

Abstract
Deoxy-functionalization of alcohols represents a class of reactions that has had a profound impact on modern medicine. In particular, deoxyfluorination is commonly employed as a means to incorporate high-value fluorine atoms into drug-like molecules. Recently, the trifluoromethyl (CF3) group has garnered attention from medicinal chemists due to its ability to markedly improve the pharmaceutical properties of small-molecule drug candidates. To date, however, there remains no general means to accomplish the analogous deoxygenative trifluoromethylation of alcohols. We report herein a copper metallaphotoredox-mediated direct deoxytrifluoromethylation, wherein alcohol substrates are activated in situ by benzoxazolium salts for C(sp3)–CF3 bond formation.
The structural topology of drug candidates is inextricably linked to the state of the art of chemical synthesis.1–4 Consequently, novel synthetic methods that provide access to underexplored chemical space can enable the discovery of breakthrough therapeutics.5,6 Nowhere is this relationship more apparent than the case of fluorine in drug discovery.
While the benefit of fluoroalkyl groups in medicinal chemistry has long been understood,7 it was not until robust synthetic methods for the construction of C–F bonds emerged that these motifs were viewed as feasible synthetic targets. Indeed, a stark increase in the number of fluorinated FDA-approved drugs occurred in the years following the first disclosure of deoxyfluorination reagents such as diethylamino-sulfur trifluoride (DAST) (Figure 1a).8
Figure 1.

Deoxytrifluoromethylation of alcohols.
In recent years, the trifluoromethyl group has become one of the most widely utilized fluoroalkyl groups in drug discovery, due to its ability to increase drug potency and oral bioavailability while decreasing the rate of oxidative clearance.9,10 In 2020 alone, nearly 10% of top selling small-molecule drugs contained at least one trifluoromethyl group. However, only 3% of these top selling drugs contain an aliphatic trifluoromethyl group [C(sp3)–CF3].11 This discrepancy represents an opportunity for the development of novel C(sp3)–CF3 bond forming reactions.
Among limited examples to date, copper has emerged as the metal of choice for catalytic C(sp3)–CF3 bond formation. Facile reductive elimination from formal Cu(III) centers has enabled significant advancements in the trifluoromethylation of aliphatic radical precursors such as carboxylic acids,12,13 alkyl bromides,14,15 alkyl iodides,16 xanthate esters,17,18 and C–H bonds.19–21 At present, however, the largest reservoir of aliphatic building blocks—the alcohol—remains underutilized for the construction of C(sp3)–CF3 bonds.
Alcohols are among the most abundant sources of functional C(sp3) carbon atoms (Figure 1b).22–25 Chemical transformations, such as deoxyfluorination, that make use of this feedstock material have already proven critical to the treatment of human disease (vida supra), making alcohols the ideal precursors for aliphatic trifluoromethyl groups. By analogy, we anticipate that such a deoxytrifluoromethylation reaction would enable unprecedented access to fluorinated organic frameworks that are of paramount importance to global health. To this end, two pioneering methods have been developed. However, both protocols require activating the alcohols as xanthate esters in a separate synthetic step and either employ expensive CF3 sources17 or are limited in scope.18 Consequently, there remains no general method for the direct conversion of native alcohols to aliphatic trifluoromethyl groups.26
Our group recently reported that N-heterocyclic carbene (NHC) precursors can condense with alcohols under mild conditions to form adducts susceptible to metallaphotoredox activation without any purification or workup. This discovery led to the development of a robust nickel-mediated deoxyarylation protocol.27 Given the diversity of alcohol chemical matter, and the importance of trifluoromethyl groups in medicinal chemistry, we recognized an opportunity to exploit this activation mode for the deoxytrifluoromethylation of alcohols via copper metallaphotoredox catalysis (Figure 1c).
Our mechanistic design is detailed in Figure 2. We envisioned that aliphatic alcohol 1 would first be activated in situ by condensation with benzoxazolium salt 2, forming NHC–alcohol adduct 3. Excitation of photocatalyst 4 [Ir(dF(OMe)ppy)2(5,5′(CF3)bpy)PF6] by blue light is known to produce a highly oxidizing excited state (5, E1/2red[*IrIII/IrII] = +1.60 V vs SCE in MeCN)14 that could be quenched by 3 via single-electron transfer (SET). Subsequent deprotonation of the now acidified methine C–H (pKa ~10)27 would provide α-amino radical 7, which can undergo exothermic β-scission28 of the alcohol C–O bond to afford alkyl radical 9 and inert byproduct 8. Concurrently, formal reduction of electrophilic CF3 source 10 in the presence of Cu(I) is known to give rise to Cu(II)–CF3 species19 12, capable of trapping the newly generated alkyl radical 9 at near diffusion controlled rates.29 Reductive elimination from the resulting putative alkyl–Cu(III)–CF3 complex 13 would furnish the desired aliphatic trifluoromethylated product 14.30
Figure 2.

Plausible mechanism for deoxytrifluoromethylation.
Following an extensive optimization campaign, we identified the conditions outlined in Table 1 as optimal. Alcohol 15 was condensed with NHC salt 2 under mildly basic conditions, then subjected to irradiation with blue light, along with 1 mol % photocatalyst 4, 5 mol % Cu(terpy)Cl2 (17), 1.5 equiv of dMesSCF3 (10), 1.6 equiv of quinuclidine, and 2 equiv of tetrabutylammonium chloride (TBACl) in DMSO. After 8 h, the trifluoromethylated product 16 was obtained in 84% yield. The presence of exogenous chloride anion (Cl−) proved critical to the overall success of this transformation. Control experiments revealed that this effect is unique to soluble chloride sources and not general for other X-type ligands (see the SI for details). Prior work from our lab has shown that chloride anions can modulate the coordination sphere of copper complexes, resulting in a proposed shift in redox properties and reactivity.19 While the exact role of Cl− in the present transformation remains under investigation, preliminary data suggest this X-type ligand is suppressing off-cycle reduction of dMesSCF3 (10) to fluoroform (CHF3) by low-valent copper. This proposal is supported by four key observations: (1) in the absence of chloride anion, consumption of dMesSCF3 is rapid and causes the reaction to stall; (2) unproductive consumption of dMesSCF3 is mediated by both copper and light; (3) Cl− modulates the ligand sphere of CuI(terpy)Cl as measured by UV–vis spectroscopy; (4) Cl− suppresses reduction of dMesSCF3 by CuI(terpy)Cl. For a more detailed discussion, see Supporting Information Section 5.
Table 1.
Control Reactions of Optimized Conditionsa

| ||
|---|---|---|
| entry | deviation | yieldb |
| 1 | none | 84% |
| 2 | NaCl instead of TBACl | 74% |
| 3 | CuCl2 instead of Cu(terpy)Cl2 | 80% |
| 4 | no TBACl | 41% |
| 5 | no photocatalyst | 27% |
| 6 | no copper catalyst | 0% |
| 7 | no photocatalyst, no TBACl | 12% |
| 8 | no light | 0% |
Reactions performed with alcohol (1.0 equiv), Cu(terpy)Cl2 (5 mol %), dMesSCF3 (1.5 equiv), TBACl (2 equiv), quinuclidine (1.6 equiv), DMSO (0.025M), integrated photoreactor (450 nm, 100% light intensity).
Yields determined by 19F NMR analysis using 1,4-difluorobenzene as internal standard. See the SI for experimental details.
Curiously, control experiments revealed that a modest yield of 27% was obtained in the absence of a photocatalyst but in the presence of 450 nm light (entry 5). This result suggests a minor background reaction possibly mediated by a photoactive copper species.31
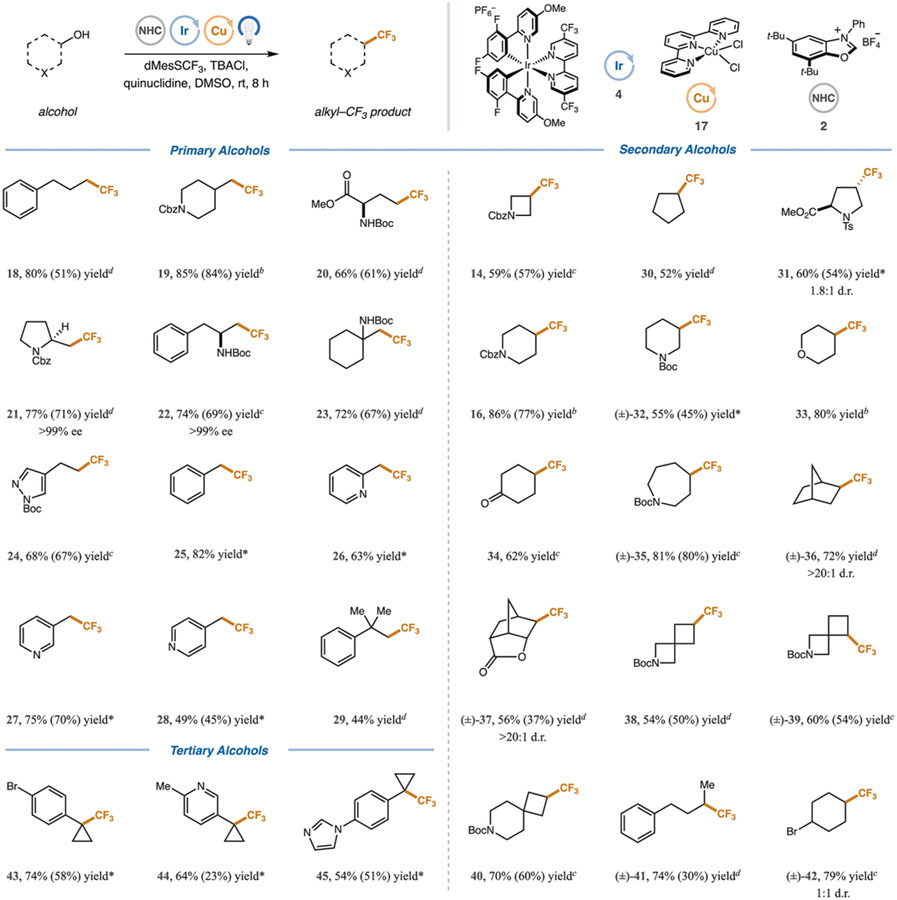
With optimized conditions in hand, we sought to evaluate the scope of the present transformation. We were pleased to find that a variety of structurally diverse primary alcohols readily underwent deoxytrifluoromethylation in good to excellent yields (Table 2). Unactivated primary alcohols were trifluoromethylated to afford 18, 19, and 20 in 80%, 85%, and 66% yield, respectively. Alcohols proximal to cyclic (21, 77% yield) and acyclic (22 and 23) amines also underwent efficient bond formation (72–77% yield). Notably, a primary alcohol containing a coordinating pyrazole moiety was trifluoromethylated to afford 24 in 68% yield.
Table 2.
Scope of Metallaphotoredox-Enabled Deoxygenative Trifluoromethylation Reactiona
|
Performed on a 0.5 mmol scale with alcohol (1.0 equiv), 2 (1.2 equiv), pyridine (1.2 equiv), tBuOMe (0.1 M), Ir(dF(OMe)ppy)2(5,5′(CF3)bpy)PF6 (1 mol %), dMesSCF3 (1.5 equiv), TBACl (2 equiv), quinuclidine (1.6 equiv), DMSO (0.025 M), integrated photoreactor (450 nm, 100% light intensity). Due to volatility of products, yields were determined by 19F NMR analysis of the crude reaction mixture using 1,4-difluorobenzene as an internal standard. Isolated yields are in parentheses.
With Cu(terpy)Cl2 (5 mol %).
With Cu(terpy)Cl2 (7.5 mol %).
With Cu(terpy)Cl2 (10 mol %).
Reaction performed under modified conditions; see the SI for details.
In addition, activated, benzylic alcohols were trifluoromethylated in good to high yields (25, 82% yield) including those bearing a pyridinyl nitrogen in the 2- (26, 63% yield), 3- (27, 75% yield), and 4- (28, 49% yield) position. Gratifyingly, a hindered neopentyl alcohol was trifluoromethylated to deliver 29 in a synthetically useful 44% yield.
We next turned our attention to secondary alcohols. A variety of cyclic substrates of different ring sizes were well tolerated in this transformation, allowing construction of the desired C(sp3)–CF3 bond in good to excellent yields (14, 16, 30–35, 52–86% yield). Bicyclic and spirocyclic ring systems, often used as bioisosteres for saturated heterocycles,32 were trifluoromethylated in 56–72% yield (36 and 37) and 54–70% yield (38–40). Acyclic secondary phenyl butanol underwent deoxytrifluoromethylation to give 41 in 74% yield. Notably, alcohols bearing reactive functional groups, such as alkyl bromides, were amenable to trifluoromethylation, delivering product 42 (79% yield), which is poised for orthogonal functionalization. Lastly, it is important to note that the carboxylic acids and alkyl bromides corresponding to Cbz-prolinol (21), bicyclic lactone (37), and [3.3]spirocycle (38) either are not commercially available or are prohibitively expensive, highlighting the practical utility of this new alcohol-based cross-coupling protocol.33
Quaternary trifluoromethylaryl cyclopropanes are of particular interest to medicinal chemists for their ability to function as bioisosteres for aryl tert-butyl groups.34,35 Traditionally, these motifs are prepared through a multistep synthetic sequence requiring the use of hazardous reagents and forcing temperatures.36 Although significant progress has been made in the last 5 years,37 the synthesis of these fluoroalkyl groups remains a significant challenge. To this end, we subjected a series of arylcyclopropanols to a modified set of reaction conditions (see Supporting Information Section 7) and were delighted to observe that the desired trifluoromethylated quaternary center was formed in good to high yields (43–45, 54–74% yield). For additional examples and limitations see the Supporting Information, Table S9.
From the outset, we sought to develop a deoxytrifluoromethylation protocol that would be compatible with the structural idiosyncrasies of drug discovery campaigns.6 Accordingly, we subjected a series of “drug-like” alcohols to this deoxytrifluoromethylation protocol (Table 3). We were pleased to find pyrazole and isoxazole sulfonamides delivered the desired products (46 and 47) in 86% and 72% yield, respectively. The successful synthesis of isoxazole 47 is of particular significance, given the propensity for the N–O bond to be cleaved via oxidative addition by low-valent metals.38 Additionally, aryltriazole 48 and triazolopyrazine 49 were obtained from the corresponding alcohols in modest to good yields (44% and 54% yield, respectively).
Table 3.
Application of Deoxygenative Trifluoromethylation to Complex Heterocyclic Alcoholsa

|
Performed on a 0.5 mmol scale with alcohol (1.0 equiv), 2 (1.2 equiv), pyridine (1.2 equiv), tBuOMe (0.1 M), Ir(dF(OMe)ppy)2(5,5′(CF3)-bpy)PF6 (1 mol %), dMesSCF3 (1.5 equiv), TBACl (2 equiv), quinuclidine (1.6 equiv), DMSO (0.025 M), integrated photoreactor (450 nm, 100% light intensity). Assay yields were determined by 19F NMR analysis of the crude reaction mixture using 1,4-difluorobenzene as an internal standard. Isolated yields are in parentheses.
With Cu(terpy)Cl2 (10 mol %).
With Cu(terpy)Cl2 (7.5 mol %).
Reaction performed under modified conditions, see the SI for details.
Initial attempts to synthesize aminopyrimidine 50 under the standard protocol were beset by poor yields and observation of side products by UPLC/MS resulting from oxidation of the piperidine nitrogen.39 By simply changing to a less oxidizing photocatalyst ([Ir(F(Me)ppy)2(dtbbpy)PF6], E1/2red[*IrIII/IrII] = +0.77 V vs saturated calomel electrode (SCE) in MeCN)40 we were able to suppress the formation of these oxidative byproducts and forge the desired C(sp3)–CF3 bond in 62% yield. Using these same modified conditions, chloropyridazine 51 was also obtained in 50% yield.
Installation of small trifluoromethylated alkyl groups on complex heteroarenes is often accomplished through Negishi coupling.41 Although highly effective, this protocol requires that the corresponding organometallic reagent must first be made from an alkyl halide. An orthogonal strategy that harnesses widely available alcohols as coupling partners would greatly expand synthetic accessibility to this chemical space. Accordingly, we investigated the use of small diols as precursors to esoteric trifluoromethylated alkyl groups. We adopted an iterative functionalization strategy to synthesize complex pyrazolopyridine 54 in two steps from commercially available materials. Initial arylation of diol 52 under conditions previously reported by our group27 delivered the monoarylated intermediate 53 in 47% yield while leaving the second alcohol untouched. Exposure of this alcohol to deoxytrifluoromethylation delivered 54 in 68% yield.
Monosaccharides serve as building blocks for biologically important macromolecules, and fluorination of their highly oxygenated skeletons has the potential to greatly alter their physical properties.42,43 As shown in Table 4, protected glucose 55 and furanose 56 were obtained via deoxytrifluoromethylation in 65% and 79% yield, respectively. Additionally, deoxyribose 57 was obtained in a synthetically useful 32% yield. Although modest in yield, we anticipate this building block can serve as a precursor to a library of synthetically challenging trifluoromethylated nucleoside analogues (vide infra).
Table 4.
Direct Functionalization of Monosaccharides and Nucleosidesa

|
Performed on a 0.5 mmol scale with alcohol (1.0 equiv), 2 (1.2 equiv), pyridine (1.2 equiv), tBuOMe (0.1 M); Ir(dF(OMe)ppy)2(5,5′(CF3)-bpy)PF6 (1 mol %), dMesSCF3 (1.5 equiv), TBACl (2 equiv), quinuclidine (1.6 equiv), DMSO (0.025 M), integrated photoreactor (450 nm, 100% light intensity). Assay yields were determined by 19F NMR analysis of the crude reaction mixture using 1,4-difluorobenzene as an internal standard. Isolated yields are in parentheses.
With Cu(terpy)Cl2 (10 mol %).
With Cu(terpy)Cl2 (5 mol %).
Reaction performed under modified conditions, see the SI for details.
At this stage we turned our attention to the long-standing challenge of synthesizing trifluoromethylated nucleoside derivatives. There are few published examples of nucleoside analogues with trifluoromethyl groups at the 3′ position of deoxyribose. Traditionally, these molecules require up to 11 synthetic steps to access.44–49 Recently, Cook et al. reported a two-step approach to a 3′-(CF3)-thymidine derivative, a major advancement in this field.17 We were interested in further accelerating the synthesis of these targets by employing our one-step deoxytrifluoromethylation protocol. Pleasingly, direct trifluoromethylation of the 3′ hydroxyl group in dimethoxytrityl (DMT)-protected thymidine could be achieved in 38% yield (58). Two additional nucleosides, DMT-adenosine and DMT-5-methylcytidine, were also trifluoromethylated in 31% (59 and 60) yield. Although these products are formed in modest yield, the protocol described herein dramatically reduces the amount of time, effort, and resources required to access these elusive structures. Finally, we demonstrated the utility of our deoxytrifluoromethylation protocol in the context of late-stage functionalization of pharmaceutical agents. To our delight, a derivative of the cardiovascular drug Ticagrelor (61) was trifluoromethylated to deliver 62 in 63% yield.
In summary, we describe herein an efficient protocol for the direct deoxytrifluoromethylation of alcohols. A wide range of substrates are amenable to this transformation, including primary, secondary, and tertiary alcohols, monosaccharides, nucleosides, and complex drug-like molecules. We anticipate that this reaction will be of value to the medicinal chemistry community and will serve to accelerate the discovery of novel trifluoromethyl-containing therapeutics.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful for financial support provided by the National Institute of General Medical Sciences (NIGMS), the NIH (under Award R35GM134897-02), the Princeton Catalysis Initiative, and kind gifts from Pfizer, MSD, Janssen, Bristol-Myers Squibb, and Genentech. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIGMS. N.E.I. and D.L.D. thank Princeton Universty, E. Taylor, and the Taylor family for an Edward C. Taylor Fellowship. A.M. acknowledges support from BioLEC (Bioinspired Light-Escalated Chemistry), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award #DE-SC0019370. The authors thank Z. Dong, P. Sarver, and V. Bacauanu for helpful scientific discussions, B. Li and H. Sakai for graphic design support, and R. Lambert for assistance in preparing the manuscript.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c04807
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c04807.
Additional experimental details, including procedures, photographs of experimental setup, characterization data, and spectra (PDF)
The authors declare the following competing financial interest(s): D.W.C.M. declares a competing financial interest with respect to the integrated photoreactor.
Contributor Information
Nicholas E. Intermaggio, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States.
Agustin Millet, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States.
Dali L. Davis, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States
David W. C. MacMillan, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States
REFERENCES
- (1).Brown DG; Boström J Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone?: Miniperspective. J. Med. Chem 2016, 59 (10), 4443–4458. [DOI] [PubMed] [Google Scholar]
- (2).Walters WP; Green J; Weiss JR; Murcko MA What Do Medicinal Chemists Actually Make? A 50-Year Retrospective. J. Med. Chem 2011, 54 (19), 6405–6416. [DOI] [PubMed] [Google Scholar]
- (3).Wang Y; Haight I; Gupta R; Vasudevan A What Is in Our Kit? An Analysis of Building Blocks Used in Medicinal Chemistry Parallel Libraries. J. Med. Chem 2021, 64 (23), 17115–17122. [DOI] [PubMed] [Google Scholar]
- (4).Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52 (21), 6752–6756. [DOI] [PubMed] [Google Scholar]
- (5).Dombrowski AW; Gesmundo NJ; Aguirre AL; Sarris KA; Young JM; Bogdan AR; Martin MC; Gedeon S; Wang Y Expanding the Medicinal Chemist Toolbox: Comparing Seven C(sp2)-C(sp3) Cross-Coupling Methods by Library Synthesis. ACS Med. Chem. Lett 2020, 11 (4), 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nat. Chem 2018, 10 (4), 383–394. [DOI] [PubMed] [Google Scholar]
- (7).Meanwell NA Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem 2018, 61 (14), 5822–5880. [DOI] [PubMed] [Google Scholar]
- (8).Hagmann WK The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 2008, 51 (15), 4359–4369. [DOI] [PubMed] [Google Scholar]
- (9).Furet P; Guagnano V; Fairhurst RA; Imbach-Weese P; Bruce I; Knapp M; Fritsch C; Blasco F; Blanz J; Aichholz R; Hamon J; Fabbro D; Caravatti G Discovery of NVP-BYL719 a Potent and Selective Phosphatidylinositol-3 Kinase Alpha Inhibitor Selected for Clinical Evaluation. Bioorg. Med. Chem. Lett 2013, 23 (13), 3741–3748. [DOI] [PubMed] [Google Scholar]
- (10).Zhou Y; Wang J; Gu Z; Wang S; Zhu W; Aceña JL; Soloshonok VA; Izawa K; Liu H Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II-III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev 2016, 116 (2), 422–518. [DOI] [PubMed] [Google Scholar]
- (11).McGrath NA; Brichacek M; Njardarson JT A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ 2010, 87 (12), 1348–1349. [Google Scholar]
- (12).Kautzky JA; Wang T; Evans RW; MacMillan DWC Decarboxylative Trifluoromethylation of Aliphatic Carboxylic Acids. J. Am. Chem. Soc 2018, 140 (21), 6522–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tan X; Liu Z; Shen H; Zhang P; Zhang Z; Li C Silver-Catalyzed Decarboxylative Trifluoromethylation of Aliphatic Carboxylic Acids. J. Am. Chem. Soc 2017, 139 (36), 12430–12433. [DOI] [PubMed] [Google Scholar]
- (14).Kornfilt DJP; MacMillan DWC Copper-Catalyzed Trifluoromethylation of Alkyl Bromides. J. Am. Chem. Soc 2019, 141 (17), 6853–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhao X; MacMillan DWC Metallaphotoredox Perfluoroalkylation of Organobromides. J. Am. Chem. Soc 2020, 142 (46), 19480–19486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chen Y; Ma G; Gong H Copper-Catalyzed Reductive Trifluoromethylation of Alkyl Iodides with Togni’s Reagent. Org. Lett 2018, 20 (15), 4677–4680. [DOI] [PubMed] [Google Scholar]
- (17).Liu Z-Y; Cook SP Interrupting the Barton-McCombie Reaction: Aqueous Deoxygenative Trifluoromethylation of O-Alkyl Thiocarbonates. Org. Lett 2021, 23 (3), 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhu L; Liu S; Douglas JT; Altman RA Copper-Mediated Deoxygenative Trifluoromethylation of Benzylic Xanthates: Generation of a C-CF3 Bond from an O-Based Electrophile. Chem.—Eur. J 2013, 19 (38), 12800–12805. [DOI] [PubMed] [Google Scholar]
- (19).Sarver PJ; Bacauanu V; Schultz DM; DiRocco DA; Lam Y; Sherer EC; MacMillan DWC The Merger of Decatungstate and Copper Catalysis to Enable Aliphatic C(sp3)-H Trifluoromethylation. Nat. Chem 2020, 12 (5), 459–467. [DOI] [PubMed] [Google Scholar]
- (20).Guo S; AbuSalim DI; Cook SP Aqueous Benzylic C-H Trifluoromethylation for Late-Stage Functionalization. J. Am. Chem. Soc 2018, 140 (39), 12378–12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Xiao H; Liu Z; Shen H; Zhang B; Zhu L; Li C Copper-Catalyzed Late-Stage Benzylic C(sp3)-H Trifluoromethylation. Chem. 2019, 5 (4), 940–949. [DOI] [PubMed] [Google Scholar]
- (22).Ertl P; Schuhmann T A Systematic Cheminformatics Analysis of Functional Groups Occurring in Natural Products. J. Nat. Prod 2019, 82 (5), 1258–1263. [DOI] [PubMed] [Google Scholar]
- (23).Ertl P An Algorithm to Identify Functional Groups in Organic Molecules. J. Cheminformatics 2017, 9 (1), 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Henkel T; Brunne RM; Müller H; Reichel F Statistical Investigation into the Structural Complementarity of Natural Products and Synthetic Compounds. Angew. Chem., Int. Ed 1999, 38 (5), 643–647. [DOI] [PubMed] [Google Scholar]
- (25).Reaxys search from January 2022 of commercially available alkyl fragments: alcohols (177 700), carboxylic acids (98, 451), alkyl bromides (26 966), alkyl iodides (3232).
- (26).While limited examples of nucleophilic deoxytrifluoromethylation have been reported, the weak nucleophilicity of CF3− limits the scope of these transformations to activated alcohols.; (a) de Azambuja F; Lovrien SM; Ross P; Ambler BR; Altman RA Catalytic One-Step Deoxytrifluoromethylation of Alcohols. J. Org. Chem 2019, 84 (4), 2061–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang W; Lin J; Wu W; Cao Y; Xiao J Dehydroxylative Trifluoromethylthiolation, Trifluoromethylation, and Difluoromethylation of Alcohols. Chin. J. Chem 2020, 38 (2), 169–172. [Google Scholar]; (c) Li J-L; Yang X-J; Wang Y; Liu J-T Synthesis of Trifluoromethylated Compounds from Alcohols via Alkoxydiphenyl-phosphines. J. Fluor. Chem 2015, 178, 254–259. [Google Scholar]
- (27).Dong Z; MacMillan DWC Metallaphotoredox-Enabled Deoxygenative Arylation of Alcohols. Nature 2021, 598 (7881), 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Sakai HA; MacMillan DWC Nontraditional Fragment Couplings of Alcohols and Carboxylic Acids: C(sp3)–C(sp3) Cross-Coupling via Radical Sorting. J. Am. Chem. Soc 2022, 144 (14), 6185–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Creutz SE; Lotito KJ; Fu GC; Peters JC Photoinduced Ullmann C-N Coupling: Demonstrating the Viability of a Radical Pathway. Science 2012, 338 (6107), 647–651. [DOI] [PubMed] [Google Scholar]
- (30).Liu S; Liu H; Liu S; Lu Z; Lu C; Leng X; Lan Y; Shen Q C(sp3)-CF 3 Reductive Elimination from a Five-Coordinate Neutral Copper(III) Complex. J. Am. Chem. Soc 2020, 142 (21), 9785–9791. [DOI] [PubMed] [Google Scholar]
- (31).This minor pathway may be mediated by CF3 or Cl radicals activating the NHC adduct via hydrogen atom transfer (HAT) of the hydridic methine C–H bond in 3. High-valent LnCu(CF3)x species are known to absorb blue light and photoeliminate CF3 radicals, which can subsequently abstract strong C–H bonds.17,31a Furthermore, Cu–Cl bonds are also known to photolyze and release Cl radical, which can subsequently perform HAT from strong C–H bonds, including hydridic methine C–H’s similar to 3.31b–d; (a) Choi G; Lee GS; Park B; Kim D; Hong SH Direct C(sp3)-H Trifluoromethylation of Unactivated Alkanes Enabled by Multifunctional Trifluoromethyl Copper Complexes. Angew. Chem., Int. Ed 2021, 60 (10), 5467–5474. [DOI] [PubMed] [Google Scholar]; (b) Kang YC; Treacy SM; Rovis T Iron-Catalyzed Photoinduced LMCT: A 1° C-H Abstraction Enables Skeletal Rearrangements and C(sp3)-H Alkylation. ACS Catal. 2021, 11 (12), 7442–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Treacy SM; Rovis T Copper Catalyzed C(sp3)-H Bond Alkylation via Photoinduced Ligand-to-Metal Charge Transfer. J. Am. Chem. Soc 2021, 143 (7), 2729–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kariofillis SK; Shields BJ; Tekle-Smith MA; Zacuto MJ; Doyle AG Nickel/Photoredox-Catalyzed Methylation of (Hetero)Aryl Chlorides Using Trimethyl Orthoformate as a Methyl Radical Source. J. Am. Chem. Soc 2020, 142 (16), 7683–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Carreira EM; Fessard TC Four-Membered Ring-Containing Spirocycles: Synthetic Strategies and Opportunities. Chem. Rev 2014, 114 (16), 8257–8322. [DOI] [PubMed] [Google Scholar]
- (33).SciFinder search from April 2022. “Prohibitively expensive” defined as >5× the cost of alcohol building block per 1 g of material.
- (34).Barnes-Seeman D; Jain M; Bell L; Ferreira S; Cohen S; Chen X-H; Amin J; Snodgrass B; Hatsis P Metabolically Stable tert -Butyl Replacement. ACS Med. Chem. Lett 2013, 4 (6), 514–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Johnson BM; Shu Y-Z; Zhuo X; Meanwell NA Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem 2020, 63 (12), 6315–6386. [DOI] [PubMed] [Google Scholar]
- (36).Milgram BC; Marx IE; Stellwagen J; Zhao W; Cherney AH Cyclopropyl Dihydroquinoline Sulfonamide Compounds. U.S. Patent US 2021/0387977 A1, Dec 16, 2021.
- (37).Phelan JP; Lang SB; Compton JS; Kelly CB; Dykstra R; Gutierrez O; Molander GA Redox-Neutral Photocatalytic Cyclopropanation via Radical/Polar Crossover. J. Am. Chem. Soc 2018, 140 (25), 8037–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Akiba K; Kashiwagi K; Ohyama Y; Yamamoto Y; Ohkata K Ring Transformation Equilibrium (Bond Switch) in 5-(2Aminovinyl)Isothiazole System via Hypervalent Sulfurane. Synthesis, Structure Determination, and Kinetic Study. J. Am. Chem. Soc 1985, 107 (9), 2721–2730. [Google Scholar]
- (39).McManus JB; Onuska NPR; Nicewicz DA Generation and Alkylation of α-Carbamyl Radicals via Organic Photoredox Catalysis. J. Am. Chem. Soc 2018, 140 (29), 9056–9060. [DOI] [PubMed] [Google Scholar]
- (40).Liu W; Lavagnino MN; Gould CA; Alcázar J; MacMillan DWC A Biomimetic S H 2 Cross-Coupling Mechanism for Quaternary Sp3-Carbon Formation. Science 2021, 374 (6572), 1258–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).(a) Milgram BC; Marx IE; Wang H; Cherney AH Cyclobutyl Dihydroquinoline Sulfonamide Compounds. U.S. Patent US 2021/0387978 A1, Dec 16, 2021.; (b) Goldberg S; Martin CL; Fennema EG; Kummer DA; Nishimura RT; Tanis VM; Woods CR; Fourie AM; Xue X Phenyl and Pyridinyl Substituted Imidazoles as Modulators of RORγt. U.S. Patent US 2019/0382354 A1, Dec 19, 2019.
- (42).Miethchen R Modified Natural Substances-Fluorinated and Fluoroalkylated Monosaccharides and Inositols. J. Fluor. Chem 2004, 125 (6), 895–901. [Google Scholar]
- (43).Bilska-Markowska M; Szwajca A; Marciniak B Design, Properties and Applications of Fluorinated and Fluoroalkylated N-Containing Monosaccharides and Their Analogues. J. Fluor. Chem 2019, 227, 109364. [Google Scholar]
- (44).Johnson CR; Bhumralkar DR; De Clercq E 3′-C-Trifluoromethyl Ribonucleosides. Nucleosides Nucleotides Nucleic Acids 1995, 14 (1–2), 185–194. [Google Scholar]
- (45).Serafinowski PJ; Brown CA New Method for the Preparation of Some 2′- and 3′-Trifluoromethyl-2′,3′-Dideoxyuridine Derivatives. Tetrahedron 2000, 56, 333. [Google Scholar]
- (46).Jeannot F; Mathé C; Gosselin G Synthesis and Antiviral Evaluation of 3′-C-trifluoromethyl Nucleoside Derivatives Bearing Adenine as the Base. Nucleosides Nucleotides Nucleic Acids 2001, 20 (4–7), 755–758. [DOI] [PubMed] [Google Scholar]
- (47).Jeannot F Synthesis and Studies of 3′-C-Trifluoromethyl Nucleoside Analogues Bearing Adenine or Cytosine as the Base. Bioorg. Med. Chem 2002, 10 (10), 3153–3161. [DOI] [PubMed] [Google Scholar]
- (48).Sharma PK; Nair V Synthesis of 3′-Trifluoromethyl Nucleosides as Potential Antiviral Agents. Nucleosides Nucleotides Nucleic Acids 2000, 19 (4), 757–774. [DOI] [PubMed] [Google Scholar]
- (49).Serafinowski PJ; Brown CA; Barnes CL Synthesis of Some 2′- and 3′-fluoroalkyl Substituted Nucleosides and Oligonucleotides. Nucleosides Nucleotides Nucleic Acids 2001, 20 (4–7), 921–925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.