

Abstract
Radiation therapy (RT) is commonly used to treat solid tumors of the breast, lung, and esophagus; however, the heart is an unintentional target of ionizing radiation (IR). IR exposure to the heart results in chronic toxicities including heart failure. We hypothesize that the circadian system plays regulatory roles in minimizing IR-induced cardiotoxicity. We treated mice in control (Day Shift), environmentally-disrupted (Rotating Shift) and genetically-disrupted (Per 1/2 mutant) circadian conditions with 18 Gy of IR to the heart. Compared to control mice, circadian clock disruption significantly exacerbated post-IR systolic dysfunction (by ultrasound echocardiography) and increased fibrosis in mice. At the cellular level, Bmal1 protein bound to Atm, Brca1, and Brca2 promoter regions and its expression level was inversely correlated with DNA damage levels based on the state of the clock. Further studies with circadian synchronized cardiomyocytes revealed that Bmal1 depletion increased IR-induced DNA damage and apoptosis. Collectively, these findings suggest that the circadian clock protects from IR-induced toxicity and potentially impacts RT treatment outcome in cancer patients through IR-induced DNA damage responses.
Keywords: Circadian clock, radiation, heart, toxicity, Bmal1
INTRODUCTION
Radiation therapy (RT) is widely used in neoadjuvant and/or adjuvant treatment for most breast, esophageal, and lung tumors, with over 1300 clinical trials as of this writing (1). Ionizing radiation (IR) used in RT induces lethal DNA double strand breaks (DSBs) to cause tumor cell death; however there are off-target effects to the heart, given its anatomical proximity (2). Consequently, occurrences of chronic RT-induced cardiotoxicity are common in patients and manifest in life-threatening disease states such as heart failure, myocarditis, cardiomyopathy, fibrosis, ischemic and valvular heart disease, constrictive pericarditis and pericardial effusion (3–5). In particular, breast cancer patients typically receive fractionated RT ranging between 10 Gy and 27 Gy, and a recent study involving over 2000 patients showed a 7.4 % increase in coronary events with each 1 Gray (Gy) of mean IR dose to the heart (3, 6).
As a protective strategy, we explored the role of the circadian (~24 hour) clock to minimize RT-induced cardiotoxicity. The circadian clock is an endogenous genetically encoded time-keeping machinery that drives molecular and physiological functions of an organism (9). The molecular clockwork is generated by transcriptional-translational feedback loops (TTFL) where the heterodimeric transcription factor complex CLOCK-BMAL1 positively drives the expression of target clock-controlled genes (CCGs) transcriptionally by binding to E-box elements. Two core clock genes, Cryptochrome (CRY1-2) and Period (PER1-3), form the negative arm of the primary feedback loop by translationally inhibiting the activity of CLOCK-BMAL1 (9–12). In addition to the primary feedback loop, there is a secondary feedback loop to regulate the circadian clock by the activation or repression of ARNTL (which encodes BMAL1) transcription via RAR-related orphan receptor (ROR) and REV-ERB elements, respectively (13).
Recent studies have suggested that the expression levels of up to 43% and 50% of mouse and human genes respectively across different tissues are regulated by the circadian clock mechanism mostly in a tissue-specific manner (14, 15). Consequently, biological processes such as cell cycle, DNA repair, blood pressure, heart rate, platelet aggregation, thrombus formation, and sleep-wake cycle are regulated by the circadian clock to maintain heart homeostasis (16–18). Nevertheless, circadian rhythms are disrupted with modern living conditions such as chronic jetlag or shift work (conditions characterized by repeated misalignment of the internal clock to the external light/dark cycle), which subsequently desynchronize the expression of CCGs in the body, giving rise to deleterious cellular and systemic effects on health (19–21). Furthermore, circadian clock disruption has been associated with cardiovascular diseases such as myocardial infarction, and increase in coronary events (22, 23).
We hypothesize that the mechanistic framework of the circadian clock can be harnessed to improve RT efficacy and reduce toxicity. Previous studies on both rodent models and human studies have demonstrated time-of-day dependent physiological responses to RT (24–27). However, the underlying mechanism(s) associating circadian disruption and radiation sensitivity is/are not well known. Therefore, in this study, we demonstrate cellular and physiological connections of how clock disruption, environmentally and genetically, affects radiation sensitivity in order to minimize cardiotoxicity.
MATERIALS AND METHODS
Animal housing and irradiation experiments
All animal procedures were in accordance with the National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee of Washington State University. Female, 8- to 12-week old, wild-type and Per1/2−/− C57BL/6 mice were obtained from the Jackson Laboratories. The circadian clock-disrupted mPer1, mPer2 mutant mice were originally developed and characterized by Dr. David Weaver’s group (28). These Per1/2−/− mice are well-studied in the circadian clock field and are viable, fertile, and without any gross abnormalities except for the lack of normal circadian behavior due to the deleted regions of the mPer1 and mPer2 gene sequence which interacts with the putative bHLH and PAS domains of the CLOCK-BMAL1 heterodimeric complex during transcriptional-translational feedback loop formation (28, 29). The mice were maintained under a 12-hour light/12-hour dark (LD 12:12) cycle (light on at 7 AM, ZT0, and lights off at 7 PM, ZT12) at least 4 weeks before and through the duration of the study. Rotating Shift was initiated with weekly alternating LD cycles 15 days before any treatments. Our Rotating Shift model involves subjecting the mice to weekly alternating 12-hour light/12-hour dark (LD 12:12) cycles as previously reported (21).
At sacrifice, to collect blood samples at different time points across a 24-hour time period, mice were placed in complete dark conditions at least 24 hours prior. This was done to enable collection of samples based on the circadian times (CT2, 6, 10, 14, 18, 22), without the influence of a light as a zeitgeber. Blood was collected by cardiac puncture from mice kept under 3% isoflurane anesthesia.
For radiation treatments, C57BL/6 mice were anesthetized under 1–2% isoflurane. Whole heart (thorax region) irradiation treatments were performed with a light field 2 × 1.5 cm square collimator (Fig. S1), similar to a previously published measurement (30). Though the exposed area covered about 50–75% of the thorax and lungs, 100% of the heart was irradiated. Treatments were administered in a X-RAD 320 machine (Precision X-Ray, Inc), comissioned as previously described (31). The dose output was 1.3 Gy/min using a 1.5 mm Al 0.25 mm Cu 0.75 mm Sn filter for a total dose of 18 Gy. Cells were treated with using a 2 mm Al filter.
Tissue collections for all IR-treated animals were done between ZT 9 and ZT12, to align with treatment time by ZT. Liver samples were harvested and rinsed at least once in PBS, flash frozen, and stored in −80°C. Heart samples were rinsed twice in PBS and divided into equal halves longitudinally: one half was flash frozen and stored in −80°C for immunoblot and the other half was fixed in 10% formalin for IHC analysis.
Cell line, circadian synchronization, chemical inhibition, and knockdown experiments
Rat cardiomyocytes (H9c2) (ATCC; Cat # CRL-1446 (32) were cultured in DMEM supplemented with 10% FBS and synchronized by serum shock as previously described (33). Briefly, cells were grown to approximately 100% confluency, at which point DMEM supplemented with 10% FBS was replaced with DMEM supplemented with 50% horse serum and kept in a 37°C incubator with 5% CO2 for two hours. Following serum shock synchronization, cells were gently washed twice with 1X PBS, and given serum-free DMEM containing either vehicle control (DMSO) or 25 μM Rev-Erbα agonist SR9011 (Cayman chemical; Cat # 1930) and returned to 37°C incubator with 5% CO2 for 24 hours.
For knockdown experiments, cells were grown to approximately 70% confluency, and transfected with rat-specific Bmal1 SiRNA (Dharmacon; Cat # L-087911-02-0005) alongside non-target siRNA (Dharmacon; Cat # D-001810-10-20) with a final working concentration of 25 nM. Knockdown efficiency was confirmed to be 85–90% after 48 hours. Afterwards, cells were synchronized by serum shock as described above.
Echocardiography
Heart function of mice was measured by echocardiography using the Vevo 2100 Imaging System (VisualSonics) as previously described (34). Commercially available Nair hair removal lotion was applied using Kimwipes to the chest region of the mice prior to measurement. Mice were kept on a heated, bench-mounted adjustable rail system under 2–3% isoflurane and an ms550S probe was used to visualize heart movement. Images were captured on short axis view, and wall thickness and chamber dimensions were determined from M-mode tracings using the Vevo 2100 analysis software. Left ventricular (LV) wall thickness was measured from the interventricular septum (IVS) and posterior wall (LVPW) while the internal volume was assessed using the left ventricular inner dimension (LVID) at maximum heart contraction (diastole) and relaxation (systole). Collectively, these parameters were used to calculate the percent ejection fraction and fractional shortening, which is the measure of heart function in pumping out blood and muscular contractility.
Histological analyses
Heart tissue specimens were fixed in 10% neutral buffered formalin for 24 hours, and preserved in 80% ethanol. Embedding and sectioning were performed by the Histology Core at WSU Spokane. Tissues sections were deparaffinized using Xylene and stained with Masson’s trichrome stain according to the manufacturer’s protocol (ScyTek Laboratories; Cat # TRM-2-IFU). Preheated Bouin’s Fluid was added to the tissue sections for 1 hour, then rinsed under tap water followed by distilled water. A working mixture of equal parts of Weigart’s (A) and Weigart’s (B) iron hematoxylin was used to stain the sections for 5–10 minutes, then rinsed under tap water for 2–5 minutes. Biebrich Scarlet/Acid Fuchsin Solution was added to the tissue sections for 5–15 minutes and then rinsed with distilled water. The stained tissue was differentiated using Phosphomolybdic/Phosphotungstic Acid Solution for 10–15 minutes or until collagen is not red. Without rinsing, Aniline Blue Solution was added to sections for 5–10 minutes and then rinsed in distilled water. Finally, 1% Acetic Acid Solution was added for 3–5 minutes followed by quick dehydration in 2 changes of 95% alcohol and absolute alcohol.
Microscopy
Tissue slides were mounted onto a Zeiss AxioImager M2 microscope (Zeiss) under bright field illumination using 20X objective magnification. The images were captured by the ZEN Imaging software (Zeiss).
Immunoblotting
Frozen heart tissue samples were homogenized in liquid nitrogen using a mortar and pestle. Protein lysate was extracted from tissues using 1X RIPA lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Nonidet P-40, and 1% sodium deoxycholate), and from cells using 1X lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Tween-20). Conventional immunoblotting procedures were used to determine the levels of selected proteins involved in DDR signaling and the circadian clock. The following antibodies were used: Actin (Santa Cruz Biotechnology; Cat #s sc-1616), Bmal1 (Bethyl Laboratories; Cat #s A302-616A), c-Caspase 3, pH2a.x, H2a, and Rev-erbα (Cell Signaling Technology; Cat #s 9664P, 9718S, 12349S and 13418S respectively). The appropriate anti-mouse, anti-rabbit, and anti-goat HRP-conjugated secondary antibodies (Sigma-Aldrich) were used for detection with Clarity Western ECL chemiluminescent (Bio-Rad Laboratories) and/or SuperSignal West Femto (Thermo Fisher Scientific) reagent methods with Bio-Rad ChemiDoc XRS+ imager.
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed, and buffers were prepared as previously described (29, 35). Heart and liver tissues collected from C56BL/6 mice at ZT8 were initially homogenized in liquid nitrogen using a mortar and pestle, approximately 20–30 mg (heart) or 50–100 mg (liver) of which were added to a 15 ml tube and fixed in 5 ml of 1% formaldehyde in PBS (v/v) with protease inhibitor cocktail (PIC) (Roche) for 10 to 20 minutes while shaking at room temperature. Glycine was then added to a final 0.1 M concentration to quench unreacted formaldehyde followed by centrifugation (4000 RPM for 3 minutes) and 2 washes with ice cold PBS with PIC. The resulting cell pellet was lysed by adding 1 ml of cold cell lysis buffer (50 mM Tris-Cl, 85 mM KCl, 0.5% NP-40, 1x PIC added prior to use), briefly vortexed, and incubated on ice for 10 minutes. Lysis was then repeated, this time with a 5-minute incubation on ice followed by addition of an equal volume of ChIP lysis buffer (10 mM Tris-Cl, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-Deoxycholate, 0.5% N-Laurylsarcosine, 1x PIC added prior to use), and an additional 5-minute incubation on ice. This was then sonicated on ice water with a total of 1500 Joules (Misonix Sonicator 3000 with microtip), the resulting lysate was centrifuged (4000 RPM, 5 minutes, 4°C) after which the supernatant was transferred to new microcentrifuge tubes and pre-cleared with 20 ul of Protein A/G beads (Santa Cruz Biotechnology) per ml of lysate for 30 minutes at 4°C on a tube rotator. These were centrifuged (3000 RPM, 5 minutes, 4°C) and aliquoted (1 ml for antibody and control, 20 μl for input) for immunoprecipitation. Appropriate antibodies were added to these aliquots and incubated overnight with rotation in a cold room. 40 μl of Protein A/G beads were washed with 1 ml ChIP buffer, then an additional 500 μl ChIP buffer, 2 μl of BSA (100 mg/ml) and 4 μl of Salmon sperm DNA (10 mg/ml) were added for blocking and overnight incubation with rotation in the cold room. Blocked Protein A/G beads were then washed with 1 ml ChIP buffer and resuspended in 300 μl of ChIP buffer as a stock for use. 40 μl of the blocked Protein A/G bead stock was added to each immunoprecipitation tube and allowed to incubate with rotation in a cold room overnight. These were centrifuged (4000 RPM, 5 minutes, 4°C) and the beads were washed 4 to 6 times with washing buffer (5 minutes each) by rotation in a cold room followed by centrifugation and an additional 2 washes with 1 ml TE buffer. Supernatant was removed and DNA was eluted twice by adding 100 μl ChIP elution buffer (1% SDS, 0.1 NaHCO3) to beads, allowing to rotate at room temperature for 15 minutes, centrifuging (7000 RPM, 5 minutes), and collecting supernatant to new microcentrifuge tube. 8 μl of 5M NaCl was added to each tube of 200 μl eluate, wrapped with parafilm to prevent sample loss and incubated at 65°C overnight. For input, 180 μl of elution buffer was added to make equal volume and 8 μl of 5 M NaCl was added and incubation was done as with samples. 4 μl of 0.5M EDTA, 8 μl of 1 M Tris-HCl pH 6.5, and 1 μl of Proteinase K (20mg/ml) were added, mixed well, and allowed to incubate at 50°C for 1 hour. DNA was purified using a QIAquick Gel extraction kit following the manufacturer’s instructions. BMAL1 (Bethyl Laboratories; Cat # A302-616A) and rabbit IgG (Santa Cruz Biotechnology; Cat # sc-2027) antibodies were used for immunoprecipitation reactions, and the interacting gene regions were enriched using the primers shown in Table 1A.
Table 1A.
Primer sets for ChIP-PCR for mouse liver samples
| Name | Direction | Sequence (−1 to −1500 bp) | Sequence (−1501 to −3000 bp) |
|---|---|---|---|
| Atm | 5’ | TACACCGAGGAGCTTCGACT | GAACACAGGGGGAAAGGAAT |
| Atm | 3’ | CAGCTCGGATCATGCTGTTA | GCCTCGATGGAATTTTTCAA |
| Brca1 | 5’ | AGCTGGTTGGATTTCCCTTT | GCATAGCTGTGGGGTTCATT |
| Brca1 | 3’ | CAGCGTTCTGTGTTCGTGTT | TCCAGCCCTACAGGCTCTTA |
| Brca2 | 5’ | ACCTCTCTCCTCCGACTTCC | CCGACCTAGCAATCCCATTA |
| Brca2 | 3’ | GGCCAGAGAGACAGCTGAAC | AGCCTTTTGGGTTCTGGAAT |
| Per2 | 5’ | ACAGGAGCCTGAACGCTTTA | GAAGGAGACTCTGCCAGGTG |
| Per2 | 3’ | TAACCAAATGCGGTGGTGTA | GCACCTCTGGTTCCTCTGAC |
ChIP-Seq analysis
Previously published mouse ChIP-Seq data of liver tissue using Bmal1 antibody (36) was obtained from the Gene Expression Omnibus (GEO) with accession number GSE39860. The BigWig (BW) file was downloaded and visualized using the Integrated Genomics viewer (IGV) software 2.3. The genome was set to mouse mm9 and the Bmal1-binding sites were identified to be strongest at ZT8, and hence ZT8 dataset was used for analysis. For each gene of interest, the gene region information was visualized and the Bmal1 enriched tags counts were identified around the promoter-exon1 regions. The summit normalized tag counts, positions of enrichment, and distribution regions are reported. This analysis used a Bmal1 knockout mouse model as a negative control and normalization was achieved by down-sampling to the lowest uniquely mapped reads number. The validity of the generated graphical tags on the IGV software were checked against the numerical values, for Per2 and Cry1 as positive controls. Subsequently, the amino acid sequences for these regions were extracted and canonical and noncanonical E-Boxes for Bmal1 binding and activity as previously reported (37, 38) were identified in these sequences.
qRT-PCR and cosine-wave analysis
Briefly, freshly collected whole blood samples were immediately centrifuged at 5000 × g for 3 mins and the upper plasma layer was transferred to a separate tube. To the remaining blood samples, RBCs were lysed using 1ml RBC lysis buffer (150mM NH4Cl, 10mM NaHCO3, 1mM EDTA) twice, followed by two washes in 1X PBS (centrifugation at 12000 × g for 2 minutes). The WBCs were collected, and total RNA was isolated using TRIZOL reagent with the Zymo whole blood RNA mini prep kit (Cat # R1021). The RNA concentration was measured using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific), and 1μg of total RNA was used to synthesize cDNA using iScript RT Supermix (Bio-RAD). Real-time quantitative PCR was performed using iTaq Universal SYBR Green Supermix (Bio-RAD) and appropriate primers (Table 1B) in a StepOnePlus Real-Time-PCR System (Applied Biosystems) following the manufacturer’s protocol. RT-PCR cycle thresholds of individual genes were normalized to the corresponding actin mRNA expression values. The time point values were fit by a nonlinear regression equation: . Significant oscillations were determined by F test using the r2 value from nonlinear regression wave fit (39).
Table 1B.
Primer sets for qRT-PCR for mouse blood samples
| Name | Direction | Sequence |
|---|---|---|
| Arntl | 5’ | ACCTCGCAGAATGTCACAGGCA |
| Arntl | 3’ | CTGAACCATCGACTTCGTAGCG |
| Dbp | 5’ | CTTTTCCACTCCTTGCTTCG |
| Dbp | 3’ | CACGACTCACATGTCCTGCT |
Quantification and statistics
All experiments were performed with at least 3 separate animals/samples, unless otherwise noted, and are represented as Mean ± SEM. Signals from imaged blots and histology were quantified using ImageJ. Two-tailed student’s t test, Two-way ANOVA (with Repeated Measures for time-course analysis) were performed using Prism version 6.01 (GraphPad software). Dunnett’s and Tukey’s multiple comparison tests were used for post-hoc comparisons where appropriate.
RESULTS
The circadian clock preserves heart function from the chronic effects of IR treatment
We used the well-documented C57BL/6 strain to investigate how the circadian clock modulates the physiology of the heart in response to IR treatment (40, 41). Wild-type mice were randomly grouped into control/Day Shift and Rotating Shift (environmental clock disruption) as previously reported (20). In addition, we used a genetically disrupted Per1/2−/− mouse model which lacks endogenous circadian rhythmicity (28). Initially, we wanted to see if our environmental clock-disruption model was sufficient to impact gene expression. To this end, we performed a time course qRT-PCR using white blood cells to investigate core clock gene Arntl and clock controlled gene Dbp with environmentally clock disrupted (Rotating Shift) and control/Day shift C57BL/6 mice. We found that in the Rotating Shift mice, the rhythmicity of Dbp is completely lost, and though Arntl showed a significant rhythm, its peak time is delayed and amplitude is well dampened over 24 hours (Fig. S2) (36, 42). This result demonstrates that our rotating shift model indeed disrupts the core clock and clock-controlled gene expression.
To test how the circadian clock modulates heart function in response to IR treatment, we irradiated Day Shift, Rotating Shift, and Per1/2−/− mice with 18 Gy of IR targeted to the heart on experimental day 0 (between zeitgeber time (ZT) 9 to 12, where ZT0 is lights on and ZT12 is lights off) and monitored heart function over a period of 6 weeks (Fig. 1A) (30, 41). We measured fractional shortening (FS) and left ventricular ejection fraction (LVEF) as indicators of cardiac function, and histology to examine fibrosis within the myocardium. In clock-disrupted mice, there was greater widening of the systolic left ventricle inner dimension from −IR to +IR by 6 weeks (Fig. 1B), which translated to severe systolic dysfunction with significantly decreased LVEF and FS (Fig. 1C–D) compared to Day Shift controls. There were 25% and 17% differences in LVEF and 19% and 13% differences in FS among Rotating Shift and Per1/2−/− mice, respectively, compared to the Day Shift controls. Expectedly, there was significantly increased fibrosis within the myocardium of clock-disrupted mice compared to Control/Day Shift mice, with an untreated negative control which had no signs of fibrosis (Fig. 1E–F). Meanwhile, untreated mice had no heart dysfunction based on clock status (Fig. 1C–D). Collectively, these findings suggest that the circadian clock offers protection from IR-induced cardiotoxicity by preserving cardiac function.
Fig. 1. The circadian clock preserves heart function from the chronic effects of IR treatment.
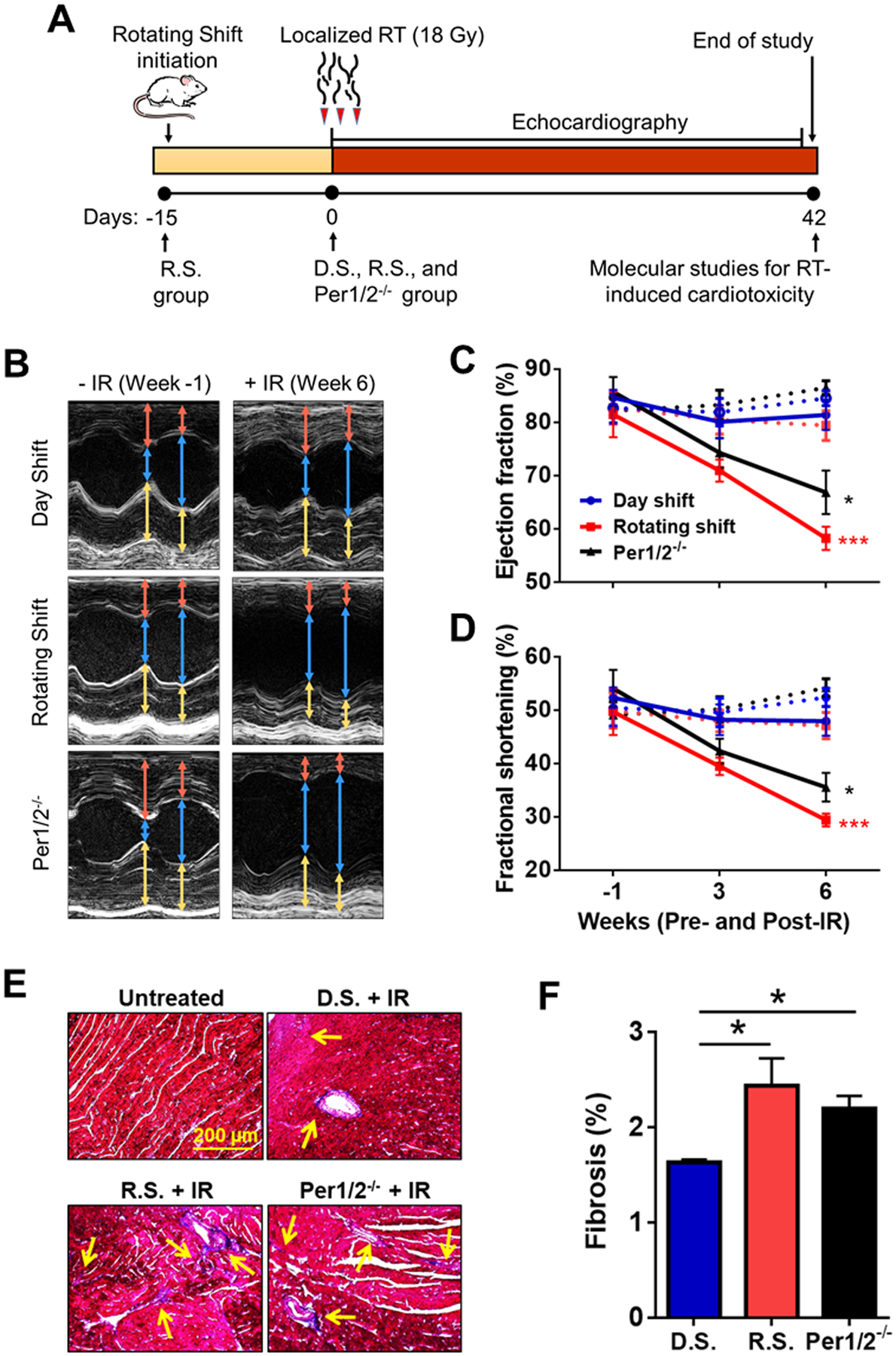
C57BL/6 female mice aged 8 to 12-weeks were used in all groups. Day Shift (D.S.) is the control group and represents the intact clock, while Rotating Shift (R.S.) and Per1/2−/− groups are environmentally- and genetically-clock disrupted groups, respectively. (A) Rotating shift was initiated by subjecting wild-type mice to weekly alternating Light/Dark cycles beginning 15 days before IR treatment. These mice remained in this condition throughout the study. On Day 0, all groups received a single 18 Gy dose of IR, localized to target the heart region. (B) Heart function was measured by echocardiography under anesthesia pre- (Week −1) and post-IR treatment with representative images captured on short axis views. There were also measurements for untreated mice. The captured images were analyzed using the sizes of the interventricular septum (IVS, red arrow), left ventricle inner dimension (LVID, blue arrow), and left ventricular posterior wall (LVPW, yellow arrow) parameters during diastole and systole. Collectively, these parameters determine the (C) left ventricular ejection fraction (LVEF), the measure of heart function in pumping out blood, and (D) fractional shortening (FS), the degree of shortening of the left ventricular diameter between end-diastole and end-systole, as percentages. Dotted lines represent untreated while solid lines represent IR-treated mice. (E) Representative images of Masson’s trichrome stain to indicate fibrosis (yellow arrows showing blue stains) in heart tissue sections with quantification in (F). Statistical analysis was done using Repeated Measures Two-way ANOVA for heart function (n=6 per group) and student’s t test for fibrosis (n=3 per group). *=p<0.05, ***=p<0.001. Error bars = S.E.M.
Reduction in Bmal1 levels exacerbates IR-induced DNA damage in the heart tissue
To explore the mechanism underpinning our physiological findings, we looked into Bmal1 because it is a transcriptional factor of the core clock mechanism, potentially regulates DNA damage response (DDR) genes, and has decreased transcript expression levels over a 24 hour time course in our rotating shift model and the brain of mPer2 deficient mice previously reported (Fig. S2A) (28). We measured the protein expression levels of Bmal1 in the whole heart tissues of untreated and IR-treated control/Day Shift and clock-disrupted mice collected between ZT9 to ZT12 timepoints. In untreated mice, we found significantly decreased levels of Bmal1 with clock-disruption (Figs. 2A–B, S3). Remarkably, the levels of Bmal1 were inversely correlated with endogenous DNA damage levels as measured by H2a.x phosphorylation at Serine 139, corresponding with a previous report on increased DNA damage by ROS levels in Bmal1−/− clock disrupted mice (Fig. 2A, C) (44). The total levels of Histone 2a (H2a) however remained unchanged. We hypothesize that the decrease of Bmal1 in clock disrupted mice increases endogenous DNA damage and potentially creates a more genetically unstable environment that is susceptible to exogenous DNA damage such as from IR. To assess the chronic effect of IR treatment, heart tissues collected 6 weeks post-IR showed an increase in Bmal1 protein levels in all groups compared to untreated conditions. These increases may be due to IR causing phase advances in Bmal1 oscillation as previously reported (45). Interestingly, Per1/2−/− mice consistently had significantly less Bmal1 levels relative to Day Shift mice. Nevertheless, the measure of DNA damage by H2a.x phosphorylation was not different across all groups post-IR. This observation could be indicative of saturation in accumulated DNA damage over time as heart tissues collected at 24 hours post-IR showed less damage in Day Shift mice compared to clock disrupted mice (Fig. S3). The increased presence of Bmal1 and the inverse relationship of Bmal1 and DNA damage levels in untreated mice suggest that Bmal1 may play an important role in regulating IR-induced DDR in mouse heart.
Fig. 2. The clock regulates IR-induced DNA damage and Bmal1 levels in heart tissues.
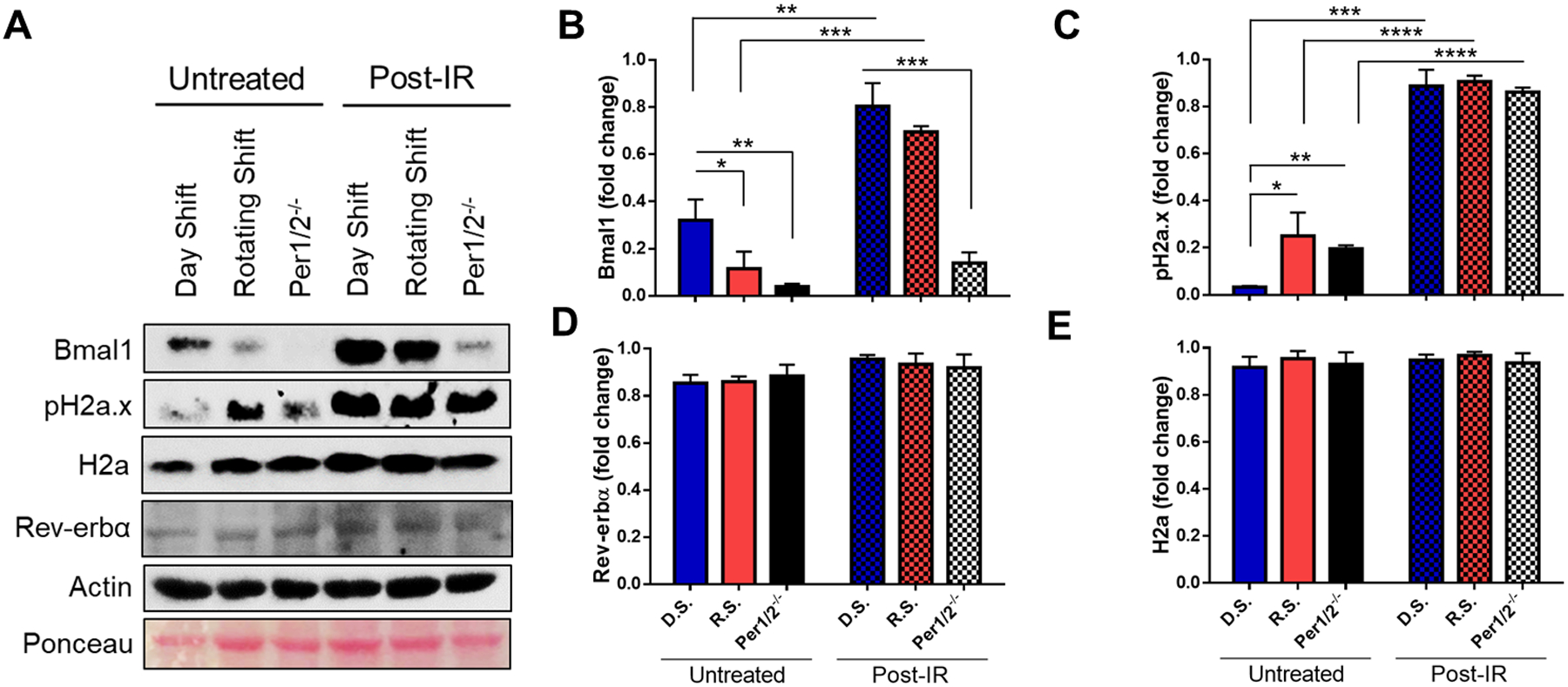
(A) Protein lysates from whole heart tissue samples collected from untreated and 6 weeks post-IR treated mice were probed by immunoblotting for Bmal1, pH2a.x, Histone 2A (H2a), and Rev-erbα expression. (B-E) Densitometry analysis, normalized to actin, of immunoblot images. D.S. means Day Shift, R.S. means Rotating Shift, and Per1/2−/− means Per1/2 mutant mice. Statistical analysis was done using Two-way ANOVA. *=p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001. Error bars = S.E.M. (n=3 mice per group).
Bmal1 depletion sensitizes cardiomyocytes to IR-induced damage and apoptosis
To further understand the Bmal1-related mechanism on a cellular level, we used synchronized rat cardiomyocytes (H9c2), a cell line derived from rat heart. Cardiomyocytes have been shown to exhibit robust circadian rhythms (18, 19), and recent studies showed that antagonism of the Bmal1 repressor protein, Rev-erbα, offered cardioprotection to cardiac surgery patients from perioperative myocardial ischaemia-reperfusion injury and reduced fibrosis in dystrophic muscle (46–48). Hence, we reasoned that decrease in Bmal1 via increased Rev-erbα will be deleterious in cardiomyocytes. To test this, we treated H9c2 cells with 25 μM of the Rev-erbα agonist, SR9011, to inhibit Bmal1 compared to DMSO controls, with maximum effect seen at 48 hours (Fig. S4D). This dose has been demonstrated not to cause toxicity or affect cell viability (49, 50). Next, we irradiated cells with 18 Gy of IR and detected time-dependent increased DNA damage levels indicated by H2a.x phosphorylation with inhibited Bmal1, compared to the no IR control (Fig. S4E). Note that at the 8 h time point, most IR-induced DNA damage has been repaired (51), so that in the Bmal1-effective cells, the signal of pH2a.x was insignificantly increased over the mock-irradiated control. Furthermore, there was increased apoptosis as seen by cleaved Caspase-3 levels in the IR-treated cells with degraded Bmal1, compared to the no IR treated (control) cells (Fig. S4F). The increase in pH2a.x congruent with apoptotic markers after the repair of IR-induced DSBs likely reflects DNA fragmentation in apoptotic cells. Juxtaposing this result with our heart tissue data, the stability of Rev-erbα across our clock disrupted models suggests that other factors contribute to reduced Bmal1 expression levels, which increases IR sensitivity (Fig. 2A, D–E).
In an alternative approach, small interfering RNA was used to silence Bmal1 in understanding IR sensitivity. Remarkably, the downregulation of Bmal1 induced maximum damage and triggered apoptosis as early as 8 hours post-IR, compared to the control (non-target) group (Fig. S4A–C). Again, Bmal1-effective cells demonstrate better protection from IR-induced damage and apoptosis compared to Bmal1-deficient cells. Taken together, the core clock protein, Bmal1, may offer protective regulatory mechanisms against IR-induced heart damage by regulating DDR signaling and apoptosis in cardiomyocytes.
Transcriptional control of IR-induced DDR signaling genes by core clock protein, Bmal1
To understand the nature of the regulation of Bmal1 in IR-induced DDR mechanisms, we performed a chromatin immunoprecipitation (ChIP) assay to see if this regulation was transcriptional. Using mouse heart nuclear extracts collected at ZT10 (Fig. 3A–B), we found that the promoter regions of important homologous recombination (HR) genes, Brca1 and Brca2, as well as the key DSB detection and signaling gene, Atm, were bound by the core circadian clock protein, Bmal1. Brca1, Brca2, and Atm play important roles in DSB repair, and deficiency in any of these factors is associated with severe radiation sensitivity (52, 53). To further confirm our ChIP experimental data, we performed an in silico analysis of previously published mouse ChIP-Seq data that showed Bmal1-DNA interaction in mouse liver tissues (36). Analyzing data collected at ZT8, the time-point with the most prominent Bmal1-associated interactions, we identified peak normalized tag counts of up to 24.0, 7.0, 3.0, and 2.0 at the promoter-exon1 regions of Per2, Atm, Brca1, and Brca2 genes, respectively (Table 2) with the core clock gene Per2 used as a positive control (54). Further, we pulled sequences of these enriched regions to identify canonical (CACGTG) and noncanonical (CACGTT; AACGTG) E-Boxes required for Bmal1 binding and activity, as previously reported (Fig. 3C) (37, 38). Additionally, we used mouse liver nuclear extracts to validate the previously published ChIP-Seq data using our ChIP assay and demonstrated Bmal1 binding to promoters of DDR signaling genes (Fig. S5). These data strongly suggest that the circadian clock, through Bmal1-related mechanisms, potentially influences HR, or DSB repair in general, and therefore affect RT-induced DDR signaling and cytotoxicity.
Fig. 3. The core clock protein, Bmal1, transcriptionally binds to pro-survival genes of DNA repair in mouse heart.

(A) BMAL1 binds to the promoter region of its targeted genes from C57BL/6 mouse heart nuclear extracts in ChIP assay at −1500 bp (region 1) and −3000 bp (region 2) upstream of the transcription start site. (B) Quantification of ChIP experiment. (C) From ChIP-seq analysis in Table 1, the identification of canonical and noncanonical E-Boxes, the binding sites for Bmal1, found in the enriched promoter region sequences, along with their distances from transcriptional start sites. Statistical analysis was done using two-tailed student’s t test. *=p<0.05, **=p<0.01, ***=p<0.001. Error bars = S.E.M. (n=3 mice per group).
Table 2.
ChIP-seq analysis of mouse liver nuclear extracts.
| Gene | Chromosome | Summit enrichment position | Summit enrichment tag count | Distribution region | |
|---|---|---|---|---|---|
| Start | End | ||||
| Per2 | 1 | 93355770 | 93356400 | 24.0 | Promoter, Exon 1 |
| Atm | 9 | 53344671 | 53344929 | 7.0 | Promoter, Exon 1 |
| Brca1 | 11 | 101413161 | 101413320 | 3.0 | Promoter, Exon 1 |
| Brca2 | 5 | 151325616 | 151325872 | 2.0 | Promoter |
Previously published ChIP-seq dataset was re-analyzed using IGV genome browser software to identify specific regions of Bmal1 enrichment on target DDR signaling genes Atm, Brca1, and Brca2, with Per2 as a positive control. We report the Bmal1 summit enrichment position, tag count, and regions of distribution.
DISCUSSION
We were able to demonstrate both mechanistically and physiologically how radiation-induced loss of cardiac function are exacerbated with clock disruption, either environmentally or genetically. In our society today, the major cause of circadian disruption is through shift work, affecting about 15 million workers in the United States (55). Interestingly, the circadian clock can be a strategic tool to optimize radiation treatments, especially in breast cancer patients who suffer from chronic cardiotoxicities. Hence, our study employed the use of female mouse models, even though we acknowledge that the results may vary based on sex. Nevertheless, our study has made important findings to understanding cardiotoxicity in the context of circadian clock function. Our findings are summarized in the model in Fig. 4.
Fig. 4. Schematic of the influence of the circadian clock on radiation-induced toxicity and treatment efficacy in cancers.

Circadian clock protects against IR-induced cardiotoxicity by regulating DDR signaling-mediated pro-survival mechanisms, but a disrupted clock by silencing or inhibiting Bmal1 via increased Rev-erbα activity leads to increased toxicities.
Exposure of the heart to IR leads to long-term cardiovascular diseases such as heart failure (5). However, the cellular mechanisms of RT-mediated cardiotoxicity are yet to be fully understood. Here, our study focused on clock-controlled DDR signaling mechanisms that regulate cardiotoxicity, which would also potentially inform investigations into understanding cellular mechanisms in RT-mediated cardiotoxicity. The circadian clock influences physiological and pathophysiological parameters such as heart rate and timing of onset of arrhythmias, respectively, and circadian disruption has been associated with cardiovascular diseases such as hypertension (17, 22). It has also been demonstrated that DNA repair activity, particularly NER, is high in mouse heart tissues, possibly due to the need for cardiac tissue remodeling after injury (56, 57).
Upon IR treatment, there is the induction of DNA damage, which initiates a cascade of cellular events such as DNA repair, inflammatory responses, and apoptosis (58). DNA-dependent kinases, Atm and Chk2, are activated and phosphorylate p53 at N-terminal sites (59, 60). Accordingly, p53 through p21, a CCG, was demonstrated to play an important role in preventing IR-induced myocardial injury by increasing cell cycle arrest (30). Interestingly, it was shown that the clock protein, Per2, associates with cytosolic p53 and Mdm2 to form a trimeric stable complex which, in response to genotoxic stress such as IR, disassembles to release p53 and coordinate downstream genes involved in cell cycle arrest, DNA repair, and apoptosis (61). In the Per2−/− mouse model in vivo, the induction of p53 is reduced, resulting in increased radiosensitivity (61, 62). Similarly, we observed the effects of IR exposure in the heart through DNA damage marker, pH2a.x, and fibrosis, which ultimately affects heart function significantly in our genetically (Per1/2−/−) as well as environmentally (Rotating Shift) clock-disrupted models (Fig. 1). These can also be linked to DNA repair mechanisms as a recent study showed that the Atm kinase-dependent function of the nuclear isoform of the Receptor for Advanced Glycation End-products (nRAGE) plays a critical role in DSB repair to prevent pulmonary fibrosis (63). Our clock-disrupted models were more sensitive to IR-induced loss of cardiac function possibly through p53- and Atm-mediated DDR mechanisms. To confirm this mechanism, our experimental ChIP assay and ChIP-Seq results showed Bmal1, the positive regulator of Per1/2, transcriptionally binds to the promoters of Atm, as well as Brca1 and Brca2 genes, and Bmal1 protein levels were significantly reduced in heart samples of clock-disrupted mice (Fig. 2A–B, 3, and Table 2).
Given that cardiomyocytes possess an autonomous robust circadian clock mechanism that drives gene transcription and protein activity involved in mitochondrial metabolism, signal transduction, cardiac remodeling, and electrophysiology, all geared towards maintaining cardiac homeostasis (64, 65), it is important to understand the role of the clock in IR-induced DDR signaling in cardiomyocytes. Studies utilizing cardiomyocyte-specific Bmal1 knockout mice saw a decrease in heart function (ejection fraction), and causation of heart failure and early mortality (66, 67). Interestingly, we demonstrated that the depletion of Bmal1 either genetically (through siRNA) or pharmacologically (through Rev-erbα agonism), sensitized cardiomyocytes to DNA damage and apoptosis in the presence of IR (Fig. S4), and supports a study which showed increased sensitivity of Bmal1−/− mice to ultraviolet radiation (44). A few recent studies have shown that the pharmacological activation of Rev-erbs is lethal to cancer cells by inhibiting DNA repair at damage sites (50, 68). Taken together, the depletion of Bmal1 impairs the CLOCK-BMAL1 complex, which in turn represses transcriptional activation of the downstream DDR signaling genes. Collectively, these mechanisms shed light on the importance of clock proteins in regulating IR-induced DDR signaling for therapeutic applications.
Supplementary Material
Acknowledgements/funding:
We thank Drs. William K. Kaufmann and Bala Koritala for providing helpful comments, the Histology Core and the Veterinarian, Dr. Daniel Montonye, at WSU Spokane for processing the tissues samples and offering guidance on IHC analysis, and Dr. Yi-Ying Chiou of National Chung Hsing University for information on ChIP-seq analysis. This work was supported by grants from the National Institutes of Health CA227381, ES022640 (to S.G.) and in part by the Congressionally Directed Medical Research Program Award CA171123 (to S.G.), and HL119605 (to Z.C.), and the American Heart Association Pre-doctoral Fellowship (to P.D.); and WSU College of Pharmacy and Pharmaceutical Sciences start-up funds (to S.G.).
Nonstandard Abbreviations
- RT
radiation therapy
- IR
ionizing radiation
- CCGs
clock-controlled genes
- DDR
DNA damage response
- DSBs
Double strand breaks
- BMAL1
Brain and muscle ARNT-like 1
- CLOCK
Circadian locomotor output cycles kaput
- PER
Period
- CRY
Cryptochrome
Footnotes
Conflict of Interest Disclosure: The authors declare that they have no conflicts of interest.
REFERENCES
- 1.National Library of Medicine (U.S.). (2019) ClinicalTrials.gov linking patients to medical research. U.S. National Library of Medicine, National Institutes of Health, Dept. of Health & Human Services,, Bethesda, MD [Google Scholar]
- 2.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, and Linn S (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73, 39–85 [DOI] [PubMed] [Google Scholar]
- 3.Darby SC, Ewertz M, McGale P, Bennet AM, Blom-Goldman U, Bronnum D, Correa C, Cutter D, Gagliardi G, Gigante B, Jensen MB, Nisbet A, Peto R, Rahimi K, Taylor C, and Hall P (2013) Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med 368, 987–998 [DOI] [PubMed] [Google Scholar]
- 4.Boekel NB, Schaapveld M, Gietema JA, Russell NS, Poortmans P, Theuws JC, Schinagl DA, Rietveld DH, Versteegh MI, Visser O, Rutgers EJ, Aleman BM, and van Leeuwen FE (2016) Cardiovascular Disease Risk in a Large, Population-Based Cohort of Breast Cancer Survivors. Int J Radiat Oncol Biol Phys 94, 1061–1072 [DOI] [PubMed] [Google Scholar]
- 5.Puukila S, Lemon JA, Lees SJ, Tai TC, Boreham DR, and Khaper N (2017) Impact of Ionizing Radiation on the Cardiovascular System: A Review. Radiat Res 188, 539–546 [DOI] [PubMed] [Google Scholar]
- 6.(2000) Favourable and unfavourable effects on long-term survival of radiotherapy for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet 355, 1757–1770 [PubMed] [Google Scholar]
- 7.Fitzgerald TJ, Jodoin MB, Tillman G, Aronowitz J, Pieters R, Balducci S, Meyer J, Cicchetti MG, Kadish S, McCauley S, Sawicka J, Urie M, Lo YC, Mayo C, Ulin K, Ding L, Britton M, Huang J, and Arous E (2008) Radiation therapy toxicity to the skin. Dermatol Clin 26, 161–172, ix [DOI] [PubMed] [Google Scholar]
- 8.Ryan JL (2012) Ionizing radiation: the good, the bad, and the ugly. J Invest Dermatol 132, 985–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shearman LP, Sriram S, Weaver DR, Maywood ES, Chaves I, Zheng B, Kume K, Lee CC, van der Horst GT, Hastings MH, and Reppert SM (2000) Interacting molecular loops in the mammalian circadian clock. Science 288, 1013–1019 [DOI] [PubMed] [Google Scholar]
- 10.Xu H, Gustafson CL, Sammons PJ, Khan SK, Parsley NC, Ramanathan C, Lee HW, Liu AC, and Partch CL (2015) Cryptochrome 1 regulates the circadian clock through dynamic interactions with the BMAL1 C terminus. Nat Struct Mol Biol 22, 476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Partch CL, Green CB, and Takahashi JS (2014) Molecular architecture of the mammalian circadian clock. Trends Cell Biol 24, 90–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, Panda S, and Hogenesch JB (2009) Harmonics of circadian gene transcription in mammals. PLoS Genet 5, e1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, Chong LW, DiTacchio L, Atkins AR, Glass CK, Liddle C, Auwerx J, Downes M, Panda S, and Evans RM (2012) Regulation of circadian behaviour and metabolism by REV-ERB-alpha and REV-ERB-beta. Nature 485, 123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang R, Lahens NF, Ballance HI, Hughes ME, and Hogenesch JB (2014) A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci U S A 111, 16219–16224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruben MD, Wu G, Smith DF, Schmidt RE, Francey LJ, Lee YY, Anafi RC, and Hogenesch JB (2018) A database of tissue-specific rhythmically expressed human genes has potential applications in circadian medicine. Sci Transl Med 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sancar A, Lindsey-Boltz LA, Gaddameedhi S, Selby CP, Ye R, Chiou YY, Kemp MG, Hu J, Lee JH, and Ozturk N (2015) Circadian clock, cancer, and chemotherapy. Biochemistry 54, 110–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martino TA, and Young ME (2015) Influence of the cardiomyocyte circadian clock on cardiac physiology and pathophysiology. J Biol Rhythms 30, 183–205 [DOI] [PubMed] [Google Scholar]
- 18.Dakup P, and Gaddameedhi S (2017) Impact of the Circadian Clock on UV-Induced DNA Damage Response and Photocarcinogenesis. Photochem Photobiol 93, 296–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bass J, and Lazar MA (2016) Circadian time signatures of fitness and disease. Science 354, 994–999 [DOI] [PubMed] [Google Scholar]
- 20.Van Dycke KC, Rodenburg W, van Oostrom CT, van Kerkhof LW, Pennings JL, Roenneberg T, van Steeg H, and van der Horst GT (2015) Chronically Alternating Light Cycles Increase Breast Cancer Risk in Mice. Curr Biol 25, 1932–1937 [DOI] [PubMed] [Google Scholar]
- 21.Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutkar A, Bartlebaugh J, Vander Heiden MG, and Jacks T (2016) Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab 24, 324–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morris CJ, Purvis TE, Hu K, and Scheer FA (2016) Circadian misalignment increases cardiovascular disease risk factors in humans. Proc Natl Acad Sci U S A 113, E1402–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vetter C, Devore EE, Wegrzyn LR, Massa J, Speizer FE, Kawachi I, Rosner B, Stampfer MJ, and Schernhammer ES (2016) Association Between Rotating Night Shift Work and Risk of Coronary Heart Disease Among Women. JAMA 315, 1726–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lappenbusch WL (1972) Effect of circadian rhythm on the radiation response of the Chinese hamster (Cricetulus griseus). Radiat Res 50, 600–610 [PubMed] [Google Scholar]
- 25.Plikus MV, Vollmers C, de la Cruz D, Chaix A, Ramos R, Panda S, and Chuong CM (2013) Local circadian clock gates cell cycle progression of transient amplifying cells during regenerative hair cycling. Proc Natl Acad Sci U S A 110, E2106–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan S, Zhang L, Rowbottom L, McDonald R, Bjarnason GA, Tsao M, Barnes E, Danjoux C, Popovic M, Lam H, DeAngelis C, and Chow E (2017) Effects of circadian rhythms and treatment times on the response of radiotherapy for painful bone metastases. Ann Palliat Med 6, 14–25 [DOI] [PubMed] [Google Scholar]
- 27.Ijiri K, and Potten CS (1990) The circadian rhythm for the number and sensitivity of radiation-induced apoptosis in the crypts of mouse small intestine. Int J Radiat Biol 58, 165–175 [DOI] [PubMed] [Google Scholar]
- 28.Bae K, Jin X, Maywood ES, Hastings MH, Reppert SM, and Weaver DR (2001) Differential functions of mPer1, mPer2, and mPer3 in the SCN circadian clock. Neuron 30, 525–536 [DOI] [PubMed] [Google Scholar]
- 29.Ye R, Selby CP, Chiou YY, Ozkan-Dagliyan I, Gaddameedhi S, and Sancar A (2014) Dual modes of CLOCK:BMAL1 inhibition mediated by Cryptochrome and Period proteins in the mammalian circadian clock. Genes Dev 28, 1989–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee CL, Moding EJ, Cuneo KC, Li Y, Sullivan JM, Mao L, Washington I, Jeffords LB, Rodrigues RC, Ma Y, Das S, Kontos CD, Kim Y, Rockman HA, and Kirsch DG (2012) p53 functions in endothelial cells to prevent radiation-induced myocardial injury in mice. Sci Signal 5, ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Azimi R, Alaei P, and Hui S (2012) SU-E-T-272: Commissioning an Orthovoltage Unit Used for Radiobiology Research. Med Phys 39, 3766. [DOI] [PubMed] [Google Scholar]
- 32.Kimes BW, and Brandt BL (1976) Properties of a clonal muscle cell line from rat heart. Exp Cell Res 98, 367–381 [DOI] [PubMed] [Google Scholar]
- 33.Balsalobre A, Damiola F, and Schibler U (1998) A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 93, 929–937 [DOI] [PubMed] [Google Scholar]
- 34.Cheng Z, DiMichele LA, Rojas M, Vaziri C, Mack CP, and Taylor JM (2014) Focal adhesion kinase antagonizes doxorubicin cardiotoxicity via p21(Cip1.). J Mol Cell Cardiol 67, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye R, Selby CP, Ozturk N, Annayev Y, and Sancar A (2011) Biochemical analysis of the canonical model for the mammalian circadian clock. J Biol Chem 286, 25891–25902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, and Takahashi JS (2012) Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoo SH, Ko CH, Lowrey PL, Buhr ED, Song EJ, Chang S, Yoo OJ, Yamazaki S, Lee C, and Takahashi JS (2005) A noncanonical E-box enhancer drives mouse Period2 circadian oscillations in vivo. Proc Natl Acad Sci U S A 102, 2608–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hatanaka F, Matsubara C, Myung J, Yoritaka T, Kamimura N, Tsutsumi S, Kanai A, Suzuki Y, Sassone-Corsi P, Aburatani H, Sugano S, and Takumi T (2010) Genome-wide profiling of the core clock protein BMAL1 targets reveals a strict relationship with metabolism. Mol Cell Biol 30, 5636–5648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiessling S, Beaulieu-Laroche L, Blum ID, Landgraf D, Welsh DK, Storch KF, Labrecque N, and Cermakian N (2017) Enhancing circadian clock function in cancer cells inhibits tumor growth. BMC Biol 15, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janko M, Ontiveros F, Fitzgerald TJ, Deng A, DeCicco M, and Rock KL (2012) IL-1 generated subsequent to radiation-induced tissue injury contributes to the pathogenesis of radiodermatitis. Radiat Res 178, 166–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mezzaroma E, Di X, Graves P, Toldo S, Van Tassell BW, Kannan H, Baumgarten C, Voelkel N, Gewirtz DA, and Abbate A (2012) A mouse model of radiation-induced cardiomyopathy. Int J Cardiol 156, 231–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gachon F, Olela FF, Schaad O, Descombes P, and Schibler U (2006) The circadian PAR-domain basic leucine zipper transcription factors DBP, TEF, and HLF modulate basal and inducible xenobiotic detoxification. Cell Metab 4, 25–36 [DOI] [PubMed] [Google Scholar]
- 43.Luther DJ, Thodeti CK, and Meszaros JG (2013) Injury models to study cardiac remodeling in the mouse: myocardial infarction and ischemia-reperfusion. Methods Mol Biol 1037, 325–342 [DOI] [PubMed] [Google Scholar]
- 44.Geyfman M, Kumar V, Liu Q, Ruiz R, Gordon W, Espitia F, Cam E, Millar SE, Smyth P, Ihler A, Takahashi JS, and Andersen B (2012) Brain and muscle Arnt-like protein-1 (BMAL1) controls circadian cell proliferation and susceptibility to UVB-induced DNA damage in the epidermis. Proc Natl Acad Sci U S A 109, 11758–11763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papp SJ, Huber AL, Jordan SD, Kriebs A, Nguyen M, Moresco JJ, Yates JR, and Lamia KA (2015) DNA damage shifts circadian clock time via Hausp-dependent Cry1 stabilization. Elife 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Welch RD, Billon C, Valfort AC, Burris TP, and Flaveny CA (2017) Pharmacological inhibition of REV-ERB stimulates differentiation, inhibits turnover and reduces fibrosis in dystrophic muscle. Sci Rep 7, 17142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montaigne D, Marechal X, Modine T, Coisne A, Mouton S, Fayad G, Ninni S, Klein C, Ortmans S, Seunes C, Potelle C, Berthier A, Gheeraert C, Piveteau C, Deprez R, Eeckhoute J, Duez H, Lacroix D, Deprez B, Jegou B, Koussa M, Edme JL, Lefebvre P, and Staels B (2018) Daytime variation of perioperative myocardial injury in cardiac surgery and its prevention by Rev-Erbalpha antagonism: a single-centre propensity-matched cohort study and a randomised study. Lancet 391, 59–69 [DOI] [PubMed] [Google Scholar]
- 48.Kuznetsov AV, Javadov S, Sickinger S, Frotschnig S, and Grimm M (2015) H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim Biophys Acta 1853, 276–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trump RP, Bresciani S, Cooper AW, Tellam JP, Wojno J, Blaikley J, Orband-Miller LA, Kashatus JA, Boudjelal M, Dawson HC, Loudon A, Ray D, Grant D, Farrow SN, Willson TM, and Tomkinson NC (2013) Optimized chemical probes for REV-ERBalpha. J Med Chem 56, 4729–4737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sulli G, Rommel A, Wang X, Kolar MJ, Puca F, Saghatelian A, Plikus MV, Verma IM, and Panda S (2018) Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 553, 351–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gu Q, Feng T, Cao H, Tang Y, Ge X, Luo J, Xue J, Wu J, Yang H, Zhang S, and Cao J (2013) HIV-TAT mediated protein transduction of Cu/Zn-superoxide dismutase-1 (SOD1) protects skin cells from ionizing radiation. Radiat Oncol 8, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abbott DW, Thompson ME, Robinson-Benion C, Tomlinson G, Jensen RA, and Holt JT (1999) BRCA1 expression restores radiation resistance in BRCA1-defective cancer cells through enhancement of transcription-coupled DNA repair. J Biol Chem 274, 18808–18812 [DOI] [PubMed] [Google Scholar]
- 53.Abbott DW, Freeman ML, and Holt JT (1998) Double-strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J Natl Cancer Inst 90, 978–985 [DOI] [PubMed] [Google Scholar]
- 54.Dakup PP, Porter KI, Little AA, Gajula RP, Zhang H, Skornyakov E, Kemp MG, Van Dongen HPA, and Gaddameedhi S (2018) The circadian clock regulates cisplatin-induced toxicity and tumor regression in melanoma mouse and human models. Oncotarget 9, 14524–14538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McMenamin TM (2007) A time to work: recent trends in shift work and flexible schedules. Monthly Labor Review, 1–15 [Google Scholar]
- 56.Latimer JJ, Majekwana VJ, Pabon-Padin YR, Pimpley MR, and Grant SG (2015) Regulation and disregulation of mammalian nucleotide excision repair: a pathway to nongermline breast carcinogenesis. Photochem Photobiol 91, 493–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uygur A, and Lee RT (2016) Mechanisms of Cardiac Regeneration. Dev Cell 36, 362–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bray FN, Simmons BJ, Wolfson AH, and Nouri K (2016) Acute and Chronic Cutaneous Reactions to Ionizing Radiation Therapy. Dermatol Ther (Heidelb) 6, 185–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vogelstein B, Lane D, and Levine AJ (2000) Surfing the p53 network. Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 60.Tripathi K, Hussein UK, Anupalli R, Barnett R, Bachaboina L, Scalici J, Rocconi RP, Owen LB, Piazza GA, and Palle K (2015) Allyl isothiocyanate induces replication-associated DNA damage response in NSCLC cells and sensitizes to ionizing radiation. Oncotarget 6, 5237–5252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gotoh T, Vila-Caballer M, Liu J, Schiffhauer S, and Finkielstein CV (2015) Association of the circadian factor Period 2 to p53 influences p53’s function in DNA-damage signaling. Mol Biol Cell 26, 359–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu L, Pelicano H, Liu J, Huang P, and Lee C (2002) The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 111, 41–50 [DOI] [PubMed] [Google Scholar]
- 63.Kumar V, Fleming T, Terjung S, Gorzelanny C, Gebhardt C, Agrawal R, Mall MA, Ranzinger J, Zeier M, Madhusudhan T, Ranjan S, Isermann B, Liesz A, Deshpande D, Haring HU, Biswas SK, Reynolds PR, Hammes HP, Peperkok R, Angel P, Herzig S, and Nawroth PP (2017) Homeostatic nuclear RAGE-ATM interaction is essential for efficient DNA repair. Nucleic Acids Res 45, 10595–10613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Durgan DJ, Hotze MA, Tomlin TM, Egbejimi O, Graveleau C, Abel ED, Shaw CA, Bray MS, Hardin PE, and Young ME (2005) The intrinsic circadian clock within the cardiomyocyte. Am J Physiol Heart Circ Physiol 289, H1530–1541 [DOI] [PubMed] [Google Scholar]
- 65.Kohsaka A, Das P, Hashimoto I, Nakao T, Deguchi Y, Gouraud SS, Waki H, Muragaki Y, and Maeda M (2014) The circadian clock maintains cardiac function by regulating mitochondrial metabolism in mice. PLoS One 9, e112811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Young ME, Brewer RA, Peliciari-Garcia RA, Collins HE, He L, Birky TL, Peden BW, Thompson EG, Ammons BJ, Bray MS, Chatham JC, Wende AR, Yang Q, Chow CW, Martino TA, and Gamble KL (2014) Cardiomyocyte-specific BMAL1 plays critical roles in metabolism, signaling, and maintenance of contractile function of the heart. J Biol Rhythms 29, 257–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bray MS, Shaw CA, Moore MW, Garcia RA, Zanquetta MM, Durgan DJ, Jeong WJ, Tsai JY, Bugger H, Zhang D, Rohrwasser A, Rennison JH, Dyck JR, Litwin SE, Hardin PE, Chow CW, Chandler MP, Abel ED, and Young ME (2008) Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am J Physiol Heart Circ Physiol 294, H1036–1047 [DOI] [PubMed] [Google Scholar]
- 68.Ka NL, Na TY, Na H, Lee MH, Park HS, Hwang S, Kim IY, Seong JK, and Lee MO (2017) NR1D1 Recruitment to Sites of DNA Damage Inhibits Repair and Is Associated with Chemosensitivity of Breast Cancer. Cancer Res 77, 2453–2463 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.