

Summary
The biosynthesis of thousands of proteins requires targeting a signal sequence or transmembrane segment (TM) to the endoplasmic reticulum (ER). These hydrophobic α-helices must localize to the appropriate cellular membrane and integrate in the correct topology to maintain a high-fidelity proteome. Here, we show that the P5A-ATPase ATP13A1 prevents the accumulation of mislocalized and misoriented proteins which are eliminated by different ER-associated degradation (ERAD) pathways in mammalian cells. Without ATP13A1, mitochondrial tail-anchored proteins mislocalize to the ER through the ER membrane protein complex and are cleaved by signal peptide peptidase for ERAD. ATP13A1 also facilitates the topogenesis of a subset of proteins with an N-terminal TM or signal sequence that should insert into the ER membrane with a cytosolic N terminus. Without ATP13A1, such proteins accumulate in the wrong orientation and are targeted for ERAD by distinct ubiquitin ligases. Thus, ATP13A1 prevents ERAD of diverse proteins capable of proper folding.
eTOC
McKenna et al. show that corrective quality control by the P5A-ATPase ATP13A1 at the ER prevents wasteful ER-associated degradation of mislocalized and misoriented proteins capable of folding properly.
Graphical Abstract
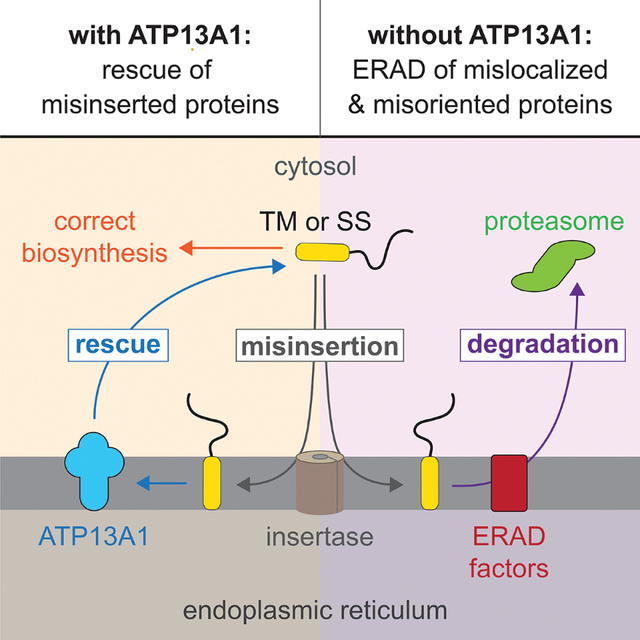
Introduction
The endoplasmic reticulum (ER) is the first step of trafficking mechanisms that provide proteins access to the extracellular space, plasma membrane, and endo-secretory compartments. Hence, the ER is the site of biosynthesis for thousands of transmembrane, secreted, and soluble organellar proteins that comprise over 30% of the human proteome, and defects in protein biosynthesis at the ER are associated with a wide range of human diseases (Dubnikov et al., 2017; Hegde and Keenan, 2022; Olzmann et al., 2013). Information for protein targeting to the ER is often encoded by an N-terminal signal sequence that is cleaved upon protein translocation into the ER lumen. Alternatively, for membrane proteins that lack a signal sequence, ER targeting is triggered by the first transmembrane segment (TM) that emerges from the ribosome. Although ER-targeting signal sequences and TMs all form hydrophobic α-helices that eventually insert into the ER membrane, their amino acid sequences are highly diverse and degenerate. Moreover, different signal sequences and TMs are not equally robust in mediating ER targeting or membrane insertion, and TM-mediated protein localization is error-prone (Chen et al., 2014b; Conti et al., 2015; Krumpe et al., 2012; McKenna et al., 2020; Okreglak and Walter, 2014; Rane et al., 2010). However, the mechanisms that maintain ER homeostasis despite enormous substrate diversity and inefficiencies at various steps of protein biosynthesis remain incompletely defined.
All proteins with a signal sequence or TM face at least two biosynthetic challenges in addition to protein folding. First, they must localize to the correct cellular membrane. Second, their signal sequence or TM must insert into the lipid bilayer in the correct orientation (Hegde and Keenan, 2022). Most nascent proteins are cotranslationally targeted to the ER by the signal recognition particle (SRP), which engages and delivers ribosomes synthesizing nascent proteins with a signal sequence or TM to the ER (Walter and Blobel, 1981; Zhang and Shan, 2014). At the ER, ribosomes dock onto the SEC61 complex, which forms an aqueous channel with a lateral gate that allows soluble protein domains to translocate into the ER lumen and TMs to partition into the ER membrane (van den Berg et al., 2004; Park and Rapoport, 2012). For proper protein topogenesis, signal sequences and ‘type II’ TMs must engage the SEC61 complex with their N terminus facing the cytosol (referred to as Ncyto topology) (Rapoport et al., 2017). The ER membrane protein complex (EMC) also mediates the topogenesis of some N-terminal TMs that assume the opposite (NER) orientation (Chitwood et al., 2018; O’Keefe et al., 2021; Wu and Rapoport, 2021). Although most proteins target to the ER cotranslationally in mammals, tail-anchored (TA) proteins are an exception. Because TA proteins contain a single C-terminal TM which precludes cotranslational recognition, all TA proteins insert into target membranes post-translationally with their N terminus in the cytosol (Chio et al., 2017; Farkas and Bohnsack, 2021). TA proteins can insert into the ER via the EMC or the GET1/GET2 (also known as WRB/CAMLG) complex, with EMC clients generally possessing a less hydrophobic TM (Guna et al., 2018; Schuldiner et al., 2008; Wang et al., 2014; Yamamoto and Sakisaka, 2012). The SEC61 complex, EMC, and GET1/GET2 comprise the entire set of known TM insertases at the mammalian ER with functions validated by reconstitutions with purified factors (Görlich and Rapoport, 1993; Guna et al., 2018; Mariappan et al., 2011).
Protein biosynthesis is tightly integrated with quality control (QC) mechanisms that must promptly recognize and resolve or eliminate aberrant protein intermediates produced by errors in biosynthesis. At the ER, the best-studied form of protein QC is ER-associated degradation (ERAD). ERAD is characterized by the recognition, ubiquitination, and extraction of aberrant proteins from the ER for proteasomal degradation (Olzmann et al., 2013; Wu and Rapoport, 2018). In the bulk ER of yeast, two E3 ubiquitin ligase complexes mediate ERAD of most proteins. Generally, Hrd1 targets proteins with misfolding lesions in lumenal or membrane domains, while Doa10 targets proteins with misfolding lesions in cytosolic domains (Carvalho et al., 2006; Vashist and Ng, 2004). In contrast, mammalian cells possess approximately thirty ER-resident ubiquitin ligases, and numerous additional factors have been implicated in ERAD (Leto et al., 2019; Olzmann et al., 2013). Although ERAD is usually associated with the degradation of misfolded proteins, ERAD also eliminates proteins that fail to assemble into the correct protein complex or that are mislocalized despite being able to fold properly (Matsumoto et al., 2019; Natarajan et al., 2020; Ruggiano et al., 2016). The factors that mediate such instances of context-dependent ERAD and the clientele of mammalian ERAD factors remain poorly defined.
We recently identified the ER-resident P5A-ATPase ATP13A1 (Spf1 in yeast) as a QC factor which dislocates mislocalized mitochondrial TA proteins from the ER to provide clients additional opportunities to insert into the mitochondrial outer membrane (McKenna et al., 2020). In cells lacking ATP13A1, mitochondrial TA proteins such as OMP25 (outer membrane protein of 25 kDa, also known as synaptojanin-2 binding protein or SYNJ2BP) aberrantly accumulate at the ER and become destabilized. However, how mitochondrial TA proteins mislocalize to the ER and how they are degraded in the absence of ATP13A1 is unknown. Proteomics data and other studies have also linked the P5A-ATPase to the stability of a subset of ER-targeted proteins that possess an N-terminal ‘type II’ TM or signal sequence which should assume an Ncyto topology (Feng et al., 2020; Li et al., 2021; McKenna et al., 2020; Qin et al., 2020). However, it is not clear why ATP13A1 is required for the stability of these proteins. In this study, we systematically follow the fates of different types of ATP13A1-dependent proteins. We show that the EMC is responsible for mitochondrial TA mislocalization and that the signal peptide peptidase (SPP) mediates mislocalized mitochondrial TA protein ERAD. We also show that loss of ATP13A1 function results in the accumulation of type II and signal sequence-containing proteins inserted in the incorrect topology at the ER, and that such misoriented proteins are targeted for ERAD by different ubiquitin ligases. Our findings reveal a general role for ATP13A1 in mediating correct protein localization and topogenesis, thereby averting wasteful ERAD of a diverse set of proteins that retain the potential to fold properly.
Results
Mitochondrial TA proteins mislocalize to the ER via the EMC
Although it is established that ATP13A1 dislocates mitochondrial TA proteins from the ER, how mitochondrial TA proteins initially mislocalize to the ER is not clear. To address this question, we used immunofluorescence to determine how depleting the two known ER TA protein insertases, the GET1/GET2 complex or the EMC, affects the localization of F-OMP25, a FLAG-tagged reporter containing the TM of the mitochondrial TA protein OMP25. As we previously observed, F-OMP25 localizes to mitochondria in wildtype (WT) cells but is mislocalized to the ER and destabilized in ATP13A1 knockout (KO) cells (Figures 1A and 1B) (McKenna et al., 2020). Knocking down EMC2 or EMC6, which destabilizes the entire EMC (Figure S1A) (Guna et al., 2018; Volkmar et al., 2018), reduces ER mislocalization and partially rescues mitochondrial localization of F-OMP25 in ATP13A1 KO cells (Figures 1A, 1B, and S1B). In contrast, knocking down GET1 does not prevent F-OMP25 mislocalization in ATP13A1 KO cells. Depleting core EMC components also rescued mitochondrial localization of TA proteins containing the BAK1 or MAVS TM in ATP13A1 KO cells (Figures S1C–S1E). Thus, the EMC is responsible for the mislocalization of multiple mitochondrial TA proteins.
Figure 1. Mitochondrial TA proteins mislocalize to the ER via the EMC.
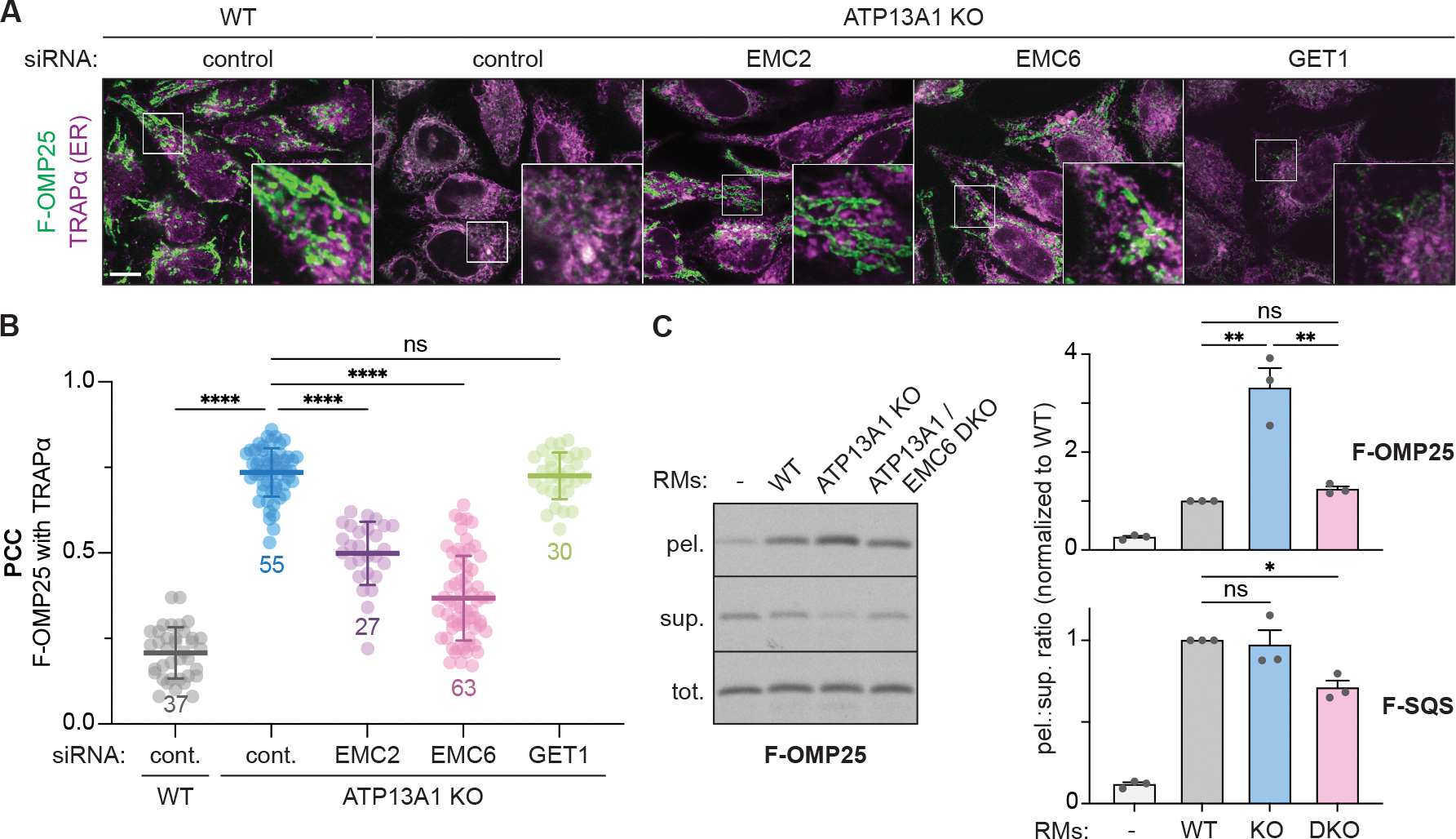
(A) EMC depletion rescues the localization of FLAG-tagged OMP25 (F-OMP25), a mitochondrial TA protein, in ATP13A1 knockout (KO) cells. Immunofluorescence of F-OMP25 (green) and the ER marker TRAPα (magenta) in wildtype (WT) and ATP13A1 KO Flp-In HeLa T-REx cells treated with control siRNAs or siRNAs targeting EMC2, EMC6 or GET1. Scale bar, 10 μm.
(B) Pearson’s correlation coefficients (PCC; mean ± sd and individual points for indicated sample size) measuring colocalization of F-OMP25 and TRAPα. ****, p<0.0001; ns, not significant.
(C) Knocking out the EMC reduces F-OMP25 insertion into ER-derived rough microsomes (RMs). SDS-PAGE and autoradiography of total (tot.), membrane-inserted (pel.), and soluble (sup.) radiolabeled F-OMP25 from insertion reactions with RMs obtained from WT, ATP13A1 KO, or ATP13A1 and EMC6 double knockout (DKO) Flp-In 293 T-REx cells (left). Ratios of pelleted to soluble (pel.:sup.) F-OMP25 (top right) or a matched reporter containing the TM of the ER TA protein SQS (F-SQS; bottom right) were normalized to values obtained with WT RMs (mean + sem) for 3 replicates. **, p<0.01; *, p<0.05.
See also Figure S1.
To directly assay how the EMC affects ER insertion of F-OMP25, we performed in vitro insertion assays of radiolabeled F-OMP25 with ER-derived rough microsomes (RMs) isolated from WT, ATP13A1 KO, or ATP13A1 and EMC6 double-knockout (DKO) cells (Figures 1C and S1F). As we observed before (McKenna et al., 2020), F-OMP25 accumulates to a higher degree in ATP13A1 KO RMs than in WT RMs. In comparison, RMs that lack both ATP13A1 and EMC6 do not accumulate F-OMP25 relative to WT RMs (Figure 1C). This result indicates that EMC depletion prevents the accumulation of F-OMP25 at the ER in the absence of ATP13A1 dislocation activity. In contrast, the insertion of F-SQS, a matched ER-targeted TA protein reporter of the EMC insertion client squalene synthase (Guna et al., 2018), was not affected by knocking out ATP13A1 but significantly decreased with ATP13A1 and EMC6 DKO RMs (Figure 1C). These data suggest that mitochondrial TA proteins such as F-OMP25 misinsert into the ER through the EMC.
SPP cleaves mislocalized mitochondrial TA proteins for ERAD
Next, we investigated how mislocalized F-OMP25 is degraded at the ER. Inhibiting proteasome activity selectively stabilized a F-OMP25 fragment in ATP13A1 KO cells (Figure 2A), while only low levels of this fragment were observed in WT cells treated with proteasome inhibitors. We observed a similar F-OMP25 fragment in cell-free insertion reactions with ATP13A1 KO RMs (Figure S1F). Cellular fractionation showed that the proteasome-sensitive F-OMP25 fragment is cytosolic (Figure 2B), and comparisons with in vitro synthesized F-OMP25 fragments of defined lengths indicated that the F-OMP25 fragment is truncated within the TM (Figure S2C). These results indicate that F-OMP25 may be cleaved within the plane of the ER membrane prior to proteasomal degradation in ATP13A1 KO cells.
Figure 2. SPP cleaves mislocalized mitochondrial TA proteins during ERAD.
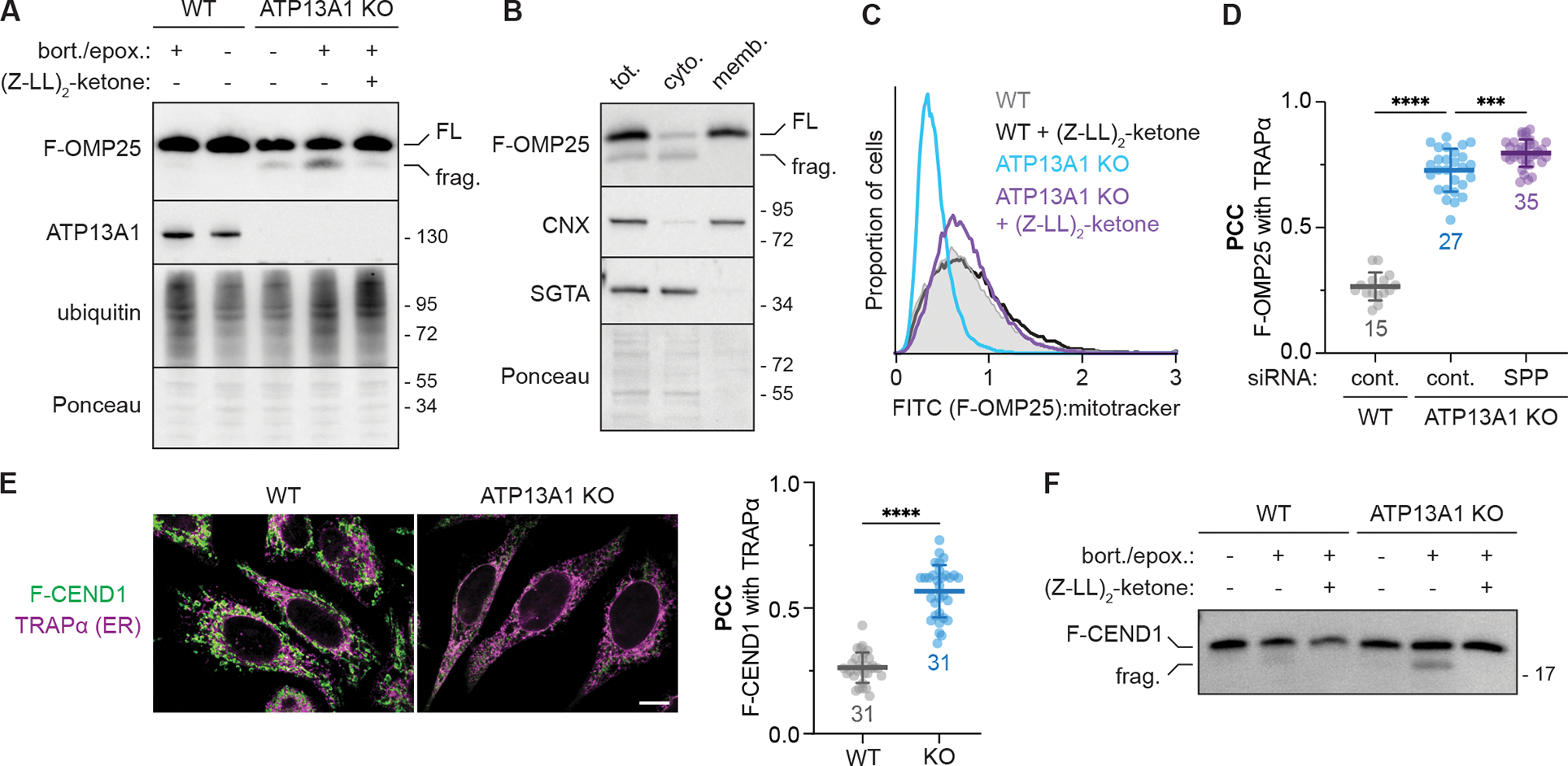
(A) Proteasome inhibition stabilizes an SPP-dependent fragment of mislocalized F-OMP25. SDS-PAGE and immunoblotting for the indicated factors in wildtype (WT) and ATP13A1 knockout (KO) cells treated without or with proteasome inhibitors (0.5 μM bortezomib and 0.5 μM epoxomicin; bort./epox.) and/or an inhibitor [5 μM (Z-LL)2-ketone] of the signal peptide peptidase (SPP). FL, full-length F-OMP25; frag., F-OMP25 fragment.
(B) SPP-cleaved F-OMP25 is cytosolic. Immunoblotting of lysates (tot.) of ATP13A1 KO cells expressing F-OMP25 treated with bort./epox. and separated into cytosolic (cyto.) and membrane-bound (memb.) fractions.
(C) SPP inhibition stabilizes mislocalized F-OMP25. Fluorescent flow cytometry of F-OMP25 levels normalized to mitotracker staining in WT or ATP13A1 KO cells treated without or with 5 μM (Z-LL)2-ketone.
(D) SPP depletion does not rescue mitochondrial TA protein localization. Pearson’s correlation coefficients (PCC; mean ± sd and individual points for indicated sample size) measuring the colocalization of F-OMP25 and the ER marker TRAPα in WT or ATP13A1 KO cells treated with control (cont.) or SPP siRNAs as in Figure S2G. ****, p<0.0001; ***, p<0.0003. Cont. siRNA samples are a subset of Figure 1B.
(E) CEND1 is an ATP13A1-dependent mitochondrial TA protein. Immunofluorescence (left) and PCC (right; mean ± sd and individual points for indicated sample size) showing colocalization of a FLAG-tagged reporter containing the CEND1 TM (F-CEND1, green) and the ER marker TRAPα (magenta) in WT or ATP13A1 KO Flp-In HeLa T-REx cells. Scale bar, 10 μm; ****, p<0.0001.
(F) Mislocalized CEND1 is cleaved by SPP. SDS-PAGE and immunoblotting of soluble fractions from semi-permeabilized WT or ATP13A1 KO cells expressing F-CEND1 treated without or with proteasome inhibitors (bort./epox.) and/or (Z-LL)2-ketone.
See also Figures S2, S3, and Supplemental Table 1.
A candidate for this cleavage activity is signal peptide peptidase (SPP), an ER-resident intramembrane aspartyl protease. SPP processes N-terminal signal sequences that have been cleaved off translocating proteins and mediates the degradation of some ER-targeted TA proteins, including SQS, to regulate their abundance (Boname et al., 2014; Heidasch et al., 2021; Yücel and Lemberg, 2020). Indeed, we observed F-SQS cleavage in cell-free insertion reactions that was inhibited by the SPP inhibitor (Z-LL)2-ketone (Figure S2A) (Weihofen et al., 2000). The extent of F-SQS cleavage did not differ between WT and ATP13A1 KO RMs but decreased significantly in ATP13A1 and EMC6 DKO RMs (Figures S2A and S2B). In comparison, the level of F-OMP25 fragment was higher in ATP13A1 KO RMs compared to both WT and DKO RMs (Figures S1F and S2B). Knocking down SPP (Figure S2D) or treating ATP13A1 KO cells with (Z-LL)2-ketone (Figures 2A and 2C), which does not alter SPP levels (Figure S2E), also eliminated the F-OMP25 fragment and stabilized full-length F-OMP25 levels. Consistent with our in vitro results (Figure S2B), in WT cells, (Z-LL)2-ketone stabilized F-SQS but not F-OMP25 (Figures 2C and S2F), indicating that SPP selectively acts on F-OMP25 in the absence of ATP13A1. In contrast to EMC depletion (Figures 1A and 1B), depleting SPP does not rescue mitochondrial localization of F-OMP25 in ATP13A1 KO cells (Figures 2D and S2G), consistent with a model in which EMC acts upstream of F-OMP25 mislocalization while SPP functions downstream. These results indicate that SPP specifically cleaves mislocalized F-OMP25 for ERAD.
The degradation of other SPP clients has been linked to two ER-resident E3 ubiquitin ligases, TRC8 and MARCH6 (Chen et al., 2014a; Heidasch et al., 2021; Stefanovic-Barrett et al., 2018). Knocking down MARCH6 but not TRC8 slightly stabilized endogenous OMP25 (Figures S2H), although it remains unclear how SPP interfaces with other ERAD factors. Quantitative profiling of the proteomes of WT and ATP13A1 KO cells without and with SPP inhibition confirmed known SPP clients such as SQS and heme oxygenase 1 (HMOX1) (Figure S3A) (Boname et al., 2014; Heidasch et al., 2021). 46 proteins, including endogenous OMP25, were destabilized by knocking out ATP13A1 and restabilized by SPP inhibition (Figures S3B and S3C). One TA protein in this group, CEND1 (cell cycle exit and neuronal differentiation 1), is a poorly characterized protein linked to neural development (Politis et al., 2007). We found that F-CEND1, a reporter containing the CEND1 TM, localizes to mitochondria in WT cells (Figures 2E and S3D). Like OMP25, F-CEND1 becomes mislocalized and is specifically cleaved by SPP upon ATP13A1 depletion (Figures 2E and 2F). Our data suggest that CEND1 is a mitochondrial TA protein whose localization and abundance is regulated by ATP13A1-mediated dislocation from the ER, which prevents ERAD through SPP.
Unlike mitochondrial TA proteins, TA proteins resident to other cellular membranes are indirectly affected by ATP13A1 and SPP. The levels of STX1B (syntaxin-1B), a SNARE protein involved in synaptic vesicle docking (Schubert et al., 2014), were sensitive to ATP13A1 depletion and SPP inhibition in cells. However, SPP cleaves the STX1B TM equally efficiently in both WT and ATP13A1 KO RMs (Figure S3C and S3E), suggesting that nascent STX1B is not directly dislocated by ATP13A1. Instead, STX1B stability may be influenced by indirect effects of ATP13A1 depletion on post-ER vesicular trafficking. The peroxisomal TA protein PLAT3 is also destabilized in ATP13A1 KO cells and stabilized by SPP inhibition, but we could not detect SPP-dependent PLAT3 fragments (Figure S3C and S3F). Although there were no significant differences in peroxisome number in WT and ATP13A1 KO cells (Figure S3G), ATP13A1 and SPP may have pleiotropic effects on peroxisome biogenesis or function, which are closely linked to the ER (Mayerhofer, 2016). Thus, additional considerations influence how ATP13A1 and SPP regulate TA proteins resident to other organelles.
A reporter system to assay protein topology at the ER
Loss of ATP13A1 function also destabilizes a set of ER-targeted type II membrane proteins with an N-terminal TM that should assume an Ncyto topology (McKenna et al., 2020). Like mitochondrial TA proteins, these type II proteins, such as CHST10 (carbohydrate sulfotransferase 10), are destabilized by knocking out ATP13A1 and rescued by acutely re-expressing WT but not catalytically inactive ATP13A1 (McKenna et al., 2020). Type II proteins and mitochondrial TA proteins have in common the presence of a TM near one terminus of the protein. Based on this similarity, we hypothesize that type II proteins require dislocation by ATP13A1 if they are initially inserted in the opposite (i.e. NER) orientation.
Testing this hypothesis requires a sensitive assay of TM topology. To address this, we established a split fluorescent protein reporter system to assay membrane protein topology at the ER. First, we generated Flp-In 293 T-REx cells that constitutively express the first ten β-strands of mNeonGreen3K [referred to as GFP(1–10)cyto] in the cytosol and the first ten β-strands of mCherry3V [referred to as RFP(1–10)ER] (Feng et al., 2019; Zhou et al., 2020) localized to the ER lumen by the preprolactin signal sequence and a C-terminal ER retention signal (Figure 3A). We then incorporated into the Flp-In locus reporters that contain the 11th β-strands of mNeonGreen3K [GFP(b11)] and mCherry3V [RFP(b11)] in tandem. Each reporter is also separated by a P2A ribosomal skipping sequence from TagBFP (BFP) encoded in the same transcript. Using fluorescent flow cytometry, we can quantitatively assay cytosolic mNeonGreen3K fluorescence (referred to as GFPcyto) and ER-localized mCherry3V fluorescence (referred to as RFPER), which should only occur if the complementary 11th β-strand of the fluorescent protein resides in the corresponding cellular compartment. Normalizing these values to BFP fluorescence additionally provides a readout for protein stability by reporting the relative proportion of reporter localized to each compartment compared to the total amount of reporter synthesized (Figure 3B).
Figure 3. A reporter system to assay ER membrane protein topology and stability.
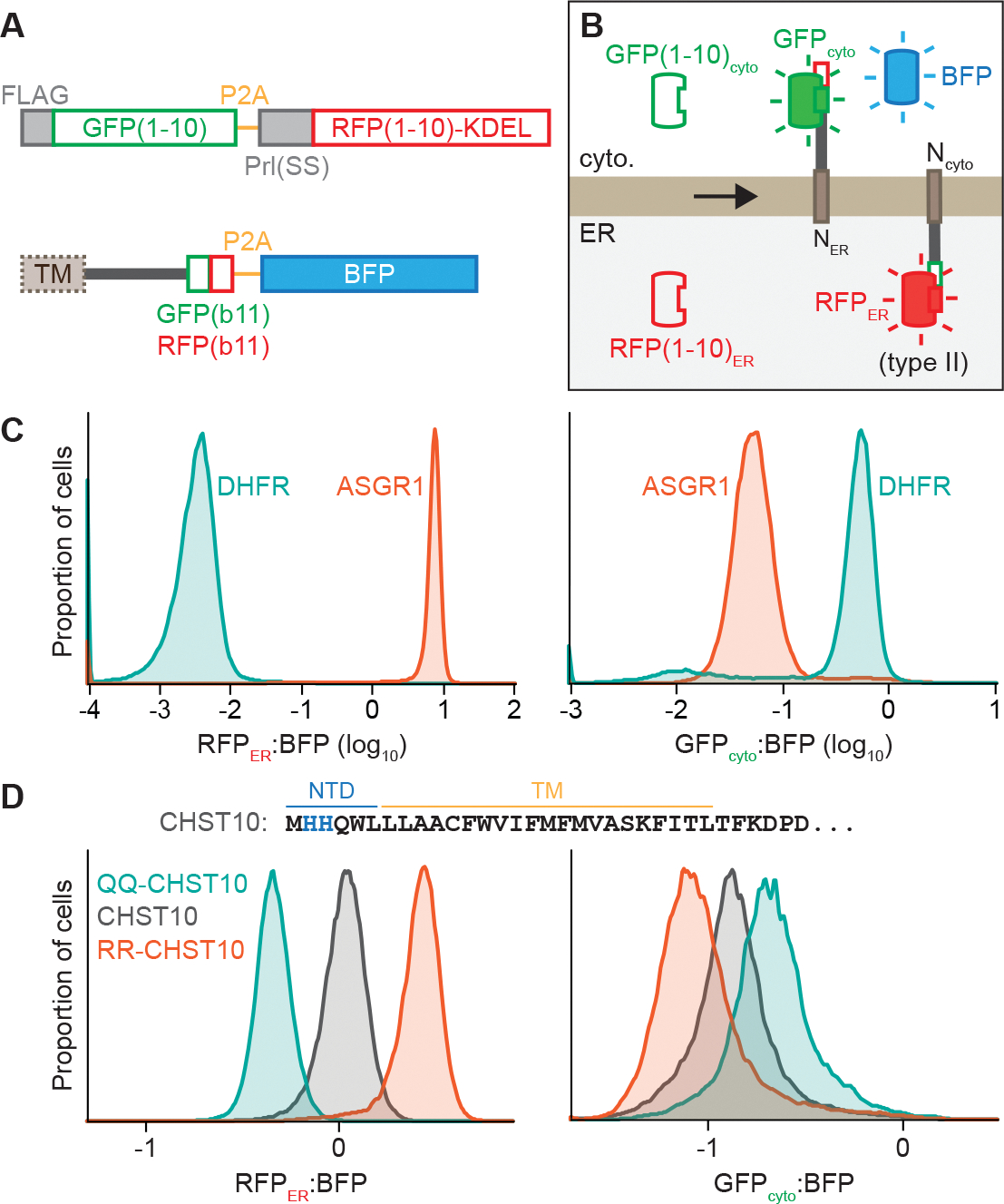
(A) Scheme of split fluorescent reporter system to assay protein topology. Flp-In 293 T-REx cells were engineered to express the first 10 β-strands mNeonGreen3K [GFP(1–10)] in the cytosol and the first 10 β-strands of mCherry3V [RFP(1–10)] localized to the ER by the bovine preprolactin signal sequence and a C-terminal KDEL ER retention signal. Reporters incorporated into the Flp-In locus express proteins without or with an N-terminal ER targeting signal sequence (SS) or type II TM followed by the distinct 11th β-strands of mNeonGreen3K (GFPb11) and mCherry3V (RFPb11) and separated by a P2A ribosome skipping sequence from TagBFP (BFP).
(B) Protein products of the reporter system in (A). GFPcyto and RFPER fluorescence depends on reporter protein topology.
(C) RFPER:BFP (left) and GFPcyto:BFP (right) ratios of topology reporters of the type II protein ASGR1 (dark orange) or the cytosolic protein DHFR (teal).
(D) RFPER:BFP (left) or GFPcyto:BFP (right) ratios of topology reporters of wildtype CHST10 (dark gray) or CHST10 in which His2 and His3 are mutated to glutamines (QQ-CHST10; teal) or arginines (RR-CHST10; dark orange). The N-terminal and TM sequence of CHST10 is shown with His2 and His3 in blue.
See also Figure S4.
We validated this system with two control reporters: the cytosolic protein dihydrofolate reductase (DHFR) and asialoglycoprotein receptor 1 (ASGR1), a type II membrane protein with a relatively large 40-residue N-terminal domain (NTD) preceding the TM that facilitates topogenesis (Wahlberg and Spiess, 1997). Correct type II TM topology should place the tandem GFP(b11)-RFP(b11) tag of ASGR1 in the ER to complement RFP(1–10)ER. Fluorescence microscopy (Figure S4A) and fluorescent flow cytometry (Figure 3C) validated that ER-localized ASGR1 produced a strong RFPER signal and minimal GFPcyto signal while cytosolic DHFR produced a strong GFPcyto signal and minimal RFPER signal. In both cases, the BFP signal remained cytosolic (Figure S4A). In comparison, placing the tandem b11 tag in the cytosolic domain before the TM of the ER-targeted TA protein SEC61β resulted in a high GFPcyto signal colocalized to the ER (Figure S4B). Fluorescent flow cytometry after selective permeabilization of the plasma membrane to release cytosolic contents additionally supported membrane integration of the ASGR1 and SEC61β reporter proteins, while DHFR and BFP were released (Figure S4C). Thus, this system accurately reports the localization of the tandem GFP(b11)-RFP(b11) tag in the cytosol or ER based on GFPcyto or RFPER signal, respectively.
Next, we used the topology reporter system to analyze CHST10 (Figure 3D). Unlike ASGR1, CHST10 has a short 6-residue NTD which may be more likely to translocate into the ER lumen, resulting in TM insertion in the incorrect topology (Wahlberg and Spiess, 1997). Another feature that may contribute to CHST10 misinsertion is the lack of basic residues in the NTD (Wahlberg and Spiess, 1997), as positive charges are preferentially retained in the cytosol according to the ‘positive-inside’ rule (von Heijne, 1986, 1989). To test this possibility, we mutated two weakly basic histidine residues in the CHST10 NTD to either arginines (H2R/H3R, referred to as RR-CHST10) or glutamines (H2Q/H3Q, referred to as QQ-CHST10) to increase or decrease the positive charge of the NTD, respectively. Relative to WT CHST10, RR-CHST10 produced a higher RFPER:BFP ratio and a lower GFPcyto:BFP ratio (Figure 3D), suggesting that a higher proportion of RR-CHST10 assumes the correct type II topology. QQ-CHST10 produced the opposite effect, resulting in a lower RFPER:BFP ratio and a higher GFPcyto:BFP ratio. Thus, increasing the positive charge of the NTD enhances CHST10 TM insertion in the correct type II topology. These results establish that the topology reporter system can detect relative changes in type II protein topogenesis and that CHST10 has weak topology-dictating features.
CHST10 topogenesis requires ATP13A1
Supporting our quantitative proteomics results (McKenna et al., 2020), knocking down ATP13A1 reduced the RFPER:BFP ratio of WT CHST10 corresponding to correct type II topology by ~50% (Figure 4A). In comparison, the RFPER:BFP ratio of RR-CHST10 and ASGR1 changed by less than 10% with ATP13A1 depletion. To further investigate how ATP13A1 influences CHST10 insertion, we synthesized radiolabeled CHST10 using an in vitro translation system in the presence of ER-derived RMs isolated from WT or ATP13A1 KO cells (Figure 4B). The C-terminal domain of CHST10 following the TM contains three N-linked glycosylation sites which should be modified if CHST10 assumes the correct type II topology. Mirroring the cellular reporters, the majority of CHST10 was glycosylated with WT RMs, while CHST10 synthesized in the presence of ATP13A1 KO RMs was almost entirely non-glycosylated. In contrast, RR-CHST10 and ASGR1 were similarly glycosylated with both WT and ATP13A1 KO RMs. Thus, CHST10 biosynthesis is selectively impaired by ATP13A1 depletion, and this defect can be circumvented by stronger topology-dictating features.
Figure 4. ATP13A1 facilitates type II membrane protein topogenesis.
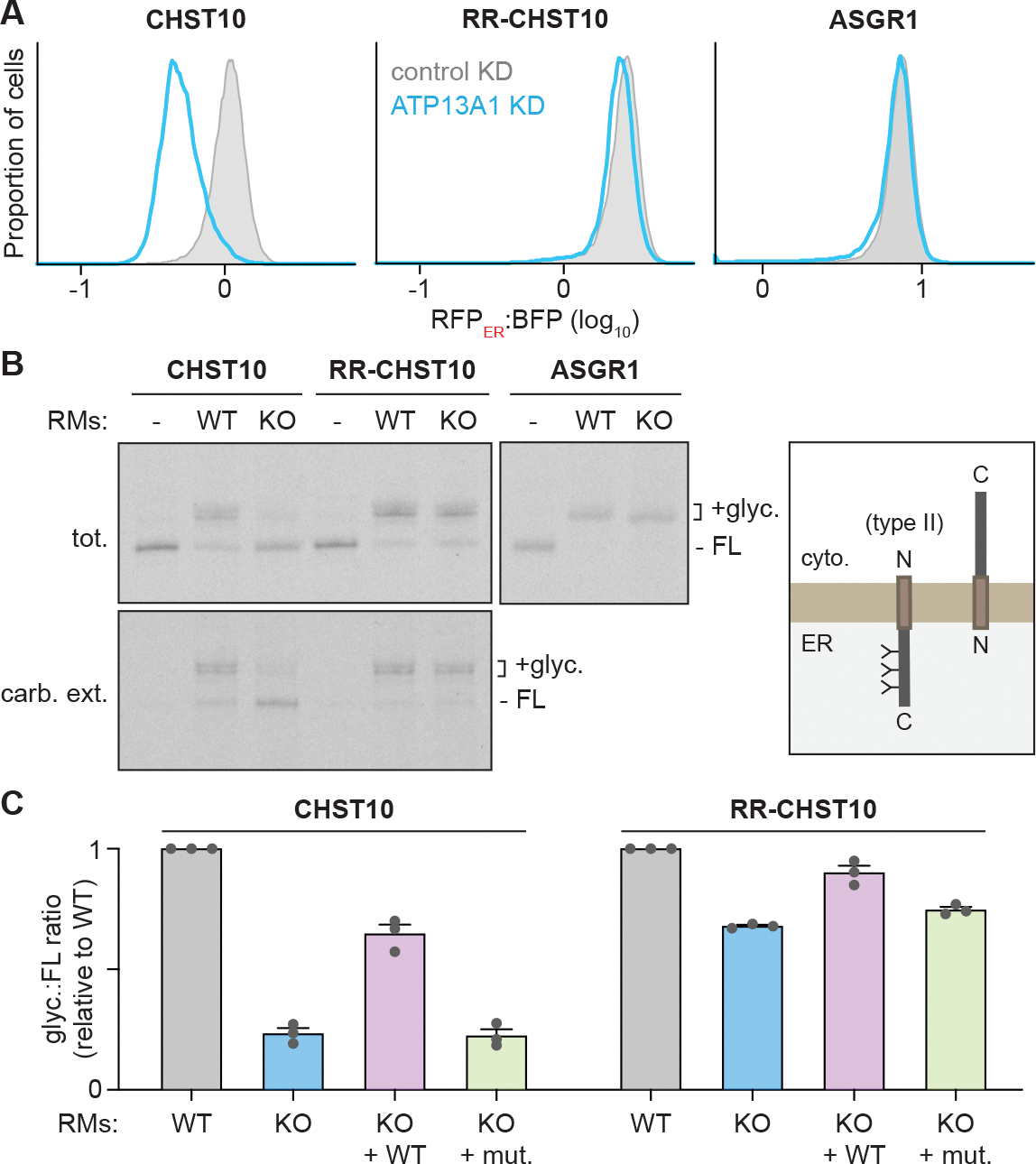
(A) ATP13A1 is required for CHST10 stability. RFPER:BFP ratios of wildtype CHST10 (left), RR-CHST10 (middle), or ASGR1 (right) type II protein topology reporters in wildtype cells treated with control siRNAs (control KD; gray; same samples as Figure 3D) or siRNAs against ATP13A1 (ATP13A1 KD; light blue).
(B) Reconstitution of ATP13A1-dependent CHST10 topogenesis. SDS-PAGE and autoradiography (left) of radiolabeled CHST10, RR-CHST10, or ASGR1 synthesized in vitro without or with ER-derived rough microsomes (RMs) isolated from wildtype (WT) or ATP13A1 knockout (KO) cells before (tot.) or after carbonate extraction (carb. ext.) to isolate membrane-embedded populations. All proteins contain N-glycosylation sites that are modified in the correct type II topology (right) and a C-terminal FLAG tag used for denaturing immunoprecipitations. FL, full-length substrate; +glyc, glycosylated substrate.
(C) Glyc.:FL ratios (mean ± sem) of CHST10 (left) or RR-CHST10 (right) in insertion assays as in (B) containing RMs derived from WT, ATP13A1 KO, or ATP13A1 KO cells re-expressing WT or catalytically inactive (D533A; mut.) ATP13A1, normalized to reactions containing WT RMs for 3 replicates.
See also Figure S5.
Non-glycosylated CHST10 observed in insertion reactions with ATP13A1 KO RMs was resistant to carbonate extraction (Figure 4B). This result suggests that ATP13A1 depletion does not appreciably impair TM insertion, but rather leads to a larger proportion of CHST10 inserted into the ER membrane in the incorrect topology. Consistent with CHST10 being a direct client of ATP13A1, the defect in CHST10 topogenesis with ATP13A1 KO RMs is rescued by re-expressing WT but not catalytically inactive ATP13A1 (Figures 4C and S5A). However, in contrast to our findings with OMP25 (Figure 1), knocking down the EMC did not affect CHST10 topology in ATP13A1 KO cells (Figure S5B), and insertion reactions with ATP13A1 and EMC6 DKO RMs did not change the amount of membrane-integrated, non-glycosylated CHST10 compared to ATP13A1 KO RMs (Figure S5C). Thus, the EMC does not seem to be the primary route for CHST10 misinsertion. Altogether, our findings indicate that ATP13A1 facilitates topogenesis of type II proteins like CHST10 which contain an N-terminal TM and weak topology-guiding features.
ATP13A1 mediates signal sequence topogenesis
The largest class of proteins destabilized by the loss of P5A-ATPase function contain cleavable N-terminal signal sequences (McKenna et al., 2020). Notably, the biosynthesis of reporters containing the signal sequence of WNT1 or the C. elegans homolog EGL-20 have been shown to require P5A-ATPase activity (Li et al., 2021). Although WNT1 is normally secreted and not detected in our proteomics datasets, the levels of endogenous β-catenin, which is stabilized by WNT1 signaling (Nusse and Clevers, 2017), decrease in ATP13A1 KO cells and is selectively rescued by re-expressing WT ATP13A1 (McKenna et al., 2020) (Figure S6A and S6B). Since signal sequences must engage SEC61 in the same orientation as type II TMs, we hypothesized that ATP13A1 also facilitates the topogenesis of certain signal sequences. Supporting this idea, mutations that make the EGL-20 signal sequence more hydrophobic (referred to as EGL-20H), and therefore more likely to insert in the incorrect, NER, topology (Goder and Spiess, 2003; Sakaguchi et al., 1992), are more destabilized by P5A-ATPase depletion (Li et al., 2021). To further test this possibility, we incorporated WNT1 or a reporter containing the EGL-20 or EGL-20H signal sequence (Li et al., 2021) into the topology reporter system (Figure S6C). ATP13A1 depletion resulted in a strong reduction in the RFPER:BFP ratio of these proteins (Figure 5A). In comparison, the RFPER:BFP ratio of a reporter containing the robust signal sequence of bovine preprolactin (referred to as PRL) did not change with ATP13A1 depletion. These results support the findings that ATP13A1 mediates the biogenesis of certain signal sequence-containing proteins.
Figure 5. ATP13A1 facilitates signal sequence topogenesis.
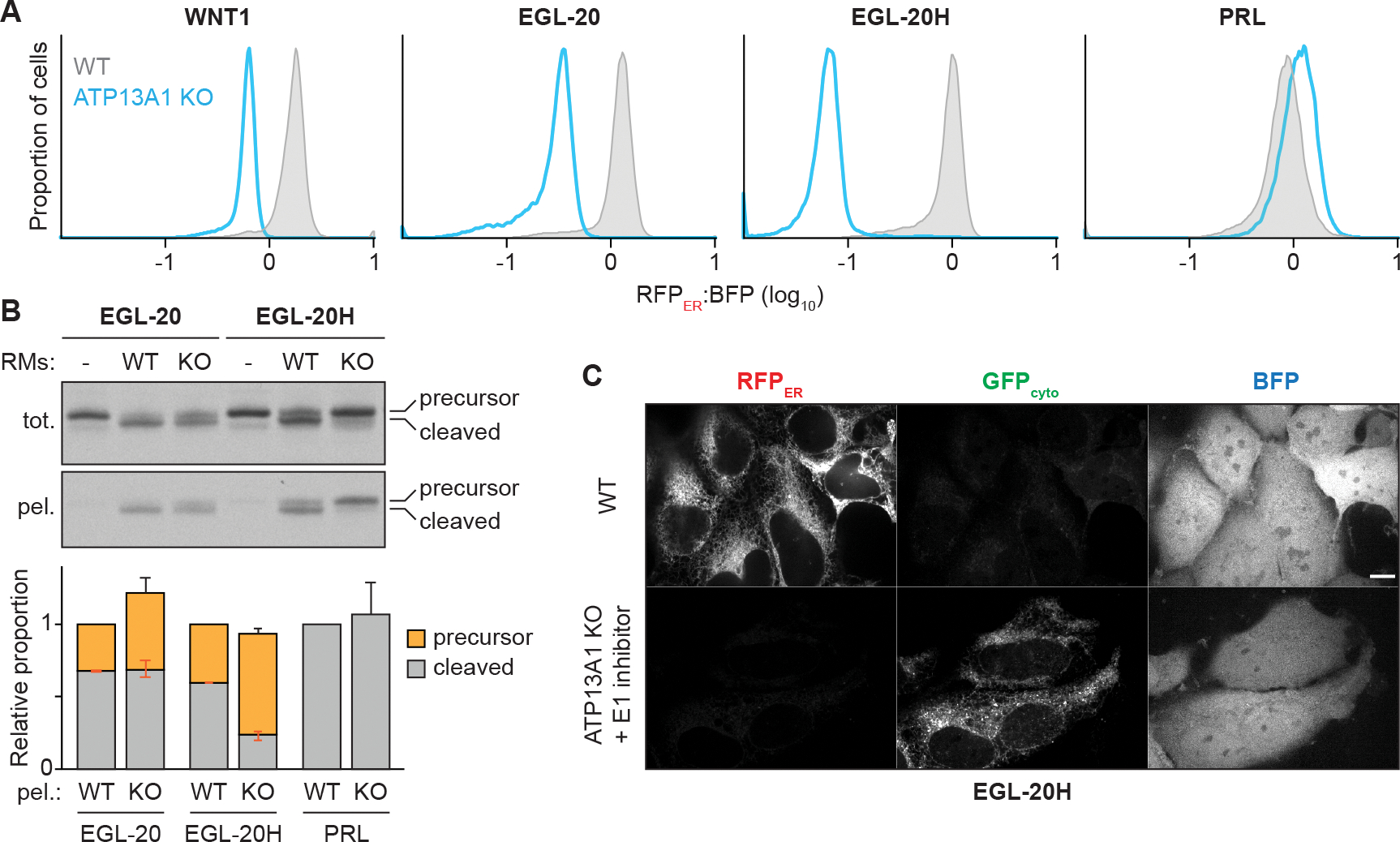
(A) ATP13A1 is required for the stability of WNT1 homologs. RFPER:BFP ratios of topology reporters of the signal sequence-containing proteins WNT1, its C. elegans homolog EGL-20, an EGL-20 variant with a more hydrophobic signal sequence (EGL-20H), or preprolactin (PRL) in wildtype (WT; gray) or ATP13A1 knockout (KO; light blue) cells.
(B) Reconstitution of ATP13A1-dependent signal sequence topogenesis. SDS-PAGE and autoradiography (top) of radiolabeled EGL-20 or EGL-20H synthesized in vitro without or with ER-derived rough microsomes (RMs) isolated from WT or ATP13A1 KO cells before (tot.) or after pelleting (tot.) to enrich RM-associated proteins. All proteins contain a C-terminal FLAG tag used for denaturing immunoprecipitations. Precursor and signal-cleaved substrates were quantified from the pelleted samples (bottom) and normalized to the average WT signal for each substrate. Shown are mean +/− sem of precursor (orange with negative orange error bar), signal-cleaved (gray with positive orange error bar), and total (black error bar) values for 3 replicates.
(C) Fluorescence microscopy showing RFPER, GFPcyto, and BFP signal of the EGL-20H topology reporter in untreated WT cells or ATP13A1 KO cells treated with 1 μM MLN7243. Scale bar, 10 μm.
See also Figure S6.
Cell-free insertion reactions revealed efficient cleavage of the EGL-20 and EGL-20H signal sequences with WT RMs, while uncleaved precursor proteins accumulated in reactions with ATP13A1 KO RMs (Figure 5B). Pelleting RMs efficiently recovered precursor EGL-20H with ATP13A1 KO RMs, indicating that a substantial proportion of the uncleaved protein resides at the ER in the absence of ATP13A1. In comparison, cleavage of the PRL signal sequence was unaffected by ATP13A1 KO (Figures 5B and S6D). Misorientation of EGL-20H at ER lacking ATP13A1 was further supported by fluorescence microscopy. In WT cells, the EGL-20H topology reporter produced a reticular RFPER pattern and very low GFPcyto signal. In ATP13A1 KO cells treated with an inhibitor of the E1 ubiquitin activating enzyme (see below), RFPER was no longer detectable while GFPcyto was observed in a reticular pattern distinct from the diffuse signal of cytosolic BFP (Figure 5C). These findings suggest that ATP13A1 mediates the topogenesis of signal sequences prone to misinserting in the NER orientation.
ATP13A1 prevents ERAD of misoriented proteins
Our topology reporters provide the distinct ability to specifically assay the stability of misoriented proteins. Consistent with increased incorporation in the incorrect topology, fluorescent flow cytometry revealed a higher GFPcyto:BFP ratio of the CHST10 type II protein reporter in ATP13A1 KO cells than in WT cells (Figure 6A). The GFPcyto:BFP ratio of the CHST10 reporter further increased with inhibitors of proteasomal activity, the E1 ubiquitin activating enzyme, or the AAA-ATPase p97, but not with SPP inhibition (Figure 6A), consistent with the substrate preference of SPP for transmembrane helices in the Ncyto orientation (Yücel and Lemberg, 2020). We observed similar results with the WNT1 and EGL-20H signal sequence reporters (Figures 6A and S7A) but not with the PRL reporter (Figure S7B). Importantly, the same inhibitors did not alter RFPER:BFP ratios corresponding to correctly inserted or translocated reporters (Figures 6A and S7A). In addition, the GFPcyto signal stabilized in ATP13A1 KO cells treated with ubiquitin-proteasome inhibitors persisted after semi-permeabilization (Figure S7C), suggesting membrane integration of the reporters in the incorrect topology. Thus, ERAD mechanisms distinct from those that act on mislocalized mitochondrial TA proteins specifically degrade misoriented ATP13A1-dependent proteins.
Figure 6. ATP13A1 protects misoriented proteins from ERAD.
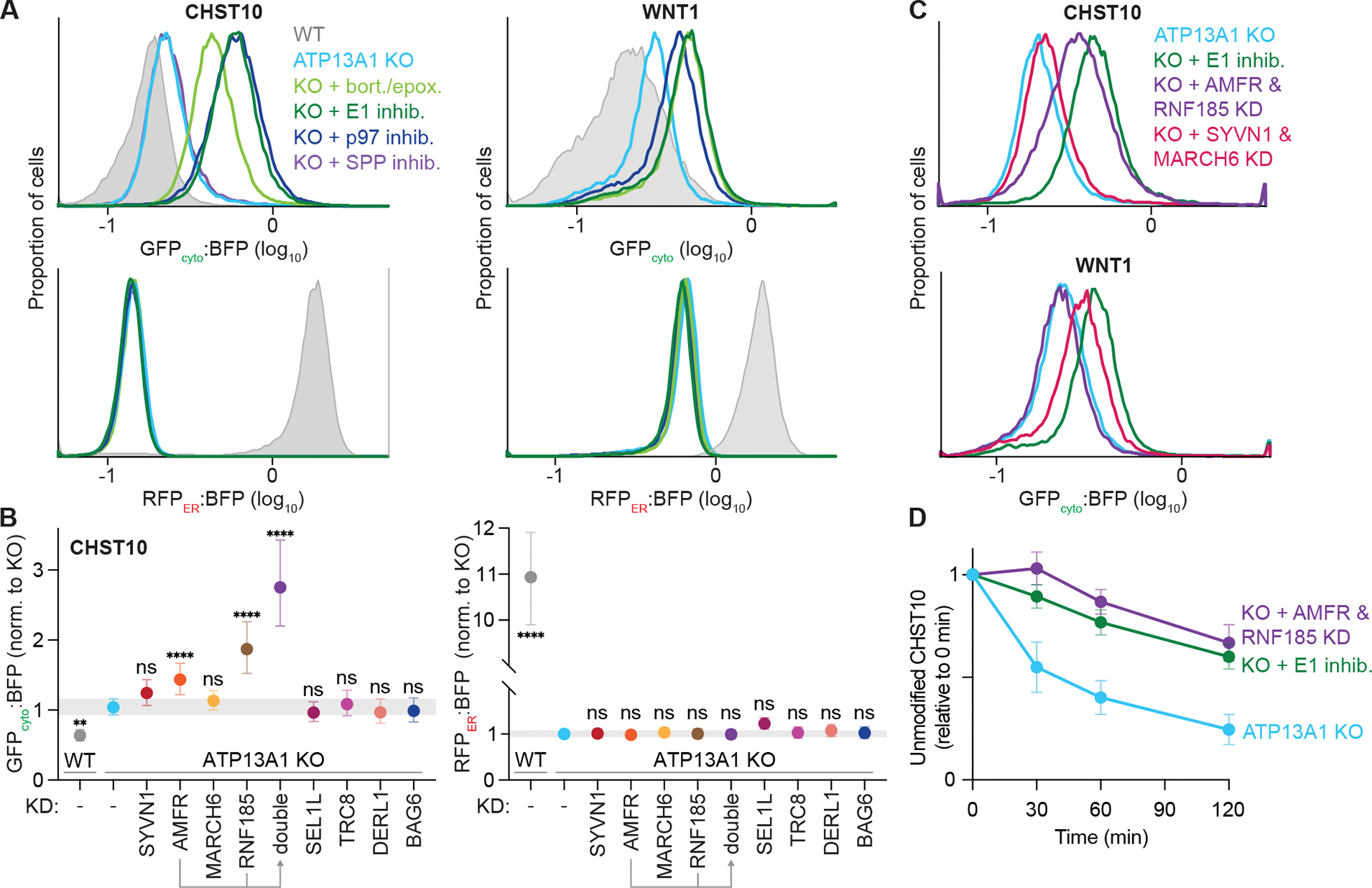
(A) Inhibiting the ubiquitin-proteasome system stabilizes misoriented proteins. GFPcyto:BFP (top) and RFPER:BFP (bottom) ratios of the CHST10 type II (left) or WNT1 signal sequence (right) topology reporter in wildtype (WT; gray) or ATP13A1 knockout (KO; light blue) cells treated without or with inhibitors (inhib.) of SPP (purple), proteasomal activity (bort./epox.; light green), the E1 ubiquitin activating enzyme (1 μM MLN7243; dark green), or the p97 AAA-ATPase (1 μM CB-5083; dark blue).
(B) Distinct ubiquitin ligases mediate misoriented CHST10 ERAD. GFPcyto:BFP (left) and RFPER:BFP (right) ratios of the CHST10 topology reporter in WT (gray) ATP13A1 KO cells treated with siRNAs to knock down (KD) the indicated factors. Shown are median values and interquartile range of at least two biological replicates of n≥5,000 each.
(C) Specific but redundant ubiquitin ligases mediate misoriented protein ERAD. GFPcyto:BFP ratios of the CHST10 (top) or WNT1 (bottom) topology reporter in ATP13A1 KO cells treated without (light blue) or with E1 inhibitor (dark green), or with siRNAs to knock down either AMFR and RNF185 (purple) or SYVN1 and MARCH6 (pink).
(D) Timecourse of misoriented CHST10 ERAD. Translation shut-off reactions of ATP13A1 KO cells expressing the CHST10 topology reporter treated without (light blue) or with siRNAs against AMFR and RNF185 (purple) or E1 inhibitor (dark green). Samples taken at the indicated timepoints were analyzed by immunoblotting for unmodified CHST10, normalized to time = 0 min, and the mean +/− sem for 3 replicates plotted.
See also Figure S7.
Investigating a panel of mammalian ERAD factors (Figures 6B and S7D) revealed that knocking down the ER-resident ubiquitin ligases AMFR (also known as RNF45 or gp78) (Ninagawa et al., 2020; Olzmann et al., 2013) or RNF185 (Kaneko et al., 2016; Khouri et al., 2013; van de Weijer et al., 2020) stabilized the GFPcyto:BFP ratio of the CHST10 reporter in ATP13A1 KO cells without affecting the RFPER:BFP ratio (Figure 6B). Knocking down both AMFR and RNF185 stabilized the GFPcyto:BFP ratio of the CHST10 reporter to a similar extent as E1 inhibition (Figures 6B and 6C), suggesting that these ligases may have partially redundant functions (Wang et al., 2022) in misoriented CHST10 ERAD. In contrast, the GFPcyto:BFP ratio of the WNT1 reporter did not change after knocking down AMFR or RNF185 individually or in combination (Figures 6C and S7E). Instead, knocking down MARCH6 selectively stabilized misoriented WNT1 but not WNT1 in the correct orientation or CHST10 in either orientation (Figures 6B, 6C, and S7E). Immunoblotting and translation shut-off experiments measuring the rate of misoriented protein degradation supported the redundant roles of AMFR and RNF185 in misoriented CHST10 ERAD (Figures 6D, S7F, and S7G) and the distinct role of MARCH6 in misoriented WNT1 ERAD (Figure S7H). Thus, multiple ERAD pathways act to eliminate different misoriented type II and signal sequence-containing proteins. Altogether, our findings establish that accurate protein topogenesis mediated by ATP13A1 protects misoriented type II and signal sequence-containing proteins from ERAD.
Discussion
We have shown that the P5A-ATPase ATP13A1 prevents ERAD of mislocalized and misoriented proteins (Figure 7). ATP13A1 clients theoretically retain the ability to fold properly but fail another step of protein biosynthesis such as localization or topogenesis. Client selection by ATP13A1 occurs primarily through biophysical features, specifically a C- or N-terminal transmembrane helix, as only a short lumenal domain is compatible with dislocation. An acidic cavity at the substrate-binding site of the P5A-ATPase confers additional preference for proteins with basic residues near the lumenal side of the membrane, which may help select mitochondrial TA proteins and enforce the positive-inside rule. The eligibility of proteins to become ATP13A1 clients also may be influenced by the binding of complex partners to properly inserted proteins that may prevent recognition or dislocation by ATP13A1. Similar models involving the recognition of ‘orphan’ subunits apply to protein QC mediated by Msp1/ATAD1, a protein dislocase at the outer mitochondrial membrane and peroxisomes (Weir et al., 2017), and by the Asi complex, an E3 ligase complex that safeguards the integrity of the inner nuclear membrane in yeast (Natarajan et al., 2020).
Figure 7. ATP13A1 prevents wasteful ERAD of mislocalized and misoriented proteins.
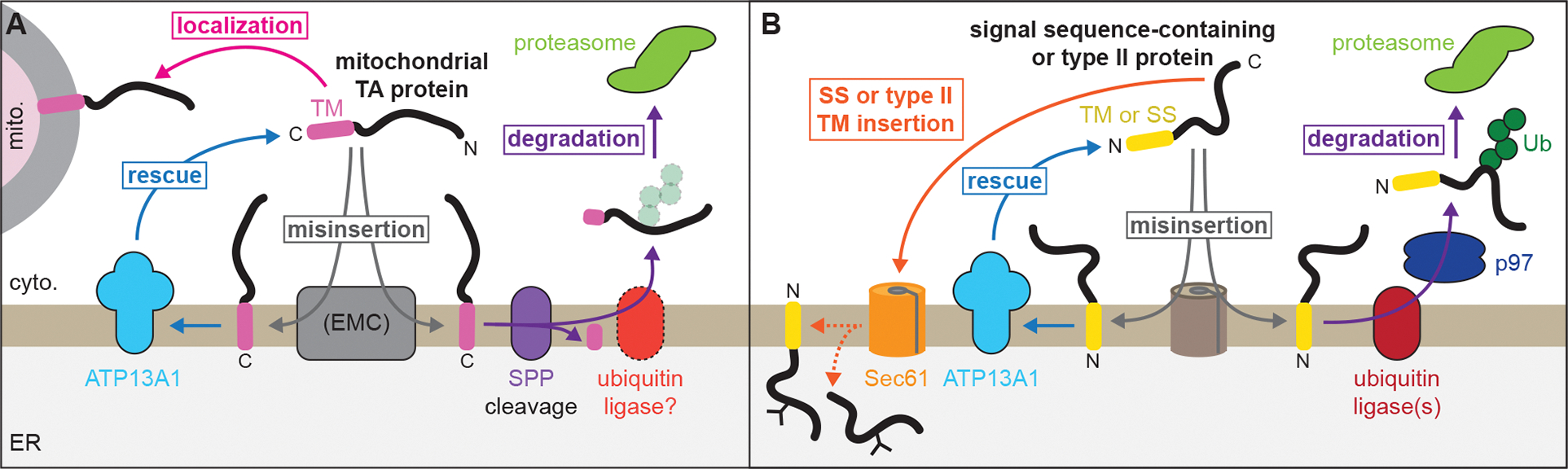
(A) Dislocation by ATP13A1 provides mislocalized mitochondrial TA proteins additional opportunities to insert into the outer membrane of mitochondria (mito.) and protects against SPP-mediated ERAD.
(B) Correct topogenesis of certain signal sequences and type II TMs requires ATP13A1, which prevents ERAD of misoriented substrates mediated by ER-resident ubiquitin ligases. Ribosomes present during cotranslational protein translocation and TM insertion are omitted for clarity.
Although ATP13A1 operates on both mislocalized and misoriented proteins, different ATP13A1 clients engage distinct biosynthetic and ERAD factors. Our results indicate that multiple mitochondrial TA proteins mislocalize to the ER via the EMC (Figure 1) (Roboti et al., 2021), although we cannot rule out the possibility that some misinsert through GET1/GET2. This observation is consistent with reports that TA protein clients of the EMC have a moderately hydrophobic TM (Chitwood et al., 2018; Guna et al., 2018), a general characteristic of mitochondrial TA proteins (Fry et al., 2021). A reported interaction between the P5A-ATPase and EMC in yeast (Shurtleff et al., 2018) may reflect a coupled mechanism for ensuring the integrity of TA protein insertion. Without ATP13A1, mislocalized mitochondrial TA proteins in mammalian cells undergo ERAD mediated by SPP (Figures 2 and 7A). The possible involvement of MARCH6 also may be consistent with observations that its yeast homolog, Doa10, mediates the degradation of mislocalized TA proteins (Dederer et al., 2019; Matsumoto et al., 2019). Both SPP and MARCH6 also regulate the levels of an ER TA protein (Stefanovic-Barrett et al., 2018), but how SPP cooperates with ubiquitin ligase(s) during ERAD remains to be determined.
The EMC has been reported to insert N-terminal TMs in the NER topology (Chitwood et al., 2018; O’Keefe et al., 2021) but does not appear to misinsert the type II protein CHST10 (Figure S5C). Considering these results, the most likely candidate for CHST10 misinsertion is the SEC61 complex. Signal sequences and TMs that are more hydrophobic and that have fewer basic N-terminal residues are more likely to insert in the wrong topology (Goder and Spiess, 2003; Sakaguchi et al., 1992; Wahlberg and Spiess, 1997) and to depend on ATP13A1 (Li et al., 2021). In the simplest model, ATP13A1 acts independently downstream of mistakes that generate type II proteins in the incorrect orientation (Figure 7B). Alternatively, our observations also may support direct cooperation between ATP13A1 and SEC61 to achieve correct type II TM and signal sequence topogenesis. Notably, kinetic constraints of cotranslational protein import by SEC61 may limit the window for ATP13A1 function in topogenesis, and coordination may be especially important if ATP13A1 functions on multi-spanning membrane proteins such as yeast Hmg2 (Shearer and Hampton, 2004). Indeed, prior studies have shown that some signal sequences and type II TMs initially insert headfirst before inverting to achieve correct type II topology at the SEC61 complex (Devaraneni et al., 2011; Goder and Spiess, 2003); our findings suggest that such inversions may involve ATP13A1. The function of ATP13A1 in protein topogenesis may explain its far-reaching effects on diverse ER-targeted proteins and functions (Feng et al., 2020; Li et al., 2021; McKenna et al., 2020; Qin et al., 2020; Shearer and Hampton, 2004; Tipper and Harley, 2002).
Without ATP13A1 function, misoriented type II and signal sequence-containing proteins engage traditional ERAD processes involving ubiquitination, p97, and proteasomal degradation (Figures 6A and 7B). However, different misoriented proteins are targeted by distinct ER-resident ubiquitin ligases. RNF185 and AMFR appear to have specific but redundant roles in degrading misoriented populations of the type II protein CHST10 but have no effect on cytosolic or misoriented populations of the signal sequence-containing protein WNT1. How specificity is determined when multiple mammalian ubiquitin ligases function in the ERAD of misoriented proteins remains an important question. Tools such as the topology reporter system established in this study which simultaneously reports the stability of correctly inserted and misoriented protein populations will be instrumental for investigating such questions.
Considered together, our study supports a paradigm for protein QC in which correction is prioritized over degradation to deal with errors of protein biosynthesis. Prioritization for ATP13A1 function over ERAD may be facilitated by the apparent ability of ATP13A1 to operate without obligate cofactors, in contrast to multi-step ERAD pathways that require multiple components (Olzmann et al., 2013; Wu and Rapoport, 2018). Protein dislocation by ATP13A1 also seems to be rapid relative to ERAD processes. For example, ~50% of F-OMP25 is dislocated by ATP13A1 in WT RMs within 15 min in vitro (McKenna et al., 2020), while only ~10% of F-OMP25 is cleaved by SPP in ATP13A1 KO RMs after 45 min (Figure S1F). Moreover, the destabilization of diverse proteins by ATP13A1 depletion indicates that errors in protein biosynthesis occur more often than commonly appreciated. ATP13A1 therefore represents an early defense mechanism against mistakes in protein localization and topogenesis at the ER that do not irretrievably lead to protein misfolding. As protein synthesis is one of the most energy-demanding cellular processes, such corrective mechanisms may apply to many aspects of protein biosynthesis to prevent the wasteful degradation of nascent protein intermediates capable of being rescued.
Limitations of the study
Our topology reporter system uses all common selection markers and expresses three fluorescent proteins, which limits the feasibility of rescue assays. Partial knockdown efficiencies of ERAD factors also may limit interpretations. Thus, future work is required to determine the precise role of identified factors in misoriented protein ERAD.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to the lead contact, Sichen Shao (sichen_shao@hms.harvard.edu).
Materials availability
Renewable materials generated in this study are available upon request to the lead contact.
Data and code availability
Proteomics data have been deposited at MassIVE and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FLAG M2 | Sigma | F1804 (RRID: AB_262044) |
| FLAG M2-HRP | Sigma | A8592 (RRID: AB_439702) |
| CHST10 | Sigma | PA5-92187 (RRID: AB_2806403) |
| SEL1L | Sigma | PA5-88333 (RRID: AB_2804837) |
| DERL1 | Abcam | ab176732 (RRID: AB_2858187) |
| BAG6 | Gift from R.S. Hegde | N/A |
| TRAPα | Gift from R.S. Hegde | N/A |
| TOM20 | Abcam | ab186735 (RRID: AB_2889972) |
| ATP13A1 | Proteintech | 16244-1-AP (RRID: AB_2290293) |
| CNX | Enzo Life Sciences | ADI-SPA-865 (RRID: AB_10618434) |
| EMC2 | Proteintech | 25443-1-AP (RRID: AB_2750836) |
| EMC6 | Abcam | Ab84902 (RRID: AB_1925516) |
| GET1 | Synaptic Systems | 324 002 (RRID: AB_2620063) |
| Ubiquitin | Santa Cruz Biotechnology | sc-8017 (RRID: AB_628423) |
| SQLE | Proteintech | 12544-1-AP (RRID: AB_2195888) |
| OMP25 | Proteintech | 15666-1-AP (RRID: AB_2201149) |
| PLAT3 | Abcam | Ab88447 (RRID: AB_2041691) |
| SPP | Abcam | Ab247061 |
| AMFR | Novus Biologicals | NBP2-15374 |
| SYVN1 | Abcam | Ab170901 (RRID: AB_2833021) |
| RNF185 | Abcam | Ab181999 |
| PEX14 | Proteintech | 10594-1-AP (RRID: AB_2252194) |
| SGTA | Mateja et al., 2015 | |
| β-catenin | Cell Signaling Technology | 9562 (RRID: AB_331149) |
| WNT1 | Invitrogen | PIMA515544 |
| FITC-conjugated FLAG M2 | Sigma | F4049 (RRID: AB_439701) |
| Peroxidase-conjugated anti-HA antibody | Cell Signaling Technology | 2999 (RRID: AB_1264166) |
| Peroxidase-conjugated Goat Anti-Mouse | Jackson ImmunoResearch | 115-035-003 (RRID: AB_10015289) |
| Peroxidase-conjugated Goat Anti-Rabbit | Jackson ImmunoResearch | 111-035-003 (RRID: AB_2313567) |
| Alexa Fluor 488-conjugated Goat Anti-Mouse | Jackson ImmunoResearch | 115-545-003 (RRID: AB_2338840) |
| Alexa Fluor 594-conjugated Goat Anti-Rabbit | Jackson ImmunoResearch | 111-585-003 (RRID: AB_2338059) |
| Bacterial and virus strains | ||
| DH5α E. coli | Homemade | N/A |
| Biological samples | ||
| RRL-based in vitro translation system | Feng and Shao, 2018 | N/A |
| In vitro transcription system | Feng and Shao, 2018 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Acetonitrile | Millipore Sigma | 34851 |
| Doxycycline hyclate | Sigma | D9891 |
| Hygromycin B | Millipore Sigma | 31282-04-9 |
| Blasticidin S HCl | Gibco | A11139-03 |
| Puromycin | Gibco | A11138-03 |
| Bortezomib | LC laboratories | B-1408 |
| Epoxomicin | ApexBio | 134381-21-8 |
| (Z-LL)2-ketone | Millipore Sigma | 313664-40-3 |
| E1 inhibitor (MLN7243) | Cayman Chemical | 1450833-55-2 |
| P97 inhibitor (CB-5083) | Cayman Chemical | 1542705-92-9 |
| Cycloheximide | Sigma | 01810 |
| Paraformaldehyde | Electron Microscopy Sciences | 100503-917 |
| MitoTracker CMXRos | Invitrogen | M7512 |
| RNase-free DNase | QIAGEN | 79254 |
| Lys-C protease, MS grade | ThermoFisher Scientific | 90051 |
| Trypsin Gold, MS grade | Promega | V5280 |
| Urea | Millipore Sigma | 51456 |
| Hydroxylamine, 50% | ThermoFisher Scientific | 90115 |
| Trifluoroacetic acid, LC-MS grade | ThermoFisher Scientific | 85183 |
| Formic acid | Sigma | 27001 |
| cOmplete EDTA-free protease inhibitor cocktail | Roche | 05056489001 |
| CaptivA PriMAB Protein A affinity resin | Repligen | CA-PRI-1000 |
| Commercial assays | ||
| Pierce BCA protein assay kit | ThermoFisher Scientific | 23225 |
| Pierce quantitative colorimetric peptide assay | ThermoFisher Scientific | 23275 |
| TMTpro 18-plex label reagent set | ThermoFisher Scientific | A52045 |
| Deposited data | ||
| Proteomics data | MassIVE repository | MSV000088575 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-3216 (RRID: CVCL_0063) |
| Flp-In 293 T-REx | Invitrogen | R78007 (RRID: CVCL_U427) |
| Flp-In HeLa T-REx | Gift from B. Raught | N/A |
| ATP13A1 KO Flp-In HeLa T-REx | McKenna, Sim et al. 2020 | N/A |
| ATP13A1 KO Flp-In 293 T-REx | McKenna, Sim et al. 2020 | N/A |
| ATP13A1 KO Flp-In 293 T-REx: WT ATP13A1 | McKenna, Sim et al. 2020 | N/A |
| ATP13A1 KO Flp-In 293 T-REx HEK293: D533A ATP13A1 | McKenna, Sim et al. 2020 | N/A |
| ATP13A1 KO, EMC6 KO Flp-In 293 T-REx | This study | N/A |
| Flp-In HeLa T-REx: F-OMP25 | McKenna, Sim et al. 2020 | N/A |
| ATP13A1 KO Flp-In HeLa T-REx: F-OMP25 | McKenna, Sim et al. 2020 | N/A |
| Flp-In HeLa T-REx: F-BAK1 | McKenna, Sim et al. 2020 | N/A |
| Flp-In HeLa T-REx: F-CEND1 | This study | N/A |
| ATP13A1 KO Flp-In HeLa T-REx: F-CEND1 | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: ASGR1-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx: ASGR1-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: DHFR-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: BFP-P2A-CNb11-SEC61β | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: CHST10-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx: CHST10-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: QQ-CHST10-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: RR-CHST10-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: WNT1-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx: WNT1-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: EGL-20-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx: EGL-20-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: EGL-20H-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx: EGL-20H-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: PRL-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx: PRL-NCb11-P2A-BFP | This study | N/A |
| Oligonucleotides | ||
| SRP14 fwd | GAGAGCGAGCAGTTCCTGAC | |
| SRP14 rev | GTTTGGTTCGACCGTCATACT | |
| TRC8 fwd | CAAAGCCGGTTCTGCATCG | |
| TRC8 rev | AGTTGCAGCTAACAGAAAGGC | |
| MARCH6 fwd | GTTCCCTTGGATCAGACTCC | |
| MARCH6 rev | CAATGTTCCGGATGCCATTTG | |
| EMC6 sg1 fwd | CACCGGCCGCCTCGCTGATGAACGG | |
| EMC6 sg1 rev | AAACCCGTTCATCAGCGAGGCGGCc | |
| Recombinant DNA | ||
| pHDM-G | PlasmID | EvNO00061606 |
| pHDM-HGPM2 | PlasmID | EvNO00061607 |
| pHDM-tat1B | PlasmID | EvNO00061608 |
| pRC-CMV-rev1B | PlasmID | EvNO00061616 |
| pHAGE-FLAG-mNG3K(1-10)-P2A-Prl(SS)-mCh3V(1-10)-KDEL | This study | N/A |
| pX459 | Addgene | 62988 |
| pcDNA5 FRT/TO F-OMP25 | McKenna, Sim et al. 2020 | N/A |
| pcDNA5 FRT/TO F-CEND1 | This study | N/A |
| pcDNA5 FRT/TO GFP-OMP25 | This study | N/A |
| pcDNA5 FRT/TO GFP-MAVS | This study | N/A |
| pcDNA5 FRT/TO ASGR1-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO DHFR-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO BFP-P2A-NCb11-SEC61β | This study | N/A |
| pcDNA5 FRT/TO CHST10-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO QQ-CHST10-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO RR-CHST10-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO WNT1-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO EGL-20-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO EGL-20H-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO PRL-NCb11-P2A-BFP | This study | N/A |
| pSP64 F-OMP25 | McKenna, Sim et al. 2020 | N/A |
| pSP64 F-SQS | This study | N/A |
| pSP64 F-STX1B | This study | N/A |
| pSP64 CHST10-FLAG | This study | N/A |
| pSP64 RR-CHST10-FLAG | This study | N/A |
| pSP64 EGL-20-FLAG | This study | N/A |
| pSP64 EGL-20H-FLAG | This study | N/A |
| pSP64 PRL-FLAG | This study | N/A |
| pDONR ASGR1 | DNASU | HsCD00005525 |
| pDONR CHST10 | Gift from the Harper lab | N/A |
| pRK5-HA-ubiquitin-WT | Addgene | 17608 |
| pTAG-BiP(SS)-BFP-KDEL | Friedman et al., 2011 | N/A |
| Software and algorithms | ||
| ImageJ | NIH | N/A |
| Prism 9 | GraphPad | N/A |
| FlowJo v10 | FlowJo | N/A |
| Illustrator | Adobe | N/A |
| Photoshop | Adobe | N/A |
| ImageQuant TL | Cytiva | N/A |
| In-house MS data analysis software | Huttlin et al., 2010 | N/A |
| Other | ||
| Control siRNA | Horizon | D-001810-10-05 |
| EMC2 siRNA | Horizon | L-010631-00-0005 |
| EMC5 siRNA | Horizon | L-018365-00-0005 |
| EMC6 siRNA | Horizon | L-014711-02-0005 |
| WRB siRNA | Horizon | L-011898-01-0005 |
| ATP13A1 siRNA | Horizon | L-020426-00-0005 |
| SPP siRNA | Horizon | L-005896-00-0005 |
| TRC8 siRNA | Horizon | L-006942-00-0005 |
| MARCH6 siRNA | Horizon | L-006925-00-0005 |
| AMFR siRNA | Horizon | L-006522-00-0005 |
| SYVN1 siRNA | Horizon | L-007090-00-0005 |
| RNF185 siRNA | Horizon | L-007107-00-0005 |
| SEL1L siRNA | Horizon | L-004885-00-0005 |
| BAG6 siRNA | Horizon | L-005062-01-0005 |
| DERL1 siRNA | Horizon | L-010733-02-0005 |
| DMEM + GlutaMAX | Gibco | 10569-010 |
| FluoroBrite DMEM | Gibco | A1896701 |
| Trypsin-EDTA | Gibco | 25200-056 |
| PBS | Gibco | 10010-023 |
| DMSO | Sigma | 472301 |
| OPTI-MEM | Gibco | 31985-062 |
| TransIT-293 | Mirus | MIR 2706 |
| Lipofectamine 3000 | Invitrogen | L3000-015 |
| Lipofectamine RNAiMAX | Invitrogen | 13778-150 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | ThermoFisher Scientific | 34580 |
| Ponceau S solution | Sigma | P7170 |
| EasyTag L-[35S]-Methionine | Perkin Elmer | NEG709A005MC |
| Polypropylene Microfuge Tubes | Beckman Coulter | 357448 |
| Digitonin | Millipore Sigma | 11024-24-1 |
| Triton X-100 | Sigma | T9284 |
| Tween 20 | Sigma | P7949 |
| 10% SDS | Sigma | 71736 |
| 40% acrylamide (29:1) | Bio Rad | 1610146 |
| Nitrocellulose membrane | Bio Rad | 1620112 |
| Multi-well glass-bottom cell culture plates | MatTek | P24G-1.5-13-F |
| BioMax MR film | Carestream | 894 1114 |
| Buffer RLT | QIAGEN | 79216 |
| RNeasy Mini Kit | QIAGEN | 74106 |
| SuperScript III First-Strand Synthesis System | Invitrogen | 18080051 |
| SYBR Green Master Mix | Applied Biosystems | 4309155 |
| Pierce C18 tips, 100 μL | ThermoFisher Scientific | 87784 |
| Sep-Pak C18 cartridge, 100 mg | Waters | Wat036935 |
| 300Extend C18 column | Agilent | custom |
| Accucore 150 resin | ThermoFisher Scientific | 16126-000 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
All cell lines were maintained in DMEM with high glucose, GlutaMAX, and sodium pyruvate supplemented with 10% fetal bovine serum at 37°C and 5% CO2. Parent HEK293T, Flp-In 293 T-REx and HeLa T-REx cell lines were authenticated by short tandem repeat analysis and tested for mycoplasma yearly. To knock out EMC6 for RM isolation, ATP13A1 KO Flp-In 293 T-REx cells were transfected with px459 with target gRNA sequences inserted with standard molecular biology techniques using TransIT 293 (Mirus 2706) according to manufacturer’s instructions. 48 hr after transfection, cells were placed under selection with 2 μg/mL puromycin for 48 hr. Clonal lines were isolated and validated with Western blotting and genotyping.
Cells constitutively expressing mNeonGreen3K(1–10) [GFP(1–10)cyto] and ER lumenal mCherry3V(1–10)-KDEL [RFP(1–10)ER] were generated using lentiviral transduction. To generate lentiviral particles, HEK293T cells were transfected with a 2:1:1:1:10 ratio of pHDM-VSVG, pHDM-HGPM2, pHDM-tat1B, pRC-CMV-rev1B and pHAGE containing the desired insert using TransIT according to the manufacturer’s instructions. The media was harvested 48 hr after transfection and used to transduce Flp-In 293 T-REx cells in the presence of 10 μg/mL polybrene. 48 hr after transduction, cells were placed under selection with 1 μg/mL puromycin for 48 hr. Clonal lines expressing the desired product were initially confirmed by immunoblotting for FLAG-tagged mNeonGreen3K(1–10) and validated by complementation of the two split fluorescent proteins by transiently transfecting cells with plasmids encoding proteins containing tandem β11 stands of mNeonGreen3K and mCherry3V (NCb11) in the cytosol or ER lumen. Knockout of ATP13A1 in this reporter cell line was performed as described above, except clonal lines were isolated in the absence of puromycin selection.
All reporters were integrated into the doxycycline-inducible Flp-In locus by cotransfecting a 1:1 ratio of pOG44 and pcDNA5/FRT/TO containing the gene of interest using TransIT 293 (for Flp-In 293 T-Rex cells) or Lipofectamine 3000 (for Flp-In HeLa T-Rex cells; Invitrogen L3000) according to manufacturer’s instructions. Cells were selected with 10 μg/mL blasticidin and 50 μg/mL (Flp-In 293 T-Rex cells) or 300 μg/mL (Flp-In HeLa T-Rex cells) hygromycin for 2–3 weeks, and expression was validated by induction with 100 ng/mL doxycycline for 24 hr followed by blotting, fluorescence microscopy, or fluorescent flow cytometry.
METHOD DETAILS
Plasmids and antibodies
pONDR-ASGR1 (HsCD00005525) was obtained from DNASU and pDONR-CHST10 was a gift from the Harper lab. pRK5-HA-ubiquitin-WT (Addgene) was a gift from the Finley lab. Flag-tagged reporter proteins were incorporated into pcDNA5/FRT/TO for cell line generation or an SP64-based vector for in vitro transcription as previously described (Feng and Shao, 2018; McKenna et al., 2020) using standard molecular biology techniques. The gRNA sequence (GCCGCCTCGCTGATGAACGGCGG) to knock out EMC6 was inserted into pX459 using standard molecular biology techniques. The complete sequences for mNeonGreen3K and mCherry3V were ordered as gBlocks and the sequences corresponding to GFP(1–10) and RFP(1–10) as described in (Feng et al., 2017, 2019; Zhou et al., 2020) lacking stop codons were PCR amplified with overhangs for Gibson assembly (see below). Sequences encoding the P2A ribosome skipping sequence and the preprolactin signal sequence were PCR amplified from described plasmids (Chu et al., 2021; Shao and Hegde, 2011) with overhangs for assembly. A 5-piece Gibson assembly reaction was initially performed to incorporate the following in this order into pcDNA5/FRT/TO: GFP(1–10), P2A, preprolactin signal sequence, RFP(1–10). The KDEL retention sequence followed by a stop codon was included as an overhang in a reverse primer and incorporated using full plasmid PCR followed by blunt end ligation. The entire open reading frame was then transferred into pHAGE behind an N-terminal 3xFLAG tag by restriction enzyme digestion and ligation. pHAGE and helper plasmids (pHDM-G, pHDM-HGPM2, pHDM-tat1B, pRC-CMV-rev1B) for lentivirus production were from PlasmID. Topology reporter constructs encoding proteins with the tandem β11 tag separated from BFP by a P2A sequence were generated in a pcDNA5/FRT/TO-based vector using standard molecular biology techniques by combining the individual β11, P2A, BFP, and reporter sequences amplified from the gBlocks and plasmids described above.
Anti-FLAG M2 (Sigma F1804, 1:5000 for IB, 1:500 for IF), HRP-conjugated anti-FLAG M2 (Sigma A8592, 1:5000 for IB), FITC-conjugated anti-FLAG M2 (Sigma F4049, 1:100 for flow cytometry), anti-CHST10 (Sigma PA5–92187, 1:2000 for IB, 1:150 for IP), anti-SEL1L (Sigma PA5–88333, 1:1000 for IB), anti-DERL1 (Abcam ab176732, 1:2000 for IB), anti-TOM20 (Abcam ab186735, 1:300 for IF), anti-ATP13A1 (Proteintech 16244–1-AP, 1:2500 for IB), anti-CNX (Enzo Life Sciences ADI-SPA-865, 1:5000 for IB), anti-EMC2 (Proteintech 25443–1-AP, 1:2500 for IB), anti-EMC6 (Abcam ab84902, 1:1000 for IB), anti-GET1 (Synaptic Systems 324 002, 1:1000 for IB), anti-ubiquitin (Santa Cruz Biotechnology sc-8017, 1:200 for IB), anti-SQLE (Proteintech 12544–1-AP, 1:1000 for IB), anti-OMP25 (Proteintech 15666–1-AP, 1:1000 for IB), anti-PLAT3 (Abcam ab88447, 1:1000 for IB), anti-SPP (Abcam ab247061, 1:2000 for IB), anti-AMFR (Novus Biologicals NB-15374, 1:1000 for IB), anti-SYVN1 (Abcam ab170901, 1:2000 for IB), anti-RNF185 (Abcam ab181999, 1:5000 for IB), anti-PEX14 (Proteintech 10594–1-AP, 1:500 for IF), anti-β-catenin (Cell Signaling Technology 9562, 1:1000 for IB), HRP-conjugated anti-HA (Cell Signaling Technology, 1:1000 for IB), HRP-conjugated goat anti-mouse (Jackson ImmunoResearch 115–035-003, 1:5000 for IB), HRP-conjugated goat anti-rabbit (Jackson ImmunoResearch 111–035-003, 1:5000 for IB), Alexa Fluor 488-conjugated goat anti-mouse (Jackson ImmunoResearch 115–545-003, 1:500 for IF), and Alex Fluor 594-conjugated goat anti-rabbit (Jackson ImmunoResearch 111–585-003, 1:500 for IF) antibodies were all purchased. Antibodies against SGTA (1:2500 for IB) are as previously described (Mateja et al., 2015), and antibodies against TRAPα (1:300 for IF) and BAG6 (1:5000 for IB) were gifts from the Hegde lab.
Cell treatments, siRNA-mediated knockdowns, and RT-qPCR
Unless otherwise noted, reporter protein expression was induced with 100 ng/mL doxycycline for 24 hr. Cells were treated with 5 μM (Z-LL)2-ketone (Millipore Sigma 313664–40-3) for 24 hr to inhibit SPP activity, 0.5 μM bortezomib (LC laboratories B-1408) and 0.5 μM epoxomicin (ApexBio 134381–21-8) for 4 hr to inhibit proteasomal activity, 1 μM MLN7423 (Cayman Chemical 1450833–55-2) for 4 hr to inhibit E1 activity, or 1 μM CB-5083 (Cayman Chemical 1542705–92-9) for 4 hr to inhibit p97 activity. For translation shutoff assays, cells were treated with 50 μg/mL cycloheximide (Sigma 01810) for the indicated amount of time prior to analysis. For most siRNA-mediated knockdowns, cells were seeded in 6-well plates (for flow cytometry) or in 24-well glass-bottom plates (for immunofluorescence) 72 hr prior to analysis. Cells at 30% confluence were transfected with 30 pmol (6-well) or 10 pmol (24-well) siRNA 48 hr prior to analysis using Lipofectamine RNAiMAX (Invitrogen 13778–150) according to manufacturer’s instructions. Reporter protein expression was induced 24 hours after siRNA transfection. To knock down ERAD factors (Figures S2H, 6B–D and S7E–H), reverse transfection was performed: trypsinized cells were plated at 30% confluence into 6-well plates containing 30 pmol siRNA 48 hr prior to analysis with Lipofectamine RNAiMAX according to manufacturer’s instructions. For double knockdowns, 30 pmol of each siRNA was used. To detect ubiquitination (Figure S7F), ERAD factors were knocked down in ATP13A1 knockout cells by reverse transfection as described above. 24 hr prior to analysis, cells were simultaneously treated with 100 ng/mL doxycycline to induce expression of the CHST10 reporter protein, and transiently transfected with pRK5-HA-ubiquitin-WT using TransIT-293 according to manufacturer’s instructions. Cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% Sodium Deoxycholate, 0.1% SDS) plus 20 mM NEM and cOmplete protease inhibitor cocktail (Roche) for 1 hr at 4°C, and clarified by centrifuging at 13,000g for 20 min. Soluble material was incubated with anti-CHST10 antibody and Protein A resin for 2 hr, washed with RIPA buffer, eluted with protein sample buffer, and directly analyzed.
For RT-qPCR, cells were collected in cold PBS, pelleted by centrifugation at 1,000g for 5 min at 4°C, snap frozen in liquid nitrogen, and stored at −80°C. Cell pellets were lysed by adding Buffer RLT (QIAGEN 79216) with 1% β-mercaptoethanol and shaking in a thermomixer at 1400 rpm for 15 min. RNA extraction was performed using the Rneasy Mini kit (QIAGEN 74106). The eluted RNA was treated with Rnase-free Dnase (QIAGEN 79254) and re-isolated using the Rneasy Mini kit. Reverse transcription of 0.5 μg RNA was performed using oligo(dT)20 primers and the SuperScript III First-Strand Synthesis System (Invitrogen 18080051) according to manufacturer’s instructions, except that DTT was omitted from the reactions. The resulting cDNA was diluted 1:200 and mixed with SYBR Green Master Mix (Applied Biosystems 4309155) and 300 nM each of forward and reverse primers. RT-qPCR was performed using the QuantStudio 7 Pro real-time PCR system. Amplification efficiencies for each primer set were assessed using a 4-fold dilution series. For each biological replicate, the average CT (threshold cycle) was calculated from three technical replicates. Fold changes were calculated using the ΔΔCT method, correcting for amplification efficiency (Livak and Schmittgen, 2001; Pfaffl, 2001). SRP14 was used as the reference gene. Data from three biological replicates were analyzed using GraphPad Prism 9.
Fluorescence microscopy
All images were collected with a Yokagawa CSU-X1 spinning-disk confocal microscope with Spectral Applied Research Aurora Borealis modification on a Nikon Ti motorized inverted microscope equipped with Plan Apo 100×/1.4 numerical aperture oil-immersion objective.
For immunofluorescence experiments, Flp-In HeLa T-REx cells in 24-well #1.5 coverslip-bottom plates (MatTek P24G-1.5–13-F) expressing the desired reporter protein and treated as described above were washed twice with PBS and fixed in 4% formaldehyde in PBS for 15 min. After fixation, cells were washed with PBS, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and then incubated with blocking solution (10% FBS/PBS) for 30 min, followed by primary antibodies in blocking solution for 1 hr. Cells were washed three times in PBS, incubated with secondary antibodies in blocking solution for 1 hr, and washed three more times with PBS before imaging. Fluorescence was excited with solid-state lasers at 488 nm (for Alexa Fluor 488) and 561 nm (for Alexa Fluor 594) and collected using ET525/50m or ET620/60m emission filters, respectively. Images were acquired with a Hamamatsu ORCA-R2 cooled CCD camera using Metamorph software. Z-series optical sections were collected with a step size of 0.5 μm and single sections were selected from approximately the center of the cell.
For live cell imaging, Flp-In 293 T-REx cells expressing topology reporters (Figures S4A, S4B, and 5C) were cultured in 6-well #1.5 coverslip-bottomed plates. For experiments shown in Figure S1C, WT, ATP13A1 KO, or ATP13A1 and EMC6 double-KO cells on 24-well #1.5 coverslip-bottomed plates were transiently transfected with pcDNA5/FRT/TO containing the OMP25 or MAVS TM fused to the C-terminus of sfGFP, plus pTagBFP-C containing TagBFP attached to the N-terminal BiP ER signal sequence and the C-terminal KDEL ER retention sequence using TransIT-293 according to manufacturer’s instructions. Prior to analysis, cells were changed into FluoroBrite DMEM (Gibco A1896701) containing 10% FBS. Cells were imaged in an Okolab stage top incubation chamber set to 37°C with humidified 5% CO2. For experiments shown in Figures S4A and S4B, BFP (TagBFP) fluorescence was excited using a SOLA395 light source and collected using an ET460/50m emission filter. GFPcyto (mNeonGreen3K) and RFPER (mCherry3V) were excited with solid-state lasers at 488 nm and 561 nm, respectively, and data collection was as described above. For experiments shown in Figures S1D and 5C, BFP (TagBFP) fluorescence was excited with a solid-state laser at 405 nm and collected using an ET455/50m emission filter. GFPcyto (mNeonGreen3K) and RFPER (mCherry3V) were excited and collected as described above. Images were acquired with a Hamamatsu ORCA-Fusion BT cooled CCD camera using Elements software.
Cellular fractionations and rough microsome preparation
Whole cell lysates were obtained by lysing cells in 1% digitonin in PSB [50 mM Hepes (pH 7.5), 100 mM KOAc, 2.5 mM Mg(Oac)2] and 1x cOmplete protease inhibitor cocktail (PIC; Roche 05056489001) on ice for 10 min before centrifuging at 21,000g for 5 min. The supernatant was collected, and protein concentrations were normalized prior to gel loading. To separate cytosolic and organellar membrane fractions, cells were first lysed in 0.02% digitonin in PSB containing 250 mM sucrose and 1x PIC, incubated on ice for 10 min and centrifuged at 1,000g for 5 min. The supernatant (cytosol) was collected, and the pellet (organellar membranes) was washed once in 250 mM sucrose in PSB. For immunoblotting, the washed pellet was solubilized in 1% digitonin in PSB and 1x PIC, incubated on ice for 10 min, and centrifuged at 21,000g for 5 min before collecting the supernatant (organellar membranes). For flow cytometry, the washed pellet (obtained after solubilization in 0.02% digitonin, as described above) was resuspended in PBS and directly analyzed.
Rough microsomes (RMs) were isolated from WT, ATP13A1 KO, ATP13A1 and EMC6 double KO, and ATP13A1 KO Flp-In 293 T-REx cells stably re-expressing FLAG-tagged WT or D533A ATP13A1 as previously described (Chitwood et al., 2018; McKenna et al., 2020). Briefly, cells were harvested and washed twice in cold PBS and centrifuged at 500g for 5 min. The cell pellet was resuspended in three volumes of 10 mM Hepes (pH 7.4), 250 mM sucrose, 2 mM MgCl2, and 1x PIC, and lysed by 27 passages through a 27-gauge needle. Lysates were centrifuged twice at 3,800g for 30 min, and the resulting supernatant was centrifuged at 75,000g for 1 hr at 4°C in a TLA-55 rotor. The RM pellet was resuspended in hRM buffer [10 mM Hepes (pH 7.4), 250 mM sucrose, 1 mM MgCl2, 0.5 mM DTT], adjusted and normalized to an absorbance at 280 nm (A280) of roughly 50 as measured by solubilization in 1% SDS.
In vitro translation and insertion assays
In vitro transcription reactions were performed with SP6 polymerase as previously described for 1 hr at 37°C and used directly in translation reactions (Feng and Shao, 2018). In vitro translation reactions were performed at 32°C. For CHST10, ASGR1, EGL-20, EGL-20H and PRL, translation reactions were performed for 45 min in the presence of RMs or hRM buffer. For FLAG-tagged TA proteins (F-OMP25, F-SQS and F-STX1B), translations were performed in the absence of RMs for 20 min before translation was stopped by the addition of 50 μg/mL Rnase A. RMs or hRM buffer were then added and samples were incubated at 32°C for another 45 min. Membrane-inserted material was isolated by diluting reactions 1:4 in PSB, layering onto 15% sucrose in PSB, and centrifuging at 186,000g for 20 min in a TLA55 rotor. The supernatant containing uninserted protein was collected. For F-OMP25, CHST10, and ASGR1, the pellet was subjected to carbonate extraction to isolate membrane-integrated proteins. Briefly, the pellet was resuspended in 10 reaction volumes of cold 100 mM NaCO3 and incubated on ice for 15 min, followed by centrifugation at 186,000g for 20 min in a TLA55 rotor. For F-OMP25 and F-SQS, protein sample buffer was added directly to total, supernatant, and pellet fractions prior to SDS-PAGE and autoradiography analysis. Reactions used to calculate pellet:supernatant ratios of F-SQS (Figure 1C) contained 5 μM (Z-LL)2-ketone to inhibit SPP cleavage. For CHST10 and ASGR1, total and pellet samples were subjected to denaturing FLAG immunoprecipitations. Briefly, the samples were denatured in 1% SDS in 0.1 M Tris (pH 8) and heated to 95°C for 5 min. Ten volumes of IP buffer and 10 μL of packed M2 resin were added and the samples were incubated at 4°C for 1 hr on a rotating platform. The resin was washed three times in IP buffer before protein sample buffer was added. For EGL-20, EGL-20H and PRL, the pellet obtained following centrifugation through a 15% sucrose cushion was washed once with PSB and then resuspended directly into 1% SDS in 0.1 M Tris (pH 8). Samples were then subjected to denaturing FLAG immunoprecipitations as described above.
Fluorescent flow cytometry
For whole cell flow cytometry, cells were detached using 0.25% trypsin-EDTA, collected in complete growth medium, centrifuged at 500g for 3 min. Pelleted cells were washed in PBS and filtered through a 35 μm mesh strainer. Data were collected on an Attune NxT flow cytometer and data analysis was performed using FlowJo. Membrane fractions were directly analyzed after semi-permeabilization and resuspension as described above. To analyze FLAG-tagged TA proteins by flow cytometry, cells were incubated with 200 mM MitoTracker CMXRos (Invitrogen M7512) for 30 min, washed with PBS, and detached and isolated as above. Pelleted cells were washed twice with PBS, fixed in 4% formaldehyde in PBS for 15 min, and permeabilized in 0.7% Tween20 in PBS for 5 min. Cells were washed with PBS, incubated with blocking solution (10% FBS/PBS) for 30 min, followed by FITC-conjugated M2 FLAG antibodies in blocking solution for 1 hr in the dark. Cells were washed three times in PBS, filtered through a 35 μm mesh strainer and data was collected on an Attune NxT flow cytometer. Fluorescence was excited with lasers at 405 nm (for TagBFP), 488 nm (for FITC-conjugated M2 FLAG antibodies and mNeonGreen3K), and 561 nm (for MitoTracker CMXRos and mCherry3V), and collected using 440/50, 530/30, and 615/20 emission filters, respectively. Samples were gated for live and single cells. All flow cytometry experiments are representative of at least 2 independent replicates.
Proteomics sample preparation and data acquisition
Cells were washed twice with cold PBS, collected using a cell scraper, and pelleted. Cells were then lysed in 100 μl denaturing lysis buffer [8M Urea, 50 mM NaCl] with 1x PIC. Lysates were sonicated with ten 1 sec pulses and then centrifuged at 17,000g for 5 min at room temperature. The supernatant was normalized to 100 μg using a BCA assay (Pierce 23225) per the manufacturer’s instructions and adjusted to 100 μl using lysis buffer. The samples were reduced with 15 mM DTT for 1 hr at room temperature with shaking, alkylated with 20 mM iodoacetamide for 20 min in the dark at room temperature with shaking, quenched with 10 mM DTT for 15 min in the dark at room temperature with shaking, and then methanol-chloroform precipitated. Four parts of methanol were added to each sample, followed by one part chloroform, vortexing, and three parts water. Samples were vortexed and centrifuged at 14,000g for 5 min at room temperature and washed two times methanol. Samples were then resuspended in 100 μl 200 mM 3-[4-(2-Hydroxyethyl)piperazin-1-yl]propane-1-sulfonic acid (EPPS) pH 8.0 and digested with 2 μg LysC protease (Thermo Fisher Scientific 90051) at room temperature for 4 hr followed by overnight digestion with 2 μg trypsin at 37°C. Digested samples were quantified using the Pierce quantitative colorimetric peptide assay (Thermo Fisher Scientific 23275). 50 μg of peptides was brought to 100 μl in 200 mM EPPS pH 8.0, 25% acetonitrile (ACN). Dried TMTpro 18-plex labels (Thermo Fisher Scientific A52045) were reconstituted to a stock concentration of 20 μg/μl in anhydrous ACN. From this stock, 5 μl was added to the appropriate sample, briefly vortexed, and incubated for 1 hr at room temperature. Labeled samples were then quenched with 0.3% hydroxylamine for 15 min at room temperature. Samples were then combined, dried in a speedvac, and cleaned using C18 solid-phase extraction with a 100 mg Sep-Pak cartridge (Waters 036935) according to the manufacturer’s instructions.
Eluted samples were resuspended in 100 μl 10 mM ammonium bicarbonate pH 8.0 and fractionated via basic pH reversed-phase HPLC. Across a 60 min gradient, samples were fractionated offline into a 96 well plate by high pH reverse-phase HPLC (Agilent LC1260) through a 300Extend C18 column (3.5 μm particles, 4.5 mm ID × 220 mm length) with a mobile phase A of 5% ACN and 10 mM ammonium bicarbonate pH 8.0 and a mobile phase B of 90% ACN 10 mM ammonium bicarbonate pH 8.0. Resulting fractions were combined into 24 fractions as previously described in Figure S5 of (Paulo et al., 2016), dried in a speedvac, resuspended in 0.5% trifluoroacetic acid, 1% ACN, and cleaned using Pierce C18 tips (Thermo Fisher Scientific 87784) according to the manufacturer’s instructions. Eluted samples were dried in a speedvac and resuspended in 5% ACN, 5% formic acid.
Mass spectrometric data were collected on an Orbitrap Lumos mass spectrometer coupled to a Proxeon NanoLC-1200 UHPLC. The 100 μm capillary column was packed with 35 cm of Accucore 150 resin (2.6 μm, 150Å; Thermo Fisher Scientific 12126–000). The scan sequence began with an MS1 spectrum (Orbitrap analysis, resolution 60,000, 400–1600 Th, automatic gain control (AGC) target 4 ×105, maximum injection time 50 ms). Data were acquired ~90 min per fraction. MS2 analysis consisted of collision-induced dissociation (CID), quadrupole ion trap analysis, automatic gain control (AGC) 1 ×104, NCE (normalized collision energy) 35, q-value 0.25, maximum injection time 35 ms), isolation window at 0.6 Th, and TopSpeed set at 3 sec. RTS was enabled and quantitative SPS-MS3 scans (resolution of 50,000; AGC target 2.5×105; max injection time of 250 ms) were processed through Orbiter with a real-time false discovery rate filter implementing a modified linear discriminant analysis. For FAIMS, the dispersion voltage (DV) was set at 5,000V, the compensation voltages (CVs) used were −40V, −60V, and −80V, and the TopSpeed parameter was set at 1.25 sec.
QUANTIFICATION AND STATISTICAL ANALYSIS
Colocalization analysis
Background subtraction was performed using the Otsu thresholding method (Otsu, 1979). A region of interest (ROI) was drawn around each cell in the FLAG channel and colocalization was assessed in 3D using the ImageJ Coloc2 plugin with a point spread of 2 and 25 randomizations. Manders’ colocalization coefficients (MCC) (Manders et al., 1993) indicate the contribution of FLAG signal above 0 that overlaps with mitochondria relative to total FLAG signal in the ROI. Pearson’s correlation coefficients (PCC) were used to assess the correlation of FLAG signal with the ER marker TRAPα because of the influence of unreliable thresholding of the diffuse ER signal on MCC values. MCC and PCC values were averaged and reported with standard deviations and individual values using GraphPad Prism 9. Where applicable, unpaired t-tests were applied. Sample sizes are listed in the figures.
Quantification of autoradiography experiments
Quantification of autoradiography experiments was performed using phosphorimaging on a Typhoon 5 biomolecular imager (GE) and ImageQuant TL (Cytiva), or MR film scans in ImageJ. To calculate pellet:supernatant ratios, the volumes of the different fractions were normalized to input volumes prior to SDS-PAGE. After background subtraction, the intensity ratios were calculated and normalized to the values obtained with WT RMs. Normalized ratios from independent experiments were averaged and reported with the sem and individual values in all plots for the sample size indicated in the figure legends.
Fluorescent flow cytometry analysis
For the analysis of ERAD factor knockdowns (Figures 6B and S7E), outlier values for each condition were removed using the ROUT method with a false discovery rate of 1% (Motulsky and Brown, 2006). All values in each experiment were normalized to the median value of ATP13A1 KO cells treated with scrambled siRNAs in the same experiment to account for different instrumentation settings between experiments. Replicate values were then combined such that each condition is represented by at least two biological replicates, each containing at least 5,000 to over 30,000 measurements (see below) representing individual cells, and subjected to nested one-way ANOVA analysis. The number of cells (n) analyzed before outlier removal are as follows (semicolons separate replicates): CHST10 in WT – 28,040; 28,832; 27,855; 5,174; 30,555; 26,638; ATP13A1 KO – 28,300; 20,773; 26,257; 6,777; 30,698; 25,721; 28,270; KO + SYVN1 KD – 27,675; 26,501; 7,397; 30,760; 25,279; KO + AMFR KD – 27,757; 27,499; 6,987; 30,203; 25,820; KO + MARCH6 KD – 27,273; 7,078; 30,309; 27,860; KO + RNF185 KD – 27,327; 27,840; KO + RNF185 & AMFR double KD – 26,603; 27,962; KO + SEL1L KD – 27,474; 6,457; 30,381; KO + TRC8 KD – 6,874; 30,439; KO + DERL1 KD – 7,017; 30,326; KO + BAG6 KD – 7,183; 30,279; and WNT1 in WT – 18,765; 27,467; KO – 28,747; 23,785; 28,994; 29,449; KO + SYVN1 KD – 28,135; 28,037; KO + AMFR KD – 28,027; 26,683; KO + MARCH6 KD – 16,813; 27,860; 29,243; KO + RNF185 KD – 27,830; 29,054; KO + RNF185 & AMFR double KD – 28,808; 28,854; KO + SEL1L KD – 28,004; 26,023; KO + TRC8 KD – 28,725; 28,885; KO + DERL1 KD – 28,060; 28,985; KO + BAG6 KD – 28,063; 28,824.
Quantitative proteomics data analysis
Mass spectra were processed using a Comet-based proprietary software pipeline developed by the Gygi lab (Eng et al., 2013; Huttlin et al., 2010). Spectra were converted to mzXML using a modified version of ReAdW.exe. Database searching included all entries from the Human Reference Proteome (2018–12) as well as a curated list of contaminants. This was concatenated to a database composed of all protein sequences in the reversed order. A maximum of two missed cleavages was allowed and the minimum peptide length was set to 7 amino acids. Searches used a 20 ppm precursor ion tolerance for total protein level analysis. Product ion parameters were set to a tolerance of 1.0005, a fragment bin offset of 0.4, and theoretical fragment ions set to 1. TMTpro tag labeling on peptide N-termini and lysine residues (+304.207145 Da) and carbamidomethylation of cysteine residues (+57.0214637236 Da) were set as a static modification, while oxidation of methionine residues (+15.9949 Da) was set as a variable modification. Peptide-spectrum matches (PSMs) were set to a false discovery rate of 2% and PSMs were filtered using a linear discriminant analysis (Huttlin et al., 2010) considering the following parameters: Comet Log Expect, Diff Seq. Delta Log Expect, missed cleavages, peptide length, charge state, and precursor mass accuracy.
For TMT reporter ion quantifications, the summed signal to noise ratio for each TMT channel was extracted and the closest matching centroid to the expected mass of the TMT reporter ion was found, with an integration tolerance of 0.003 Daltons. For protein level quantification comparisons, PSMs were subjected to a 2% false discovery rate and the reporter ion counts were summed across all matching PSMs using in-house software as previously described (Huttlin et al., 2010). MS3 spectra with more than 6 TMTpro reporter ion channels missing, isolation specificities of less than 0.6, or with TMT reporter summed signal to noise ratios of less than 100 were excluded from quantification.
Protein quantification values were median normalized to assume equal loading across all channels. The resulting dataset was curated to remove contaminant proteins. Intensity values were subjected to an unpaired two tailed t-test and Log2 transformation fold change analysis. The resulting dataset was imported into R for further subsetting and plotting. Raw spectral data are deposited on MassIVE (MSV000088575).
Supplementary Material
Supplemental Table 1. Quantitative profiling of WT and ATP13A1 treated without or with (Z-LL)2-ketone, Related to Figure 2.
Highlights.
ATP13A1 dislocates mitochondrial TA proteins that mislocalize to the ER via the EMC
SPP cleaves mislocalized mitochondrial TA proteins for ERAD
ATP13A1 mediates signal sequence and type II transmembrane protein topogenesis
Distinct ubiquitin ligases target misoriented proteins for ERAD
Acknowledgements
Microscopy was performed at the Nikon Imaging Center (NIC) at Harvard Medical School (HMS). qPCR was performed at the HMS ICCB-Longwood Screening Facility. We thank S.P. Gygi and the HMS Taplin Mass Spectrometry Facility for use of their mass spectrometers and software, and Shao lab members and David Ron for useful discussions. This work was supported by the Smith Family Foundation, the Vallee Foundation, a Packard fellowship, NIH DP2GM137415, and NIH R01AG073277 (to S.S.), and NIH R01GM132129 (to J.A.P.). M.J.M. was supported in part by AHA postdoctoral fellowship 19POST34400009.
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boname JM, Bloor S, Wandel MP, Nathan JA, Antrobus R, Dingwell KS, Thurston TL, Smith DL, Smith JC, Randow F, et al. (2014). Cleavage by signal peptide peptidase is required for the degradation of selected tail-anchored proteins. J Cell Biol 205, 847–862. 10.1083/jcb.201312009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Goder V, and Rapoport TA (2006). Distinct Ubiquitin-Ligase Complexes Define Convergent Pathways for the Degradation of ER Proteins. Cell 126, 361–373. [DOI] [PubMed] [Google Scholar]
- Chen C, Malchus NS, Hehn B, Stelzer W, Avci D, Langosch D, and Lemberg MK (2014a). Signal peptide peptidase functions in ERAD to cleave the unfolded protein response regulator XBP1u. EMBO J 33, 2492–2506. 10.15252/embj.201488208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-C, Umanah GKE, Dephoure N, Andrabi SA, Gygi SP, Dawson TM, Dawson VL, and Rutter J (2014b). Msp1/ATAD1 maintains mitochondrial function by facilitating the degradation of mislocalized tail-anchored proteins. EMBO J 33, 1548–1564. 10.15252/embj.201487943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio US, Cho H, and Shan S (2017). Mechanisms of Tail-Anchored Membrane Protein Targeting and Insertion. Annu Rev Cell Dev Biol 33, 417–438. 10.1146/annurev-cellbio-100616-060839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitwood PJ, Juszkiewicz S, Guna A, Shao S, and Hegde RS (2018). EMC Is Required to Initiate Accurate Membrane Protein Topogenesis. Cell 175, 1507–1519.e16. 10.1016/j.cell.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu V, Feng Q, Lim Y, and Shao S (2021). Selective destabilization of polypeptides synthesized from NMD-targeted transcripts. Mol Biol Cell 32, ar38. 10.1091/mbc.e21-08-0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti BJ, Devaraneni PK, Yang Z, David LL, and Skach WR (2015). Cotranslational stabilization of Sec62/63 within the ER Sec61 translocon is controlled by distinct substrate-driven translocation events. Mol Cell 58, 269–283. 10.1016/j.molcel.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dederer V, Khmelinskii A, Huhn AG, Okreglak V, Knop M, and Lemberg MK (2019). Cooperation of mitochondrial and ER factors in quality control of tail-anchored proteins. Elife 8, e45506. 10.7554/elife.45506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraneni PK, Conti B, Matsumura Y, Yang Z, Johnson AE, and Skach WR (2011). Stepwise Insertion and Inversion of a Type II Signal Anchor Sequence in the Ribosome-Sec61 Translocon Complex. Cell 146, 134–147. 10.1016/j.cell.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnikov T, Ben-Gedalya T, and Cohen E (2017). Protein Quality Control in Health and Disease. CSH Perspect Biol 9, a023523. 10.1101/cshperspect.a023523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, Jahan TA, and Hoopmann MR (2013). Comet: An open-source MS/MS sequence database search tool. Proteomics 13, 22–24. 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- Farkas Á, and Bohnsack KE (2021). Capture and delivery of tail-anchored proteins to the endoplasmic reticulum. J Cell Biol 220, e202105004. 10.1083/jcb.202105004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, and Shao S (2018). In vitro reconstitution of translational arrest pathways. Methods 137, 20–36. 10.1016/j.ymeth.2017.12.018. [DOI] [PubMed] [Google Scholar]
- Feng S, Sekine S, Pessino V, Li H, Leonetti MD, and Huang B (2017). Improved split fluorescent proteins for endogenous protein labeling. Nat Commun 8, 370. 10.1038/s41467-017-00494-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Varshney A, Villa DC, Modavi C, Kohler J, Farah F, Zhou S, Ali N, Müller JD, Hoven MKV, et al. (2019). Bright split red fluorescent proteins for the visualization of endogenous proteins and synapses. Commun Biology 2, 344. 10.1038/s42003-019-0589-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Zhao Y, Li T, Nie W, Yang X, Wang X, Wu J, Liao J, and Zou Y (2020). CATP-8/P5A ATPase Regulates ER Processing of the DMA-1 Receptor for Dendritic Branching. Cell Rep 32, 108101. 10.1016/j.celrep.2020.108101. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, and Voeltz GK (2011). ER Tubules Mark Sites of Mitochondrial Division. Science 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry MY, Saladi SM, Cunha A, and Clemons WM (2021). Sequence-based features that are determinant for tail-anchored membrane protein sorting in eukaryotes. Traffic 22, 306–318. 10.1111/tra.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goder V, and Spiess M (2003). Molecular mechanism of signal sequence orientation in the endoplasmic reticulum. EMBO J 22, 3645–3653. 10.1093/emboj/cdg361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görlich D, and Rapoport TA (1993). Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell 75, 615–630. [DOI] [PubMed] [Google Scholar]
- Guna A, Volkmar N, Christianson JC, and Hegde RS (2018). The ER membrane protein complex is a transmembrane domain insertase. Science 359, 470–473. 10.1126/science.aao3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde RS, and Keenan RJ (2022). The mechanisms of integral membrane protein biogenesis. Nat Rev Mol Cell Bio 23, 107–124. 10.1038/s41580-021-00413-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidasch R, Avci D, Lüchtenborg C, Kale D, Beard H, Mentrup T, Barniol-Xicota M, Schröder B, Verhelst S, Brügger B, et al. (2021). Intramembrane protease SPP defines a cholesterol-regulated switch of the mevalonate pathway. Biorxiv 2021.07.19.452877. 10.1101/2021.07.19.452877. [DOI] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Iwase I, Yamasaki Y, Takai T, Wu Y, Kanemoto S, Matsuhisa K, Asada R, Okuma Y, Watanabe T, et al. (2016). Genome-wide identification and gene expression profiling of ubiquitin ligases for endoplasmic reticulum protein degradation. Sci Rep 6, 30955. 10.1038/srep30955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khouri EE, Pavec GL, Toledano MB, and Delaunay-Moisan A (2013). RNF185 Is a Novel E3 Ligase of Endoplasmic Reticulum-associated Degradation (ERAD) That Targets Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). J Biol Chem 288, 31177–31191. 10.1074/jbc.m113.470500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumpe K, Frumkin I, Herzig Y, Rimon N, Özbalci C, Brügger B, Rapaport D, and Schuldiner M (2012). Ergosterol content specifies targeting of tail-anchored proteins to mitochondrial outer membranes. Mol Biol Cell 23, 3927–3935. 10.1091/mbc.e11-12-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto DE, Morgens DW, Zhang L, Walczak CP, Elias JE, Bassik MC, and Kopito RR (2019). Genome-wide CRISPR Analysis Identifies Substrate-Specific Conjugation Modules in ER-Associated Degradation. Mol Cell 73, 377–389.e11. 10.1016/j.molcel.2018.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Yang X, Feng Z, Nie W, Fang Z, and Zou Y (2021). P5A ATPase controls ER translocation of Wnt in neuronal migration. Cell Rep 37, 109901. 10.1016/j.celrep.2021.109901. [DOI] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔC T Method. Methods 25, 402–408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, and Aten JA (1993). Measurement of co-localization of objects in dual-colour confocal images. J Microsc 169, 375–382. 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- Mariappan M, Mateja A, Dobosz M, Bove E, Hegde RS, and Keenan RJ (2011). The mechanism of membrane-associated steps in tail-anchored protein insertion. Nature 477, 61–66. 10.1038/nature10362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateja A, Paduch M, Chang H-Y, Szydlowska A, Kossiakoff AA, Hegde RS, and Keenan RJ (2015). Protein targeting. Structure of the Get3 targeting factor in complex with its membrane protein cargo. Science 347, 1152–1155. 10.1126/science.1261671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto S, Nakatsukasa K, Kakuta C, Tamura Y, Esaki M, and Endo T (2019). Msp1 Clears Mistargeted Proteins by Facilitating Their Transfer from Mitochondria to the ER. Mol Cell 76, 191–205.e10. 10.1016/j.molcel.2019.07.006. [DOI] [PubMed] [Google Scholar]
- Mayerhofer PU (2016). Targeting and insertion of peroxisomal membrane proteins: ER trafficking versus direct delivery to peroxisomes. Biochim Biophys Acta 1863, 870–880. 10.1016/j.bbamcr.2015.09.021. [DOI] [PubMed] [Google Scholar]
- McKenna MJ, Sim SI, Ordureau A, Wei L, Harper JW, Shao S, and Park E (2020). The endoplasmic reticulum P5A-ATPase is a transmembrane helix dislocase. Science 369, eabc5809. 10.1126/science.abc5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motulsky HJ, and Brown RE (2006). Detecting outliers when fitting data with nonlinear regression – a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 7, 123. 10.1186/1471-2105-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan N, Foresti O, Wendrich K, Stein A, and Carvalho P (2020). Quality Control of Protein Complex Assembly by a Transmembrane Recognition Factor. Mol Cell 77, 108–119.e9. 10.1016/j.molcel.2019.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninagawa S, George G, and Mori K (2020). Mechanisms of productive folding and endoplasmic reticulum-associated degradation of glycoproteins and non-glycoproteins. Biochim Biophys Acta 1865, 129812. 10.1016/j.bbagen.2020.129812. [DOI] [PubMed] [Google Scholar]
- Nusse R, and Clevers H (2017). Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 169, 985–999. 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- O’Keefe S, Zong G, Duah KB, Andrews LE, Shi WQ, and High S (2021). An alternative pathway for membrane protein biogenesis at the endoplasmic reticulum. Commun Biology 4, 828. 10.1038/s42003-021-02363-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okreglak V, and Walter P (2014). The conserved AAA-ATPase Msp1 confers organelle specificity to tail-anchored proteins. Proc Natl Acad Sci 111, 8019–8024. 10.1073/pnas.1405755111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Kopito RR, and Christianson JC (2013). The mammalian endoplasmic reticulum-associated degradation system. CSH Perspect Biol 5, a013185–a013185. 10.1101/cshperspect.a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu N (1979). A Threshold Selection Method from Gray-Level Histograms. IEEE Transact Syst Man Cybern 9, 62–66. 10.1109/tsmc.1979.4310076. [DOI] [Google Scholar]
- Park E, and Rapoport TA (2012). Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu Rev Biophys 41, 21–40. 10.1146/annurev-biophys-050511-102312. [DOI] [PubMed] [Google Scholar]
- Paulo JA, O’Connell JD, Everley RA, O’Brien J, Gygi MA, and Gygi SP (2016). Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J Proteomics 148, 85–93. 10.1016/j.jprot.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW (2001). A new mathematical model for relative quantification in real-time RT–PCR. Nucl Acids Res 29, e45–e45. 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politis PK, Makri G, Thomaidou D, Geissen M, Rohrer H, and Matsas R (2007). BM88/CEND1 coordinates cell cycle exit and differentiation of neuronal precursors. Proc Natl Acad Sci 104, 17861–17866. 10.1073/pnas.0610973104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Zhao T, Zou W, Shen K, and Wang X (2020). An Endoplasmic Reticulum ATPase Safeguards Endoplasmic Reticulum Identity by Removing Ectopically Localized Mitochondrial Proteins. Cell Rep 33, 108363. 10.1016/j.celrep.2020.108363. [DOI] [PubMed] [Google Scholar]
- Rane NS, Chakrabarti O, Feigenbaum L, and Hegde RS (2010). Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein. J Cell Biol 188, 515–526. 10.1083/jcb.200911115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport TA, Li L, and Park E (2017). Structural and Mechanistic Insights into Protein Translocation. Annu Rev Cell Dev Biol 33, 369–390. 10.1146/annurev-cellbio-100616-060439. [DOI] [PubMed] [Google Scholar]
- Roboti P, Lawless C, and High S (2021). Mitochondrial antiviral-signalling protein is a client of the BAG6 protein quality control complex. Biorxiv 2021.12.02.470791. 10.1101/2021.12.02.470791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiano A, Mora G, Buxó L, and Carvalho P (2016). Spatial control of lipid droplet proteins by the ERAD ubiquitin ligase Doa10. EMBO J 35, 1644–1655. 10.15252/embj.201593106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi M, Tomiyoshi R, Kuroiwa T, Mihara K, and Omura T (1992). Functions of signal and signal-anchor sequences are determined by the balance between the hydrophobic segment and the N-terminal charge. Proc Natl Acad Sci 89, 16–19. 10.1073/pnas.89.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert J, Siekierska A, Langlois M, May P, Huneau C, Becker F, Muhle H, Suls A, Lemke JR, de Koval CGF, et al. (2014). Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat Genet 46, 1327–1332. 10.1038/ng.3130. [DOI] [PubMed] [Google Scholar]
- Schuldiner M, Metz J, Schmid V, Denic V, Rakwalska M, Schmitt HD, Schwappach B, and Weissman JS (2008). The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell 134, 634–645. 10.1016/j.cell.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, and Hegde RS (2011). A calmodulin-dependent translocation pathway for small secretory proteins. Cell 147, 1576–1588. 10.1016/j.cell.2011.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer AG, and Hampton RY (2004). Structural Control of Endoplasmic Reticulum-associated Degradation: Effect of Chemical Chaperones on 3-Hydroxy-3-Methylglutaryl-CoA Reductase. J Biol Chem 279, 188–196. 10.1074/jbc.m307734200. [DOI] [PubMed] [Google Scholar]
- Shurtleff MJ, Itzhak DN, Hussmann JA, Oakdale NTS, Costa EA, Jonikas M, Weibezahn J, Popova KD, Jan CH, Sinitcyn P, et al. (2018). The ER membrane protein complex interacts cotranslationally to enable biogenesis of multipass membrane proteins. Elife 7, e37018. 10.7554/elife.37018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic-Barrett S, Dickson AS, Burr SP, Williamson JC, Lobb IT, Boomen DJ, Lehner PJ, and Nathan JA (2018). MARCH6 and TRC8 facilitate the quality control of cytosolic and tail-anchored proteins. EMBO Rep 19. 10.15252/embr.201745603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipper DJ, and Harley CA (2002). Yeast Genes Controlling Responses to Topogenic Signals in a Model Transmembrane Protein. Mol Biol Cell 13, 1158–1174. 10.1091/mbc.01-10-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Weijer ML, Krshnan L, Liberatori S, Guerrero EN, Robson-Tull J, Hahn L, Lebbink RJ, Wiertz EJHJ, Fischer R, Ebner D, et al. (2020). Quality Control of ER Membrane Proteins by the RNF185/Membralin Ubiquitin Ligase Complex. Mol Cell 79, 768–781.e7. 10.1016/j.molcel.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg B, Clemons WM, Collinson I, Modis Y, Hartmann E, Harrison SC, and Rapoport TA (2004). X-ray structure of a protein-conducting channel. Nature 427, 36–44. 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- Vashist S, and Ng DTW (2004). Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol 165, 41–52. 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmar N, Thezenas M-L, Louie SM, Juszkiewicz S, Nomura DK, Hegde RS, Kessler BM, and Christianson JC (2018). The ER membrane protein complex promotes biogenesis of sterol-related enzymes maintaining cholesterol homeostasis. J Cell Sci 132, jcs223453. 10.1242/jcs.223453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G (1986). Net N-C charge imbalance may be important for signal sequence function in bacteria. J Mol Biol 192, 287–290. 10.1016/0022-2836(86)90365-7. [DOI] [PubMed] [Google Scholar]
- von Heijne G (1989). Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature 341, 456–458. 10.1038/341456a0. [DOI] [PubMed] [Google Scholar]
- Wahlberg JM, and Spiess M (1997). Multiple Determinants Direct the Orientation of Signal–Anchor Proteins: The Topogenic Role of the Hydrophobic Signal Domain. J Cell Biol 137, 555–562. 10.1083/jcb.137.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, and Blobel G (1981). Translocation of proteins across the endoplasmic reticulum III. Signal recognition protein (SRP) causes signal sequence-dependent and site-specific arrest of chain elongation that is released by microsomal membranes. J Cell Biol 91, 557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Chan C, Weir NR, and Denic V (2014). The Get1/2 transmembrane complex is an endoplasmic-reticulum membrane protein insertase. Nature 512, 441–444. 10.1038/nature13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Li J, Wang Q, Ge M-X, Ji J, Liu D, Wang Z, Cao Y, Zhang Y, and Zhang Z-R (2022). TMUB1 is an endoplasmic reticulum-resident escortase that promotes the p97-mediated extraction of membrane proteins for degradation. Mol Cell 10.1016/j.molcel.2022.07.006. [DOI] [PubMed] [Google Scholar]
- Weihofen A, Lemberg MK, Ploegh HL, Bogyo M, and Martoglio B (2000). Release of Signal Peptide Fragments into the Cytosol Requires Cleavage in the Transmembrane Region by a Protease Activity That Is Specifically Blocked by a Novel Cysteine Protease Inhibitor. J Biol Chem 275, 30951–30956. 10.1074/jbc.m005980200. [DOI] [PubMed] [Google Scholar]
- Weir NR, Kamber RA, Martenson JS, and Denic V (2017). The AAA protein Msp1 mediates clearance of excess tail-anchored proteins from the peroxisomal membrane. ELife 6, 13004. 10.7554/elife.28507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, and Rapoport TA (2018). Mechanistic insights into ER-associated protein degradation. Curr Opin Cell Biol 53, 22–28. 10.1016/j.ceb.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, and Rapoport TA (2021). Translocation of Proteins through a Distorted Lipid Bilayer. Trends Cell Biol 31, 473–484. 10.1016/j.tcb.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, and Sakisaka T (2012). Molecular Machinery for Insertion of Tail-Anchored Membrane Proteins into the Endoplasmic Reticulum Membrane in Mammalian Cells. Mol Cell 48, 387–397. 10.1016/j.molcel.2012.08.028. [DOI] [PubMed] [Google Scholar]
- Yücel SS, and Lemberg MK (2020). Signal Peptide Peptidase-Type Proteases: Versatile Regulators with Functions Ranging from Limited Proteolysis to Protein Degradation. J Mol Biol 432, 5063–5078. 10.1016/j.jmb.2020.05.014. [DOI] [PubMed] [Google Scholar]
- Zhang X, and Shan S (2014). Fidelity of Cotranslational Protein Targeting by the Signal Recognition Particle. Annu Rev Biophys 43, 381–408. 10.1146/annurev-biophys-051013-022653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Feng S, Brown D, and Huang B (2020). Improved yellow-green split fluorescent proteins for protein labeling and signal amplification. Plos One 15, e0242592. 10.1371/journal.pone.0242592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Quantitative profiling of WT and ATP13A1 treated without or with (Z-LL)2-ketone, Related to Figure 2.
Data Availability Statement
Proteomics data have been deposited at MassIVE and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FLAG M2 | Sigma | F1804 (RRID: AB_262044) |
| FLAG M2-HRP | Sigma | A8592 (RRID: AB_439702) |
| CHST10 | Sigma | PA5-92187 (RRID: AB_2806403) |
| SEL1L | Sigma | PA5-88333 (RRID: AB_2804837) |
| DERL1 | Abcam | ab176732 (RRID: AB_2858187) |
| BAG6 | Gift from R.S. Hegde | N/A |
| TRAPα | Gift from R.S. Hegde | N/A |
| TOM20 | Abcam | ab186735 (RRID: AB_2889972) |
| ATP13A1 | Proteintech | 16244-1-AP (RRID: AB_2290293) |
| CNX | Enzo Life Sciences | ADI-SPA-865 (RRID: AB_10618434) |
| EMC2 | Proteintech | 25443-1-AP (RRID: AB_2750836) |
| EMC6 | Abcam | Ab84902 (RRID: AB_1925516) |
| GET1 | Synaptic Systems | 324 002 (RRID: AB_2620063) |
| Ubiquitin | Santa Cruz Biotechnology | sc-8017 (RRID: AB_628423) |
| SQLE | Proteintech | 12544-1-AP (RRID: AB_2195888) |
| OMP25 | Proteintech | 15666-1-AP (RRID: AB_2201149) |
| PLAT3 | Abcam | Ab88447 (RRID: AB_2041691) |
| SPP | Abcam | Ab247061 |
| AMFR | Novus Biologicals | NBP2-15374 |
| SYVN1 | Abcam | Ab170901 (RRID: AB_2833021) |
| RNF185 | Abcam | Ab181999 |
| PEX14 | Proteintech | 10594-1-AP (RRID: AB_2252194) |
| SGTA | Mateja et al., 2015 | |
| β-catenin | Cell Signaling Technology | 9562 (RRID: AB_331149) |
| WNT1 | Invitrogen | PIMA515544 |
| FITC-conjugated FLAG M2 | Sigma | F4049 (RRID: AB_439701) |
| Peroxidase-conjugated anti-HA antibody | Cell Signaling Technology | 2999 (RRID: AB_1264166) |
| Peroxidase-conjugated Goat Anti-Mouse | Jackson ImmunoResearch | 115-035-003 (RRID: AB_10015289) |
| Peroxidase-conjugated Goat Anti-Rabbit | Jackson ImmunoResearch | 111-035-003 (RRID: AB_2313567) |
| Alexa Fluor 488-conjugated Goat Anti-Mouse | Jackson ImmunoResearch | 115-545-003 (RRID: AB_2338840) |
| Alexa Fluor 594-conjugated Goat Anti-Rabbit | Jackson ImmunoResearch | 111-585-003 (RRID: AB_2338059) |
| Bacterial and virus strains | ||
| DH5α E. coli | Homemade | N/A |
| Biological samples | ||
| RRL-based in vitro translation system | Feng and Shao, 2018 | N/A |
| In vitro transcription system | Feng and Shao, 2018 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Acetonitrile | Millipore Sigma | 34851 |
| Doxycycline hyclate | Sigma | D9891 |
| Hygromycin B | Millipore Sigma | 31282-04-9 |
| Blasticidin S HCl | Gibco | A11139-03 |
| Puromycin | Gibco | A11138-03 |
| Bortezomib | LC laboratories | B-1408 |
| Epoxomicin | ApexBio | 134381-21-8 |
| (Z-LL)2-ketone | Millipore Sigma | 313664-40-3 |
| E1 inhibitor (MLN7243) | Cayman Chemical | 1450833-55-2 |
| P97 inhibitor (CB-5083) | Cayman Chemical | 1542705-92-9 |
| Cycloheximide | Sigma | 01810 |
| Paraformaldehyde | Electron Microscopy Sciences | 100503-917 |
| MitoTracker CMXRos | Invitrogen | M7512 |
| RNase-free DNase | QIAGEN | 79254 |
| Lys-C protease, MS grade | ThermoFisher Scientific | 90051 |
| Trypsin Gold, MS grade | Promega | V5280 |
| Urea | Millipore Sigma | 51456 |
| Hydroxylamine, 50% | ThermoFisher Scientific | 90115 |
| Trifluoroacetic acid, LC-MS grade | ThermoFisher Scientific | 85183 |
| Formic acid | Sigma | 27001 |
| cOmplete EDTA-free protease inhibitor cocktail | Roche | 05056489001 |
| CaptivA PriMAB Protein A affinity resin | Repligen | CA-PRI-1000 |
| Commercial assays | ||
| Pierce BCA protein assay kit | ThermoFisher Scientific | 23225 |
| Pierce quantitative colorimetric peptide assay | ThermoFisher Scientific | 23275 |
| TMTpro 18-plex label reagent set | ThermoFisher Scientific | A52045 |
| Deposited data | ||
| Proteomics data | MassIVE repository | MSV000088575 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-3216 (RRID: CVCL_0063) |
| Flp-In 293 T-REx | Invitrogen | R78007 (RRID: CVCL_U427) |
| Flp-In HeLa T-REx | Gift from B. Raught | N/A |
| ATP13A1 KO Flp-In HeLa T-REx | McKenna, Sim et al. 2020 | N/A |
| ATP13A1 KO Flp-In 293 T-REx | McKenna, Sim et al. 2020 | N/A |
| ATP13A1 KO Flp-In 293 T-REx: WT ATP13A1 | McKenna, Sim et al. 2020 | N/A |
| ATP13A1 KO Flp-In 293 T-REx HEK293: D533A ATP13A1 | McKenna, Sim et al. 2020 | N/A |
| ATP13A1 KO, EMC6 KO Flp-In 293 T-REx | This study | N/A |
| Flp-In HeLa T-REx: F-OMP25 | McKenna, Sim et al. 2020 | N/A |
| ATP13A1 KO Flp-In HeLa T-REx: F-OMP25 | McKenna, Sim et al. 2020 | N/A |
| Flp-In HeLa T-REx: F-BAK1 | McKenna, Sim et al. 2020 | N/A |
| Flp-In HeLa T-REx: F-CEND1 | This study | N/A |
| ATP13A1 KO Flp-In HeLa T-REx: F-CEND1 | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: ASGR1-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx: ASGR1-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: DHFR-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: BFP-P2A-CNb11-SEC61β | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: CHST10-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx: CHST10-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: QQ-CHST10-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: RR-CHST10-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: WNT1-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx: WNT1-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: EGL-20-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx: EGL-20-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: EGL-20H-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx: EGL-20H-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER Flp-In 293 T-REx: PRL-NCb11-P2A-BFP | This study | N/A |
| GFP(1-10)cyto RFP(1-10)ER ATP13A1 KO Flp-In 293 T-REx: PRL-NCb11-P2A-BFP | This study | N/A |
| Oligonucleotides | ||
| SRP14 fwd | GAGAGCGAGCAGTTCCTGAC | |
| SRP14 rev | GTTTGGTTCGACCGTCATACT | |
| TRC8 fwd | CAAAGCCGGTTCTGCATCG | |
| TRC8 rev | AGTTGCAGCTAACAGAAAGGC | |
| MARCH6 fwd | GTTCCCTTGGATCAGACTCC | |
| MARCH6 rev | CAATGTTCCGGATGCCATTTG | |
| EMC6 sg1 fwd | CACCGGCCGCCTCGCTGATGAACGG | |
| EMC6 sg1 rev | AAACCCGTTCATCAGCGAGGCGGCc | |
| Recombinant DNA | ||
| pHDM-G | PlasmID | EvNO00061606 |
| pHDM-HGPM2 | PlasmID | EvNO00061607 |
| pHDM-tat1B | PlasmID | EvNO00061608 |
| pRC-CMV-rev1B | PlasmID | EvNO00061616 |
| pHAGE-FLAG-mNG3K(1-10)-P2A-Prl(SS)-mCh3V(1-10)-KDEL | This study | N/A |
| pX459 | Addgene | 62988 |
| pcDNA5 FRT/TO F-OMP25 | McKenna, Sim et al. 2020 | N/A |
| pcDNA5 FRT/TO F-CEND1 | This study | N/A |
| pcDNA5 FRT/TO GFP-OMP25 | This study | N/A |
| pcDNA5 FRT/TO GFP-MAVS | This study | N/A |
| pcDNA5 FRT/TO ASGR1-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO DHFR-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO BFP-P2A-NCb11-SEC61β | This study | N/A |
| pcDNA5 FRT/TO CHST10-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO QQ-CHST10-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO RR-CHST10-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO WNT1-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO EGL-20-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO EGL-20H-NCb11-P2A-BFP | This study | N/A |
| pcDNA5 FRT/TO PRL-NCb11-P2A-BFP | This study | N/A |
| pSP64 F-OMP25 | McKenna, Sim et al. 2020 | N/A |
| pSP64 F-SQS | This study | N/A |
| pSP64 F-STX1B | This study | N/A |
| pSP64 CHST10-FLAG | This study | N/A |
| pSP64 RR-CHST10-FLAG | This study | N/A |
| pSP64 EGL-20-FLAG | This study | N/A |
| pSP64 EGL-20H-FLAG | This study | N/A |
| pSP64 PRL-FLAG | This study | N/A |
| pDONR ASGR1 | DNASU | HsCD00005525 |
| pDONR CHST10 | Gift from the Harper lab | N/A |
| pRK5-HA-ubiquitin-WT | Addgene | 17608 |
| pTAG-BiP(SS)-BFP-KDEL | Friedman et al., 2011 | N/A |
| Software and algorithms | ||
| ImageJ | NIH | N/A |
| Prism 9 | GraphPad | N/A |
| FlowJo v10 | FlowJo | N/A |
| Illustrator | Adobe | N/A |
| Photoshop | Adobe | N/A |
| ImageQuant TL | Cytiva | N/A |
| In-house MS data analysis software | Huttlin et al., 2010 | N/A |
| Other | ||
| Control siRNA | Horizon | D-001810-10-05 |
| EMC2 siRNA | Horizon | L-010631-00-0005 |
| EMC5 siRNA | Horizon | L-018365-00-0005 |
| EMC6 siRNA | Horizon | L-014711-02-0005 |
| WRB siRNA | Horizon | L-011898-01-0005 |
| ATP13A1 siRNA | Horizon | L-020426-00-0005 |
| SPP siRNA | Horizon | L-005896-00-0005 |
| TRC8 siRNA | Horizon | L-006942-00-0005 |
| MARCH6 siRNA | Horizon | L-006925-00-0005 |
| AMFR siRNA | Horizon | L-006522-00-0005 |
| SYVN1 siRNA | Horizon | L-007090-00-0005 |
| RNF185 siRNA | Horizon | L-007107-00-0005 |
| SEL1L siRNA | Horizon | L-004885-00-0005 |
| BAG6 siRNA | Horizon | L-005062-01-0005 |
| DERL1 siRNA | Horizon | L-010733-02-0005 |
| DMEM + GlutaMAX | Gibco | 10569-010 |
| FluoroBrite DMEM | Gibco | A1896701 |
| Trypsin-EDTA | Gibco | 25200-056 |
| PBS | Gibco | 10010-023 |
| DMSO | Sigma | 472301 |
| OPTI-MEM | Gibco | 31985-062 |
| TransIT-293 | Mirus | MIR 2706 |
| Lipofectamine 3000 | Invitrogen | L3000-015 |
| Lipofectamine RNAiMAX | Invitrogen | 13778-150 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | ThermoFisher Scientific | 34580 |
| Ponceau S solution | Sigma | P7170 |
| EasyTag L-[35S]-Methionine | Perkin Elmer | NEG709A005MC |
| Polypropylene Microfuge Tubes | Beckman Coulter | 357448 |
| Digitonin | Millipore Sigma | 11024-24-1 |
| Triton X-100 | Sigma | T9284 |
| Tween 20 | Sigma | P7949 |
| 10% SDS | Sigma | 71736 |
| 40% acrylamide (29:1) | Bio Rad | 1610146 |
| Nitrocellulose membrane | Bio Rad | 1620112 |
| Multi-well glass-bottom cell culture plates | MatTek | P24G-1.5-13-F |
| BioMax MR film | Carestream | 894 1114 |
| Buffer RLT | QIAGEN | 79216 |
| RNeasy Mini Kit | QIAGEN | 74106 |
| SuperScript III First-Strand Synthesis System | Invitrogen | 18080051 |
| SYBR Green Master Mix | Applied Biosystems | 4309155 |
| Pierce C18 tips, 100 μL | ThermoFisher Scientific | 87784 |
| Sep-Pak C18 cartridge, 100 mg | Waters | Wat036935 |
| 300Extend C18 column | Agilent | custom |
| Accucore 150 resin | ThermoFisher Scientific | 16126-000 |