1995. Characterization of sunburn cells after exposure to ultraviolet light. Photodermatol Photoimmunol Photomed 11(4):149–154. [DOI] [PubMed] [Google Scholar]
- Bertolotto C, Busca R, Abbe P, Bille K, Aberdam E, Ortonne JP, Ballotti R. 1998. Different cis-acting elements are involved in the regulation of TRP1 and TRP2 promoter activities by cyclic AMP: pivotal role of M boxes (GTCATGTGCT) and of microphthalmia. Mol Cell Biol 18(2):694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogh MK, Schmedes AV, Philipsen PA, Thieden E, Wulf HC. 2011. Vitamin D production depends on ultraviolet-B dose but not on dose rate: a randomized controlled trial. Exp Dermatol 20(1):14–18. [DOI] [PubMed] [Google Scholar]
- Bohm M, Wolff I, Scholzen TE, Robinson SJ, Healy E, Luger TA, Schwarz T, Schwarz A. 2005. alpha-Melanocyte-stimulating hormone protects from ultraviolet radiation-induced apoptosis and DNA damage. J Biol Chem 280(7):5795–5802. [DOI] [PubMed] [Google Scholar]
- Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, Boyle J, Oh KS, Imoto K, Inui H, Moriwaki S, Emmert S, Pike KM, Raziuddin A, Plona TM, DiGiovanna JJ, Tucker MA, Kraemer KH. 2011. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet 48(3):168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash DE. 2015. UV signature mutations. Photochem Photobiol 91(1):15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem R, Guven M, Karran P. 2017. Oxidatively-generated damage to DNA and proteins mediated by photosensitized UVA. Free Radic Biol Med 107:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem R, Karran P. 2012. Multiple forms of DNA damage caused by UVA photoactivation of DNA 6-thioguanine. Photochem Photobiol 88(1):5–13. [DOI] [PubMed] [Google Scholar]
- Brown TM, Krishnamurthy K. 2020. Histology, Dermis. StatPearls. Treasure Island (FL). [PubMed] [Google Scholar]
- Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. 2005. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 19(9):1040–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinali G, Bolasco G, Aspite N, Lucania G, Lotti LV, Torrisi MR, Picardo M. 2008. Melanosome transfer promoted by keratinocyte growth factor in light and dark skin-derived keratinocytes. J Invest Dermatol 128(3):558–567. [DOI] [PubMed] [Google Scholar]
- Choi K, Lazovich D, Southwell B, Forster J, Rolnick SJ, Jackson J. 2010. Prevalence and characteristics of indoor tanning use among men and women in the United States. Arch Dermatol 146(12):1356–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40(2):179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaver JE. 1968. Defective repair replication of DNA in xeroderma pigmentosum. Nature 218(5142):652–656. [DOI] [PubMed] [Google Scholar]
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J. 2003. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17(10):1195–1214. [DOI] [PubMed] [Google Scholar]
- Cui R, Widlund HR, Feige E, Lin JY, Wilensky DL, Igras VE, D’Orazio J, Fung CY, Schanbacher CF, Granter SR, Fisher DE. 2007. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell 128(5):853–864. [DOI] [PubMed] [Google Scholar]
- Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Brocker EB, LeBoit PE, Pinkel D, Bastian BC. 2005. Distinct sets of genetic alterations in melanoma. N Engl J Med 353(20):2135–2147. [DOI] [PubMed] [Google Scholar]
- Dakup PP, Porter KI, Little AA, Gajula RP, Zhang H, Skornyakov E, Kemp MG, Van Dongen HPA, Gaddameedhi S. 2018. The circadian clock regulates cisplatin-induced toxicity and tumor regression in melanoma mouse and human models. Oncotarget 9(18):14524–14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daya-Grosjean L. 2008. Xeroderma pigmentosum and skin cancer. Adv Exp Med Biol 637:19–27. [DOI] [PubMed] [Google Scholar]
- de Gruijl FR, van der Leun JC. 2000. Environment and health: 3. Ozone depletion and ultraviolet radiation. CMAJ 163(7):851–855. [PMC free article] [PubMed] [Google Scholar]
- de Laat WL, Jaspers NG, Hoeijmakers JH. 1999. Molecular mechanism of nucleotide excision repair. Genes Dev 13(7):768–785. [DOI] [PubMed] [Google Scholar]
- Dedon PC. 2008. The chemical toxicology of 2-deoxyribose oxidation in DNA. Chem Res Toxicol 21(1):206–219. [DOI] [PubMed] [Google Scholar]
- Denat L, Kadekaro AL, Marrot L, Leachman SA, Abdel-Malek ZA. 2014. Melanocytes as instigators and victims of oxidative stress. J Invest Dermatol 134(6):1512–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing SW, Roth SI. 1974. The derivation of the cells of the epidermal strata of the boa constrictor (Constrictor constrictor). J Invest Dermatol 62(4):450–457. [DOI] [PubMed] [Google Scholar]
- Drobetsky EA, Turcotte J, Chateauneuf A. 1995. A role for ultraviolet A in solar mutagenesis. Proc Natl Acad Sci U S A 92(6):2350–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Widlund HR, Horstmann MA, Ramaswamy S, Ross K, Huber WE, Nishimura EK, Golub TR, Fisher DE. 2004. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell 6(6):565–576. [DOI] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D. 2015. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol Cell 59(6):998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisinger M, Marko O. 1982. Selective proliferation of normal human melanocytes in vitro in the presence of phorbol ester and cholera toxin. Proc Natl Acad Sci U S A 79(6):2018–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ghissassi F, Baan R, Straif K, Grosse Y, Secretan B, Bouvard V, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V, Group WHOIAfRoCMW. 2009. A review of human carcinogens--part D: radiation. Lancet Oncol 10(8):751–752. [DOI] [PubMed] [Google Scholar]
- EPA. 2014. Radiation From Solar Activity.
- Fitzpatrick TB, Breathnach AS. 1963. [the Epidermal Melanin Unit System]. Dermatol Wochenschr 147:481–489. [PubMed] [Google Scholar]
- Freeman SE, Hacham H, Gange RW, Maytum DJ, Sutherland JC, Sutherland BM. 1989. Wavelength dependence of pyrimidine dimer formation in DNA of human skin irradiated in situ with ultraviolet light. Proc Natl Acad Sci U S A 86(14):5605–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E. 2007. Scratching the surface of skin development. Nature 445(7130):834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E, Raghavan S. 2002. Getting under the skin of epidermal morphogenesis. Nat Rev Genet 3(3):199–209. [DOI] [PubMed] [Google Scholar]
- Gaddameedhi S, Kemp MG, Reardon JT, Shields JM, Smith-Roe SL, Kaufmann WK, Sancar A. 2010. Similar nucleotide excision repair capacity in melanocytes and melanoma cells. Cancer Res 70(12):4922–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaddameedhi S, Sancar A. 2011. Melanoma and DNA damage from a distance (farstander effect). Pigment Cell Melanoma Res 24(1):3–4. [DOI] [PubMed] [Google Scholar]
- Gaddameedhi S, Selby CP, Kaufmann WK, Smart RC, Sancar A. 2011. Control of skin cancer by the circadian rhythm. Proc Natl Acad Sci U S A 108(46):18790–18795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaddameedhi S, Selby CP, Kemp MG, Ye R, Sancar A. 2015. The circadian clock controls sunburn apoptosis and erythema in mouse skin. J Invest Dermatol 135(4):1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Borron JC, Abdel-Malek Z, Jimenez-Cervantes C. 2014. MC1R, the cAMP pathway, and the response to solar UV: extending the horizon beyond pigmentation. Pigment Cell Melanoma Res 27(5):699–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloster HM Jr., Neal K. 2006. Skin cancer in skin of color. J Am Acad Dermatol 55(5):741–760; quiz 761-744. [DOI] [PubMed] [Google Scholar]
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. 2010. ATM activation by oxidative stress. Science 330(6003):517–521. [DOI] [PubMed] [Google Scholar]
- Guy GP Jr., Machlin SR, Ekwueme DU, Yabroff KR. 2015. Prevalence and costs of skin cancer treatment in the U.S., 2002–2006 and 2007–2011. Am J Prev Med 48(2):183–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaban R, Ghosh S, Duray P, Kirkwood JM, Lerner AB. 1986. Human melanocytes cultured from nevi and melanomas. J Invest Dermatol 87(1):95–101. [DOI] [PubMed] [Google Scholar]
- Halaban R, Langdon R, Birchall N, Cuono C, Baird A, Scott G, Moellmann G, McGuire J. 1988. Basic fibroblast growth factor from human keratinocytes is a natural mitogen for melanocytes. J Cell Biol 107(4):1611–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144(5):646–674. [DOI] [PubMed] [Google Scholar]
- Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, Arany Z, Widlund HR. 2013. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 23(3):302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings MH, Reddy AB, Maywood ES. 2003. A clockwork web: circadian timing in brain and periphery, in health and disease. Nat Rev Neurosci 4(8):649–661. [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DS, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L. 2012. A landscape of driver mutations in melanoma. Cell 150(2):251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH. 2009. DNA damage, aging, and cancer. N Engl J Med 361(15):1475–1485. [DOI] [PubMed] [Google Scholar]
- Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R. 2013. TERT promoter mutations in familial and sporadic melanoma. Science 339(6122):959–961. [DOI] [PubMed] [Google Scholar]
- Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. 2013. Highly recurrent TERT promoter mutations in human melanoma. Science 339(6122):957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyter S, Coleman DJ, Ganguli-Indra G, Merrill GF, Ma S, Yanagisawa M, Indra AK. 2013. Endothelin-1 is a transcriptional target of p53 in epidermal keratinocytes and regulates ultraviolet-induced melanocyte homeostasis. Pigment Cell Melanoma Res 26(2):247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imokawa G. 2004. Autocrine and paracrine regulation of melanocytes in human skin and in pigmentary disorders. Pigment Cell Res 17(2):96–110. [DOI] [PubMed] [Google Scholar]
- Imokawa G, Kawai M, Mishima Y, Motegi I. 1986. Differential analysis of experimental hypermelanosis induced by UVB, PUVA, and allergic contact dermatitis using a brownish guinea pig model. Arch Dermatol Res 278(5):352–362. [DOI] [PubMed] [Google Scholar]
- Imokawa G, Yada Y, Miyagishi M. 1992. Endothelins secreted from human keratinocytes are intrinsic mitogens for human melanocytes. J Biol Chem 267(34):24675–24680. [PubMed] [Google Scholar]
- Iyengar B, Misra RS. 1987. Reaction of dendritic melanocytes in vitiligo to the substrates of tyrosine metabolism. Acta Anat (Basel) 129(3):203–205. [DOI] [PubMed] [Google Scholar]
- Iyer DR, Rhind N. 2017. The Intra-S Checkpoint Responses to DNA Damage. Genes (Basel) 8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett SG, Carter KM, Bautista RM, He D, Wang C, D’Orazio JA. 2018. Sirtuin 1-mediated deacetylation of XPA DNA repair protein enhances its interaction with ATR protein and promotes cAMP-induced DNA repair of UV damage. J Biol Chem 293(49):19025–19037. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Jarrett SG, Carter KM, D’Orazio JA. 2017. Paracrine regulation of melanocyte genomic stability: a focus on nucleotide excision repair. Pigment Cell Melanoma Res 30(3):284–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett SG, Wolf Horrell EM, Christian PA, Vanover JC, Boulanger MC, Zou Y, D’Orazio JA. 2014. PKA-mediated phosphorylation of ATR promotes recruitment of XPA to UV-induced DNA damage. Mol Cell 54(6):999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Jiang Y, Rabbi M, Kim M, Ke C, Lee W, Clark RL, Mieczkowski PA, Marszalek PE. 2009. UVA generates pyrimidine dimers in DNA directly. Biophys J 96(3):1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimbow K, Roth SI, Fitzpatrick TB, Szabo G. 1975. Mitotic activity in non-neoplastic melanocytes in vivo as determined by histochemical, autoradiographic, and electron microscope studies. J Cell Biol 66(3):663–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson O, Liu PY, Bondesson L, Nordlind K, Olsson MJ, Lontz W, Verhofstad A, Liang Y, Gangi S. 1998. A serotonin-like immunoreactivity is present in human cutaneous melanocytes. J Invest Dermatol 111(6):1010–1014. [DOI] [PubMed] [Google Scholar]
- Johnson DB, Sosman JA. 2015. Therapeutic Advances and Treatment Options in Metastatic Melanoma. JAMA Oncol 1(3):380–386. [DOI] [PubMed] [Google Scholar]
- Joshi PG, Nair N, Begum G, Joshi NB, Sinkar VP, Vora S. 2007. Melanocyte-keratinocyte interaction induces calcium signalling and melanin transfer to keratinocytes. Pigment Cell Res 20(5):380–384. [DOI] [PubMed] [Google Scholar]
- Kadekaro AL, Kavanagh R, Kanto H, Terzieva S, Hauser J, Kobayashi N, Schwemberger S, Cornelius J, Babcock G, Shertzer HG, Scott G, Abdel-Malek ZA. 2005. alpha-Melanocortin and endothelin-1 activate antiapoptotic pathways and reduce DNA damage in human melanocytes. Cancer Res 65(10):4292–4299. [DOI] [PubMed] [Google Scholar]
- Kadekaro AL, Leachman S, Kavanagh RJ, Swope V, Cassidy P, Supp D, Sartor M, Schwemberger S, Babcock G, Wakamatsu K, Ito S, Koshoffer A, Boissy RE, Manga P, Sturm RA, Abdel-Malek ZA. 2010. Melanocortin 1 receptor genotype: an important determinant of the damage response of melanocytes to ultraviolet radiation. FASEB J 24(10):3850–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HY, Kang WH, Lee C. 1998. Endothelin-B receptor-mediated Ca2+ signaling in human melanocytes. Pflugers Arch 435(3):350–356. [DOI] [PubMed] [Google Scholar]
- Kang TH, Lindsey-Boltz LA, Reardon JT, Sancar A. 2010. Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc Natl Acad Sci U S A 107(11):4890–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TH, Reardon JT, Kemp M, Sancar A. 2009. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc Natl Acad Sci U S A 106(8):2864–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TH, Reardon JT, Sancar A. 2011. Regulation of nucleotide excision repair activity by transcriptional and post-transcriptional control of the XPA protein. Nucleic Acids Res 39(8):3176–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WK. 2010. The human intra-S checkpoint response to UVC-induced DNA damage. Carcinogenesis 31(5):751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami A, Fisher DE. 2017. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab Invest 97(6):649–656. [DOI] [PubMed] [Google Scholar]
- Kemp MG. 2017. DNA damage-induced ATM- and Rad-3-related (ATR) kinase activation in non-replicating cells is regulated by the XPB subunit of transcription factor IIH (TFIIH). J Biol Chem 292(30):12424–12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp MG, Lindsey-Boltz LA, Sancar A. 2011. The DNA damage response kinases DNA-dependent protein kinase (DNA-PK) and ataxia telangiectasia mutated (ATM) Are stimulated by bulky adduct-containing DNA. J Biol Chem 286(22):19237–19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp MG, Sancar A. 2016. ATR Kinase Inhibition Protects Non-cycling Cells from the Lethal Effects of DNA Damage and Transcription Stress. J Biol Chem 291(17):9330–9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AQ, Travers JB, Kemp MG. 2018. Roles of UVA radiation and DNA damage responses in melanoma pathogenesis. Environ Mol Mutagen 59(5):438–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer KH, Lee MM, Andrews AD, Lambert WC. 1994. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol 130(8):1018–1021. [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, Capatana A, Holman EC, Bosenberg M, Sznol M, Kluger HM, Brash DE, Stern DF, Materin MA, Lo RS, Mane S, Ma S, Kidd KK, Hayward NK, Lifton RP, Schlessinger J, Boggon TJ, Halaban R. 2012. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 44(9):1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latonen L, Laiho M. 2005. Cellular UV damage responses--functions of tumor suppressor p53. Biochim Biophys Acta 1755(2):71–89. [DOI] [PubMed] [Google Scholar]
- Lee TH, Park JM, Leem SH, Kang TH. 2014. Coordinated regulation of XPA stability by ATR and HERC2 during nucleotide excision repair. Oncogene 33(1):19–25. [DOI] [PubMed] [Google Scholar]
- Lehmann AR, McGibbon D, Stefanini M. 2011. Xeroderma pigmentosum. Orphanet J Rare Dis 6:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Musich PR, Cartwright BM, Wang H, Zou Y. 2013. UV-induced nuclear import of XPA is mediated by importin-alpha4 in an ATR-dependent manner. PLoS One 8(7):e68297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Fisher DE. 2007. Melanocyte biology and skin pigmentation. Nature 445(7130):843–850. [DOI] [PubMed] [Google Scholar]
- Lin KK, Kumar V, Geyfman M, Chudova D, Ihler AT, Smyth P, Paus R, Takahashi JS, Andersen B. 2009. Circadian clock genes contribute to the regulation of hair follicle cycling. PLoS Genet 5(7):e1000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo JA, Fisher DE. 2014. The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science 346(6212):945–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, Hughes TM, Thompson JF, Scolyer RA, Kefford RF. 2011. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol 29(10):1239–1246. [DOI] [PubMed] [Google Scholar]
- Lucas RM, Norval M, Neale RE, Young AR, de Gruijl FR, Takizawa Y, van der Leun JC. 2015. The consequences for human health of stratospheric ozone depletion in association with other environmental factors. Photochem Photobiol Sci 14(1):53–87. [DOI] [PubMed] [Google Scholar]
- Madison KC. 2003. Barrier function of the skin: “la raison d’etre” of the epidermis. J Invest Dermatol 121(2):231–241. [DOI] [PubMed] [Google Scholar]
- Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T, Ono T, Albertson DG, Pinkel D, Bastian BC. 2003. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst 95(24):1878–1890. [DOI] [PubMed] [Google Scholar]
- Manzella N, Bracci M, Strafella E, Staffolani S, Ciarapica V, Copertaro A, Rapisarda V, Ledda C, Amati M, Valentino M, Tomasetti M, Stevens RG, Santarelli L. 2015. Circadian Modulation of 8-Oxoguanine DNA Damage Repair. Sci Rep 5:13752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. 2014. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15(7):465–481. [DOI] [PubMed] [Google Scholar]
- Matsui MS, Pelle E, Dong K, Pernodet N. 2016. Biological Rhythms in the Skin. Int J Mol Sci 17(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K, Tajima H, Nakamura T. 1991. Hepatocyte growth factor is a potent stimulator of human melanocyte DNA synthesis and growth. Biochem Biophys Res Commun 176(1):45–51. [DOI] [PubMed] [Google Scholar]
- McGill GG, Horstmann M, Widlund HR, Du J, Motyckova G, Nishimura EK, Lin YL, Ramaswamy S, Avery W, Ding HF, Jordan SA, Jackson IJ, Korsmeyer SJ, Golub TR, Fisher DE. 2002. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell 109(6):707–718. [DOI] [PubMed] [Google Scholar]
- Medrano EE, Farooqui JZ, Boissy RE, Boissy YL, Akadiri B, Nordlund JJ. 1993. Chronic growth stimulation of human adult melanocytes by inflammatory mediators in vitro: implications for nevus formation and initial steps in melanocyte oncogenesis. Proc Natl Acad Sci U S A 90(5):1790–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AJ, Mihm MC Jr. 2006. Melanoma. N Engl J Med 355(1):51–65. [DOI] [PubMed] [Google Scholar]
- Morgan MD, Pairo-Castineira E, Rawlik K, Canela-Xandri O, Rees J, Sims D, Tenesa A, Jackson IJ. 2018. Genome-wide study of hair colour in UK Biobank explains most of the SNP heritability. Nat Commun 9(1):5271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouret S, Baudouin C, Charveron M, Favier A, Cadet J, Douki T. 2006. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc Natl Acad Sci U S A 103(37):13765–13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouret S, Forestier A, Douki T. 2012. The specificity of UVA-induced DNA damage in human melanocytes. Photochem Photobiol Sci 11(1):155–162. [DOI] [PubMed] [Google Scholar]
- Mouret S, Philippe C, Gracia-Chantegrel J, Banyasz A, Karpati S, Markovitsi D, Douki T. 2010. UVA-induced cyclobutane pyrimidine dimers in DNA: a direct photochemical mechanism? Org Biomol Chem 8(7):1706–1711. [DOI] [PubMed] [Google Scholar]
- NASA. 2008. Solar Irradiance.
- Neville JA, Welch E, Leffell DJ. 2007. Management of nonmelanoma skin cancer in 2007. Nat Clin Pract Oncol 4(8):462–469. [DOI] [PubMed] [Google Scholar]
- Nikkola V, Gronroos M, Huotari-Orava R, Kautiainen H, Ylianttila L, Karppinen T, Partonen T, Snellman E. 2018. Circadian Time Effects on NB-UVB-Induced Erythema in Human Skin In Vivo. J Invest Dermatol 138(2):464–467. [DOI] [PubMed] [Google Scholar]
- Nordlund JJ. 2007. The melanocyte and the epidermal melanin unit: an expanded concept. Dermatol Clin 25(3):271–281, vii. [DOI] [PubMed] [Google Scholar]
- Okano-Mitani H, Ikai K, Imamura S. 1997. Human melanoma cells generate leukotrienes B4 and C4 from leukotriene A4. Arch Dermatol Res 289(6):347–351. [DOI] [PubMed] [Google Scholar]
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR, Ye K, Alipaz J, Bauer MJ, Beare D, Butler A, Carter RJ, Chen L, Cox AJ, Edkins S, Kokko-Gonzales PI, Gormley NA, Grocock RJ, Haudenschild CD, Hims MM, James T, Jia M, Kingsbury Z, Leroy C, Marshall J, Menzies A, Mudie LJ, Ning Z, Royce T, Schulz-Trieglaff OB, Spiridou A, Stebbings LA, Szajkowski L, Teague J, Williamson D, Chin L, Ross MT, Campbell PJ, Bentley DR, Futreal PA, Stratton MR. 2010. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463(7278):191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premi S, Wallisch S, Mano CM, Weiner AB, Bacchiocchi A, Wakamatsu K, Bechara EJ, Halaban R, Douki T, Brash DE. 2015. Photochemistry. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science 347(6224):842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proksch E, Brandner JM, Jensen JM. 2008. The skin: an indispensable barrier. Exp Dermatol 17(12):1063–1072. [DOI] [PubMed] [Google Scholar]
- Rana BK, Hewett-Emmett D, Jin L, Chang BH, Sambuughin N, Lin M, Watkins S, Bamshad M, Jorde LB, Ramsay M, Jenkins T, Li WH. 1999. High polymorphism at the human melanocortin 1 receptor locus. Genetics 151(4):1547–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Sancar A. 2005. Nucleotide excision repair. Prog Nucleic Acid Res Mol Biol 79:183–235. [DOI] [PubMed] [Google Scholar]
- Rees JL. 2000. The melanocortin 1 receptor (MC1R): more than just red hair. Pigment Cell Res 13(3):135–140. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. 2001. Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol 63:647–676. [DOI] [PubMed] [Google Scholar]
- Ruben MD, Wu G, Smith DF, Schmidt RE, Francey LJ, Lee YY, Anafi RC, Hogenesch JB. 2018. A database of tissue-specific rhythmically expressed human genes has potential applications in circadian medicine. Sci Transl Med 10(458). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthemann P, Balbo Pogliano C, Naegeli H. 2016. Global-genome Nucleotide Excision Repair Controlled by Ubiquitin/Sumo Modifiers. Front Genet 7:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sample A, He YY. 2018. Mechanisms and prevention of UV-induced melanoma. Photodermatol Photoimmunol Photomed 34(1):13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. 2004. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73:39–85. [DOI] [PubMed] [Google Scholar]
- Sandu C, Dumas M, Malan A, Sambakhe D, Marteau C, Nizard C, Schnebert S, Perrier E, Challet E, Pevet P, Felder-Schmittbuhl MP. 2012. Human skin keratinocytes, melanocytes, and fibroblasts contain distinct circadian clock machineries. Cell Mol Life Sci 69(19):3329–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarasin A. 1999. The molecular pathways of ultraviolet-induced carcinogenesis. Mutat Res 428(1–2):5–10. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Gaddameedhi S. 2018. UV-B-Induced Erythema in Human Skin: The Circadian Clock Is Ticking. J Invest Dermatol 138(2):248–251. [DOI] [PubMed] [Google Scholar]
- Sato M, Nishigori C, Zghal M, Yagi T, Takebe H. 1993. Ultraviolet-specific mutations in p53 gene in skin tumors in xeroderma pigmentosum patients. Cancer Res 53(13):2944–2946. [PubMed] [Google Scholar]
- Satyanarayana A, Kaldis P. 2009. A dual role of Cdk2 in DNA damage response. Cell Div 4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer OD. 2013. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol 5(10):a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuch AP, Garcia CC, Makita K, Menck CF. 2013. DNA damage as a biological sensor for environmental sunlight. Photochem Photobiol Sci 12(8):1259–1272. [DOI] [PubMed] [Google Scholar]
- Schuch AP, Moreno NC, Schuch NJ, Menck CFM, Garcia CCM. 2017. Sunlight damage to cellular DNA: Focus on oxidatively generated lesions. Free Radic Biol Med 107:110–124. [DOI] [PubMed] [Google Scholar]
- Scott MC, Wakamatsu K, Ito S, Kadekaro AL, Kobayashi N, Groden J, Kavanagh R, Takakuwa T, Virador V, Hearing VJ, Abdel-Malek ZA. 2002. Human melanocortin 1 receptor variants, receptor function and melanocyte response to UV radiation. J Cell Sci 115(Pt 11):2349–2355. [DOI] [PubMed] [Google Scholar]
- Scott TL, Christian PA, Kesler MV, Donohue KM, Shelton B, Wakamatsu K, Ito S, D’Orazio J. 2012. Pigment-independent cAMP-mediated epidermal thickening protects against cutaneous UV injury by keratinocyte proliferation. Exp Dermatol 21(10):771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane M, Buhs S, Iglesias P, Strauss J, Puller AC, Muller J, Gerull H, Feldhaus S, Alawi M, Brandner JM, Eggert D, Du J, Thomale J, Wild PJ, Zimmermann M, Sternsdorf T, Schumacher U, Nollau P, Fisher DE, Horstmann MA. 2019. Lineage-specific control of TFIIH by MITF determines transcriptional homeostasis and DNA repair. Oncogene 38(19):3616–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain AH, Bastian BC. 2016. The Genetic Evolution of Melanoma. N Engl J Med 374(10):995–996. [DOI] [PubMed] [Google Scholar]
- Shenenberger DW. 2012. Cutaneous malignant melanoma: a primary care perspective. Am Fam Physician 85(2):161–168. [PubMed] [Google Scholar]
- Shtivelman E, Davies MQ, Hwu P, Yang J, Lotem M, Oren M, Flaherty KT, Fisher DE. 2014. Pathways and therapeutic targets in melanoma. Oncotarget 5(7):1701–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh CK, Mintie CA, Ndiaye MA, Chhabra G, Dakup PP, Ye T, Yu M, Ahmad N. 2019. Chemoprotective Effects of Dietary Grape Powder on UVB Radiation-Mediated Skin Carcinogenesis in SKH-1 Hairless Mice. J Invest Dermatol 139(3):552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slominski A, Tobin DJ, Shibahara S, Wortsman J. 2004. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev 84(4):1155–1228. [DOI] [PubMed] [Google Scholar]
- Society AC. 2019. Cancer Facts & Figures 2019. Atlanta. American Cancer Society. [Google Scholar]
- Society AC. 2020. Cancer Facts and Figures 2020.
- Strub T, Giuliano S, Ye T, Bonet C, Keime C, Kobi D, Le Gras S, Cormont M, Ballotti R, Bertolotto C, Davidson I. 2011. Essential role of microphthalmia transcription factor for DNA replication, mitosis and genomic stability in melanoma. Oncogene 30(20):2319–2332. [DOI] [PubMed] [Google Scholar]
- Sugasawa K. 2010. Regulation of damage recognition in mammalian global genomic nucleotide excision repair. Mutat Res 685(1–2):29–37. [DOI] [PubMed] [Google Scholar]
- Suzuki I, Im S, Tada A, Scott C, Akcali C, Davis MB, Barsh G, Hearing V, Abdel-Malek Z. 1999. Participation of the melanocortin-1 receptor in the UV control of pigmentation. J Investig Dermatol Symp Proc 4(1):29–34. [DOI] [PubMed] [Google Scholar]
- Swope V, Alexander C, Starner R, Schwemberger S, Babcock G, Abdel-Malek ZA. 2014. Significance of the melanocortin 1 receptor in the DNA damage response of human melanocytes to ultraviolet radiation. Pigment Cell Melanoma Res 27(4):601–610. [DOI] [PubMed] [Google Scholar]
- Swope VB, Starner RJ, Rauck C, Abdel-Malek ZA. 2019. Endothelin-1 and alpha-melanocortin have redundant effects on global genome repair in UV-irradiated human melanocytes despite distinct signaling pathways. Pigment Cell Melanoma Res. [DOI] [PubMed] [Google Scholar]
- Szabo G. 1967. The Regional Anatomy of the Human Integument with Special Reference to the Distribution of Hair Follicles, Sweat Glands and Melanocytes. Philosophical Transactions of the Royal Society of London Series B 252:447–485. [Google Scholar]
- Tadokoro T, Yamaguchi Y, Batzer J, Coelho SG, Zmudzka BZ, Miller SA, Wolber R, Beer JZ, Hearing VJ. 2005. Mechanisms of skin tanning in different racial/ethnic groups in response to ultraviolet radiation. J Invest Dermatol 124(6):1326–1332. [DOI] [PubMed] [Google Scholar]
- Takahashi JS. 2017. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet 18(3):164–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas NE, Berwick M, Cordeiro-Stone M. 2006. Could BRAF mutations in melanocytic lesions arise from DNA damage induced by ultraviolet radiation? J Invest Dermatol 126(8):1693–1696. [DOI] [PubMed] [Google Scholar]
- Tsatmali M, Ancans J, Thody AJ. 2002. Melanocyte function and its control by melanocortin peptides. J Histochem Cytochem 50(2):125–133. [DOI] [PubMed] [Google Scholar]
- Tsatmali M, Graham A, Szatkowski D, Ancans J, Manning P, McNeil CJ, Graham AM, Thody AJ. 2000. alpha-melanocyte-stimulating hormone modulates nitric oxide production in melanocytes. J Invest Dermatol 114(3):520–526. [DOI] [PubMed] [Google Scholar]
- U.S. Department of health and human services Codcap, and National Cancer Institute. 2019. U.S. Cancer statistics working group. U.S. Cancer statistics Data Visualization Tool, based on November 2018 submission data (1999–2016). [Google Scholar]
- Unsal-Kacmaz K, Mullen TE, Kaufmann WK, Sancar A. 2005. Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol 25(8):3109–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viros A, Fridlyand J, Bauer J, Lasithiotakis K, Garbe C, Pinkel D, Bastian BC. 2008. Improving melanoma classification by integrating genetic and morphologic features. PLoS Med 5(6):e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Koschembahr AM, Swope VB, Starner RJ, Abdel-Malek ZA. 2015. Endothelin-1 protects human melanocytes from UV-induced DNA damage by activating JNK and p38 signalling pathways. Exp Dermatol 24(4):269–274. [DOI] [PubMed] [Google Scholar]
- Wakamatsu K, Graham A, Cook D, Thody AJ. 1997. Characterisation of ACTH peptides in human skin and their activation of the melanocortin-1 receptor. Pigment Cell Res 10(5):288–297. [DOI] [PubMed] [Google Scholar]
- Wakasugi M, Sasaki T, Matsumoto M, Nagaoka M, Inoue K, Inobe M, Horibata K, Tanaka K, Matsunaga T. 2014. Nucleotide excision repair-dependent DNA double-strand break formation and ATM signaling activation in mammalian quiescent cells. J Biol Chem 289(41):28730–28737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HT, Choi B, Tang MS. 2010. Melanocytes are deficient in repair of oxidative DNA damage and UV-induced photoproducts. Proc Natl Acad Sci U S A 107(27):12180–12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Digiovanna JJ, Stern JB, Hornyak TJ, Raffeld M, Khan SG, Oh KS, Hollander MC, Dennis PA, Kraemer KH. 2009. Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Proc Natl Acad Sci U S A 106(15):6279–6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk J, Brenneisen P, Meewes C, Wlaschek M, Peters T, Blaudschun R, Ma W, Kuhr L, Schneider L, Scharffetter-Kochanek K. 2001. UV-induced oxidative stress and photoaging. Curr Probl Dermatol 29:83–94. [DOI] [PubMed] [Google Scholar]
- Whiteman DC, Parsons PG, Green AC. 1999. Determinants of melanocyte density in adult human skin. Arch Dermatol Res 291(9):511–516. [DOI] [PubMed] [Google Scholar]
- Wolf Horrell EM, Boulanger MC, D’Orazio JA. 2016. Melanocortin 1 Receptor: Structure, Function, and Regulation. Front Genet 7:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Horrell EM, Jarrett SG, Carter KM, D’Orazio JA. 2017. Divergence of cAMP signalling pathways mediating augmented nucleotide excision repair and pigment induction in melanocytes. Exp Dermatol 26(7):577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood RD. 2010. Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ Mol Mutagen 51(6):520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Hemesath TJ, Takemoto CM, Horstmann MA, Wells AG, Price ER, Fisher DZ, Fisher DE. 2000. c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes Dev 14(3):301–312. [PMC free article] [PubMed] [Google Scholar]
- Wu X, Shell SM, Liu Y, Zou Y. 2007. ATR-dependent checkpoint modulates XPA nuclear import in response to UV irradiation. Oncogene 26(5):757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M, Chen K, Yao X, Xu Y, Yao J, Yan J, Shao Z, Wang G. 2017. Mediator MED23 Links Pigmentation and DNA Repair through the Transcription Factor MITF. Cell Rep 20(8):1794–1804. [DOI] [PubMed] [Google Scholar]
- Yaar M, Gilchrest BA. 1991. Human melanocyte growth and differentiation: a decade of new data. J Invest Dermatol 97(4):611–617. [DOI] [PubMed] [Google Scholar]
- Yada Y, Higuchi K, Imokawa G. 1991. Effects of endothelins on signal transduction and proliferation in human melanocytes. J Biol Chem 266(27):18352–18357. [PubMed] [Google Scholar]
- Yamaguchi Y, Hearing VJ. 2009. Physiological factors that regulate skin pigmentation. Biofactors 35(2):193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasumoto K, Yokoyama K, Shibata K, Tomita Y, Shibahara S. 1994. Microphthalmia-associated transcription factor as a regulator for melanocyte-specific transcription of the human tyrosinase gene. Mol Cell Biol 14(12):8058–8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohn JJ, Morelli JG, Walchak SJ, Rundell KB, Norris DA, Zamora MR. 1993. Cultured human keratinocytes synthesize and secrete endothelin-1. J Invest Dermatol 100(1):23–26. [DOI] [PubMed] [Google Scholar]
- Zanello SB, Jackson DM, Holick MF. 2000. Expression of the circadian clock genes clock and period1 in human skin. J Invest Dermatol 115(4):757–760. [DOI] [PubMed] [Google Scholar]
- Zhang R, Lahens NF, Ballance HI, Hughes ME, Hogenesch JB. 2014. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci U S A 111(45):16219–16224. [DOI] [PMC free article] [PubMed] [Google Scholar]