

Abstract
Alcohols and carboxylic acids are among the most commercially abundant, synthetically versatile, and operationally convenient functional groups in organic chemistry. Under visible light photoredox catalysis, these native synthetic handles readily undergo radical activation, and the resulting open-shell intermediates can subsequently participate in transition metal catalysis. In this report, we describe the C(sp3)–C(sp3) cross-coupling of alcohols and carboxylic acids through the dual combination of N-heterocyclic carbene (NHC)-mediated deoxygenation and hypervalent iodine-mediated decarboxylation. This mild and practical Nicatalyzed radical-coupling protocol was employed to prepare a wide array of alkyl–alkyl cross-coupled products, including highly congested quaternary carbon centers from the corresponding tertiary alcohols or tertiary carboxylic acids. We demonstrate the synthetic applications of this methodology to alcohol C1-alkylation and formal homologation, as well as to the late-stage functionalization of drugs, natural products, and biomolecules.
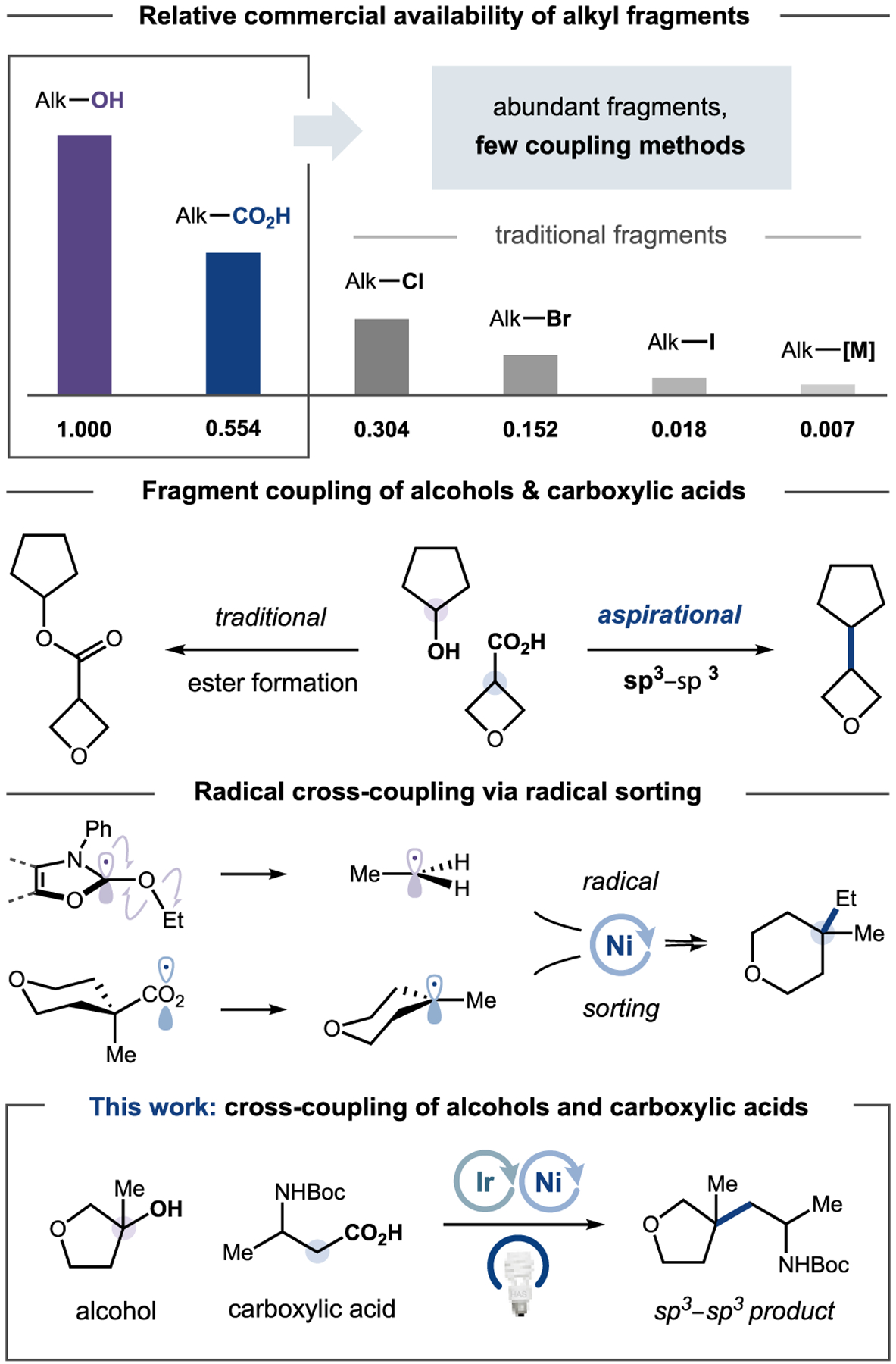
Alcohols and carboxylic acids are ubiquitous, native functional groups with unparalleled structural diversity, wide-ranging synthetic applicability, and broad representation among both natural and commercial sources.1,2 These two structural motifs are most commonly coupled via the venerable esterification reaction, reported in its first iteration by Fischer and Speier over 125 years ago.3 The widespread adoption of this disconnection can be attributed at least in part to the desirability of alcohols and carboxylic acids as highly abundant organic fragments. By contrast, the direct coupling of alcohols and carboxylic acids to forge new C(sp3)–C(sp3) bonds has remained an appealing yet elusive goal.4 Recently, our group has harnessed both carboxylic acids and alcohols as alkylating agents in visible-light-driven processes.5 We questioned whether these activation modes could be combined in a unified metallaphotoredox strategy that could achieve the longstanding goal of alcohol–carboxylic acid C(sp3)–C(sp3) cross-coupling.6 This technology would leverage the versatility, stability, and convenience of alcohols and carboxylic acids, thus offering a modern, orthogonal approach to well-known esterification protocols.
In recent years, metallaphotoredox catalysis has transformed organic synthesis by enabling the activation and subsequent cross-coupling of previously inert alkyl fragments, such as alcohols, carboxylic acids, and C(sp3)–H bonds.7 In particular, alkyl carboxylic acids are highly amenable to light-induced redox activation, participating in a diverse array of transformations, including arylation, alkylation, and amination, among others.5a,8 In a similar fashion, the radical deoxygenative functionalization of alcohols has been achieved through a variety of mechanisms.9–13 These approaches often entail preactivation of the alcohol substrate, requiring additional chemical steps and purifications.14,15 Moreover, the homolytic cleavage event can liberate byproducts that are incompatible with transition metal catalysis.16 To overcome these challenges, our group recently disclosed an alternative technology that leverages an N-heterocyclic carbene (NHC)-based reagent to achieve the deoxyarylation of an extensive array of complex, structurally distinct alcohols.5b The NHC reagent reacts with the alcohol substrate to generate an electron-rich intermediate that is poised to undergo in situ oxidative fragmentation, ejecting an alkyl radical that can be subsequently captured by a metal catalyst.5b
While reports of decarboxylation and deoxygenation have been described in separate contexts, the main challenge for a nontraditional C(sp3)–C(sp3) fragment coupling is ensuring the cross-compatibility of activation modes in combination with a suitable transition metal catalyst. We sought to merge NHC-promoted oxidative radical formation with a reductive strategy for decarboxylation8d to enable a redox-neutral coupling protocol. However, the proposed transformation involves transient generation of two alkyl radicals that must be differentiated in order to achieve efficient cross-coupling.17 As a design principle, we recognized that the relative instability of more highly substituted metal–alkyl species should favor formation of the desired product via catalyst-controlled radical sorting mechanisms.18 Nickel,19 with its well-established ability to efficiently capture and stabilize alkyl radicals, was selected to mediate bond formation. We hypothesized that the nickel catalyst would preferentially bind and stabilize the less-substituted alkyl species in the form of a more persistent metal–alkyl complex,20 directing its cross-coupling with the more highly substituted free radical (vide infra). If successful, the ability to directly couple two of the most abundant and versatile alkyl sources—alcohols and carboxylic acids—would permit broad combinatorial access to sp3-rich products in a single-step process,21 thereby facilitating a practical synthesis of aliphatic motifs encompassing an expansive region of chemical space (Figure 1).22
Figure 1.
Cross-coupling of alcohols and carboxylic acids.
We envisioned achieving dual nickel/photoredox-catalyzed cross-coupling of alcohols and carboxylic acids via the design plan depicted in Figure 2. First, carboxylic acid 1 is premixed with iodomesitylene diacetate to afford the activated iodonium dicarboxylate 2, which can be prepared directly on a rotary evaporator without additional purification.8d,23 The alcohol substrate 3 condenses with benzoxazolium salt NHC-1 to form the activated NHC–alcohol adduct (4) under mildly basic conditions.24 Visible-light excitation of the photocatalyst [Ir(dF(Me)ppy)2(dtbbpy)]PF6 (5) [dF(Me)ppy = 2-(2,4-difluorophenyl)-5-(methyl)-pyridinyl; dtbbpy = 4,4′-bis(tert-butyl)-2,2′-bipyridine] generates a long-lived, oxidizing triplet excited state (6, τ = 1.2 μs, E1/2red [*Ir(III)/Ir(II)] = +0.77 V vs saturated calomel electrode (SCE) in MeCN).25 The excited state complex 6 can readily undergo reductive quenching by 4 (in preference to oxidative quenching by 2; see Figures S8 and S15 for emission quenching studies) via single-electron transfer (SET) to provide the reduced Ir(II) photocatalyst 7. Rapid deprotonation of the transient amine radical cation26 generates a carbon-centered radical adjacent to three heteroatoms (8). At this stage, subsequent β-scission27,28 (ΔG‡ < 12 kcal/mol by density functional theory; see Table S17) liberates an aromatized byproduct29 (9) and alkyl radical 10, which can be rapidly trapped by the nickel catalyst 12 to form Ni–alkyl intermediate 13. Concurrently, reduction of the preformed iodonium dicarboxylate (2, Epc = −1.00 V vs SCE in 1:1 DMSO/MTBE) by 7 (E1/2red [Ir(III)/Ir(II)] = −1.25 V vs SCE in in 1:1 DMSO/MTBE) should afford, upon fragmentation and CO2 extrusion, the acid-derived radical 11 along with the regenerated Ir(III) photocatalyst (5). Finally, nickel-catalyzed bond formation30,31 would deliver the desired C(sp3)–C(sp3) coupled product (14) and reconstitute the nickel catalyst 12.
Figure 2.
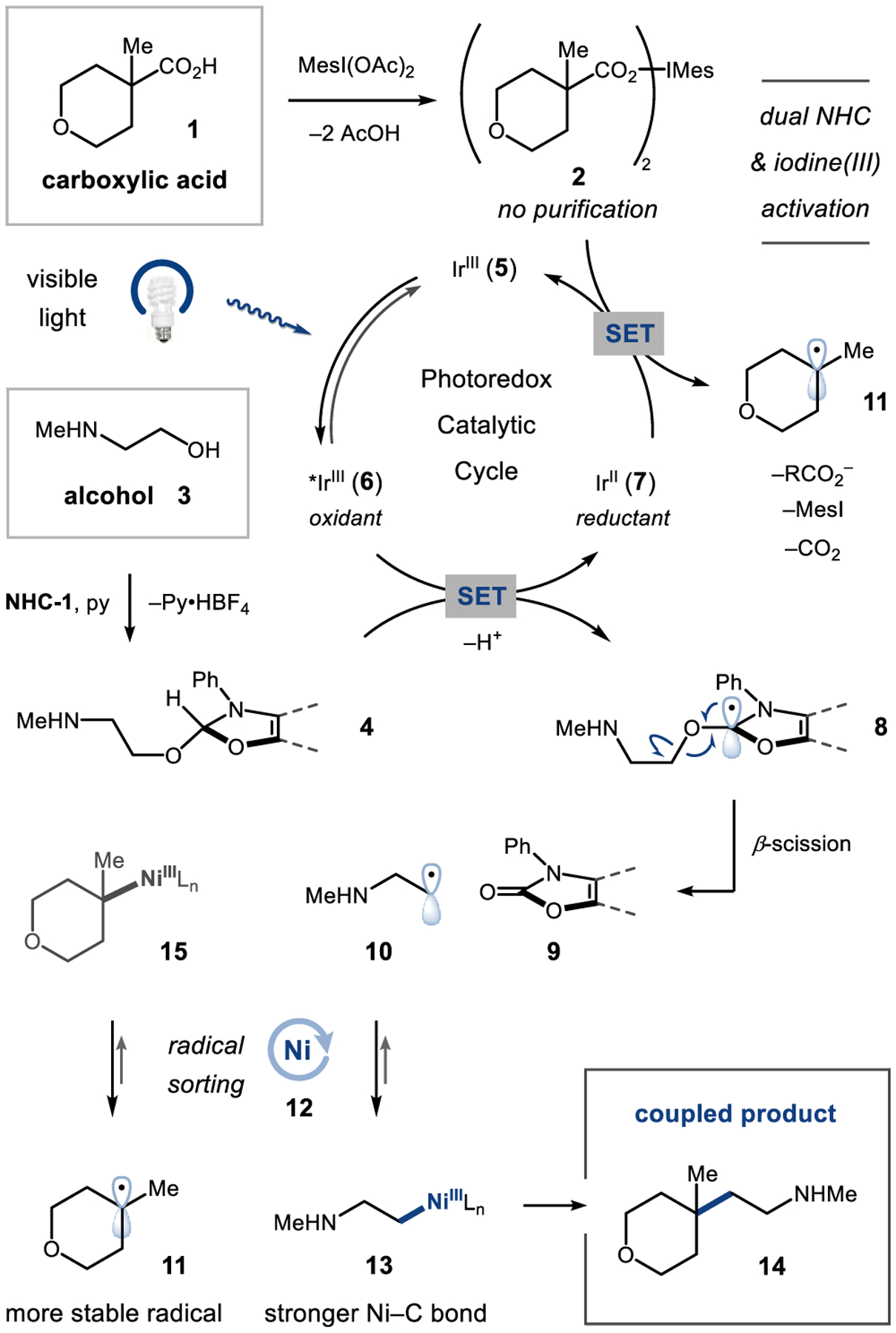
Proposed reaction design.
Although alternative sequences of radical capture and bond formation are possible, we postulated that the nickel catalyst should effectively distinguish between the two radical species and direct their productive cross-coupling as a combined consequence of (i) the differing relative stabilities of alkyl radicals,17 (ii) differences in nickel–carbon bond strengths,20 and (iii) the reversibility of radical capture for hindered alkyl radicals.32 Literature precedent and preliminary computational studies suggest that nickel catalyst 12 should preferentially bind and sequester the less-substituted alkyl radical, 10 (ΔG = −12.4 kcal/mol by DFT; Figure S23), thereby promoting the buildup of the more-substituted radical, 11, in solution. Under steady-state reaction conditions, we postulated that this “radical sorting” mechanism should favor the accumulation of species 11 and 13 (over 10 and 15), from which bond formation would provide the desired cross-coupled product.18
We first explored this idea in the context of the model deoxymethylation shown in Table 1. Following an extensive evaluation of reaction conditions (Tables S1–S10), we ultimately found that the alcohol substrate underwent efficient in situ condensation with NHC-1 (1.10 equiv) and pyridine (1.05 equiv) in MTBE (0.10 M), followed by cross-coupling with iodomesitylene diacetate (2.0 equiv) in the presence of photocatalyst 5 (1 mol %) and nickel catalyst 12 (10 mol %)30 in MTBE/DMSO (1:1, 0.02 M) to afford the desired product in excellent yield (Table 1, 76% yield) after 1 h of visible light irradiation (450 nm) in an integrated photoreactor.33 This protocol permits the direct in situ activation of alcohol substrates, representing a highly practical and exceptionally mild procedure for alkyl cross-couplings. Diminished reaction performance was observed with related nickel salts, such as acetylacetonate- or bipyridine-ligated systems (entries 2 and 3, 73% and 24% yield, respectively). While the commercially available carboxylate precursor MesI(OAc)2 remains optimal under these conditions, the related reagent, phenyliodine(III) diacetate (PIDA),34 can be used with minimal reduction in yield (entry 4, 71% yield). Reduced stoichiometric excess of the carboxylate is well-tolerated (entry 5, 67% yield) and may be desirable for structurally complex or high-value coupling partners. Both the NHC- and iodine(III)-mediated radical generation pathways are exceptionally facile, and the vast majority of product formation occurs in a matter of minutes (entry 6, 67% yield). Control experiments indicate that iridium, light, and nickel are each essential for optimal efficiency of product formation (entries 7–11), although small amounts of cross-coupled product are formed through background radical coupling in the absence of 12 (entry 8).
Table 1.
Control Reactions of Optimized Conditionsa
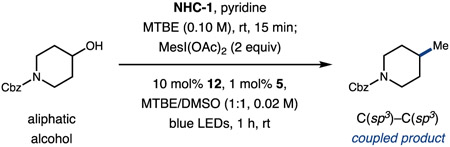
| ||
|---|---|---|
| entry | deviation from above | yieldb |
| 1 | none | 76% |
| 2 | Ni(acac)2 instead of 12 | 73% |
| 3 | NiCl2·dtbbpy instead of 12 | 24% |
| 4 | PhI(OAc)2 instead of MesI(OAc)2 | 71% |
| 5 | 1 equiv of MesI(OAc)2 | 67% |
| 6 | irradiation for 5 min | 67% |
| 7 | no Ir catalyst | <5% |
| 8 | no Ni catalyst | 12% |
| 9 | no Ir catalyst, no light | 0% |
| 10 | no Ir catalyst, no Ni catalyst | 0% |
| 11 | no light, 50 °C | 0% |
Performed with alcohol (0.05 mmol, 1.0 equiv), NHC precursor (1.10 equiv), pyridine (1.05 equiv), and iodomesitylene dicarboxylate (2.0 equiv).
Yields determined by HPLC analysis with acetanilide as internal standard. See Supporting Information for experimental details.
With optimized conditions in hand, we set out to explore the scope of our reaction (Table 2). Using a β-alanine derivative (16) as the carboxylic acid coupling partner, primary, secondary, and tertiary alcohols could be successfully cross-coupled under the reaction conditions.35 Secondary aliphatic alcohols containing saturated scaffolds of pharmaceutical relevance were competent substrates in our protocol, affording alkylated products incorporating pyrrolidine (17, 51% yield), tetrahydropyran (18, 62% yield), piperidine (19 and 20, 62% and 63% yield, respectively), dioxane (21, 78% yield), and azepane (22, 68% yield) motifs.36 Rotationally unconstrained secondary acyclic substrates could also be successfully utilized to access the desired products in good yield (23 and 24, 79% and 73% yield, respectively), and sterically encumbered polycyclic alcohols such as 2-adamantanol and exo-norborneol were employed without appreciable decrease in reaction performance (25 and 26, 47% and 79% yield, respectively). Significant homocoupling is observed in the cross-coupling of two primary radicals, as the nickel catalyst is less able to effectively differentiate between these two active species. Nonetheless, using 2 equiv of the activated acid component, primary aliphatic alcohols could be employed to provide C(sp3)–C(sp3) coupled products in synthetically useful yields, demonstrating tolerance of functional groups such as primary alkyl chlorides (27, 50% yield), as well as ethers and protected amines (28–30, 52–75% yield). Notably, tertiary alcohols underwent successful deoxygenative alkylation to afford products with hindered alkyl quaternary carbon centers—a longstanding challenge in the field of alkyl–alkyl cross-coupling.18,37,38 This protocol was successfully applied to both cyclic and acyclic tertiary alcohols, including tert-butanol, illustrating the power of this method to deliver previously elusive products from readily available starting materials (31–35, 57–75% yield).
Table 2.
Scope of Metallaphotoredox C(sp3)–C(sp3) Cross-Coupling of Carboxylic Acids and Alcoholsa
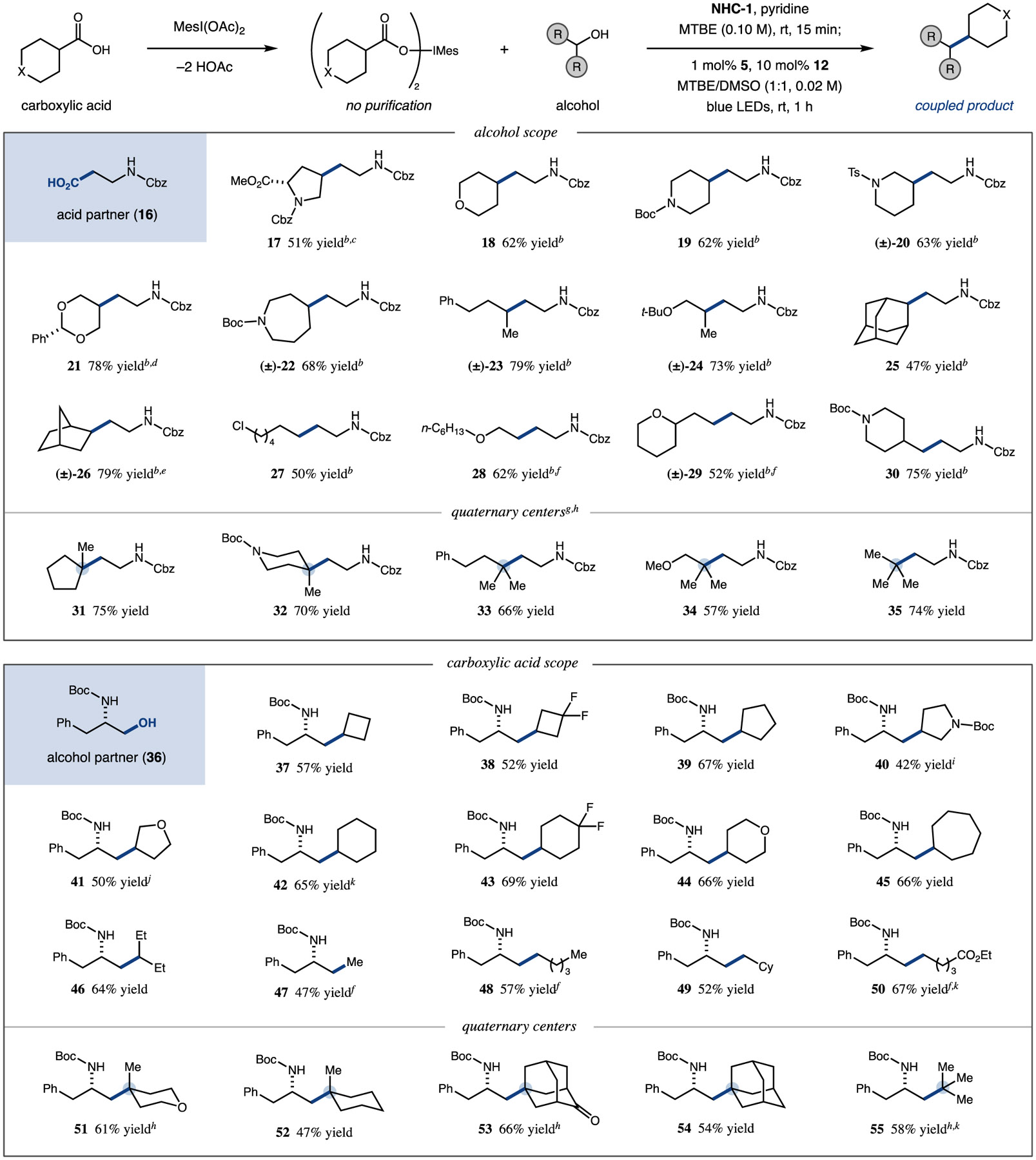
|
Iodonium dicarboxylate formed with MesI(OAc)2 (2 equiv) and carboxylic acid (4 equiv) in toluene (0.05 M) at 55 °C over 10 min. Coupling performed with alcohol substrate (0.50 mmol, 1.0 equiv), NHC-1 (1.10 equiv), and pyridine (1.05 equiv) in MTBE (0.10 M) for 15 min at room temperature, then iridium photocatalyst 5 (1.0 mol %), nickel catalyst 12 (10 mol %), and preformed iodomesitylene dicarboxylate (2.0 equiv, added over 5 min) in MTBE/DMSO (1:1, 0.02 M) with blue LED irradiation for 1 h at 23 °C. Homodimerization of the limiting alcohol substrate is typically 5–10%. Yields are isolated unless otherwise noted. See SI for experimental details.
1.30 equiv of NHC-1, 1.25 equiv of pyridine.
2.7:1 dr.
8:1 dr.
>20:1 dr.
Yield by 1H NMR.
NHC-2 in PhCF3, −20 to 0 °C for 4 h; 2 mol % 5, 20 mol % 12.
1.5 equiv iodomesitylene dicarboxylate.
1.1:1 dr.
1:1 dr.
>99% ee by HPLC.
With respect to the carboxylic acid coupling partner, we selected phenylalanine-derived alcohol substrate 36 to interrogate the performance of a range of primary, secondary, and tertiary alkyl acids under our reaction conditions. An array of secondary carbocyclic substrates performed well using this technology, affording a host of alkyl coupled products. Small ring systems, such as cyclobutane (37 and 38, 57% and 52% yield, respectively), cyclopentane (39, 67% yield), pyrrolidine (40, 42% yield), and tetrahydrofuran (41, 50% yield), were found to be viable coupling partners, as were larger cyclohexane (42 and 43, 65% and 69% yield, respectively), tetrahydrofuran (44, 66% yield), and cycloheptane (45, 66% yield) scaffolds. Commercially available fluoroalkyl moieties could be readily incorporated into these cross-coupled products (38 and 43)—an important objective in the synthesis of medicinal agents where the ability of fluorine to modulate physicochemical properties is well-recognized.39 Secondary acyclic carboxylic acids could be subjected to our reaction conditions, affording alkylated products in good yields (e.g., 46, 64% yield). A range of acetic and primary carboxylic acids underwent successful cross-coupling, including substrates with α-branching and electrophilic groups, such as carboxylate esters (47–50, 47–67% yield). Of note, tertiary carboxylic acids could be effectively utilized for the preparation of fully C(sp3)-substituted quaternary carbon centers, including those arising from monocyclic (51 and 52, 61% and 47% yield, respectively) and polycyclic (53 and 54, 66% and 54% yield, respectively) tertiary acid substrates. The sterically hindered, planarized tert-butyl radical derived from pivalic acid was successfully employed to generate the corresponding product in good yield (55, 58% yield).
To further demonstrate the value of this method, we next sought to deploy our protocol in the context of deoxygenative C1-alkylation.40 Acetic acid derived C1-alkylating reagents bearing isotopic or heteroatom substituents were successfully employed to prepare products with deuteromethyl (56, 78% yield), aminomethyl (57, 52% yield), and aryloxymethyl (58, 63% yield) functionality (Table 3). In addition, using readily available α-hydroxy acids as highly convenient and versatile homologation reagents,41 we accessed alcohol homologation products bearing benzyl (59, 70% yield), acetoxy (60, 58% yield), and p-methoxybenzyl (61, 59% yield) protecting groups. Finally, to illustrate the practical advantages of this technology in the context of late-stage functionalization of drugs and biomolecules, we subjected complex alcohol and acid substrates to our reaction conditions. We were excited to obtain synthetically useful quantities of alkyl coupled products, demonstrating the applicability of this synthetic technology to the late-stage derivatization of drugs, natural products, and biomolecules (Table 4, 62–65, 34–80% yield).
Table 3.
Deoxygenative C1-Alkylationa
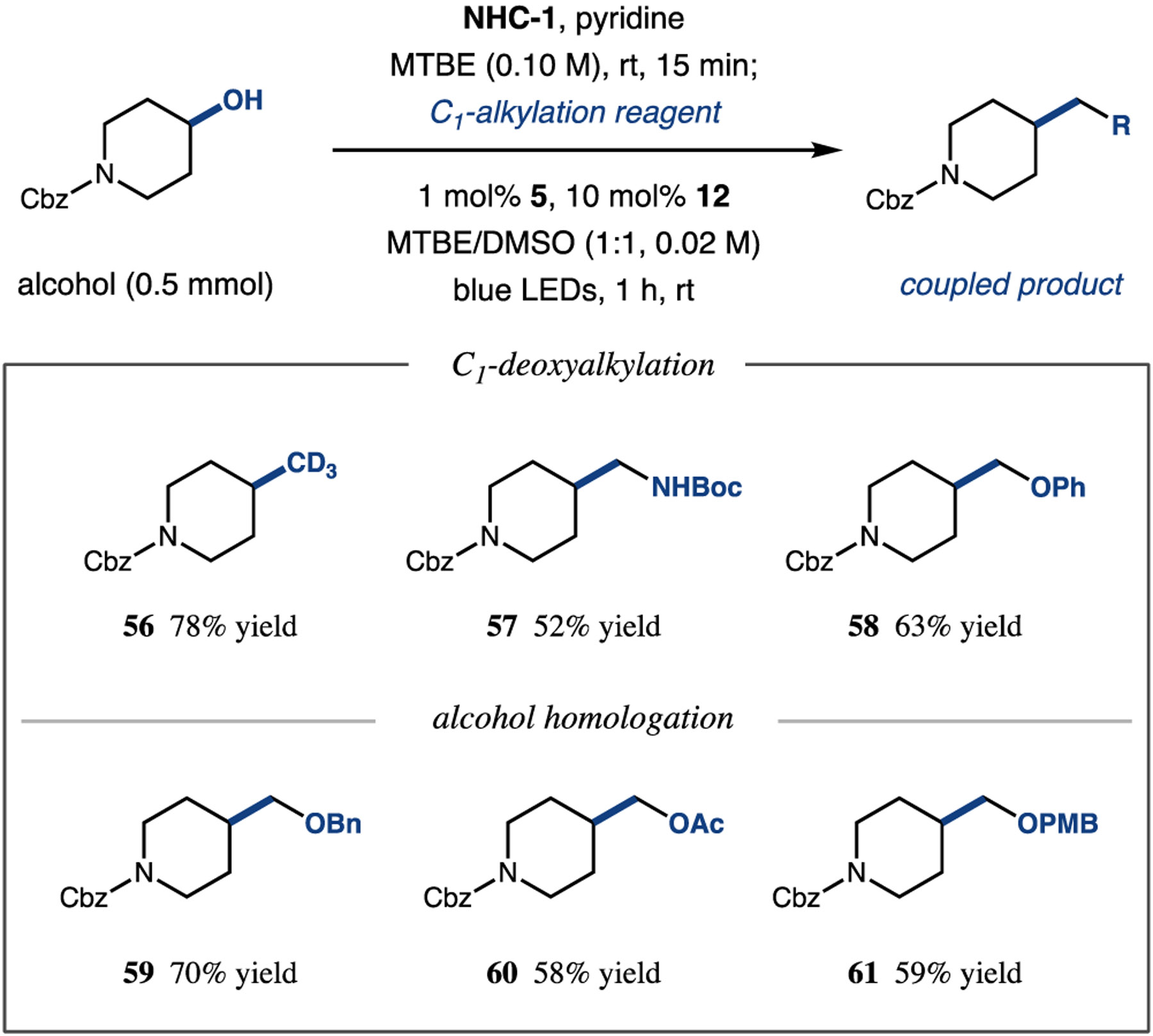
|
See SI for experimental details. All yields are isolated.
Table 4.
Application to Late-Stage Functionalizationa
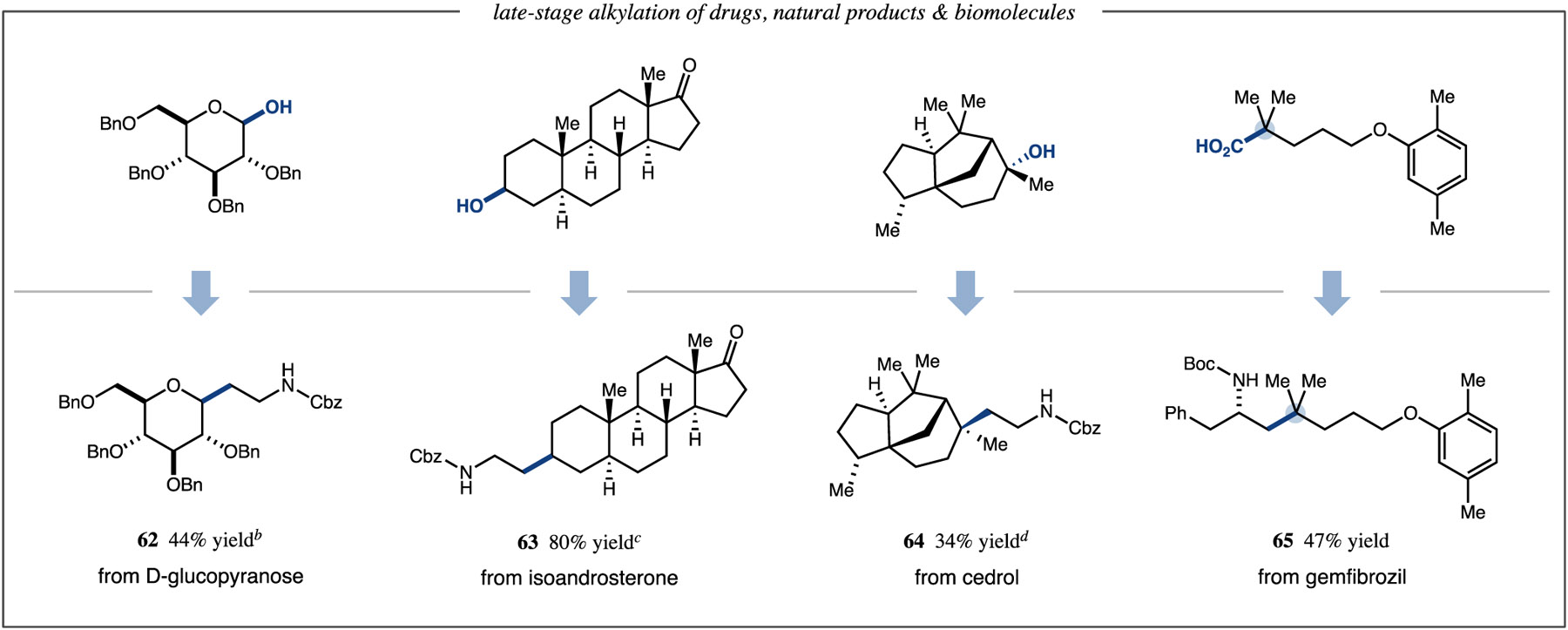
|
In summary, we introduce here the merger of alcohols and carboxylic acids via C(sp3)–C(sp3) cross-coupling as an orthogonal fragment coupling to the traditional esterification reaction. By combining NHC-mediated deoxygenation with hypervalent iodine-mediated decarboxylation, we have successfully developed a dual nickel/photoredox-catalyzed technology applicable to a wide range of aliphatic alcohols and carboxylic acids. We demonstrate the utility of this methodology for quaternary carbon center synthesis, alcohol homologation, and late-stage derivatization. Additional studies probing the nature of the bond formation and its application to new synthetic contexts are underway and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful for financial support provided by the National Institute of General Medical Sciences (NIGMS), the NIH (under Award No. R35GM134897-03), the Princeton Catalysis Initiative, and kind gifts from Merck, Janssen, BMS, Genentech, Celgene, and Pfizer. H.A.S. thanks Princeton University, E. Taylor, and the Taylor family for an Edward C. Taylor Fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIGMS. The authors thank Z. Dong, W. Liu, and V. Bacauanu for helpful scientific discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c02062.
Additional experimental and computational results, characterization data and spectra (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c02062
The authors declare no competing financial interest.
Contributor Information
Holt A. Sakai, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States
David W. C. MacMillan, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States.
REFERENCES
- (1).(a) Ertl P An Algorithm to Identify Functional Groups in Organic Molecules. J. Cheminform 2017, 9, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ertl P; S T A Systematic Cheminformatics Analysis of Functional Groups Occurring in Natural Products. J. Nat. Prod 2019, 82, 1258–1263. [DOI] [PubMed] [Google Scholar]; (c) Henkel T; Brunne RM; Müller H; Reichel F Statistical Investigation into the Structural Complementarity of Natural Products and Synthetic Compounds. Angew. Chem., Int. Ed 1999, 38, 643–647. [DOI] [PubMed] [Google Scholar]
- (2).Reaxys search from January 2022 of commercially available alkyl fragments: alcohols (177,700), carboxylic acids (98,451), alkyl chlorides (54,009), alkyl bromides (26,966), alkyl iodides (3,232), alkyl boronates/zincates/Grignards/lithiums (1,297).
- (3).Esterification is estimated to account for ~10% of all reactions in medicinal chemistry.; (a) Brown DG; Boström J Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem 2016, 59, 4443–4458. [DOI] [PubMed] [Google Scholar]; (b) Fischer E; Speier A Darstellung der Ester. Chem. Ber 1895, 28, 3252–3258. [Google Scholar]
- (4).Choi J; Fu GC Transition Metal–Catalyzed Alkyl-Alkyl Bond Formation: Another Dimension in Cross-Coupling Chemistry. Science 2017, 356, eaaf7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Merging Photoredox with Nickel Catalysis: Coupling of α-Carboxyl sp3-Carbons with Aryl Halides. Science 2014, 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dong Z; MacMillan DWC Metallaphotoredox-Enabled Deoxygenative Arylation of Alcohols. Nature 2021, 598, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For early examples of traditional nucleophile–electrophile sp3–sp3 cross-couplings:; (a) Devasagayaraj A; Stüdemann T; Knochel P A New Nickel-Catalyzed Cross-Coupling Reaction between sp3 Carbon Centers. Angew. Chem., Int. Ed. Engl 1996, 34, 2723–2725. [Google Scholar]; (b) Giovannini R; Stüdemann T; Dussin G; Knochel P An Efficient Nickel-Catalyzed Cross-Coupling Between sp3 Carbon Centers. Angew. Chem., Int. Ed 1998, 37, 2387–2390. [DOI] [PubMed] [Google Scholar]; (c) Terao J; Watanabe H; Ikumi A; Kuniyasu H; Kambe N Nickel-Catalyzed Cross-Coupling Reaction of Grignard Reagents with Alkyl Halides and Tosylates: Remarkable Effect of 1,3-Butadienes. J. Am. Chem. Soc 2002, 124, 4222–4223. [DOI] [PubMed] [Google Scholar]; (d) Zhou J; Fu GC Cross-Couplings of Unactivated Secondary Alkyl Halides: Room-Temperature Nickel-Catalyzed Negishi Reactions of Alkyl Bromides and Iodides. J. Am. Chem. Soc 2003, 125, 14726–14727. [DOI] [PubMed] [Google Scholar]
- (7).(a) Chan AY; Perry IB; Bissonnette NB; Buksh BF; Edwards GA; Frye LI; Garry OL; Lavagnino MN; Li BX; Liang Y; Mao E; Millet A; Oakley JV; Reed NL; Sakai HA; Seath CP; MacMillan DWC Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev 2022, 122, 1485–1542. [DOI] [PubMed] [Google Scholar]; (b) Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Levin MD; Kim S; Toste FD Photoredox Catalysis Unlocks Single-Electron Elementary Steps in Transition Metal Catalyzed Cross-Coupling. ACS Cent. Sci 2016, 2, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Zuo Z; Cong H; Li W; Choi J; Fu GC; MacMillan DWC Enantioselective Decarboxylative Arylation of α -Amino Acids via the Merger of Photoredox and Nickel Catalysis. J. Am. Chem. Soc 2016, 138, 1832–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Johnston CP; Smith RT; Allmendinger S; MacMillan DWC Metallaphotoredox-Catalysed sp3–sp3 Cross-Coupling of Carboxylic Acids with Alkyl Halides. Nature 2016, 536, 322–325. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kautzky JA; Wang T; Evans RW; MacMillan DWC Decarboxylative Trifluoromethylation of Aliphatic Carboxylic Acids. J. Am. Chem. Soc 2018, 140, 6522–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liang Y; Zhang X; MacMillan DWC Decarboxylative sp3 C–N Coupling via Dual Copper and Photoredox Catalysis. Nature 2018, 559, 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhao W; Wurz RP; Peters JC; Fu GC Photoinduced, Copper-Catalyzed Decarboxylative C–N Coupling to Generate Protected Amines: An Alternative to the Curtius Rearrangement. J. Am. Chem. Soc 2017, 139, 12153–12156. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Mao R; Frey A; Balon J; Hu X Decarboxylative C(sp3)–N cross-coupling via synergetic photoredox and copper catalysis. Nat. Catal 2018, 1, 120–126. [Google Scholar]
- (9). References 10–13 refer to radical-based deoxygenation strategies in a noncross-coupling context. See references 14–16 for applications to cross-coupling.
- (10).For Barton–McCombie-type xanthate-based approaches:; (a) Lopez RM; Hays DS; Fu GC Bu3SnH-Catalyzed Barton-McCombie Deoxygenation of Alcohols. J. Am. Chem. Soc 1997, 119, 6949–6950. [Google Scholar]; (b) Zard SZ On the Trail of Xanthates: Some new Chemistry form an Old Functional Group. Angew. Chem., Int. Ed. Engl 1997, 36, 672–685. [Google Scholar]; (c) Togo H; Matsubayashi S; Yamazaki O; Yokoyama M J. Org. Chem 2000, 65, 2816–2819. [DOI] [PubMed] [Google Scholar]; (d) Spiegel DA; Wiberg KB; Schacherer LN; Medeiros MR; Wood JL Deoxygenation of Alcohols Employing Water as the Hydrogen Atom Source. J. Am. Chem. Soc 2005, 127, 12513–12515. [DOI] [PubMed] [Google Scholar]; (e) Chenneberg L; Baralle A; Daniel M; Fensterbank L; Goddard J-P; Ollivier C Visible Light Photocatalytic Reduction of O-Thiocarbamates: Development of a Tin-Free Barton–McCombie Deoxygenation Reaction. Adv. Synth. Catal 2014, 356, 2756–2762. [Google Scholar]; (f) Friese FW; Studer A Deoxygenative Borylation of Secondary and Tertiary Alcohols. Angew. Chem., Int. Ed 2019, 58, 9561–9564. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wu J; Bär RM; Guo L; Noble A; Aggarwal VK Photoinduced Deoxygenative Borylations of Aliphatic Alcohols. Angew. Chem., Int. Ed 2019, 58, 18830–18834. [DOI] [PubMed] [Google Scholar]
- (11).For oxalate-based approaches:; (a) Lackner GL; Quasdorf KW; Overman LE Direct Construction of Quaternary Carbons from Tertiary Alcohols via Photoredox-Catalyzed Fragmentation of tert-Alkyl N-Phthalimidoyl Oxalates. J. Am. Chem. Soc 2013, 135, 15342–15345. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nawrat CC; Jamison CR; Slutskyy Y; MacMillan DWC; Overman LE Oxalates as Activating Groups for Alcohols in Visible Light Photoredox Catalysis: Formation of Quaternary Centers by Redox Neutral Fragment Coupling. J. Am. Chem. Soc 2015, 137, 11270–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For phosphorus-based deoxygenation:; (a) Zhang L; Koreeda M Radical Deoxygenation of Hydroxyl Groups via Phosphites. J. Am. Chem. Soc 2004, 126, 13190–13191. [DOI] [PubMed] [Google Scholar]; (b) Stache EE; Ertel AB; Rovis T; Doyle AG Generation of Phosphoranyl Radicals via Photoredox Catalysis Enables Voltage-Independent Activation of Strong C–O Bonds. ACS Catal. 2018, 8, 11134–11139. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guo H-M; Wu X Selective deoxygenative alkylation of alcohols via photocatalytic domino radical fragmentations. Nat. Commun 2021, 12, 5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).For low-valent titanium-based approaches:; (a) Xie H; Guo J; Wang Y-Q; Wang K; Guo P; Su P-F; Wang X; Shu X-Z Radical Dehydroxylative Alkylation of Tertiary Alcohols by Ti Catalysis. J. Am. Chem. Soc 2020, 142, 16787–16794. [DOI] [PubMed] [Google Scholar]; (b) Xie H; Wang S; Wang Y; Guo P; Shu X-Z Ti-Catalyzed Reductive Dehydroxylative Vinylation of Tertiary Alcohols. ACS Catal. 2022, 12, 1018–1023. [Google Scholar]; (c) Suga T; Takahashi Y; Miki C; Ukaji Y Direct and Unified Access to Carbon Radicals from Aliphatic Alcohols by Cost-Efficient Titanium-Mediated Homolytic C–OH Bond Cleavage. Angew. Chem., Int. Ed 2022, 61, e202112533. [DOI] [PubMed] [Google Scholar]
- (14).For direct or in situ radical activation of stabilized allylic or benzylic alcohols in cross-coupling:; (a) Suga T; Ukaji Y Nickel-Catalyzed Cross-Electrophile Coupling between Benzyl Alcohols and Aryl Halides Assisted by Titanium Co-reductant. Org. Lett 2018, 20, 7846–7850. [DOI] [PubMed] [Google Scholar]; (b) Guo P; Wang K; Jin W-J; Xie H; Qi L; Liu X-Y; Shu X-Z Dynamic Kinetic Cross-Electrophile Arylation of Benzylic Alcohols by Nickel Catalysis. J. Am. Chem. Soc 2021, 143, 513–523. [DOI] [PubMed] [Google Scholar]; (c) Ma W-Y; Han G-Y; Kang S; Pang X; Liu X-Y; Shu X-Z Cobalt-Catalyzed Enantiospecific Dynamic Kinetic Cross-Electrophile Vinylation of Allylic Alcohols with Vinyl Triflates. J. Am. Chem. Soc 2021, 143, 15930–15935. [DOI] [PubMed] [Google Scholar]
- (15).For in situ bromination of alcohols in cross-coupling:; (a) Lin Q; Ma G; Gong H Ni-Catalyzed Formal Cross-Electrophile Coupling of Alcohols with Aryl Halides. ACS Catal. 2021, 11, 14102–14109. [Google Scholar]; (b) Li Z; Sun W; Wang X; Li L; Zhang Y; Li C Electrochemically Enabled, Nickel-Catalyzed Dehydroxylative Cross-Coupling of Alcohols with Aryl Halides. J. Am. Chem. Soc 2021, 143, 3536–3543. [DOI] [PubMed] [Google Scholar]; (c) Chi BJ; Widness JK; Gilbert MM; Salgueiro DC; Garcia KJ; Weix DJ In-Situ Bromination Enables Formal Cross-Electrophile Coupling of Alcohols with Aryl and Alkenyl Halides. ACS Catal. 2022, 12, 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).For redox activation of preactivated alcohols compatible with transition metal catalysis:; (a) Zhang X; MacMillan DWC Alcohols as Latent Coupling Fragments for Metallaphotoredox Catalysis: sp3–sp2 Cross-Coupling of Oxalates with Aryl Halides. J. Am. Chem. Soc 2016, 138, 13862–13865. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vara BA; Patel NR; Molander GA O-Benzyl Xanthate Esters under Ni/Photoredox Dual Catalysis: Selective Radical Generation and Csp3–Csp2 Cross-Coupling. ACS Catal. 2017, 7, 3955–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gao M; Sun D; Gong H Ni-Catalyzed Reductive C–O Bond Arylation of Oxalates Derived from α-Hydroxy Esters with Aryl Halides. Org. Lett 2019, 21, 1645–1648. [DOI] [PubMed] [Google Scholar]; (d) Ye Y; Chen H; Sessler JL; Gong H Zn-Mediated Fragmentation of Tertiary Alkyl Oxalates Enabling Formation of Alkylated and Arylated Quaternary Carbon Centers. J. Am. Chem. Soc 2019, 141, 820–824. [DOI] [PubMed] [Google Scholar]; (e) Mills LR; Monteith JJ; Gomes G; dos Passos Gomes G; Aspuru-Guzik A; Rousseaux SAL The Cyclopropane Ring as a Reporter of Radical Leaving-Group Reactivity for Ni-Catalyzed C(sp3)–O Arylation. J. Am. Chem. Soc 2020, 142, 13246–13254. [DOI] [PubMed] [Google Scholar]; (f) Wei Y; Ben-zvi B; Diao T Diastereoselective Synthesis of Aryl C-Glycosides from Glycosyl Esters via C–O Bond Homolysis. Angew. Chem., Int. Ed 2021, 60, 9433–9438. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Cai A; Yan W; Liu W Aryl Radical Activation of C–O Bonds: Copper-Catalyzed Deoxygenative Difluoromethylation of Alcohols. J. Am. Chem. Soc 2021, 143, 9952–9960. [DOI] [PubMed] [Google Scholar]; (h) Kariofillis SK; Jiang S;Żurański AM; Gandhi SS; Alvarado JIM; Doyle AG Using Data Science To Guide Aryl Bromide Substrate Scope Analysis in a Ni/Photoredox-Catalyzed Cross-Coupling with Acetals as Alcohol-Derived Radical Sources. J. Am. Chem. Soc 2022, 144, 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Leifert D; Studer A The Persistent Radical Effect in Organic Synthesis. Angew. Chem., Int. Ed 2020, 59, 74–108. [DOI] [PubMed] [Google Scholar]; (b) Sun Z; Tang B; Liu KKC; Zhu HY Direct Photochemical Cross-Coupling Between Aliphatic Acids and BF3K Salts. Chem. Commun 2020, 56, 1294–1297. [DOI] [PubMed] [Google Scholar]; (c) Vasilopoulos A; Krska SW; Stahl SS C(sp3)–H Methylation Enabled by Peroxide Photosensitization and Ni-Mediated Radical Coupling. Science 2021, 372, 398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).For a recent conceptualization of catalyst-mediated radical sorting in metallaphotoredox catalysis, see:; Liu W; Lavagnino MN; Gould CA; Alcázar J; MacMillan DWC A Biomimetic SH2 Cross-Coupling Mechanism for Quaternary sp3-Carbon Formation. Science 2021, 374, 1258–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).For a general review on nickel catalysis, see:; Tasker SZ; Standley EA; Jamison TF Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Simões JAM; Beauchamp JL Transition Metal–Hydrogen and Metal–Carbon Bond Strengths: The Keys to Catalysis. Chem. Rev 1990, 90, 629–688. [Google Scholar]
- (21).(a) Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]; (b) Lovering F Escape from Flatland 2: Complexity and Promiscuity. MedChemComm 2013, 4, 515–519. [Google Scholar]
- (22).(a) Burke MD; Schreiber SL A Planning Strategy for Diversity-Oriented Synthesis. Angew. Chem., Int. Ed 2004, 43, 46–58. [DOI] [PubMed] [Google Scholar]; (b) Lipinski C; Hopkins A Navigating Chemical Space for Biology and Medicine. Nature 2004, 432, 855–861. [DOI] [PubMed] [Google Scholar]
- (23).(a) Minisci F; Vismara E; Fontana F; Barbosa MCN A New General Method of Homolytic Alkylation of Protonated Heteroaromatic Bases by Carboxylic Acids and Iodosobenzene Diacetate. Tetrahedron Lett. 1989, 30, 4569–4572. [Google Scholar]; (b) Kiyokawa K; Watanabe T; Fra L; Kojima T; Minakata S Hypervalent Iodine(III)-Mediated Decarboxylative Ritter-Type Amination Leading to the Production of α-Tertiary Amine Derivatives. J. Org. Chem 2017, 82, 11711–11720. [DOI] [PubMed] [Google Scholar]; (c) Zhang X; Smith RT; Le C; McCarver SJ; Shireman BT; Carruthers NI; MacMillan DWC Copper-Mediated Synthesis of Drug-Like Bicyclopentanes. Nature 2020, 580, 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).(a) Coulembier O; Lohmeijer BGG; Dove AP; Pratt RC; Mespouille L; Culkin DA; Benight SJ; Dubois P; Waymouth RM; Hedrick JL Alcohol Adducts of N-Heterocyclic Carbenes: Latent Catalysts for the Thermally-Controlled Living Polymerization of Cyclic Ethers. Macromolecules 2006, 39, 5617–5628. [Google Scholar]; (b) Bellemin-Laponnaz S Synthesis of N,O-Heterocyclic Carbene and Coordination to Rhodium (I) and Copper (I). Polyhedron 2010, 29, 30–33. [Google Scholar]
- (25).(a) Lowry MS; Goldsmith JI; Slinker JD; Rohl R; Pascal RA; Malliaras GG; Bernhard S Single-Layer Electroluminescent Devices and Photoinduced Hydrogen Production from an Ionic Iridium(III) Complex. Chem. Mater 2005, 17, 5712–5719. [Google Scholar]; (b) Ladouceur S; Fortin D; Zysman-Colman E Enhanced Lumniscent Iridium(III) Complexes Bearing Aryltriazole Cyclometallated Ligands. Inorg. Chem 2011, 50, 11514–11526. [DOI] [PubMed] [Google Scholar]
- (26).(a) McNally A; Prier CK; MacMillan DWC Discovery of an α-Amino C–H Arylation Reaction Using the Strategy of Accelerated Serendipity. Science 2011, 334, 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dinnocenzo JP; Banach TE Deprotonation of Tertiary Amine Radical Cations. A Direct Experimental Approach. J. Am. Chem. Soc 1989, 111, 8646–8653. [Google Scholar]
- (27).For selected examples of C–O homolysis via β-scission from nonxanthate precursors:; (a) Kuhn LP; Wellman C Reaction of t-Butyl Peroxide with Acetals. J. Org. Chem 1957, 22, 774–776. [Google Scholar]; (b) Hartzell GE; Huyser ES Generation of Methyl Radicals by Decomposition of Bibenzyl Compounds Containing α-Methoxy Substituents. J. Org. Chem 1964, 29, 3341–3344. [Google Scholar]; (c) Kariofillis SK; Shields BJ; Tekle-Smith MA; Zacuto MJ; Doyle AG Nickel/Photoredox-Catalyzed Methylation of (Hetero)aryl Chlorides Using Trimethyl Orthoformate as a Methyl Radical Source. J. Am. Chem. Soc 2020, 142, 7683–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).For a conceptually distinct alcohol activation involving β-scission from alkoxy radicals in metal-catalyzed cross-coupling:; (a) Huang L; Ji T; Rueping M Remote Nickel-Catalyzed Cross-Coupling Arylation via Proton-Coupled Electron Transfer-Enabled C–C Bond Cleavage. J. Am. Chem. Soc 2020, 142, 3532–3539. [DOI] [PubMed] [Google Scholar]; (b) Cong F; Lv X-Y; Day CS; Martin R Dual catalytic Strategy for Forging sp2–sp3 and sp3–sp3 Architectures via β-scission of Aliphatic Alcohol Derivatives. J. Am. Chem. Soc 2020, 142, 20594–20599. [DOI] [PubMed] [Google Scholar]; (c) Chen Y; Wang X; He X; An Q; Zuo Z Photocatalytic Dehydroxymethylative Arylation by Synergistic Cerium and Nickel Catalysis. J. Am. Chem. Soc 2021, 143, 4896–4902. [DOI] [PubMed] [Google Scholar]
- (29).Bhunia A; Studer A Recent Advances in Radical Chemistry Proceeding Through Pro-Aromatic Radicals. Chem. 2021, 7, 2060–2100. [Google Scholar]
- (30).While inner-sphere reductive elimination remains possible, Molander and Gutierrez have computationally interrogated a related system based on the same catalyst we employ; their results are consistent with an “outer-sphere reductive elimination” to form quaternary carbon centers:; Yuan M; Song Z; Badir SO; Molander GA; Gutierrez O On the Nature of C(sp3)–C(sp2) Bond Formation in Nickel-Catalyzed Tertiary Radical Cross-Couplings: A Case Study of Ni/Photoredox Catalytic Cross-Coupling of Alkyl Radicals and Aryl Halides. J. Am. Chem. Soc 2020, 142, 7225–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Under sufficiently oxidizing conditions, Sanford has also invoked SH2-type reductive elimination from high-valent nickel complexes:; Bour JR; Ferguson DM; McClain EJ; Kampf JW; Sanford MS Connecting Organometallic Ni(III) and Ni(IV): Reactions of Carbon Centered Radicals with High-Valent Organonickel Complexes. J. Am. Chem. Soc 2019, 141, 8914–8920. [DOI] [PubMed] [Google Scholar]
- (32).Homolytic equilibria with reversible radical capture at nickel–dialkyl species has been previously invoked in; Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Le CC; Wismer MK; Shi Z-C; Zhang R; Conway DV; Li G; Vachal P; Davies I; MacMillan DWC A General Small-Scale Reactor to Enable Standardization and Acceleration of Photocatalytic Reactions. ACS Cent. Sci 2017, 3, 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).(a) Yoshimura A; Zhdankin VV Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev 2016, 116, 3328–3435. [DOI] [PubMed] [Google Scholar]; (b) Zhdankin VV; Stang PJ Chemistry of Polyvalent Iodine. Chem. Rev 2008, 108, 5299–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35). β-amino substrates (16 and 36) display excellent reactivity as coupling partners in this transformation; we hypothesize that the amine (or its protecting group) can support recognition by nickel and stabilize the resulting Ni–alkyl intermediate via chelation. This stabilization may also help the catalyst to differentiate the β-amino radical from other alkyl radicals, particularly in primary–primary combinations, allowing for improved selectivity. However, we note that the N–H moiety is not required for efficient cross-coupling, as indicated by examples in Tables 1 and S14.
- (36).(a) Taylor RD; MacCoss M; Lawson ADG Rings in Drugs. J. Med. Chem 2014, 57, 5845–5859. [DOI] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (37).For general reviews:; (a) Quasdorf KW; Overman LE Catalytic Enantioselective Synthesis of Quaternary Carbon Stereocenters. Nature 2014, 516, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Talele TT Opportunities for Tapping into Three-Dimensional Chemical Space through a Quaternary Carbon. J. Med. Chem 2020, 63, 13291–13315. [DOI] [PubMed] [Google Scholar]
- (38).For select examples of all-aliphatic quaternary carbon center formation via cross-coupling:; (a) Tsuji T; Yorimitsu H; Oshima K Cobalt-Catalyzed Coupling Reaction of Alkyl Halides with Allylic Grignard Reagents. Angew. Chem., Int. Ed 2002, 41, 4137–4139. [DOI] [PubMed] [Google Scholar]; (b) Yang C-T; Zhang Z-Q; Liang J; Liu J-H; Lu X-Y; Chen H-H; Liu L Copper-Catalyzed Cross-Coupling of Nonactivated Secondary Alkyl Halides and Tosylates with Secondary Alkyl Grignard Reagents. J. Am. Chem. Soc 2012, 134, 11124–11127. [DOI] [PubMed] [Google Scholar]; (c) Iwasaki T; Takagawa H; Singh SP; Kuniyasu H; Kambe N Co-Catalyzed Cross-Coupling of Alkyl Halides with Tertiary Alkyl Grignard Reagents Using a 1,3-Butadiene Additive. J. Am. Chem. Soc 2013, 135, 9604–9607. [DOI] [PubMed] [Google Scholar]; (d) Green SA; Huffman TR; McCourt RO; van der Puyl V; Shenvi RA Hydroalkylation of Olefins to Form Quaternary Carbons. J. Am. Chem. Soc 2019, 141, 7709–7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).(a) Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]; (b) Liang T; Neumann CN; Ritter T Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem., Int. Ed 2013, 52, 8214–8264. [DOI] [PubMed] [Google Scholar]
- (40).Incorporation of small alkyl substituents, in particular methyl groups, is of significant interest in medicinal chemistry:; Schönherr H; Cernak T Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions. Angew. Chem., Int. Ed 2013, 52, 12256–12267. [DOI] [PubMed] [Google Scholar]
- (41).Alcohol homologation has been historically achieved using syngas and Co- or Ru-catalysis under forcing conditions:; (a) Wender I; Levine R; Orchin M Homologation of Alcohols. J. Am. Chem. Soc 1949, 71, 4160–4161. [Google Scholar]; (b) Andrianary P; Jenner G; Libs S; Teller G Homogeneous Catalysis of CO–H2 Reactions: Homologation of C3 Alcohols. J. Mol. Catal 1987, 39, 93–103. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.