

The term “proteoform” was first introduced in 2013, and was quickly adopted by the research community.1 A proteoform is a defined form of a protein from a given gene with a specific amino acid sequence and localized post-translational modifications. Proteoforms are thus the collection of diverse protein molecules (in many forms) that derive from each gene in the genome. The importance of proteoforms in biology and medicine reflects the fact that understanding of biological systems relies upon knowledge of their elements. Proteoforms are the ultimate molecular effectors of function in biology, and as such are central to understanding that function.
Proteomics as it is practiced today is almost exclusively accomplished by mass spectrometry. It comes in three main flavors, referred to as “bottom-up”, “top-down”, and “middle-down.” Bottom-up, in which proteins are digested into peptides, is by far the most well-developed and widely used approach; it provides deeper proteome coverage than top-down or middle-down, but at the cost of loss of molecular context – you cannot determine what proteoforms are present from their component peptides. Top-down, in contrast, skips the digestion step and seeks to identify the proteoforms in the mass spectrometer directly. It does allow complete characterization of the intact proteoform, but at the cost of decreased sensitivity, which translates into lower proteome coverage. Middle-down is between the two, seeking to reduce protein sizes to manageable levels by means of controlled and limited digestion, but this approach remains a bit artisanal in nature, and is not well-suited for comprehensive proteome analysis.
One powerful way to conceptualize proteomics at the proteoform level is through “proteoform families” (Figure 1).2,3 A proteoform family is the set of proteoforms derived from a given gene. For the ~20,000 genes in human, for example, there would thus be ~20,000 proteoform families, and a comprehensive proteoform-level analysis in human would reveal the members and their abundances detected in the sample for each family. There is much to be done to actualize this vision. Advances in mass spectrometry and other emerging platforms such as nanopore sequencing and cryo-EM are in active development worldwide, and the technological progress is breathtaking.
Figure 1.
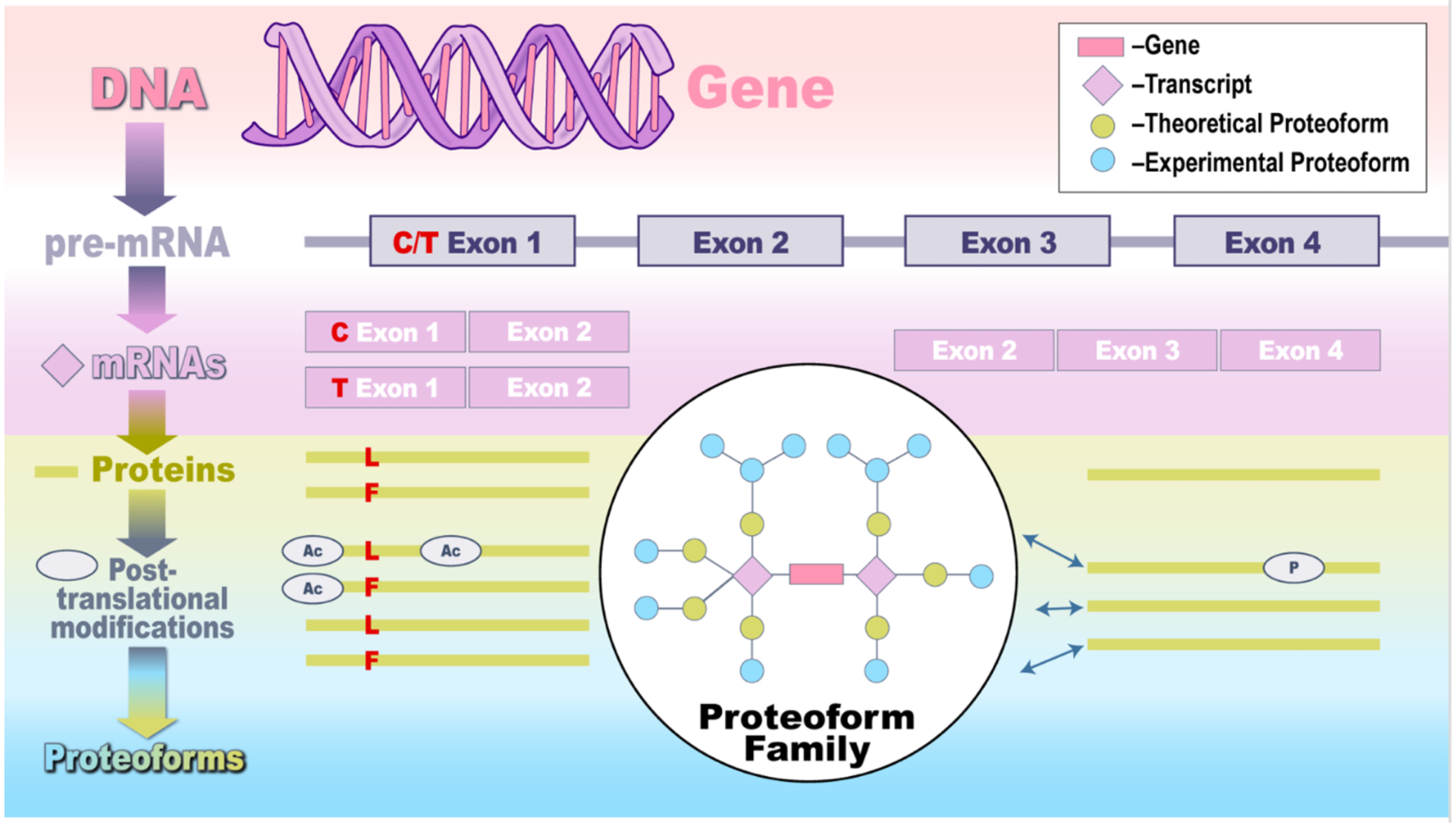
Illustration of a proteoform family, encompassing sources of protein variation at genomic, transcriptomic, and post-translational levels.
A concept that may help to drive progress concerns the distinction and transition between “discovery” technologies, and “scoring” technologies. A prominent example of this occurred in the Human Genome Project, where the early large-scale sequencing efforts (discovery technology) revealed millions of single-nucleotide polymorphisms that were compiled in public databases such as dbSNP (https://www.ncbi.nlm.nih.gov/snp/); once compiled however, simpler and less expensive approaches such as hybridization arrays (scoring technology) could be used to screen large numbers of samples, enabling deep analysis across populations in genome-wide association studies (GWAS). This kind of possibility motivates the Human Proteoform Project, an ambitious international effort to accelerate proteoform technology development and to establish comprehensive atlases of proteoforms for humans and model organisms.4 Such atlases can enable the transition to a scoring technology, by, for example, allowing a relatively simple proteoform mass measurement to serve as a proteoform identifier.2,3 Sample-specific databases built on a foundation of multiple data types, such as genomic and transcriptomic nucleic acid sequence data and deep bottom-up proteomic data, will play an important role in establishing and employing proteoform atlases, as they will allow proteoform databases to be built that correctly reflect the particular individual and tissue under study.5,6
This volume presents a compendium of cutting-edge emerging tools for proteoform analysis, in a form designed to allow others to be able to practice them. Chapters describe emerging approaches to proteoform separation, data handling and interpretation, and important application areas.
Separations
Separations are central to proteomics analyses of complex mixtures due to the fundamental mismatch between the complexity of a proteome and the much more limited ability of a mass spectrometer to resolve mixtures. While a proteome-wide study might seek to reveal thousands or tens of thousand of peptides or proteins, a decipherable high quality mass spectrum of peptide or protein mixtures will generally have far fewer components, arguably in the range of 1–100. While separation strategies for comprehensive peptide analysis are fairly mature and widely practiced, separations of proteins and their proteoforms are much less developed. New strategies and materials for proteoform separations by capillary electrophoresis, size exclusion chromatography, reverse-phase chromatography, and isoelectric focusing are described.
Bioinformatic tools
Bioinformatic tools are similarly critical to proteoform analysis. The complexity of the information required for proteoform-level analysis can be daunting. Complete sequence coverage of even a single proteoform relies upon the acquisition of very complex data; for example, if a proteoform had 500 amino acids, corresponding to a molecular weight of around 50 kDa, uniform cleavage at every peptide bond would produce 1000 b and y ions, each of which would have only 1/1000th of the ion intensity of the parent ion. In reality cleavage is not uniform, and the spectrum will be further complicated by internal ions, missing ions, possible co-eluting interferences, and so on. In the case of a proteome-wide analysis complex data of this sort needs to be obtained for thousands or tens of thousands of components. Powerful bioinformatic tools are clearly needed to rapidly and reliably process these massive data streams, providing the needed information on identities and abundances of the proteoforms present. Development of new algorithms and software tools to this end is a rich and vibrant area of current research, and several such state-of-the art capabilities are presented in this volume.
Applications
Applications comprise the third and final set of chapters presented here. In many ways, such applications are “where the rubber hits the road” in state-of-the-art proteoform analysis. Tools are only as good as the results they are able to provide when faced with real-world problems. Three concluding chapters present the application of proteoform analysis to histone proteoforms, monoclonal antibody characterization, and the discovery of proteoforms containing new post-translational modifications.
Taken together, this volume presents a beautiful snapshot of the status of emerging technologies in the exciting frontier of proteoform analysis. Readers interested in learning about and applying the latest approaches will find it well worth their time.
References
- 1.Smith LM, Kelleher NL, Consortium for Top-down Proteomics. 2013. Proteoform: a single term describing protein complexity. Nat. Methods 10 (3) 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shortreed MR, Frey BL, Scalf M, Knoener RA, Cesnik A, Smith LM 2016. Elucidating Proteoform Families from Proteoform Intact Mass and Lysine Count Measurements. J. Proteome Res, 15(4), 1213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cesnik A, Shortreed M, Schaffer L, Knoener R, Frey B, Scalf M, Solntsev S, Dai Y, Gasch A, Smith L, 2018. Proteoform Suite: Software for Constructing, Quantifying and Visualizing Proteoform Families. J Proteome Res, 17(1) 568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith L; Agar J; Chamot-Rooke J; Danis P; Ge Y; Loo J; Pasa-Tolic L; Tsybin Y; Kelleher N The Human Proteoform Project: A Plan to Define the Human Proteome. Preprints 2020, 2020100368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai Y, Shortreed M, Scalf M, Frey B, Cesnik A, Solntsev S, Schaffer L, Smith L, 2017. Elucidating E. coli Proteoform Families Using Intact-mass Proteomics and a Global PTM Discovery Database. J. Proteome Res, 16, 4156–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cesnik AJ, Miller RM, Ibrahim K, Lu L, Millikin RJ, Shortreed MR, Frey BL, Smith LM 2020. Spritz: A Proteogenomic Database Engine. J Proteome Res. DOI: 10.1021/acs.jproteome.0c00407 [DOI] [PMC free article] [PubMed] [Google Scholar]