

Abstract
Autoimmune lymphoproliferative syndrome (ALPS) is a disorder of abnormal lymphocyte homeostasis resulting from mutations in the Fas apoptotic pathway. Clinical manifestations include non-infectious and non-malignant lymphadenopathy, splenomegaly and autoimmune pathology, most commonly autoimmune cytopenias. Rarely, and associated with specific genetic mutations, patients with ALPS may go on to develop secondary lymphoid malignancies. Though ALPS is a rare disorder, it should be suspected and ruled out in children presenting with chronic and refractory multilineage cytopenias associated with nonmalignant lymphoproliferation. Revised diagnostic criteria and insights into disease biology have improved both diagnosis and treatment. Sirolimus and mycophenolate mofetil (MMF) are best-studied and most effective corticosteroid sparing therapies for ALPS, and they should be considered first-line therapy for patients who need chronic treatment. This review will highlight practical clinical considerations for the diagnosis and management of ALPS.
1. Background
Autoimmune lymphoproliferative syndrome (ALPS) is a disorder of abnormal lymphocyte survival caused by a dysregulation of the Fas apoptotic pathway [1–3]. As a result of disrupted lymphocyte homeostasis, autoreactive cells persist leading to development of chronic nonmalignant lymphadenopathy, splenomegaly, multi-lineage cytopenias and increased risk of lymphoma [4–5]. Approximately two-thirds of ALPS patients have identified germline or somatic mutations, which are now incorporated into diagnostic criteria [6]. Although improved clinical and molecular criteria have resulted in consensus based diagnostic criteria, the management of ALPS varies significantly.
2. Pathophysiology
ALPS is a disorder caused by a dysregulation of the Fas apoptotic pathway. Extrinsic lymphocyte apoptosis is initiated by Fas ligand binding to Fas leading to conformational changes of the intracellular portion of the Fas protein. This, in turn, results in recruitment of the Fas-associated death domain (FADD) to the homologous Fas domain. Therein procaspases 8 or 10 are recruited through interaction of their death effector domains with the amino terminus of FADD resulting in intrinsic apoptosis. This triggers the caspase cascade ending in cell death, DNA degradation and proteolysis (Figure 1) [2–4]. An abnormality in this cascade, as seen in ALPS, results in the accumulation of lymphocytes.
Figure 1:
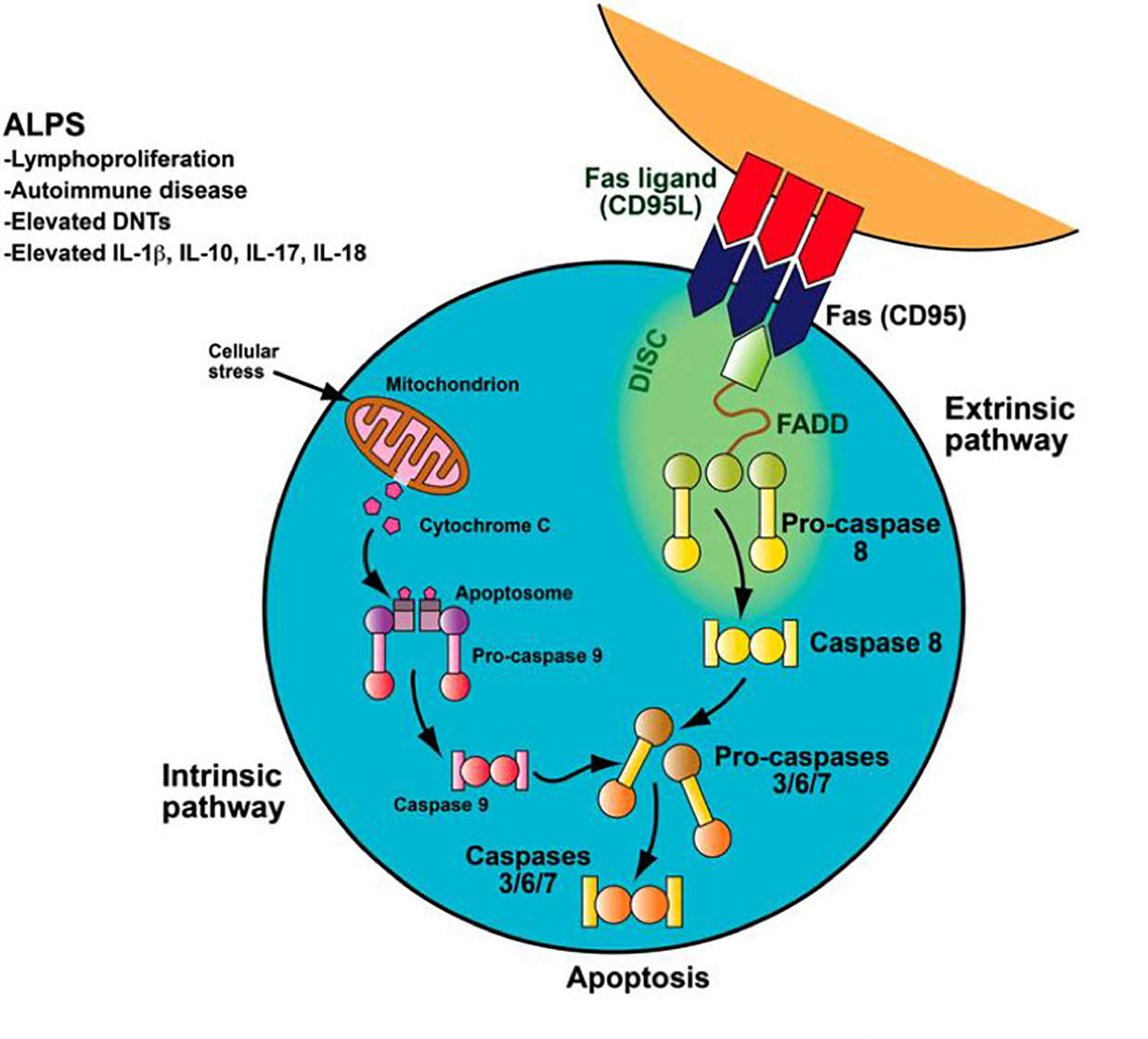
Fas apoptotic pathway.
Fas apoptotic pathway. Normally, as part of the downregulation of the immune response, activated B and T lymphocytes upregulate FAS and activated T lymphocytes upregulate FAS-ligand. These lymphocytes interact and trigger the caspase cascade, leading to proteolysis, DNA degradation, and apoptosis. This FAS-mediated pathway is part of the extrinsic apoptotic pathway. In contrast, mitochondrial-induced apoptosis after cellular stress is part of the intrinsic apoptotic pathway. Patients with ALPS have a defect in the FAS apoptotic pathway, leading to abnormal lymphocyte survival.
Reproduced with permission © Sue Seif.
The role of the Fas antigen in lymphocyte homeostasis and peripheral tolerance via negative selection of autoreactive T cells was initially reported in lymphoproliferation (lpr) mice that carry loss of function homozygous Fas mutations. Like ALPS patients, these mice developed lymphadenopathy, autoimmunity and possessed expansion double negative T cells (DNT) (phenotype: TCRαβ + CD4−/CD8− T cells) [5]. Animal model findings were translated to characterization of a human disease state, i.e. ALPS, wherein patients with lymphoproliferation were determined to have a mutation of the Fas apoptotic pathway resulting in failure of lymphocyte apoptosis and elevated DNTs [2, 3]. The exact role of DNTs in ALPS is not confirmed; however, recent studies suggest they are no longer considered an epiphenomenon and support they are the pathogenic cell driving the disease. Further these studies suggest DNTs are not thymic derived, but rather are terminally differentiated effector T cells that have lost CD8+ expression [7, 8]
Autoimmunity is a result of a failure of self-tolerance. Self-tolerance can be divided into central and peripheral tolerance. Central tolerance is achieved by the elimination of autoreactive lymphocytes by induction of apoptotic cell death in the thymus or marrow. Peripheral tolerance mechanisms include anergy, suppression by T regulatory cells and apoptotic deletion with the ultimate goal of avoiding autoimmunity and subsequent tissue damage.
Extrinsic apoptosis, cell death, is triggered by cell surface receptor-ligand interactions while intrinsic apoptosis is achieved by activation of mitochondrial proteins that process and activate intracellular caspases. Lymphocyte apoptosis is mediated by the cell surface receptor Fas, which is a member of the tumor necrosis family receptor superfamily of proteins. Activated B and T lymphocytes upregulate Fas-ligand expression to down regulate a normal immune response. Fas directly triggers lymphocyte apoptosis to maintain lymphocyte homeostasis and peripheral immune tolerance thereby preventing autoimmunity [1].
3. Genetics
Molecular characterization of ALPS has translated to a paradigm shift in the diagnosis of ALPS. Revised international consensus criteria now incorporate gene-based classification into ALPS diagnosis and nomenclature (Table 1 and 2) [6]. The vast majority of ALPS patients (~70%) have an identifiable mutation in the Fas pathway. Although ALPS is well characterized molecularly, approximately 20–30% of ALPS patients do not have an identifiable mutation [9–12].
Table 1:
ALPS genetics
| Prior Nomenclature | Revised Nomenclature | Mutation | Definition | Incidence |
|---|---|---|---|---|
|
| ||||
| 0 | ALPS-FAS | FAS | meet ALPS diagnostic criteria germline homozygous FAS mutations |
70% |
| Ia | ALPS-FAS |
FAS TNFRSF6 germline |
meet ALPS diagnostic criteria germline heterozygous FAS mutations |
|
| Ib | ALPS-FASLG |
FAS
TNFSF6 |
meet ALPS diagnostic criteria germline mutations FAS ligand |
<1% |
| Im | ALPS-sFAS |
FAS YNFRSF6 somatic |
meet ALPS diagnostic criteria somatic FAS mutations |
10% |
| IIa | ALPS-CASP10 | Caspase 10 | meet ALPS diagnostic criteria germline Caspase 10 mutations |
2% |
| III | ALPS-U | not known | meet ALPS diagnostic criteria no mutation in FAS, FASL or CASP10 |
20% |
ALPS= autoimmune lymphoproliferative disorder; sFasL= soluble Fas ligand; FasL= Fas ligand; CASP10= Caspase 10
Table 2:
ALPS Diagnostic Criteria
| a. Diagnostic Criteria for ALPS defined by 1999 NIH Consensus | ||
| Required | Chronic non malignant lymphadenopathy and/or splenomegaly Increased peripheral DNT cells Lymphocyte apopotic defect |
|
| Supporting | ALPS family history Histopathology consistent with ALPS Autoimmune manifestations |
|
| b. Diagnostic Criteria for ALPS defined by 2009 NIH Consensus | ||
| Required | 1. Chronic (>6 months) non-infectious, non-malignant lymphadenopathy and/or splenomegaly 2. Elevated DNT (≥1.5% of total lymphocytes or ≥ 2.5% of CD3+ lymphocytes) |
|
| Accessory | Primary | 1. Defective lymphocyte apopotosis (in 2 separate assays) 2. Somatic or gemeline mutation in FAS, FASLG or CASP10 |
| Secondary | 1. Elevated plasma sFasL (>200 pg/mL) or interleukin-10 (>20 pg/mL) or interleukin-18 (>500 pg/mL) or elevated plasma or serum Vitamin B12 (>1500 ng/L) 2. Immunohistological findings consistent with ALPS 3. Autoimmune cytopenias (thrombocytopenia, hemolytic anemia or neutropenia) and elevated immunoglobulin G (polyclonal hypergammaglobinemia) 4. family history of non-infectious/non-malgnant lymphoproliferation ± autoimmunity |
|
| Definitive Diagnosis | Required plus one primary accessory criteria | |
| Probable Diagnosis | Required plus one secondary accessory criteria | |
ALPS= autoimmune lymphoproliferative disorder; DNT= double negative T-cells (CD3+, CD4−, CD8−, T-cell receptor αβ+); sFasL= soluble Fas ligand
The majority of patients have germline (~ 60%) or somatic (~ 10%) mutations in FAS, also known as TNFRSF6. Germline FAS mutations are typically inherited in an autosomal dominant fashion [11]. These mutations are most commonly dominant negative, heterozygous, missense mutations in the intracellular death domain (exon 7–9) whereby the mutated allele inhibits the function of the wild-type allele resulting in absent Fas activity. Approximately 30% of ALPS causative mutations affect the extracellular domain. Among these mutations, the majority are nonsense or frameshift mutations causing haploinsufficiency resulting in decreased, but not absent, Fas activity [12].
Somatic FAS mutations primarily affect nonthymic DNTs, which are a subset of T cells typically only present at small percentages in peripheral blood but are markedly elevated in ALPS. As expected, patients with somatic mutations typically present later than patients with germline mutations; however, disease manifestations are similar [13, 14]. Importantly germline mutations in the gene that encodes Fas ligand (FASL) (<1%) and the gene that encodes Caspase 10 (CASP10) (2%) are well described; however, somatic mutations in CASP10 or FASL have not been described [9].
Although FAS mutations have variable penetrance, clinical manifestations have a higher penetrance in families with dominant negative mutations. Importantly, dominant negative intracellular mutations are an exception to the general principle of poor genotype-phenotype correlation in ALPS and are associated with a high risk of secondary lymphoid malignancies [12]. The remaining ALPS mutations associated with poor genotype-phenotype predictability suggests a second ‘hit’ (e.g. additional genetic mutation and/or an environmental trigger) may be required for clinical disease. This hypothesis is supported by recent work that characterized seven ALPS patients with inherited heterozygote FAS mutations. In addition to their underlying mutations, analysis confirmed they also acquired somatic alterations in the second FAS allele [15].
4. Clinical Manifestations
Clinical manifestations of ALPS include lymphadenopathy, splenomegaly, and propensity to develop autoimmune disease and, rarely, secondary lymphoid malignancy [4, 16]. Importantly, although disease manifestations can result in morbidity and rarely mortality, ALPS patients have a near-normal life span and disease manifestations improve with age.
Due to the diagnostic challenge presented by ALPS, the National Institutes of Health outlined an initial ALPS diagnostic criterion in 1999 (Table 2a). Thereafter, further understanding of ALPS disease pathology and molecular characterization informed revised 2010 international consensus diagnostic guidelines that incorporated both phenotypic manifestations and genotype (Table 2b) [6].
Among disease symptoms, lymphoproliferation is the most common clinical finding and includes lymphadenopathy, splenomegaly and, less commonly, hepatomegaly [10, 11]. All ALPS patients develop lymphoproliferation consisting of lymphadenopathy and/or splenomegatly at some point in their disease course, but it is most commonly initially observed at a young age (median age of onset: 11.5 months). In its’ most severe form, lymphoproliferation can result in complications such as respiratory compromise secondary to abdominal competition or upper airway obstruction. Lymphadenopathy is not accompanied by constitutional symptoms, and if it is, should raise concern for other etiology or disease evolution, e.g. malignancy. Hepatomegaly, while present in approximately 40% of ALPS patients, is not part of diagnostic criteria. Although symptoms can be quite impressive, lymphoproliferation typically resolves in early adulthood [10,11].
Greater than 70% of patients develop autoimmune disease, most commonly immune mediated cytopenias, affecting erythrocytes (autoimmune hemolytic anemia), platelets (immune thrombocytopenia) or neutrophils (autoimmune neutropenia). Immune mediated cytopenias vary from asymptomatic laboratory abnormalities to multi-lineage life threatening illness. Unlike lymphoproliferation, autoimmune hematological manifestations are unlikely to resolve with adulthood, but can improve. Autoimmune disease typically presents after lymphoproliferation and often worsens during puberty [10,11].
Autoimmunity outside cytopenias is less frequent, but occurs in approximately 10–20% of ALPS patients. Pathology can affect nearly every organ system. The most non-hematologic autoimmune manifestation is uritcaria. Described autoimmune manifestations include nephritis, hepatitis, arthritis, colitis, infiltrative lung lesions, pancreatitis, uveitis, pancreatitis, transverse myelitis, cerebellar ataxia, and myocarditis [10, 17].
In addition to autoimmunity, risk of lymphoma is a major cause of morbidity and mortality among ALPS patients. Among ALPS patient’s Non-Hodgkin Lymphoma (NHL) risk is approximately 60X and Hodgkin Lymphoma is nearly 150X greater than the general population [18]. Importantly, risk of malignancy does not appear to be evenly distributed among ALPS patients and is most prevalent in patients with germline FAS mutant ALPS [9]. Although ALPS patients have a higher risk of lymphoma, the vast majority of patients respond to conventional multi-agent chemotherapy [9, 10]. This is consistent with findings that FAS family gene mutations are prevalent in lymphomas in the general population [18, 19]. Lastly, family members with an FAS mutation, but without clinical manifestations of ALPS, are also predisposed to malignancy [19].
5. Diagnosis
Revised NIH consensus diagnostic criteria outlined in Table 2b are based on 2 required and 6 additional criteria. Definitive diagnosis includes required criteria and one primary accessory criterion, while a probable diagnosis meets required criteria and one secondary accessory criteria [6]. Required criteria includes non-infectious and non-malignant lymphadenopathy and/or splenomegaly (>6 months), and elevated DNTs (phenotype: TCRαβ + CD3+/CD4−/CD8− T cells) [6, 7, 20]. Of importance, the difference between definitive and probable diagnosis is semantic and meant for research in order to distinguish patients with genetically or biologically proven defects in Fas. Clinically, patients with definitive and probable ALPS should be treated the same and patients and families with “probable” ALPS should be counseled that they have ALPS.
DNTs are evaluated by flow cytometry and should be completed at an experienced laboratory that includes testing for the TCRα/β receptor. This is because TCRαβ−CD3+/CD4−/CD8− T cells are nonpathogenic and evaluation must be able to distinguish this cell type from ALPS DNTs. Abnormal elevation of DNTs is defined by flow cytometry gating parameters wherein aberrant elevation of DNTs is ≥1.5% of total lymphocytes or ≥ 2.5% of T cells (CD3+ lymphocytes) [6, 13]. Knowing how the reference laboratory gates for DNT analysis will be important for the interpretation of flow cytometry results. Lastly, patients that are lymphopenic and/or on lymphotoxic therapy, e.g. steroids or sirolimus, should not undergo DNT evaluation due to risks of false negative or false positive evaluations [10].
The 2010 revised ALPS consensus diagnosis guidelines additionally outline accessory criteria. In vitro Fas mediated apoptosis was previously required and is now an accessory criteria (Table 2). This assay is time and labor intensive assay and only offered at select laboratories thereby making its routine use impractical. Peripheral blood is drawn from the patient and an unaffected control from which peripheral blood mononuclear cells (PBMCs) are isolated. Isolated PBMCs are then activated with mitogen and expanded in the presence of interleukin-2 in culture for approximately 3–4 weeks [2, 10]. Typically mitogen stimulation upregulates the Fas pathway such that when cells are then exposed to anti-Fas immunoglobulin M they undergo rapid apoptosis. Unlike normal cells in the assay, ALPS cells do not die following exposure [2,10]. Importantly, and informative as to why this is no longer essential for diagnosis, patients with somatic FAS mutations can present with normal in vitro Fas-induced apoptosis [21, 22]. DNTs do not survive in normal culture systems and the apoptosis assay tests FAS function on the non-DNT T cell population.
Additional accessory criteria represent the improved understanding of underlying physiology and molecular characterization of ALPS. Recent study identified elevated Interleukin-10, Interleukin-18, Vitamin B12 and soluble Fas ligand as ALPS biomarkers [23]. Further the characterized genetic mutations outlined in Table 1 are incorporated as accessory criteria outlined in Table 2b. Gene sequencing for ALPS is available at select commercial labs; however, FAS polymorphisms are common and a diagnostic mutation should be based on either prior description of ALPS mutations and/or with known functional consequence. Pathogenic ALPS mutations are available at the National Institute of Health ALPS website: http://www3.niaid.nih.gov/topics/ALPS/ [9].
6. Differential Diagnosis
Clinical findings observed in ALPS are heterogenous resulting in a broad differential including, autoimmune cytopenias, immunodeficiency syndromes, malignancy, infection and other lymphoproliferative disorders. Most of the conditions in the differential diagnosis can be readily distinguished from ALPS by a careful history, physical exam, laboratory evaluation and, rarely, nodal biopsy. A few conditions have considerable overlap with ALPS and warrant particular consideration, including Evan’s Syndrome (ES), common variable immunodeficiency (CVID), Rosai-Dorfman (RD), Ras-associated leukoproliferative disorder (RALD), Caspase 8 deficiency syndrome (CEDS), mutations in cytotoxic T-lymphocyte antigen 4 (CTLA4) and phosphoinositide 3-kinase catalytic domain (PI3Kc). There are additional overlapping pathologies that that are beyond the scope of this article [24–26]. We will focus on select pathologies that, at our center, most commonly overlapped with patients seen either in evaluation for ALPS or for whom an ALPS diagnosis was made.
A limited, but general outline for work up of a patient with suspected ALPS and the corresponding overlapping diseases of interest includes: DNT (ALPS), anti-nuclear antibody (systemic lupus erythematosus), quantitative immunoglobulins and specific antibody titers (CVID), T cell subsets including CD3/4 and CD3/8 and mitogens (T cell immune deficiency), plasma cytokine analysis, vitamin B12, sFASL, and HIV testing. Importantly, the intervention strategies for this diagnosis may vary greatly; thus, careful attention to appropriate diagnostic work up is important.
Evan’s Syndrome (ES) may be a phenotypic expression of disorders of immune dysregulation and should be a diagnosis of exclusion. In particular, ES shares many characteristics with ALPS. The treatment modalities for both pathologies are quite different, including a relative contraindication for splenectomy in ALPS patients, thereby making the distinction between ALPS and ES important. Evan’s Syndrome has variable definitions ranging from autoimmune destruction of only red cells and platelets, to a broader application including idiopathic autoimmune destruction of multiple cell lineages or autoimmune disease affecting any 2-cell lineages [27]. Although variable in definition, ES manifestations closely mimic ALPS related cytopenias; thus, it is important that any patient with ES be screened for ALPS. Single and multi-institutional study by our group observed that 30–40% of children diagnosis with ES ultimately met criteria for ALPS diagnosis. Among children diagnosed with ES, those with increased immunoglobulin levels, increased Vitamin B12 and isolated lymphadenopathy without splenomegaly were predictive of an underlying ALPS diagnosis [27, 28]. In contrast, a recent French national observational study of childhood autoimmune hemolytic anemia (AIHA) included 99 patients with ES, but did not find a high incidence of ALPS among their reported ES cohort [29]. Importantly, not all subjects in the study were tested for ALPS and an assigned ES diagnosis was restricted to patients diagnosed with AIHA; thus, the heterogeneity of ES definitions may contribute to these conflicting findings [27–29]. Additionally because ALPS is a genetic disease, there is likely heterogeneity in disease frequency among different patient populations [27–29].
Like ALPS, patients with CVID often present with lymphadenopathy and autoimmune disease, but can be distinguished by evaluation of immunoglobulin and vaccine titer response [30]. However, it is notable that a small percentage of ALPS patients have co-morbid CVID. Similarly, although RD is a histiocytic disorder whose histopathological finding of emperiolesis (lymphophagocytosis) is thought to be pathognomonic, similar findings can also be observed in nodal biopsies of ALPS patients [31]. Thus, among any patients with multi-lineage or chronic autoimmune cytopenias, CVID or RD, we recommend an ALPS evaluation.
Due to the similar functions of caspase 8 and 10, patients with mutations in CASP8 were previously classified as having ALPS. Like ALPS phenotypic manifestations, patients with CASP8 mutations demonstrate lymphadenopathy, defective Fas-mediated apoptosis and defects in T lymphocyte apoptosis. However, apoptotic defects in CASP8 mutations extend beyond findings in ALPS patients and include profound apoptotic defects in B, T and NK lymphocytes [32]. As a result, patients with CASP8 mutations are predisposed to significant herpes virus mucocutaneous infections [32]. Because of these differences, CASP8 mutations are now considered to have a distinct disease termed CEDS.
In the process of investigating ALPS pathophysiology, conditions with overlapping features were identified that include RALD, CTLA4 deficiency and PASLI (p110 delta activating mutation causing senescent T cells, lymphadenopathy and immunodeficiency). Importantly, while these pathologies share phenotypic similarities to ALPS, they are separate disease states that should be treated and approached differently than ALPS. RALD is due to gain of function acquired or somatic mutations in the RAS family genes (KRAS and NRAS). These mutations result in impaired cytokine withdrawal-induced apoptosis in T cells [33–36]. Clinical findings include lymphoproliferation, autoimmune cytopenias and hypergammaglobulinemia. Distinguishing RALD from juvenile myelomonocytic leukemia (JMML), a rare childhood cancer, can be difficult. Additionally, DNTs may be elevated in RALD, but are typically normal. Among the approximate 10 patients that have been described with RALD, they have followed an indolent course suggesting RALD is a non-malignant disease; however, the possible clonal nature of these abnormal cells has not been confirmed or disproved. Secondary cancers and unexplained sudden death have been described in RALD and somatic gain-of-function mutations in RAS family genes are extremely common in cancer. Thus, more studies are needed to determine if RALD is a benign lymphoproliferative disorder, a pre-malignant condition, or a malignant state.
PASLI disease is caused by gain of function mutations in PIK3CD that encodes the p110δ catalytic subunit of phosphatidylinositol-3-OH kinase (PI(3)K), which is predominantly expressed in leukocytes and is critical for adaptive immunity. Among patients PASLI disease phenotypic manifestations include lymphoproliferation and an immunodeficiency state characterized by recurrent sinopulmonary infections, chronic cytomegalovirus and Epstein-Barr virus infections with an increased risk of EBV related lymphoma [37–40]. Further evaluation of these patients revealed deficiency of naïve T cells, excess of senescent T effector cells and disrupted T and B cell development. In vitro evaluation of T effector cells demonstrated increased mTOR activity. Treatment of patients with PIK3CD mutations with rapamycin partially restored naïve T cells, rescued the observed in vitro T cell defects and resulted in an improved clinical course [10, 37].
Cytotoxic T-lymphocyte antigen 4 (CTLA-4), is a homologue of the T-cell co-stimulatory molecule CD28. CTLA-4 is an essential negative regulator of immune responses wherein CTLA-4 engagement inhibits the up-regulation of early T-cell activation and CTLA-4 inhibition enhances T-cell responses [41–42]. CTLA-4 haploinsufficiency results in disrupted T and B cell homeostasis and a complex immune dysregulation syndrome characterized by hypogammaglobulinemia, recurrent infections and multiple autoimmune clinical features [42]. There is no clear therapeutic recommendation for CTLA-4 aberrations; however, recent pre-clinical and clinical reports support the possible efficacy of rapamycin and abatecept (a fusion protein composed of IgG1 Fc fused with the extracellular domain of CTLA-4) [43, 44].
7. Management
It is our general practice to treat definitive and probable ALPS the same clinically. Figure 2 outlines the general schematic of our approach to the treatment of ALPS. Lymphadenopathy alone, outside of causing respiratory or organ compromise, does not necessitate treatment. Autoimmune disease manifestations, particularly cytopenias, most commonly require intervention with immunosuppression therapy. Figure 2 provides a general outline of immunosuppression agents used for the treatment of ALPS. Given that ALPS is caused by defects in the Fas apoptotic pathway, agents that can induce lymphocyte apoptosis are efficacious. Importantly management strategies such as splenectomy and rituximab are commonly employed for autoimmune disease, including cytopenias, but may be relatively contraindicated in ALPS patients while less common therapies (i.e. rapamycin) appear to be very effective in ALPS management [25].
Figure 2:
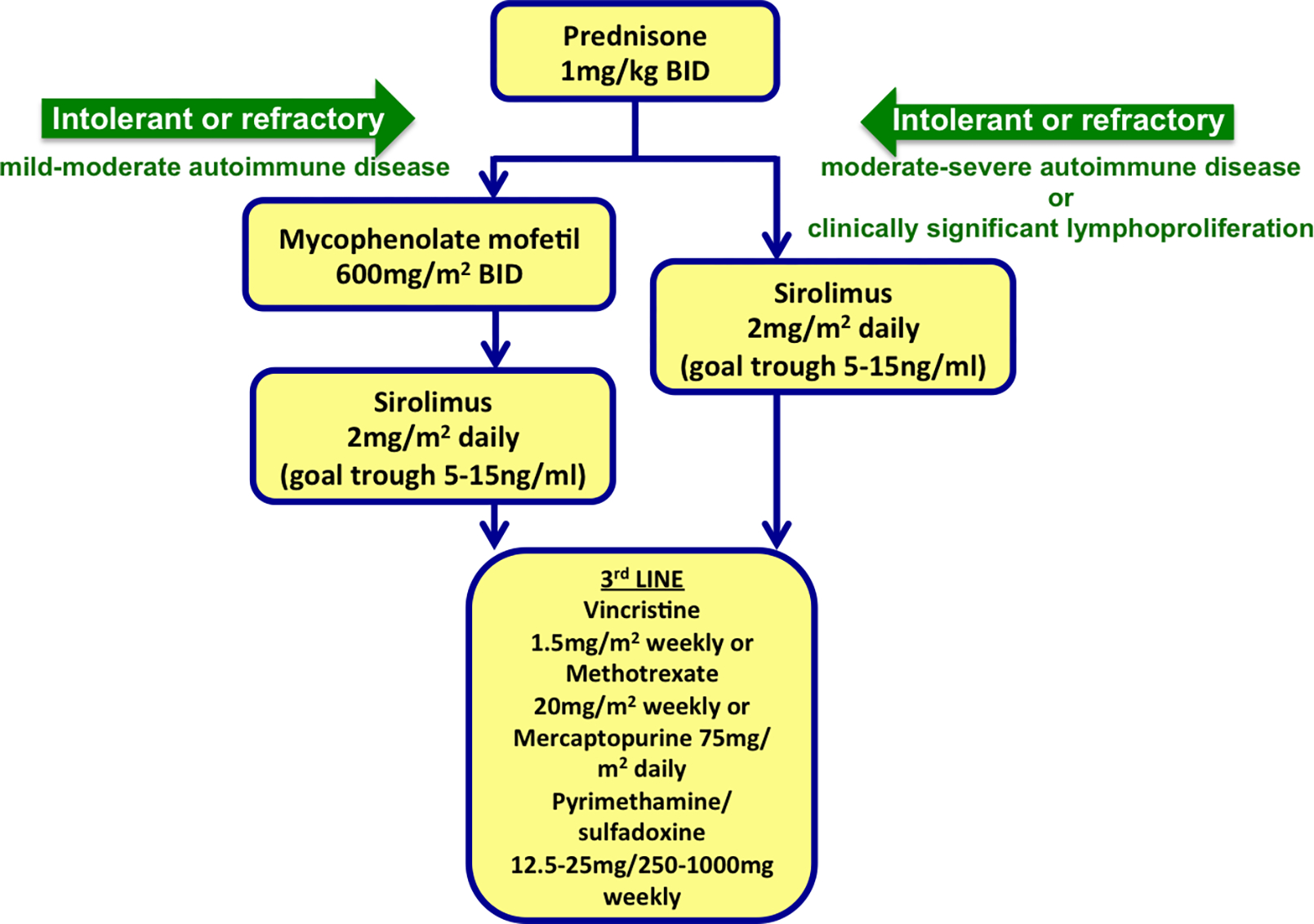
A general schematic of our approach to the treatment of ALPS. BID= twice daily
7.1. Acute Exacerbations
Acute ALPS exacerbations may be effectively treated with corticosteroids. However, given the side effect burden of prolonged steroid use, a low threshold for early transition to steroid sparing maintenance medications or second line therapies is encouraged. Immune suppression with pulse intravenous methylprednisolone (5–10 mg/kg) for 1–3 days followed by low-dose oral prednisone (1–2 mg/kg) maintenance tapered over the course of several weeks is a suggested first line therapy for ALPS presentation and/or acute exacerbations [9].
Corticosteroid therapy combined with intravenous immunoglobulin (1–2 gram/kg) may benefit patients with severe and refractory cytopenias. However, intravenous immunoglobulin is typically ineffective as a single agent therapy, except in the setting of a single-lineage autoimmune cytopenia [9]. The use of WinRho for isolated thrombocytopenia is generally avoided because many patients may be DAT positive and go on to developed exaggerated hemolysis with WinRho administration [9, 21]. Isolated neutropenia that is either acute or chronic, when not managed on maintenance therapy, may benefit from G-CSF. Suggested starting doses are 1–2 μg/kg administered two to three times weekly [9].
7.2. 2nd line Therapies
Second line therapies may be considered with persistent cytopenias (generally hemoglobin <8 g/dL, platelets <20,000/dL or an absolute neutrophil count <200 cells with a history of soft tissue infections). Many patients with autoimmune neutropenia do not develop infections even with very low ANCs as they can mount a neutrophil response to infection. Thus, we recommend only treating autoimmune neutropenia in the rare patients who develop infections. If tolerated, second line therapies should be initiated while patients are on steroids with appropriate overlap to allow for therapeutic levels of the second line therapy to be achieved prior to cessation of corticosteroids [9, 21].
7.2a. Mycophenolate Mofetil
Mycophenolate mofetil (MMF) inactivates inosine monophosphate dehydrogenase, which is important in purine synthesis and essential for lymphocyte proliferation. Use of MMF given twice-daily 600mg/m2 per dose for cytopenias was initially described in 2005. The preliminary report demonstrated steroid sparing efficacy in 12 of 13 children [45]. Subsequent expansion confirmed response in 56/61 ALPS patients; however, several patients did not have a sustained response [9]. Additional data from Italy evaluated 58 children with autoimmune cytopenias, including 12 ALPS and 24 ALPS-related subjects. Among the 39 subjects who received therapy beyond steroids, 11 were definitely diagnosed with ALPS and 13 had an ALPS related syndrome. All 11 subjects with ALPS and 5 of 10 with ALPS-related syndrome demonstrated clinical improvement with MMF therapy [46].
Importantly, however, MMF does not reduce lymphoproliferation or DNTs. While MMF should not be considered for up-front therapy and has limitations, it is an attractive agent given that it does not require monitoring and has a mild side effect profile. The most commonly observed side effects are diarrhea and neutropenia, and importantly, there has been no evidence of increased infectious risk when administered as monotherapy to ALPS patients. As such, it is generally our practice to offer MMF as a first line for chronic treatment in patients with mild-moderate autoimmune disease who do not have clinically significant lymphoproliferation (organ compromise or marked hypersplenism). If patients fail chronic MMF therapy, have moderate to severe autoimmune disease, or have clinically significant lymphoproliferation, sirolimus is most commonly the chronic therapy trialed.
7.2b. Sirolimus (rapamycin, Rapamune)
Sirolimus belongs to the class of drugs that inhibit the mammalian target of rapamycin (mTOR), which may induce apoptosis in abnormal B and T lymphocytes [47, 48]. Dysfunction of the Fas apoptosis in ALPS results in abnormal activation of PI3K/Akt/mTOR signaling pathway that may be driving disease. Thus, sirolimus may be a direct disease-targeting therapy. This hypothesis and the use of sirolimus was evaluated in a murine model of ALPS wherein sirolimus was found to dramatically reduce disease manifestations and supported superior efficacy over MMF [49].
In 2009, we published the first use of sirolimus in patients with ALPS. We demonstrated that treatment with the sirolimus led to a complete response (CR) in a small retrospective cohort of 5 children with corticosteroid refractory autoimmune lymphoproliferative syndrome (ALPS) [50]. We recently published the results of a multi-institutional prospective clinical, treating 30 patients with autoimmune cytopenias with sirolimus. Twelve patients with ALPS were included in this trial. All 12 subjects with ALPS had a complete and durable and rapid response with resolution of autoimmune disease and lymphoproliferation [51]. The majority of patients had elimination of detectable DNTs [51]. In addition to the children we have treated on our clinical trial, we have also guided clinicians in the treatment of over 30 additional children with ALPS from 18 different countries on 6 continents. The vast majority achieved a complete remission. Among this population, the majority of patients failed previous treatment, including many who had failed MMF (unpublished data).
In addition to treating ALPS autoimmunity, it is hypothesized sirolimus may actually decrease risk of malignancy by eliminating ALPS clones. Aberrant activation of FAS results in up-regulation of mTOR signaling. Therefore in effect, by inhibiting mTOR signaling, sirolimus reduces aberrant FAS activation and may represent a targeted therapy (unpublished data). Additional hypothesis include recognition that sirolimus is a T-regulatory cell (TReg) sparing immunosuppression agent. Thus, sirolimus may serve to adjunctively encourage underlying mechanisms of lymphocyte homeostasis by sparing TRegs. However, neither the contribution of TReg dysfunction in modulating self-tolerance in ALPS pathology nor the role of sirolimus on TReg function is clear.
In our practice and congruent with recently published prospective clinical trial results, if patients have severe disease or a large degree of lymphoproliferation it is generally our recommendation to use sirolimus as the first choice among chronic therapies [51]. Otherwise, sirolimus is typically employed in patients who fail MMF.
Like MMF, sirolimus when used as a single immunosuppressive agent has not been associated with an increased infectious risk [9, 21]. Of particular note, we monitored immune function in a subset of patients with ALPS treated on our clinical trial [51]. We found B and T cell number and function did not decrease in ALPS patients treated with chronic sirolimus. Thus, the risk of infection in this population when using sirolimus appears trivial and antimicrobial prophylaxis is not warranted. In contrast, B and T cell number and function often decreased significantly in ALPS patients treated with MMF (unpublished data). We encourage monitoring of B and T cell number and function in any patient with any autoimmune disease treated with chronic immune suppression, including corticosteroids. We do recommend avoidance of live vaccines on sirolimus or MMF, but encourage killed vaccine administration.
Sirolimus dosing is targeted to achieve a trough of 5–15ng/ml, which commonly approximates to a dose of 2.5 mg/m2 daily [50, 51]. It has not been our experience that targeted trough values correlate with side effect findings. Although sirolimus is generally well tolerated, it is prudent to monitor for hyperlipidemia, decreased renal function and myelosuppression. One of the most common side effects of sirolimus is oral ulceration mucositis that is most pronounced in the first month and, if mild, typically resolves on therapy. Additionally, multiple medications alter sirolimus pharmacokinetics. As such it is advised that any concurrent medication is cross-referenced for potential to alter sirolimus metabolism. General recommendations for sirolimus monitoring are outlined in table 3.
Table 3:
Sirolimus monitoring
| Lab Monitoring | Baseline-6 months | Long Term |
|---|---|---|
|
| ||
| CBC, reticulocyte | Baseline, every 2 weeks until improvement, then every 4–8 weeks | Every 3 months or as indicated |
| BMP, LFTs | Baseline, every 2 weeks initially, then monthly | Every 3 months or as indicated |
| Cholesterol, | Baseline, monthly | Every 3 months or as indicated |
| Sirolimus Trough | Day 3–5, Day 7–10, twice weekly until therapeutic, then monthly | Every 3 months AND with any change in medication |
| Quantitative Immunoglobins (IgG, A, M) | Baseline, every 2–3 months | Every 6 months |
| T cell subsets | Baseline, every 2–3 months | Every 6 months |
| T cell mitogens | Baseline, every 2–3 months | Every 6 months |
CBC= complete blood count; BMP=basic metabolic panel; LFTs=liver function tests
7.2c. Rituximab and splenectomy
Until all other immunosuppression agents have been exhausted, it is generally recommended to avoid both rituximab and splenectomy in ALPS patients [9–11]. Use of rituximab in ALPS patients has been minimally effective for AIHA and thrombocytopenia manifestations. Likely due to the underlying lymphocyte disorder, ALPS patients appear particularly prone to traditional toxicities related to rituximab. Noted prior toxicities include profound and persistent hypogammaglobulinemia requiring immunoglobulin replacement and failure to respond to polysaccharide vaccines. Such toxicities and associated infectious risk, particularly in patients who have undergone splenectomy, warrant caution in the use of rituximab [9, 10]. If rituximab therapy is ultimately employed, standard dosing consisting of 375 mg/m2 for 4 total doses is recommended [52, 53].
Splenectomy is generally contraindicated in ALPS patients. Despite antibiotic and vaccine prophylaxis, post splenectomy ALPS patients have a markedly increased risk of sepsis, particularly pneumococcal bacteremia (30%). Rao et al reported 34 ALPS patients who underwent splenectomy, most of which were done for immune mediated cytopenias before a diagnosis of ALPS was made. Among this group approximately 1/3 experienced sepsis, many of which were recurrent and 2 cases resulted in mortality. Further, approximately half of this splenectomized ALPS cohort experienced relapse of their cytopenias [52].
7.3. 3rd Line agents:
The vast majority of ALPS patients demonstrate a favorable response to 2nd line or chronic treatment options. As such, the short and long-term prognosis for the vast majority of ALPS patients is very good. Although use of MMF and sirolimus for chronic disease manifestations of ALPS are the most commonly employed and have been the most extensively studied, there have been reports using multi-agent therapy or additional single agent therapies including mercaptopurine, dapsone and azathioprine [2, 9, 10]. Use of pediatric acute lymphoblastic leukemia-like maintenance regimen consisting of vincristine, mercaptopurine and methotrexate as outlined in Figure 2 has been trialed with anecdotal success (personal communication, DT Teachey). Reports of these regimens are too few to make consistent recommendations. In addition to immunosuppressive agents, the antimalarial drug, fansidar, has been trialed in ALPS patients. Preliminary findings observed a clinical response, but subsequent follow up and study demonstrated fansidar ultimately failed to ameliorate lymphoproliferation and demonstrate efficacy [54, 55]. As such, fansidar should not be considered for treatment of ALPS.
There have been reports in patients with refractory cytopenias or other autoimmune pathology undergoing allogeneic bone marrow transplantation (BMT). The risks associated with an unrelated donor transplant appear too high to justify such a procedure except for rare patients who fail immune suppression. If patients with ALPS related autoimmunity are likely to require lifelong extensive immunosuppression with multiple agents a matched sibling transplant may be considered. In this rare scenario caution should be executed to ensure the sibling does not harbor the same mutation with incomplete or absent phenotypic penetrance.
7.4. Potential future therapies: Interleukin-17 (IL-17) Blockade
The potential use of IL-17 inhibition therapy has not yet been explored. Recent work by Boggio et al demonstrated that IL-17 is an important mediator of and can inhibit Fas-induced apoptosis in normal T cells [56]. They further demonstrated that IL-17 was abnormally elevated in the sera of ALPS patients. Their investigation showed that use of targeted therapy to block IL-17 could increase Fas induced apoptosis in T cells collected from ALPS patients and improved autoimmune disease observed in a murine model of ALPS [5]. The implications of this work are not yet known, but raise the possibility that IL-17 blockade may be an effective therapy for ALPS patients.
7.5. Other management considerations: Lymphoma Risk
Unfortunately, underlying ALPS related lymphadenopathy complicates diagnosis of lymphoma in ALPS patients. ALPS and lymphoma related lymphoproliferation appear the same on imaging, including positron emission tomography and therefore may not reliably be employed in malignancy evaluation [57]. Unlike many solid tumors, early diagnosis does not alter the outcome of lymphoma. As such it is not our practice to perform routine screening imaging. Instead, development of constitutional symptoms, changes in the degree or areas of lymphadenopathy or ascribed ALPS related pathology should trigger further evaluation for underlying malignancy. Importantly, this differs from other expert opinion that does recommend routine lymphoma surveillance by serial CT and PET imaging every 2–3 years [7]. ALPS is PET avid disease. Thus, it is expected patients with ALPS will have abnormal CT and PET scans and frequent imaging is likely to lead to unnecessary biopsies and stress for families, patients, and physicians as the results of CT and PET imaging are difficult to interpret.
8. Summary:
ALPS can have a variety of clinical manifestations complicating diagnosis, management, and treatment. The past approximate 2 decades have brought advances in the understanding of disease pathophysiology and molecular characterization resulting in a revised diagnostic criteria that has improved ALPS diagnosis and management. Current long-term management success in ALPS has been reported with both MMF and sirolimus. Recent findings from a multicenter prospective clinical trial of sirolimus in ALPS demonstrated a complete and durable response in a majority of children with refractory multi-lineage autoimmune cytopenias. In the future, it is likely the genomic era will lead to greater disease understanding that may both help characterize normal and abnormal lymphocyte biology as well as allow for rational selection of targeted ALPS therapies.
Key Points:
Autoimmune lymphoproliferative syndrome (ALPS) is a disorder of disrupted lymphocyte homeostasis that can have a variety of clinical manifestations including, lymphoproliferation and autoimmune pathology.
Molecular characterization of ALPS has resulted in a revised diagnostic criteria aiding in the improvement of ALPS diagnosis and treatment.
Sirolimus and mycophenolate mofetil have been studied for long-term management of patients with ALPS.
Acknowledgements:
This work is supported by grants from the National Blood Foundation (LAG) and National Hemophilia Foundation (LAG), Cures within Reach (DTT), the United States Immunodeficiency Network (USIDNET) through the National Institute of Allergy and Infectious Diseases (NIAID) grant number N01-A1-30070 (DTT), NIAID grants R56A1091791 and R21A1099301 (DTT), a Foerderer-Murray Award (DTT) the Goldman Philanthropic Partnerships and the Rockefeller Brothers Fund (DTT), the Partnership for Cures Patient Impact Initiative (DTT), and the Barbara Brodsky Foundation (DTT), as well as, generous donations by families cared for at the Children’s Hospital of Philadelphia (DTT).
Footnotes
Conflict of Interests:
No commercial funding was provided for writing this manuscript. Drs. George and Teachey have no conflicts of interest.
References:
- 1.Siegel RM, Chan FK, Chun HJ, Lenardo MJ. The multifaceted role of Fas signaling in immune cell homeostasis and autoimmunity. Nat Immunol. 2000;1:469–474. [DOI] [PubMed] [Google Scholar]
- 2.Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–946. [DOI] [PubMed] [Google Scholar]
- 3.Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fisher A, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–1349. [DOI] [PubMed] [Google Scholar]
- 4.Sneller MC, Straus SE, Jaffe ES, Fleisher TA, Stetler-Stevenson M, Strober W. A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. J Clin Invest. 1992;90:334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. [DOI] [PubMed] [Google Scholar]
- 6.Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, Rieux-Laucat F, Siegel RM, Su HC, Teachey DT. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116:e35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rensing-Ehl A, Völkl S, Speckmann C, Lorenz MR, Ritter J, Janda A, et al. Abnormally differentiated CD4+ or CD8+ T cells with phenotypic and genetic features of double negative T cells in human Fas deficiency. Blood. 2014;124:851–60. [DOI] [PubMed] [Google Scholar]
- 8.Grishkan IV1, Ntranos A, Calabresi PA, Gocke AR. Helper T cells down-regulate CD4 expression upon chronic stimulation giving rise to double-negative T cells. Cell Immunol. 2013;284:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao VK1, Oliveira JB. How I treat autoimmune lymphoproliferative syndrome. Blood. 2011;118:5741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teachey DT. New advances in the diagnosis and treatment of autoimmune lymphoproliferative syndrome. Curr Opin Pediatr. 2012;24:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teachey DT, Seif AE, Grupp SA. Advances in the management and understanding of autoimmune lymphoproliferative syndrome (ALPS). Br J Haematol. 2010;148:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuehn HS, Caminha I, Niemela JE, Rao VK, Davis J, Fleisher TA, Oliveira JB. FAS haploinsufficiency is a common disease mechanism in the human autoimmune lymphoproliferative syndrome. J Immunol. 2011;186:6035–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magerus-Chatinet A, Stolzenberg MC, Loffredo MS, Neven B, Schaffner C, Ducrot N, et al. FAS-L, IL-10, and double-negative CD4− CD8− TCR alpha/beta+ T cells are reliable markers of autoimmune lymphoproliferative syndrome (ALPS) associated with FAS loss of function. Blood. 2009;113:3027–3030. [DOI] [PubMed] [Google Scholar]
- 14.Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F, Prieur AM, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. 2004;351:1409–1418. [DOI] [PubMed] [Google Scholar]
- 15.Magerus-Chatinet A, Neven B, Stolzenberg MC, Daussy C, Arkwright PD, Lanzarotti N, et al. Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation. J Clin Invest. 121:106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le Deist F, Emile JF, Rieux-Laucat F, Benkerrou M, Roberts I, Brousse N, et al. Clinical, immunological, and pathological consequences of Fas-deficient conditions. Lancet. 1996;348:719–723. [DOI] [PubMed] [Google Scholar]
- 17.Sneller MC, Wang J, Dale JK, Strober W, Middelton LA, Choi Y, et al. Clincial, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997; 89:1341–1348. [PubMed] [Google Scholar]
- 18.Price S1, Shaw PA, Seitz A, Joshi G, Davis J, Niemela JE, Perkins K, Hornung RL, Folio L, Rosenberg PS, Puck JM, Hsu AP, Lo B, Pittaluga S, Jaffe ES, Fleisher TA, Rao VK, Lenardo MJ. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. 2014;123:1989–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poppema S, Maggio E, van den Berg A. Development of lymphoma in Autoimmune Lymphoproliferative Syndrome (ALPS) and its relationship to Fas gene mutations. Leuk Lymphoma. 2004;45:423–431. [DOI] [PubMed] [Google Scholar]
- 20.Bleesing JJ, Brown MR, Novicio C, Guarraia D, Dale JK, Stauss SE et al. A composite picture of TcR alpha/beta(+) CD4(−)CD8(−) T cells (alpha/beta-DNTCs) in humans with auto- immune lymphoproliferative syndrome. Clin Immunol. 2002;104:21–30. [DOI] [PubMed] [Google Scholar]
- 21.Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, Lynch D, et al. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 2000;288:2354–2357. [DOI] [PubMed] [Google Scholar]
- 22.Martin DA, Zheng L, Siegel RM, Huang B, Fisher GH, Wang J, et al. Defective CD95/APO-1/Fas signal complex formation in the human autoimmune lymphoproliferative syndrome, type Ia. Proc Natl Acad Sci. 1999; 96(8):4552–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caminha I, Fleisher TA, Hornung RL, Dale JK, Niemela JE, Price S, et al. Using biomarkers to predict the presence of FAS mutations in patients with features of the autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. 2010;125:946–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gámez-Díaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol. 2016;137:223–30. [DOI] [PubMed] [Google Scholar]
- 25.Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, et al. Spectrum of Phenotypes Associated with Mutations in LRBA. J Clin Immunol. 2016;36:33–45. [DOI] [PubMed] [Google Scholar]
- 26.Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. 2015;125(4):591–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seif AE, Manno CS, Sheen C, Grupp SA, Teachey DT. Identifying autoimmune lymphoproliferative syndrome in children with Evans syndrome: a multi-institutional study. Blood. 2010. Mar 18;115:2142–5. [DOI] [PubMed] [Google Scholar]
- 28.Teachey DT, Manno CS, Axsom KM, Andrews T, Choi JK, Greenbaum BH, McMann JM, Sullivan KE, Travis SF, Grupp SA. Unmasking Evans syndrome: T-cell phenotype and apoptotic response reveal autoimmune lymphoproliferative syndrome (ALPS). Blood. 2005;105:2443–8. [DOI] [PubMed] [Google Scholar]
- 29.Aladjidi N, Leverger G, Leblanc T, Picat MQ, Michel G, Bertrand Y et al. New insights into childhood autoimmune hemolytic anemia: a French national observational study of 265 children. Haematologica. 2011; 96:655–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savasan S, Warrier I, Buck S, Kaplan J, Ravindranath Y. Increased lymphocyte Fas expression and high incidence of common variable immunodeficiency disorder in childhood Evans’ syndrome. Clin Immunol. 2007;125:224–229. [DOI] [PubMed] [Google Scholar]
- 31.Maric I, Pittaluga S, Dale JK, Niemela JE, Delsol G, Diment J, et al. Histologic features of sinus histiocytosis with massive lymphadenopathy in patients with autoimmune lymphoproliferative syndrome. Am J Surg Pathol. 2005;29:903–11. [DOI] [PubMed] [Google Scholar]
- 32.Chun HJ, Zheng L, Ahmad M, Wang J, Speirs CK, Siegel RM, et al. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 2002;419:395–399. [DOI] [PubMed] [Google Scholar]
- 33.Niemela J, Kuehn HS, Kelly C, Zhang M, Davies J, Melendez J, et al. Caspase-8 Deficiency Presenting as Late-Onset Multi-Organ Lymphocytic Infiltration with Granulomas in two Adult Siblings. J Clin Immunol. 2015. May;35(4):348–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niemela JE, Lu L, Fleisher TA, Davis J, Caminha I, Natter M, et al. Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood. 2010;117:2883–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oliveira JB, Bidere N, Niemela JE, Zheng L, Sakai K, Nix CP, et al. NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci U S A. 2007; 104:8953–8958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takagi M, Shinoda K, Piao J, Mitsuiki N, Matsuda K, Muramatsu H, et al. Autoimmune lymphoproliferative syndrome-like disease with somatic KRAS mutation. Blood. 2010;117:2887–2890. [DOI] [PubMed] [Google Scholar]
- 37.Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol. 2014;15:88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science 2013. 342 (6160): 866–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, Cavazzana M, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest. 2014;124:3923–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, Butrick M, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med. 2014;211:2537–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. [DOI] [PubMed] [Google Scholar]
- 42.Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014. Dec;20(12):1410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pedicord VA, Cross JR, Montalvo-Ortiz W, Miller ML, Allison JP. Friends not foes: CTLA-4 blockade and mTOR inhibition cooperate during CD8+ T cell priming to promote memory formation and metabolic readiness. J Immunol. 2015;194:2089–98. [DOI] [PubMed] [Google Scholar]
- 44.Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Autoimmune Disease. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015;349:436–440. [DOI] [PubMed] [Google Scholar]
- 45.Rao VK, Dugan F, Dale JK, Davis J, Tretler J, Hurley JK, et al. Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome. Br J Haematol. 2005;129:534–538. [DOI] [PubMed] [Google Scholar]
- 46.Miano M, Scalzone M, Perri K, Palmisani E, Caviglia I, Micalizzi C, et al. Mycophenolate mofetil and Sirolimus as second or further line treatment in children with chronic refractory Primitive or Secondary Autoimmune Cytopenias: a single centre experience. Br J Haematol. 2015;171:247–253. [DOI] [PubMed] [Google Scholar]
- 47.Brown VI, Fang J, Alcorn K, Barr R, Kim JM, Wasserman R et al. Rapamycin is active against B-precursor leukemia in vitro and in vivo, an effect that is modulated by IL-7- mediated signaling. Proc Natl Acad Sci U S A. 2003;100:15113–15118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strauss G, Osen W, Debatin KM. Induction of apoptosis and modulation of activation and effector function in T cells by immunosuppressive drugs. Clin Exp Immunol. 2002;128:255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teachey DT1, Obzut DA, Axsom K, Choi JK, Goldsmith KC, Hall J, Hulitt J, Manno CS, Maris JM, Rhodin N, Sullivan KE, Brown VI, Grupp SA. Rapamycin improves lymphoproliferative disease in murine autoimmune lymphoproliferative syndrome (ALPS). Blood. 2006;108:1965–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teachey DT, Greiner R, Seif A, et al. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol. 2009;145:101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bride KL, Vincent T, Smith-Whitley K, Lambert MP, Bleesing JJ, Seif AE, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood 2016;127:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rao A, Kelly M, Musselman M, Ramadas J, Wilson D, Grossman W. et al. Safety, efficacy, and immune reconstitution after rituximab therapy in pediatric patients with chronic or refractory hematologic autoimmune cytopenias. Pediatr Blood Cancer. 2008;50:822–825. [DOI] [PubMed] [Google Scholar]
- 53.Neven B, Magerus-Chatinet A, Florkin B, Gobert D, Lambotte O, De Somer L, et al. A survey of 90 patients with auto- immune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood 2011;118:4798–807. [DOI] [PubMed] [Google Scholar]
- 54.van der Werff Ten Bosch J, Schotte P, Ferster A, Azzi N, Boehler T, Laurey G, et al. Reversion of autoimmune lymphoproliferative syndrome with an antimalarial drug: preliminary results of a clinical cohort study and molecular observations. Br J Haematol. 2002;117:176–88. [DOI] [PubMed] [Google Scholar]
- 55.Rao VK, Dowdell KC, Dale JK, Dugan F, Pesnicak L, Bi LL, et al. Pyrimethamine treatment does not ameliorate lymphoproliferation or autoimmune disease in MRL/lpr−/− mice or in patients with autoimmune lymphoproliferative syndrome. American J Hematol. 2007;82:1049–55. [DOI] [PubMed] [Google Scholar]
- 56.Boggio E, Clemente N, Mondino A, Cappellano G, Oriellieri E, Gigliotti CL, et al. IL-17 protects T cells from apoptosis and contributes to development of ALPS-like phenotypes. Blood. 2014;123:1178–1186. [DOI] [PubMed] [Google Scholar]
- 57.Rao VK, Carrasquillo JA, Dale JK, Bacharach SL, Whatley M, Dugan F, et al. Fluorodeoxyglucose positron emission tomography (FDG-PET) for monitoring lymphadenopathy in the autoimmune lymphoproliferative syndrome (ALPS). Am J Hematol. 2006;81:81–85. [DOI] [PubMed] [Google Scholar]