

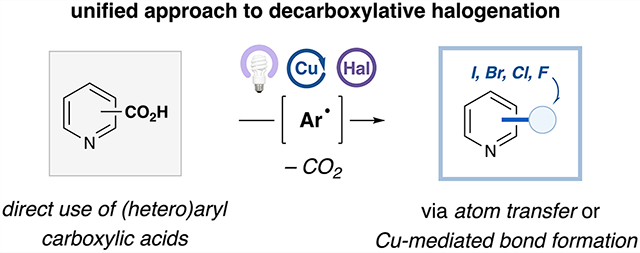
Abstract
Aryl halides are a fundamental motif in synthetic chemistry, playing a critical role in metal-mediated cross-coupling reactions and serving as important scaffolds in drug discovery. Although thermal decarboxylative functionalization of aryl carboxylic acids has been extensively explored, the scope of existing halodecarboxylation methods remains limited, and there currently exists no unified strategy that provides access to any type of aryl halide from an aryl carboxylic acid precursor. Herein, we report a general catalytic method for direct decarboxylative halogenation of (hetero)aryl carboxylic acids via ligand-to-metal charge transfer. This strategy accommodates an exceptionally broad scope of substrates. We leverage an aryl radical intermediate toward divergent functionalization pathways: (1) atom transfer to access bromo- or iodo(hetero)arenes or (2) radical capture by copper and subsequent reductive elimination to generate chloro- or fluoro(hetero)arenes. The proposed ligand-to-metal charge transfer mechanism is supported through an array of spectroscopic studies.
Graphical Abstract
INTRODUCTION
The aryl halide functional group is of prime importance in organic synthesis. In particular, (hetero)aryl chlorides, bromides, and iodides serve as versatile coupling partners and precursors to organometallic species in metal-mediated cross-coupling reactions, providing facile access to diverse aromatic scaffolds.1,2 Moreover, aryl fluorides and chlorides are ubiquitous in drug discovery due to their tendency to dramatically alter the biological properties of bioactive molecules.3,4 As such, the ability to selectively access diverse aryl halides from common arene functionalities through operationally simple methods is of vital interest to synthetic chemists in fields as varied as medicinal chemistry, agro-chemistry, materials, and imaging.5,6
Along these lines, aryl carboxylic acids represent attractive aryl halide precursors, as they are abundant, structurally diverse, and readily accessible from feedstock chemicals, such as tolyl groups and esters. In fact, the carboxylic acid motif has been harnessed to achieve site-specific aryl halide installation via halodecarboxylation; these methods rely on electrophilic halogenation reagents or metal mediators and often benefit from thermal activation.7 However, the generality of such strategies is limited by dual difficulties associated with aryl carboxylic acid decarboxylation and the poor selectivity of reactive halogenation reagents.7–10 For instance, bromo- and iododecarboxylation protocols often require careful substrate selection due to a propensity for overhalogenation,11 and no reliable and general chlorodecarboxylation method exists that can be applied to a broad scope of aryl carboxylic acids.7 Indeed, no current halodecarboxylation platform is capable of transforming aryl carboxylic acids to bromo-, iodo-, chloro-, and fluoroarenes. As such, the development of an operationally simple, general, and unified method to access every type of aryl halide from a diverse range of aryl carboxylic acids remains an important synthetic challenge.
The development of a unified halodecarboxylation strategy presents several challenges. First, the method must generate a common aryl intermediate capable of traversing distinct mechanistic pathways toward halogenation. Additionally, given that a C(sp2)–I bond has dramatically different properties—such as bond strength and polarization—compared to a C(sp2)–F bond, different approaches to functionalization are likely required.12 Along these lines, we recognized that an aryl radical intermediate could be harnessed via one of two pathways: (1) atom transfer or (2) radical capture by a metal species followed by metal-mediated bond formation (Figure 2). Given the diverse nature of C(sp2)–halogen bonds and the varied reactivity of existing halogenation reagents, we anticipated that these divergent reaction pathways should enable a general and practical platform for accessing aryl halides from aryl carboxylic acids.
Figure 2.
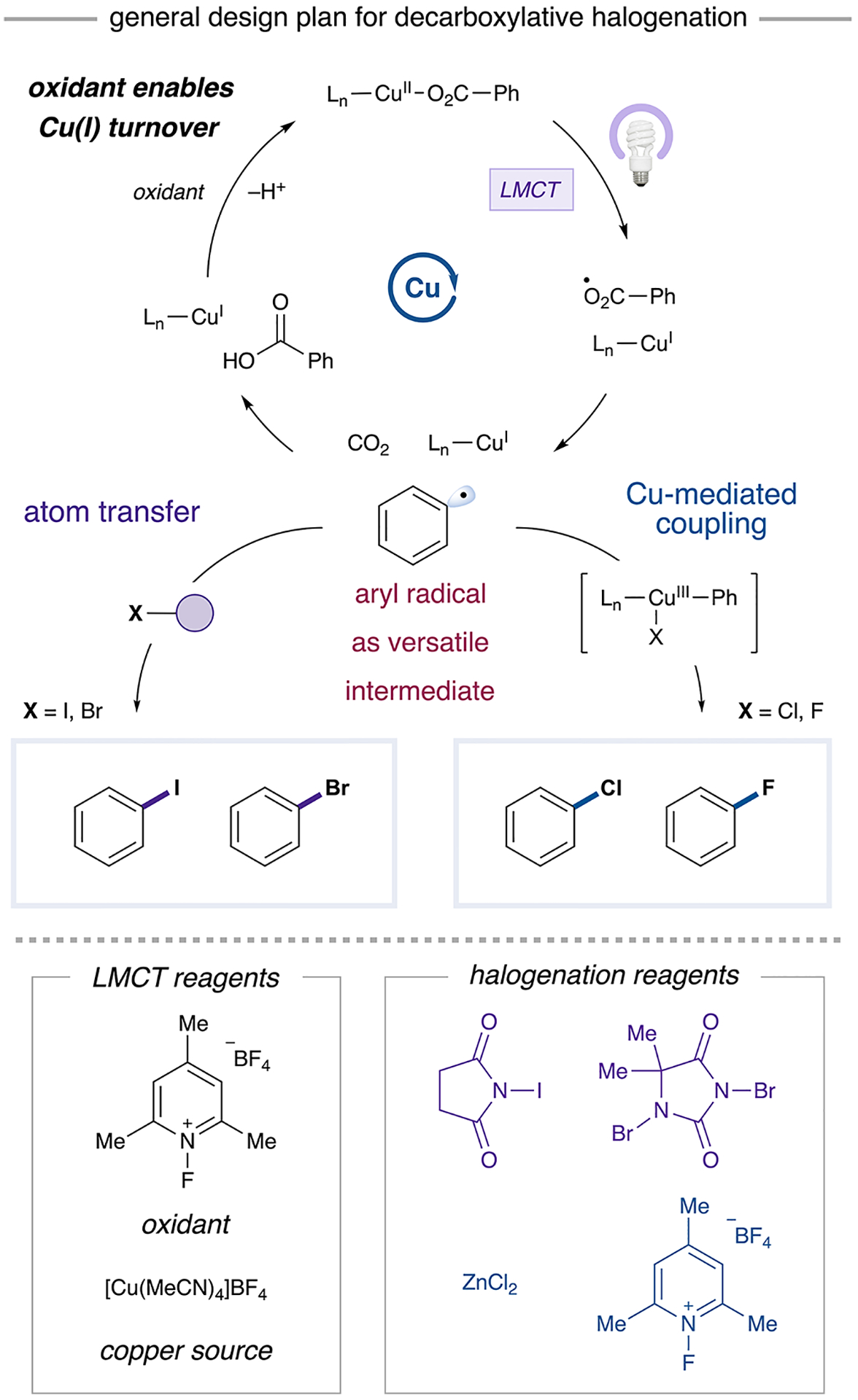
General design plan for decarboxylative halogenation of (hetero)aryl carboxylic acids.
A unified, general halodecarboxylation method would require the use of a mild decarboxylation protocol. Recently, the Rovis, Yoon, and Ritter groups demonstrated that copper ligand-to-metal charge transfer (LMCT) activation is a viable approach for generating reactive open-shell intermediates.13,14 Accordingly, we anticipated that LMCT would afford an O-centered aryl carboxylate radical that could undergo loss of CO2 to yield an aryl radical intermediate (Figure 2).15 This proposed open-shell mechanism contrasts with thermal copper-mediated decarboxylation methods, which are accepted to proceed through simultaneous cleavage of the aryl–CO2 bond and formation of a copper–aryl bond at a ligated Cu(I) center via a single transition state (Figure 1).9,10 Furthermore, by including a single-electron oxidant to convert Cu(I) to a photoactive carboxylate-ligated Cu(II) state (Figure 2), we could achieve catalytic decarboxylation at room temperature, thus solving an additional outstanding challenge in this area.16
Figure 1.
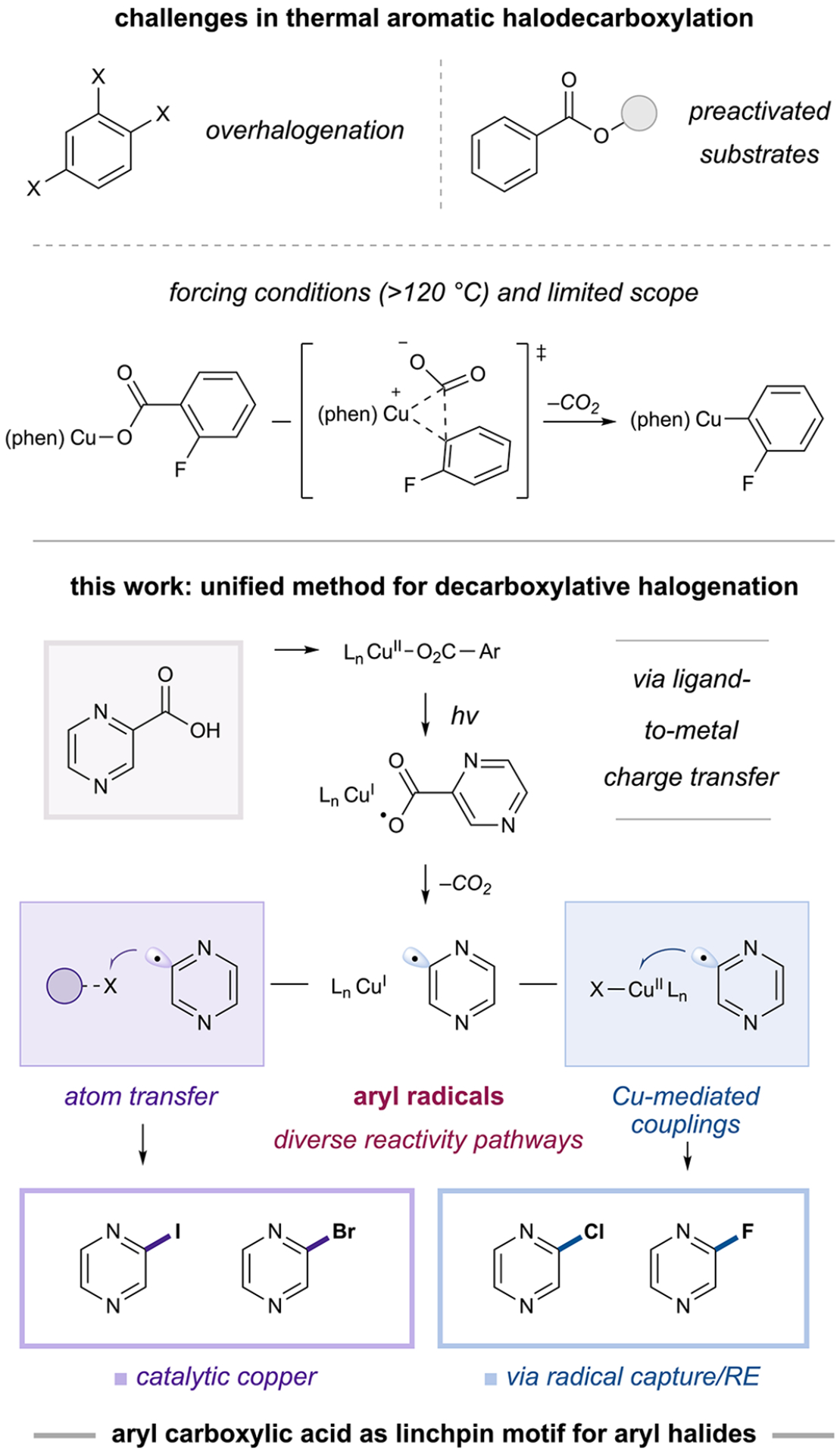
Direct decarboxylative halogenation of (hetero)aryl carboxylic acids via Cu–LMCT. RE, reductive elimination.
RESULTS AND DISCUSSION
We set out to develop a general, catalytic method for halodecarboxylation of (hetero)aryl carboxylic acids via the combination of a copper salt, a 365 nm LED light source, a stoichiometric oxidant, and an atom transfer reagent. We envisioned that diverse radical trapping reagents could engage the aryl radical intermediate in halogen atom transfer and anticipated that separating aryl radical formation from the subsequent atom transfer would facilitate the use of different halogenation reagents with minimal impact on reaction efficiency.
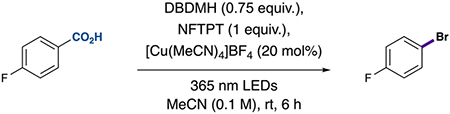
We first explored this idea in the context of a bromodecarboxylation. A mixture of 4-fluorobenzoic acid, [Cu(MeCN)4]BF4 (20 mol %), 1-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate (NFTPT) (1 equiv), and the bromination reagent, 1,3-dibromo-5,5-dimethylhydantoin (DBDMH, 0.75 equiv), in CH3CN was exposed to 365 nm LED irradiation, and the desired bromodecarboxylated product was obtained in 72% yield (Table 1, entry 1). Control reactions support the proposed mechanism of this transformation. No conversion was observed in the absence of light irradiation, copper, or oxidant (Table 1, entries 2–4). As expected, omission of the halogenation reagent led to protodecarboxylation of the aryl carboxylic acid via hydrogen atom transfer (HAT) (Table 1, entry 5; Table S6). Further, deuterodecarboxylation via deuterium atom transfer was observed when the reaction was conducted in the absence of halogenation reagent with CD3CN as solvent (Table S7). These results support the intermediacy of aryl radicals in this transformation and, furthermore, provide a facile strategy for the proto- or deuterodecarboxylation of directing groups in arenes.17,18 For additional results supporting aryl radical intermediacy, see Tables S8 and S9. Additional evaluation of the bromination reagent revealed that N-brominated reagents generated the highest yields for this electron-deficient substrate (Table 1, entry 6; Table S10). Moreover, a variety of oxidants could be leveraged in this reaction with good efficiencies under catalytic copper conditions; these oxidants are believed to facilitate the oxidation of Cu(I) to Cu(II) (Table 1, entry 7; for additional examples, see Table S12). In support of this hypothesis, the use of stoichiometric Cu(II) in the absence of oxidant leads to decarboxylative bromination in yields similar to those observed using catalytic copper and oxidant (Table 1, entry 8; Table S24).
Table 1.
Control Experiments for LMCT Decarboxylative Bromination
| ||
|---|---|---|
| entrya | deviations | yieldb |
| 1 | none | 72% |
| 2 | no light | 0% |
| 3 | no Cu | 0% |
| 4 | no oxidant | 0% |
| 5 | no bromination reagent | 0%c |
| 6 | with NBS (1 equiv) instead of DBDMH | 58% |
| 7 | with DCP (1 equiv) instead of NFTPT | 57% |
| 8 | with 1 equiv Cu(II) + no oxidantd | 73% |
0.1 mmol scale.
Yields determined by 1H NMR analysis.
24% yield fluorobenzene as determined by 1H NMR analysis.
Performed with 1 equiv Cu(OTf)2 and 1 equiv of 2,2,6,6-tetramethylpiperidine as base.
NBS, N-bromosuccinimide. DCP, dicumylperoxide. See Supporting Information for further details.
With optimized conditions in hand, we evaluated the generality of the reaction scope in the context of both electron-rich and electron-deficient substrates. In substrates bearing electron-donating substituents, replacement of DBDMH with bromotrichloromethane (BrCCl3) served to effectively suppress over-halogenation via electrophilic aromatic substitution pathways (see Table S26 for additional examples). As shown in Figure 3, we were gratified to find that a range of electron-deficient and electron-rich substrates were readily functionalized (2–5, 69–80% yield), overcoming a limitation of existing direct aryl bromodecarboxylation strategies.7
Figure 3.
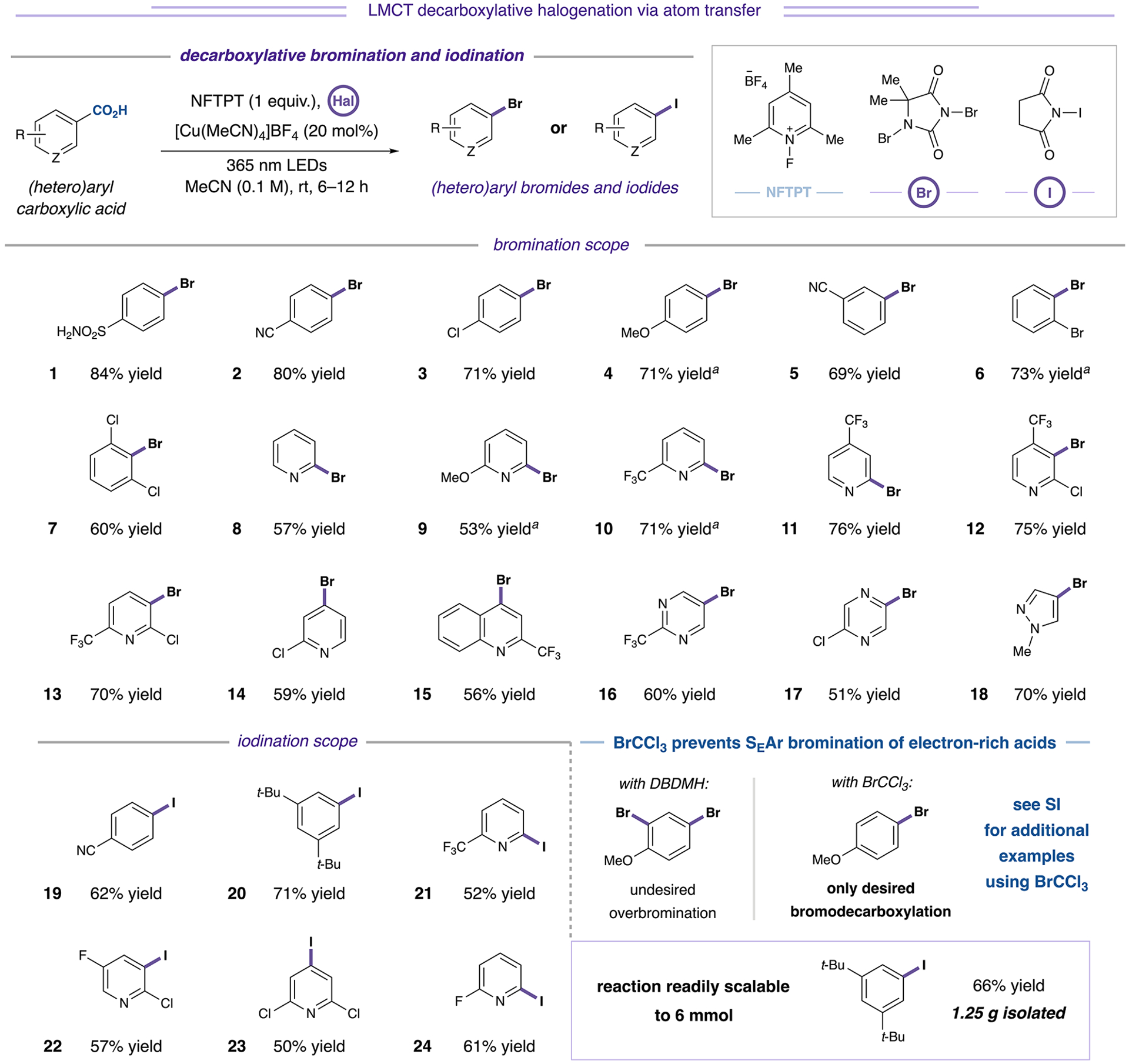
Decarboxylative bromination and iodination of (hetero)aryl carboxylic acids. Bromination conducted with 1,3-dibromo-5,5-dimethylhydantoin (0.75 equiv) unless otherwise specified. Iodination conducted with N-iodosuccinimide (1 equiv). aWith BrCCl3 (3 equiv) as bromination reagent. See Supporting Information for additional examples and experimental details.
Furthermore, the reaction was agnostic with respect to the substitution pattern; in contrast to previous thermal decarboxylation platforms, the presence of a destabilizing ortho-substituent was not required for reactivity. Both meta- and para-substituted acids underwent reaction with similar efficiency compared to sterically encumbered benzoic acids (1–7, 60–84% yield).11 With respect to the scope of heterocyclic carboxylic acids, all possible regioisomers of pyridine carboxylic acids, including picolinic acids (8–11, 53–76% yield), nicotinic acids (12 and 13, 75% and 70% yield, respectively), and isonicotinic acids (14, 59% yield), exhibited excellent efficiency. In addition, complex heterocyclic acids (15–17, 51–60% yield) furnished the corresponding brominated products in good yield, as did 5-membered heterocycles, such as pyrazole (18, 70% yield).
We similarly found that iododecarboxylation could be achieved through the use of N-iodosuccinimide as the halogenation reagent (Figure 3). As shown, iododecarboxylated products of benzoic acids (19 and 20, 62% and 71% yield, respectively) and all corresponding pyridine acid regioisomers (21–24, 50–61% yield) could be obtained efficiently under standard reaction conditions. Furthermore, we were pleased to find that gram-scale decarboxylative iodination of 3,5-di-tert-butylbenzoic acid proceeded in good efficiency to deliver 1.25 grams of 20, highlighting the scalability of LMCT decarboxylative halogenation reactions. For additional examples of bromo- or iododecarboxylation using these protocols, see Table S27.
In contrast to the bromination and iodination protocols, we found N-chlorosuccinimide (NCS) and other chlorine atom transfer reagents to be far less effective at promoting the desired chlorodecarboxylation reaction (Figure 4). We attribute this result to the greater bond dissociation energy of the N–Cl bond relative to N–Br or N–I and the larger barrier to chlorine atom transfer.19 Indeed, utilizing 1 equivalent of NCS as the chlorination reagent under the standard reaction conditions generated a combination of the chlorodecarboxylated product (15% yield), the parent arene resulting from protodecarboxylation (18% yield), and a competitive aryl carboxylic acid decarboxylative C(sp2)–O bond formation byproduct (24% yield, relative to equivalents of starting material consumed) (Figure 4). The formation of the parent arene demonstrates that HAT from solvent to the aryl radical is more efficient than chlorine atom transfer from NCS. Furthermore, the formation of the homocoupling byproduct supports the existence of a rapid bond formation pathway proceeding via aryl radical capture and subsequent C(sp2)–O reductive elimination from copper (see Tables S18 and S25 for optimized conditions). In line with our proposed hypothesis, the latter byproducts were not observed in the bromo- and iododecarboxylation reactions. Moreover, chlorodecarboxylation was observed in moderate yields using stoichiometric CuCl or CuCl2 as the sole copper source and chlorinating reagent (Table S15).
Figure 4.
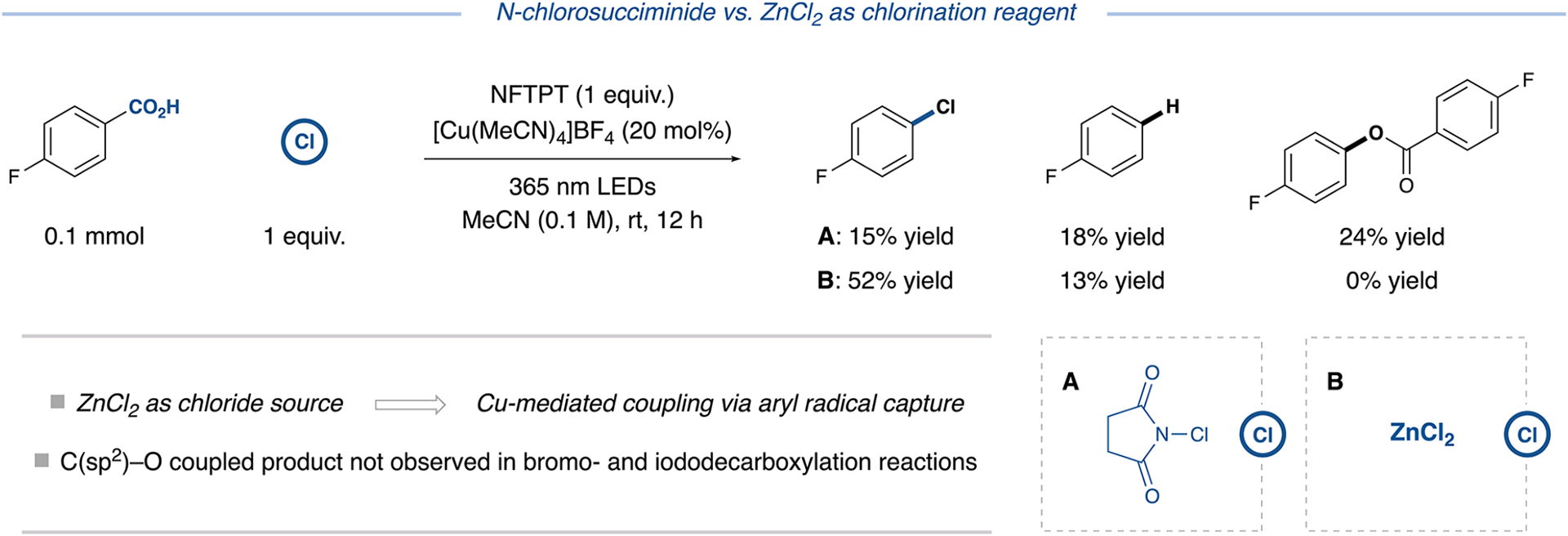
Chlorodecarboxylation reaction profiles using N-chlorosuccinimide or ZnCl2 as chlorination reagent. Analytical yields via NMR analysis. See Supporting Information for experimental details.
Based on these findings, we conceived of an alternative chlorodecarboxylation pathway that would bypass sluggish chlorine atom transfer to the aryl radical.20 Namely, we envisioned in situ generation of the copper chloride intermediate,21 coupled with radical capture by copper, and subsequent C(sp2)–Cl reductive elimination from the high-valent copper species to generate the desired aryl chloride product.22 For a more detailed outline of plausible pathways that may be undertaken by the aryl radical intermediate, see Figure S3. Importantly, reductive elimination from high-valent copper complexes is known to be facile for a variety of coupling partners,23 and copper-mediated radical capture/reductive elimination sequences have been demonstrated for challenging couplings.24,25 Thus, this general approach could exploit the unique bond-forming capabilities of copper and its ability to facilitate LMCT to broadly achieve more challenging decarboxylative halogenations.
Indeed, this strategy proved successful. A survey of chloride salts revealed ZnCl2 as the optimal chloride source (Figure 4, entry B). Under these conditions, formation of the C(sp2)–O coupled byproduct was precluded, and the propensity toward hydrodecarboxylation was curtailed. Other chloride salts resulted in significantly lower yields of the desired chlorodecarboxylation product (up to 27% yield, Table S15). Interestingly, electrophilic chlorination reagents, such as trichloroisocyanuric acid (trichlor) or 1,3-dichloro-5,5-dime-thylhydantoin, provided no more than 20% yield (Table S15). Upon investigating the scope of this reaction, we were delighted to find that chlorodecarboxylation could be readily achieved with both electron-rich and electron-deficient benzoic acids (25–28, 53–70% yield, Figure 5). Moreover, the protocol accommodates 2-, 3-, and 4-substituted pyridine acids (29–33, 46–98% yield), dinitrogen-containing heterocyclic acids, such as pyridazine (34, 59% yield) and pyrazine acids (35, 48% yield), and five-membered scaffolds such as thiazole-4-carboxylic acid (36, 54% yield). Direct decarboxylative chlorination of aryl carboxylic acids has posed a general synthetic challenge, with existing methods displaying limited scope due to the harshness of chlorination conditions.7,20 This protocol expands the state-of-the-art for decarboxylative chlorination of aryl carboxylic acids considerably by enabling access to diverse (hetero)aryl chloride pharmacophores and coupling handles from readily available aryl carboxylic acid substrates.
Figure 5.
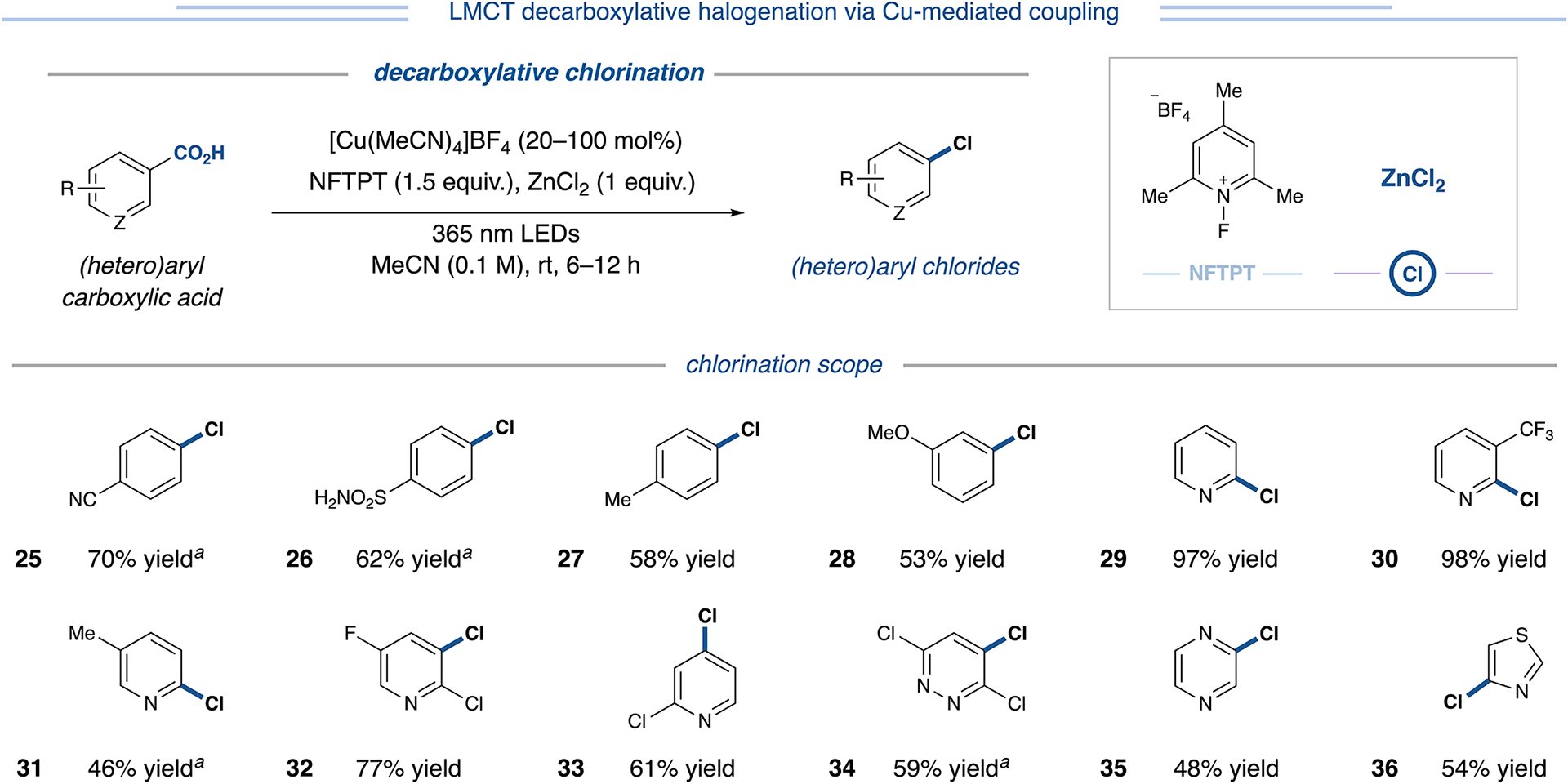
Decarboxylative chlorination of (hetero)aryl carboxylic acids. Analytical yields via 1H NMR analysis unless otherwise specified. aIsolated yield. See Supporting Information for experimental details.
A similar strategy was employed to accomplish fluorodecarboxylation of aryl carboxylic acids. The use of 3 equivalents of [Cu(MeCN)4]BF4 led to both LMCT decarboxylation and subsequent radical capture and C(sp2)–F bond formation. While decarboxylative fluorination using nucleophilic fluoride has been reported,13b our approach uses the NFTPT oxidant as a convenient, bench-stable electrophilic fluorine source and thus represents a complementary strategy to previous methods. This approach proved competent for the fluorodecarboxylation of electron-rich and electron-deficient benzoic acids with differentiated substitution patterns (37–42, 52–71% yield, Figure 6). Most notably, this copper-mediated protocol was effective across diverse classes of heterocyclic substrates, expediently furnishing fluorinated derivatives of pyridine (43–46, 49–81% yield), pyrimidine (47 and 48, 56% and 40% yield, respectively), pyrazine (49–51, 72–77% yield), pyridazine (52, 55% yield), and bicyclic quinoxaline and isoquinoline (53 and 54, 66% and 50% yield, respectively) scaffolds. For additional examples of chloro- or fluorodecarboxylation using these protocols, see Table S27.
Figure 6.
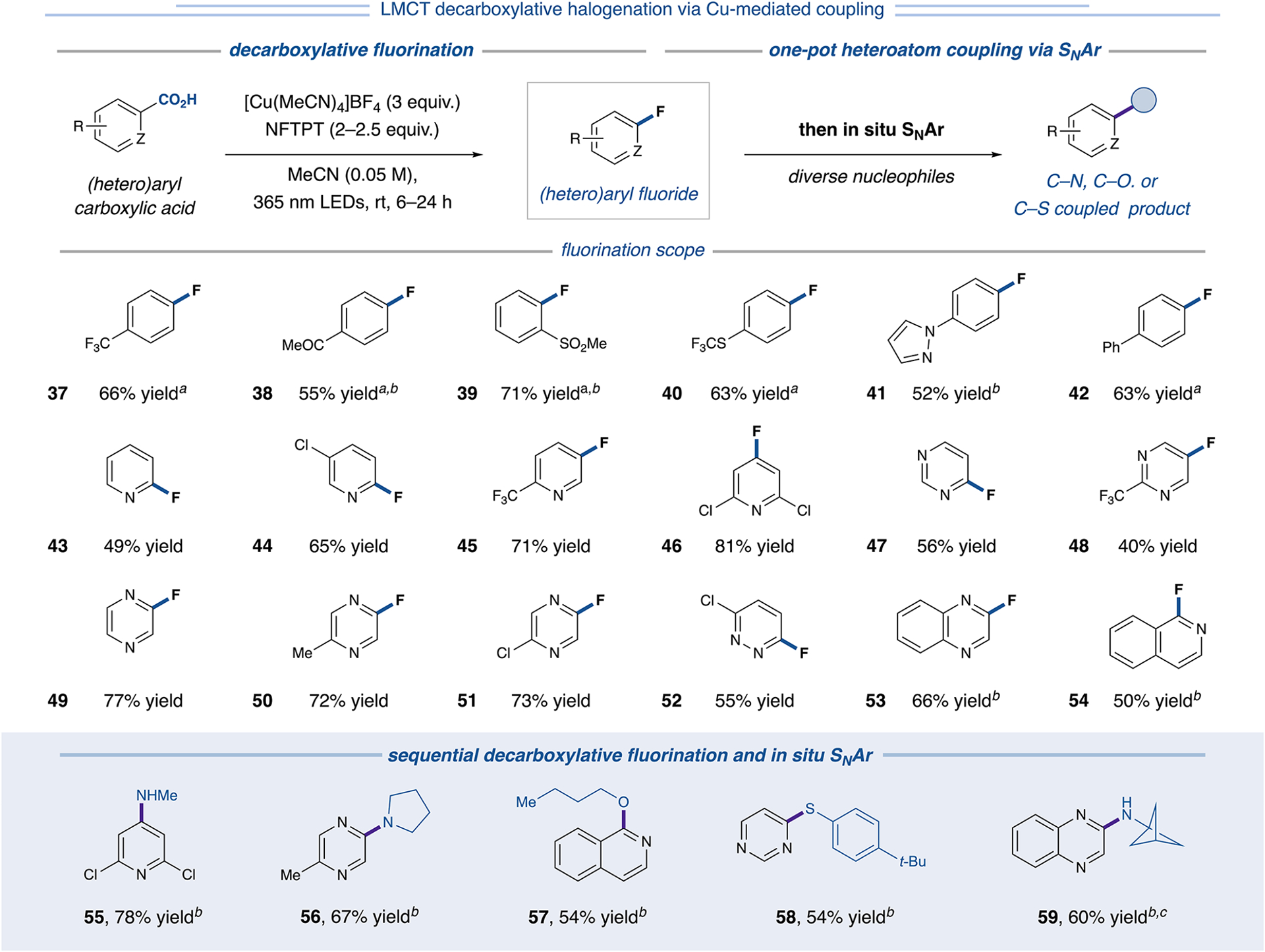
Decarboxylative fluorination and sequential SNAr functionalization of (hetero)aryl carboxylic acids. Analytical yields via NMR analysis unless otherwise specified. aWith 1.0 equiv CsF. bIsolated yield. cSNAr performed after aqueous workup. See Supporting Information for experimental details.
We recognized that the expanded heteroaryl scope of the fluorodecarboxylation reaction could provide opportunities to rapidly access structural complexity via SNAr.26 Delightfully, in situ SNAr of fluorinated (hetero)aryl products successfully furnished a diverse library of C(sp2)–nucleophile-coupled derivatives (55–59, 54–78% yield). This protocol enables operationally facile access to valuable C(sp2)–O, C(sp2)–S, or C(sp2)–N coupled products directly from commercially available and bench-stable heteroaryl carboxylic acids.
By simply altering the halogenation reagent and modifying the loading of both the oxidant and copper source, this approach provides a unified strategy for accessing all four decarboxylative halogenation reactions via a versatile aryl radical intermediate generated using a common set of conditions. As summarized in Figure 7a, this provides an operationally simple and mild protocol for the rapid diversification of a common, bench-stable, and readily accessible aryl carboxylic acid starting material to any desired aryl halide. For additional examples, see Table S27.
Figure 7.
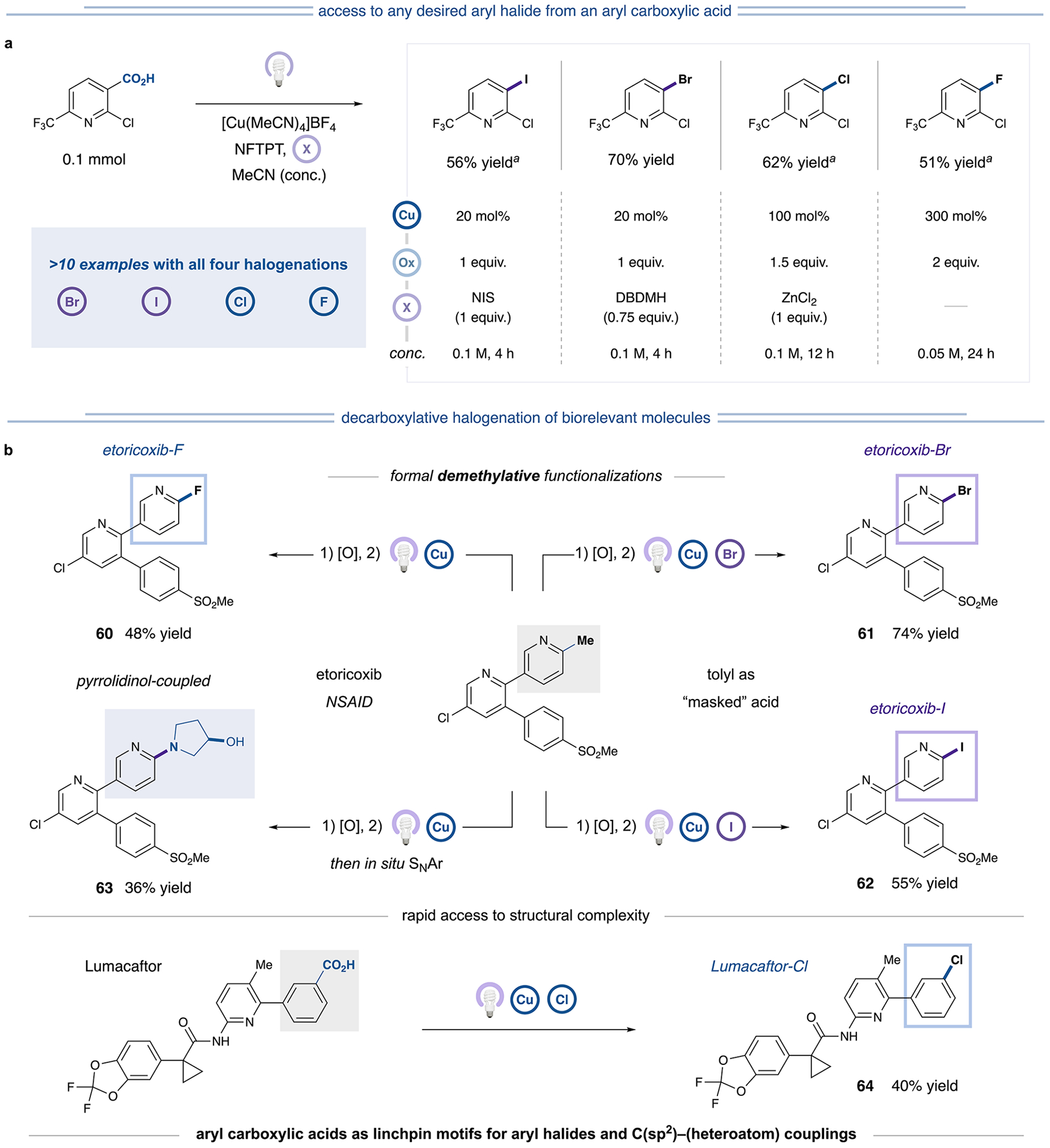
(a) Comparison of all four halogenation reactions on one substrate. See Table S27 for an additional 10 examples. aAnalytical yields via NMR spectroscopy. (b) Late-stage decarboxylative halogenations of biorelevant molecules. All reported yields are isolated unless otherwise specified. See Supporting Information for experimental details.
Benzoic acids can be “masked” as a variety of broadly available and bench-stable feedstock functionalities. For example, tolyl moieties in complex molecules such as etoricoxib can be revealed as aryl carboxylic acids upon oxidation. We envisioned that our protocol could be leveraged to transform such an intermediate into any desired aryl halide product, allowing access to a library of diversified compounds via a formal demethylative halogenation pathway. As shown in Figure 7b, etoricoxib-CO2H, readily obtained upon etoricoxib oxidation, was subjected to halodecarboxylation conditions to furnish the fluorinated, brominated, and iodinated derivatives in useful efficiencies (60–62, 48–74% yield). Beyond demethylative halogenation, this sequence enables rapid access to structural complexity. Notably, the etoricoxib-F product can be further functionalized via in situ SNAr reactions to generate C(sp2)–pyrrolidinol coupled product 63 in 36% yield. Furthermore, the drug lumacaftor was subjected to chlorodecarboxylation conditions to yield 64 (40% yield), bearing a useful chloride functional handle or pharmacophore. These examples highlight the broad tolerance of this strategy to structural complexity and the unique advantages of aryl carboxylic acids as synthons in a programmable synthetic sequence, potentially enabling the synthesis of valuable libraries of haloarenes that can be utilized as pharmacophores or subjected to further derivatization.27
Finally, to investigate the key LMCT event (see Figure 2), we conducted static UV–vis and transient absorption (TA) spectroscopy experiments (Figure 8). In static UV–vis spectra, Cu(II) coordination complexes possess a diagnostic absorption band between 600 and 1000 nm assigned to ligand-field (d–d) transitions, a feature that is not observed in Cu(I) d10 species.28,29 Monitoring the steady-state absorption of the reaction mixture irradiated with 370 nm light would thus elucidate the oxidation state of copper.
Figure 8.
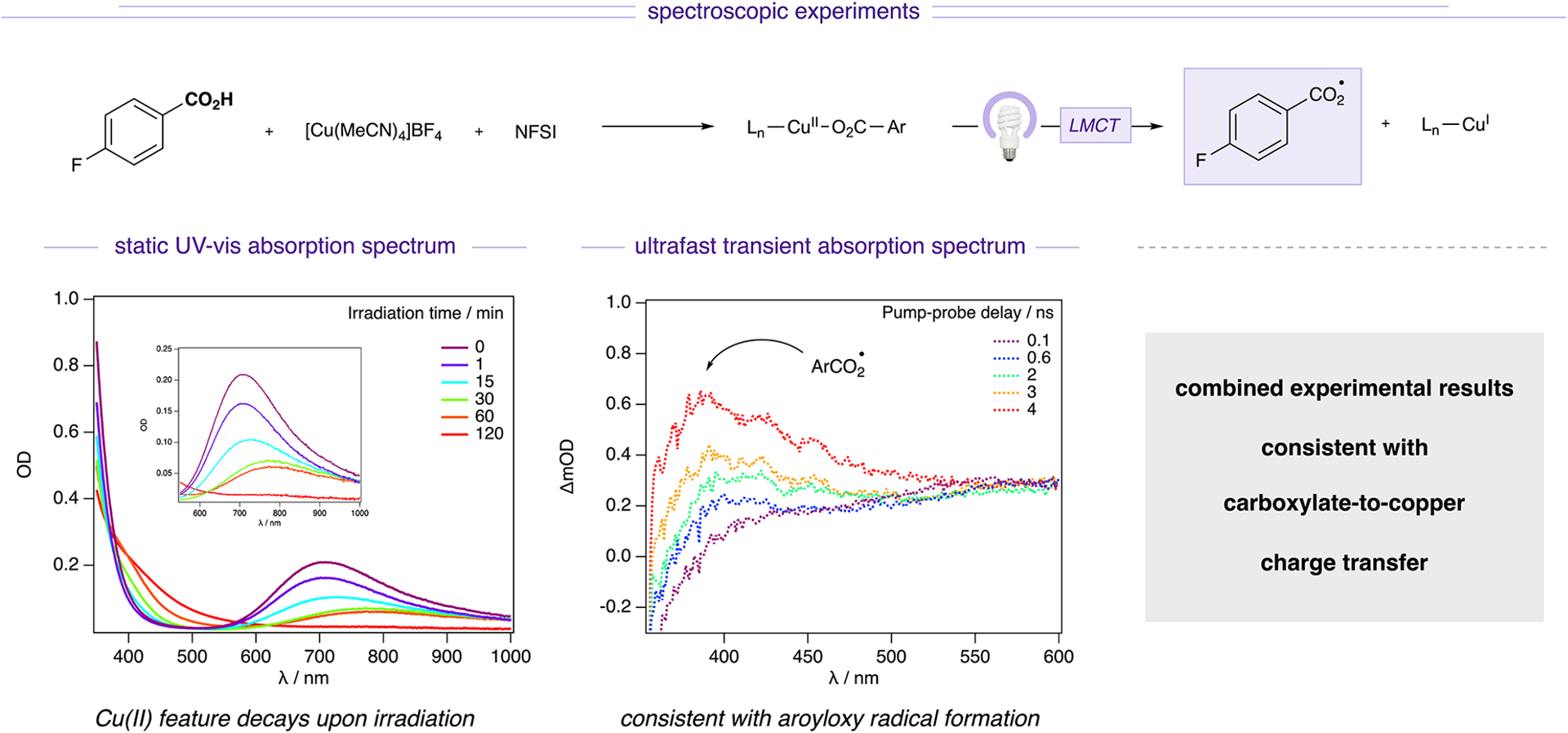
Spectroscopic support for Cu(II)–LMCT activation. Combined static UV–vis absorption spectroscopy and ultrafast transient absorption spectroscopy experimental results are consistent with carboxylate-to-copper charge transfer. See Supporting Information for experimental details. (m)OD, (milli)optical density.
Upon addition of N-fluorobenzenesulfonimide (NFSI), a model oxidant (see Table S2), to [Cu(MeCN)4]BF4 and prior to illumination (t = 0), a strong absorption band at 700 nm was observed, indicating the immediate formation of Cu(II) via oxidation of Cu(I). During 2 h of continuous irradiation, this absorption band gradually red-shifted to 800 nm and eventually decayed, indicating photoreduction of Cu(II) to Cu(I) via LMCT over the course of the reaction. In the absence of irradiation, no change in the absorption spectrum was observed over the 2 h experimental time window, demonstrating that light is needed for reduction of Cu(II).
Additionally, we used TA spectroscopy to probe the intermediacy of the proposed photogenerated aroyloxy radical (Figure 8). Benzoyloxy radicals, readily formed from their respective peroxides, feature a broad absorption band in the visible region between 500 and 800 nm, a sharper absorption component with a maximum near 400 nm, and a strong UV absorption centered at 320 nm.30 When conducted in flow, the reaction solution generates a difference spectrum that closely resembles the benzoyloxy radical spectrum previously reported in the literature31 and which appeared in the prompt spectrum in this experiment, consistent with benzoyloxy radical formation within the laser pulse. The benzoyloxy radical was monitored using ultrafast TA spectroscopy (λex = 350 nm, 1.9 mJ/pulse, 100 fs fwhm) and formed over the 4 ns time delay window of the experiment. Notably, these features are not observed in the absence of the aryl carboxylic acid. In combination, these spectroscopic studies are consistent with copper–aryl carboxylate bond homolysis resulting from LMCT, supporting the mechanistic basis of this manifold as a general strategy for activating previously elusive aryl carboxylic acid building blocks toward all categories of halodecarboxylation.
CONCLUSIONS
We describe herein a unified strategy for iodo-, bromo-, chloro-, and fluorodecarboxylation that is compatible with an expansive scope of (hetero)aryl carboxylic acids. This platform exploits the diverse reactivity of aryl radicals generated via an LMCT mechanism, as supported by spectroscopic studies. The intermediacy of an aryl radical enables divergent mechanistic pathways, namely, atom transfer or copper-mediated bond formation, which can be leveraged toward any type of halodecarboxylation. We further expanded the fluorodecarboxylation method to integrate a robust one-pot, in situ SNAr protocol en route to C(sp2)–N-, C(sp2)–O-, and C(sp2)–S-coupled products. We demonstrated that a series of formal demethylation halogenations could be achieved with etoricoxib, showcasing the late-stage utility of this method. We anticipate that these protocols will expand the utility of (hetero)aryl carboxylic acids as a synthetic building block and enable new scaffolds and strategies for late-stage functionalization.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful for financial support provided by the National Institute of General Medical Sciences (NIGMS), the NIH (under Award R35GM134897-03), the Princeton Catalysis Initiative, BioLec, an Energy Frontier Research Center (U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award DE-SC0019370), and kind gifts from Pfizer, Merck, Janssen, Bristol-Myers Squibb, and Genentech. T.Q.C. thanks Bristol-Myers Squibb for a graduate fellowship. P.S.P. thanks the NSF for a predoctoral fellowship (Award DGE-1656466). N.W.D. thanks Princeton University, E.C. Taylor and the Taylor family for support via the Edward C. Taylor fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIGMS. This work was performed in part at the Laboratory for Imaging and Kinetic Spectroscopy at North Carolina State University. The authors thank Rebecca Lambert for assistance in the preparation of this manuscript.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c02392.
Additional experimental and characterization data and spectra (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c02392
The authors declare the following competing financial interest(s): D.W.C.M. declares a competing financial interest with respect to the Integrated Photoreactor.
Contributor Information
Tiffany Q. Chen, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States.
P. Scott Pedersen, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States.
Nathan W. Dow, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States
Remi Fayad, Department of Chemistry, North Carolina State University, Raleigh, North Carolina 27695, United States.
Cory E. Hauke, Department of Chemistry, North Carolina State University, Raleigh, North Carolina 27695, United States
Michael C. Rosko, Department of Chemistry, North Carolina State University, Raleigh, North Carolina 27695, United States;.
Evgeny O. Danilov, Department of Chemistry, North Carolina State University, Raleigh, North Carolina 27695, United States;.
David C. Blakemore, Worldwide Research and Development, Pfizer, Inc., Groton, Connecticut 06340, United States
Anne-Marie Dechert-Schmitt, Worldwide Research and Development, Pfizer, Inc., Groton, Connecticut 06340, United States.
Thomas Knauber, Worldwide Research and Development, Pfizer, Inc., Groton, Connecticut 06340, United States;.
Felix N. Castellano, Department of Chemistry, North Carolina State University, Raleigh, North Carolina 27695, United States;.
David W. C. MacMillan, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States;.
REFERENCES
- (1).Buchwald SL; Mauger C; Mignani G; Scholz U Industrial-Scale Palladium-Catalyzed Coupling of Aryl Halides and Amines – A Personal Account. Adv. Synth. Catal 2006, 348, 23–39. [Google Scholar]
- (2).Joseph PJA; Priyadarshini S Copper-Mediated C–X Functionalization of Aryl Halides. Org. Process Res. Dev 2017, 21, 1889–1924. [Google Scholar]
- (3).Wang J; Sánchez-Roselló M; Aceña JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev 2014, 114, 2432–2506. [DOI] [PubMed] [Google Scholar]
- (4).Wilcken R; Zimmermann MO; Lange A; Joerger AC; Boeckler FM Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem 2013, 56, 1363–1388. [DOI] [PubMed] [Google Scholar]
- (5).Sharninghausen LS; Brooks AF; Winton WP; Makaravage KJ; Scott PJH; Sanford MS NHC-Copper Mediated Ligand-Directed Radiofluorination of Aryl Halides. J. Am. Chem. Soc 2020, 142, 7362–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Evano G; Nitelet A; Thilmany P; Dewez DF Metal-Mediated Halogen Exchange in Aryl and Vinyl Halides: A Review. Front. Chem 2018, 6, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Varenikov A; Shapiro E; Gandelman M Decarboxylative Halogenation of Organic Compounds. Chem. Rev 2021, 121, 412–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hu X-Q; Liu Z-K; Hou Y-X; Gao Y Single Electron Activation of Aryl Carboxylic Acids. iScience 2020, 23, No. 101266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gooßen LJ; Rodriguez N; Linder C; Lange PP; Fromm A Comparative Study of Copper- and Silver-Catalyzed Protodecarboxylations of Carboxylic Acids. ChemCatChem 2010, 2, 430–442. [Google Scholar]
- (10).Hoover JM Mechanistic Aspects of Copper-Catalyzed Decarboxylative Coupling Reactions of (Hetero)Aryl Carboxylic Acids. Comments Inorg. Chem 2017, 37, 169–200. [Google Scholar]
- (11).For representative examples of bromo- and iododecarboxylation of aryl carboxylic acids, see; (a) Perry GJP; Quibeil JM; Panigrahi A; Larrosa I Transition-Metal-Free Decarboxylative Iodination: New Routes for Decarboxylative Oxidative Cross-Couplings. J. Am. Chem. Soc 2017, 139, 11527–11536. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Quibeil JM; Perry GJP; Cannas DM; Larrosa I Transition-metal-free decarboxylative bromination of aromatic carboxylic acids. Chem. Sci 2018, 9, 3860–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fu Z; Li Z; Song Y; Yang R; Liu Y; Cai H Decarboxylative Halogenation and Cyanation of Electron-Deficient Aryl Carboxylic Acids via Cu Mediator as Well as Electron-Rich Ones through Pd Catalyst under Aerobic Conditions. J. Org. Chem 2016, 81, 2794–2803. [DOI] [PubMed] [Google Scholar]; (d) Cason J; Walba DM Reaction Pathway in the Modified Hunsdiecker Reaction. J. Org. Chem 1972, 37, 669–671. [Google Scholar]; (e) Barton DHR; Lacher B; Zard SZ Radical Decarboxylative Bromination of Aromatic Acids. Tetrahedron Lett. 1985, 26, 5939–5942. [Google Scholar]
- (12).Blanksby SJ; Ellison GB Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- (13).(a) Treacy SM; Rovis T Copper Catalyzed C(sp3)–H Bond Alkylation via Photoinduced Ligand-to-Metal Charge Transfer. J. Am. Chem. Soc 2021, 143, 2729–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu P; Lopez-Rojas P; Ritter T Radical Decarboxylative Carbometalation of Benzoic Acids: A Solution to Aromatic Decarboxylative Fluorination. J. Am. Chem. Soc 2021, 143, 5349–5354. [DOI] [PubMed] [Google Scholar]; (c) Su W; Xu P; Ritter T Decarboxylative Hydroxylation of Benzoic Acids. Angew. Chem., Int. Ed 2021, 60, 24012–24017. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li QY; Gockel SN; Lutovsky GA; DeGlopper KS; Baldwin NJ; Bundesmann MW; Tucker JW; Bagley SW; Yoon TP Decarboxylative cross-nucleophile coupling via ligand-to-metal charge transfer photoexcitation of Cu(II) carboxylates. Nat. Chem 2022, 14, 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For additional examples of applications of LMCT in organic synthesis, see; (a) Guo J-J; Hu A; Chen Y; Sun J; Tang H; Zuo Z Photocatalytic C–C Bond Cleavage and Amination of Cycloalkanols by Cerium(III) Chloride Complex. Angew. Chem., Int. Ed 2016, 55, 15319–15322. [DOI] [PubMed] [Google Scholar]; (b) Abderrazak Y; Bhattacharyya A; Reiser O Visible-Light-Induced Homolysis of Earth-Abundant Metal-Substrate Complexes: A Complementary Activation Strategy in Photoredox Catalysis. Angew. Chem., Int. Ed 2021, 60, 21100–21115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kochi JK Photolyses of Metal Compounds: Cupric Chloride in Organic Media. J. Am. Chem. Soc 1962, 84, 2121–2127. [Google Scholar]; (d) Shields BJ; Doyle AG Direct C(sp3)–H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc 2016, 2016, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xia S; Hu K; Lei C; Jin J Intramolecular Aromatic C–H Acyloxylation Enabled by Iron Photocatalysis. Org. Lett 2020, 22, 1385–1389. [DOI] [PubMed] [Google Scholar]; (f) Feng G; Wang X; Jin J Decarboxylative C-C and C-N Bond Formation by Ligand-Accelerated Iron Photo-catalysis: Decarboxylative C-C and C-N Bond Formation by Ligand-Accelerated Iron Photocatalysis. Eur. J. Org. Chem 2019, 6728–6732. [Google Scholar]; (g) Morimoto JY; DeGraff BA Photochemistry of the copper(II)-malonate system. Sensitized reaction. J. Phys. Chem 1972, 76, 1387–1388. [Google Scholar]; (h) Natarajan P; Ferraudi G Photochemical properties of copper(II)-amino acid complexes. Inorg. Chem 1982, 20, 3708–3712. [Google Scholar]
- (15).Misawa H; Sawabe K; Takahara S; Sakuragi H; Tokumaru K Decarboxylation Rates of Benzoyloxyl Radicals as Determined by Laser Flash Photolysis. Further Insight into the Mechanism for Photodecomposition of Dibenzoyl Peroxides. Chem. Lett 1988, 17, 357–360. [Google Scholar]
- (16).Gooßen LJ; Deng G; Levy LM Synthesis of Biaryls via Catalytic Decarboxylative Coupling. Science 2006, 313, 662–664. [DOI] [PubMed] [Google Scholar]
- (17).(a) Gooßen LJ; Linder C; Rodríguez N; Lange PP; Fromm A Silver-catalysed protodecarboxylation of carboxylic acids. Chem. Commun 2009, 7173–7175. [DOI] [PubMed] [Google Scholar]; (b) Lu P; Sanchez C; Cornella J; Larrosa I Silver-Catalyzed Protodecarboxylation of Heteroaromatic Carboxylic Acids. Org. Lett 2009, 11, 5710–5713. [DOI] [PubMed] [Google Scholar]; (c) Gooßen LJ; Thiel WR; Rodríguez N; Linder C; Melzer B Copper-Catalyzed Protodecarboxylation of Aromatic Carboxylic Acids. Adv. Synth. Catal 2007, 349, 2241–2246. [Google Scholar]
- (18).Font M; Quibell JM; Perry GJP; Larrosa I The use of carboxylic acids as traceless directing groups for regioselective C–H bond functionalisation. Chem. Commun 2017, 53, 5584–5597. [DOI] [PubMed] [Google Scholar]
- (19).(a) Tuaillon J; Couture Y; Lessard J Radical reactions of N-haloamides with olefins. XIII. Quantum yields for the photochemical addition of N-halo-N-hydroamides and N-chlorosuccinimide to 3,3-methyl-1-butene. Can. J. Chem 1987, 65, 2194–2197. [Google Scholar]; (b) Cottrell TL The strengths of chemical bonds, 2nd ed.; Butterworths: London, 1958. [Google Scholar]
- (20).For representative examples of chlorodecarboxylation of aryl carboxylic acids, see; (a) Becker K; Geisel M; Grob C; Kuhnen F Improved Preparation of Tertiary Chlorides by Halodecarboxylation. Synthesis 1973, 1973, 493–494. [Google Scholar]; (b) Peng XF; Shao XF; Liu ZQ Pd(II)-Catalyzed Bromo- and Chlorodecarboxylation of Electron-Rich Arenecarboxylic Acids. Tetrahedron Lett. 2013, 54, 3079–3081. [Google Scholar]; (c) Deacon GB; Farquharson GJ Permercurated Arenes. Iii. Syntheses of Periodoarenes and Perchloroarenes by Iodo- and Chloro-Demercuration of Some Permercurated Arenes. Aust. J. Chem 1977, 30, 1701–1703. [Google Scholar]
- (21).Ramette RW; Fan G Copper(II) Chloride Complex Equilibrium Constants. Inorg. Chem 1983, 22, 3323–3326. [Google Scholar]
- (22).(a) Casitas A; King AE; Parella T; Costas M; Stahl SS; Ribas X Direct observation of CuI/CuIII redox steps relevant to Ullmann-type coupling reactions. Chem. Sci 2010, 1, 326–330. [Google Scholar]; (b) King AE; Huffman LM; Casitas A; Costas M; Ribas X; Stahl SS Copper-Catalyzed Aerobic Oxidative Functionalization of an Arene C–H Bond: Evidence for an Aryl-Copper(III) Intermediate. J. Am. Chem. Soc 2010, 132, 12068–12073. [DOI] [PubMed] [Google Scholar]
- (23).Hickman AJ; Sanford MS High-valent organometallic copper and palladium in catalysis. Nature 2012, 484, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kryger RG; Lorand JP; Stevens NR; Herron NR Radicals and scavengers. 7. Diffusion controlled scavenging of phenyl radicals and absolute rate constants of several phenyl radical reactions. J. Am. Chem. Soc 1977, 99, 7589–7600. [Google Scholar]
- (25).Le C; Chen TQ; Liang T; Zhang P; MacMillan DWC A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science 2018, 360, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Boström J; Brown DG; Young RJ; Keserü GM Expanding the medicinal chemistry synthetic toolbox. Nat. Rev. Drug Discovery 2018, 17, 709–727. [DOI] [PubMed] [Google Scholar]
- (27).Kutchukian PS; Dropinski JF; Dykstra KD; Li B; DiRocco DA; Streckfuss EC; Campeau L-C; Cernak T; Vachal P; Davies IW; et al. Chemistry informer libraries: a chemoinformatics enabled approach to evaluate and advance synthetic method. Chem. Sci 2016, 7, 2604–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Garakyaraghi S; McCusker CE; Khan S; Koutnik P; Bui AT; Castellano FN Enhancing the Visible-Light Absorption and Excited-State Properties of Cu(I) MLCT Excited States. Inorg. Chem 2018, 57, 2296–2307. [DOI] [PubMed] [Google Scholar]
- (29).Fayad R; Engl S; Danilov EO; Hauke CE; Reiser O; Castellano FN Direct Evidence of Visible Light-Induced Homolysis in Chlorobis(2,9-dimethyl-1,10-phenanthroline)copper(II). J. Phys. Chem. Lett 2020, 11, 5345–5349. [DOI] [PubMed] [Google Scholar]
- (30).Chateauneuf J; Lusztyk J; Ingold KU Spectroscopic and kinetic characteristics of aroyloxyl radicals. 1. The 4-methoxybenzoyloxyl radical. J. Am. Chem. Soc 1988, 110, 2877–2885. [Google Scholar]
- (31).Chateauneuf J; Lusztyk J; Ingold KU Spectroscopic and kinetic characteristics of aroyloxyl radicals. 2. Benzoyloxyl and ring-substituted aroyloxyl radicals. J. Am. Chem. Soc 1988, 110, 2886–2893. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.