

Abstract
To study the role of surface-associated proteins in the virulence of Streptococcus pneumoniae, we used two serotype 3 strains, ATCC 6303 and WU2, and two PspA-negative mutants of WU2, an encapsulated one, JY1123 (Caps+/PspA−), and an unencapsulated one, DW3.8 (Caps−/PspA−). ATCC 6303 and WU2 were highly virulent in mice, while the virulence of JY1123 was slightly decreased (50% lethal doses [LD50s], 24, 6, and 147 CFU/mouse, respectively); DW3.8 was avirulent (LD50, 2 × 108 CFU). In vitro, ATCC 6303, WU2, and JY1123 (Caps+/PspA−) strongly resisted complement activation and complement-dependent opsonophagocytosis, whereas DW3.8 (Caps−/PspA−) was easily phagocytized in fresh serum. Trypsin treatment of ATCC 6303, WU2, and JY1123 (Caps+/PspA−) resulted in enhanced complement activation and complement-dependent opsonophagocytosis. Trypsin had no deleterious effect on the polysaccharide capsule. In addition, trypsin pretreatment of ATCC 6303 strongly reduced virulence upon intraperitoneal challenge in mice. This indicated that surface proteins play a role in the resistance to complement activation and opsonophagocytosis and contribute to the virulence of type 3 pneumococci. In subsequent experiments, we could show that the modulation of complement activation was associated with surface components that bind complement regulator factor H; binding is trypsin sensitive and independent of prior complement activation. Immunoblotting of cell wall proteins of the virulent strain ATCC 6303 with anti-human factor H antibody revealed three factor H-binding proteins of 88, 150, and 196 kDa. Immunogold electron microscopy showed a close association of factor H-binding components with the outer surface of the cell wall. The role of these factor H-binding surface proteins in the virulence of pneumococci is interesting and warrants further investigation.
Despite the availability of effective antibiotics and a multivalent capsular polysaccharide vaccine, Streptococcus pneumoniae remains an important pathogen to humans of all ages (4). This microorganism is the predominant causative agent of otitis media, community-acquired pneumonia, septicemia, and meningitis. Infections due to pneumococci are most frequently seen in infants, elderly persons, and immunocompromised patients (13, 25, 33).
It has been established that the polysaccharide capsule of the pneumococcus is an important virulence factor (1, 44). Watson and Musher (43) reported the production of pneumococcal mutant strains, lacking detectable polysaccharide capsule, from an encapsulated serotype 3 strain by Tn916 transposon insertion; the unencapsulated mutants had greatly reduced virulence in mice compared to the parental strain. However, it is clearly evident that among the approximately 90 known capsular serotypes, the degree of virulence varies widely and only a minority of the pneumococcal serotypes cause infections in humans (15, 42). Additionally, there is considerable difference in virulence among pneumococcal strains with identical capsular serotypes.
Several studies have demonstrated the existence of noncapsular virulence factors in pneumococci (6, 18, 31, 34). Briles and McDaniel established that the expression of a surface protein, pneumococcal surface protein A (PspA), is associated with the virulence of pneumococci in mice (8, 9, 26–28). In addition, strong evidence for an independent role of pneumolysin in the virulence of pneumococci has been presented (5, 24, 35). Thus, to date PspA and pneumolysin are the only well-characterized noncapsular virulence factors of pneumococci. It is also evident, however, that the spectrum of noncapsular virulence factors is still unknown, and their quantitative contribution to virulence is therefore poorly defined. Opsonophagocytosis is thought to play an important role in host defense against pneumococci (11, 12, 20, 40, 46). This process is initiated by complement activation via either antibody-dependent or antibody-independent pathways (11). Pneumococcal strains differ in their ability to activate the complement cascade (14, 39). The determinants for these differences, however, remain unclear, although the type of pathway and the extent of interaction of complement with the various pneumococcal capsular polysaccharides may, in part, explain these differences.
Hostetter previously showed that although both cell wall and capsular polysaccharide of type 3 pneumococci activate complement, leading to C3b deposition on both cell wall and capsule, type 3 pneumococci strongly resist phagocytosis (21). Angel et al. subsequently demonstrated that type 3 pneumococci express C3-degrading activity associated with the cell wall (3). The underlying mechanism, however, was not further explored. The purpose of the present paper is to define the role of surface-associated proteins of type 3 pneumococci in resistance to complement activation and opsonophagocytosis and to identify the mechanisms involved in this resistance.
MATERIALS AND METHODS
Animals.
Male outbred Swiss mice were used for 50% lethal dose (LD50) determinations. They were obtained from Harlan CPB (Zeist, The Netherlands), maintained in the animal facilities of Utrecht University, and used at 8 to 14 weeks of age.
LD50 determination.
Groups of five mice were injected intraperitoneally (i.p.) with 0.5 ml of a 10-fold dilution series of bacterial suspensions (1 to 109 CFU/ml/strain) in saline. Deaths were recorded over an 8-day period. LD50 values were calculated by the method of Reed and Muench (32).
Buffers.
Phosphate (20 mM)-buffered saline (PBS) (pH 7.4) was used for washing bacteria. Veronal (5 mM)-buffered saline (pH 7.4) containing 0.15 mM Ca2+ and 0.5 mM Mg2+ (VSB2+) and veronal (5 mM)-buffered saline containing 10 mM EDTA (EDTA-VB) or 8 mM EGTA and 2.5 mM magnesium (EGTA-VB) were used as incubation buffers in complement assays. All buffers were prepared from a 5× stock solution (41).
For the trypsin treatment of bacteria, VSB2+ was used. Hanks balanced salt solution containing 0.1% gelatin (GHBSS) was used for the dilution of serum and bacteria in the phagocytosis assay. Alsever’s old solution (114 mM citrate, 27 mM glucose, 72 mM sodium chloride [pH 6.1]) served to store chicken blood.
Bacterial strains.
Streptococcus pneumoniae serotype 3 (ATCC 6303) was obtained from the American Type Culture Collection (Rockville, Md.). Wild-type 3 strain WU2 and its encapsulated PspA-negative mutant JY1123 was provided by L. S. McDaniel (Birmingham, Ala.). Strain DW3.8 was generated by conjugative transfer of transposon Tn916 from donor strain Enterococcus faecalis CG110 to the genome of WU2 (43). The pneumococci were grown to mid-logarithmic phase at 37°C in Todd-Hewitt broth (Difco, Detroit, Mich.) supplemented with 0.5% yeast extract in a 5% CO2 atmosphere. After incubation, the bacteria were washed three times with PBS.
Pneumococci were heat killed by being incubated for 60 min at 56°C. Subsequently, the bacteria were washed three times in PBS and stored until use.
The strains are designated according to their phenotypic characteristics with respect to the presence or absence of capsule and PspA; i.e., ATCC 6303 and WU2 are designated (Caps+/PspA+), DW3.8 is designated (Caps−/PspA−), and JY1123 is designated (Caps+/PspA−).
Enzyme treatment of pneumococci.
A total of 109 CFU of heat-killed pelleted bacteria was suspended in 1 ml of VSB2+ containing 1 mg of trypsin (Boehringer GmbH, Mannheim, Germany) purified by cristallyzation. The bacterial suspensions were incubated at 37°C for 1 h. After being washed, the bacteria were resuspended in VSB2+ to 109 CFU per ml.
Choline chloride treatment of pneumococci.
A total of 109 pelleted pneumococci were resuspended for 30 min at room temperature on a rotator in 1 ml of PBS containing 2% choline chloride (Riedel-de Haen, Seelze, Germany). After treatment, the bacteria were washed three times and subsequently resuspended to the original concentration in test buffer (either Veronal buffer [VSB2+] or GHBSS) (109/ml).
Normal human serum.
Human serum was collected from 30 healthy adults and pooled. The pool is hereafter referred to as normal human serum (NHS). NHS was stored in 1-ml aliquots at −70°C until use.
Erythrocytes.
Chicken blood, diluted 1:1 in Alsever’s old solution, was obtained from bioTrading (Wilnis, the Netherlands) and used as the source of chicken erythrocytes. Before use, the cells were washed three times with saline (0.85%).
PMN.
Polymorphonuclear leukocytes (PMN) were isolated from heparinized human blood. A volume of blood was mixed 1:1 with PBS and layered on a Ficoll-Histopaque gradient consisting of 10 ml of Ficoll-Paque (Pharmacia, Uppsala, Sweden) on top of 12 ml of Histopaque (density, 1.119; Sigma Diagnostics, St. Louis, Mo.). After centrifugation at 400 × g and room temperature for 20 min, PMN were collected from the upper part of the Histopaque and ice-cold RPMI was added. The cells were centrifuged at 250 × g, and the residual erythrocytes were lysed by resuspending the cells in 9 ml of ice-cold distilled water. After 30 s, the cell suspension was rendered isotonic with 10× PBS. Next, RPMI was added. The PMN fraction was centrifuged and resuspended in RPMI at 107/ml. The cell concentration consisted of >95% PMN. Viability always exceeded 98% as determined by propidium iodide exclusion.
ELISA for capsular polysaccharide content.
A sandwich enzyme-linked immunosorbent assay (ELISA), performed in polyvinyl chloride microtiter plates (Flow Laboratories, McLean, Va.) was used (36). Polyclonal goat anti-serotype 3 polysaccharide immunoglobulin G (IgG) (100 μl from a 1-μg/ml solution) was used as the capturing antibody (the coating conditions were 37°C for 1 h and then 4°C overnight). PBS containing 0.1% Tween 20 was used as washing buffer after each coating or incubation step. Bovine skim milk powder (100 μl; 4% in PBS–0.1% Tween 20) was added (37°C for 1 h) to prevent nonspecific binding. A 50-μl volume of either a suspension of pneumococci (5 × 106 CFU/ml in PBS) or purified type 3 capsular polysaccharide (a gift of A. F. M. Verheul, Eijkman-Winkler Institute, Utrecht, The Netherlands) at a starting concentration of 1.25 μg/ml, both serially diluted in saline, was added to the microtiter plates before incubation at 37°C for 1 h. Mouse polyclonal IgG against serotype 3 type strain ATCC 6303 polysaccharide (50 μl; 1:500 in PBS–0.1% Tween 20) was used for the detection of serotype 3 capsular polysaccharide (incubation at 37°C for 1 h). The plates were developed by adding 50 μl of peroxidase-labeled goat anti-mouse IgG (heavy plus light chains; H+L) (Nordic, Tilburg, The Netherlands), diluted 1:6,000 in PBS–0.1% Tween 20, at 37°C for 1 h followed by extensive washing and then by the addition to each well of a mixture of tetramethylbenzidine (Sigma) and hydrogen peroxide (incubation at 37°C for 10 min). The substrate consisted of 0.1 M sodium acetate–citric acid buffer (pH 5.5) containing 200 μl of a stock solution of tetramethylbenzidine (6.0 mg/ml of dimethyl sulfoxide) and 2 μl of a 30% hydrogen peroxide solution per 12 ml of buffer. Substrate conversion was stopped by the addition of 50 μl of 1 M sulfuric acid. Absorbance values at 450 nm were determined by using a ELISA reader (no. 3550; Bio-Rad, San Francisco, Calif.). The calibration curve obtained by plotting optical density values against the concentration of purified type 3 capsular polysaccharide (in micrograms per milliliter) was used to calculate the amount of capsule per pneumococcal cell (in femtograms/bacterial cell) from the optical density values obtained with the bacterial suspension.
Complement activation microassay.
The complement activation assay was performed as described by Geelen et al. (17). Briefly, in the wells of U-shaped microtiter plates, 20 μl of pneumococcal suspension in VSB2+ (6 × 106 pneumococci) was mixed with 80 μl of 10% NHS in VSB2+ and incubated at 37°C for 10 min. During this step, the bacteria activate the complement cascade, resulting in C5 convertase formation. Subsequently, 50 μl of chicken erythrocyte suspension (2 × 108 cells/ml) in EDTA-VB was added, and the mixtures were incubated at 37°C for 60 min to allow C5b6-mediated lysis of the erythrocytes (bystander hemolysis). The unlysed cells and cell ghosts were centrifuged in a microtiter plate centrifuge (2,000 × g for 5 min), and 50-μl volumes of the supernatants were transferred to the wells of flat-bottom microtiter plates containing 200 μl of water. The absorbance at 405 nm, estimated by using the ELISA reader, was taken as the measure of hemoglobin release. The percentage of lysis was determined by the formula
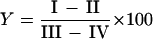 |
in which I refers to the absorbance of the samples in NHS, II refers to the absorbance of the samples in heat-inactivated (56°C for 30 min) serum, and III and IV refer to the absorbances of controls for 100% lysis (water) and 0% lysis (buffer), respectively. All tests were performed in duplicate. Complement activity was expressed as a Z value representing the number of active hemolytic sites/erythrocyte. Z values were derived from Y by the formula Z= −ln (1 − Y).
Phagocytosis assay.
The phagocytosis assay was adapted from those of Alonso de Velasco et al. (2) and Sanders et al. (37). In short, dilutions of fresh human serum were made in bovine serum albumin (BSA)-HBSS and pipetted into round-bottom microtiter plates. Samples of 5 × 106 fluorescein isothiocyanate (FITC)-labeled pneumococci were added to each well. Opsonization, in a total volume of 50 μl, was performed at 37°C on a microtiter plate agitator. After 10 min of opsonization, the plates were placed on ice and 2.5 × 105 PMN in 50 μl of BSA-HBSS were added to each well. Phagocytosis was performed for 12 min at 37°C with shaking. After a wash with ice-cold BSA-HBSS, the cells were transferred to fluorescence-activated cell sorter tubes, fixed with paraformaldehyde (2%) in PBS, and analyzed in a flow cytometer (FACScan; Becton Dickinson, Mountain View, Calif.). The percentage of FITC-positive PMN was used as a measure of the phagocytic activity of serum by determining the serum dilution resulting in 50% FITC-positive PMN (50% phagocytosis titer). The results are expressed as the opsonic index, which is the inverse of the 50% phagocytosis titer. For example, if 50% FITC-positive PMN are found at a serum dilution of 1:100, i.e., 1% serum, the opsonic index is 100.
ELISA for soluble TCC.
Essentially, the technique described by Mollnes et al. (29) was used for ELISA for soluble terminal complement complexes (TCC). Briefly, 20-μl volumes of pneumococcal suspensions in VSB2+ were mixed with 80-μl volumes of 10% NHS in VSB2+ in the wells of U-shaped microtiter plates and incubated at 37°C for 10 min. During the incubation, the bacteria activated the complement cascade and gave rise to the generation of soluble TCC [(S)C5b-9]. Complement activation was stopped by the addition of 50 μl of EDTA-VB, and the mixture was centrifuged (1,500 × g) at 4°C to remove the bacteria. The supernatant was tested for TCC by a sandwich ELISA with a monoclonal antibody (aE11) recognizing a C9 neo-epitope in TCC as the capturing antibody and rabbit anti-human C5 IgG (Dakopatts A/S, Glostrup, Denmark) as the detecting agent (30). As a positive control for TCC generation, 400 mg of zymosan was added to 40 ml of pooled human serum and incubated for 1 h at 37°C. Next, the suspension was centrifuged at 43,000 × g and the supernatant was stored at −70°C until use.
Fluorescence-activated cell sorter assay for PspA expression.
Eppendorf vials (1.5 ml) were coated with BSA (Boseral; Organon Teknika, Boxtel, The Netherlands) by incubation with a 1% solution for 1 h at 37°C. The vials were washed three times with PBS, and 200 μl of pneumococcal suspension (5 × 108 CFU/ml) was added. The vials were centrifuged at 15,000 × g for 7 min. the pelleted bacteria were mixed with 200 μl of PspA-specific mouse monoclonal antibody XiR 278 (kindly provided by L. S. McDaniel) diluted 1:2,000 in PBS. The mixture was rinsed and incubated at 37°C for 60 min. The bacteria were centrifuged, washed twice, suspended in 40 μl of FITC-labeled goat-anti-mouse IgG plus IgM (1:25; no. M35201, Caltag Laboratories, San Francisco, Calif.), and incubated at 4°C for 30 min. The bacteria were washed three times, pelleted, and resuspended in 300 μl of PBS. The bacterial suspension was subsequently examined by flow cytometry with a FACScan cytometer (Becton Dickinson; filter 530/30). The results were expressed as mean fluorescence values.
FACS assay for complement factor H binding.
Eppendorf vials (1.5 ml) were coated with 1% BSA (Sigma) by incubation for 1 h at 37°C. The vials were washed three times with PBS, and 200 μl of a pneumococcal suspension (5 × 108 CFU/ml) and 800 μl of 10% NHS or purified factor H (45) (kindly provided by J. van Strijp, Eijkman-Winkler Institute, Utrecht, The Netherlands) were added (45). The mixtures were incubated at 37°C for 10 min. Complement activation in NHS was stopped by the addition of 500 μl of ice-cold EDTA-VB, and the vials were centrifuged at 13,200 × g for 7 min. The pelleted bacteria were washed twice, and 200 μl of polyclonal rabbit anti-human factor H antiserum (CLB KH40P [Central Laboratory of the Blood Transfusion Service, Amsterdam, The Netherlands], diluted 1:500 in PBS) was added. The vials were then incubated at 37°C for 1 h and centrifuged. The pellet was washed twice with PBS, resuspended in 40 μl of FITC-labeled goat anti-rabbit IgG (1:40; Sigma F-1262), and incubated at 4°C for 30 min. The bacteria were washed three times, pelleted, and resuspended in 300 μl of PBS. The labeled bacteria were examined by flow cytometry with the FACScan cytometer (filter 530/30; Beckton Dickinson), and the results were expressed as mean fluorescence values.
Preparation of CWE from pneumococci.
A modification of the method described by Lacks and Neuberger (22) for autoplast formation was used to produce a cell wall extract (CWE) of S. pneumoniae. Bacteria were cultured overnight, washed in PBS, pelleted, and resuspended in 3 ml of PBS containing 30% sucrose and 0.05 M MgCl2. The suspension was incubated for 24 h at room temperature and subsequently centrifuged for 4,000 × g for 10 min at 4°C to remove protoplasts. The supernatant was dialyzed against three changes of PBS at 4°C and centrifuged for 10 min at 10,000 × g to remove insoluble material. This preparation was designated the CWE. The amount of protein in the CWE was determined by a Bradford assay. The CWE was stored in aliquots at −20°C until use. The whole isolation procedure was performed in the presence of the protease inhibitor phenylmethylsulfonyl fluoride (2 mM).
SDS-PAGE and immunoblotting of factor H-binding proteins of pneumococci.
Proteins from pneumococcal CWE were separated under nonreducing conditions by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (12.5% polyacrylamide) and blotted onto a nitrocellulose membrane. The membrane was blocked by incubation with 3% gelatin for 1 h at 37°C and subsequently washed three times with PBS–0.1% Tween 20. Next, the membrane was incubated with 1% NHS for 1 h at 37°C. After three washes, the membrane was incubated for 1 h at 37°C with 1:500 anti-factor H antibody (polyclonal rabbit anti-human factor H, CLB KH40P; Central Laboratory of the Blood Transfusion Service). Subsequently, the membrane was washed and incubated with 1:5,000-diluted goat anti-rabbit IgG (H + L) labeled with peroxidase. After being washed, the membrane was stained with 40 ml of a solution containing 20 mg of diaminobenzidine (Sigma) and 100 μl of H2O2 in PBS. The reaction was stopped by adding a large amount of tap water.
Ultracryotomy and immunoelectron microscopy of factor H binding by pneumococci.
Heat-inactivated pneumococci were incubated with NHS (10 min at 37°C) and washed extensively. Next, pneumococci were resuspended in 2% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). After 2 h, the fixative was removed and the bacteria were washed three times with PBS–0.15 M glycine and finally pelleted in 10% gelatin in phosphate buffer. The gelatin was allowed to solidify, and small cubes were cut at 4°C and infused with 2.3 M sucrose for at least 2 h at 4°C. The blocks were mounted on a copper specimen holder and frozen in liquid nitrogen.
Ultrathin cryosections were prepared at −120°C on an Ultracut S instrument (Leica, Vienna, Austria) with a diamond knife (Drukker International, Cuyk, The Netherlands) by the method of Liou et al. (23). Immunogold labelling was performed by the method of Slot et al. (38) by sequential incubation of pneumococci with a polyclonal rabbit anti-human factor H serum (1:500 dilution in PBS) and protein A coupled to 10-nm-diameter gold particles (Department of Cell Biology, University Medical Centre, Utrecht, The Netherlands) in PBS–1% BSA. The specificity of the procedure was checked by performing immunogold labelling with heat-inactivated pneumococci incubated without NHS or by using a nonsense polyclonal rabbit antibody. Both controls were negative.
Statistical analysis.
Data are expressed as the mean ± standard errors of the mean of n independent observations. Correlation coefficients were calculated by the least-squares method. The significance of differences between means of experiments was calculated by either Student’s t test or χ2 analysis. Differences with P < 0.05 were considered significant.
RESULTS
Virulence of pneumococcal strains in mice.
The virulence of the various pneumococcal strains was estimated by determining LD50s in mice upon i.p. injection. The results are shown in Table 1. Strains ATCC 6303 and WU2 (both Caps+/PspA+) were highly virulent, with LD50s of 24 and 6 CFU/mouse, respectively. DW3.8 (Caps−/PspA−) was nonvirulent, with an LD50 of 2 × 108 CFU/mouse. The (Caps+/PspA−) mutant of WU2, JY1123, showed intermediate virulence, with an LD50 of 147 CFU/mouse. Finally, by using a critical dose of 20 CFU of ATCC 6303 per mouse, approximately equal to the LD50 (24 CFU/mouse), we could show that untreated pneumococci caused a 70% mortality whereas trypsin-treated live pneumococci of this strain had a significantly diminished lethality for mice of only 20% (Fig. 1).
TABLE 1.
Virulence of S. pneumoniae type 3 (ATCC 6303 and WU2) and type 3 mutant strains (JY1123 and DW3.8) in micea
| Pneumococcal strain | LD50 (CFU/mouse) |
|---|---|
| ATCC 6303 (Caps+/PspA+) | 24 |
| WU2 (Caps+/PspA+) | 6 |
| JY1123 (Caps+/PspA−) | 147 |
| DW3.8 (Caps−/PspA−) | 2.24 × 108 |
The virulence was determined by i.p. injection (see Materials and Methods).
FIG. 1.

Survival of mice after intraperitoneal challenge with S. pneumoniae ATCC 6303 (Caps+/PspA+) pretreated with trypsin or buffer as a control (see Materials and Methods). The curves show survival time (days) of mice after an i.p. injection of 20 CFU/mouse (15 mice per group). Trypsin-treated pneumococci were significantly different (P < 0.01) from pneumococci treated with buffer as estimated by Kaplan-Meier survival analysis.
Opsonophagocytosis of pneumococcal strains.
Phagocytosis of (Caps−/PspA−) mutant DW3.8 proceeded efficiently in fresh NHS (Fig. 2). Figure 2 also shows that the fully encapsulated virulent strains ATCC 6303 and WU2 had considerable resistance to opsonophagocytosis in fresh serum. The (Caps+/PspA−) mutant JY1123, however, was significantly better phagocytized than the parent strain, WU2 (Caps+/PspA+).
FIG. 2.

Opsonophagocytosis of fully encapsulated type 3 pneumococci and unencapsulated and PspA-deficient mutants in NHS. Opsonization, determined by flow cytometry, is expressed as opsonic index (see Materials and Methods for details). Bars represent means ± standard errors of three separate experiments. S3 (ATCC 6303) and WU2 are fully encapsulated parent strains (Caps+/PspA+), JY1123 is a PspA-deficient mutant of WU2 (Caps+/PspA−), and DW3.8 is an unencapsulated mutant of WU2 (Caps−/PspA−). JY1123 and DW3.8 significantly different from WU2 (∗∗∗, P < 0.002; ∗∗∗∗, P < 0.001 [Student’s t test]).
The results of these experiments confirm the resistance of virulent wild-type pneumococci to complement-mediated phagocytosis. Moreover, they indicate that PspA is not a major determinant of this resistance to opsonophagocytosis, since the PspA− mutant JY1123 was almost as poorly opsonized as its parent, WU2.
Complement activation by pneumococcal strains.
The ability of the different strains to activate complement in human serum is shown in Table 2. The virulent strains ATCC 6303 and WU2 activated the complement cascade poorly. In contrast, the avirulent (Caps−/PspA−) mutant DW3.8 and the intermediately virulent (Caps+/PspA−) mutant of WU2, JY1123, were powerful and intermediate complement activators, respectively.
TABLE 2.
Complement activation induced by S. pneumoniae type 3 (ATCC 6303 and WU2) and type 3 mutant strains (JY1123 and DW3.8)a
| Pneumococcal strain | Z (no. of active sites/erythrocyte)b |
|---|---|
| ATCC 6303 (Caps+/PspA+) | 0.11 ± 0.03 |
| WU2 (Caps+/PspA+) | 0.13 ± 0.01 |
| JY1123 (Caps+/PspA−) | 0.24 ± 0.02** |
| DW 3.8 (Caps−/PspA−) | 0.51 ± 0.03**** |
Complement activation was measured as lysis of bystander cells (chicken erythrocytes) and is expressed as the Z value, representing the number of active hemolytic sites per erythrocyte (mean ± standard errors of three separate experiments). Hemolysis was measured in the presence of 6 × 106 pneumococci in a reaction volume of 150 μl in a microtiter plate (see Materials and Methods).
JY1123 and DW3.8 were significantly different from WU2 (**, P < 0.01; ****, P < 0.001 [Student’s t test]).
The role of the classic and alternative pathways in complement activation by the different pneumococcal strains was investigated by using EGTA- and EDTA-containing buffers during the activation step. Complement activation by all strains was completely blocked in the presence of either Mg2+-EGTA or EDTA-serum (results not shown).
To investigate at which level the virulent strains interfered with complement activation, the generation of soluble TCCs in 8% serum was estimated after incubation with the strains. Figure 3 shows that the formation of TCC was high in serum incubated with (Caps−/PspA−) mutant DW3.8 while significantly fewer TCC were generated in serum after incubation with ATCC 6303, WU2 (both Caps+/PspA+), and JY1123 (Caps+/PspA−).
FIG. 3.

Formation of TCC by the type 3 pneumococcal strains S3 (ATCC 6303) and WU2 (both Caps+/PspA+), the unencapsulated mutant DW3.8 (Caps−/PspA−) of WU2, and the PspA-deficient mutant JY1123 (Caps+/PspA−) of WU2. TCC were determined by ELISA after incubation of pneumococci with NHS for 10 min at 37°C, and TCC formation was expressed as the ELISA titer (means ± standard errors of three independent experiments [horizontal top axis]). Control, no pneumococci added; Zymosan, positive control for TCC formation. DW3.8 was significantly different from ATCC 6303, WU2, and JY1123 (∗ P < 0.05 [Student’s t test]).
Trypsin treatment of pneumococcal strains.
To investigate whether resistance to complement activation and opsonophagocytosis was mediated by proteinaceous surface components, the various strains were treated with trypsin prior to complement and opsonophagocytosis experiments. Figure 4 shows that the pretreatment of ATCC 6303, WU2 (both Caps+/PspA+), or JY1123 (Caps+/PspA−) with trypsin considerably increased complement activation by these strains whereas trypsin treatment of DW3.8 (Caps−/PspA−) only moderately increased complement activation. Table 3 demonstrates that the capsule content of ATCC 6303, WU2, and JY1123 was not affected by trypsin pretreatment.
FIG. 4.

Effect of trypsin treatment of pneumococci on complement activation by type 3 strains ATCC 6303 and WU2 (both Caps+/PspA+) and unencapsulated and PspA-deficient mutants of WU2. Complement activation, determined by the hemolytic assay, is expressed as a Z value, representing the number of active hemolytic sites per erythrocyte (y axis). Bars represent mean is ± standard errors of three independent experiments. S3 (ATCC 6303) and WU2 are fully encapsulated parent strains, JY1123 is a PspA-deficient mutant of WU2 (Caps+/PspA−), and DW3.8 is an unencapsulated mutant of WU2 (Caps−/PspA−). Trypsin-treated bacteria (open bars) were significantly different from untreated bacteria (hatched bars), and both buffer-treated JY1123 and DW3.8 were significantly different from buffer-treated WU2 (∗∗, P < 0.01: ∗∗∗∗, P < 0.001 [Student’s t test]).
TABLE 3.
Capsular polysaccharide content of S. pneumoniae type 3 (ATCC 6303 and WU2) and type 3 mutant strains (JY1123 and DW3.8) before and after trypsin treatment
| Pneumococcal strain | PS3 content (fg/bacterium) after pretreatmenta:
|
|
|---|---|---|
| None (VSB++) | Trypsin | |
| ATCC 6303 (Caps+/PspA+) | 17.2 ± 3.2 | 34.7 ± 6.2 |
| WU2 (Caps+/PspA+) | 124 ± 14.5 | 213 ± 27.6 |
| JY1123 (Caps+/PspA−) | 149 ± 29.4 | 156 ± 27.6 |
| DW 3.8 (Caps−/PspA−) | <0.004b | <0.004b |
The type 3 capsular polysaccharide content (PS3) was measured by ELISA and is expressed as femtograms per bacterial cell (see Materials and methods).
Below detection limit (means ± standard errors of three separate experiments).
The effect of trypsin pretreatment on opsonophagocytosis of the virulent strain ATCC 6303 is shown in Fig. 5. Trypsin treatment strongly enhanced the phagocytosis of ATCC 6303 by human PMN after opsonization in human serum. Similar results were obtained after trypsin treatment of strains WU2 and JY1123, while trypsin treatment did not affect the opsonophagocytosis of DW3.8 (data not shown).
FIG. 5.

Effect of trypsin treatment on opsonophagocytosis of type 3 strain S3 (ATCC 6303) in NHS. Opsonization, determined by flow cytometry, is expressed as the opsonic index (see Materials and Methods for details). Bars represent means ± standard errors of three separate experiments. S3/VSB++, S3 (ATCC 6303) pretreated with buffer; S3/trypsin, S3 (ATCC 6303) pretreated with trypsin. Trypsin-treated bacterial were significantly different from untreated bacteria (∗∗, P < 0.01 [Student’s t test]).
Factor H binding by pneumococcal strains.
The above experiments indicated that virulent pneumococcal strains ATCC 6303, WU2, and the (Caps+/PspA−) mutant of WU2, JY1123, interfered with complement activation at the level of the generation of either the C3 or C5 convertase. For this reason, we investigated whether the pneumococcal strains bound one of the known complement-regulatory proteins. The experiments showed that the encapsulated pneumococcal strains, upon incubation in human serum, did not react with polyclonal antibodies against human decay-accelerating factor or complement receptor 1 or with antibodies against the soluble inhibitors C1 inhibitor and C4-binding protein (results not shown). However, significant reactivity with polyclonal anti-factor H serum was observed after incubation of strain ATCC 6303, WU2, or JY1123 with NHS or purified factor H under conditions similar to those in the bystander hemolysis assay (17) (Fig. 6). Figure 6 demonstrates that considerably less factor H bound to the surface of DW3.8 (Caps−/PspA−). This was the case not only under conditions permitting complement activation but also in the presence of EDTA. Upon pretreatment of bacteria with trypsin, factor H binding by WU2, ATCC 6303, and JY1123 was strongly decreased (Fig. 6). It should be noted, however, that trypsin treatment also removed PspA from strains ATCC 6303 and WU2. Figure 7 also confirms the virtual absence of PspA from JY1123, which vigorously bound factor H (Fig. 6). Moreover, selective removal of PspA from WU2 and ATCC 6303 by pretreatment of the bacteria with choline chloride did not influence factor H binding (data not shown).
FIG. 6.

Effect of trypsin treatment on the binding of complement-regulatory protein factor H by type 3 pneumococcal strains S3 (ATCC 6303) and WU2 (both Caps+/PspA+), the unencapsulated mutant DW3.8 (Caps−/PspA−) of WU2 and the PspA-deficient mutant JY1123 (Caps+/PspA−) of WU2. Factor H binding was determined by flow cytometry and is expressed as fluorescence units (means ± standard errors of three independent experiments) on the y axis. DW3.8 was significantly different from S3 (ATCC 6303), WU2, and JY1123 (∗∗, P < 0.01 [Student’s t test]). Trypsin-treated bacteria were significantly different from buffer-treated bacteria (∗, P < 0.05; ∗∗∗∗, P < 0.001 [Student’s t test]).
FIG. 7.

Effect of trypsin treatment on the PspA content of pneumococci. The PspA content of pneumococcal strains ATCC 6303 (S3) (Caps+/PspA+), WU2 (Caps+/PspA+), JY1123 (Caps+/PspA+), and DW3.8 (Caps+/PspA+) was determined by FACS analysis with monoclonal antibody XiR 278 (see Materials and Methods). Results are expressed as arbitrary fluorescence units. Bars denote means ± standard errors of three independent experiments. Trypsin-treated S3 and WU2 were significantly different from bacteria treated with buffer, and JY1123 and DW3.8 were significantly different from buffer-treated WU2 (∗∗∗∗, P < 0.001 [Student’s t test]).
Immunoblotting of factor H-binding proteins of pneumococci.
The results of the immunoblotting experiments with cell wall proteins from S. pneumoniae ATCC 6303 are presented in Fig. 8. After SDS-PAGE the electrophoresed proteins were blotted onto a nitrocellulose membrane and subsequently probed for factor H-binding proteins by sequential incubation with NHS, polyclonal rabbit anti-human factor H, and, finally, horseradish peroxidase-labeled goat anti-rabbit IgG antibody.
FIG. 8.

Immunoblotting of cell wall proteins of type 3 pneumococcal strain ATCC 6303 with anti-human factor H antibody. Pneumococcal proteins blotted onto a nitrocellulose membrane were sequentially incubated with NHS, rabbit anti-human factor H antibody, and peroxidase-labeled goat anti-rabbit antibody. Left lane, high-range molecular mass markers (47 to 208 kDa); middle lane, pneumococcal proteins reactive with factor H (88, 150, and 196 kDa); right lane, low-range molecular mass markers (34.6 to 104 kDa). The molecular masses of low- and high-range marker proteins are indicated on right and left of the immunoblot, respectively.
The immunoblot clearly shows two sharply demarcated bands of 88 and 196 kDa. In addition, a somewhat fainter band with an apparent molecular mass of 150 kDa appeared.
Immunoelectron microscopy of factor H-binding sites on the pneumococcal cell wall.
The results of immunoelectron microscopy of pneumococci after sequential incubation with human serum, anti-human factor H antibody, and gold-labeled protein A are depicted in Fig. 9. The pneumococcal cells appeared morphologically intact, with clearly distinguishable plasma membranes and cell walls. The mild fixation in combination with ultrathin cryosectioning of the bacteria allowed a very accurate localization of factor H binding as visualized by gold particle deposition. It is evident that gold particles were associated almost exclusively with the outer surface of the pneumococcal cell wall, either directly adjacent to it or embedded in or superimposed on the “fuzzy” layer covering the cell wall (Fig. 9). In some instances, there appeared to be an association of gold particles with structures extending from the cell wall (Fig. 9). However, these structures could not be visualized any better, despite attempts at higher magnification.
FIG. 9.

Immunogold electron microscopy of factor H binding by the pneumococcal strain ATCC 6303. Distribution of gold particles (10 nm in diameter) representing factor H binding on ultrathin sections of pneumococcal cells is clearly restricted to the immediate vicinity of the cell wall and the fuzzy coat covering the cell surface. Arrow, gold particle directly bound to the cell wall; asterisks, particles associated with projections extending from the cell wall. Bar, 250 nm. Magnification, ×55,000.
DISCUSSION
The results of the present study show that virulent type 3 S. pneumoniae strains bind the complement-regulatory protein factor H. In addition, our findings strongly suggest that the binding of factor H by virulent type 3 pneumococci is a key factor in the resistance of these strains to complement activation and complement-dependent opsonophagocytosis, which is an essential part of the host defense against these pathogenic bacteria (10, 13, 20, 40, 46). This is the first study that shows that virulent pneumococci are capable of binding a complement-regulatory protein, in this case factor H, independent of the previous activation of complement.
The binding of complement-regulatory proteins is a natural process during complement activation by autologous cells to protect these cells, notably blood cells, from complement-induced lysis. In this process, fixing of complement-regulatory proteins such as factor H is strictly complement activation dependent. Factor H binding occurs to both C3 and C5 convertases, strongly restricting the formation of lytic C5b-C9 complexes. Indeed, we could show that the generation of these terminal complement complexes by the virulent type 3 strains ATCC 6303 and WU2 was greatly diminished compared with that by the nonvirulent mutant strain DW3.8, which showed diminished binding of factor H and was therefore a potent activator of complement.
It has been shown previously that in type 7 pneumococci both capsule and cell wall may fix factor H, but this binding was found to be strictly C3b dependent (10). In contrast, in both ATCC 6303 and WU2, factor H binding was EDTA resistant and therefore independent of C3b deposition on either cell wall or capsule.
Thus, our data indicate that virulent type 3 pneumococci carry specific receptors or binding sites for factor H. Factor H binding was strongly decreased by the pretreatment of strains ATCC 6303 and WU2 with trypsin, indicating that the putative factor H receptors are proteinaceous. In addition, trypsin pretreatment strongly enhanced complement activation and complement-dependent phagocytosis by ATCC 6303 and WU2. Finally, trypsin pretreatment strongly decreased the virulence of ATCC 6303 upon i.p. challenge in mice. Together, these data indicate that the binding of factor H, complement activation, opsonophagocytosis, and the virulence of type 3 pneumococci are modulated by the same surface-associated proteins. These findings also exclude the capsule of type 3 pneumococci as a major target of factor H binding, since the extent of the capsule was not affected by trypsin treatment. In this respect, i.e., by the binding of factor H, the putative protein receptors on three pneumococci act similarly to M protein, an important virulence factor of group A streptococci (16, 19). Collectively, these data suggest that the putative factor H-binding proteins constitute an independent virulence factor of type 3 pneumococci and, possibly, other serotypes as well.
This conclusion is further supported by the fact that the binding of factor H appears to be independent of the well-established virulence factor PspA. PspA is an important determinant of pneumococcal virulence independent of capsular type. It is interesting that PspA has structural properties similar to streptococcal M protein (47). However, the PspA-deficient mutant of WU2, JY1123, bound similar or even larger amounts of factor H than its parent strain did. Together, these findings clearly indicate that PspA is not a major candidate for the binding of factor H by virulent type 3 pneumococci. Nevertheless, a minor role of PspA in interfering with complement activation and opsonophagocytosis may be based on a mechanism different from factor H binding.
The factor H-dependent inactivation of C3 appears to be independent of the C3 degradation activity displayed by pneumococcal serotypes 3, 4, and 14 as described by Angel et al. (3). These investigators showed that the degradation of purified C3 by these pneumococcal serotypes occurred in the absence of any other serum or complement proteins, notably in the absence of complement-regulatory proteins.
Hypothetically, C3 inactivation and degradation by type 3 pneumococci could be a two-stage process. First, bound C3 is inactivated by receptor-bound factor H, and subsequently, bound C3 may be degraded by the putative C3 proteinase as described by Angel et al. (3). Finally, the immunoblotting and ultrastructural studies revealed that there are probably multiple factor H-binding proteins present on the pneumococcal cell wall. Specifically, the immunoblotting experiments clearly showed three bands of 88, 150, and 196 kDa. Whether these bands represent different polymerizations of the same basic protein molecule or completely different proteins must await structural analysis. Sequence analysis should also indicate whether the factor H-binding proteins of type 3 pneumococci are structurally related to the M proteins of group A streptococci, which possess factor H-binding domains (16, 19).
The ultrastructural studies involving immunogold electron microscopy revealed two basic modes of factor H binding to the pneumococcal cell wall, either directly adjacent to the outer surface of the cell wall or apparently associated with projections extending from the cell wall. The structural features of these apparent surface projections could not be analyzed any further in the present study despite attempts at better visualization at higher magnification. Together, these observations suggest that factor H binding by type 3 pneumococci is probably not dependent on a single molecule but is a result of the action of a variety of proteins present on different sites on the pneumococcal surface. Apparently, these factor H-binding proteins are all sensitive to trypsin, and in this connection we made the interesting observation that trypsin treatment of ATCC 6303 considerably reduced virulence upon i.p. infection in mice. This is possibly explained by assuming that the synthesis of surface proteins is apparently sufficiently delayed in the peritoneal cavity to permit efficient complement activation and opsonophagocytosis to proceed.
In conclusion, our results indicate that the virulence of the type 3 pneumococcal strains ATCC 6303 and WU2 is strongly related to their ability to resist complement activation and complement-mediated phagocytosis. This modulation of complement activation appears to be associated with trypsin-sensitive surface components that are responsible for the complement activation-independent binding of factor H to the bacterial surface. Immunoblotting and immunogold electron microscopic studies suggested that factor H binding may be dependent on several proteins, which may be expressed on the pneumococcal surface in two different ways, i.e., either directly adjacent to the outer surface of the cell wall or associated with projections extending from the cell wall. The role of these interesting putative factor H-binding surface proteins in the virulence of pneumococci warrants further investigation.
REFERENCES
- 1.Alonso De Velasco E, Verheul A F, Verhoef J, Snippe H. Streptococcus pneumoniae: virulence factors, pathogenesis, and vaccines. Microbiol Rev. 1995;59:591–603. doi: 10.1128/mr.59.4.591-603.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alonso De Velasco E, Dekker B A, Verheul A F, Feldman R G, Verhoef J, Snippe H. Antipolysaccharide immunoglobulin isotype levels and opsonic activity of antisera: relationships with protection against Streptococcus pneumoniae infection in mice. J Infect Dis. 1995;172:562–565. doi: 10.1093/infdis/172.2.562. [DOI] [PubMed] [Google Scholar]
- 3.Angel C S, Ruzek M, Hostetter M K. Degradation of C3 by Streptococcus pneumoniae. J Infect Dis. 1994;170:600–608. doi: 10.1093/infdis/170.3.600. [DOI] [PubMed] [Google Scholar]
- 4.Austrian R. Some observations on the pneumococcus and on the current status of pneumococcal disease and its prevention. In: Quie P G, Kass E H, editors. The pneumococcus and the pneumococcal vaccine. Chicago, Ill: University of Chicago Press; 1982. pp. 191–207. [Google Scholar]
- 5.Berry A M, Yother J, Briles D E, Hansman D, Paton J C. Reduced virulence of a defined pneumolysin negative mutant of Streptococcus pneumoniae. Infect Immun. 1989;57:2037–2042. doi: 10.1128/iai.57.7.2037-2042.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berry A M, Paton J C. Sequence heterogenity of PsaA, a putative adhesin essential for virulence of Streptococcus pneumoniae. Infect Immun. 1996;64:5255–5262. doi: 10.1128/iai.64.12.5255-5262.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyle M D P, Gee A P, Okada M, Borsos T. Differences in cell bound C8 sites on chicken erythrocytes measured by their reactivity with guinea pig and human C9. J Immunol. 1980;125:2818–2822. [PubMed] [Google Scholar]
- 8.Briles D E, Yother J, McDaniel L S. Role of pneumococcal surface protein A in the virulence of Streptococcus pneumoniae. Rev Infect Dis. 1988;10:S372–S374. doi: 10.1093/cid/10.supplement_2.s372. [DOI] [PubMed] [Google Scholar]
- 9.Briles D E, King J D, Gray M A, McDaniel L S, Swiatlo E, Benton K A. PspA, a protection-eliciting pneumococcal protein: immunogenicity of isolated native PspA in mice. Vaccine. 1996;14:858–867. doi: 10.1016/0264-410x(96)82948-3. [DOI] [PubMed] [Google Scholar]
- 10.Brown E, Joiner K, Gaither T, Hammer C, Frank M M. The interaction of C3b bound to pneumococci with factor H, factor I and properdin factor B of the human complement system. J Immunol. 1983;131:409–415. [PubMed] [Google Scholar]
- 11.Brown E J, Hosea S W, Hammer C H, Burch C G, Frank M M. A quantitative analysis of the interactions of antipneumococcal antibody and complement in experimental pneumococcal bacteremia. J Clin Investig. 1982;69:85–98. doi: 10.1172/JCI110444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown E J, Hosea S W, Frank M M. The role of antibody and complement in the reticuloendothelial clearance of pneumococci from the blood stream. Rev Infect Dis. 1983;5:S797–S805. doi: 10.1093/clinids/5.supplement_4.s797. [DOI] [PubMed] [Google Scholar]
- 13.Burman L A, Norrby R, Trollfors B. Invasive pneumococcal infections: incidence, predisposing factors, and prognosis. Rev Infect Dis. 1985;7:133–142. doi: 10.1093/clinids/7.2.133. [DOI] [PubMed] [Google Scholar]
- 14.Fine D P. Pneumococcal type-associated variability in alternate complement pathway activation. Infect Immun. 1975;12:772–778. doi: 10.1128/iai.12.4.772-778.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finland M, Barnes M W. Changes in the occurrence of capsular serotypes of Streptococcus pneumoniae at Boston City Hospital during selected years between 1935 and 1974. J Clin Microbiol. 1977;5:154–166. doi: 10.1128/jcm.5.2.154-166.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischetti V A. Streptococcal M protein: molecular design and biological behavior. Clin Microbiol Rev. 1989;2:285–314. doi: 10.1128/cmr.2.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geelen S P M, Aerts P C, Verhoef J, Fleer A, van Dijk H. Functional microassay of complement activation by pneumococci. J Microbiol Methods. 1992;14:257–265. [Google Scholar]
- 18.Hammerschmidt S, Talay S R, Brandtzaeg P, Ghhatwal G S. SpsA, a novel pneumococcal surface protein with specific binding to secretory immunoglobulin A and secretory component. Mol Microbiol. 1997;25:1113–1124. doi: 10.1046/j.1365-2958.1997.5391899.x. [DOI] [PubMed] [Google Scholar]
- 19.Horstmann R, Sievertsen H J, Knobloch J, Fischetti V A. Antiphagocytic activity of streptococcal M-protein: selective binding of complement control protein factor H. Proc Natl Acad Sci USA. 1988;85:1657–1661. doi: 10.1073/pnas.85.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hosea S W, Brown E J, Frank M M. The critical role of complement in experimental pneumococcal sepsis. J Infect Dis. 1980;142:903–909. doi: 10.1093/infdis/142.6.903. [DOI] [PubMed] [Google Scholar]
- 21.Hostetter M K. Serotypic variations among virulent pneumococci in deposition and degradation of covalently bound C3b. Implications for phagocytosis and antibody production. J Infect Dis. 1986;153:682–693. doi: 10.1093/infdis/153.4.682. [DOI] [PubMed] [Google Scholar]
- 22.Lacks S, Neuberger M. Membrane localization of a deoxyribonuclease implicated in the genetic transformation of Diplococcus pneumoniae. J Bacteriol. 1975;124:11321–11329. doi: 10.1128/jb.124.3.1321-1329.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liou W, Geuze H J, Slot J W. Improving structural integrity of cryosections for immunogold labeling. Histochem Cell Biol. 1996;106:41–58. doi: 10.1007/BF02473201. [DOI] [PubMed] [Google Scholar]
- 24.Lock R A, Paton J C, Hansman D. Comparative efficacy of pneumococcal neuraminidase and pneumolysin as immunogens protective against Streptococcus pneumoniae. Microb Pathog. 1988;5:461–467. doi: 10.1016/0882-4010(88)90007-1. [DOI] [PubMed] [Google Scholar]
- 25.Klein J O. The epidemiology of pneumococcal disease in infants and children. Rev Infect Dis. 1981;3:246–253. doi: 10.1093/clinids/3.2.246. [DOI] [PubMed] [Google Scholar]
- 26.McDaniel L S, Scott G, Kearney J F, Briles D E. Monoclonal antibodies against protease sensitive pneumococcal antigens protect mice from lethal infections with Streptococcus pneumoniae. J Exp Med. 1984;160:386–397. doi: 10.1084/jem.160.2.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McDaniel L S, Scott G, Widenhofer K, Caroll J M, Briles D E. Analysis of a surface protein of Streptococcus pneumoniae recognized by protective monoclonal antibodies. Microb Pathog. 1986;1:519–531. doi: 10.1016/0882-4010(86)90038-0. [DOI] [PubMed] [Google Scholar]
- 28.McDaniel L S, Sheffield J S, Delucchi P, Briles D E. PspA, a surface protein of Streptococcus pneumoniae, is capable of eliciting protection against pneumococci of more than one capsular type. Infect Immun. 1991;59:222–228. doi: 10.1128/iai.59.1.222-228.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mollnes T E, Lea T, Froland S S, Harboe M. Quantification of the terminal complement complex in human plasma by an enzyme-linked immunosorbent assay based on monoclonal antibodies against a neoantigen of the complex. Scand J Immunol. 1985;22:197–202. doi: 10.1111/j.1365-3083.1985.tb01871.x. [DOI] [PubMed] [Google Scholar]
- 30.Mollnes T E, Lea T, Harboe M, Tschopp J. Monoclonal antibodies recognizing a neoantigen of poly (C9) detect the human terminal complement complex in tissue and plasma. Scand J Immunol. 1985;22:183–195. doi: 10.1111/j.1365-3083.1985.tb01870.x. [DOI] [PubMed] [Google Scholar]
- 31.Paton J C, Berry A M, Lock R A. Molecular analysis of putative pneumococcal virulence proteins. Microb Drug Resist. 1997;3:1–10. doi: 10.1089/mdr.1997.3.1. [DOI] [PubMed] [Google Scholar]
- 32.Reed L J, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg. 1938;27:493–497. [Google Scholar]
- 33.Riley I D, Douglas R M. An epidemiologic approach to pneumococcal disease. Rev Infect Dis. 1981;3:233–245. doi: 10.1093/clinids/3.2.233. [DOI] [PubMed] [Google Scholar]
- 34.Rosonow C, Ryan P, Weiser J N, Johnson S, Fontan P, Ortqvist A, Masure H R. Contribution of novel choline-binding proteins to adherence, colonisation and immunogenicity of Streptococcus pneumoniae. Mol Microbiol. 1997;25:819–829. doi: 10.1111/j.1365-2958.1997.mmi494.x. [DOI] [PubMed] [Google Scholar]
- 35.Rubins J B, Janoff E N. Pneumolysin: a multifunctional pneumococcal virulence factor. J Lab Clin Med. 1998;131:21–27. doi: 10.1016/s0022-2143(98)90073-7. [DOI] [PubMed] [Google Scholar]
- 36.Rijkers G T. Anti-pneumococcal polysaccharide ELISA. J Immunol Methods. 1986;88:185–186. doi: 10.1016/0022-1759(86)90017-7. [DOI] [PubMed] [Google Scholar]
- 37.Sanders A M, Feldman R G, Voorhorst-Ogink M M, de Haas M, Rijkers G T, Capel P J A, Zegers B J M, van de Winkel J G J. Human immunoglobulin G (IgG) Fc receptor IIA (CD32) polymorphism and IgG2-mediated bacterial phagocytosis by neutrophils. Infect Immun. 1995;63:73–81. doi: 10.1128/iai.63.1.73-81.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slot J W, Geuze H J, Gigengack S, Lienhard G E, James D E. Immunolocalisation of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol. 1991;113:123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stephens C G, Williams R C, Reed W P. Classical and alternative complement pathway activation by pneumococci. Infect Immun. 1977;17:296–302. doi: 10.1128/iai.17.2.296-302.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tuomanen E, Hengstler B, Zak O, Tomasz A. The role of complement in inflammation during experimental pneumococcal meningitis. Microb Pathog. 1986;1:15–32. doi: 10.1016/0882-4010(86)90028-8. [DOI] [PubMed] [Google Scholar]
- 41.Van Dijk H, Rademaker P M, Willers J M N. Estimation of the classical pathway of mouse complement activity by use of sensitized rabbit erythrocytes. J Immunol Methods. 1980;39:257–268. doi: 10.1016/0022-1759(80)90060-5. [DOI] [PubMed] [Google Scholar]
- 42.Vinther Nielsen S, Henrichsen J. Capsular serotypes of Streptococcus pneumoniae isolated from blood and CSF during 1982–1987. Clin Infect Dis. 1992;15:794–798. doi: 10.1093/clind/15.5.794. [DOI] [PubMed] [Google Scholar]
- 43.Watson D A, Musher D M. Interruption of capsule production in Streptococcus pneumoniae serotype 3 by insertion of transposon Tn916. Infect Immun. 1990;58:3135–3138. doi: 10.1128/iai.58.9.3135-3138.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watson D A, Musher D M, Verhoef J. Pneumococcal virulence factors and host responses to them. Eur J Clin Microbiol Infect Dis. 1995;14:479–490. doi: 10.1007/BF02113425. [DOI] [PubMed] [Google Scholar]
- 45.Whaley K. Purification of complement components and preparation of antisera. In: Whaley K, editor. Methods in complement for clinical immunologists. Edinburgh, United Kingdom: Churchill-Livingstone; 1985. pp. 721–764. [Google Scholar]
- 46.Winkelstein J A. The role of complement in the host’s defense against Streptococcus pneumoniae. Rev Infect Dis. 1981;3:289–298. doi: 10.1093/clinids/3.2.289. [DOI] [PubMed] [Google Scholar]
- 47.Yother J, White J M. Novel surface attachment mechanisms of the Streptococcus pneumoniae protein PspA. J Bacteriol. 1994;176:2976–2985. doi: 10.1128/jb.176.10.2976-2985.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]