

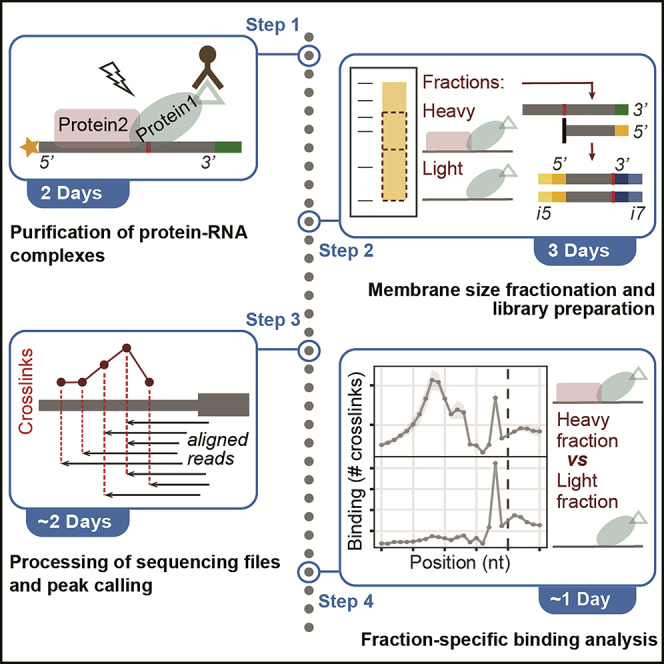
Summary
Thousands of RNA-binding proteins orchestrate RNA processing and altered protein-RNA interactions frequently lead to disease. Here, we present experimental and computational analysis pipelines of fractionated eCLIP-seq (freCLIP-seq), a modification of enhanced UV-crosslinking and RNA immunoprecipitation followed by sequencing. FreCLIP-seq allows transcriptome-wide analysis of protein-RNA interactions at single-nucleotide level and provides an additional level of resolution by isolating binding signals of individual RNA-binding proteins within a multicomponent complex. Binding occupancy can be inferred from read counts and crosslinking events.
For complete details on the use and execution of this protocol, please refer to Biancon et al. (2022).
Subject areas: Bioinformatics, Cell Biology, Molecular Biology, Protein Biochemistry, Protein expression and purification, RNAseq, Sequencing
Graphical abstract
Highlights
-
•
Generation of crosslinked cell pellets and immunoprecipitation
-
•
Isolation of distinct RBP-RNA contacts within a complex by membrane size fractionation
-
•
Identification of binding peaks by a transcriptome-wide or region-specific approach
-
•
Annotation and differential analysis of binding occupancies
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Thousands of RNA-binding proteins orchestrate RNA processing and altered protein-RNA interactions frequently lead to disease. Here, we present experimental and computational analysis pipelines of fractionated eCLIP-seq (freCLIP-seq), a modification of enhanced UV-crosslinking and RNA immunoprecipitation followed by sequencing. FreCLIP-seq allows transcriptome-wide analysis of protein-RNA interactions at single-nucleotide level and provides an additional level of resolution by isolating binding signals of individual RNA-binding proteins within a multicomponent complex. Binding occupancy can be inferred from read counts and crosslinking events.
Before you begin
The protocol below describes the specific steps for performing freCLIP-seq on inducible human erythroleukemia (HEL) cell lines expressing FLAG-tagged wild-type or mutant U2AF1 splicing factor. Together, U2AF1 and U2AF2 form the U2AF complex that recognizes the 3′ splice site (3′SS) and recruits U2 small nuclear ribonucleoproteins (snRNPs) for spliceosome activation. U2AF1 hotspot mutations affecting the S34 or Q157 residues alter RNA binding and are associated with myeloid malignancies and solid cancers. The application of freCLIP-seq allowed us to distinguish U2AF1 from U2AF2 binding signals and to identify distinct targets of the two U2AF subunits. Moreover, we were able to dissect changes in binding occupancy and targets under mutant (S34F or Q157R) vs wildtype conditions.
FreCLIP-seq is based on previously published iCLIP-seq and eCLIP-seq protocols (Hafner et al., 2021; König et al., 2010; Van Nostrand et al., 2016), with the introduction of the membrane size fractionation step that allows to separate RNA binding signals of individual proteins within a complex. The experimental protocol described here has been successfully applied with and without membrane size fractionation in several cellular models, including inducible and standard cell lines for U2AF1 and U2AF2 binding analysis (Biancon et al., 2022; Glasser et al., 2022), and cytoplasmic fractions derived from primary mouse cells for AGO2 binding analysis (Griffin et al., 2022).
We also provide two possible computational approaches for the analysis of freCLIP-seq and eCLIP-seq data to address different research questions. Both approaches are based on read counts (coverage) and crosslinking-induced termination events to identify and quantify protein-RNA interactions. The first approach focuses on the transcriptome-wide identification of protein-RNA interactions, using the PureCLIP peak calling pipeline (Krakau et al., 2017). The second approach focuses on the comparison of binding occupancies on specific transcript regions, i.e., intron-exon junctions.
Our comprehensive protocol is highly adaptable at both experimental and computational levels. It provides suggestions and alternatives to handle different experimental conditions and to easily process obtained sequencing data. Critical steps are sufficiently detailed to allow researchers with different backgrounds and expertise to successfully follow the protocol.
Institutional permissions
Any experiments on live vertebrates or higher invertebrates must be performed in accordance with relevant institutional and national guidelines and regulations.
Cell preparation
Timing: ∼2 weeks
Note: Make sure to grow sufficient cells for at least 2 crosslinked biological replicates and 1 non-crosslinked control sample (Van Nostrand et al., 2016). To validate antibody specificity, we suggest to initially run the immunoprecipitation (IP) with 2 additional control samples: IP with anti-target antibody in cells lacking that target (in our case study, mouse anti-FLAG antibody in non-induced HEL cells lacking FLAG-tagged proteins) and IP with IgG control according to the antibody source (in our case study, mouse IgG).
-
1.
Grow HEL suspension cells in T175 flasks up to 50% confluence in 100 mL RPMI 1640 supplemented with 9% tetracycline-negative FBS, 1% L-glutamine and 1% penicillin-streptomycin. Cells are cultured in 5% CO2 at 37°C.
-
2.
Add 1 μg/mL doxycycline to the cells for 48 h to induce the expression of FLAG-tagged U2AF1 proteins.
Note: The concentration of doxycycline may have to be titrated and optimized for each new model to avoid excessive protein expression in comparison to endogenous levels.
Note: Perform a comparative analysis between two induced conditions (in our case study, FLAG-tagged U2AF1 wildtype vs FLAG-tagged U2AF1 S34F or Q157R) to remove any potential induction-related bias.
UV-crosslinking
-
3.
Count cells and spin down 30 × 106 cells per pellet at 300 × g, 4°C, 5 min (consider preparing: biological replicates, n≥2; technical replicates, n≥2).
-
4.
Each 30 × 106 cell pellet will be resuspended in 25 mL ice-cold PBS. Using a 10 mL serological pipet gently pipet up and down to generate a single cell suspension.
CRITICAL: Do not use a 25 mL pipet as it does not allow for single cell suspension. Optimal crosslinking occurs when HEL cells are at a density of 1.2 × 106 per mL hence the volumes are kept as mentioned above.
-
5.
Add 10 mL of the cells in ice cold PBS per 10 cm Petri dish. For 30 × 106 cell pellet in 25 mL ice-cold PBS, use 3 Petri dishes (10 mL + 10 mL + 5 mL). Ideally, process 60 × 106 cells and plate 50 mL in 5 Petri dishes (10 mL in each).
-
6.
Crosslink 5 Petri dishes at a time on ice. Remove the lid and make sure the layer of PBS covers the surface evenly.
-
7.
Irradiate for 200 mJ/cm2 at 254 nm (if using UV Stratalinker 2400 by Stratagene, press: Energy → 2000 → Start). Swirl the cells and irradiate again for 200 mJ/cm2.
-
8.
Collect the crosslinked HEL cells from the 5 Petri dishes into two 50 mL conical tubes. Use 6 mL ice-cold PBS to wash all 5 Petri dishes and distribute equally into the two 50 mL conical tubes. Spin down at 300 × g, 4°C, 5 min.
-
9.
Prepare dry ice-absolute ethanol bath.
-
10.
Aspirate the supernatant and snap-freeze the pellet in the dry ice-absolute ethanol bath. Pellets can be stored at −80°C until further use.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-FLAG (clone M2) | Sigma-Aldrich | Cat#F1804; RRID: AB_262044 |
| Chemicals, peptides, and recombinant proteins | ||
| RNAse Away Reagent | Thermo Fisher Scientific | Cat#10328011 Note: For removal of RNAse contamination from surfaces, tools and gloves! |
| H2O, RNAse and Endotoxin Tested | AmericanBio | Cat#AB02128-00500; CAS: 7732-18-5 Note: For buffer preparation. |
| EtOH, pure alcohol, 200 proof | AmericanBio | Cat#AB00515-00500; CAS: 64-17-5 Note: For buffer preparation and bead washing steps. |
| RPMI 1640 | Thermo Fisher Scientific | Cat#11875093 |
| Tetracycline-negative FBS | Gemini Bio-Products | Cat#100-800 |
| L-glutamine | Thermo Fisher Scientific | Cat#25030081 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat#15140122; CAS: 3810-74-0 |
| Doxycycline | Sigma-Aldrich | Cat#D3447; CAS: 10592-13-9 |
| Tris HCl, 1 M solution, pH 7.4 | AmericanBio | Cat#AB14044-01000; CAS: 7732-18-5, 77-86-1 |
| Sodium chloride, 5 M solution | AmericanBio | Cat#AB13198-01000; CAS: 7732-18-5, 7647-14-5 |
| NP-40 Surfact-Amps™ Detergent Solution | Thermo Fisher Scientific | Cat#85124; CAS: 9016-45-9 |
| SDS, 10% Solution, RNase-free | Thermo Fisher Scientific | Cat#AM9822; CAS: 151-21-3 |
| Sodium deoxycholate | Sigma-Aldrich | Cat#D6750-10G; CAS: 302-95-4 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#11836170001; CAS: 25322-68-3, 30827-99-7 |
| SUPERase⋅In™ RNase Inhibitor (20 U/μL) | Thermo Fisher Scientific | Cat#AM2696; CAS: 56-81-5 |
| EDTA, 0.5 M Solution, pH 8.0 | AmericanBio | Cat#AB00502-01000; CAS: 7732-18-5, 6381-92-6 |
| Magnesium chloride, 1 M solution | AmericanBio | Cat#AB09006-00100; CAS: 7732-18-5, 7791-18-6 |
| TWEEN 20 | AmericanBio | Cat#AB02038-00500; CAS: 9005-64-5 |
| Tris, 2 M solution, pH 7.5 | AmericanBio | Cat#AB14116-01000; CAS: 7732-18-5, 77-86-1 |
| Potassium chloride solution | Sigma-Aldrich | Cat#60135; CAS: 7732-18-5, 7447-40-7 |
| Triton X-100 Detergent | Bio-Rad | Cat#1610407; CAS: 9002-93-1 |
| Tris-HCl 1 M, pH 6.5 | Teknova | Cat#T1065; CAS: 77-86-1 |
| DTT (5 g) | DOT Scientific | Cat#DSD11000-5; CAS: 3483-12-3 |
| UltraPure™ 1 M Tris-HCI Buffer, pH 7.5 | Thermo Fisher Scientific | Cat#15567027 |
| UltraPure™ Urea | Thermo Fisher Scientific | Cat#15505035 |
| NuPAGE™ MOPS SDS Running Buffer (20×) | Thermo Fisher Scientific | Cat#NP0001; CAS: 151-21-3, 68-12-2 |
| NuPAGE™ Transfer Buffer (20×) | Thermo Fisher Scientific | Cat#NP00061 |
| Methanol ACS reagent, ≥99.8% | Sigma-Aldrich | Cat#179337; CAS: 67-56-1 |
| Novex™ TBE Running Buffer (5×) | Thermo Fisher Scientific | Cat#LC6675 |
| Dynabeads™ Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat#10004D; CAS: 26628-22-8 |
| Ambion™ RNase I (100 U/μL) | Thermo Fisher Scientific | Cat#AM2295; CAS: 56-81-5 |
| TURBO™ DNase (2 U/μL) | Thermo Fisher Scientific | Cat#AM2239; CAS: 56-81-5 |
| FastAP Thermosensitive Alkaline Phosphatase (1 U/μL) | Thermo Fisher Scientific | Cat#EF0651; CAS: 56-81-5, 9002-93-1 |
| AffinityScript Multiple Temperature Reverse Transcriptase | Agilent Technologies | Cat#600107; CAS: 56-81-5, 7447-40-7 |
| T4 Polynucleotide Kinase | New England Biolabs | Cat#M0201L; CAS: 56-81-5, 60-00-4 |
| 100% DMSO | New England Biolabs | Cat#B0515AVIAL; CAS: 67-68-5 |
| T4 RNA Ligase 1 (ssRNA Ligase), High Concentration | New England Biolabs | Cat#M0437M; CAS: 60-00-4 |
| NuPAGE™ LDS Sample Buffer (4×) | Thermo Fisher Scientific | Cat#NP0007; CAS: 56-81-5 |
| ATP, [γ-32P] | PerkinElmer | Cat#BLU002Z250UC |
| NuPAGE™ 4%–12%, Bis-Tris, 1.0–1.5 mm 10 W, Mini Protein Gels | Thermo Fisher Scientific | Cat#NP0321BOX |
| Precision Plus Protein Dual Color Standards | Bio-Rad | Cat#1610374 |
| Nitrocellulose Membrane, Roll, 0.45 μm, 30 cm × 3.5 m | Bio-Rad | Cat#1620115; CAS: 9004-70-0 |
| Mini Trans-Blot® Filter Paper, Pkg of 50 | Bio-Rad | Cat#1703932 |
| HyBlot CL® Autoradiography Film | Thomas Scientific | Cat#1141J51 |
| Proteinase K Solution (20 mg/mL), RNA grade | Thermo Fisher Scientific | Cat#25530049; CAS: 39450-01-6 |
| Acid-Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1) | Thermo Fisher Scientific | Cat#AM9722; CAS: 108-95-2, 67-66-3, 123-51-3 |
| Chloroform | J.T.Baker | Cat#9180-01; CAS: 67-66-3 |
| Dynabeads™ MyOne™ Silane | Thermo Fisher Scientific | Cat#37002D |
| Buffer RLT | QIAGEN | Cat#79216; CAS: 593-84-0 |
| ATP (10 mM) | New England Biolabs | Cat#P0756S; CAS: 1310-73-2 |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | Cat#N0447S |
| ExoSAP-IT™ PCR Product Cleanup Reagent | Thermo Fisher Scientific | Cat#78200.200.UL |
| Sodium hydroxide solution (1.0 N, BioReagent, suitable for cell culture) | Sigma-Aldrich | Cat#S2770-100ML; CAS: 1310-73-2 |
| Hydrochloric acid solution (1.0 N, BioReagent, suitable for cell culture) | Sigma-Aldrich | Cat#H9892-100ML; CAS: 7647-01-0 |
| iQ™ SYBR® Green Supermix | Bio-Rad | Cat#1708880; CAS: 56-81-5, 67-68-5 |
| Q5® High-Fidelity 2× Master Mix | New England Biolabs | Cat#M0492S |
| AMPure XP Reagent | Beckman Coulter | Cat#A63880 |
| Novex™ TBE-Urea Gels, 6%, 12 well | Thermo Fisher Scientific | Cat#EC68652BOX |
| GelStar™ Nucleic Acid Gel Stain, 10,000× | Lonza | Cat#50535; CAS: 67-68-5 |
| Critical commercial assays | ||
| RNA Clean & Concentrator-5 kit | Zymo Research | Cat#R1016 |
| Deposited data | ||
| Sequencing data |
Biancon et al. (2022),Griffin et al. (2022) |
GEO: GSE195620; GSE181264 |
| freCLIP-seq and eCLIP-seq analysis code |
Biancon et al. (2022) This paper |
GitHub: https://github.com/TebaldiLab/eCLIP_seq https://doi.org/10.5281/zenodo.7121228 |
| Original Images | This paper | Mendeley Data: https://doi.org/10.17632/m7g92hkwmh.1 |
| Experimental models: Cell lines | ||
| Human: HEL | ATCC | TIB-180 |
| Experimental models: Organisms/strains | ||
| Mouse: Ago2 conditional knockout males (Ddx4-Cre/+;Ago2flox/Δ) | Griffin et al. (2022) | N/A |
| Oligonucleotides | ||
| RNA oligos (X1A/B, X2A/B, RiL19) and DNA oligos (AR17, rand3Tr3) | Van Nostrand et al. (2016) | eCLIP Standard Operating Procedure v1.P 20151108 |
| Illumina Adapters (D501-508, D701-D712) | Illumina Adapter Sequences | https://support-docs.illumina.com/SHARE/AdapterSeq/illumina-adapter-sequences.pdf Pages 50–51. Remember to order the reverse complement of the sequence reported for D701–D712. |
| Recombinant DNA | ||
| CS-TRE-U2AF1-PRE-Ubc-tTA-I2G plasmids |
Yamaguchi et al., 2012, Yoshida et al., 2011 |
N/A |
| psPAX2 | Addgene | Cat#12260 |
| VSV.G | Addgene | Cat#14888 |
| Software and algorithms | ||
| FastQC | N/A | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| FastUniq | Xu et al. (2012) | https://sourceforge.net/projects/fastuniq/ |
| Cutadapt | Martin (2011) | https://cutadapt.readthedocs.io/en/stable/ |
| STAR v2.7.0f | Dobin et al. (2013) | https://github.com/alexdobin/STAR/releases |
| PureCLIP | Krakau et al. (2017) | https://github.com/skrakau/PureCLIP |
| Bedtools | Quinlan and Hall (2010) | https://github.com/arq5x/bedtools2 |
| edgeR | Robinson et al. (2010) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Tidyverse | CRAN | https://www.tidyverse.org/ |
| data.table | CRAN | https://cran.r-project.org/web/packages/data.table/index.html |
| Ggseqlogo | CRAN | https://cran.r-project.org/web/packages/ggseqlogo/index.html |
| GenomicAlignments | Bioconductor | https://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html |
| GenomicRanges | Bioconductor | https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html |
| Bamsignals | Bioconductor | https://bioconductor.org/packages/release/bioc/html/bamsignals.html |
| BSgenome | Bioconductor | https://bioconductor.org/packages/release/bioc/html/BSgenome.html |
| Other | ||
| RNAse-free filter tips | USA Scientific | Cat#1121-3810, 1120-1810, 1120-8810, 1126-7810 |
| Low-binding microcentrifuge tubes | Sorenson BioScience | 1.7 mL tubes, Cat#11700 |
| Phase Lock Gel Heavy Tubes | Quantabio | 2 mL tubes, Cat#2302830 |
| UV crosslinker | Stratagene | UV Stratalinker 2400 |
| Tube rotator | Any brand/model available in the lab | N/A |
| Sonicator | Branson | Digital Sonifier 450 |
| Thermomixer | Benchmark Scientific | H5000-HC |
| Magnetic separation rack | Any brand/model available in the lab | N/A |
| Laboratory cold room | Yale University | Note: For IP and washing steps. |
| Radioactivity room (only if using γ-32P-ATP, not mandatory) | Yale University | Note: Specific training courses are required. Refer to the safety guidelines of your institution. |
Materials and equipment
| Oligonucleotide | Stock concentration | Working concentration |
|---|---|---|
| X1A/B, X2A/B | 200 μM | 20 μM |
| RiL19 | 200 μM | 40 μM |
| AR17 | 200 μM | 20 μM |
| rand3Tr3 | 200 μM | 80 μM |
| D501-508, D701-D712 | 100 μM | 20 μM |
Lysis Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.4 (1 M) | 50 mM | 5 mL |
| NaCl (5 M) | 100 mM | 2 mL |
| NP-40 (10% solution) | 1% | 10 mL |
| SDS (10% solution) | 0.1% | 1 mL |
| Sodium deoxycholate (10% solution) | 0.5% | 5 mL |
| H2O | N/A | 77 mL |
| Total | N/A | 100 mL |
High-salt Wash Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.4 (1 M) | 50 mM | 5 mL |
| NaCl (5 M) | 1 M | 20 mL |
| EDTA (0.5 M) | 1 mM | 0.2 mL |
| NP-40 (10% solution) | 1% | 10 mL |
| SDS (10% solution) | 0.1% | 1 mL |
| Sodium deoxycholate (10% solution) | 0.5% | 5 mL |
| H2O | N/A | 58.8 mL |
| Total | N/A | 100 mL |
PNK Wash Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.4 (1 M) | 20 mM | 2 mL |
| MgCl2 (1 M) | 10 mM | 1 mL |
| Tween 20 (20% solution) | 0.2% | 1 mL |
| H2O | N/A | 96 mL |
| Total | N/A | 100 mL |
1× FastAP Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris, pH 7.5 (2 M) | 10 mM | 0.25 mL |
| MgCl2 (1 M) | 5 mM | 0.25 mL |
| KCl (3 M solution) | 100 mM | 1.67 mL |
| Triton X-100 | 0.02% | 10 μL |
| H2O | N/A | 47.82 mL |
| Total | N/A | 50 mL |
5× PNK pH 6.5 Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 6.5 (1 M) | 350 mM | 17.5 mL |
| MgCl2 (1 M) | 50 mM | 2.5 mL |
| DTT (100 mM) | 5 mM | 2.5 mL |
| H2O | N/A | 27.5 mL |
| Total | N/A | 50 mL |
10× RNA Ligase Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.5 (1 M) | 500 mM | 25 mL |
| MgCl2 (1 M) | 100 mM | 5 mL |
| H2O | N/A | 20 mL |
| Total | N/A | 50 mL |
Dilute 1:10 in H2O for 1× RNA Ligase Buffer.
Proteinase K Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.5 (1 M) | 100 mM | 10 mL |
| NaCl (5 M) | 50 mM | 1 mL |
| EDTA (0.5 M) | 10 mM | 2 mL |
| H2O | N/A | 87 mL |
| Total | N/A | 100 mL |
Proteinase K/Urea Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.5 (1 M) | 100 mM | 10 mL |
| NaCl (5 M) | 50 mM | 1 mL |
| EDTA (0.5 M) | 10 mM | 2 mL |
| Urea | 7 M | 42 g |
| H2O | N/A | 87 mL |
| Total | N/A | 100 mL |
All buffers contain harmful or toxic reagents. Wear protective gloves/eye protection/face protection.
1× SDS Running Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| NuPAGE™ MOPS SDS Running Buffer (20×) | 1× | 50 mL |
| deionized H2O | N/A | 950 mL |
| Total | N/A | 1,000 mL |
We suggest to prepare it fresh for each experiment. It can be stored at 23°C.
1× Transfer Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| NuPAGE™ Transfer Buffer (20×) | 1× | 50 mL |
| Methanol (100%) | 10% | 100 mL |
| deionized H2O | N/A | 850 mL |
| Total | N/A | 1,000 mL |
We suggest to prepare it fresh for each experiment.
1× TBE Running Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Novex™ TBE Running Buffer (5×) | 1× | 200 mL |
| deionized H2O | N/A | 800 mL |
| Total | N/A | 1,000 mL |
We suggest to prepare it fresh for each experiment. It can be stored at 23°C.
Step-by-step method details
Purification of protein-RNA complexes
The experimental pipeline has two major steps. This first step allows to purify protein-RNA complexes, constituted by the protein(s) of interest and bound fragmented RNAs from crosslinked cell lysates.
Day 1
-
1.Cell lysis.
-
a.Prepare Lysis Mix: to 10 mL Lysis Buffer, add 400 μL Proteinase Inhibitor Cocktail (1 tablet dissolved in 400 μL H2O) and 20 μL SUPERase⋅In™ RNase Inhibitor.
-
b.Thaw crosslinked cell pellets on ice. Resuspend each pellet in 1.2 mL Lysis Mix.
-
a.
-
2.Bead preparation (Dynabeads™ Protein G).
-
a.Vortex Protein G beads, supplied in a solution containing sodium azide as preservative, for 30 s to obtain a homogeneous suspension.
-
b.Per each sample, transfer 50 μL of bead suspension to a labeled Low Binding tube.Note: The volume of Protein G beads can be scaled up or down according to the experimental conditions. Standard iCLIP-seq and eCLIP-seq suggest to use 100–125 μL beads/sample (König et al., 2010; Van Nostrand et al., 2016).
-
c.Place Low Binding tubes on the magnetic rack, which will collect beads on the side of the tubes.
-
d.Wait 30 s until the solution is clear.
-
e.Keeping the tubes on the magnet, remove the supernatant without disturbing beads.
-
f.Wash Protein G beads 2× with 500 μL Lysis Mix.
-
i.Remove the tubes from the magnetic rack.
-
ii.Resuspend beads in 500 μL Lysis Mix.
-
iii.Replace tubes onto magnetic rack.
-
iv.Wait 30 s until the solution is clear and discard supernatant.
-
v.Repeat for a second time.
-
i.
-
g.Resuspend beads in 500 μL Lysis Mix.
-
h.Add 12 μg of anti-FLAG antibody (12 μL of 1 mg/mL stock stored at −20°C) to the bead slurry.Note: The amount of antibody depends on the antibody quality, purity, binding strength, and specificity. This should be optimized in preliminary experiments following manufacturer’s instructions for IP application.
-
i.Rotate tubes at 23°C for 60 min. In the meantime, go to 3. Partial RNA digestion.
-
j.When cell lysates are ready at the end of step 2, wash Protein G beads with bound antibody 2× with 500 μL Lysis Mix.
-
k.Resuspend beads in 500 μL Lysis Mix and keep on ice.
-
a.
-
3.Partial RNA digestion.Optional: Sonicate samples (not required for cytoplasmic fraction as indicated in Brugiolo et al., 2017):
- Clean sonicator tip with RNAse Away Reagent before using it.
- Transfer each lysate to a 15 mL tube and place the tube in an ice beaker.CRITICAL: Avoid foaming. Keep the probe approximately 0.5 cm from the bottom of the tube without touching tube sides.
- Sonicate with 6 pulses each 10 s at 30% amplitude, 20 s pause.Note: We use the Branson Digital Sonifier 450. Sonication conditions may need to be converted to suit another instrument.
- Transfer 1.2 mL lysate to Low Binding tube.
- Spin down at 16,000 × g, 4°C, 10 min.
- Transfer the supernatant into a fresh Low Binding tube (leave some to prevent carryover).
-
a.Prepare a dilution of RNAse I at 1:30 in H2O [Low RNase].Note: Several RNAse dilutions should be initially tested with the help of P32-labeling in step 9 (Huppertz et al., 2014). With a 1:50 High RNase dilution, immunoprecipitated proteins are bound to short RNAs migrating as a less diffuse band ∼5 kD above the expected molecular weight of the protein (this also helps in determining the actual protein size). With Low RNase conditions the band shifts up and becomes more diffuse.
-
b.Add 10 μL of diluted RNAse I and 5 μL of TURBO™ DNase to each lysate.
-
c.Incubate at 37°C exactly for 3 min while shaking at 1,100 rpm.
-
d.Place immediately on ice for at least 3 min.
-
4.Immunoprecipitation.
-
a.For each cell lysate, save 24 μL (2%) as pre-IP input sample for library preparation, and other 24 μL as pre-IP control for western blot. Store at 4°C.Note: Paired pre-IP input samples are sequenced to allow normalization for background signal and potential differences in transcript abundances between conditions.
-
b.Magnetically remove the Lysis Mix from the Protein G beads with bound antibody.
-
c.Add the remaining cell lysate to the beads.
-
d.Rotate beads/lysate mix for 1 h at 4°C in the cold room.
-
e.Wash beads with bound protein-RNA complexes 3× with 800 μL High-salt Wash Buffer.
-
i.Short spin and magnetic separation (let sit for 2 min in the cold room).
-
ii.Discard supernatant.
-
iii.Add 800 μL High-salt Wash Buffer, vortex for few seconds until the beads are fully resuspended, 2 min rotation in the cold room.
-
iv.Repeat for other 2 times.
-
i.
-
f.Wash 1× with 800 μL PNK Wash Buffer for 2 min rotation in the cold room as in 4.e.
-
g.Wash 1× with 800 μL 1× FastAP Buffer for 2 min rotation in the cold room as in 4.e. At the end, remove residual buffer with fine tip.
-
a.
-
5.3′ end dephosphorylation.
-
a.FastAP treatment. Prepare FastAP Master Mix for n+1 samples as follows:
Reagent Amount 10× FastAP Buffer 10 μL SUPERase·In™ RNase Inhibitor 2 μL TURBO™ DNase 1 μL FastAP enzyme 8 μL H2O 79 μL Mix, add 100 μL/sample, incubate in Thermomixer at 1,200 rpm, 37°C, 15 min. Then, short spin and magnetic separation to remove Master Mix. -
b.PNK treatment. Prepare PNK Master Mix for n+1 samples as follows:
Reagent Amount 5× PNK pH 6.5 Buffer 60 μL 100 mM DTT 3 μL SUPERase·In™ RNase Inhibitor 5 μL TURBO™ DNase 1 μL T4 PNK enzyme 7 μL H2O 224 μL Mix, add 300 μL/sample, incubate in Thermomixer at 1,200 rpm, 37°C, 20 min. Then, short spin and magnetic separation to remove Master Mix.
-
a.
-
6.Wash beads with bound protein-RNA complexes as in 4.e.
-
a.Wash 1× with 800 μL PNK Wash Buffer for 2 min rotation in the cold room.
-
b.Wash 1× with 800 μL High-salt Wash Buffer for 2 min rotation in the cold room.
-
c.Wash 2× with 800 μL PNK Wash Buffer for 2 min rotation in the cold room.
-
d.Wash 2× with 300 μL 1× RNA Ligase Buffer for 2 min rotation in the cold room. At the end, remove residual buffer with fine tip.
-
a.
-
7.3′ linker ligation.
-
a.Prepare 3′ Ligation Master Mix for n+1 samples as follows:
Reagent Amount 10× RNA Ligase Buffer 3 μL 100 mM ATP 0.3 μL 100% DMSO 0.8 μL SUPERase⋅In™ RNase Inhibitor 0.4 μL T4 RNA Ligase High Concentration 2.5 μL 50% PEG 8000 9 μL H2O 9 μL Mix carefully, add 25 μL/sample, flick. -
b.To each sample, add 5 μL of a different barcoded RNA adapter at 20 μM: X1A, X1B, X2A, X2B.
-
c.Incubate at 23°C for 75 min flicking every 10 min.
 Pause point: Keep at 4°C for 12–16 h.
Pause point: Keep at 4°C for 12–16 h.
-
a.
Day 2
-
8.Wash beads with bound protein-RNA complexes as in 4.e.
-
a.Magnetically separate (let sit for 5 min) and remove supernatant.
-
b.Wash beads 1× with 800 μL PNK Wash Buffer for 2 min rotation in the cold room.
-
c.Wash 2× with 800 μL High-salt Wash Buffer for 2 min rotation in the cold room.
-
d.Wash 2× with 800 μL PNK Wash Buffer for 2 min rotation in the cold room.
-
e.Resuspend beads in 1 mL PNK Wash Buffer and keep on ice.
-
f.Dilute 4× NuPAGE™ Loading Buffer to 1.5× in H2O.
-
g.Add 20 μL 1.5× NuPAGE™ Buffer to pre-IP controls for western blot saved on Day 1.
-
h.Take out 100 μL of the 1 mL beads in PNK Wash Buffer, perform magnetic separation, discard supernatant and resuspend beads in 20 μL 1.5× NuPAGE™ Buffer as post-IP controls for western blot. Keep on ice.
-
a.
-
9.Optional: P32-labeling.Note: We usually perform radiolabeling because it helps with visualization and purification of cross-linked protein-RNA complexes after SDS-PAGE and membrane transfer, in the optimization of RNA digestion conditions and in the identification of potential non-specific signal. However, as reported in Van Nostrand et al. (2016), this is not a mandatory step.
-
a.Take out another 100 μL beads of the remaining 900 μL beads in PNK Wash Buffer.Prepare PNK Master Mix for n+1 samples as follows:
Reagent Amount T4 PNK enzyme 0.2 μL 10× PNK buffer 0.4 μL H2O 3 μL Discard the buffer from the beads, add 3.6 μL/sample. -
b.Add 0.4 μL γ-32P-ATP (corresponding to 4 μCi).CRITICAL: The half-life of γ-32P-ATP is 14.3 days. Adjust γ-32P-ATP and H2O volumes accordingly (e.g., if the use date is 2 weeks after the calibration date, double γ-32P-ATP volume using 0.8 μL and reduce H2O to 2.6 μL https://www.perkinelmer.com/tools/calculatorrad/product).
-
c.Mix well by pipetting up and down and incubate in Thermomixer at 1,100 rpm, 37°C, 5 min.
-
d.Perform short spin and magnetic separation.
-
e.Discard the buffer after magnetic separation and resuspend radioactive beads in 20 μL 1.5× NuPAGE™ Buffer.
-
f.Remove the buffer from the remaining cold beads (800 μL).
-
g.Collect NuPAGE™ Buffer + radioactive beads and combine with corresponding cold beads.
-
a.
-
10.Prepare samples for gel loading.
-
a.Denature experimental samples and pre-/post-IP controls for western blot in Thermomixer at 70°C, 10 min, without shaking.
-
b.Collect eluates and keep on ice. Store controls for western blot at −80°C.
-
a.
-
11.SDS-PAGE and membrane transfer.
-
a.Have ready the prepared 1× SDS Running Buffer and 1× Transfer Buffer.
-
b.Use 10-well NuPAGE™ 4%–12% Bis-Tris gels. Peel off the tape and place the gel cassette into the electrophoresis tank with the lower cassette side facing you.Note: We use the Thermo Fisher Scientific Mini Gel Tank that can allocate up to 2 gels per run. Remove the cassette clamp from the unused chamber to run only one gel. See manufacturer’s instructions available here: https://www.thermofisher.com/order/catalog/product/A25977.
-
c.Fill the tank with 1× SDS Running Buffer.
-
d.Load 20 μL eluted samples and 6 μL pre-stained protein ladder.CRITICAL: Leave one lane free for cutting (max 4 samples/gel). Immediately before loading, use a pipette or syringe to rinse the wells 3 times with 1× SDS Running Buffer.
-
e.Run at 180 V, 55 min.
-
f.After the gel run, cut off wells and dye front (after P32-labeling, it contains free radioactive ATP).
-
g.Soak sponge pads and filter papers in pre-cold 1× Transfer Buffer.
-
h.Use 0.45 μm nitrocellulose membranes.Note: Nitrocellulose membranes can represent a source of bacterial contamination (Van Nostrand et al., 2017; Rosenberg et al., 2021). Using Bio-Rad membranes (Cat#1620115) we consistently obtained mapping rates between 80%–90%. If low mapping rates are encountered, consider changing membranes.
-
i.Assemble the transfer stack as follows:
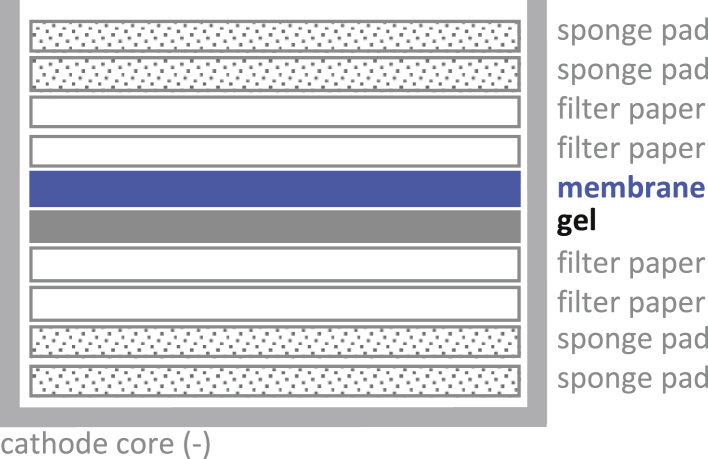 CRITICAL: Use a roller to remove air bubbles trapped in the stack!Note: We use the Thermo Fisher Scientific XCell II™ Blot Module in the XCell SureLock system. See manufacturer’s instructions available here: https://www.thermofisher.com/order/catalog/product/EI9051.
CRITICAL: Use a roller to remove air bubbles trapped in the stack!Note: We use the Thermo Fisher Scientific XCell II™ Blot Module in the XCell SureLock system. See manufacturer’s instructions available here: https://www.thermofisher.com/order/catalog/product/EI9051. -
j.Fill the blot module with 1× Transfer Buffer.
-
k.Transfer at 30 V, 90 min.
-
l.After the transfer, rinse the membrane in 1× PBS and wrap it in plastic wrap.Pause point: Store the membrane at −80°C for 12–16 h.Optional: If you performed P32-labeling: Tape the wrapped membrane to the cassette with intensifying screen (we use Kodak BioMax Cassette with BioMax MS Intensifying Screen). Use luminescent stickers to help aligning the developed film after exposure to the membrane.Pause point: Expose the membrane to X-ray film for 12–16 h.
-
a.

Fractionation and library preparation
This second major step of the experimental pipeline extracts RNAs from protein-RNA complexes and converts isolated RNAs into cDNA high-throughput sequencing libraries.
Day 3
-
12.Membrane cutting (with or without fractionation).
-
a.Optional if you performed P32-labeling: after exposure for 12–16 h, proceed with autoradiographic visualization. Use the signal on the developed film as a mask for cutting out regions of interest. Remember that you are working with radioactive RNA!Note: In case of experimental samples with absent or faint signal, see troubleshooting 1.
-
b.Cut each sample (and each fraction) with a fresh sterile scalpel blade.Note: The average molecular weight of 70 nt long RNA is ∼20 kD. The 3′ RNA linker is 34 nt long, ∼10 kD. Cut the membrane considering the size of the protein with the crosslinked fragmented RNA (Huppertz et al., 2014; Van Nostrand et al., 2016). For U2AF1 freCLIP-seq (Biancon et al., 2022), we cut a “light fraction” between 37 kD (U2AF1 size) and 65 kD, containing U2AF1 monomer plus bound RNA, and a “heavy fraction” between 65 kD (U2AF2 size) and 110 kD, containing U2AF2 and U2AF heterodimer plus bound RNA (Figure 1 top panel). To apply our pipeline without membrane size fractionation, cut a lane from the protein size to approximately 80 kD above it. For AGO2 eCLIP-seq (Griffin et al., 2022), according to our western blot results and to the signal on the developed film, we cut a lane starting between 75-100 kD (AGO2 size is 97 kD) and ending approximately at 250 kD (Figure 1 bottom panel).Note: Control samples should show absent or significantly reduced signal in comparison to experimental samples (Figure S1), resulting in insufficient material for library preparation and sequencing and therefore confirming insignificant non-specific binding.
-
c.Place each membrane region on a fresh glass slide and slice it into 1–2 mm pieces. Transfer all the pieces to a Low Binding 1.7 mL tube.CRITICAL: Use tweezers to remove the plastic wrap and handle the membrane but don’t forget to clean them with 70% ethanol and RNAse Away Reagent between each sample (and each fraction).
-
a.
-
13.RNA extraction.
-
a.Submerge the membrane pieces in 200 μL Proteinase K Buffer. Add 10 μL Proteinase K. Incubate in a Thermomixer at 1,100 rpm, 37°C, 20 min.
-
b.Add 200 μL Proteinase K/Urea Buffer. Incubate in Thermomixer at 1,100 rpm, 37°C, 20 min.
-
c.Spin 2 mL Phase Lock Gel tubes at 16,000 × g, 23°C, 1 min before use.
-
d.Transfer the supernatant (without membrane slices) to a Phase Lock Gel tube.
-
e.Add 400 μL phenol/chloroform. Incubate in Thermomixer at 1,100 rpm, 30°C, 5 min.
-
f.Spin down at 16,000 × g, 23°C, 5 min.
-
g.Add 400 μL chloroform. Incubate in Thermomixer at 1,100 rpm, 30°C, 5 min.
-
h.Spin down at 16,000 × g, 23°C, 5 min.
-
i.Transfer the upper aqueous layer (350 μL, avoid the gel barrier) to 15 mL conical tube.
-
j.RNA Cleanup (RNA Clean & Concentrator-5 kit. All centrifugation steps are carried out at 13,000 × g, 23°C, 30 s).
-
i.Add 700 μL RNA Binding Buffer.
-
ii.Add 1,050 μL ethanol 100% and mix.
-
iii.Transfer 750 μL sample to Zymo-Spin IC Column. Spin down and discard flow-through. Repeat 2× to transfer all sample volume.
-
iv.Add 400 μL RNA Prep Buffer. Spin down and discard flow-through.
-
v.Add 700 μL RNA Wash Buffer. Spin down and discard flow-through.
-
vi.Add 400 μL RNA Wash Buffer. Spin down and discard flow-through. Centrifuge for additional 2 min.
-
vii.Transfer column to a Low Binding tube. Add 12 μL DNase/RNase-Free Water to column, let sit for 1 min and spin down.
-
i.
-
a.
-
14.Processing of pre-IP input samples.
-
a.Take out 10 μL from the pre-IP input samples stored at 4°C on Day 1.
-
b.FastAP treatment. Prepare FastAP Master Mix for n+1 samples as follows:
Reagent Amount 10× FastAP Buffer 2.5 μL SUPERase⋅In™ RNase Inhibitor 0.5 μL FastAP enzyme 2.5 μL H2O 10 μL Mix, add 15.5 μL/sample, incubate in Thermomixer at 1,200 rpm, 37°C, 15 min. -
c.PNK treatment. Prepare PNK Master Mix for n+1 samples as follows:
Reagent Amount 5× PNK pH 6.5 Buffer 20 μL 100 mM DTT 1 μL SUPERase⋅In™ RNase Inhibitor 1 μL TURBO™ DNase 1 μL T4 PNK enzyme 7 μL H2O 45 μL Mix, add 75 μL/sample, incubate in Thermomixer at 1,200 rpm, 37°C, 20 min. Then, short spin and magnetic separation. -
d.Silane cleanup of input RNA (Dynabeads™ MyOne™ Silane).
-
i.Vortex Silane beads for 30 s. Magnetically separate 20 μL beads per sample in Low Binding tubes, remove supernatant.
-
ii.Wash beads with 900 μL Buffer RLT (let sit 30 s).
-
iii.Resuspend beads in 300 μL Buffer RLT and add them to sample, mix.
-
iv.Add 10 μL 5 M NaCl.
-
v.Add 615 μL 100% EtOH.
-
vi.Mix and rotate at 23°C for 15 min.
-
vii.Magnetically separate and remove supernatant.
-
viii.Resuspend beads in 1 mL 75% EtOH and move suspension to a new Low Binding tube.
-
ix.After 30 s, magnetically separate and remove supernatant.
-
x.Wash beads 2× with 75% EtOH (let sit 30 s).
-
xi.Quick spin and perform magnetic separation to remove residual liquid.
-
xii.Air-dry 5 min (do not over-dry!).
-
xiii.Resuspend beads in 10 μL H2O, let sit 5 min.
-
xiv.Magnetically separate and transfer supernatant to new Low Binding tubes: 5 μL for proceeding with library preparation (keep on ice), 5 μL as backup sample (store at −80°C).
-
i.
-
e.3′ linker ligation to input RNA.
-
i.Add 1.5 μL 100% DMSO.
-
ii.Add 0.5 μL RiL19 barcoded RNA adapter at 40 μM.
-
iii.Incubate in Thermomixer at 65°C, 2 min, no shaking. Place immediately on ice for at least 1 min.
-
iv.Prepare 3′ Ligation Master Mix for n+1 samples as follows:
Reagent Amount 10× T4 RNA Ligase Reaction Buffer 2 μL 10 mM ATP 2 μL 100% DMSO 0.3 μL SUPERase·In™ RNase Inhibitor 0.2 μL T4 RNA Ligase High Concentration 1.3 μL 50% PEG 8000 8 μL Mix carefully, add 13.8 μL/sample, flick, incubate at 23°C for 75 min (flick to mix every 15 min).
-
i.
-
f.Silane cleanup of linker-ligated input RNA (Dynabeads™ MyOne™ Silane).
-
i.Vortex Silane beads for 30 s. Magnetically separate 20 μL beads per sample in Low Binding tubes, remove supernatant.
-
ii.Wash beads with 900 μL Buffer RLT (let sit 30 s).
-
iii.Resuspend beads in 61.6 μL Buffer RLT and add them to sample, mix.
-
iv.Add 61.6 μL 100% EtOH. Mix every 5 min for 15 min.
-
v.Magnetically separate and remove supernatant.
-
vi.Resuspend beads in 1 mL 75% EtOH and move suspension to a new Low Binding tube.
-
vii.After 30 s, magnetically separate and remove supernatant.
-
viii.Wash beads 2× with 75% EtOH (let sit 30 s).
-
ix.Quick spin and perform magnetic separation to remove residual liquid.
-
x.Air-dry 5 min (do not over-dry!).
-
xi.Resuspend beads in 10 μL H2O, let sit 5 min.
-
xii.Magnetically separate and transfer supernatant to a new Low Binding tube.Pause point: Store the extracted INPUT RNA at −80°C. CLIP and INPUT RNA samples are now all synchronized.
-
i.
-
a.
Figure 1.

Autoradiographic visualization and membrane size-selection
Top: representative U2AF1 freCLIP-seq membrane cutting to obtain U2AF1 light and heavy fractions in U2AF1 wildtype and mutant (S34F or Q157R) conditions. Figure adapted and reprinted with permission from (Biancon et al., 2022). Bottom: representative AGO2 eCLIP-seq membrane cutting to obtain AGO2-RNA complexes. See also Figure S1.
Day 4
-
15.Reverse transcription.
-
a.Primer annealing.
-
i.Thaw stored 10 μL CLIP and INPUT RNA samples.Note: If you performed P32-labeling, CLIP samples are still radioactive.
-
ii.Transfer all the samples to 8-tube PCR strips. Make a RTneg tube with 10 μL H2O.
-
iii.Add 0.5 μL AR17 primer at 20 μM.
-
iv.Place at 65°C for 2 min in pre-heated PCR block. Place immediately on ice for at least 1 min.
-
i.
-
b.Prepare Reverse Transcription Mix for n+1 samples as follows:
Reagent Amount 10× AffinityScript RT buffer 2 μL 100 mM DTT 2 μL dNTPs (10 mM each) 2 μL SUPERase⋅In™ RNase Inhibitor 0.3 μL AffinityScript Multi-Temp RT 0.9 μL H2O 2.8 μL Mix, add 10 μL/sample, incubate in pre-heated PCR block at 55°C, 45 min.
-
a.
-
16.RT cleanup.
-
a.Add 3.5 μL ExoSAP-IT™ to each sample, pipette-mix.
-
b.Incubate in pre-heated PCR block at 37°C, 15 min.
-
c.Add 1 μL 0.5 M EDTA.
-
d.Add 3 μL 1 M NaOH, pipette-mix.
-
e.Incubate in pre-heated PCR block at 70°C, 12 min.
-
f.Add 3 μL 1 M HCl, pipette-mix.
-
a.
-
17.Silane cleanup of cDNA (Dynabeads™ MyOne™ Silane).
-
a.Vortex Silane beads for 30 s. Magnetically separate 10 μL beads per sample in Low Binding tubes, remove supernatant.
-
b.Wash beads with 500 μL Buffer RLT (let sit 30 s).
-
c.Resuspend beads in 93 μL Buffer RLT and add them to sample, mix.
-
d.Add 111.6 μL 100% EtOH. Mix, let sit for 5 min, mix, let sit for other 5 min. Then, magnetically separate and remove supernatant.
-
e.Resuspend beads in 1 mL 80% EtOH and move suspension to a new Low Binding tube.Note: If performed P32-labeling, at this point samples are not radioactive anymore.
-
f.After 30 s, magnetically separate and remove supernatant.
-
g.Wash beads 2× with 80% EtOH (let sit 30 s). Quick spin and perform magnetic separation to remove residual liquid.
-
h.Air-dry 5 min (do not over-dry!).
-
i.Resuspend beads in 5 μL 5 mM Tris-HCl pH 7.5, let sit 5 min (do not remove from beads).
-
a.
-
18.3′ linker ligation to cDNA (5′ ligation considering the original RNA fragment).
-
a.Add 0.8 μL rand3Tr3 ssDNA adapter at 80 μM.Note: Added to single strand cDNA fragments, this adapter works as a randomer-UMI because it contains an inline randomer of 10 nt that allows to recognize and remove PCR duplicates.
-
b.Add 1 μL 100% DMSO.
-
c.Incubate in Thermomixer at 75°C, 2 min, no shaking. Place immediately on ice for at least 1 min.
-
d.Prepare 5′ Ligation Master Mix for n+1 samples as follows:
Reagent Amount 10× T4 RNA Ligase Reaction Buffer 2 μL 10 mM ATP 2 μL T4 RNA Ligase High Concentration 0.5 μL 50% PEG 8000 9 μL Mix carefully, add 13.5 μL/sample, mix by stirring contents. -
e.Add another 1 μL T4 RNA Ligase High Concentration to each sample, flick to mix.
-
f.Incubate in Thermomixer at 1,200 rpm, 23°C, 30 s. Place tubes in a rack on your bench.Pause point: Flick ideally every hour, at least twice, before incubating at 23°C for 12–16 h.
-
a.
Day 5
-
19.Silane cleanup of linker-ligated cDNA (Dynabeads™ MyOne™ Silane).
-
a.Vortex Silane beads for 30 s. Magnetically separate 5 μL beads per sample in Low Binding tubes, remove supernatant.
-
b.Wash beads with 500 μL Buffer RLT (let sit 30 s).
-
c.Resuspend beads in 60 μL Buffer RLT and add them to sample, mix.
-
d.Add 60 μL 100% EtOH. Mix, let sit 5 min, mix, let sit other 5 min. Then, magnetically separate and remove supernatant.
-
e.Resuspend beads in 1 mL 75% EtOH and move suspension to a new Low Binding tube.
-
f.After 30 s, magnetically separate and remove supernatant.
-
g.Wash beads 2× with 75% EtOH (let sit 30 s). Quick spin and perform magnetic separation to remove residual liquid.
-
h.Air-dry 5 min (do not over-dry!).
-
i.Resuspend beads in 27 μL 10 mM Tris-HCl pH 7.5, let sit 5 min.
-
j.Magnetically separate, transfer 25 μL supernatant to a new Low Binding tube and place on ice.
-
a.
-
20.cDNA quantification by qPCR.
-
a.Dilute cDNA samples 1:10 in H2O. As controls, use RT H2O (RTneg) and qPCR H2O. Samples and controls are quantified in triplicate.
-
b.Prepare qPCR Master Mix for n+3 samples as follows:
Reagent Amount iQ™ SYBR® Green Supermix 5 μL qPCR primer mix (10 μM each D502/D702) 0.4 μL H2O 3.6 μL Mix, add 9 μL/sample in 96 well qPCR plate, add 1 μL diluted cDNA or controls, mix, seal. -
c.Run qPCR (“Quick Plate_96 wells_SYBR Only” 2-step protocol in the BIO-RAD CFX96 Touch Real-Time PCR Detection System) with the following cycling conditions:
Steps Temperature Time Cycles Initial Denaturation 95°C 3 min 1 Denaturation 95°C 15 s 39 cycles Annealing/Extension 60°C 30 s Melt/Dissociation 55°C 5 s 1 95°C 50 s -
d.Use automatically calculated Cq values. The number of cycles for final PCR amplification will be 3 cycles less than the Cq of the diluted CLIP sample.Note: The “3 cycles less” rule may be adjusted considering the type of sample (CLIP or INPUT), final library concentration and possible adapter bands.
-
a.
-
21.cDNA amplification.
-
a.Organize 8-tube PCR strips according to the Cq values of each sample.
-
b.Transfer 12.5 μL CLIP samples or 10 μL INPUT samples in the assigned strips. For each PCR strip, make a PCRneg tube with 10 μL H2O. Store the remaining 12.5 μL CLIP samples and 15 μL INPUT samples at −80°C for troubleshooting 2 (pre-AMP libraries).
-
c.Dispense PCR Master Mix in 8-tube PCR strips as follows:
Reagent Amount Q5® High-Fidelity 2× Master Mix 25 μL H2O 7.5 μL CLIP or 10 μL INPUT 20 μM right primer D50× 2.5 μL 20 μM left primer D70× 2.5 μL Note: For adapter pairs and sample pooling, we follow Illumina guidelines reported in Table S1 (and previously available as document #770-2017-004-B). Refer to Illumina website for current guidelines: https://support.illumina.com/downloads/index-adapters-pooling-guide-1000000041074.html. Illumina adapters were purchased from Integrated DNA Technologies, IDT (100 nmole DNA Oligos, PAGE purification).For PCRneg tubes, use adapter pair of one of the library samples in the strip. -
d.Mix and run PCR with the following cycling conditions:
Steps Temperature Time Cycles Initial Denaturation 98°C 30 s 1 Denaturation 98°C 15 s 6 cycles Annealing 68°C 30 s Extension 72°C 40 s Denaturation 98°C 15 s X cycles (-3 rule) Annealing/Extension 72°C 1 min Final extension 72°C 1 min 1 Hold 4°C forever Note: INPUT samples are typically amplified with 9 total cycles (6+3); CLIP samples, especially freCLIP fractions, typically required 16–18 total cycles (6+10–12). If Cq value=22, keep 18 total cycles; if Cq value ≥22 but lower than H2O Cq, use 19 total cycles, do not increase further! As reported in Van Nostrand et al. (2016), 18 cycles will yield ∼30%–50% PCR duplicate reads.
-
a.
-
22.SPRI cleanup of amplified libraries (AMPure XP beads).
-
a.Vortex AMPure XP beads for 30 s.
-
b.Add 90 μL beads to 50 μL PCR products, mix well and incubate at 23°C, 10 min (mix 2× during incubation).
-
c.Magnetically separate and remove supernatant.
-
d.Wash beads 2× with 75% EtOH (let sit 30 s).
-
e.Short spin and magnetic separation to remove residual liquid.
-
f.Air-dry 5 min (do not over-dry!).
-
g.Resuspend beads in 20 μL H2O, let sit 5 min.
-
h.Magnetically separate, transfer 18 μL supernatant to a new Low Binding tube (post-AMP libraries).
-
a.
-
23.Optional: Library visualization by TBE-Urea gel.
-
a.Have ready the prepared 1× TBE Running Buffer.
-
b.Take out 6 μL purified post-AMP libraries and mix with 1 μL 6× loading dye.
-
c.Run libraries on 6% TBE-Urea gel at 180 V, 40 min.
-
d.Stain gel with GelStar™ (3 μL GelStar™ in 50 mL 1× TBE Running Buffer). Gentle shaking at 23°C, 10 min.
-
e.Visualize gel (Figure 2A). Library size should be between 175–350 bp.
-
a.
Note: Primer excess (<100 bp) does not affect sequencing because these short fragments cannot bind to the flow cell. Consider buying new AMPure XP beads!
Figure 2.

Quality control of representative U2AF1 freCLIP-seq libraries before sequencing
(A) Visualization by TBE-Urea gel. Yellow lines mark the expected library size.
(B) Visualization by Agilent 2100 Bioanalyzer. Peak size is reported. Lower and upper marker peaks are indicated, respectively, in green and purple. These libraries are ready for sequencing: 175–350 bp range (yellow box), good concentration, no adapter dimers (<10%), primer excess can be ignored.
Expected outcomes
Steps 1–22 of the experimental pipeline will allow to obtain freCLIP-seq or eCLIP-seq libraries that are ready for sequencing.
Right after SPRI cleanup, purified post-AMP libraries can be visualized by TBE-Urea gel (Figure 2A). This step is useful to quickly verify the presence of libraries within the correct bp range and possible adapter contamination. However, we discourage to perform this step with low concentration libraries (high Cq values) to avoid wasting precious sample.
Before sequencing, purified post-AMP libraries need to be subjected to quality control and quantification through the steps listed below. These steps were performed by the Yale Center for Genome Analysis (YCGA).
Quality control (QC) by automated electrophoresis. Run purified post-AMP libraries on Agilent 2100 Bioanalyzer using the High Sensitivity DNA Assay (Cat#5067-4626). Assess library size (175–350 bp), concentration, primer excess (80 bp peak) and adapter dimers (150 bp peak) (Figure 2B). For libraries with over 10% adapter dimers (troubleshooting 2), proceed to step 2.
Optional: Size-selection by gel purification. Run libraries on 2% Invitrogen™ E-Gel™ EX Agarose Gels (Cat#A42346), and gel-extract using QIAGEN QIAquick Gel Extraction Kit (Cat#28706). Repeat QC.
Library quantification. To quantify adapter-ligated fragments in the libraries, use Roche KAPA Library Quantification Kit optimized for Illumina/LightCycler 480 (Cat#07960298001). Required concentration: 1.15 nM (for NovaSeq).
Our freCLIP-seq and eCLIP-seq libraries were deep-sequenced on Illumina HiSeq 2500 system paired-end 75 bp, and on Illumina HiSeq 4000 or NovaSeq 6000 system paired-end 100 bp, at 80 million reads/sample.
Alternative strategy
Single-end CLIP (seCLIP). As reported in Van Nostrand et al. (2017), seCLIP exploits a new 3′ linker ligation strategy that allows for single-end sequencing thus reducing sequencing costs. In the paired-end eCLIP, the crosslinked binding site corresponds to the nucleotide right before the 5′ of the second paired-end read (R2). seCLIP protocol inverts this read structure removing the need of paired-end sequencing. Moreover, the new released seCLIP protocol (Blue et al., 2022) introduced biotin labeling for RNA visualization overcoming the absence of visualization methods or the use of radioactivity. Our pipeline can be easily adapted for seCLIP, at both the experimental and computational level.
Quantification and statistical analysis
We here describe our computational pipeline for the analysis of freCLIP-seq and eCLIP-seq data. Sample datasets to repeat our approach are available in GEO under the accession numbers GSE195620 (U2AF1 freCLIP-seq) and GSE181264 (AGO2 eCLIP-seq).
Pre-processing of next-generation sequencing data
This section lists all the computational steps necessary to obtain aligned eCLIP-seq reads (BAM files), starting from sequencing raw data (FASTQ files). The details are specified for paired-end eCLIP-seq libraries, but can be easily adapted to single-end libraries. An implementation of these steps is available as a bash script (https://github.com/TebaldiLab/eCLIP_seq). For any step, alternative computational tools can be used, with additional care optionally necessary to optimize the compatibility of input/output file formats.
-
1.
Quality control with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). For multiple samples, QC can be visually inspected with MultiQC (https://multiqc.info/). Output: quality control reports.
-
2.
Deduplication with FastUniq, a tool for removal of duplicates in paired short reads, not requiring complete genome sequences as prerequisites (Xu et al., 2012). Output: FASTQ files with unique reads that are used in the following step.
-
3.
Adapter removal with Cutadapt (Martin, 2011). In case of paired-end libraries, this step is performed on R2, removing the first 10 nucleotides (randomer-UMI) and removing the sequence of the 3′ RNA linker (Table S2) from the end of the read. Only reads longer than 10 nucleotides after linker removal are kept. Output: FASTQ files with trimmed reads, to be used in the following step.
-
4.
Alignment to a reference genome with a splice-aware aligner. In our case studies, we used the STAR aligner (Dobin et al., 2013) with default options, and we aligned reads to human and mouse reference genomes using annotations provided by GENCODE. Output: BAM files with aligned reads.
U2AF1 freCLIP-seq and AGO2 eCLIP-seq metrics are available in Table S3.
Identification of transcriptome-wide protein-RNA interactions with a peak calling algorithm
This section lists all the computational steps necessary to identify and compare protein-RNA interaction locations, starting from aligned reads (BAM files). In the simplest experimental design, CLIP and INPUT libraries with ideally 2 or more biological replicates are expected. An implementation of these steps is available as bash and R scripts (https://github.com/TebaldiLab/eCLIP_seq). The case study shown is based on a dataset where AGO2 binding to cytoplasmic RNAs was quantified in mouse whole testis cells (Griffin et al., 2022). For any step, alternative computational tools can be used, with additional care optionally necessary to optimize the compatibility of input/output file formats.
-
5.
Peak calling with PureCLIP (Krakau et al., 2017). PureCLIP uses a Hidden Markov Model-based approach, and performs peak calling using both read coverage and crosslinking-induced termination events. Output: BED files with the locations of the predicted interaction peaks.
-
6.Downstream analysis of PureCLIP peaks. This analysis is performed within the R environment and includes the following steps:
-
a.Comparison with controls (INPUT and/or IgG) and selection of CLIP specific peaks. This comparison requires to collect coverage for each peak in each CLIP and INPUT replicate, with bedtools (Quinlan and Hall, 2010) coverage function (alternatively, featureCounts or similar tools). The selection of CLIP specific peaks is performed using differential expression tools, edgeR (Robinson et al., 2010) in our example, by comparing CLIP vs INPUT (and optionally IgG) samples.
-
b.Location analysis (distribution of CLIP specific peaks on transcript regions, Figure 3A).
-
c.Sequence analysis (identification of consensus sequences in CLIP specific peaks, Figures 3B and 3C).
-
d.Differential analysis (optional, if multiple CLIPs can be compared).
-
e.Functional enrichment analysis (based on the genes with CLIP specific peaks).
-
a.
Figure 3.

Characterization of transcriptome-wide protein-RNA interactions
(A) Distribution of AGO2 eCLIP-seq peaks in genome regions.
(B) Distribution of crosslinked nucleotides in AGO2 eCLIP-seq peaks.
(C) Sequence logo of AGO2 eCLIP-seq peak regions (10 nucleotide on each side from the crosslinked nucleotide).
Comparison of binding occupancy between different conditions in specific transcript regions
This section lists all the computational steps necessary to quantify and compare occupancy profiles of specific transcript regions, starting from aligned reads (BAM files) and an annotation listing the regions of interest. Binding profiles are based on the position and the number of crosslinking sites, calculated from R2 5′ end. The case study shown is based on a dataset where RNA binding of the splicing factor U2AF1 was quantified in HEL cells and a comparison between wildtype and mutant U2AF1 was performed, specifically looking at intron-exon junctions (Biancon et al., 2022). An implementation of these steps is available as bash and R scripts (https://github.com/TebaldiLab/eCLIP_seq). For any step, alternative computational tools can be used, with additional care optionally necessary to optimize the compatibility of input/output file formats.
-
7.
Conversion of reads into putative crosslinking sites. This step converts a BAM file with aligned reads into a BAM file with mapped putative crosslinking sites. First the input BAM file is converted into a BED file, with bedtools bamtobed function. Next, the crosslinking site is calculated for each read as the upstream nucleotide with respect to the read 5′ end. Finally, the bed file with putative crosslinking sites can be converted back to a BAM file with the bedToBam function.
-
8.Downstream analysis of crosslinking sites in specific regions. This analysis is performed within the R environment and includes the following steps:
-
a.Quantification of binding across each region of interest (in the case study, the genomic coordinates of each intron-exon junction are stored in a GRanges object). Coverage on each nucleotide of each region is collected from the crosslink BAM files generated in the previous step with the bamProfile function.
-
b.Generation of metaprofiles (cumulative signal on all intron-exon junctions). Regions considered to generate the metaprofile can be selected according to a coverage filter. Normalization across different CLIP libraries can be achieved by representing the occupancy at each nucleotide as the fraction, or percentage, of signal with respect to the total signal across the regions. Metaprofile can be generated for specific subsets of junctions, i.e., considering only internal exons (Figure 4).
-
c.Sequence analysis (performed on all the intron-exon junctions used to generate the metaprofile). It can be used to split metaprofiles according to sequence motifs (e.g., introns ending in CAG vs UAG).
-
d.Differential analysis (identification of regions where binding occupancy is significantly different when comparing two conditions, e.g., wildtype vs mutant U2AF1).
-
e.Functional enrichment analysis (based on the genes with differentially bound intron-exon junctions).
-
a.
Figure 4.

Comparison of freCLIP-seq occupancies between different conditions in specific transcript regions
(A) Metaprofile (mean±SEM) of U2AF1 freCLIP-seq occupancies in all intron-exon junctions, comparing wildtype with the S34F mutant samples.
(B) Profile (mean±SEM) of U2AF1 freCLIP-seq occupancies in a selected intron-exon junction, comparing wildtype with the S34F mutant samples.
Limitations
FreCLIP-seq, as other CLIP-seq protocols, requires a significant number of cells limiting the application on primary patient-derived cells. We suggest to use at least 10 × 106 cells for standard eCLIP-seq, and 20–30 × 106 cells for freCLIP-seq.
Please note that eCLIP-seq technologies do not directly measure RNA binding affinity. By mapping and counting the end of eCLIP-seq reads we can quantify the binding occupancy with single-nucleotide resolution.
Troubleshooting
Problem 1
Experimental samples with absent or faint signal on autoradiogram after IP (step 12). Absent signal may represent an antibody-related issue during IP. Faint signal may represent an issue during P32-labeling and visualization (e.g., P32 signal may not be proportional to the amount of RNA and film development issues may produce a dark film) (Figure 5).
Figure 5.

Issues during autoradiographic visualization
Representative U2AF1 freCLIP-seq samples obtained from not-infected HEL cells.
Potential solution
In case of absent signal, perform western blot on saved pre-IP and post-IP samples to evaluate IP efficiency of the used antibody and adjust IP conditions. In case of faint signal, as in Figure 5, proceed with the experimental pipeline.
Problem 2
Adapter dimers (step 23). Post-AMP libraries can present adapter bands (Figure 6). Adapter dimers cause index hopping and demultiplexing issues and need to be removed.
Figure 6.

Presence and removal of adapter dimers
(A) Visualization by TBE-Urea gel of representative U2AF1 freCLIP-seq libraries. Yellow lines mark the expected library size. Purple stars indicate adapter dimers around 150 bp in post-AMP libraries. Gray-labeled samples are the purple-labeled samples after re-amplification changing PCR cycles or adapters.
(B) Visualization by Agilent 2100 Bioanalyzer of representative U2AF1 freCLIP-seq libraries obtained from not-infected HEL cells. Yellow boxes mark the expected library size. Peak size is reported. Purple stars indicate adapter dimers’ peak around 150 bp in post-AMP libraries. Black-labeled samples are the respective purple-labeled samples after gel purification.
Potential solution
We typically manage adapter troubleshooting starting from TBE-Urea gel visualization (Figure 6A). In case of a bright adapter band on the TBE-Urea gel, use the stored pre-AMP library and repeat final PCR with another adapter pair (always following pooling guidelines). In case of a visible but faint adapter band, use the stored pre-AMP library and repeat final PCR with the same adapter pair but 1 cycle less.
However, changing PCR conditions may be not sufficient to get rid of adapters, especially in low concentrated libraries usually containing high adapter levels. For libraries with over 10% adapter dimers after QC by Bioanalyzer (Figure 6B), perform gel extraction (QIAGEN QIAquick Gel Extraction Kit, Cat#28706) and repeat QC.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Stephanie Halene (stephanie.halene@yale.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
We thank Brooke Sullivan, Emma Sykes, Christopher Castaldi, and the Yale Center for Genome Analysis for library sequencing and troubleshooting, Benjamin Walters (Yale University) for the preparation of AGO2 lysates, and Valentina Botti, Karla Neugebauer, and Jun Lu (Yale University) for helpful suggestions. This study was funded in part by the Edward P. Evans Foundation, the Frederick A. DeLuca Foundation, the NIH/NIDDK R01DK102792, and the NIH/NIDDK R01DK124788 (to S.H.). T.T. was supported by AIRC under MFAG 2020 (ID. 24883 project). G.B. was supported by NIH/NIDDK Cooperative Centers of Excellence in Hematology U24DK126127. B.J.L. was supported by the NIH R01HD098128.
Author contributions
Conceptualization: G.B., T.T., and S.H.; methodology: G.B., P.J., and S.H.; investigation: G.B. and P.J.; formal analysis: G.B., E.B., and T.T.; resources: B.J.L. and S.H.; validation: E.B., P.J., and B.J.L.; writing: G.B., T.T., and S.H.; visualization: G.B. and T.T.; supervision: T.T. and S.H.; project administration: S.H.; funding acquisition: G.B., T.T., and S.H.
Declaration of interests
S.H., consultancy, Forma Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101823.
Contributor Information
Giulia Biancon, Email: giulia.biancon@yale.edu.
Stephanie Halene, Email: stephanie.halene@yale.edu.
Toma Tebaldi, Email: toma.tebaldi@unitn.it.
Supplemental information
Data and code availability
This study did not generate any unique datasets. Raw and analyzed U2AF1 freCLIP-seq sequencing data are available in GEO under the accession number GSE195620 (Biancon et al., 2022). Raw and analyzed AGO2 eCLIP-seq sequencing data are available in GEO under the accession number GSE181264 (Griffin et al., 2022). Original autoradiogram and gel images have been deposited in Mendeley Data at https://doi.org/10.17632/m7g92hkwmh.1.
Software used for the analyses are described and referenced in the quantification and statistical analysis subsections and are listed in the key resources table.
Analysis scripts in bash and R, with all the computational steps described in the quantification and statistical analysis subsections, are available on GitHub (https://github.com/TebaldiLab/eCLIP_seq https://doi.org/10.5281/zenodo.7121228).
Any additional information required to perform the experimental part or run the computational part of the here presented pipeline is available upon request from the lead contact.
References
- Biancon G., Joshi P., Zimmer J.T., Hunck T., Gao Y., Lessard M.D., Courchaine E., Barentine A.E.S., Machyna M., Botti V., et al. Precision analysis of mutant U2AF1 activity reveals deployment of stress granules in myeloid malignancies. Mol. Cell. 2022;82:1107–1122.e7. doi: 10.1016/j.molcel.2022.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blue S.M., Yee B.A., Pratt G.A., Mueller J.R., Park S.S., Shishkin A.A., Starner A.C., Van Nostrand E.L., Yeo G.W. Transcriptome-wide identification of RNA-binding protein binding sites using seCLIP-seq. Nat. Protoc. 2022;17:1223–1265. doi: 10.1038/s41596-022-00680-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugiolo M., Botti V., Liu N., Müller-McNicoll M., Neugebauer K.M. Fractionation iCLIP detects persistent SR protein binding to conserved, retained introns in chromatin, nucleoplasm and cytoplasm. Nucleic Acids Res. 2017;45:10452–10465. doi: 10.1093/nar/gkx671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser E., Maji D., Biancon G., Puthenpeedikakkal A.M.K., Cavender C.E., Tebaldi T., Jenkins J.L., Mathews D.H., Halene S., Kielkopf C.L. Pre-mRNA splicing factor U2AF2 recognizes distinct conformations of nucleotide variants at the center of the pre-mRNA splice site signal. Nucleic Acids Res. 2022;50:5299–5312. doi: 10.1093/nar/gkac287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin K.N., Walters B.W., Li H., Wang H., Biancon G., Tebaldi T., Kaya C.B., Kanyo J., Lam T.T., Cox A.L., Halene S., Chung J.J., Lesch B.J. Widespread association of the Argonaute protein AGO2 with meiotic chromatin suggests a distinct nuclear function in mammalian male reproduction. Genome Res. 2022;32:1655–1668. doi: 10.1101/gr.276578.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M., Katsantoni M., Köster T., Marks J., Mukherjee J., Staiger D., Ule J., Zavolan M. CLIP and complementary methods. Nat. Rev. Methods Prim. 2021;1:20. [Google Scholar]
- Huppertz I., Attig J., D’Ambrogio A., Easton L.E., Sibley C.R., Sugimoto Y., Tajnik M., König J., Ule J. iCLIP: protein–RNA interactions at nucleotide resolution. Methods. 2014;65:274–287. doi: 10.1016/j.ymeth.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D.J., Luscombe N.M., Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakau S., Richard H., Marsico A. PureCLIP: capturing target-specific protein–RNA interaction footprints from single-nucleotide CLIP-seq data. Genome Biol. 2017;18:240. doi: 10.1186/s13059-017-1364-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10. [Google Scholar]
- Van Nostrand E.L., Pratt G.A., Shishkin A.A., Gelboin-Burkhart C., Fang M.Y., Sundararaman B., Blue S.M., Nguyen T.B., Surka C., Elkins K., et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP) Nat. Methods. 2016;13:508–514. doi: 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand E.L., Nguyen T.B., Gelboin-Burkhart C., Wang R., Blue S.M., Pratt G.A., Louie A.L., Yeo G.W. Robust, cost-effective profiling of RNA binding protein targets with single-end enhanced crosslinking and immunoprecipitation (seCLIP) Methods Mol. Biol. 2017;1648:177–200. doi: 10.1007/978-1-4939-7204-3_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg M., Levy V., Maier V.K., Kesner B., Blum R., Lee J.T. Denaturing cross-linking immunoprecipitation to identify footprints for RNA-binding proteins. STAR Protoc. 2021;2:100819. doi: 10.1016/j.xpro.2021.100819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Luo X., Qian J., Pang X., Song J., Qian G., Chen J., Chen S. FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One. 2012;7 doi: 10.1371/journal.pone.0052249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T., Hamanaka S., Kamiya A., Okabe M., Kawarai M., Wakiyama Y., Umino A., Hayama T., Sato H., Lee Y.S., et al. Development of an all-in-one inducible lentiviral vector for gene specific analysis of reprogramming. PLoS One. 2012;7 doi: 10.1371/journal.pone.0041007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K., Sanada M., Shiraishi Y., Nowak D., Nagata Y., Yamamoto R., Sato Y., Sato-Otsubo A., Kon A., Nagasaki M., et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets. Raw and analyzed U2AF1 freCLIP-seq sequencing data are available in GEO under the accession number GSE195620 (Biancon et al., 2022). Raw and analyzed AGO2 eCLIP-seq sequencing data are available in GEO under the accession number GSE181264 (Griffin et al., 2022). Original autoradiogram and gel images have been deposited in Mendeley Data at https://doi.org/10.17632/m7g92hkwmh.1.
Software used for the analyses are described and referenced in the quantification and statistical analysis subsections and are listed in the key resources table.
Analysis scripts in bash and R, with all the computational steps described in the quantification and statistical analysis subsections, are available on GitHub (https://github.com/TebaldiLab/eCLIP_seq https://doi.org/10.5281/zenodo.7121228).
Any additional information required to perform the experimental part or run the computational part of the here presented pipeline is available upon request from the lead contact.