

Abstract
Background:
Zone 1 resuscitative endovascular balloon occlusion of the aorta has been recommended for refractory shock after a dismounted complex blast injury for the austere combat scenario. While resuscitative endovascular balloon occlusion of the aorta should enhance coronary perfusion, there is a potential risk of secondary brain injury due to loss of cerebral autoregulation. We developed a combat casualty relevant dismounted complex blast injury swine model to evaluate the effects of resuscitative endovascular balloon occlusion of the aorta zone I on intracranial pressure and cerebral edema. We hypothesized that zone 1 aortic occlusion with resuscitative endovascular balloon occlusion of the aorta would increase mean arterial pressure transmitted in excessive intracranial pressure, thereby worsening brain injury.
Methods:
50 kg male Yorkshire swine were subjected to a combination dismounted complex blast injury model consisting of blast traumatic brain injury (50 psi, ARA Mobile Shock Laboratory), tissue injury (bilateral femur fractures), and hemorrhagic shock (controlled bleeding to a base deficit goal of 10 mEq/L). During the shock phase, pigs were randomized to no aortic occlusion (n = 8) or to 30 minutes of zone 1 resuscitative endovascular balloon occlusion of the aorta (zone 1 aortic occlusion group, n = 6). After shock, pigs in both groups received a modified Tactical Combat Casualty Care–based resuscitation and were monitored for an additional 240 minutes until euthanasia/death for a total of 6 hours. Intracranial pressure was monitored throughout, and brains were harvested for water content. Linear mixed models for repeated measures were used to compare mean arterial pressure and intracranial pressure between zone 1 aortic occlusion and no aortic occlusion groups.
Results:
After dismounted complex blast injury, the zone 1 group had a significantly higher mean arterial pressure during hemorrhagic shock compared to the control group (41.2 mm Hg vs 16.7 mm Hg, P = .002). During balloon occlusion, intracranial pressure was not significantly elevated in the zone 1 aortic occlusion group vs control, but intracranial pressure was significantly lower in the zone 1 group at the end of the observation period. In addition, the zone 1 aortic occlusion group did not have increased brain water content (zone 1 aortic occlusion: 3.95 ± 0.1g vs no aortic occlusion: 3.95 ± 0.3 g, P = .87). Troponin levels significantly increased in the no aortic occlusion group but did not in the zone 1 aortic occlusion group.
Conclusion:
Zone 1 aortic occlusion using resuscitative endovascular balloon occlusion of the aorta in a large animal dismounted complex blast injury model improved proximal mean arterial pressure while not significantly increasing intracranial pressure during balloon inflation. Observation up to 240 minutes postresuscitation did not show clinical signs of worsening brain injury or cardiac injury. These data suggest that in a dismounted complex blast injury swine model, resuscitative endovascular balloon occlusion of the aorta in zone 1 may provide neuro- and cardioprotection in the setting of blast traumatic brain injury. However, longer monitoring periods may be needed to confirm that the neuroprotection is lasting.
Introduction
Hemorrhagic shock (HS) and traumatic brain injury (TBI) are leading causes of death after trauma in both civilian1,2 and military settings.3 In the military setting, explosions caused by improvised explosive devices (IEDs) are the most common mechanism of injury and account for 63% of deaths in modern combat.4 The resulting injury profile caused by IEDs is termed dismounted complex blast injury (DCBI) and is characterized by an amputation of at least 1 lower extremity, junctional (pelvic/abdominal/truncal) injury that is associated with noncompressible hemorrhage, and lastly moderate to severe TBI.5 Uncontrolled bleeding from the junctional (especially pelvic) injuries is the leading cause of death after DCBI.4
Resuscitative endovascular balloon occlusion of the aorta (REBOA) is a promising endovascular technique to provide temporary hemorrhage control until definitive management can be performed. Advanced endovascular technologies have allowed REBOA to be deployed in austere combat environments to treat HS on the battlefield, which is identified as the leading cause of preventable deaths.6 Currently, the majority of prehospital REBOA cases in austere combat environments are for DCBI casualties.7 However, despite the promising results of REBOA to provide life-saving hemorrhage control, its effect on concurrent TBI is unclear. DCBI is also the most common cause of TBI in warfighters,8 and thus the recommendation to employ REBOA for this type of injury mechanism should consider the implications on primary and secondary brain injury. TBI progression and ultimately death following REBOA use has been reported.9 HS swine models have already shown that REBOA can cause supraphysiologic intracranial pressures (ICP),10 and it can increase systolic blood pressures (SBP) to the 200s and increase carotid blood flow 300%.11 Clinically, TBI outcomes have a U-shaped relationship between SBP and mortality, with worse outcomes associated not only with hypotension but also elevated pressures.12,13 Preclinical studies focusing on evaluating the effect of REBOA in animal models with TBI and HS have employed cortical impact devices to create TBI; however, this injury is not representative of blast TBI (bTBI). Using our large animal DCBI model,14 we hypothesized that zone 1 aortic occlusion (aortic oclusion [AO]) with REBOA would increase mean arterial pressure (MAP) transmitted in excessive intracranial pressure (ICP), thereby worsening brain injury.
Methods
Animal setting
The University of Colorado Institutional Animal Care and Use Committee approved this animal study under protocol #1050. The facility where the research occurred is fully accredited with the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Research was conducted in compliance with the Animal Welfare Act, implementing Animal Welfare Regulations, and the principles of the Guide for the Care and Use of Laboratory Animals, National Research Council, and results are reported in accordance with the ARRIVE guidelines. Healthy adolescent Yorkshire swine weighing between 45 and 56 kg were acclimated in the facilities for a minimum of 3 days before the experimental protocol. All swine underwent a protocol consisting of general anesthesia, instrumentation, and a series of injuries to replicate a DCBI.14
Animal setup
Briefly, anesthesia is induced using ketamine (20.0 mg/kg), xylazine (2.0 mg/kg), and acepromazine (0.2 mg/kg) followed by intubation under direct laryngoscopy. Continuous infusion anesthesia employs propofol (3 mg/kg/h) and fentanyl (3 mcg/kg/h) throughout the model. An intracranial pressure (ICP) monitor (Integra LifeSciences, Plainsboro, NJ) is placed into the left frontal lobe 1 cm from midline and 1 cm above the brow line. Invasive pressure monitoring occurs through an arterial line established in the axillary artery using surgical cutdown (5 Fr catheter). All blood samples for processing are taken from this line. Percutaneous access to both femoral arteries and a single femoral vein is performed. The femoral arteries (one 12 Fr catheter and one 7 Fr sheath compatible with REBOA) are used for blood removal for the fixed-pressure HS phase. Fluid resuscitation occurs through a large sheath (10 Fr) inserted into the femoral vein. Swine are euthanized using pentobarbital (86.6 mg/kg) at the conclusion of the experiment.
DCBI model
The model is summarized in Figure 1. Briefly, the DCBI is composed of a blast traumatic brain injury (bTBI), tissue injury consisting of bilateral open femur fractures, and a fixed-pressure targeted HS phase. After anesthesia and instrumentation, swine are transported to the Mobile Shock Tube Laboratory (Applied Research Associates, Inc., Littleton, CO), where they undergo a 55 psi targeted Friedlander type blast wave using compressed helium. During the blast overpressure injury, swine wear a National Institute of Justice (NIJ) level II vest, ear plugs, and goggles to protect the abdomen, torso, eyes, and ears from the blast. This allows for a primary bTBI while minimizing damage to the other organs. After the bTBI, the tissue injury and noncompressible hemorrhage component associated with DCBI pelvic trauma is created by femoral artery bleeding combined with severe, open complex comminuted fractures of the femurs. A captive bolt stunner (Blitz-Kerner, Turbocut JOBB GmbH, Germany) is fired directly onto surgically exposed femurs, inducing complex fractures that are confirmed by digital palpation and visual inspection. Fixed-pressure HS is performed by bilateral bleeding from both femoral arteries into citrated 450 mL blood transfusion bags (Jorgensen Laboratories, Inc., Loveland, CO). The target mean arterial pressure (MAP) is 20 mm Hg within 5 minutes; once this MAP is achieved, further blood removal is titrated to maintain a MAP of 15 mm Hg with a corresponding end-tidal carbon dioxide (EtCO2) goal of 20 mm Hg. Base excess (BE, measured by iSTAT-1 point of care analyzers, Abbott Point of Care, Inc., Princeton, NJ) is used to monitor for adequate depth of shock,15 with the final BE target of −10 mEq/L.
Figure 1.
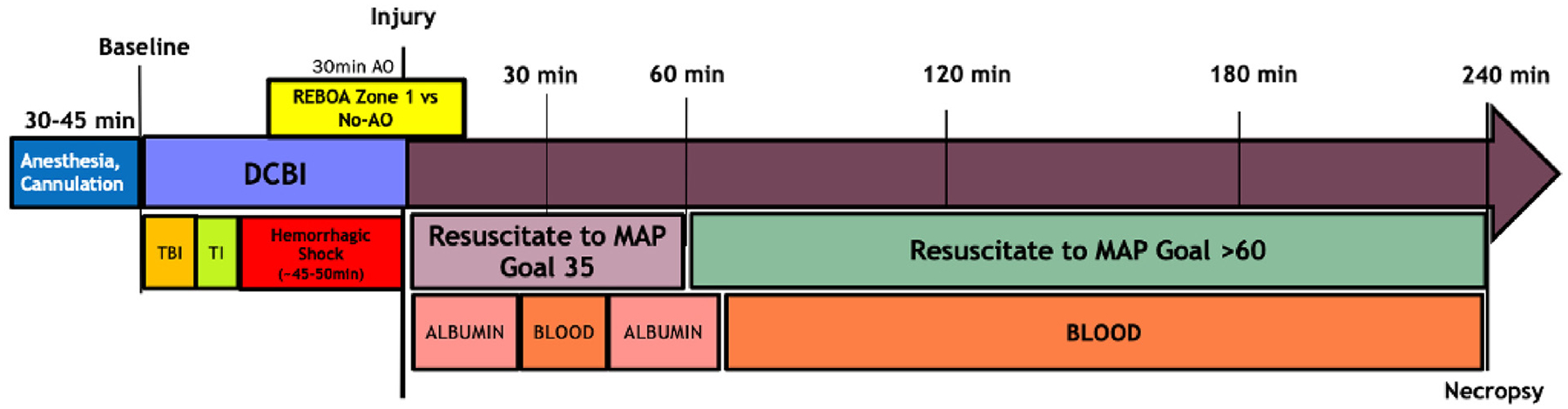
A Dismounted complex blast injury (DCBI) followed by 30 minutes resuscitative endovascular balloon occlusion of the aorta (REBOA) in zone 1 or 30 minutes of no aortic occlusion.
Randomization
Randomization to groups (zone 1 AO or no AO) is performed during experimental setup by a single investigator using Microsoft Excel RAND function. The results are sealed until presented to the principal investigator and surgical research staff during the HS phase before the deployment of the ER-REBOA catheter for AO.
Intervention
Zone 1 AO is performed using the ER-REBOA Catheter (Prytime Medical Devices, Inc., Boerne, TX). The catheter is inserted into the 7 Fr sheath placed in the femoral artery and is advanced to 50 cm and inflated using 6 mL of normal saline. Zone 1 AO is maintained for the final 30 minutes of the HS phase.
Resuscitation
At the end of HS, resuscitation is initiated following a modified combat casualty care protocol. For the zone 1 AO group, the REBOA catheter balloon is slowly deflated during the first 15 minutes of resuscitation. Fluid replacement is initiated with 500 mL of 5% human albumin (Grifols Biologicals, Inc., Los Angeles, CA), followed by 1 unit of shed blood. A bolus of 1 g 10% calcium chloride (International Medication Systems Ltd, South El Monte, CA) is given with the first unit of blood. After the first unit of shed blood, another 500 mL of 5% human albumin is infused, followed by the remaining shed blood. The animals are observed for 240 minutes after the conclusion of HS.
Data collection
Vital signs and intracranial pressure (ICP) are monitored continuously and recorded every 5 minutes. Blood samples are collected at baseline and then postinjury 60 minutes and postinjury 240 minutes for measuring troponin levels to monitor cardiac perfusion. Brain water content is calculated using a validated wet-to-dry protocol. At necropsy, whole brains are immediately removed and weighed (Adventurer Pro Precision Electronic Balance, Ohaus, Pine Brook, NJ). Each sample is dried at 100°C for 48 hours and reweighed for the dry weight. The brain water content is calculated using the formula: water content = (wet weight – dry weight)/(dry weight).16,17
Statistical analysis
Data were analyzed using SAS Studio 2021 and SAS v. 9.4 (SAS Institute, Cary, NC). A power analysis for detecting changes in ICP was performed. Based on reported values,18 we assumed a baseline mean ICP of 11.5 mm Hg with standard deviation of 2.9 mm Hg; thus a total sample size of 14 would allow the detection of a minimum difference in ICP of 4.7 mm Hg with a power of 80% and alpha level of .05. Linear mixed models for repeated measures were used to compare the temporal trends of the groups, with contrasts between groups adjusted by using the false discovery rate method. For variables not normally distributed, a Box-Cox power transformation was performed to approximate normality. Numerical data are presented as mean ± standard deviation or median (interquartile range) unless otherwise noted.
Results
Eight swine were randomly assigned to the DCBI model without AO (no AO) and 6 to the model with zone 1 REBOA for aortic occlusion (zone 1 AO). At baseline, the groups had similar vital signs and electrolyte panels (Table I).
Table I.
Physiologic variables and laboratory values of each model at baseline; variables are represented as means with standard deviations (SD)
| No AO | Zone 1 AO | |
|---|---|---|
| MAP, mm Hg | 77 (13.7) | 66.5 (17.9) |
| EtCO2, mm Hg | 36.6 (4.1) | 37.0 (5.6) |
| HR, bpm | 73.5 (18.2) | 84.5 (17.2) |
| ICP, mm Hg | 11.1 (5.6) | 7.8 (2.7) |
| BE, mmol/L | 10.3 (2.2) | 9.8 (2.5) |
| Ionized calcium, mmol/L | 1.3 (0.1) | 1.2 (0.3) |
| Na, mmol/L | 142 ± 3.4 | 141.8 ± 2.3 |
| K, mmol/L | 4.1 ± 0.2 | 4.2 ± 0.1 |
| Hct, %pcv | 28.9 (1.8) | 28.8 (2.5) |
| Hgb, g/dL | 9.8 (0.6) | 9.8 (0.8) |
| Glucose, mg/dL | 121.4 (20.0) | 136.2 (26.9) |
The groups were clinically similar at baseline.
EtCO2, end-tidal carbon dioxide; ICP, intracranial pressure; MAP, mean arterial pressure.
One swine in each group died before the conclusion of the experiment (mortality 12% for no AO vs 16.7% zone 1 AO, P = 1.00). The no AO group required an average blood loss of 46.3 ± 0.1% of total blood volume, while the zone 1 AO required 47.9 ± 0.1% of total blood volume removal to reach the targeted MAP pressure for shock (P = .80).
Hemorrhagic shock phase
The average time to achieve adequate shock based on BE levels for the no AO group was 56.3 ± 27.4 minutes vs 50 ± 7.4 minutes for the zone 1 AO (P = .65). During the first 15 minutes of the preAO HS phase, both groups achieved a similar MAP (no AO MAP 18.4 ± 1.5 mm Hg vs zone 1 AO MAP 18.8 ± 1.6 mm Hg, P = .70) and EtCO2 level (no AO EtCO2 21.3 ± 5.0 mm Hg vs zone 1 AO EtCO2 19.4 ± 6.6 mm Hg, P = .59). Once REBOA was inflated during the last 30 minutes of HS, the SBP and MAP increase significantly during balloon inflation, P < .05 (Figure 2). After balloon deflation, the MAP and SBP were similar between the 2 groups until model conclusion at 240 minutes.
Figure 2.
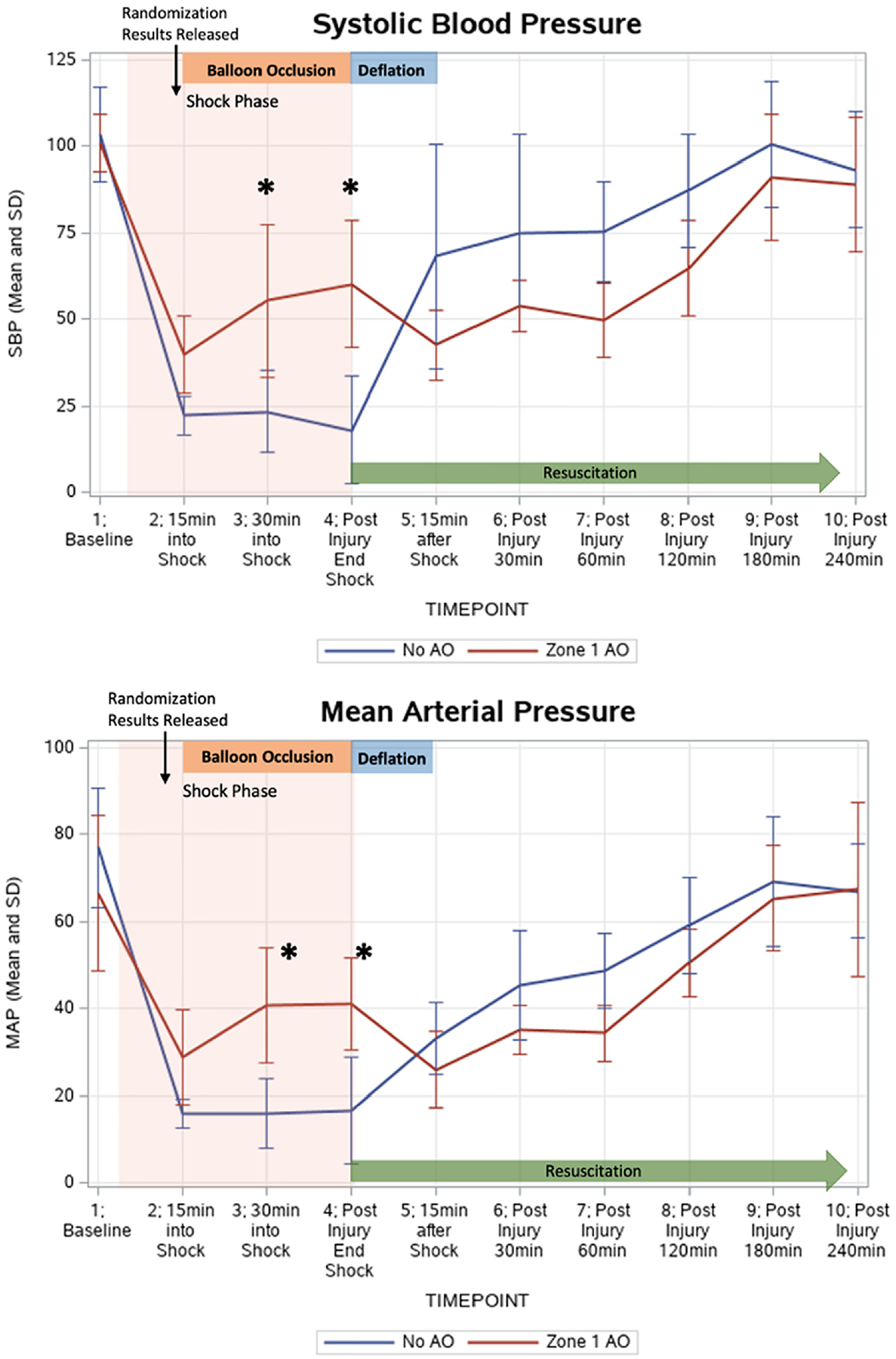
Mean arterial pressure (MAP) and systolic blood pressure (SBP) both increased significantly following zone 1 aortic occlusion (AO) during shock. The zone 1 AO group had a mean MAP of 40.8 ± 13.3 mm Hg at 30 minutes into HS (compared to no AO mean of 16.0 ± 7.9 mm Hg, P = .002) and a mean of 41.2 ± 10.6 mm Hg at completion of shock (compared to 16.7 ± 12.2 mm Hg, P = .002). The zone 1 AO group had a mean SBP of 55.3 ± 22.2 mm Hg at 30 minutes into HS shock (compared to 23.4 ± 11.8 mm Hg, P = .01) and a mean 60.2 ± 18.5 mm Hg at completion of shock (compared to 18.0 ± 15.5 mm Hg, P = .001).
Intracranial pressure
Both groups had increasing ICP after the HS phase and at the start of resuscitation using volume replacement (Figure 3). For both groups, the ICP reached significantly elevated levels above baseline as early as 60 minutes postinjury, with the no AO group exhibiting a mean ICP of 15.2 ± 4.1 mm Hg compared to its baseline of 11.1 mg Hg (P = .04) and the zone 1 AO group showing an ICP of 12.3 ± 6.0 mm Hg compared to the baseline of 7.7 mm Hg (P = .03). For the zone 1 AO, ICPs continued to trend upward until the completion of the model, but beyond postinjury 60 minutes the increases did not reach statistical significance. For the no AO group, the ICP continued to increase significantly until 180 minutes postinjury (P = .002). The increasing ICPs in the no AO model resulted in significantly elevated ICPs in the no AO group compared to the zone 1 AO at timepoints 180 minutes postinjury (22.0 ± 5.0 mm Hg vs 15.0 ± 3.9 mm Hg, respectively, P = .04) and 240 minutes postinjury (23.5 ±7.7 mm Hg vs 17.0 ± 5.2 mm Hg, respectively, P = .04).
Figure 3.
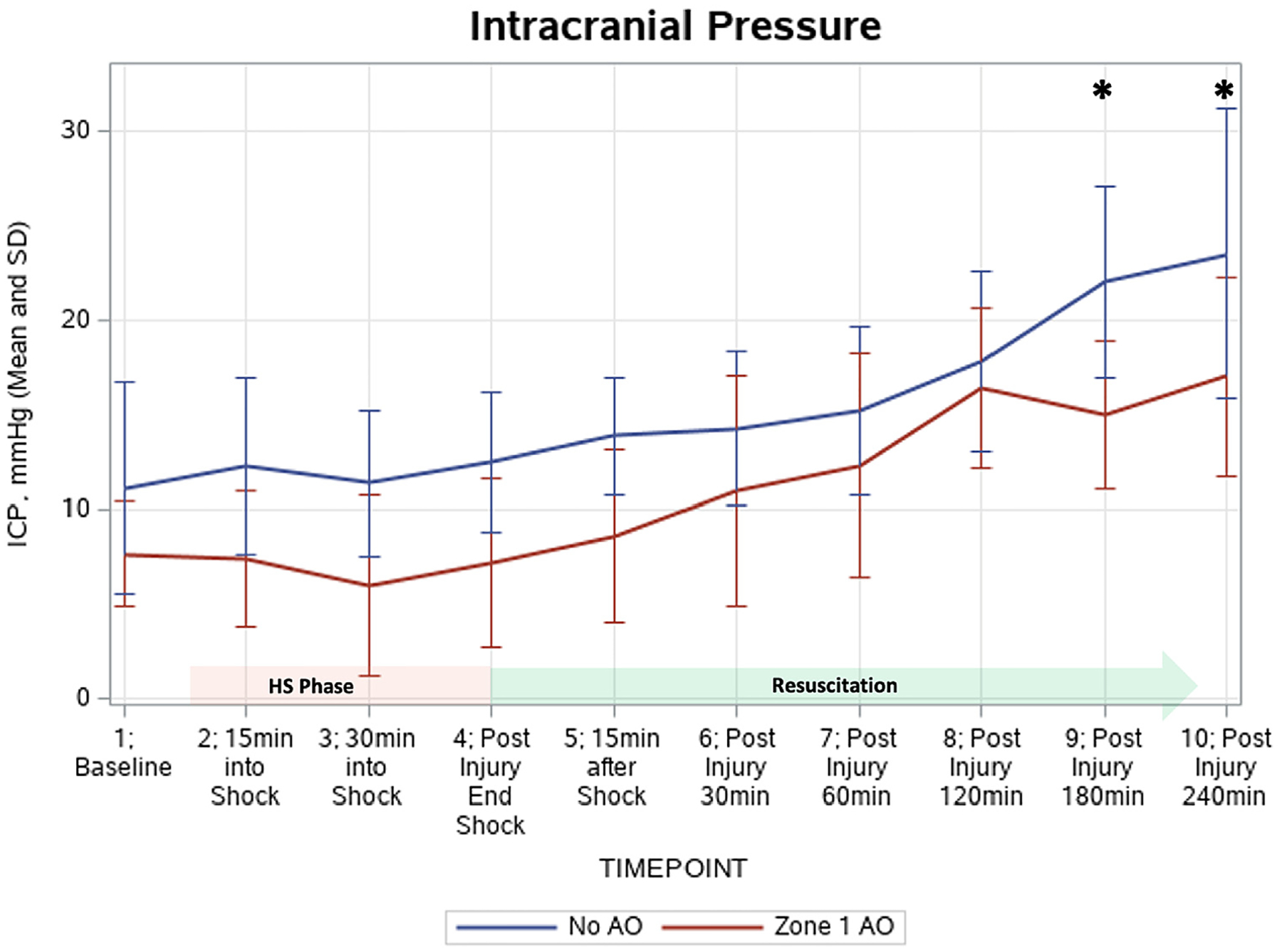
Intracranial pressure (ICP) changes throughout the experimental model. At 180 minutes postinjury and 240 minutes postinjury, the ICPs in the no aortic occlusion (AO) model were significantly elevated compared to the zone 1 AO: 22.0 ± 5.0 mm Hg vs 15.0 ± 3.9 mm Hg (P = .04) and 23.5 ±7.7 mm Hg vs 17.0 ± 5.2 mm Hg (P = .04), respectively.
Cerebral perfusion and edema
Cerebral perfusion pressures (Figure 4) were significantly higher in the zone 1 AO group during the 30 minutes of balloon occlusion only (P < .05). During the last 60 minutes of the model (180 and 240 minutes postinjury), the zone 1 AO group achieved a cerebral perfusion pressure similar to the baseline value (P = .25). For the no AO group, once the HS phase was initiated, the cerebral perfusion pressures remained significantly below the starting level and did not return to the baseline perfusion (P < .01). Brain water content showed no significant differences between the 2 groups (no AO: 3.95 ± 0.30 g vs zone 1 AO: 3.95 ± 0.09 g, P = .87).
Figure 4.
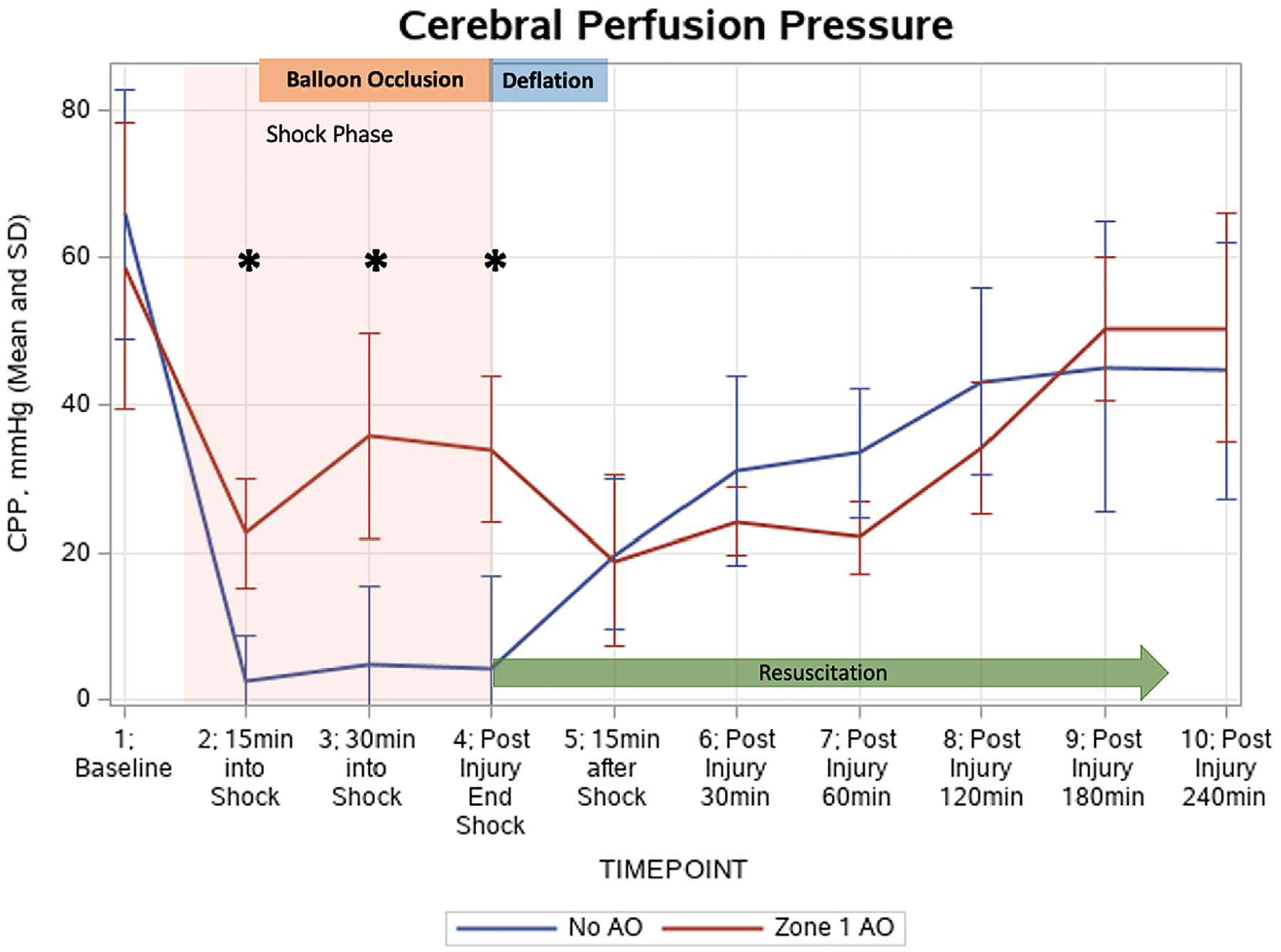
Cerebral perfusion pressure (CPP) changes throughout the model. CPP was similar at baseline. Aortic occlusion (AO) in zone 1 resulted in significantly elevated CPPimmediately after AO and throughout the duration of AO (P < .05).
Cardiac injury
At baseline, both groups had similar troponin values (P = 1.00). At the conclusion of the model, the no AO group had a significant increase in troponin (baseline: 0.03 ± 0.02 ng/mL to end: 3.5 ± 5.4 ng/mL, P = .008), while in the zone 1 AO group troponin did increase significantly (baseline: 0.03 ± 0.01 ng/mL to end: 1.7 ± 1.8 ng/mL, P = .24).
Discussion
This study examined the consequences of zone 1 REBOA in a clinically relevant animal model of DCBI with bTBI. In this study, zone 1 AO during HS provided significant elevations in the proximal blood pressure during HS. The lower troponin levels in the zone 1 AO group suggest that this strategy provides cardioprotection. Zone 1 AO via REBOA did not result in increased ICPs but provided cerebral perfusion during HS. Lastly, zone 1 AO via REBOA was not associated with increased cerebral edema after bTBI.
Military deployment of REBOA in austere environments for en route treatment protocols is occurring19 and is most often used for the treatment of HS after DCBI casualties compared to other mechanisms of injuries.7 DCBI not only results in severe hemorrhage but is the primary cause of TBIs for active military warfighters.20,21 A United Kingdom study reported that 1 of 5 combat casualties from 2002 to 2012 may have benefited from an intervention such as REBOA for treating noncompressible hemorrhage; of those patients who died during prolonged evacuation, 30% were due to TBI, and 30% later succumbed to TBI-related deaths after arrival at a military treatment facility.3 Thus, REBOA needs to be evaluated in the setting of TBI.
Cerebral perfusion pressure (CPP) is clinically recognized as the standard surrogate for measuring cerebral blood flow and brain perfusion in TBI patients by the critical care and neurosurgical guidelines.22,23 Current TBI treatment guidelines recommend that CPP should be maintained at a minimum of 60 mmHg,24 but CPP >70 mm Hg has not been shown to improve outcomes.25,26 In this study, while the CPP in the zone 1 AO grouped was significantly elevated during HS, the group with zone 1 AO did not achieve the recommended clinical threshold during HS with AO given the near 50% blood loss in this model. Another combined TBI and HS swine model with only 25% blood loss was able to achieve CPP above >60 mm Hg during the HS phase with complete AO.10
In regard to the SBP ranges, the National Brain Trauma Foundation Guidelines suggest a systolic blood pressure range of 100 to 140 mm Hg as optimal targets for CPP in treating TBI.27 Multicenter clinical studies have shown that elevated blood pressure, not just hypotension, is also associated with increased mortality12,28 and other worse outcomes13 in TBI patients. While our model incorporating 50% blood loss and the use of REBOA in isolation without concurrent transfusions was unable to achieve this specific critical blood pressure range (which was established for humans and may not be representative for pigs, in which critical thresholds are not known), this animal model did result in significant elevations in SBP and MAP. The elevations in blood pressure achieved by REBOA zone 1 AO do represent clinically meaningful increases, as evidenced by lower troponin levels representing significant cardiac perfusion. Other animal models of REBOA have actually exceeded this upper limit of 140 mm Hg SBP with supraphysiologic blood flow and MAPs.11,27,29 This is concerning because TBI causes impairment of the autoregulation mechanisms that maintain cerebral blood flow,30,31 and supraphysiologic pressures can further exacerbate intracranial hemorrhage or cerebral edema.32 Johnson et al have shown that the largest jumps in ICPs during REBOA animal models occur only during systemic supraphysiologic pressures.10
Additionally, the Brain Trauma Foundation advocates for treatment of ICPs above 22 mm Hg, as values above this threshold are associated with increased mortality.25 Our study indicated that REBOA zone 1 AO during HS was associated with a final lower ICP compared to the no AO model. In fact, the group without AO had a final ICP of 23.5 mm Hg, which is above the critical cutoff associated with increased mortality.
Unlike this study, previous TBI models with REBOA have not reached this critical ICP level and have not shown significant differences in final ICP values among REBOA versus non-REBOA groups.10,33 However, these previous models employed controlled cortical impact devices, which cause unilateral instead of global brain damage. Much less is known about the effects of blast injury on the brain, as this is a relatively new phenomenon after the rise of IEDs in military combat. Retrospective studies have identified that mild bTBI may go undiagnosed until much later because the blast overpressure (BOP) injury can cause invisible damage to the systemic vasculature and the central nervous system.20,21 Neurosurgical evaluation of US soldiers in Iraq have identified cerebral edema, intracranial hemorrhage, and prolonged vasospasms as common signs after blast exposure.34,35 The associated intracranial hypertension along with diffuse brain edema is an early hallmark of bTBI,36 and it is a significant risk factor when employing an intervention such as REBOA that has already shown to have the potential to worsen intracranial pressures.10
Animal models of bTBI show that even low levels of laboratory-controlled blast exposure results in neuroinflammatory responses.37–40 Rodent and swine models have shown evidence of immediate disruption of the blood-brain barrier permeability,37 acute cerebral vasospasm,41,42 and even enlargement of cerebral blood vessels.43 The high prevalence of microvascular pathology across animal models suggests cerebral vasculature may be particularly susceptible to blast injury,38 and thus reinforcing that bTBI and worsening brain damage may be a significant risk with REBOA. Our wet-to-dry ratio analysis evaluating brain edema found no differences in the amount of water content between REBOA and non-REBOA groups. Our previous work on this model shows that DCBI with bTBI does result in a slight increase in brain water content at 4 hours after injury (an average increase of 0.14 g of water for every gram of dry parenchyma tissue).14 This suggests that in the setting of DCBI with severe hemorrhage, REBOA zone 1 AO can be safely used to improve proximal blood pressures (and remain below supraphysiologic pressures), without causing early increases in cerebral edema. Similar findings were reported by Williams et al, who also found no differences in the amount of cerebral swelling in their TBI+HS model treated with or without partial REBOA.23 While these findings were promising, reasons for why both studies did not detect differences in cerebral edema following REBOA interventions are likely multifold. First, neither study reported supraphysiologic proximal pressures resulting in cerebral perfusion above the recommended treatment thresholds in the REBOA models. Second, both studies focused on the immediate postinjury intervention and resuscitation phase, with this study monitoring for 4 hours after HS and Williams et al monitoring for 6 hours post HS. Cerebral edema continues to manifest well beyond the initial first few hours after brain injury,44 and thus animal models evaluating cerebral edema beyond the acute phase are needed to fully understand the clinical consequences of REBOA on secondary brain injury.
There are several limitations to address for this study. First, the cost of performing such labor-intensive swine models requiring prolonged pain control makes longer monitoring timelines beyond the acute injury period challenging. Second, this study does not incorporate clinical imaging or histologic findings to further quantify the degree of brain injury progression and instead relies on measuring cerebral edema using water content. Although this is a validated method for determining cerebral edema, the clinical relevance of brain water content at 4 hours after injury is not completely understood, and findings cannot be extrapolated to survival outcomes. Third, this model analyzed complete AO instead of partial AO. As advanced catheters permitting partial AO are now available, these newer catheters need to be evaluated in future studies. Fourth, swine are imperfect surrogates for human anatomy, especially for TBI models where cranial thickness and shape differ remarkably from the humans. However, swine have gyroencephalic brains similar to humans, and Yorkshire swine of this size have vasculature similar to human anatomy, making them ideal candidates for HS and REBOA models. Yet physiologic baselines differences between species minimize the applicability of evaluating clinical guidelines and thresholds in translation models, but instead provide a general understanding of the cause and effects after treatment interventions. Lastly, the model is an imperfect adaptation of DCBI and prehospital combat casualty resuscitation. DCBI casualties can range in severity and tissue injury location regarding extremity and junctional damage, and this model imitates pelvic trauma with lower extremity damage and noncompressible hemorrhage. The proximal, open femur fractures combined with femoral bleeding are an imperfect surrogate for pelvic damage but do allow for a consistent, replicable injury across all animals. This model uses AO during the HS phase only and not during active fluid resuscitation. In clinical scenarios, fluid replacement occurs ideally before and during the use of REBOA. In order to achieve a highly lethal level of HS focusing on the effects of REBOA on brain injury, fluid resuscitation was performed after REBOA to remove confounders such as resuscitation speed and volume affecting proximal blood pressures and cerebral perfusion. Additionally, immediate resuscitation of a swine’s own fresh whole blood limits real-world applicability, so instead albumin is used to provide pressure support to minimize animal mortality while delaying the transfusion of fresh whole blood to mimic a resource-poor environment.
In conclusion, this large animal model representing a clinically relevant DCBI composed of HS and bTBI provides supporting evidence for REBOA zone 1 AO to increase proximal blood pressure while protecting cardiac and cerebral perfusion. Use of zone 1 AO was associated with lower troponin levels and resulted in improved cerebral perfusion during HS with significantly decreased ICP up to 4 hours after HS. The increased cerebral perfusion secondary to AO was not associated with increased cerebral edema.
Funding/Support
This research is funded by the Department of Defense contract number W81XWH2010205. This contract provides salary support and research support for animal and laboratory costs. Research support is also provided by the National Institute of General Medical Sciences of the National Institutes of Health (T32GM008315). The current major funding source is an RM-1grant(1RM1GM131968-01). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or Deparment of Defense.
Conflict of interest/Disclosure
E.E.M. has patents pending related to coagulation and fibrinolysis diagnostics and therapeutic fibrinolytics and is a cofounder with stock options in ThromboTherepeutics. E.E.M. has received grant support from Haemonetics, Inc., Stago, Hemosonics, Instrumentation Laboratories, Inc, and Diapharma outside the submitted work.
References
- 1.Kauvar DS, Wade CE. The epidemiology and modern management of traumatic hemorrhage: US and international perspectives. Crit Care. 2005;9(Suppl 5): S1–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rhee P, Joseph B, Pandit V, et al. Increasing trauma deaths in the United States. Ann Surg. 2014;260:13–21. [DOI] [PubMed] [Google Scholar]
- 3.Morrison JJ, Ross JD, Rasmussen TE, Midwinter MJ, Jansen JO. Resuscitative endovascular balloon occlusion of the aorta: a gap analysis of severely injured UK combat casualties. Shock. 2014;41:388–393. [DOI] [PubMed] [Google Scholar]
- 4.Cannon JW, Hofmann LJ, Glasgow SC, et al. Dismounted complex blast injuries: a comprehensive review of the modern combat experience. J Am Coll Surg. 2016;223:652–664.e8. [DOI] [PubMed] [Google Scholar]
- 5.Eastridge BJ, Mabry RL, Seguin P, et al. Death on the battlefield (2001–2011): implications for the future of combat casualty care. J Trauma Acute Care Surg. 2012;73(6 Suppl 5):S431–S437. [DOI] [PubMed] [Google Scholar]
- 6.Eastridge BJ, Holcomb JB, Shackelford S. Outcomes of traumatic hemorrhagic shock and the epidemiology of preventable death from injury. Transfusion. 2019;59(S2):1423–1428. [DOI] [PubMed] [Google Scholar]
- 7.Stokes SC, Theodorou CM, Zakaluzny SA, DuBose JJ, Russo RM. Resuscitative endovascular balloon occlusion of the aorta in combat casualties: the past, present, and future. J Trauma Acute Care Surg. 2021;91(Suppl 2):S56–S64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rafaels KA, Bass CR, Panzer MB, et al. Brain injury risk from primary blast. J Trauma Acute Care Surg. 2012;73:895–901. [DOI] [PubMed] [Google Scholar]
- 9.Uchino H, Tamura N, Echigoya R, Ikegami T, Fukuoka T. “REBOA”: is it really safe? A case with massive intracranial hemorrhage possibly due to endovascular balloon occlusion of the aorta (REBOA). Am J Case Rep. 2016;17: 810–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson MA, Williams TK, Ferencz SE, et al. The effect of resuscitative endovascular balloon occlusion of the aorta, partial aortic occlusion and aggressive blood transfusion on traumatic brain injury in a swine multiple injuries model. J Trauma Acute Care Surg. 2017;83:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoehn MR, Teeter WA, Morrison JJ, et al. Aortic branch vessel flow during resuscitative endovascular balloon occlusion of the aorta. J Trauma Acute Care Surg. 2019;86:79–85. [DOI] [PubMed] [Google Scholar]
- 12.Fuller G, Hasler RM, Mealing N, et al. The association between admission systolic blood pressure and mortality in significant traumatic brain injury: a multi-centre cohort study. Injury. 2014;45:612–617. [DOI] [PubMed] [Google Scholar]
- 13.Butcher I, Maas AI, Lu J, et al. Prognostic value of admission blood pressure in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24: 294–302. [DOI] [PubMed] [Google Scholar]
- 14.Cralley AL, Moore EE, Kissau D, et al. A combat casualty relevant dismounted complex blast injury model in swine. J Trauma Acute Care Surg. 2022. 10.1097/TA.0000000000003674. Epub ahead of print. May 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis JW, Dirks RC, Kaups KL, Tran P. Base deficit is superior to lactate in trauma. Am J Surg. 2018;215:682–685. [DOI] [PubMed] [Google Scholar]
- 16.Elliott MB, Jallo JJ, Tuma RF. An investigation of cerebral edema and injury volume assessments for controlled cortical impact injury. J Neurosci Methods. 2008;168:320–324. [DOI] [PubMed] [Google Scholar]
- 17.Keep RF, Hua Y, Xi G. Brain water content: a misunderstood measurement? Transl Stroke Res. 2012;3:263–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hawryluk GW, Phan N, Ferguson AR, et al. Brain tissue oxygen tension and its response to physiological manipulations: influence of distance from injury site in a swine model of traumatic brain injury. J Neurosurg. 2016;125:1217–1228. [DOI] [PubMed] [Google Scholar]
- 19.Northern DM, Manley JD, Lyon R, et al. Recent advances in austere combat surgery: use of aortic balloon occlusion as well as blood challenges by special operations medical forces in recent combat operations. J Trauma Acute Care Surg. 2018;85(Suppl 2):S98–S103. [DOI] [PubMed] [Google Scholar]
- 20.Wolf SJ, Bebarta VS, Bonnett CJ, Pons PT, Cantrill SV. Blast injuries. Lancet. 2009;374:405–415. [DOI] [PubMed] [Google Scholar]
- 21.Rafaels K, Bass CR, Salzar RS, et al. Survival risk assessment for primary blast exposures to the head. J Neurotrauma. 2011;28:2319–2328. [DOI] [PubMed] [Google Scholar]
- 22.Griesdale DE, Ortenwall V, Norena M, et al. Adherence to guidelines for management of cerebral perfusion pressure and outcome in patients who have severe traumatic brain injury. J Crit Care. 2015;30:111–115. [DOI] [PubMed] [Google Scholar]
- 23.Young JS, Blow O, Turrentine F, Claridge JA, Schulman A. Is there an upper limit of intracranial pressure in patients with severe head injury if cerebral perfusion pressure is maintained? Neurosurg Focus. 2003;15:E2. [DOI] [PubMed] [Google Scholar]
- 24.Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care, Carney NA. Guidelines for the management of severe traumatic brain injury. Methods. J Neurotrauma. 2007;24(Suppl 1):S3–S6. [DOI] [PubMed] [Google Scholar]
- 25.Carney N, Totten AM, O’Reilly C, et al. Guidelines for the management of severe traumatic brain injury (4th edition). Neurosurgery. 2017;80:6–15. [DOI] [PubMed] [Google Scholar]
- 26.Clifton GL, Miller ER, Choi SC, Levin HS. Fluid thresholds and outcome from severe brain injury. Crit Care Med. 2002;30:739–745. [DOI] [PubMed] [Google Scholar]
- 27.Bratton SL, Chestnut RM, Ghajar J, et al. Guidelines for the management of severe traumatic brain injury. I. Blood pressure and oxygenation. J Neurotrauma. 2007;24(Suppl 1):S7–S13. [DOI] [PubMed] [Google Scholar]
- 28.Zafar SN, Millham FH, Chang Y, et al. Presenting blood pressure in traumatic brain injury: a bimodal distribution of death. J Trauma. 2011;71:1179–1184. [DOI] [PubMed] [Google Scholar]
- 29.Russo RM, Neff LP, Lamb CM, et al. Partial resuscitative endovascular balloon occlusion of the aorta in swine model of hemorrhagic shock. J Am Coll Surg. 2016;223:359–368. [DOI] [PubMed] [Google Scholar]
- 30.Toth P, Szarka N, Farkas E, et al. Traumatic brain injury-induced autoregulatory dysfunction and spreading depression-related neurovascular uncoupling: pathomechanisms, perspectives, and therapeutic implications. Am J Physiol Heart Circ Physiol. 2016;311:H1118–H31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qasim ZA, Sikorski RA. physiologic considerations in trauma patients undergoing resuscitative endovascular balloon occlusion of the aorta. Anesth Analg. 2017;125:891–894. [DOI] [PubMed] [Google Scholar]
- 32.Sellmann T, Miersch D, Kienbaum P, et al. The impact of arterial hypertension on polytrauma and traumatic brain injury. Dtsch Arztebl Int. 2012;109:849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams AM, Bhatti UF, Dennahy IS, et al. Traumatic brain injury may worsen clinical outcomes after prolonged partial resuscitative endovascular balloon occlusion of the aorta in severe hemorrhagic shock model. J Trauma Acute Care Surg. 2019;86:415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armonda RA, Bell RS, Vo AH, et al. Wartime traumatic cerebral vasospasm: recent review of combat casualties. Neurosurgery. 2006;59:1215–1225:discussion 25. [DOI] [PubMed] [Google Scholar]
- 35.Ling G, Bandak F, Armonda R, Grant G, Ecklund J. Explosive blast neurotrauma. J Neurotrauma. 2009;26:815–825. [DOI] [PubMed] [Google Scholar]
- 36.Prima V, Serebruany VL, Svetlov A, Hayes RL, Svetlov SI. Impact of moderate blast exposures on thrombin biomarkers assessed by calibrated automated thrombography in rats. J Neurotrauma. 2013;30:1881–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabu S, Jaffer H, Petro M, et al. Blast-associated shock waves result in increased brain vascular leakage and elevated ROS levels in a rat model of traumatic brain injury. PLoS One. 2015;10:e0127971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gama Sosa MA, De Gasperi R, Janssen PL, et al. Selective vulnerability of the cerebral vasculature to blast injury in a rat model of mild traumatic brain injury. Acta Neuropathol Commun. 2014;2:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Logsdon AF, Meabon JS, Cline MM, et al. Blast exposure elicits blood-brain barrier disruption and repair mediated by tight junction integrity and nitric oxide dependent processes. Sci Rep. 2018;8:11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toklu HZ, Yang Z, Oktay S, et al. Overpressure blast injury-induced oxidative stress and neuroinflammation response in rat frontal cortex and cerebellum. Behav Brain Res. 2018;340:14–22. [DOI] [PubMed] [Google Scholar]
- 41.Bauman RA, Ling G, Tong L, et al. An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J Neurotrauma. 2009;26:841–860. [DOI] [PubMed] [Google Scholar]
- 42.Saljo A, Arrhen F, Bolouri H, Mayorga M, Hamberger A. Neuropathology and pressure in the pig brain resulting from low-impulse noise exposure. J Neurotrauma. 2008;25:1397–1406. [DOI] [PubMed] [Google Scholar]
- 43.Balaban C, Jackson RL, Liu J, Gao W, Hoffer ME. Intracranial venous injury, thrombosis and repair as hallmarks of mild blast traumatic brain injury in rats: lessons from histological and immunohistochemical studies of decalcified sectioned heads and correlative microarray analysis. J Neurosci Methods. 2016;272:56–68. [DOI] [PubMed] [Google Scholar]
- 44.Stocchetti N, Maas AI. Traumatic intracranial hypertension. N Engl J Med. 2014;371:972. [DOI] [PubMed] [Google Scholar]