

Abstract
Background & Aims
Liver contains high frequency of group 1 innate lymphoid cells (ILC), which are composed of comparable number of type 1 ILC (ILC1) and natural killer (NK) cells in steady state. Little is known about whether and how the interaction between ILC1 and NK cells affects the development of alcoholic liver disease.
Methods
A mouse model of chronic alcohol abuse plus single-binge (Gao-Binge model) was established. The levels of alanine aminotransferase/aspartate aminotransferase, hepatic lipid, and inflammatory cytokines or neutrophils were measured to evaluate the degree of liver injury, steatosis, and inflammation. Flow cytometric analysis, cell depletion, or adoptive transfer were used to interrogate the interaction between ILC1 and NK cells.
Results
Upon chronic alcohol consumption, NK cells, but not ILC1, underwent apoptosis, resulting in ILC1 dominance among group 1 ILC. Interleukin (IL) 17A expression was up-regulated, and increased IL17A was mainly derived from liver ILC1 after chronic alcohol feeding. Either depletion of ILC1 or neutralization of IL17A could significantly attenuate liver steatosis, inflammation, and injury in alcohol-fed mice. In contrast, normalization of the ILC1/NK cells ratio through NK cells transfer or expanding NK cells had a significant hepatoprotection against alcohol-induced steatohepatitis. Furthermore, NK cell-derived interferon gamma exerted a protective function via inhibiting IL17A production by liver ILC1 during alcoholic steatohepatitis.
Conclusions
This is the first study showing that the interplay between liver ILC1 and NK cells occurs and drives the development of alcoholic steatohepatitis. Our findings support further exploration of liver ILC1 or NK cells as a therapeutic target for the treatment of alcohol-associated liver disease.
Keywords: ILC1, NK Cell, Alcoholic Steatohepatitis, IFN-γ, IL17A
Abbreviations used in this paper: ALT, alanine aminotransferase; anti-AsGM1, anti-asialo GM1; AST, aspartate aminotransferase; IFN-γ, interferon gamma; IG, immunoglobulin; IL, interleukin; ILC, innate lymphoid cells; NK, natural killer; PBS, phosphate-buffered saline; polyI:C, polyriboinosinic polyribocytidylic acid; TNF-α, tumor necrosis factor alpha; TRAIL, TNF-related apoptosis-inducing ligand; WT, wild-type
Graphical abstract
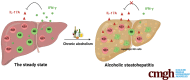
Summary.
Chronic alcohol abuse results in elevated IL17A production by liver ILC1 and NK cells apoptosis. ILC1-derived IL17A, which is constrained by IFN-γ from NK cells, plays a pathogenic role in the development of alcoholic steatohepatitis.
Innate lymphoid cells (ILC) are a heterogeneous group of innate lymphocytes that reside in barrier tissues and do not express clonally distributed diverse antigen receptors, distinguishing them from adaptive immune cells.1 These particular cells rapidly respond to a multitude of invading challenges before the initiation of the adaptive immune response and trigger immunopathology when dysregulated.2,3 Because of the specific transcription factors required for development and the different preferences for producing effector cytokines, ILC are classified into 3 subsets (group 1, group 2, and group 3).2 By secreting interferon gamma (IFN-γ)/tumor necrosis factor alpha (TNF-α) or direct cytotoxicity, group 1 ILC provide resistance to intracellular pathogens such as viruses and intracellular bacteria.4 They are composed of both type 1 ILC (ILC1, also called liver-resident NK cells) and conventional natural killer (NK) cells.4,5
Although ILC1 and NK cells share many common characteristics in terms of IFN-γ production, requirement of transcription factor T-bet, and expression of overlapping cell surface receptors,6 ILC1 are markedly different from NK cells in many aspects. NK cells are circulatory throughout the body, whereas ILC1 are tissue-resident at steady state.5 To separate ILC1 from NK cells, expression of CD49a and CD49b has often been used. In contrast to NK cells characterized as CD49a-CD49b+, ILC1 are identified as specific CD49a+CD49b- phenotype with higher expression of TNF-related apoptosis-inducing ligand (TRAIL) and CD69.7 The expression of the transcription factor Eomesodermin (Eomes) was also distinct between these 2 subsets, because NK cells are Eomes-dependent for development, whereas most of ILC1 are not.8,9 In addition, a more recent study has demonstrated that liver ILC1, but not NK cells, were developed locally from fatal liver-derived Lin-CD122+CD49a+ progenitors in an IFN-γ–dependent feed-forward loop, demonstrating the specific developmental pathway of the ILC1.10
ILC1 were originally discovered in the livers of C57BL/6 mice and subsequently found in other tissues such as salivary gland, skin, and uterus.11,12 In the liver, ILC1 and NK cells show comparable frequency within group 1 ILC and represent the predominant population of ILC family.13 It is of interest whether the imbalance in the ratio of liver ILC1 to NK cells is correlated to the progression of certain liver diseases. In addition to the phenotypic and developmental differences between ILC1 and NK cells mentioned above, ILC1 and NK cells exhibit more complexity regarding the functional roles in liver disease. For example, liver ILC1 protected mice from carbon tetrachloride-induced or acetaminophen-induced acute liver injury through the release of IFN-γ in an IL-7 and CD226-dependent mechanism, whereas the role of NK cells is minimal.14 Interestingly, ILC1 and NK cells can exert synergistic or opposite immune responses in the liver against pathogen infections or tumor metastasis.15, 16, 17, 18 Both liver ILC1 and NK cells constrained CD8+T cells proliferation by competing for local interleukin (IL) 2 availability and controlled T-cell–mediated immunopathology during chronic hepatitis B virus infection.15 Moreover, recent studies documented that ILC1 and NK cells join forces to control liver metastasis and Toxoplasma gondii, a common protozoan parasite, infection.17,18 However, ILC1 suppressed hepatic T-cell–mediated antiviral responses via the interaction between programmed death-1 and programmed death ligand 1 during both acute and chronic lymphocytic choriomeningitis virus infection. In contrast, NK cells promoted antiviral T-cell activities.16
Nevertheless, little is known about the role of ILC1 in inflammatory liver disease such as alcohol-induced steatohepatitis. More importantly, no studies have assessed whether and how the interplay between ILC1 and NK cells affects the development of liver disease. In this study, we investigated the crosstalk between ILC1 and NK cells in the pathogenesis of alcoholic steatohepatitis.
Results
Chronic Alcohol Consumption Causes Severe Imbalance in the Ratio of Liver ILC1 to NK Cells by Inducing the Apoptosis of NK Cells
Emerging evidence supports the critical role of group 2 ILC and group 3 ILC subsets in orchestrating immune response during the inflammation, metabolism, and homeostasis of their resident tissues,19, 20, 21 implying a flexible and essential function of these tissue-resident ILCs in processing signals that threaten tissue homeostasis. However, the pathophysiological role of group 1 ILC in alcohol-associated liver disease remains ill-defined. We hypothesized that group 1 ILC was involved in the pathogenesis of alcohol-induced steatohepatitis. To test this hypothesis, the mice were exposed to the Gao-binge model of ethanol-induced steatohepatitis with 10-day chronic plus single-binge ethanol feeding.22 The frequency and the absolute number of group 1 ILC in the liver decreased significantly after ethanol feeding (Figure 1A). Compared with liver group 1 ILC from control diet feeding mice, these cells from ethanol feeding mice exhibited altered phenotype with higher expression of TRAIL and CD69 (Figure 1B and C), which were closer to the characteristics of liver type 1 ILC (ILC1) in previous studies.5 To address the hypothesis that ILC1 are dominant in liver group 1 ILC after ethanol feeding, we further examined the subsets composition in group 1 ILC. We found that the liver ILC1 from ethanol-fed mice exhibited same characteristics to those from wild-type (WT) mice, showing CD45+CD3-NK1.1+CD49a+CD49b-T-bet+Eomes-RORγt- phenotype (Figure 1D). Surprisingly, the frequency of NK cells reduced significantly, leading to that the ratio of liver ILC1 to NK cells was switched from 1:1 in steady state to approximate 5:1 after ethanol feeding (Figure 1E). We observed similar ratio of liver ILC1 to NK cells after ethanol feeding in female and male mice (Figure 1E and F), suggesting that this phenomenon was not gender dependent. Furthermore, only a short-term 10-day ethanol feeding without binge is sufficient to induce the dominance of liver ILC1, indicating it has commonly occurred in various alcohol-associated liver diseases (Figure 1G).
Figure 1.

Severe imbalance in ratio of liver ILC1 to NK cells during chronic alcohol abuse. (A) Female C57BL/6 mice were exposed to the Gao-Binge model with 10-day chronic ethanol feeding plus 1 binge. Liver mononuclear cells were isolated from the mice fed with liquid diet containing ethanol at 5% (v/v) (EtOH) or pair-fed control diet (CD). Percentage and absolute number of group 1 ILCs (CD45+CD3-NK1.1+ cells) were measured by flow cytometry (n = 6–7/group). (B and C) Expression of TRAIL (B) and CD69 (C) on hepatic group 1 ILCs from CD- or ethanol-fed mice were examined by flow cytometry (n = 7–8/group). (D) Expression of T-bet, Eomes, and RORγt on liver ILC1 (CD45+CD3-NK1.1+CD49b-CD49a+) and NK cells (CD45+CD3-NK1.1+CD49b+CD49a-) from ethanol-fed mice was shown. (E) The numbers of liver ILC1 and NK cells from CD- or EtOH-fed mice were measured, and the ratios of ILC1 to NK cells were shown (n = 6/group). (F) Male C57BL/6 mice were exposed to the Gao-Binge model with 10-day chronic ethanol feeding plus 1 binge or control diet. The ratio of liver ILC1 to NK cells was shown (n = 3–4/group). (G) Female C57BL/6 mice were exposed to CD, chronic ethanol consumption for 5 days, 10 days, 10 days plus binge, or binge only. The ratios of liver ILC1 to NK cells were shown (n = 4/group). Two-tailed unpaired Student t test was performed in A–C, E, and F. One-way analysis of variance was performed in G. ns, no statistical significance. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P <.0001.
To further investigate why the liver ILC1/NK cells ratio increased dramatically after ethanol feeding, we compared the absolute numbers of ILC1 and NK cells in the liver of control diet feeding or ethanol feeding mice. Our data showed that the number of ILC1 stayed unaltered, whereas the number of NK cells, as well as other lymphocytes, decreased markedly after ethanol feeding (Figure 2A and B), suggesting that more NK cells had undergone apoptosis compared with liver ILC1 during chronic ethanol consumption. Indeed, the frequency of apoptotic NK cells increased obviously after ethanol feeding compared with control diet feeding, whereas ethanol feeding had little effect on apoptosis of liver ILC1 (Figure 2C and D). For mechanistic investigation, we compared the genome-wide transcriptional profiles of liver ILC1 and NK cells from previous study (GSE43339) by KEGG analysis.23 The data showed the genes, which are related to PI3K-Akt signaling pathway, calcium signaling pathway, and cAMP signaling pathway, are expressed at higher level in liver ILC1 than in NK cells, which may be partially associated with the anti-apoptotic feature of liver ILC1 (Figure 2E). In addition, it did not affect the proliferation of either liver ILC1 or NK cells after ethanol feeding (Figure 2F). Together, chronic alcohol consumption caused marked elevation in NK cells apoptosis but had no obvious effect on ILC1, resulting in a significant increase in the ratio of ILC1 to NK cells.
Figure 2.

Apoptosis of NK cells results in the dominance of ILC1. (A) Absolute number of liver ILC1 and NK cells in CD- or EtOH-fed mice was counted (n = 8/group). (B) Absolute number of CD4+ T, CD8+ T, and B cells in CD- or EtOH-fed mice was measured by flow cytometry (n = 4/group). (C and D) Apoptosis of liver NK cells (C) and ILC1 (D) from CD- or EtOH-fed mice was measured by using anti-annexin V antibody and 7-AAD (n = 5/group). Annexin V+ cells were identified as apoptotic cells. (E) The KEGG analysis was performed by using previously published GEO database (GSE43339). (F) Expression of Ki67 in liver ILC1 and NK cells from CD- or EtOH-fed mice was examined (n = 5/group). Two-way analysis of variance was performed in A and B. Two-tailed unpaired Student t tests were performed in C, D, and F. ns, no statistical significance. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P <.0001.
Normalization of the Ratio of Liver ILC1 to NK Cells Protects Against Alcoholic Steatohepatitis
To determine whether restoring the original ratio of liver ILC1 to NK cells during chronic ethanol feeding could attenuate alcoholic steatohepatitis, adoptive cell transfer or usage of stimuli to induce NK cell proliferation was applied to expand the number of NK cells and achieve a balanced ratio of ILC1 to NK cells in ethanol-fed mice. First, CD49b+NK cells were purified from the spleen of Rag1-/- mice and adoptively transferred into ethanol-fed mice. We counted the absolute number of NK cells and found a significant increase in liver at 6 hours after binge, whereas liver ILC1 showed no change (Figure 3A). As expected, the ratio of liver ILC1 to NK cells was normalized to 1:1 after NK cells transfer, whereas the control mice injected with phosphate-buffered saline (PBS) still showed a higher ILC1/NK cells ratio (Figure 3B). Interestingly, compared with PBS-injected control mice, mice adoptively transferred with NK cells displayed significantly decreased levels of hepatic triglyceride contents, plasma alanine aminotransferase (ALT), and aspartate aminotransferase (AST) (Figure 3C and D). Moreover, the inflammatory response was alleviated after NK cells transfer, which was evident by reduced mRNA level of IL6 and IL1β (Figure 3E). H&E and Oil Red O staining analyses demonstrated that mice receiving NK cells transfer exhibited a lower degree of steatosis when compared with PBS-injected mice that underwent the same ethanol treatment (Figure 3F).
Figure 3.

Restoration of NK cells protects mice from alcohol-induced steatohepatitis. (A–F) CD- or EtOH-fed mice were injected intravenously with PBS or WT splenic CD49b+NK cells from Rag1-/- mice at days 2, 6, and 10 after ethanol feeding (1 × 106 cells each time). Mice were killed, and liver tissue or plasma was obtained at 6 hours after binge. The number of liver ILC1 and NK cells (A) and ratio of ILC1 to NK cells (B) were shown (n = 5–6/group). (C) Hepatic triglyceride content was measured in whole liver homogenate (n = 4–6/group). (D) Plasma concentrations of ALT and AST were measured (n = 4–7/group). (E) The mRNA level of IL6 and IL1β in liver tissue was detected (n = 4–6/group). (F) H&E or Oil Red O staining of liver section were performed (scale bars, 100 μm). Representative images are shown (n = 4/group). Two-way analysis of variance was performed in A. One-way analysis of variance was performed in B–E. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P <.0001.
In addition, we made use of polyriboinosinic polyribocytidylic acid (polyI:C) to induce NK cell proliferation. The treatment of polyI:C resulted in a significant increase in the absolute number of NK cells but had no obvious effect on liver ILC1, which eventually decreased the ratio of ILC1 to NK cells in ethanol-fed mice (Figure 4A and B). Similarly, polyI:C-induced NK cells expansion could significantly ameliorate ethanol-induced steatosis and liver injury and inflammation, as demonstrated by lower hepatic triglyceride levels, less lipid accumulation in liver, reduced ALT/AST levels in plasma, and decreased mRNA level of IL6 and IL1β (Figure 4C–F). Taken together, these data revealed a protective role for NK cells against alcohol-induced steatohepatitis by normalizing liver ILC1/NK cells ratio.
Figure 4.

Injection of polyI:C protects mice from alcohol-induced steatohepatitis. (A–D) EtOH-fed mice were injected intraperitoneally with PBS or polyI:C every other day after ethanol feeding (5 mg/kg). Mice were killed, and liver tissue was obtained at 6 hours after binge. The numbers of liver ILC1 and NK cells (A) and ILC1/NK cells ratio (B) were shown (n = 3/group). (C) Hepatic triglyceride content was measured in whole liver homogenate (n = 4/group). (D) H&E staining of liver section was performed (scale bars, 100 μm). (E) Plasma levels of ALT and AST were detected (n = 5/group). (F) The mRNA level of IL1β and IL6 in liver tissue was detected (n = 4/group). Two-tailed unpaired Student t tests were performed in A and B. Representative images are shown (n = 4/group). Two-way analysis of variance was performed in A. Two-tailed unpaired Student t test was performed in B, C, E, and F. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P <.0001.
Liver ILC1 Contribute to Alcoholic Steatohepatitis via IL17 Production
We next attempted to examine the role of liver ILC1 in alcoholic steatohepatitis. Although anti-asialo GM1 (anti-AsGM1) antibody was able to deplete both liver ILC1 and NK cells in mice, the majority of depleted cells were ILC1 in liver when the antibody was administered at day 9-10 after ethanol feeding, which was due to the dominance of liver ILC1 at that time (Figure 1G, Figure 5A). Therefore, because there is a lack of effective tools to specifically deplete liver ILC1 in vivo, a single dose of anti-AsGM1 antibody at day 9 after ethanol feeding was applied to reflect the functional role of ILC1 during ethanol-induced steatohepatitis. Compared with rabbit immunoglobulin (Ig) G–treated control mice, mice receiving anti-AsGM1 antibody were resistant to the development of steatohepatitis, as demonstrated by reduced levels of hepatic triglyceride, ALT, and AST (Figure 5B and C). The decreased degree of steatosis was further confirmed by H&E and Oil Red O staining (Figure 5D). To test the effect on inflammation after depletion of ILC1, a panel of proinflammatory cytokines and neutrophils infiltration in liver were measured after ethanol feeding. Consistently, anti-AsGM1 antibody treatment significantly reduced hepatic neutrophils infiltration and several proinflammatory cytokines including IL6, IL1β, and TNF-α when compared with IgG-treated controls (Figure 5E and F). As previous study and our data showed that NK cells played a protective role in alcoholic steatohepatitis,24 the benefits of anti-AsGM1 antibody treatment further revealed the role of ILC1 as a driver in alcoholic steatohepatitis.
Figure 5.

Liver ILC1 play an important role in alcohol-induced steatohepatitis. EtOH-fed mice were injected intraperitoneally with anti-AsGM1 antibody (50 μg per mouse) or same amount of IgG as control once at 48 hours before binge. (A) The numbers of liver ILC1 and NK cells were measured at 6 hours after binge (n = 4/group). (B) Hepatic triglyceride content was measured in whole liver homogenate (n = 18–20/group). (C) Plasma concentrations of ALT and AST were measured (n = 8–12/group). (D) H&E and Oil Red O staining of liver section were performed at 6 hours after binge (scale bars, 100 μm). Representative images are shown (n = 8/group). (E) The mRNA level of IL6 (n = 6–7/group), IL1β (n = 6–8/group), and TNF-α (n = 3/group) in liver tissue was detected. (F) Female C57BL/6 mice were exposed to CD or ethanol containing diet. Some EtOH-fed mice were injected intraperitoneally with IgG or anti-AsGM1 antibody. All mice were killed at 6 hours after binge, and the percentage and absolute number of hepatic neutrophils were measured by flow cytometry (n = 6/group). Two-way analysis of variance was performed in A. Two-tailed unpaired Student t tests were performed in B, C, and E. One-way analysis of variance was performed in F. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P <.0001.
To characterize the molecular mechanism accounting for the pathogenic function of liver ILC1, we compared gene expression profiles between liver ILC1 and NK cells from naive mice by using previously published GEO database (GSE43339). We found the expression of IL13 and IL17A, which have been reported to play important roles in liver steatosis,25,26 were up-regulated in liver ILC1, suggesting that IL13 or IL17A might be responsible for ILC1-mediated alcoholic steatohepatitis (Figure 6A). We next compared the expression of IL13 or IL17A in liver tissue from control mice and ethanol-fed mice treated with IgG or anti-AsGM1 antibody. The expression of IL13 was unaltered among these groups (Figure 6B). However, we found that both mRNA and protein level of IL17A were dramatically up-regulated after ethanol feeding but abolished in anti-AsGM1 antibody–treated mice (Figure 6C and D). These data led us to the hypothesis that increased IL17A in liver during alcohol-induced steatohepatitis is mainly from liver ILC1. To test this, we measured IL17A expression in ILC1 and NK cells during ethanol feeding by using intracellular staining. As shown in Figure 6E and F, IL17A expression was significantly increased in liver ILC1, but not in NK cells, during ethanol-induced steatohepatitis. IL17A expression in hepatic CD4+ T cells was also observed, but no changes were found between controls and ethanol-fed mice (Figure 6G). Thus, the data suggested that liver ILC1 were the main source of chronic alcohol consumption–induced IL17 elevation.
Figure 6.

Liver ILC1 augment alcohol-induced steatohepatitis via IL17 production. (A) Heat map shows mRNA expression of cytokines in liver ILC1 and NK cells from RNA-seq data GSE43339. Mice were treated as described in Figure 4F. (B and C) The mRNA level of IL13 (B) and IL17A (C) in liver tissue was detected (n = 4/group). (D) Plasma concentration of IL17A was measured (n = 4–7/group). (E) Expression of IL17A in liver ILC1 from CD- or EtOH-fed mice was measured by intracellular staining (n = 6/group). (F and G) The number of hepatic IL17A+ NK cells (F) and IL17A+ CD4+ T cells (G) in CD- or EtOH-fed mice were shown (n = 4/group). (H–J) EtOH-fed mice were injected intraperitoneally with IgG as control or anti-IL17A antibody twice at days 5 and 2 before binge (400 μg per mouse, n = 4/group). Liver tissue and plasma were obtained at 6 hours after binge. Hepatic triglyceride content (H), plasma level of ALT and AST (I), and H&E or Oil Red O staining of liver section (J) were examined. Representative images are shown (scale bars, 100 μm). (K) AML-12 cell line was stimulated with PBS or free fatty acid (FFA) (1 mmol/L) with or without IL17A (40 ng/mL). After 24 hours, the mRNA level of Srebp-1c and Acc in AML-12 cells was measured by quantitative polymerase chain reaction. Data were analyzed according to 3 independent experiments. One-way analysis of variance was performed in B–D. Two-tailed unpaired Student t tests were performed in E–I. Two-way analysis of variance was performed in K. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P <.0001.
To evaluate the function of ILC1-derived IL17A in the development of alcoholic steatohepatitis, we injected intraperitoneally anti-IL17A antibody to neutralize IL17A during the period of ethanol feeding. Not surprisingly, neutralization of IL17A resulted in a significant decrease in hepatic triglyceride, plasma level of ALT and AST, and hepatic lipid accumulation after ethanol feeding (Figure 6H–J), which was consistent with previous studies.26,27 We next used an in vitro culture system to further observe the direct action of IL17A on lipid synthesis of hepatocytes. The data showed the treatment of free fatty acid increased the mRNA level of lipid synthesis genes, such as Acc and Srebp1-c, which were further up-regulated in presence of IL17A (Figure 6K). Together, these data demonstrated that in contrast to the protective function of NK cells, liver ILC1 played an important role in the pathogenesis of ethanol-induced steatohepatitis through the release of IL17A.
NK Cell-derived IFN-γ Protects Mice From Alcoholic Steatohepatitis by Dampening IL17A Production by ILC1
As demonstrated in Figures 3 and 4, normalization of the ILC1/NK cells ratio by NK cells transfer or by polyI:C injection inducing NK cells expansion without altering the number of ILC1 had significant amelioration for ethanol-induced steatohepatitis, suggesting that NK cells may mediate protection against alcoholic steatohepatitis by down-regulating the function of ILC1. Hence, we measured IL17A expression by ILC1 after NK cells transfer in ethanol-fed mice. Compared with control mice injected with PBS, mice receiving NK cells transfer showed significantly reduced IL17A+ILC1 (Figure 7A). It was reported that IFN-γ was able to inhibit Th17 differentiation in humans and mice,28,29 suggesting that IFN-γ is involved in regulating IL17A-mediated inflammatory response. We next hypothesized that if NK cell-mediated IL17A suppression was IFN-γ dependent, we would expect to observe a decreased IFN-γ producing NK cells in ethanol-fed mice. Indeed, our data showed a significant reduction in both frequency and absolute number of IFN-γ+ NK cells in ethanol-fed mice (Figure 7B). Interestingly, liver ILC1 from either control or ethanol-fed mice showed no changes in IFN-γ expression (Figure 7C). To further confirm that NK cell-derived IFN-γ was necessary to inhibit IL17A production by ILC1, we adoptively transferred IFN-γ-/- NK cells into ethanol-fed mice. Compared with WT NK cells, IFN-γ-/- NK cells failed to constrain the IL17A production by ILC1 (Figure 7A). Consistently, the mice transferred with IFN-γ-/- NK cells displayed higher levels of ALT, AST, inflammatory cytokines, lipid accumulation, and more hepatic neutrophils infiltration when compared with mice receiving WT NK cells (Figure 7D–G). These data provide strong evidence that ILC1-derived IL17A, which was regulated by IFN-γ from NK cells, is essential for the development of ethanol-induced steatohepatitis.
Figure 7.

NK cell-derived IFN-γ alleviates alcohol-induced steatohepatitis through inhibition of IL17A production by ILC1. Female C57BL/6 mice were exposed to CD or ethanol containing diet. Some EtOH-fed mice were injected intravenously with PBS or splenic CD49b+NK cells from WT or GKO mice at days 6 and 10 after ethanol feeding (1 × 106 cells each time). The number of liver IL-17A+ ILC1 was measured at 6 hours after binge by intracellular staining (n = 4–7/group). (B and C) Frequency or absolute number of liver IFN-γ–producing NK cells (B) or ILC1 (C) in CD- or EtOH-fed mice was shown (n = 8/group). (D–G) EtOH-fed mice were adoptively transferred with PBS or splenic CD49b+NK cells from WT or GKO mice at days 6 and 10 after ethanol feeding (2 × 106 cells per mouse). All mice were killed, and liver tissue or plasma was obtained at 6 hours after binge. (D) Plasma concentrations of ALT and AST were measured (n = 4/group). (E) The mRNA level of IL6 and IL1β in liver tissue was detected (n = 4/group). (F) H&E or Oil Red O staining of liver section was performed (scale bars, 100 μm). Representative images are shown (n = 4/group). (G) The percentage and absolute number of hepatic neutrophils were measured by flow cytometry (n = 4/group). One-way analysis of variance was performed in A, D, E, and G. Two-tailed unpaired Student t tests were performed in B and C. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P <.0001.
Discussion
In the present study, we provided the first evidence that the interplay between liver ILC1 and NK cells plays an important role in the development of alcohol-associated liver disease. With chronic alcohol consumption, ILC1 accounts for an increasing proportion among group 1 ILC and rises up to approximately 85% after 10 days because of the apoptosis of NK cells. ILC1, which is negatively regulated by NK cell-derived IFN-γ, contributes to the development of alcoholic steatohepatitis via the release of IL17A.
Liver acts as an innate immunity-dominant organ, which contains a variety of liver-resident or circulating innate immune cell subsets, including NK cells, NKT cells, macrophages, and neutrophils.29 Although the roles of macrophage or neutrophils in alcohol-induced liver disease are well-investigated,30 the understanding of NK cells’ involvement is in its infancy. There are very few published studies, and they focus on CD3-NK1.1+ bulk NK cells, which are now known to be group 1 ILC. A recent study, which used multiple doses of anti-AsGM1 antibody over the whole ethanol feeding period, demonstrated a protective function of bulk NK cells against alcoholic steatohepatitis by secreting IFN-γ.24 Because the treatment of anti-AsGM1 antibody causes depletion of both NK cells and ILC1, the respective roles of NK cells or ILC1 in pathogenesis of chronic alcohol-induced steatohepatitis are still blurry. On the basis of our finding that ILC1 are dominant in group 1 ILC at day 9-10 after ethanol feeding, we chose to give a single dose of anti-AsGM1 antibody at day 9 to evaluate the functional role of ILC1 and found the mice depleted with ILC1 exhibited attenuated alcoholic steatohepatitis. Moreover, adding back circulating NK cells to WT mice during ethanol feeding was advantageous, strongly suggesting that NK cells confer hepatoprotection, whereas liver ILC1 play an opposite function during alcohol-induced steatohepatitis.
Previous studies revealed that IL17A signaling plays a key role in hepatic lipogenesis, because liver steatosis was reduced significantly in the whole-body IL17A-/- or IL17RA-/- mice that underwent nonalcoholic fatty liver disease.31,32 Blockade of the IL17A axis impedes progression from steatosis to nonalcoholic steatohepatitis.32 In alcohol-induced liver disease, IL17A signaling similarly functions to promote alcohol-induced steatosis, inflammation, and even hepatocellular carcinoma via regulating macrophages or hepatocytes.26,27 The frequency of intrahepatic IL17-producing CD4+ T cells or the plasma level of IL17A is associated with the progression of nonalcoholic fatty liver disease or alcohol-induced liver disease in both human and mouse models, implying that Th17 cells may be the main source of IL17A.27,31,33 In line with these findings, we also found that the frequency of hepatic IL17A+CD4+ T cells is higher after chronic alcohol consumption (data not shown), and neutralization of IL17A inhibits development of alcoholic steatohepatitis. However, the absolute number of IL17A+CD4+ T cells was comparable between control and ethanol-fed mice, which is due to the significant decrease in total number of hepatic CD4+ T cells after chronic ethanol consumption. Unexpectedly, liver ILC1, the only cell population whose absolute number did not decline globally, expressed much higher level of IL17A after chronic alcohol consumption. Thus, we demonstrate for the first time that liver ILC1 are more likely the cellular source of increased IL17A in alcohol-induced liver disease.
Earlier studies have shown that the subsets of T-helper cells were not fixed but rather could be converted after stimulation with different cytokines.34 This plasticity was also observed among ILC subsets, whose transcription factor profiles and cytokine outputs could be modulated by environmental signals.35 For instance, after stimulation with IL12 and IL15, ILC3 was turned into ILC1-like phenotype, evident by the loss of RORγt expression but up-regulation of T-bet expression as well as IFN-γ production capacity.36 Interestingly, a human study demonstrated that CD127+ ILC1 could differentiate into ILC3, which preferentially produce IL17A and IL22, and this process was driven by IL23, IL1β, and retinoic acid.37 Our data showed the number of IL17A-producing liver ILC1 was approximately tripled after chronic alcohol feeding, suggesting the possible differentiation of liver ILC1 to ILC3. However, regardless of whether IL17A was expressed, liver ILC1 maintained the expression of transcription factors, showing the stable T-bet+Eomes-RORγt- phenotype. These findings suggest a possible new mechanism by which liver ILC1 secrete IL17A without altering the expression profile of transcription factors during chronic alcohol-induced liver disease.
Alcohol has been considered an immunosuppressive agent that leads to impaired NK cell-mediated immune response by decreasing the expression of activated receptors, blocking NK cells’ release from bone marrow, enhancing NK cells’ apoptosis, or elevating serum level of corticosterone, which inhibits NK cells’ function.37, 38, 39 In agreement, we also found chronic alcohol consumption resulted in a significant decrease in absolute number and IFN-γ expression of hepatic NK cells. In addition, other lymphocytes such as CD4+ T cells, CD8+ T cells and B cells, but not liver ILC1, also displayed a global reduction in cell number. The mechanism that liver ILC1 are able to resist alcohol-induced apoptosis warrants further investigation. Interestingly, our previous study showed that NK cells contributed to impaired hepatic CD8+ T-cell–mediated anti–hepatitis B virus immunity after alcohol consumption.40 Although the study did not define the mechanism, our present data would suggest that after chronic alcohol consumption, the bulk NK cells in liver are dominance of ILC1, which exhibit a negative regulatory feature and limit antiviral activity of hepatic T cells.16
In summary, the present study reveals a role for liver ILC1 in aggravating alcoholic steatohepatitis through IL17A production. NK cell-derived IFN-γ, which was dramatically down-regulated during chronic alcohol consumption, confers protection against alcoholic steatohepatitis by inhibiting IL17A production. Our study demonstrates the crosstalk between NK cells and ILC1 through IFN-γ/IL17A axis in alcohol-induced liver disease and provides preclinical evidence for targeting IL17A for the treatment of alcoholic steatohepatitis.
Materials and Methods
Animal Experiments
WT C57BL/6J mice and Rag1-/- mice were purchased from the GemPharmatech Co, Ltd (Nanjing, China). IFN-γ-/- (GKO) mice were obtained from Hui Peng (University of Science and Technology of China, Hefei, China). A mouse model of 10-day chronic plus single-binge was established as previously described.22 At day 11 (8:00 am) after chronic alcohol feeding, the mice were gavaged with 31.5% alcohol at 5 g/kg body weight or control diet. Mice were euthanized and harvested at 6 hours after gavage. All animals were used according to the guidelines for experimental animal use from Anhui Medical University.
To evaluate the degree of liver steatosis or injury, the levels of ALT, AST, hepatic triglyceride, and H&E or Oil Red O were examined. Plasma concentrations of ALT and AST were detected by an automatic biochemical analyzer (Mindray, Shenzhen, China). The level of triglyceride content in liver homogenate was measured by TG kit (Applygen, Beijing, China). Liver tissues were stained with H&E or Oil Red O for the examination of lipid accumulation.
For group 1 ILC depletion, mice were injected intraperitoneally once with 50 μg of anti-AsGM1 antibody (Wako Co, Tokyo, Japan) at day 9 after ethanol feeding. Control mice were injected with same dose of rabbit IgG.
To expand NK cells in vivo, mice were injected intraperitoneally with 5 mg/kg of polyI:C every other day (APExBio, Houston, TX). PBS was used as negative control.
In the adoptive transfer experiments, CD49b+NK cells were purified from the spleen of WT, Rag1-/- mice or GKO mice by negative isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). The 1 × 106 cells were injected intravenously into recipient mice each time at indicated time points.
For IL17A neutralization, mice were injected intraperitoneally with 400 μg anti-IL-17A (Selleck, Shanghai, China) antibody twice at days 6 and 9 after ethanol feeding. Control mice were injected with same dose of rabbit IgG.
Flow Cytometric Analyses
Liver mononuclear cells were isolated, blocked with anti-CD16/CD32 antibody, stained with various antibodies, and then analyzed using a BD FACS celesta flow cytometry (BD, Franklin Lakes, NJ). Live cells were defined by fixable live/dead viability dye (Thermo Fisher, Waltham, MA). For intracellular staining, liver mononuclear cells from CD- or ethanol fed mice were stimulated with 1× cell stimulation cocktail (eBioscience, San Diego, CA) in presence of monensin (MULTI, Hangzhou, China). After 4 hours, cells were fixed and permeabilized by the Fix&Perm kit (eBioscience) and then stained with anti-IFN-γ or IL17A. For detection of Ki67, T-bet, Eomes, or RORγt, liver mononuclear cells were fixed, permeabilized, and then stained without further stimulation by 1× cell stimulation cocktail ex vivo. The data were analyzed with FlowJo v10.7.1 software.
The following monoclonal antibodies were used for flow cytometry: anti-CD3, anti-CD4, anti-CD8, anti-CD19, anti-NK1.1, anti-CD45, anti-CD49a, anti-CD49b, anti-CD69, anti-annexin V, anti-Ly6G, anti-TRAIL, anti-Ki67, anti-IFN-γ, anti-IL-17A, anti-Eomes, anti-T-bet, and anti-RORγt. Antibodies are listed in Table 1.
Table 1.
Antibodies Used in This Study
| Antibody | Catalog no. | Company |
|---|---|---|
| APC-Cy7 anti-mouse CD45 | 103116 | Biolegend |
| BV510 anti-mouse CD3 | 100233 | Biolegend |
| BV421 anti-mouse NK1.1 | 108741 | Biolegend |
| PerCP-Cy5.5 anti-mouse CD3 | 100328 | Biolegend |
| PerCP-Cy5.5 anti-mouse NK1.1 | 156525 | Biolegend |
| PE anti-mouse CD49b | 103506 | Biolegend |
| APC anti-mouse CD8 | 100712 | Biolegend |
| FITC anti-mouse CD4 | 100509 | Biolegend |
| APC anti-mouse CD19 | 115512 | Biolegend |
| APC anti-mouse CD49a | 142606 | Biolegend |
| APC anti-mouse Ly6G | 127613 | Biolegend |
| PE anti-mouse IFN-γ | 505808 | Biolegend |
| APC anti-mouse IL-17A | 506816 | Biolegend |
| PE anti-mouse T-bet | 644810 | Biolegend |
| PerCP-Cy5.5 anti-mouse RoRγt | 562683 | BD Pharmingen |
| PE anti-mouse Eomes | 157705 | Biolegend |
| Purified IL-17A mAb | A2120 | Selleck |
| Purified Anti-AsGM1 | 16-6507-39 | Thermo Fisher |
In Vitro Culturing of AML-12 Cell Line
The 2 × 106 AML-12 cells were placed into 6-well plate with 2 mL AML-12 special medium (Procell, China) containing 10% fetal bovine serum, 40 ng/mL dexamethasone, insulin, transferrin, and selenium and then treated with PBS or free fatty acid (1 mmol/L) with or without IL17A (40 ng/mL). After 24-hour treatment, the cells were collected, and the mRNA level of ACC or SREBP-1c was measured by quantitative polymerase chain reaction.
Quantitative Real-Time Polymerase Chain Reaction
Total RNA was extracted from liver tissue or AML cells using RNA Extraction Kit (Solarbio, Beijing, China). For cDNA synthesis, the RNA was reverse transcribed with cDNA Synthesis Kit (Accurate, Hunan, China), and real-time polymerase chain reaction was performed by using SYBR Premix ExTaq (Accurate, Hunan, China). Results were normalized to GAPDH mRNA expression. Polymerase chain reaction primers used are listed below:
GAPDH: CAAAGTTGTCATGGATGACC (F), CCATGGAGAAGGCTGGGG (R); ACC: CTTCCTCCTGATGAGCAACTCT (F), CGTGAGTTTTCCCAAAATAAGC (R); SREBP-1c: GAGG CCAAGCTTTGGACCTGG (F), CCTGCCTTCA GGCTTCTCAGG (R); IL-1β: TTCATCTTTGAAGAAGAGCCCAT (F), TCGGAGCCTGTAGTGCAGTT (R); IL-6: TGGAAATGAGAAAAGAGTTGTGC (F), CCAGTTTGGTAGCATCCATCA (R); IL-13: AACGGCAGCATGGTATGGAGTG (F), TGGGTCCTGTAGATGGCATTGC (R); IL-17A: CCTTCACTTTCAGGGTCGAG (F), CAGTTTGGGACCCCTTTACA (R); TNF-α: ATCTACCTGGGAGGCGTCTT (F), GAGTGGCACAAGGAACTGGT (R).
Statistical Analysis
Data were presented as mean ± standard error of mean. All statistical analyses were conducted with GraphPad Prism version 8.0 (GraphPad Software Inc, San Diego, CA). Two-tailed unpaired Student t tests were used to compare the differences between 2 groups. One-way analysis of variance was used to compare values obtained from 3 or more groups with 1 independent variable. Two-way analysis of variance was used to compare groups with 2 independent variables. The data were considered statistically significant when differences in values reached P <.05.
Acknowledgments
CRediT Authorship Contributions
Chen Cheng (Data curation: Equal; Methodology: Equal; Writing – original draft: Equal)
Qian Zhang (Data curation: Equal; Methodology: Equal)
Yue Li (Data curation: Equal; Methodology: Equal)
Jiali Jiang (Data curation: Supporting)
Linxi Xie (Data curation: Supporting)
Haiyuan Shen (Data curation: Supporting)
Dongqing Wu (Methodology: Supporting)
Hejiao Zhang (Data curation: Supporting)
Huiru Zhang (Data curation: Supporting)
Xuan Wang (Data curation: Supporting)
Hongyu Wu (Methodology: Supporting)
Jingjing Xu (Methodology: Supporting)
Li Gui (Methodology: Equal)
Bao Li (Methodology: Equal)
Cynthia Ju (Supervision: Supporting)
Hui Peng (Supervision: Supporting)
Shi Yin (Methodology: Supporting; Supervision: Equal)
Long Xu, PhD (Conceptualization: Lead; Funding acquisition: Lead; Supervision: Lead; Writing – review & editing: Lead)
Footnotes
Conflicts of interest The authors disclose no conflicts.
Funding Supported by the Natural Science Foundation of China Grant 81873570, the Talent Training Program 2022YPJH101 (School of Basic Medical Sciences, Anhui Medical University), the Postgraduate Innovation Research and Practice Program of Anhui Medical University (YJS20210281), and the Scientific Research Platform and Base Upgrading Plan of Anhui Medical University (2021xkjT048).
Contributor Information
Shi Yin, Email: drshiyin@ustc.edu.cn.
Long Xu, Email: xulong@ahmu.edu.cn.
References
- 1.Artis D., Spits H. The biology of innate lymphoid cells. Nature. 2015;517:293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- 2.Colonna M. Innate lymphoid cells: diversity, plasticity, and unique functions in immunity. Immunity. 2018;48:1104–1117. doi: 10.1016/j.immuni.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kotas M.E., Locksley R.M. Why innate lymphoid cells? Immunity. 2018;48:1081–1090. doi: 10.1016/j.immuni.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weizman O.E., Adams N.M., Schuster I.S., Krishna C., Pritykin Y., Lau C., Degli-Esposti M.A., Leslie C.S., Sun J.C., O’Sullivan T.E. ILC1 confer early host protection at initial sites of viral infection. Cell. 2017;171:795–808 e12. doi: 10.1016/j.cell.2017.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng H., Tian Z. Re-examining the origin and function of liver-resident NK cells. Trends Immunol. 2015;36:293–299. doi: 10.1016/j.it.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 6.Peng H., Tian Z. Diversity of tissue-resident NK cells. Semin Immunol. 2017;31:3–10. doi: 10.1016/j.smim.2017.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Sojka D.K., Plougastel-Douglas B., Yang L., Pak-Wittel M.A., Artyomov M.N., Ivanova Y., Zhong C., Chase J.M., Rothman P.B., Yu J., Riley J.K., Zhu J., Tian Z., Yokoyama W.M. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. eLife. 2014;3 doi: 10.7554/eLife.01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daussy C., Faure F., Mayol K., Viel S., Gasteiger G., Charrier E., Bienvenu J., Henry T., Debien E., Hasan U.A., Marvel J., Yoh K., Takahashi S., Prinz I., de Bernard S., Buffat L., Walzer T. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med. 2014;211:563–577. doi: 10.1084/jem.20131560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pikovskaya O., Chaix J., Rothman N.J., Collins A., Chen Y.H., Scipioni A.M., Vivier E., Reiner S.L. Cutting edge: eomesodermin is sufficient to direct type 1 innate lymphocyte development into the conventional NK lineage. J Immunol. 2016;196:1449–1454. doi: 10.4049/jimmunol.1502396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bai L., Vienne M., Tang L., Kerdiles Y., Etiennot M., Escalière B., Galluso J., Wei H., Sun R., Vivier E., Peng H., Tian Z. Liver type 1 innate lymphoid cells develop locally via an interferon-γ-dependent loop. Science. 2021:371. doi: 10.1126/science.aba4177. [DOI] [PubMed] [Google Scholar]
- 11.Takeda K., Cretney E., Hayakawa Y., Ota T., Akiba H., Ogasawara K., Yagita H., Kinoshita K., Okumura K., Smyth M.J. TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood. 2005;105:2082–2089. doi: 10.1182/blood-2004-08-3262. [DOI] [PubMed] [Google Scholar]
- 12.Sojka D.K., Tian Z., Yokoyama W.M. Tissue-resident natural killer cells and their potential diversity. Semin Immunol. 2014;26:127–131. doi: 10.1016/j.smim.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sojka D.K., Plougastel-Douglas B., Yang L., Pak-Wittel M.A., Artyomov M.N., Ivanova Y., Zhong C., Chase J.M., Rothman P.B., Yu J., Riley J.K., Zhu J., Tian Z., Yokoyama W.M. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife. 2014;3 doi: 10.7554/eLife.01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nabekura T., Riggan L., Hildreth A.D., O’Sullivan T.E., Shibuya A. Type 1 innate lymphoid cells protect mice from acute liver injury via interferon-γ secretion for upregulating Bcl-xL expression in hepatocytes. Immunity. 2020;52:96–108. doi: 10.1016/j.immuni.2019.11.004. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fumagalli V., Venzin V., Di Lucia P., Moalli F., Ficht X., Ambrosi G., Giustini L., Andreata F., Grillo M., Magini D., Ravà M., Friedrich C., Fontenot J.D., Bousso P., Gilmore S.A., Khan S., Baca M., Vivier E., Gasteiger G., Kuka M., Guidotti L.G., Iannacone M. Group 1 ILCs regulate T cell-mediated liver immunopathology by controlling local IL-2 availability. Science Immunology. 2022;7 doi: 10.1126/sciimmunol.abi6112. [DOI] [PubMed] [Google Scholar]
- 16.Zhou J., Peng H., Li K., Qu K., Wang B., Wu Y., Ye L., Dong Z., Wei H., Sun R., Tian Z. Liver-resident NK cells control antiviral activity of hepatic T cells via the PD-1-PD-L1 axis. Immunity. 2019;50:403–417.e4. doi: 10.1016/j.immuni.2018.12.024. [DOI] [PubMed] [Google Scholar]
- 17.Ducimetière L., Lucchiari G., Litscher G., Nater M., Heeb L., Nuñez N.G., Wyss L., Burri D., Vermeer M., Gschwend J., Moor A.E., Becher B., van den Broek M., Tugues S. Conventional NK cells and tissue-resident ILC1s join forces to control liver metastasis. Proc Natl Acad Sci U S A. 2021:118. doi: 10.1073/pnas.2026271118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.López-Yglesias A.H., Burger E., Camanzo E., Martin A.T., Araujo A.M., Kwok S.F., Yarovinsky F. T-bet-dependent ILC1- and NK cell-derived IFN-γ mediates cDC1-dependent host resistance against Toxoplasma gondii. PLoS Pathogens. 2021;17 doi: 10.1371/journal.ppat.1008299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vivier E., Artis D., Colonna M., Diefenbach A., Di Santo J.P., Eberl G., Koyasu S., Locksley R.M., McKenzie A.N.J., Mebius R.E., Powrie F., Spits H. Innate lymphoid cells: 10 years on. Cell. 2018;174:1054–1066. doi: 10.1016/j.cell.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 20.Kabata H., Moro K., Koyasu S. The group 2 innate lymphoid cell (ILC2) regulatory network and its underlying mechanisms. Immunol Rev. 2018;286:37–52. doi: 10.1111/imr.12706. [DOI] [PubMed] [Google Scholar]
- 21.Zeng B., Shi S., Ashworth G., Dong C., Liu J., Xing F. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death & Disease. 2019;10:315. doi: 10.1038/s41419-019-1540-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertola A., Mathews S., Ki S.H., Wang H., Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013;8:627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng H., Jiang X., Chen Y., Sojka D.K., Wei H., Gao X., Sun R., Yokoyama W.M., Tian Z. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Invest. 2013;123:1444–1456. doi: 10.1172/JCI66381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui K., Yan G., Zheng X., Bai L., Wei H., Sun R., Tian Z. Suppression of natural killer cell activity by regulatory NKT10 cells aggravates alcoholic hepatosteatosis. Frontiers in Immunology. 2017;8:1414. doi: 10.3389/fimmu.2017.01414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gieseck R.L., 3rd, Ramalingam T.R., Hart K.M., Vannella K.M., Cantu D.A., Lu W.Y., Ferreira-Gonzalez S., Forbes S.J., Vallier L., Wynn T.A. Interleukin-13 activates distinct cellular pathways leading to ductular reaction, steatosis, and fibrosis. Immunity. 2016;45:145–158. doi: 10.1016/j.immuni.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma H.Y., Yamamoto G., Xu J., Liu X., Karin D., Kim J.Y., Alexandrov L.B., Koyama Y., Nishio T., Benner C., Heinz S., Rosenthal S.B., Liang S., Sun M., Karin G., Zhao P., Brodt P., McKillop I.H., Quehenberger O., Dennis E., Saltiel A., Tsukamoto H., Gao B., Karin M., Brenner D.A., Kisseleva T. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J Hepatol. 2020;72:946–959. doi: 10.1016/j.jhep.2019.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu J., Ma H.Y., Liu X., Rosenthal S., Baglieri J., McCubbin R., Sun M., Koyama Y., Geoffroy C.G., Saijo K., Shang L., Nishio T., Maricic I., Kreifeldt M., Kusumanchi P., Roberts A., Zheng B., Kumar V., Zengler K., Pizzo D.P., Hosseini M., Contet C., Glass C.K., Liangpunsakul S., Tsukamoto H., Gao B., Karin M., Brenner D.A., Koob G.F., Kisseleva T. Blockade of IL-17 signaling reverses alcohol-induced liver injury and excessive alcohol drinking in mice. JCI Insight. 2020;5 doi: 10.1172/jci.insight.131277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korn T., Bettelli E., Oukka M., Kuchroo V.K. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 29.Fu B., Li X., Sun R., Tong X., Ling B., Tian Z., Wei H. Natural killer cells promote immune tolerance by regulating inflammatory TH17 cells at the human maternal-fetal interface. Proc Natl Acad Sci U S A. 2013;110:E231–E240. doi: 10.1073/pnas.1206322110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z., Zhang S., Zou Z., Shi J., Zhao J., Fan R., Qin E., Li B., Li Z., Xu X., Fu J., Zhang J., Gao B., Tian Z., Wang F.S. Hypercytolytic activity of hepatic natural killer cells correlates with liver injury in chronic hepatitis B patients. Hepatology. 2011;53:73–85. doi: 10.1002/hep.23977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giles D.A., Moreno-Fernandez M.E., Stankiewicz T.E., Graspeuntner S., Cappelletti M., Wu D., Mukherjee R., Chan C.C., Lawson M.J., Klarquist J., Sünderhauf A., Softic S., Kahn C.R., Stemmer K., Iwakura Y., Aronow B.J., Karns R., Steinbrecher K.A., Karp C.L., Sheridan R., Shanmukhappa S.K., Reynaud D., Haslam D.B., Sina C., Rupp J., Hogan S.P., Divanovic S. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. Nature Medicine. 2017;23:829–838. doi: 10.1038/nm.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harley I.T., Stankiewicz T.E., Giles D.A., Softic S., Flick L.M., Cappelletti M., Sheridan R., Xanthakos S.A., Steinbrecher K.A., Sartor R.B., Kohli R., Karp C.L., Divanovic S. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology. 2014;59:1830–1839. doi: 10.1002/hep.26746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rau M., Schilling A.K., Meertens J., Hering I., Weiss J., Jurowich C., Kudlich T., Hermanns H.M., Bantel H., Beyersdorf N., Geier A. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol. 2016;196:97–105. doi: 10.4049/jimmunol.1501175. [DOI] [PubMed] [Google Scholar]
- 34.Bonelli M., Shih H.Y., Hirahara K., Singelton K., Laurence A., Poholek A., Hand T., Mikami Y., Vahedi G., Kanno Y., O’Shea J.J. Helper T cell plasticity: impact of extrinsic and intrinsic signals on transcriptomes and epigenomes. Curr Top Microbiol Immunol. 2014;381:279–326. doi: 10.1007/82_2014_371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lim A.I., Verrier T., Vosshenrich C.A., Di Santo J.P. Developmental options and functional plasticity of innate lymphoid cells. Curr Opin Immunol. 2017;44:61–68. doi: 10.1016/j.coi.2017.03.010. [DOI] [PubMed] [Google Scholar]
- 36.Vonarbourg C., Mortha A., Bui V.L., Hernandez P.P., Kiss E.A., Hoyler T., Flach M., Bengsch B., Thimme R., Hölscher C., Hönig M., Pannicke U., Schwarz K., Ware C.F., Finke D., Diefenbach A. Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt(+) innate lymphocytes. Immunity. 2010;33:736–751. doi: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernink J.H., Krabbendam L., Germar K., de Jong E., Gronke K., Kofoed-Nielsen M., Munneke J.M., Hazenberg M.D., Villaudy J., Buskens C.J., Bemelman W.A., Diefenbach A., Blom B., Spits H. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity. 2015;43:146–160. doi: 10.1016/j.immuni.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 38.Laso F.J., Madruga J.I., Girón J.A., López A., Ciudad J., San Miguel J.F., Alvarez-Mon M., Orfao A. Decreased natural killer cytotoxic activity in chronic alcoholism is associated with alcohol liver disease but not active ethanol consumption. Hepatology. 1997;25:1096–1100. doi: 10.1002/hep.510250508. [DOI] [PubMed] [Google Scholar]
- 39.Gao B., Seki E., Brenner D.A., Friedman S., Cohen J.I., Nagy L., Szabo G., Zakhari S. Innate immunity in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2011;300:G516–G525. doi: 10.1152/ajpgi.00537.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang S., Zhu Y., Cheng C., Li Y., Ma T., Peng Z., Li Q., Xu J., Xu L. NK cells contribute to hepatic CD8(+) T cell failure in hepatitis B virus-carrier mice after alcohol consumption. Virus Res. 2020;286 doi: 10.1016/j.virusres.2020.198085. [DOI] [PubMed] [Google Scholar]