

Summary
R-loops constitute a prevalent class of transcription-driven non-B DNA structures that occur in all genomes depending of both DNA sequence and topological favorability. In recent years, R-loops have been implicated in a variety of adaptive and maladaptive roles and have been linked to genomic instability in the context of human disorders. As a consequence, the accurate mapping of these structures in genomes is of high interest to many investigators. Here, we describe DRIP-seq (DNA:RNA Immunoprecipitation followed by high throughput sequencing), a robust and reproducible technique that permits accurate and semi-quantitative mapping of R-loops. We also describe a recent iteration of the method in which fragmentation is accomplished using sonication (sDRIP-seq), which allows strand-specific and high-resolution mapping of R-loops. sDRIP-seq thus addresses some of the common limitations of the DRIP-seq method in terms of resolution and strandedness, making it a method of choice for R-loop mapping.
Keywords: R-loops, DNA:RNA hybrids, S9.6, R-loop mapping, sonication, immunoprecipitation, strand-specific library
Introduction
R-loops are three-stranded nucleic acid structures composed of an RNA:DNA hybrid and a single-stranded DNA loop. These structures form primarily during transcription upon hybridization of the nascent RNA transcript to the template DNA strand. Biochemical reconstitution (Daniels and Lieber, 1995; Ginno et al., 2012; Reaban et al., 1994; Yu et al., 2003) and mathematical modeling (Stolz et al., 2019), in combination with other biophysical measurements (Carrasco-Salas et al., 2019; Duquette et al., 2004), have established that R-loops are more likely to occur over regions that exhibit specific favorable characteristics. For instance, regions that display strand asymmetry in the distribution of guanines (G) and cytosines (C) such that the RNA is G-rich, a property called positive GC skew, are favored to form R-loops when transcribed owing to the higher thermodynamic stability of the DNA:RNA hybrid compared to the DNA duplex (Huppert, 2008). Regions that have evolved positive GC skew, such as the early portions of many eukaryotic genes (Ginno et al., 2012; Green et al., 2003; Hartono et al., 2015; Polak and Arndt, 2008), are prone to forming R-loops in vitro and in vivo (Ginno et al., 2012; Malig et al., 2020; Yu et al., 2003). Negative DNA superhelical stress also greatly favors structure formation (Drolet et al., 2003; Masse et al., 1997) because R-loops efficiently absorb such topological stresses and return the surrounding DNA fiber to a favorable relaxed state (Chedin and Benham, 2020; Stolz et al., 2019).
Historically, R-loop structures were considered to result from rare, accidental entanglements of RNA with DNA during transcription. However, the development of DNA:RNA immunoprecipitation (DRIP) coupled to high-throughput DNA sequencing (DRIP-seq) allowed the first genome-wide mapping of R-loops and revealed that those structures are far more prevalent than expected in human cells (Ginno et al., 2013; Ginno et al., 2012). R-loops occur over tens of thousands of conserved, transcribed, genic hotspots in mammalian genomes, with a predilection for GC-skewed CpG islands overlapping the first intron of genes and the terminal regions of numerous genes (Sanz et al., 2016). Overall, R-loops collectively occupy 3-5% of the genome in human cells, consistent with measurements in other organisms including yeasts, plants, fly and mouse (Alecki et al., 2020; El Hage et al., 2014; Hartono et al., 2018; Wahba et al., 2016; Xu et al., 2017).
Analysis of R-loop forming hotspots in human cells revealed that such regions associate with specific chromatin signatures (Chedin, 2016). R-loops in general, are found over regions with lower nucleosome occupancy and higher RNA polymerase density. At promoters, R-loops associate with increased recruitment of two co-transcriptionally deposited histone modifications, H3K4me1 and H3K36me3 (Sanz et al., 2016). At gene termini, R-loops associate with closely arranged genes that undergo efficient transcription termination (Sanz et al., 2016), consistent with prior observations (Skourti-Stathaki et al., 2011). R-loops were also shown to participate in the initiation of DNA replication at the replication origins of bacteriophage, plasmid, mitochondrial, and the yeast genomes (Carles-Kinch and Kreuzer, 1997; Itoh and Tomizawa, 1980; Kreuzer and Brister, 2010; Lee and Clayton, 1998; Masukata and Tomizawa, 1990; Stuckey et al., 2015; Xu and Clayton, 1995). In addition, 76% of R-loop-prone human CpG island promoters function as early, constitutive replication origins (Cadoret et al., 2008; Mukhopadhyay et al., 2014; Picard et al., 2014; Sequeira-Mendes et al., 2009), further illustrating the links between R-loops and replication origins. Collectively, these studies suggest that R-loops represent a novel type of biological signal that can trigger specific biological outputs in a context-dependent manner (Chedin, 2016).
Early on, R-loops were shown to form at repeated class switch sequences during the process of immunoglobulin class switch recombination (Huang et al., 2007; Huang et al., 2006; Yu et al., 2003). Such programmed R-loops have been linked to the initiation of class switch recombination via the introduction of double-stranded DNA breaks (Yu and Lieber, 2019). Since then, “harmful” R-loop formation, generally understood to result from excessive R-loop formation, has been implicated in various processes linked to genomic instability such as hyper recombination, transcription-replication collisions, replication and transcriptional stress (for review (Costantino and Koshland, 2015; Crossley et al., 2019; Garcia-Muse and Aguilera, 2019; Santos-Pereira and Aguilera, 2015; Skourti-Stathaki and Proudfoot, 2014)). As a consequence, the accurate mapping of R-loop structures represents an exciting and important challenge to better understand the distribution and function of these structures in health and disease.
DNA:RNA immunoprecipitation (DRIP) relies on high affinity of the S9.6 monoclonal antibody for DNA:RNA hybrids (Boguslawski et al., 1986). DRIP-seq permits robust genome-wide profiling of R-loop formation (Ginno et al., 2012; Sanz and Chedin, 2019). While useful, this technique suffers from limited resolution due to the fact that restriction enzymes are used to achieve gentle DNA fragmentation. In addition, DRIP-seq does not provide information on the directionality of R-loop formation. Here we report a variant of DRIP-seq that permits the mapping of R-loops at high resolution in a strand-specific manner. This method relies on sonication to fragment the genome prior to immunoprecipitation and the method is thus called sDRIP-seq (sonication DNA:RNA immunoprecipitation coupled to high throughput sequencing) (Figure 1). The use of sonication permits an increased resolution and limits restriction enzyme-linked fragmentation biases observed in DRIP-seq approaches (Halasz et al., 2017). sDRIP-seq produces R-loop maps that are in strong agreement with results from both DRIP-seq and the previously described high-resolution DRIPc-seq method in which sequencing libraries are built from the RNA strands of immunoprecipitated R-loop structures (Sanz and Chedin, 2019).
Figure 1: Overview of the DRIP-seq and sDRIP-seq procedures.
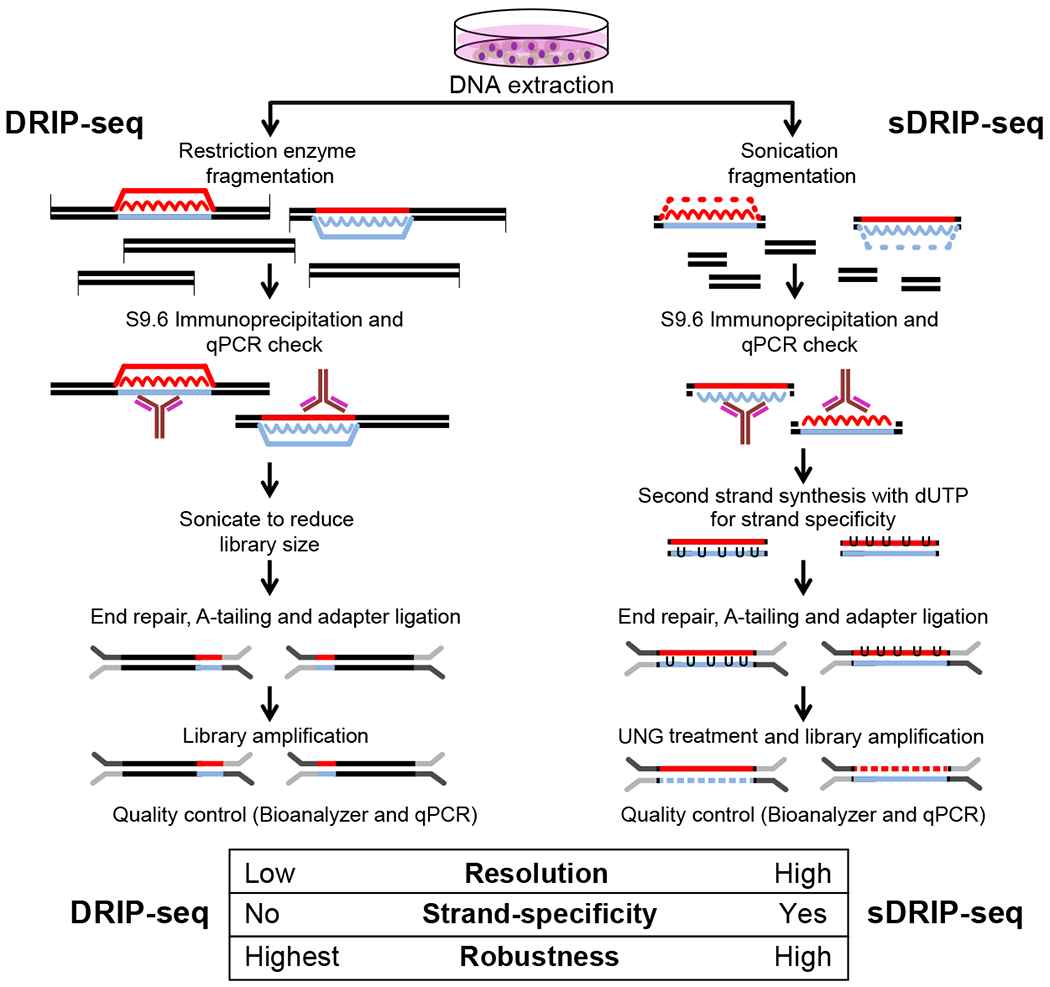
Both approaches start by the same DNA extraction steps developed to preserve R-loops (RNA strands within R-loops are represented by squiggly lines). For DRIP-seq, the genome is fragmented using restriction enzymes, often resulting in kilobase-size fragments within which shorter R-loops are embedded. For sDRIP-seq, the genome is fragmented via sonication which results in smaller fragments and the shearing and loss of the displaced single-strand of R-loops (indicated by dashed lines). Following immunoprecipitation with the S9.6 antibody, DRIP leads to the recovery of three-stranded R-loops embedded within restriction fragments, while sDRIP recovers two-stranded RNA:DNA hybrids with little flanking DNA, ensuring higher resolution. For sDRIP, a library construction step must be included to convert RNA:DNA hybrids back to duplex DNA. As shown here, this is an opportunity to build strand-specific libraries. As detailed in the protocol itself, exogenous treatment with RNase H represents a key control for the specificity of both procedures; they are not shown here.
Faced with a plethora of methods to choose from, users may wonder which particular DRIP-based approach is preferable for their needs. We offer the following advice. DRIP-seq, despite its limitations, is technically easiest and is the most robust (highest yields) of all three methods discussed here; it thus remains broadly useful. Numerous DRIP-seq datasets have been published, which provides a useful comparison point for new datasets. Finally, the bioinformatic analysis pipeline is simpler as the data is not stranded. We recommend that new users begin honing their R-loop mapping skills with DRIP followed by qPCR and DRIP-seq. sDRIP-seq represents a slightly higher degree of technical difficulty: the yields are slightly reduced due to sonication (discussed below) and the sequencing library process is slightly more complex. Yet, the gain of strandedness and higher resolution is invaluable. We note that sDRIP-seq will capture both two-stranded RNA:DNA hybrids and three-stranded R-loops. Due to the library construction steps, DRIP-seq will not capture two-stranded RNA:DNA hybrids. DRIPc-seq is the most technically demanding and requires higher amount of starting materials. In return, it offers the highest resolution and strandedness. Because sequencing libraries are built from the RNA moiety of R-loops or hybrids, DRIPc-seq may suffer from possible RNA contamination, especially since S9.6 possesses residual affinity for dsRNA (Chedin et al., 2021; Hartono et al., 2018; Phillips et al., 2013). sDRIP-seq permits strand-specific, high resolution mapping without worries about RNA contamination since sequencing libraries are derived from DNA strands. Overall, these three methods remain useful and present differing degrees of complexity and slightly different caveats. All three, however, produce highly congruent datasets (Chedin et al., 2021) and are highly sensitive to RNase H pre-treatment, which represents an essential control to ensure signal specificity (Sanz and Chedin, 2019; Smolka et al., 2021). We note that given the size selection imposed on sequencing libraries, small hybrids (estimated < 75 bp), such as those forming transiently around lagging strand DNA replication priming sites (Okazaki primers) will be excluded. Similarly, since all DRIP methods involve DNA fragmentation, unstable R-loops that require negative DNA supercoiling for their stability will be lost (Stolz et al., 2019). Thus DRIP approaches may underestimate R-loop loads, especially for short, unstable R-loops that may be best captured using “in vivo” approaches (Chedin et al., 2021; Sanz and Chedin, 2019). We note that R-loops can also be profiled in an S9.6-independent manner at deep coverage, high-resolution and in a strand-specific manner on single DNA molecules after sodium bisulfite treatment (Malig et al., 2020). Additionally, strategies using a catalytically inactive RNase H1 enzyme have been employed to map native R-loops “in vivo”, highlighting short, unstable R-loops that form primarily at paused promoters (Chen et al., 2019; Wang et al., 2021; Yan and Sarma, 2020).
Protocol
The following protocol is optimized for the human Ntera-2 cell line grown in culture but it has been successfully adapted without modification to a range of other human cell lines (HEK293, K562, HeLa, U2OS), primary cells (fibroblasts, B-cells) as well as in other organisms with small modifications (mouse, fly).
1. Cell harvest and lysis
1.1. Culture Ntera-2 cells to 75-85% confluency. To start any DRIP procedure, optimal cell count should be 5 to 6 million cells with >90% viable counts.
1.2. Wash cells once with 1x PBS and add 1.5ml of Trypsin-EDTA 1X and incubate for 2 minutes at 37°C until the cells dissociate from the dish.
1.3. Add 5 ml of warm media and, after pipetting well to resuspend cells into a single cell suspension, transfer the content in a new 15 ml tube and gently pellet the cells at 1,000 rpm for 3 minutes.
1.4. Wash the cells once with 5 ml of 1x PBS and gently pellet the cells at 1,000rpm for 3 minutes.
1.5. Fully resuspend the cells in 1.6 mL of TE buffer (10 mM Tris-Cl pH 7.5, 1 mM EDTA pH 8.0). Add 5 μL of proteinase K (20 mg/mL stock solution) and 50 μL of SDS (20% stock solution) and invert gently the tubes 5 times until solution become viscous. Do not try to pipet the solution, only mix by inversion.
1.6. Incubate overnight at 37°C.
2. DNA extraction
2.1. Pour the DNA lysate into a pre-spun 15 mL high density Maxtract phase lock gel tube and add 1 volume (1.6 mL) of Phenol/Chloroform Isoamyl alcohol (25:24:1). Gently invert 5 times and spin down at 1,500 g for 5 minutes.
2.2. Add 1/10 volume of 3M sodium acetate (NaOAc) pH 5.2 and 2.5 volumes of 100% Ethanol to a new 15 mL tube. Pour in the top aqueous phase from the phase lock gel tube and invert softly until the DNA is fully precipitated (up to 10 minutes).
2.3. Spool DNA threads using wide bore 1000 μL tip and transfer to a clean 2 mL tube while taking care of not carrying over residual supernatant.
2.4. Wash the DNA by adding 1.5 mL of ethanol 80% and gently invert the tube 5 times. Incubate for 10 minutes.
2.5. Repeat the previous step twice. Do not centrifuge during the wash steps. Carefully remove as much ethanol as possible by pipetting after the last wash while trying to not disturb DNA.
2.6. Allow the DNA to air dry completely while inverting the tube. This step can take 30 minutes to an hour depending on the amount of DNA.
2.7. Add directly on the DNA pellet 125 μL of TE if you are fragmenting the DNA through restriction enzyme digestion or 100 μL of TE if you are shearing the DNA through sonication. Keep on ice for one hour and gently resuspend DNA by pipetting a few times with a wide bore 200 μL tip. Leave on ice another hour before starting the fragmentation step.
3. DNA fragmentation
For restriction enzyme-based DRIP-seq, please follow step 3.1. For sonication-based DRIP-seq, follow skip to step 3.2.
3.1. Restriction enzyme (RE) fragmentation
3.1.1. Digest the resuspended genomic DNA (very viscous) using a cocktail of REs according to supplier’s instructions. Add 0.1 mM spermidine to the final reaction. We advise to use a cocktail of 4 to 5 enzymes with 30U of each enzyme in a total volume of 150 μL. The initial cocktail developed for DRIP-seq (HindIII, SspI, EcoRI, BsrGI, XbaI) (Ginno et al., 2012) was developed to generate an average fragment length of 5 kilobases, avoid any interference with CpG methylation, and spare GC-rich regions of the genome. Other cocktails are possible (Ginno et al., 2013)). These cocktails are suitable for both the human and mouse genomes but can be adjusted as needed. Incubate overnight at 37μC. The DNA mixture post digest should no longer be viscous. Any remaining viscosity at this step is indicative of an incomplete digestion. If observed, add an additional 10U of each enzyme and incubate for another 2-4 hours at 37°C. Note that users may not digest the entire pellet in the event they harvested more cells than recommended here.
3.1.2. Gently pipet the overnight digested DNA (150 μL) into a pre-spun 2 mL phase lock gel light tube. Add 100 μL of water and one volume (250 μL) of Phenol/Chloroform Isoamyl alcohol (25:24:1). Gently invert 5 times and spin down at 16,000 g for 10 minutes.
3.1.3. Add 1.5 μL of glycogen, 1/10 volume 3M NaOAc pH 5.2 and 2.5 volumes 100% Ethanol to a new 1.5 mL tube. Pipet the DNA from the phase lock gel tube and mix by inverting 5 times. Incubate one hour at −20°C.
3.1.4. Spin at 16,000 g for 35 minutes at 4°C. Wash DNA with 200 μL 80% ethanol and spin at 16,000 g for 10 minutes at 4°C.
3.1.5. Air dry the pellet and add 50 μL of TE buffer to the pellet. Leave the tube on ice for 30 minutes and gently resuspend the DNA.
3.1.6. Measure the concentration (OD260) of the fragmented DNA on a Nanodrop or equivalent.
3.1.7. Optional but recommended: load 1 μg of digested DNA on a 0.8% agarose gel alongside a size marker to verify that the digest is complete. If incomplete, additional enzyme can be added. Incomplete digestion can lead to loss of resolution after immunoprecipitation.
3.1.8. After this step, 10 μgs of digested DNA can be treated with 4 μL of NEB ribonuclease H (RNase H) for 1 to 2 hours at 37°C in order to control that the signal retrieved upon immunoprecipitation derives from DNA:RNA hybrids. You can then proceed to S9.6 immunoprecipitation (step 4). Note that digested DNAs can be kept frozen at −80°C for up to one month without significant loss of yield.
3.2. Sonication
3.2.1. Sonicate all or part of the extracted DNA in a 0.5 mL Eppendorf tube in 100 μL total volume. Perform 15 to 20 cycles of 30 sec ON / 30 sec OFF on a Diagenode Bioruptor NGS sonicator (spin after 5, 10 and 15 cycles to ensure homogeneous sonication).
3.2.2. Measure the concentration (OD260) of sonicated DNA on a Nanodrop or equivalent. At this step, the viscosity of the DNA should have disappeared.
3.2.3. Run an agarose gel to confirm the size distribution of sonicated DNA (300-500 bp). Over-sonicating DNA can lead to significant reduction in yield resulting from breakage and dissociation of R-loop structures.
3.2.4. After this step, 10 μgs of sonicated DNA can be treated with 4 μL of NEB RNase H for 1 to 2 hours at 37°C in order to control that the signal retrieved upon immunoprecipitation derives from DNA:RNA hybrids. You can then proceed to S9.6 immunoprecipitation (step 4).
4. S9.6 immunoprecipitation
The immunoprecipitation steps are similar regardless of whether DNA was fragmented through REs or sonication.
4.1. Prepare three tubes and aliquot 4.4 μg of fragmented DNA in a final volume of 500 μL of TE per tube. Save 50 μL (1/10 of the volume) from each tube to use later as an input.
4.2. Add 50 μL of 10X binding buffer (100 mM NaPO4 pH 7, 1.4 M NaCl, 0.5% Triton X-100) and 10 μL of S9.6 antibody (1 mg/ml) to the 450 μL of diluted DNA.
4.3. Incubate overnight at 4°C on a mini-tube rotator at 7 to 10 rpm.
4.4. For each tube, wash 50 μL of Protein A/G agarose bead slurry with 700 μL of 1X binding buffer by inverting the tubes on a mini-rotator at 7 to 10 rpm at room temperature for 10 minutes. Spin down the beads at 1,100 g for one minute and discard supernatant. Repeat this step once.
4.5. Add the DNA from step 4.3 to the 50 μL of beads and incubate for 2 hours at 4°C while inverting at 7 to 10 rpm on a mini-rotator.
4.6. Spin down the beads one minute at 1,100 g and discard supernatant.
4.7. Wash the beads with 750 μL of binding buffer 1X by inverting at 7 to 10 rpm on a mini-rotator for 15 minutes. Spin down one minute at 1,100g and discard supernatant. Repeat this step once.
4.8. Add 250 μL of elution buffer (50 mM Tris-Cl pH 8, 10 mM EDTA pH8, 0.5% SDS) and 7 μL of proteinase K (20 mg/mL stock) to the beads and incubate with rotation at 55°C for 45 minutes.
4.9. Spin down the beads one minute at 1,100 g. Transfer the supernatant to a pre-spun 2 mL phase lock gel light tube and add one volume (250 μL) of Phenol/Chloroform Isoamyl alcohol (25:24:1). Invert tubes 5 times and spin down for 10 minutes at 16,000 g at room temperature.
4.10. Add 1.5 μL of glycogen, 1/10 volume 3M NaOAc pH 5.2 and 2.5 volumes of 100% Ethanol to a new 1.5 mL tube. Pipet the DNA from the phase lock gel tube and mix by inverting 5 times. Incubate one hour at −20°C.
4.11. Spin at 16,000 g for 35 minutes at 4°C. Wash DNA with 200 μL 80% ethanol and spin at 16,000 g for 10 minutes at 4°C.
4.12. Air dry pellets and add 15 μL of 10 mM Tris-Cl pH 8 in each tube. Leave tubes on ice for 20 minutes and gently resuspend. Combine the 3 tubes in one (45 μL).
4.13. Check DRIP efficiency by qPCR using 5 μL of the 45 μL resuspended DNA (see representative results). Dilute the 5 μL in 10 μL of water and use 2 μL per reaction.
5. Pre-library step for sonicated DNA only
Sonication leads the displaced ssDNA strand of R-loops to break. Thus, three-stranded R-loop structures are converted into two-stranded DNA:RNA hybrids upon sonication. As a result, these RNA:DNA hybrids must be converted back to double-stranded DNA prior to library construction. Here we employ a second strand synthesis step. An alternative approach that has been successfully used is to instead perform a single-stranded DNA ligation followed by a second strand synthesis (Crossley et al., 2020).
5.1. To the 40 μL of DRIP’ed DNA from step 4.12, add 20 μL of 5X second strand buffer (200 mM Tris pH 7, 22 mM MgCl2, 425 mM KCl), 10 mM dNTP mix (dATP, dCTP, dGTP, and dTTT or dUTP if you are planning to achieve strand-specific DRIP sequencing), 1 μL 16 mM NAD and 32 μL water. Mix well and incubate 5 minutes on ice.
5.2. Add 1 μL of DNA polymerase I (10 units), 0.3 μL of RNase H (1.6 units) and 0.5 μL of E. coli DNA ligase. Mix and incubate at 16°C for 30 minutes.
5.3. Immediately clean up the reaction using Ampure beads with a ratio of 1.6X. Elute DNA in 40 μL of 10 mM Tris-Cl pH8.
6. Pre-library sonication step for RE DNA only.
DRIP leads to the recovery of RE fragments that are often kilobases in length and thus not suited for immediate library construction. To reduce the size of the material for library construction, sonicate the immunoprecipitated DNA in a 0.5 mL Eppendorf tube. Perform 12 cycles of 15 sec ON / 60 sec OFF on a Diagenode Bioruptor NGS sonicator (spin after 6 cycles to ensure homogeneous sonication). Proceed to step 7.
Optional step: the immunoprecipitated material still carries three-stranded R-loops which respond to sonication differently than the flanking double-stranded DNA. To even out DRIP profiles, we recommend treating the immunoprecipitated material with 1 μL of NEB RNase H in 1x RNase H buffer for 1 hour at 37°C prior to sonication.
7. Library construction
7.1. Perform end repair by adding to the 40 μL from step 4.12 (RE fragmentation) or step 5.3 (sonication shearing) 5 μL of NEB 10X end repair module buffer, 2.5 μL of 10 mM ATP and 2.5 μL NEBNext End repair module enzyme (50 μL total). Mix well and incubate for 30 minutes at room temperature. Also include 1 μg of RE-digested and sonicated (DRIP) or sonicated (sDRIP) input DNA to create control sequencing libraries corresponding to the input DNA.
7.2. Clean up the reaction using AMPure beads (1.6X ratio) and elute in 34 μL of 10 mM Tris-Cl pH8.
7.3. Perform A-tailing by adding 5 μL of NEB buffer 2, 10 μL of 1 mM dATP and 1 μL of NEB Klenow exo- (50 μL total). Mix well and incubate for 30 minutes at 37°C.
7.4. Clean up the reaction using AMPure beads (1.6X ratio) and elute in 12 μL of 10 mM Tris-Cl pH8.
7.5. Ligate adapters by adding 15 μL of NEB 2X quick ligation buffer, 1 μL of 15 μM Illumina adapters and 2 μL of NEB quick ligase (30 μL total). Mix well and incubate for 20 minutes at room temperature.
7.6. Clean up the reaction using AMPure beads (1X ratio) and elute in 20 μL of 10 mM Tris-Cl pH8.
7.7. If you used sonication shearing and dUTP in step 5.1 and want a strand-specific DRIP, add 1.5 μL (1.5 U) of AmpErase Uracil N-glycosylase. Incubate 30 minutes at 37°C.
7.8. PCR amplify 10 μL of the library from step 6.6 or 6.7. Add 1 μL of PCR primer 1.0 P5, 1 μL of PCR primer 2.0 P7, 15 μL of Phusion master mix and 3 μL of water. Mix well.
7.9. In a thermo cycler, run the following program:
| Cycle number | Duration | Temperature |
|---|---|---|
| 1 | 30 sec | 98°C |
| 2-15 | 10 sec | 98°C |
| 30 sec | 60°C | |
| 30sec | 72°C | |
| 16 | 5 min | 72°C |
| 17 | Hold | 12°C |
7.10. Proceed to a two-step clean-up of your library using AMPure. Use first a ratio of 0.65X to remove fragments over 500 bp. Keep supernatant. Proceed to a 1X ratio on the supernatant to remove fragments under 200 bp. Elute in 12 μL.
8. Quality control
8.1. Check R-loop enrichments with qPCR on two negative and three positive loci using the Pfaffl method using 1 μL of the clean-up library from step 6.10. Dilute 1 μL of the library in 10 μL of water and use 2 μL per locus.
8.2. Check the size distribution of your cleaned-up library from step 6.10 using an Agilent High sensitivity DNA 1000 kit.
Representative results
DRIP as well as sDRIP can be analyzed through qPCR (Figure 2A) and/or sequencing (Figure 2B). After the immunoprecipitation step, the quality of the experiment must be first confirmed by qPCR on positive and negative control loci, as well as with RNase H-treated controls. Primers corresponding to frequently used loci in multiple human cell lines are provided in Table 1. The results from qPCR should be displayed as a percentage of input, which corresponds to the percentage of cells carrying an R-loop at the time of the lysis for a given locus. In a successful DRIP experiment, the yield for negative loci should be less than 0.1% whereas positive loci can vary from 1% to over 10% for highly transcribed loci like RPL13A (Figure 2A). For sDRIP, yields are typically lower (20-50%) as judged by DRIP-qPCR but appear to affect recovery uniformly such that no particular subset of R-loops is affected more than another. As a result, maps derived from DRIP, sDRIP and DRIPc are in good agreement (Figure 2B). qPCR data can also be displayed as fold enrichment of the percentage of input for positive loci over negative loci, thus assessing the specificity of the experiment. Fold enrichments typically range from a minimal of 10-fold to over 200-fold depending on the loci chosen for analysis. When precise quantification across multiple samples representing gene knockdowns, knockouts, or various pharmacological treatments, is required, the use of spike in controls to normalize inter-sample experimental variation is highly encouraged. Such spike-ins can correspond to synthetic hybrids (Crossley et al., 2020) or genomes of unrelated species (Svikovic et al., 2019).
Figure 2: Result of R-loop mapping strategies.
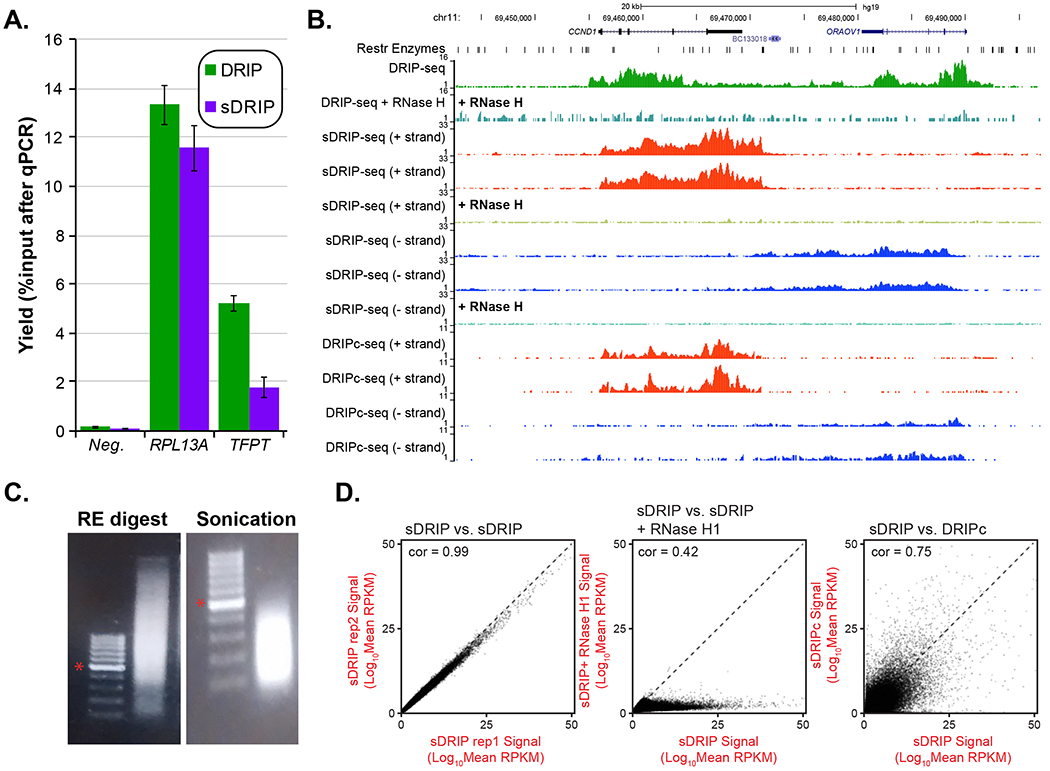
A. qPCR results from successful immunoprecipitations using the DRIP and sDRIP method (corresponding to qPCR check step 4.13). Results are from two independent experiments from human Ntera2 cells at a negative locus and two positive loci, including the highly R-loop-prone RPL13A locus and the moderately R-loop-prone locus TFPT. The y-axis indicates the yield of the immunoprecipitation as a percentage of the input DNA. Note that the recovery is slightly more robust for DRIP than sDRIP. B. The results of R-loop mapping conducted in human Ntera-2 cells are shown over a region centered around the CCND1 and neighboring ORAOV1 genes. The first two tracks correspond to DRIP-seq results, without and with RNase H treatment, respectively. The position of the restriction enzymes used to fragment the genome are shown at the top. The next six tracks represent the results of strand-specific sDRIP-seq, broken down between (+) and (−) strands (two replicates each) and pre-treated with RNase H, or not, as indicated. The last four track represents the results of R-loop mapping via the high-resolution strand-specific DRIPc-seq method (Sanz et al., 2016; Sanz and Chedin, 2019), where libraries are built from the RNA strands of R-loops. As can be clearly seen, the CCND1 and ORAOV1 genes lead to R-loop formation on the (+) and (−) strands, respectively, consistent with their directionality. RNase H treatment abolishes signal, as expected. C. Input DNA materials after restriction enzyme fragmentation (left) and sonication (right) are shown after the materials were separated by agarose gel electrophoresis. The DNA ladder corresponds to a 100 bp ladder and the 500 bp band is highlighted by an asterisk. D. XY signal correlation plots are shown to illustrate the reproducibility of sDRIP-seq (left), the overall sensitivity of sDRIP-seq to RNase H1 pre-treatment (middle), and the global correlation between sDRIP-seq and DRIPc-seq (right). All data from Ntera-2 human cells.
Table 1.
Primers used for qPCR validation in human cell lines. All sequences are listed in the 5’ to 3’ direction.
| Species | Type | Name | Sequence |
|---|---|---|---|
| human | positive control locus | RPL13A F | AGGTGCCTTGCTCACAGAGT |
| human | positive control locus | RPL13A R | GGTTGCATTGCCCTCATTAC |
| human | positive control locus | TFPT F | TCTGGGAGTCCAAGCAGACT |
| human | positive control locus | TFPT R | AAGGAGCCACTGAAGGGTTT |
| human | negative control locus | EGR1neg F | GAACGTTCAGCCTCGTTCTC |
| human | negative control locus | EGR1neg R | GGAAGGTGGAAGGAAACACA |
| human | negative control locus | SNRPNneg F | GCCAAATGAGTGAGGATGGT |
| human | negative control locus | SNRPNneg R | TCCTCTCTGCCTGACTCCAT |
DRIP and sDRIP materials can be sequenced using single or paired-end sequencing strategies. Data can be extracted and analyzed in a similar manner as most ChIP data using standard computational pipelines (see (Sanz and Chedin, 2019) for DRIP-relevant information). After adapter trimming and removal of PCR duplicates, reads can be mapped to a reference genome and uploaded to a genome browser. A typical expected output of DRIP and sDRIP is shown in Figure 2B. The DRIP output is represented by the only green track as it does not allow strand specificity whereas sDRIP shows R-loop mapping to the positive and negative strands indicated respectively in red and blue. Control tracks corresponding to a sample pre-treated with RNase H show a clear reduction of signals, confirming the specificity of the technique for RNA:DNA hybrid-derived materials. The gains in resolution permitted by sDRIP are clearly illustrated when comparing the sizes of input DNA material (Figure 2C). The reproducibility of sDRIP-seq, along with the global impact of RNase H1 pre-treatment and the correlation between sDRIP-seq and DRIPc-seq are depicted by XY plots in Figure 2D.
Discussion
We describe here two protocols to map R-loop structures in potentially any organism using the S9.6 antibody. DRIP-seq represents the first genome-wide R-loop mapping technique developed. It is an easy, robust, and reproducible technique that allows one to map the distribution of R-loops along any genome. The second technique, termed sDRIP-seq, is also robust and reproducible but achieves higher resolution and strand-specificity owing to the inclusion of a sonication step and a stranded sequencing library construction protocol. Both techniques are highly sensitive to RNase H treatment prior to immunoprecipitation, confirming that the signal is principally derived from genuine RNA:DNA hybrids. Finally, when comparing immunoprecipitation yields between R-loop positive and R-loop negative loci, both techniques offer up to a 100-fold difference in several human cell lines, providing high specificity mapping with low background.
When considering which method to implement, it is useful to consider their respective strengths and limitations. As previously noted, DRIP-seq produces maps with a lower resolution and does not give information on the strandedness of R-loop formation. The lower resolution is mainly a product of the use of REs to fragment the genome. This gentle method is best at preserving R-loops, thereby allowing unsurpassed recovery of such structures, and making DRIP-seq very robust. To circumvent the issue of limited resolution while preserving high recovery, RE cocktails can be adapted and/or maps resulting from different RE cocktails can be combined to improve resolution (Ginno et al., 2013). A technique using 4 bp cutters has been developed to improve the resolution of DRIP-seq and may achieve strand-specific mapping (Xu et al., 2017; Yang et al., 2019), although the resulting datasets have not yet been systematically compared to other human datasets. It is important to note that in RE-based approaches, larger fragments tend to be recovered more efficiently because they can carry multiple R-loop forming regions. This bias must be taken into account when analyzing DRIP-seq datasets. Similarly, peak calling for DRIP-seq data must be ultimately translated into R-loop-positive RE fragments, since it is these fragments that are immunoprecipitated and the position of R-loops within these fragments can’t be inferred. In general, we recommend that users first adopt RE-based DRIP-seq to learn the method and build their confidence in achieving the yields documented in Figure 2A. sDRIP-seq typically results in lower yields, which could result in maps with lower signal to noise ratios in untrained hands. The use of sonication as a means of fragmenting the genome offers in return a great improvement in resolution since the non-R-looped portions that typically constitute the majority of RE fragments will be broken off, allowing S9.6 to principally retrieve the R-looped portions (Figure 1). The additional benefit of strand-specificity provides numerous further benefits to the understanding of R-loop formation mechanisms, making sDRIP-seq a method of choice for the study of R-loops.
Importantly, maps obtained via DRIP-seq and sDRIP-seq represent the average distribution of R-loops through a cell population; thus, the length and position of individual R-loops cannot be addressed with those techniques. For this, an independent and complementary method termed single-molecule R-loop footprinting (SMRF-seq) (Malig et al., 2020) can be leveraged to reveal individual R-loops at high-resolution in a strand-specific manner. Assessment of R-loop formation using SMRF-seq over 20 different loci, including independently of S9.6, revealed a strong agreement between collection of individual R-loop footprints and the population average distribution gather by DRIP-based approaches (Malig et al., 2020), lending strong support to DRIP-based approaches. It is also important to consider that R-loop mapping data only provides a snapshot of R-loop genomic distribution and does not provide information on the dynamics of R-loop formation, stability, and resolution. DRIP approaches, combined with specific drug treatments and an evaluation of R-loop distributions through time series, can nonetheless be deployed to address these parameters (Crossley et al., 2020; Sanz et al., 2016). The limitations of R-loop profiling methodologies are particularly important to keep in mind when the goal is to characterize altered R-loop distributions in response to a genetic, environmental, or pharmacological perturbations. In addition to those already described above, it is key to consider any possible change to nascent transcription since these will inherently cause R-loop changes owing to the co-transcriptional nature of these structures. These issues and guidelines for developing rigorous R-loop mapping approaches have been extensively discussed (Chedin et al., 2021; Vanoosthuyse, 2018) and readers are encouraged to refer to these studies.
Materials
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
|---|---|---|---|
| S9.6 Antibody | Kerafast | ENH001 | These three sources are equivalent |
| S9.6 Antibody | Millipore/ Sigma | MABE1095 | |
| S9.6 Antibody | Abcam | ab234957 | |
| 15 mL tube High density Maxtract phase lock gel | Qiagen | 129065 | |
| 2 mL tube phase lock gel light | VWR | 10847-800 | |
| Phenol/Chloroform Isoamyl alcohol 25:24:1 | Affymetrix | 75831-400ML | |
| Agarose A/G beads | ThermoFisher Scientific | 20421 | |
| Ribonuclease H | New England BioLabs | M0297S | |
| Agencourt AMPure XP beads | Beckman Coulter | A63881 | |
| Klenow fragment (3’ to 5’ exo-) | New England BioLabs | M0212S | |
| NEBNext End repair module | New England BioLabs | E6050 | |
| Quick Ligation Kit | New England BioLabs | M2200S | |
| AmpErase Uracil N-glycosylase | ThermoFisher Scientific | N8080096 | |
| Phusion Flash High-Fidelity PCR master mix | ThermoFisher Scientific | F548S | |
| Index adapters | Illumina | Corresponds to the TrueSeq Single indexes | |
| PCR primers for library amplification | primer 1.0 P5 | ||
| (5’ AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGA 3’) | |||
| PCR primers for library amplification | PCR primer 2.0 P7 | ||
| (5’ CAAGCAGAAGACGGCATACGAGAT 3’) |
Acknowledgments
Work in the Chedin lab is supported by a grant from the National Institutes of Health (R01 GM120607).
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/62455.
Disclosure.
The authors declare no conflict of interest.
References
- Alecki C, Chiwara V, Sanz LA, Grau D, Arias Perez O, Boulier EL, Armache KJ, Chedin F, and Francis NJ (2020). RNA-DNA strand exchange by the Drosophila Polycomb complex PRC2. Nature communications 11, 1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguslawski SJ, Smith DE, Michalak MA, Mickelson KE, Yehle CO, Patterson WL, and Carrico RJ (1986). Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J Immunol Methods 89, 123–130. [DOI] [PubMed] [Google Scholar]
- Cadoret JC, Meisch F, Hassan-Zadeh V, Luyten I, Guillet C, Duret L, Quesneville H, and Prioleau MN (2008). Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc Natl Acad Sci U S A 105, 15837–15842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carles-Kinch K, and Kreuzer KN (1997). RNA-DNA hybrid formation at a bacteriophage T4 replication origin. J Mol Biol 266, 915–926. [DOI] [PubMed] [Google Scholar]
- Carrasco-Salas Y, Malapert A, Sulthana S, Molcrette B, Chazot-Franguiadakis L, Bernard P, Chedin F, Faivre-Moskalenko C, and Vanoosthuyse V (2019). The extruded non-template strand determines the architecture of R-loops. Nucleic acids research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chedin F (2016). Nascent Connections: R-Loops and Chromatin Patterning. Trends in genetics : TIG 32, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chedin F, and Benham CJ (2020). Emerging roles for R-loop structures in the management of topological stress. J Biol Chem 295, 4684–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chedin F, Hartono SR, Sanz LA, and Vanoosthuyse V (2021). Best practices for the visualization, mapping, and manipulation of R-loops. EMBO J, e106394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Zhang X, Fu XD, and Chen L (2019). R-ChIP for genome-wide mapping of R-loops by using catalytically inactive RNASEH1. Nat Protoc 14, 1661–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, and Koshland D (2015). The Yin and Yang of R-loop biology. Current opinion in cell biology 34, 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley MP, Bocek M, and Cimprich KA (2019). R-Loops as Cellular Regulators and Genomic Threats. Molecular cell 73, 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley MP, Bocek MJ, Hamperl S, Swigut T, and Cimprich KA (2020). qDRIP: a method to quantitatively assess RNA-DNA hybrid formation genome-wide. Nucleic acids research 48, e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels GA, and Lieber MR (1995). RNA:DNA complex formation upon transcription of immunoglobulin switch regions: implications for the mechanism and regulation of class switch recombination. Nucleic acids research 23, 5006–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drolet M, Broccoli S, Rallu F, Hraiky C, Fortin C, Masse E, and Baaklini I (2003). The problem of hypernegative supercoiling and R-loop formation in transcription. Front Biosci 8, d210–221. [DOI] [PubMed] [Google Scholar]
- Duquette ML, Handa P, Vincent JA, Taylor AF, and Maizels N (2004). Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev 18, 1618–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hage A, Webb S, Kerr A, and Tollervey D (2014). Genome-wide distribution of RNA-DNA hybrids identifies RNase H targets in tRNA genes, retrotransposons and mitochondria. PLoS genetics 10, e1004716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Muse T, and Aguilera A (2019). R Loops: From Physiological to Pathological Roles. Cell 179, 604–618. [DOI] [PubMed] [Google Scholar]
- Ginno PA, Lim YW, Lott PL, Korf I, and Chedin F (2013). GC skew at the 5’ and 3’ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res 23, 1590–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, and Chedin F (2012). R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Molecular cell 45, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green P, Ewing B, Miller W, Thomas PJ, and Green ED (2003). Transcription-associated mutational asymmetry in mammalian evolution. Nature genetics 33, 514–517. [DOI] [PubMed] [Google Scholar]
- Halasz L, Karanyi Z, Boros-Olah B, Kuik-Rozsa T, Sipos E, Nagy E, Mosolygo LA, Mazlo A, Rajnavolgyi E, Halmos G, et al. (2017). RNA-DNA hybrid (R-loop) immunoprecipitation mapping: an analytical workflow to evaluate inherent biases. Genome Res 27, 1063–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartono SR, Korf IF, and Chedin F (2015). GC skew is a conserved property of unmethylated CpG island promoters across vertebrates. Nucleic acids research 43, 9729–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartono SR, Malapert A, Legros P, Bernard P, Chedin F, and Vanoosthuyse V (2018). The Affinity of the S9.6 Antibody for Double-Stranded RNAs Impacts the Accurate Mapping of R-Loops in Fission Yeast. J Mol Biol 430, 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FT, Yu K, Balter BB, Selsing E, Oruc Z, Khamlichi AA, Hsieh CL, and Lieber MR (2007). Sequence dependence of chromosomal R-loops at the immunoglobulin heavy-chain Smu class switch region. Molecular and cellular biology 27, 5921–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FT, Yu K, Hsieh CL, and Lieber MR (2006). Downstream boundary of chromosomal R-loops at murine switch regions: implications for the mechanism of class switch recombination. Proc Natl Acad Sci U S A 103, 5030–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert JL (2008). Thermodynamic prediction of RNA-DNA duplex-forming regions in the human genome. Mol Biosyst 4, 686–691. [DOI] [PubMed] [Google Scholar]
- Itoh T, and Tomizawa J (1980). Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proc Natl Acad Sci U S A 77, 2450–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzer KN, and Brister JR (2010). Initiation of bacteriophage T4 DNA replication and replication fork dynamics: a review in the Virology Journal series on bacteriophage T4 and its relatives. Virology journal 7, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, and Clayton DA (1998). Initiation of mitochondrial DNA replication by transcription and R-loop processing. J Biol Chem 273, 30614–30621. [DOI] [PubMed] [Google Scholar]
- Malig M, Hartono SR, Giafaglione JM, Sanz LA, and Chedin F (2020). Ultra-deep Coverage Single-molecule R-loop Footprinting Reveals Principles of R-loop Formation. J Mol Biol 432, 2271–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masse E, Phoenix P, and Drolet M (1997). DNA topoisomerases regulate R-loop formation during transcription of the rrnB operon in Escherichia coli. J Biol Chem 272, 12816–12823. [DOI] [PubMed] [Google Scholar]
- Masukata H, and Tomizawa J (1990). A mechanism of formation of a persistent hybrid between elongating RNA and template DNA. Cell 62, 331–338. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay R, Lajugie J, Fourel N, Selzer A, Schizas M, Bartholdy B, Mar J, Lin CM, Martin MM, Ryan M, et al. (2014). Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS genetics 10, e1004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips DD, Garboczi DN, Singh K, Hu Z, Leppla SH, and Leysath CE (2013). The sub-nanomolar binding of DNA-RNA hybrids by the single-chain Fv fragment of antibody S9.6. J Mol Recognit 26, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Cadoret JC, Audit B, Arneodo A, Alberti A, Battail C, Duret L, and Prioleau MN (2014). The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLoS genetics 10, e1004282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, and Arndt PF (2008). Transcription induces strand-specific mutations at the 5’ end of human genes. Genome Res 18, 1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaban ME, Lebowitz J, and Griffin JA (1994). Transcription induces the formation of a stable RNA.DNA hybrid in the immunoglobulin alpha switch region. J Biol Chem 269, 21850–21857. [PubMed] [Google Scholar]
- Santos-Pereira JM, and Aguilera A (2015). R loops: new modulators of genome dynamics and function. Nature reviews Genetics 16, 583–597. [DOI] [PubMed] [Google Scholar]
- Sanz LA, and Chedin F (2019). High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nat Protoc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, Xu X, and Chedin F (2016). Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Molecular cell 63, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira-Mendes J, Diaz-Uriarte R, Apedaile A, Huntley D, Brockdorff N, and Gomez M (2009). Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS genetics 5, e1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti-Stathaki K, and Proudfoot NJ (2014). A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev 28, 1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti-Stathaki K, Proudfoot NJ, and Gromak N (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Molecular cell 42, 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolka JA, Sanz LA, Hartono SR, and Chedin F (2021). Recognition of RNAs by the S9.6 antibody creates pervasive artefacts when imaging RNA:DNA hybrids. Journal of Cell Biology in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz R, Sulthana S, Hartono SR, Malig M, Benham CJ, and Chedin F (2019). Interplay between DNA sequence and negative superhelicity drives R-loop structures. Proc Natl Acad Sci U S A 116, 6260–6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuckey R, Garcia-Rodriguez N, Aguilera A, and Wellinger RE (2015). Role for RNA:DNA hybrids in origin-independent replication priming in a eukaryotic system. Proc Natl Acad Sci U S A 112, 5779–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svikovic S, Crisp A, Tan-Wong SM, Guilliam TA, Doherty AJ, Proudfoot NJ, Guilbaud G, and Sale JE (2019). R-loop formation during S phase is restricted by PrimPol-mediated repriming. EMBO J 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoosthuyse V (2018). Strengths and Weaknesses of the Current Strategies to Map and Characterize R-Loops. Noncoding RNA 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Costantino L, Tan FJ, Zimmer A, and Koshland D (2016). S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev 30, 1327–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Wang H, Li C, Yin Z, Xiao R, Li Q, Xiang Y, Wang W, Huang J, Chen L, et al. (2021). Genomic profiling of native R loops with a DNA-RNA hybrid recognition sensor. Sci Adv 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, and Clayton DA (1995). A persistent RNA-DNA hybrid is formed during transcription at a phylogenetically conserved mitochondrial DNA sequence. Molecular and cellular biology 15, 580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Xu H, Li K, Fan Y, Liu Y, Yang X, and Sun Q (2017). The R-loop is a common chromatin feature of the Arabidopsis genome. Nat Plants 3, 704–714. [DOI] [PubMed] [Google Scholar]
- Yan Q, and Sarma K (2020). MapR: A Method for Identifying Native R-Loops Genome Wide. Curr Protoc Mol Biol 130, e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Liu QL, Xu W, Zhang YC, Yang Y, Ju LF, Chen J, Chen YS, Li K, Ren J, et al. (2019). m(6)A promotes R-loop formation to facilitate transcription termination. Cell Res 29, 1035–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Chedin F, Hsieh CL, Wilson TE, and Lieber MR (2003). R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol 4, 442–451. [DOI] [PubMed] [Google Scholar]
- Yu K, and Lieber MR (2019). Current insights into the mechanism of mammalian immunoglobulin class switch recombination. Crit Rev Biochem Mol Biol 54, 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]