

Abstract
Thyroid hormones are vital in metabolism, growth and development1. Thyroid hormone synthesis is controlled by thyrotropin (TSH), which acts at the thyrotropin receptor (TSHR)2. In patients with Graves’ disease, autoantibodies that activate the TSHR pathologically increase thyroid hormone activity3. How autoantibodies mimic thyrotropin function remains unclear. Here we determined cryo-electron microscopy structures of active and inactive TSHR. In inactive TSHR, the extracellular domain lies close to the membrane bilayer. Thyrotropin selects an upright orientation of the extracellular domain owing to steric clashes between a conserved hormone glycan and the membrane bilayer. An activating autoantibody from a patient with Graves’ disease selects a similar upright orientation of the extracellular domain. Reorientation of the extracellular domain transduces a conformational change in the seven-transmembrane-segment domain via a conserved hinge domain, a tethered peptide agonist and a phospholipid that binds within the seven-transmembrane-segment domain. Rotation of the TSHR extracellular domain relative to the membrane bilayer is sufficient for receptor activation, revealing a shared mechanism for other glycoprotein hormone receptors that may also extend to other G-protein-coupled receptors with large extracellular domains.
The thyroid gland regulates organ development and metabolism in all vertebrates via the thyroid hormones triiodothyronine (T3) and thyroxine (T4)1. The synthesis and secretion of the thyroid hormones is controlled by a homeostatic hypothalamic–pituitary–thyroid signalling axis2. Hypothalamic and pituitary sensing of low thyroid hormone levels induces secretion of the pituitary hormone thyrotropin—also called thyroid-stimulating hormone (TSH)—which acts at TSHR, a G-protein-coupled receptor (GPCR) located on thyroid follicles4. Activation of heterotrimeric Gs and Gq signalling pathways downstream of the TSHR leads to thyroid hormone production5, closing the negative feedback loop to set physiological thyroid hormone levels.
Dysregulation of the central hypothalamic–pituitary–thyroid signalling axis leads to inappropriately increased or decreased thyroid hormone levels, causing a disease burden that affects approximately 5% of the world population6. Hypothyroidism stems predominantly from iodine deficiency or autoimmune inflammation of the thyroid. The primary cause of hyperthyroidism in countries without iodine deficiency is Graves’ disease, an autoimmune disorder leading to inappropriate activation of the TSHR by autoantibody thyroid-stimulating immunoglobulins7,8 (TSI). Persistent direct activation of the TSHR by TSI overcomes the physiological hypothalamic–pituitary–thyroid negative feedback loop, leading to elevated thyroid hormones despite low levels of serum TSH3. Notwithstanding the central role of the TSHR in regulating thyroid hormone physiology, no currently approved medications directly target this receptor to treat thyroid diseases9. Current interventions either target thyroid hormone synthesis or, in more severe cases, destroy the thyroid gland leading to a lifelong need for thyroid hormone replacement therapy3.
A deeper understanding of how the TSHR is activated, physiologically by TSH or pathologically by TSI, would enable approaches to precisely tune the hypothalamic–pituitary–thyroid signalling axis to correct thyroid disease. A central challenge, however, has been the design of TSHR-selective molecules, in part owing to a limited understanding of the structure and dynamics of TSHR function. The TSHR shares significant similarity to two homologous receptors that are critically important in reproductive physiology: the follicle-stimulating hormone receptor (FSHR) and the luteinizing hormone–choriogonadotropin receptor (LH/CGR). Whereas the FSHR is specific for the follicle-stimulating hormone (FSH), the LH/CGR is activated by both luteinizing hormone (LH) and chorionic gonadotropin (CG). A hallmark of TSH, FSH, LH and CG—collectively termed glycoprotein hormones—is their complex N-linked glycosylation, which is required for biological activity10–12. Structures of the FSHR and LH/CGR bound to FSH and CG, respectively, have informed key aspects of glycoprotein hormone recognition and receptor activation13,14. However, these studies leave unanswered how glycosylation drives hormone activity, and for the TSHR, how pathogenic autoantibodies mimic TSH action. Here, we use a combination of cryo-electron microscopy (cryo-EM), protein engineering, signalling studies and molecular dynamics simulations to illuminate the molecular basis of action of TSH and autoantibodies at the TSHR. Our studies provide a model for physiological and pathological activation of the TSHR and a general activation mechanism for the glycoprotein hormone receptor family.
Structures of hormone-bound active TSHR
We first obtained a structure of activated TSHR bound to native human TSH (Fig. 1a). The TSHR is naturally proteolytically cleaved within the extracellular domain (ECD) leading to removal of residues 317 to 366, a region termed the C-peptide15. Although the physiological relevance of C-peptide excision remains unclear, previous biochemical studies have demonstrated that cleavage in this domain leads to lower expression levels of intact receptor16. To simplify purification of intact receptors for structural studies, we generated a TSHR construct devoid of the C-peptide; removal of the C-peptide did not affect TSH or TSI activation of Gs signalling (Extended Data Fig. 1a,d,e). To further improve expression and purification of active TSHR, we generated a construct with a C-terminal fusion of an engineered miniGαs protein. This miniGαs protein contains only the RAS-like GTPase domain of Gαs and is thermostabilized to interact with a GPCR in a nucleotide-independent manner17. We purified this construct, designated TSHR–miniGs, in complex with native TSH purified from human pituitary, recombinantly expressed Gβ1γ2, and the Gs-stabilizing nanobody Nb35 (ref. 18). The resulting preparation of TSH-activated, Gs-bound TSHR was stable enough to enable single particle cryo-EM. Because of conformational heterogeneity caused by flexibility between the ECD of the TSHR and the seven-transmembrane-segment domain (7TM) bound to Gs, we separately classified and refined these two regions of the complex to yield a 3.4 Å resolution map of the TSHR ECD bound to TSH and a 2.9 Å map of the TSHR 7TM bound to the Gs heterotrimer (Extended Data Fig. 2 and Extended Data Table 1). Although these reconstructions enabled us to build a model for the TSHR 7TM domain and the Gs heterotrimer, key interacting regions between TSH and TSHR were of poor resolution (greater than 4 Å) (Extended Data Figs. 2 and 3).
Fig. 1|. Cryo-EM structures of native human TSH and TR1402 bound to active TSHR complexed with heterotrimeric Gs.

a,b, Cryo-EM maps of the TSHR–Gs–Nb35 complex bound to TSH (a) and TR1402 superagonist (b). TR1402 and TSH bind to the ECD of TSHR. c, Model of TR1402 bound to TSHR. Resolved N-linked glycans are highlighted for both TR1402 and TSHR. NAG, N-acetylglucosamine. d, The disulfide-linked α-helices of the TSHR hinge are outlined in black. d,e, TSHβ R34 and TR1402α R16 coordinate D382 and D386, respectively, in the TSHR hinge region (d), which positions Y385 into a hydrophobic pocket at the interface of hormone α and β chains (e). f, Sequences of TSH, FSH and the CG seatbelt loop regions between the 10th and 12th cysteine residues in the hormone-specific β chains. The seatbelt loop is further divided into regions I and II by a conserved aspartate (D94 in TSHR). Net charges in region I differ between the glycoprotein hormones. Region II of the seatbelt loop is conformationally divergent among the glycoprotein hormones. g,h, Close-up views of selectivity determinants in region I (g) and region II (h), as indicated in f. i, Cryo-EM density for the lipid dipalmitoylphosphatidylcholine (DPPC) in the TSHR transmembrane pocket. A644 and A647 side chains (highlighted in red) line the lipid binding pocket. j, cAMP production assay for mutations in the lipid binding site. Data are mean ± s.d. of triplicate measurements from a representative experiment of n = 3 biological replicates.
To gain higher resolution insights into how TSH interacts with the TSHR ECD, we determined a cryo-EM structure of active TSHR bound to TR1402, a high affinity TSH ‘superagonist’19,20. Glycoprotein hormones are composed of a common glycoprotein α chain (GPHα) and hormone-specific β chains. Whereas TSH and TR1402 share an identical TSHβ chain, four arginine substitutions in the TR1402 GPHα lead to approximately 55-fold improved potency for Gs activation compared with native TSH (Extended Data Fig. 1f). Probably owing to this improved affinity, our reconstruction of TR1402-bound TSHR–miniGs was resolved to 2.7 Å for the TSHR ECD bound to TR1402 and 2.4 Å for the 7TM bound to Gs (Fig. 1b and Extended Data Fig. 4). Three-dimensional variability analysis of the TR1402 reconstruction further supported flexibility between the TSHR ECD and the 7TM domain (Supplementary Video 1). Notably, the root mean squared deviation (RMSD) for the TSHR between TR1402 and TSH-bound states is 1.26 Å, suggesting that these ligands stabilize highly similar receptor conformations.
Our reconstructions of both TSH and TR1402 resolved key glycosylation sites on the hormone, including at Asn52 of the GPHα chain previously demonstrated to be required for glycoprotein hormone signalling10–12,21 (Fig. 1c and Extended Data Fig. 5). TSH and TR1402 bind to the concave surface of the TSHR ECD leucine-rich repeat (LRR) in a similar orientation as FSH bound to FSHR and CG bound to LH/CGR13,14,22 (Extended Data Fig. 6a). Our TR1402 reconstruction revealed that the hinge region of TSHR, which connects the TSHR ECD and the 7TM domain, is ordered as two disulfide-linked α-helices that make extensive contact with both hormone chains (Fig. 1d). One of these TSHR α-helices is coordinated by ionic interactions in both the TR1402α and TSHβ chains, whereas the other interacts with the α-L1 and α-L3 loops of the TR1402 α-chain, probably via hydrophobic interactions (Fig. 1d and Extended Data Fig. 6c). The TSHR hinge-region positions Y385, a residue previously identified as important in TSH-mediated signalling23, into a hydrophobic pocket at the hormone α/β chain interface (Fig. 1e). Sulfation of Y385 in TSHR, and a homologous tyrosine in FSHR and LH/CGR, has been proposed to be critical for glycoprotein hormone action13. However, our structure suggests that a sulfate group attached to Y385 would not be compatible with TSHR binding to the hormone. Consistent with this structural prediction and contrary to previous reports23,24, we found that Y385 mutations in the TSHR that preclude sulfation (Y385F and Y385A) retained similar TSH potencies and efficacies as wild-type TSHR (Extended Data Fig. 1g).
Selective binding of glycoprotein hormones to their cognate receptors is determined by the hormone-specific β chain25,26, and more specifically, has been localized to a 16-residue segment between the 10th and 12th cysteine in the β chain, termed the ‘seatbelt’ loop27–30. A key concept from previous studies examining glycoprotein hormone selectivity is the presence of negative determinants that preclude binding of incorrectly paired hormones and receptors31. Structural comparison of the seatbelt loop between the three different glycoprotein hormones bound to their respective receptors provides insight into such negative determinants (Fig. 1f). We define two distinct regions of the seatbelt loop on the basis of a conserved aspartate residue in the hormone β-chains that is known to be critical for receptor interaction (D94 in TSH, D99 in CG and D93 in FSH): an N-terminal region (region I) and a C-terminal region (region II). The net charge of region I is negative for TSHβ and FSHβ, and positive for CGβ32 (Fig. 1f). In TSH, D91 in region I contacts TSHR residue K209. For CG bound to LH/CGR, this ionic interaction is swapped, with CGβ R95 contacting LH/CGR E206 (Fig. 1g). These swapped ionic interactions are important in determining faithful hormone–receptor pairings, as underscored by previous studies that identified enhanced CG signalling at the K209E TSHR mutant33. Comparison of region II between TSH and FSH reveals another set of opposing ionic interactions that is probably important for hormone selectivity (Fig. 1h). In this region, TSHβ presents a negatively charged E98 to contact K58 in the TSHR. By contrast, FSHβ presents a positively charged R97 to contact E76 in FSHR (Fig. 1h). Although it is likely that additional interactions are responsible for encoding hormone selectivity, these opposing ionic interactions provide a structural rationale for negative determinants that limit incorrect hormone–receptor pairing.
Our reconstruction of TR1402-bound TSHR revealed density for an endogenous phospholipid buried within the 7TM domain (Fig. 1i). A combination of mass spectrometry experiments to identify potential lipids enriched in our TSHR–Gs samples and potential candidates compatible for modelling within the observed cryo-EM density led us to tentatively assign this density as DPPC (Extended Data Fig. 6d,e). To probe whether lipid binding at this site is important for TSHR activation, we generated cell-surface expression-matched TSHR cell lines with two mutations predicted to inhibit DPPC occupancy in the 7TM domain (A644K and A647K). Both mutations significantly decreased the efficacy and potency of TSH in cAMP production (Fig. 1j), suggesting that the presence of a lipid within the TSHR transmembrane domain is important for propagating ECD binding signals to the 7TM domain. Our hormone-bound structures of the TSHR thus offer insight into the molecular determinants of receptor–hormone interactions, hormone selectivity and the presence of a lipidic conduit important for receptor activation.
Conformational changes on TSHR activation
To understand how TSH activates the TSHR, we next determined an inactive-state structure of the receptor (Fig. 2a,b). Our attempts to capture the inactive state of the TSHR stabilized by the small molecule negative allosteric modulator Org 274179-0 (ref. 34) resulted in a low-resolution reconstruction resolving the TSHR ECD and a detergent micelle (Extended Data Fig. 7). We speculated that this result may be owing to orientational flexibility of the TSHR ECD compared with the 7TM domain associated with constitutive activity of the TSHR35. To further stabilize an inactive conformation, we used a Fab fragment of the inverse-agonist CS-17 antibody, which has been previously reported to suppress constitutive activity of the TSHR36. We obtained a reconstruction of the TSHR–Org 274179-0–CS-17 complex to a global resolution of 3.1 Å, with higher resolution features for the TSHR ECD and CS-17 Fab compared with the lower resolution reconstruction of the TSHR 7TM domain (Fig. 2a, Extended Data Fig. 8). Although this map enabled modelling into the TSHR ECD and CS-17 Fab densities, the resolution in the TSHR transmembrane region limited our ability to model side chains accurately or to define the binding site for Org 274179-0.
Fig. 2|. The activation mechanism of the TSHR revealed by the inactive structure bound to the inverse agonist CS-17.
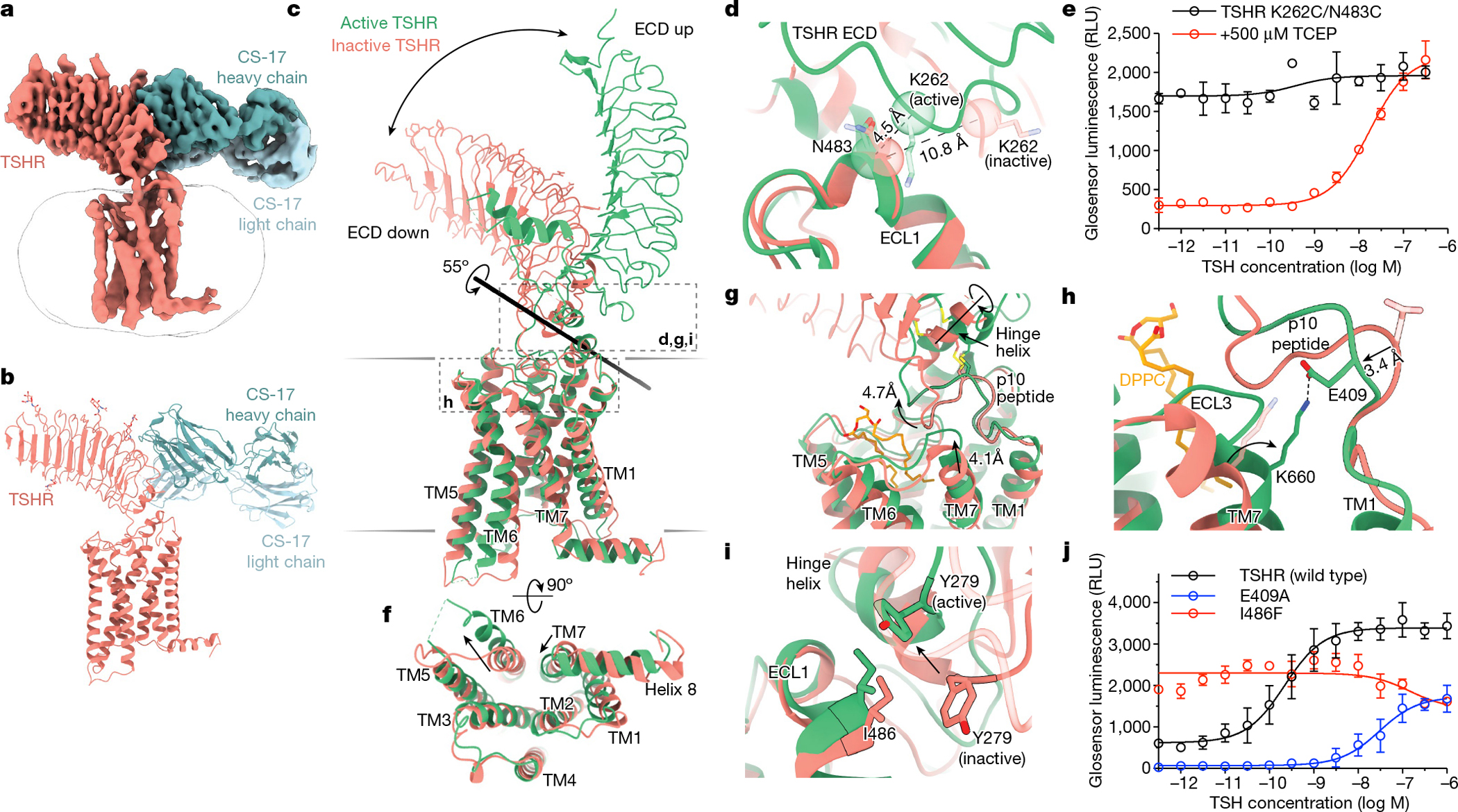
a,b, Cryo-EM map (a) and model (b) of inactive TSHR bound to the CS-17 Fab. c, Structural comparison of inactive and active TSHR with the 7TM domain aligned. In inactive TSHR, the ECD is in a down orientation close to the membrane bilayer. In active TSHR, the ECD is in an up orientation. The ECD rotates 55° along an axis, as calculated by Dyndom3D55. d, Disulfide trapping of the active TSHR ECD conformation using a K262C/N483C mutant TSHR. The Cα distance between positions 262 and 483 (indicated in parentheses) would only enable a disulfide bond when the ECD is in the active, up conformation but not in the inactive, down conformation. e, The K262C/N483C TSHR mutant is more constitutively active than wild-type TSHR (see Extended Data Fig. 1). Addition of 500 μM TCEP reduces basal activity of the K262C/N483C disulfide-locked construct. f, In the active state, TM6 of TSHR moves outward by 14 Å, and TM7 moves inward by 4 Å. g, Upon activation, rotation of the ECD leads to a rotation of the hinge helix, an extracellular displacement of the p10 peptide and an inward movement of the extracellular tip of TM7. h, E409 in the p10 peptide interacts with K660 in TM7 in active TSHR. Inactive-state side chains (transparent) are not resolved but the peptide backbone suggests that the E409–K660 interaction is not maintained. i, Y279 traverses approximately 6 Å across the ECL1–hinge helix interface directly over I486. j, Disruption of p10–TM7 interactions (E409A) and perturbation of the ECL1–hinge helix interface (I486F) affect TSH-mediated receptor activation and basal activity, respectively. Data are mean ± s.d. of triplicate measurements from a representative experiment of n = 3 biological replicates.
In the inactive state, the TSHR ECD rotates 55° towards the 7TM domain through an axis bisecting these two domains (Fig. 2c). We term this the ‘down’ state. By contrast, we label the ECD in the ‘up’ state as observed for active TSHR bound to TSH. Importantly, the internal leucine-rich-repeat structure of the ECD does not change between the inactive and active states of the TSHR (r.m.s.d. of 0.64 Å). The ECD therefore moves as a rigid body between the inactive and active states. The ECD orientation is similar between the low-resolution reconstruction of unbound, inactive TSHR and CS-17-bound, inactive TSHR (Extended Data Fig. 9), suggesting that CS-17 probably stabilizes an inactive, ECD down orientation to achieve its inverse-agonist efficacy. To test whether rigid-body motion of the TSHR ECD is sufficient to activate the receptor in the absence of other stimuli, we designed two mutations in TSHR (K262C and N483C) that would form a disulfide bond to trap the ECD in the up orientation (Fig. 2d). The K262C/N483C double mutant, but not the single cysteine substitutions, exhibited a 16-fold increase in constitutive activity compared to expression-matched wild-type TSHR (Fig. 2e and Extended Data Fig. 1b, c). Incubating cells with the disulfide reductant tris(2-carboxyethyl)phosphine (TCEP) markedly decreased constitutive activity of the K262C/N483C mutant and restored TSH-stimulated cAMP production. Rotation of the TSHR ECD to the up state is therefore sufficient to activate the receptor, even in the absence of extensive interactions between TSH and TSHR.
Comparison of these TSHR structures with previously determined structures of LH/CGR showed a similar relative orientation of the ECD in the inactive and active orientations (Extended Data Fig. 10). Given these similarities, we surmise that the activation-associated rigid-body motion of the ECD is a common feature of active-state glycoprotein hormone receptors. Additionally, the TSHR 7TM domain displays several classic hallmarks of GPCR activation. Compared with the inactive state, TM6 of active TSHR is displaced outward by 14 Å (measured from the Cα carbon of D617) to accommodate the α5 helix of miniGαs and TM7 moves about 4 Å inward relative to the transmembrane core of the receptor (measured from the Cα carbon of Y678) (Fig. 2f). These movements are similar to both the prototypical Gs-coupled family A receptor, the β2-adrenoceptor18 and to the LH/CGR14 (Extended Data Fig. 10).
A central question is how changes in the TSHR ECD orientation connect to activation of the TSHR 7TM domain. The C-terminal end of the TSHR ECD comprises two key regions of interaction with the 7TM domain—the hinge helix and the p10 peptide (Fig. 2g). The hinge helix at the base of the TSHR ECD contains two disulfide bonds that connect the last LRR β-sheet with the p10 peptide, which is a conserved 10-amino acid region connecting the ECD to transmembrane helix 1 (TM1). The p10 peptide has been proposed to be a tethered intramolecular agonist for glycoprotein hormone receptors37–39. Indeed, previous studies have found that mutation of residues within the p10 peptide are poorly tolerated in all glycoprotein hormone receptors40. We therefore aimed to understand how rotation of the ECD is coupled to conformational changes in the p10 peptide, and how this translates to activation of the TSHR 7TM. Comparison of active and inactive TSHR structures revealed that ECD transition into the up state results in rotation of the hinge helix, which makes extensive contacts with ECL1 in both active and inactive TSHR. Rotation of the hinge helix lifts the N-terminal portion of the p10 peptide around 5 Å away from the 7TM core (measured at the F409 Cα) (Fig. 2g). Upward movement of the p10 peptide produces a void into which the extracellular tip of TM7 shifts inward by approximately 4 Å (measured at the N658 Cα) (Fig. 2g). In active-state structures, we observed a well-resolved interaction between TSHR TM7 residue K660 and E409 in the p10 peptide (Fig. 2h). Although we do not resolve the E409 side chain in inactive TSHR, the peptide backbone for this residue is shifted 3 Å away from the active state, suggesting that the E409-K660 interaction stabilizes the fully active state of the TSHR. Our structures therefore reveal that TSHR-activating changes in p10 peptide couple to the 7TM domain via TM7.
We tested the importance of these interactions with structure-guided mutations. To test the relevance of the hinge helix–ECL1 interaction, we designed a substitution in ECL1 (I486F) that has been annotated as a driving mutation in thyroid follicular adenoma41. Our structures of the TSHR show that receptor activation is associated with a 6 Å movement of ECD residue Y279 relative to I486; replacing isoleucine with the bulkier phenylalanine would therefore be predicted to perturb TSHR activation (Fig. 2i). In signalling studies, I486F is constitutively active but has diminished sensitivity to TSH (Fig. 2j). A previous study demonstrated that the I486F mutant is not constitutively active if the TSHR ECD is truncated42, further highlighting that the contact between the ECD and ECL1 is a critical conduit for propagation of the ECD orientation to the 7TM domain. Although mutagenesis of the majority of the p10 peptide has been previously shown to severely compromise glycoprotein hormone receptor expression and sensitivity to glycoprotein hormones40, we tested the importance of p10 peptide–TM7 coupling by disrupting the E409–K660 interaction. We find that the E409A TSHR mutant shows diminished constitutive activity and reduced TSH potency and efficacy, supporting the importance of p10 peptide–TM7 contacts in TSHR activation (Fig. 2j). Our structural observations and mutagenesis studies, combined with extensive previous mutagenesis studies of the TSHR therefore demonstrate that rotation of the ECD is critical for receptor activation and highlight a conduit for this conformational change to impinge on the 7TM domain.
Autoantibody mimics TSH to activate TSHR
We next aimed to understand how agonistic autoantibodies activate the TSHR. Previous efforts to characterize TSI identified M22, a monoclonal antibody isolated from a patient with Graves’ disease that potently activates the TSHR43. A previous X-ray crystal structure of M22 bound to the ECD of the TSHR provided evidence of an epitope overlapping with the predicted binding site for TSH44. However, the mechanism for M22 mimicry of TSH action remains poorly understood45,46.
We prepared a complex of active TSHR–miniGs bound to the Fab fragment of the M22 autoantibody and analysed the structure and dynamics of this complex with cryo-EM. In 2D class averages, we identified two distinct orientations of the TSHR ECD bound to the M22 Fab (Fig. 3a). One class appeared consistent with a TSHR ECD orientation in the up state similar to that observed for TSH. In the other class, the TSH ECD–M22 Fab complex is distinct from either the up or down orientations of the TSHR ECD. Instead, the TSH ECD appears to embed within the side of the detergent micelle in a region that would normally occupy the lipid bilayer. Although this ‘side’ orientation of the TSHR ECD is not physiological and probably results from detergent solubilization of the TSHR, it suggests that the M22-bound TSHR ECD is more orientationally heterogeneous than when bound to glycosylated TSH. We were unable to determine a high-resolution reconstruction of this side complex. However, we successfully reconstructed two maps for the up state: the TSHR ECD bound to M22 Fab (at 3.0 Å resolution) and the TSHR 7TM domain bound to Gs (at 2.8 Å resolution) (Fig. 3b,c and Extended Data Fig. 11). In agreement with our TR1402-bound TSHR-Gs reconstruction, we also resolved a well-defined two-tailed lipid density in the transmembrane core of M22-bound TSHR, suggesting that this lipid is a common feature of activated TSHR (Fig. 3d).
Fig. 3|. Activation of the TSHR by a Graves’ disease autoantibody.
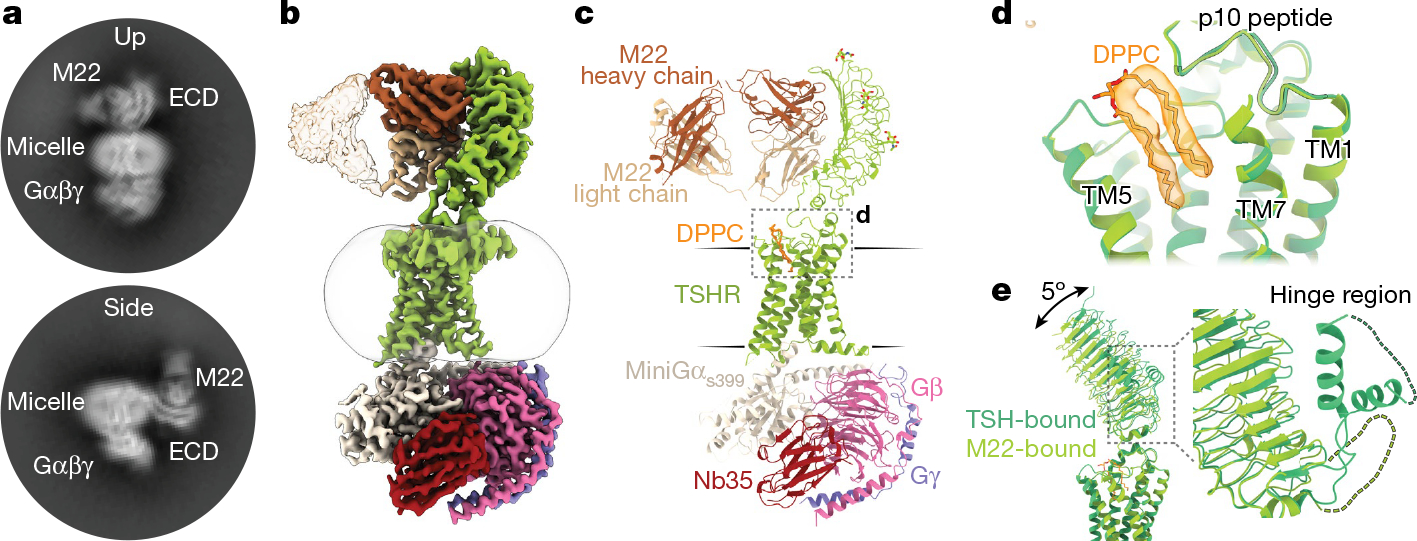
a, Selected 2D class averages of the up and side orientations of the TSHR ECD for TSHR bound to M22. b,c, Cryo-EM map (b) and model (c) of the M22 Fab–TSHR–Gs–Nb35 complex. d, DPPC (orange) is modelled into the density present in the TSHR transmembrane pocket. TM6 and ECL3 are hidden for clarity. e, Alignment of TSHR 7TM domain between TSH and M22-bound models reveals minimal (around 5°) change in the orientation of the ECD. The TSHR α-helical hinge region is not resolved in the M22-bound receptor complex.
We next compared TSH- and M22-activated TSHR (Fig. 3e). With M22 bound, the ECD up state is in an orientation that is highly similar to the TSH-activated state, with a small 5° rotation when the 7TM domains are aligned. The conformation of the 7TM domain and orientation of Gs heterotrimer are also highly similar between M22- and TSH-activated TSHR, with a r.m.s.d. of 0.87 Å. A key distinction between TSH-, TR1402- and M22-activated TSHR is that the hinge region is unresolved in M22-activated TSHR, which is consistent with previous studies showing that the hinge region is dispensable for activation of the TSHR24. The lack of a direct interaction with the hinge region is potentially responsible for the increased orientational flexibility of the TSHR ECD when bound to M22 as compared to TSH or TR1402. The structure of M22- and TSH-activated TSHR therefore demonstrate remarkable similarity in ECD orientation.
The membrane bilayer is key to TSHR activation
Our structures of the TSHR revealed that rotation of the ECD is coupled to 7TM domain activation by direct interactions between the p10 peptide and TM7. However, this does not address how binding of TSH and activating TSI lead to the up ECD orientation. From structures of CG-bound LH/CGR, it has been proposed that a clash between the CG-β chain and the membrane bilayer drives receptor activation by ‘pushing’ the LH/CGR ECD away from the membrane14. Simultaneously, an additional interaction between the hinge region and the common GPH-α chain ‘pulls’ the ECD to the up state. Whereas our structures of inactive and active TSHR revealed overall receptor conformation and ECD orientation changes similar to LH/CGR, we arrive at a distinct model for glycoprotein hormone action on the basis of our structures of TSH, the M22 antibody agonist and the CS-17 antibody inverse agonist.
To establish our model, we first used a computational approach to orient the active- and inactive-state TSHR structures within a membrane bilayer and to define hypothetical membrane bilayer boundaries47 (Fig. 4a and Supplementary Data 1). Although this static modelling approach is limited in completely describing the key receptor and membrane dynamics important in TSHR function, it provided a first order approximation to understand how TSH and antibodies exert their efficacy. Using this approach, we first modelled the binding poses of the agonist M22 and inverse-agonist CS-17 Fab fragments onto either the active (up) or inactive (down) ECD states. Modelling the M22 Fab bound to the inactive, down orientation of TSHR ECD revealed significant expected clashes between the antibody light chain and both the receptor 7TM domain and the outer membrane plane (Fig. 4b). These steric clashes suggest that M22 binding to TSHR is incompatible with the inactive TSHR. Conversely, modelling the CS-17 Fab bound to the active, up orientation of the TSHR ECD revealed clashes between the antibody constant domains and the outer membrane plane (Fig. 4c), suggesting that CS-17 binding is incompatible with the active state. Even with the limitations inherent in this modelling approach, we inferred that M22 and CS-17 exert their efficacy at TSHR by preventing the transition of ECD orientations between active and inactive states.
Fig. 4|. Membrane bilayer interactions are critical for TSHR activation.

a, Orientation of active and inactive TSHR in a mammalian plasma membrane bilayer as defined by the Orientations of Proteins in Membranes (OPM) server. b, Modelling of autoantibody agonist M22 binding to inactive TSHR with the ECD in the down orientation shows the expected clashes between the M22 light chain, the TSHR 7TM domain and the outer membrane bilayer. c, Modelling of binding of the inverse-agonist CS-17 onto active TSHR shows the expected clashes between the CS-17 constant domains and the membrane bilayer. d,e, In simulations with no antibodies present (grey traces), the TSHR ECD fluctuates between ECD up (active) and ECD down (inactive) orientations, regardless of the starting structure. M22 constrains the ECD to an ECD up orientation (brown trace), and CS-17 constrains the ECD to a down (inactive) orientation (blue trace). The ECD orientation is quantified using a projection metric as outlined in Methods. Thick traces represent smoothed values with an averaging window of 25 ns; thin traces represent unsmoothed values. Black dashed horizontal lines represent ECD orientations in cryo-EM structures. f, Modelling of TSH binding to inactive TSHR with the ECD in the down orientation. The membrane-proximal Asn52 glycan is highlighted. g, Modelling of K1-70 to inactive-state TSHR. K1-70 is compatible with binding to inactive-state TSHR. The location of an engineered membrane-proximal glycosylation site (K1-70 Q16N) and the distance to the OPM-determined static membrane plane are indicated. h, An engineered version of K1-70 with N-linked glycosylation at residue 16 (K1-70glyco) is more potent and efficacious at cAMP production than K1-70. Data are mean ± s.d. of triplicate measurements from a representative experiment of n = 3 biological replicates.
To determine whether M22 and CS-17 constrain the TSHR ECD orientation in a more realistic membrane environment, we performed all-atom molecular dynamics simulations of TSHR using both the M22-bound active structure and the CS-17-bound inactive structure as starting points. For each starting structure, we performed simulations with and without the respective antibody present. To quantify ECD orientation, we used a projection metric to define a reaction coordinate between the inactive and active ECD states from our cryo-EM models (see Methods). In simulations with the antibodies removed, the TSHR ECD fluctuates between active and inactive orientations, irrespective of the starting structure (Fig. 4d,e and Extended Data Fig. 12). By contrast, in simulations of TSHR bound to M22 or CS-17, the antibodies largely constrain the TSHR ECD to its initial state. With M22, the TSHR ECD is constrained to an active, up orientation. Conversely, CS-17 constrains the TSHR ECD to an inactive, down orientation. These simulations therefore support our static modelling, and indicate that M22 and CS-17 constrain an otherwise dynamic TSHR ECD to exert their efficacy.
We next turned to understanding how TSH activates the TSHR. We used our static modelling approach to examine how TSH would interact with the membrane bilayer in inactive TSHR with the ECD in the inactive orientation. Unlike a previous model proposed for CG activation at LH/CGR, the TSHβ chain is not predicted to clash with the membrane bilayer in inactive TSHR. Instead, glycosylation at Asn52 in the GPHα chain points directly orthogonal to the membrane bilayer (Fig. 4f). We propose that in the inactive state, the down orientation of the TSHR ECD would be unable to bind TSH owing to clashes between the Asn52 glycosylation and the membrane bilayer. Two lines of evidence support this proposal. First, our cryo-EM reconstruction of native human TSH resolves two monosaccharide moieties (Extended Data Fig. 5). At this site, a single additional glycan monosaccharide would be expected to induce a membrane clash analogous to those observed for M22 and CS-17. Typical biantennary N-linked glycans are composed of 5–12 monosaccharide units with an average size of around 9 Å per monosaccharide48. Mass spectrometry of native human TSH used to solve the cryo-EM structure identified Asn52 glycan chains with up to 16 monosaccharide units (Extended Data Fig. 5). The size of the glycosylation at Asn52 is therefore sufficiently large to be incompatible with TSH binding to inactive TSHR. Second, previous studies have demonstrated that glycosylation of TSH and CG is necessary for TSHR and LH/CGR activation, respectively. Deglycosylated TSH or CG bind to their respective receptors but function as competitive antagonists for the native hormone11,49–51. Together, these previous data and our modelling demonstrate that the conserved GPHα glycosylation at Asn52 drives TSHR activation by inhibiting the transition of the TSHR ECD to the inactive, down state.
To directly test whether glycosylation of an ECD-bound ligand is sufficient to promote full agonism at TSHR (in a manner analogous to the proposed model for TSH-mediated activation), we designed a gain-of-function experiment. Here we used an Fab fragment from the K1–70 antibody, which has previously been characterized as a TSHR neutral antagonist45. Contrary to previous reports45, we find that K1-70 is a weak partial agonist in a cAMP signalling assay (Fig. 4h). We aligned the previously solved X-ray crystal structure of K1-70 bound to the TSHR ECD with the inactive state of the TSHR from our cryo-EM structure (Fig. 4g). Notably, the TSHR ECD is highly similar in the K1-70 crystal structure and our inactive-state cryo-EM structure (r.m.s.d. 0.62 Å). This modelling suggests that, unlike M22, K1-70 is not predicted to clash with the membrane bilayer (Fig. 4g). The low efficacy agonism of K1-70 in the absence of a clear membrane interaction highlights limitations of our static modelling approach: it probably does not capture the full complement of TSHR ECD orientations or may not reflect subtle conformational changes associated with K1-70 efficacy. Using the K1-70 Fab as a starting point, we engineered a new Fab construct, K1-70glyco, with a predicted glycosylation motif in a loop that is the closest contact between K1-70 and the membrane bilayer (Fig. 4g). We predicted that the engineered glycan would mimic the effect of TSH glycosylation at Asn52 and clash with the membrane bilayer in the inactive down state of TSHR, thereby converting K1-70 from a weak partial agonist into a full agonist. We isolated a glycosylated preparation of K1-70glyco by concanavalin A affinity chromatography and confirmed its glycosylation status by native mass spectrometry (Extended Data Fig. 13a,b). Consistent with our model, K1-70glyco is significantly more efficacious at activating cAMP signalling at TSHR compared to either wild-type K1-70, a non-glycosylated control K1-70glyco(N16Q) (Fig. 4h), or a K1-70 construct with glycosylation introduced at another position distant from the membrane bilayer (Extended Data Fig. 13c,d). From these modelling exercises, molecular dynamics simulations and gain-of-function experiments, we conclude that steric interactions between ECD-interacting ligands and the membrane bilayer are a critical feature of TSHR activation.
Conclusion
We propose the following model for TSHR activation (Fig. 5). In the unliganded state, the TSHR ECD is structurally dynamic and can transition between the up and down orientations. Transient excursions to the ECD up state lead to basal signalling. TSH selects the ECD up state due to clashes between the GPHα Asn52 glycan and the membrane bilayer if the TSH ECD converts to the down state. Activation of the 7TM domain proceeds, in part, via conformational rearrangement of the p10 peptide, which is coupled to inward motion of TM7. An endogenous phospholipid also participates as a conduit for propagating activating signals from the ECD into the 7TM domain. Although interactions between the hinge region and TSH are required for potent hormone binding and signalling, they are not required for activation per se, as TSHR can be activated by the M22 autoantibody without similar contacts. Finally, orientational selection of the ECD down state by CS-17 leads to inverse-agonist activity. The orientation of the ECD relative to the membrane bilayer is therefore a critical determinant of TSHR activity.
Fig. 5|. Model for TSHR activity.
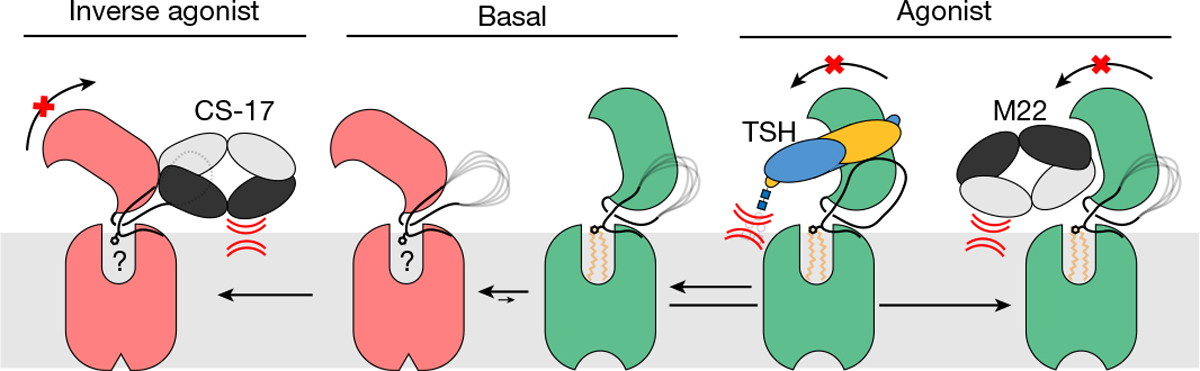
In the basal state, the TSHR ECD can spontaneously transition to the up state, leading to constitutive activity. TSH stabilizes an upright ECD because steric clashes between the GPHα N52 glycan and the membrane bilayer prevent conversion of the ECD to the down state. Agonistic autoantibodies such as M22 activate TSHR in a similar manner by preventing the ECD down state. Conversely, inverse agonistic antibodies such as CS-17 prevent the ECD from assuming the up state, thereby locking TSHR in an inactive orientation.
Our studies revise current models of glycoprotein hormone receptor activation. We demonstrate that sulfation of Tyr385 is not necessary for hormone-mediated receptor activation. On the basis of our structures of TSH- and TR1402-bound TSHR and associated modelling, we propose a structural rationale for the importance of GPHα Asn52 glycosylation. Indeed, glycosylation at the right position is sufficient to fully activate the TSHR, as illustrated by our experiments with K1-70, underscoring the importance of this post-translational modification in glycoprotein hormone action. Our structural studies with agonistic and inverse agonistic antibodies demonstrate that selection of ECD orientations is sufficient to activate or deactivate the receptor. Indeed, the lower constitutive activity of both LH/CGR and FSHR52,53 may be explained by decreased ECD dynamics in these receptors relative to TSHR. Finally, structures with TR1402 and M22 revealed a phospholipid embedded within the transmembrane domain of TSHR in a region that is overlapping with an inhibitory small molecule for LH/CGR. We therefore predict that a similar phospholipid may be a common feature that underlies glycoprotein hormone receptor activation.
The structural basis of how autoantibodies mimic the TSH hormone to activate the TSHR provides insight into the molecular pathophysiology of Graves’ disease, and more broadly highlights the unique way in which breaking of immune tolerance to self-antigens leads to aberrant GPCR signalling. For Graves’ disease in particular, our structure of the TSHR bound to M22 suggests that the progression of thyroid autoimmune diseases is largely dependent on the geometric binding behaviour of the TSHR ECD-interacting antibodies relative to the membrane bilayer. This provides some context for the presence of both TSH-blocking and TSH-agonistic autoantibodies in patients with autoimmune thyroid disease—although these antibodies probably share similar epitopes on the TSHR ECD, it is their orientation relative to the membrane bilayer that determines their efficacy. More broadly, our studies provide a structural perspective on how autoantibodies pathologically activate GPCRs, of which there are now dozens of examples in the literature54. Although the specific mechanisms for such pathological antibodies acting at various GPCRs are probably unique, recognition of GPCR extracellular domains and selection of distinct ECD orientations to mimic hormone function is likely to be a shared feature.
Our observations for the TSHR may also have broader implications for GPCR activation by other receptors containing large leucine-rich repeat extracellular domains such as LGR4, LGR5, LGR6 and the relaxin-family receptors. As the LRR domains in these receptors present static interfaces for ligand binding, we suspect that the rigid-body motions and membrane-dependent interactions described for the agonist-bound TSHR ECD are shared amongst these receptors in the transition to activated orientations. Together, our work establishes a key structural role for hormone glycosylation in receptor activation, introduces additional lipid-dependent factors for consideration in the development of small-molecules targeting the TSHR, and illuminates how both stimulatory and TSH-blocking autoantibodies can exist within a single patient with autoimmune thyroid diseases. Our structural insights therefore set the foundation for discovery of new therapies targeting the TSHR for diseases of thyroid homeostasis.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-022-05159-1.
Methods
Expression and purification of TSHR for active-state structures
The human TSHR gene with an N-terminal influenza hemagglutinin signal sequence and Flag (DYKDDDK) epitope tag was cloned into a custom pcDNA3.1 vector containing a tetracycline inducible cassette. To improve expression of intact receptor for structural studies, the sequence for the 50-residue hinge-region C-peptide (Ala317–Phe366) was removed by site-directed mutagenesis (New England Biolabs). The construct further included the miniGs399 protein17, which was fused to the C terminus of TSHR with a human rhinovirus 3C protease cleavage sequence flanked by three-residue Gly–Ser linkers. This construct (TSHR(ΔCpep)–miniGs399) was transfected into inducible Expi293F-TetR cells (unauthenticated and untested for mycoplasma contamination, Thermo Fisher) using the ExpiFectamine transfection reagent per manufacturer instructions. After 24 h, protein expression was induced with 2 μg ml−1 doxycycline hyclate, and the culture was placed in a 30 °C incubator for 36 h before collection by centrifugation. Pelleted cells were washed with 50 ml phosphate buffered saline, pH 7.5 before storage at −80 °C. For receptor purification, frozen cells were hypotonically lysed in 50 mM HEPES, pH 7.5, 1 mM EDTA, 160 μg ml−1 benzamidine, 2 μg ml−1 leupeptin for 10 min at 25 °C. The membrane fraction was collected by centrifugation, and the fusion protein was extracted with 50 mM HEPES, pH 7.5, 300 mM NaCl, 1% (w/v) glyco-diosgenin (GDN, Anatrace), 0.1% (w/v) cholesteryl hemisuccinate (CHS, Steraloids), 2 mM MgCl2, 2 mM CaCl2, 160 μg ml−1 benzamidine, 2 μg ml−1 leupeptin with dounce homogenization and incubation with stirring for one hour at 4 °C. The soluble fraction was separated from the insoluble fraction by centrifugation and was incubated in batch for 1 h at 4 °C with homemade M1–Flag antibody-conjugated Sepharose beads. Sepharose resin was then washed extensively with 50 mM HEPES, pH 7.5, 150 mM NaCl, 0.1% (w/v) GDN, 0.01% (w/v) CHS, 2 mM MgCl2, 2 mM CaCl2 and then with 50 mM HEPES, pH 7.5, 150 mM NaCl, 0.0075% (w/v) GDN, 0.00075% (w/v) CHS, 2 mM MgCl2, 2 mM CaCl2 prior to elution with 50 mM HEPES, pH 7.5, 150 mM NaCl, 0.0075% (w/v) GDN, 0.00075% (w/v) CHS, 5 mM EDTA, 0.2 mg ml−1 Flag peptide. After elution, TSHR(ΔCpep)–miniGs399 fusion protein was concentrated in a 100 kDa MWCO Amicon spin concentrator, and injected onto a Superdex200 Increase 10/300GL (Cytiva) gel filtration column equilibrated in 50 mM HEPES, pH 7.5, 150 mM NaCl, 0.005% (w/v) GDN, and 0.0005% CHS to isolate monodisperse material for further complexing with agonist, Gβ1γ2 heterodimer, and Nb35 (ref. 18).
Expression and purification of TSHR for inactive-state structures
A pcDNA3.1-containing TSHR(ΔCpep) construct was generated via site-directed mutagenesis. Expi293F-TetR cell transfection, doxycycline induction, and expression were performed as described for TSHR(ΔCpep)–miniGs399 with the additional supplement of dimethyl-sulfoxide solubilized 1 μM Org 274179-0 (ref. 34) post-induction. Purification of TSHR(ΔCpep) was performed as described for TSHR(ΔCpep)–miniGs399 with all buffers supplemented with 1 μM Org 274179-0.
Expression and purification of Gβ1γ2
Human Gβ1γ2 heterodimer was expressed in Trichoplusia ni Hi5 insect cells (unauthenticated and untested for mycoplasma contamination, Expression Systems) using a single baculovirus generated in Spodoptera frugiperda Sf9 insect cells (unauthenticated and untested for mycoplasma contamination; Expression Systems). A bicistronic pVLDual construct contained the Gβ1 subunit with a N-terminal 6× His tag, and an untagged human Gγ2 subunit. For expression, Hi5 insect cells were transduced with baculovirus at a density of ~3.0 × 106 cells per ml, grown with 27 °C shaking at 130 rpm. 48 h post-transduction, cells were collected and washed in a hypotonic buffer containing 20 mM HEPES, pH 8.0, 5 mM β-mercaptoethanol (β-ME), and protease inhibitors (20 μg ml−1 leupeptin, 160 μg ml−1 benzamidine). The membrane fraction was then separated by centrifugation and solubilized with 20 mM HEPES pH 8.0, 100 mM sodium chloride, 1.0% sodium cholate, 0.05% dodecylmaltoside (Anatrace), and 5 mM β-mercaptoethanol (β-ME). Solubilized Gβ1γ2 heterodimer was then incubated with HisPur Ni-NTA resin (Thermo Scientific) in batch. Bound Gβ1γ2 heterodimer was washed extensively and detergent was slowly exchanged to 0.1% (w/v) lauryl maltose neopentyl glycol (L-MNG, Anatrace) and 0.01% CHS before elution with 20 mM HEPES pH 7.5, 100 mM NaCl, 0.1% L-MNG, 0.01% CHS, 270 mM imidazole, 1 mM dithiothreitol (DTT), and protease inhibitors. Eluted Gβ1γ2 heterodimer was pooled and rhinovirus 3C protease was added to cleave the N-terminal 6× His tag during overnight dialysis in 20 mM HEPES pH 7.5, 100 mM NaCl, 0.02% L-MNG, 0.002% CHS, 1 mM DTT, and 10 mM imidazole. To remove uncleaved Gβ1γ2, dialysed material was incubated with HisPur Ni-NTA resin in batch. The unbound fraction was then incubated for 1 h at 4 °C with lambda phosphatase (New England Biolabs), calf intestinal phosphatase (New England Biolabs), and Antarctic phosphatase (New England Biolabs) for dephosphorylation. Final anion exchange chromatography was performed using a MonoQ 4.6/100 PE (Cytiva) column to purify only geranylgeranylated heterodimer. The resulting protein was pooled and dialysed overnight in 20 mM HEPES pH 7.5, 100 mM NaCl, 0.02% L-MNG, and 100 μM TCEP, and concentrated with a 3 kDa centrifugal concentrator to a final concentration of 162 μM. Glycerol was added to a final concentration of 20%, and the protein was flash frozen in liquid nitrogen and stored at −80 °C until further use.
Expression and purification of Nb35
A pET-26b vector containing the Nb35 sequence with a carboxy-terminal Protein C affinity tag (EDQVDPRLIDGK) was transformed into BL21 Rosetta Escherichia coli cells (UC Berkeley QB3 MacroLab) and inoculated into 8 l of Terrific Broth supplemented with 0.1% glucose, 2 mM MgCl2, and 50 μg ml−1 kanamycin. Cells were induced with 400 μM IPTG at A600 of 0.6 and allowed to express at 20 °C for 21 h. Collected cells were incubated SET Buffer (200 mM Tris pH 8.0, 500 mM sucrose, 0.5 mM EDTA) in the presence of protease inhibitors (20 μg ml−1 leupeptin, 160 μg ml−1 benzamidine) and benzonase. To initiate hypotonic lysis, two volumes of deionized water were added to the cell mixture after 30 min of SET buffer mixing. Following lysis, NaCl was added to 150 mM, CaCl2 was added to 2 mM, and MgCl2 was added to 2 mM and lysate was centrifuged to remove the insoluble fraction. Supernatant was incubated with homemade anti-Protein C antibody-coupled Sepharose. Nb35 was eluted with 20 mM HEPES pH 7.5, 100 mM NaCl, and 2 mM CaCl2, 0.2 mg ml−1 protein C-peptide, and 5 mM EDTA pH 8.0, concentrated in a 10 kDa MWCO Amicon filter and injected over a Superdex S75 Increase 10/300 GL column (Cytiva) size-exclusion chromatography column equilibrated in 20 mM HEPES pH 7.5, 100 mM NaCl. Monodisperse Nb35 fractions were pooled, concentrated, and supplemented with 20% glycerol prior to flash freezing in liquid nitrogen for storage at −80 °C until further use.
Expression and purification of M22 agonist Fab
The M22 heavy and light chains56 were cloned into pFastBac with a GP67 signal peptide and N-terminal Flag epitope and a P2A self-cleaving peptide sequence between the heavy and light chains. The construct also included a C-terminal 8× Histidine epitope tag on the heavy chain. Baculvorius was generated using the Bac-to-bac method (Thermo Fisher), and used to transduce Hi5 insect cells at a density of 2.0 × 106 cells per ml. Insect cells were cultured with shaking at 120 RPM at 25 °C for 60 h. M22 Fab was purified from the cell supernatant by first adjusting pH to 8.0 with 1 M Tris prior to a 1 h incubation with 5 mM CaCl2, 5 mM MgCl2, and 1 mM NiCl2 to precipitate chelators. After separating the insoluble fraction, the 8× His-tagged M22 Fab was captured on HisPur Ni-NTA resin, washed in a buffer comprised of 50 mM HEPES, pH 7.5, 500 mM NaCl, 20 mM imidazole, pH 7.5 and eluted with the same buffer supplemented with 500 mM imidazole. The eluate was further purified by size-exclusion chromatography over a Superdex200 Increase 10/300 GL gel filtration column equilibrated in 50 mM HEPES, 100 mM NaCl, pH 7.5. Monodisperse M22 Fab fractions were pooled, concentrated, and supplemented with 20% glycerol prior to flash freezing in liquid nitrogen for storage at −80 °C until further use.
Expression and purification of K1-70, K1-70glyco and K1-70glyco(N16Q) Fabs
Heavy and light chain sequences for K1-70 IgG46 were cloned into a pcDNA3.1 vector containing human IgG1 constant regions. Resulting constructs were transfected into Expi293F-TetR cells using a 2:1 mass ratio of heavy to light chain using the Expifectamine transfection kit, per the manufacturer’s instructions. After 5 days, cultures were collected and the supernatant purified over a 1 ml MabSelect SuRe HiTrap column (Cytiva) equilibrated in 100 mM sodium phosphate, 150 mM sodium chloride, pH 7.2. Monoclonal antibodies were eluted with 100 mM glycine, pH 3.0, neutralized with 1.5 M Tris pH 8.0, and dialysed into 20 mM sodium phosphate, 10 mM EDTA, pH 7.0 for generation of Fab fragments. Immobilized papain agarose was equilibrated in the IgG dialysis buffer with freshly added 20 mM cysteine-HCl, pH adjusted to 7.0. IgG was concentrated to ~40 mg ml−1 with a 100 kDa MWCO Amicon filter, diluted 1:1 with the IgG dialysis buffer plus cysteine-HCl, and added to the equilibrated papain agarose. The IgG-papain suspension was placed on a shaker at 37 °C for overnight digestion. After digestion, Fc fragments were removed via 1 h, room temperature batch incubation with phosphate buffered saline-equilibrated Protein A agarose (Pierce). Fab fragments were concentrated in a 10 kDa MWCO Amicon filter and injected onto a Superdex200 Increase 10/300 GL gel filtration column equilibrated in 50 mM HEPES, 100 mM NaCl, pH 7.5. Monodisperse Fab fractions were pooled, and digestion and Fc removal was confirmed by SDS–PAGE.
To produce a glycosylated version of K1-70 (K1-70glyco), a glycosylation motif was introduced at position 16 in the heavy chain in a loop region that connects the A and B strands of the VH IgG domain. The sequence KKPGQS was replaced with KKPGNGS to generate an N-linked N-x-S/T glycosylation motif. As a control for glycosylation, we also generated a version of K1-70glyco with a N16Q mutation K1-70glyco(N16Q). The resulting constructs were then purified as full-length IgG as described above for K1-70. Despite introduction of the glycosylation motif in K1-70glyco, the extent of glycosylation was not complete. To enrich for the glycosylated fraction of K1-70glyco, monodisperse K1-70glyco Fab was brought to 1 mM Mn2+ and Ca2+ and loaded over a Concanavalin A-conjugated Sepharose 4B packed HiTrap column (Cytiva) pre-equilibrated in 20 mM HEPES, 500 mM NaCl, 1 mM Mn2+, 1 mM Ca2+, pH 7.4. The glycosylated fraction K1-70glyco was eluted with 20 mM HEPES, 500 mM NaCl, 500 mM methyl α-d-mannopyranoside, pH 7.4, fractions were concentrated to 100 μM and flash frozen in liquid nitrogen for storage at −80 °C until further use.
Expression and purification of CS-17 Fab
Heavy and light chain sequences of CS-17 were determined from sequencing of CS-17 murine hybridoma cell line PTA-8174 (unauthenticated and untested for mycoplasma contamination, American Type Culture Collection) by Genscript. The resulting sequences were cloned into a pcDNA3.4-containing mouse IgG2a construct. Heavy and light chain constructs were transfected, expressed and purified as previously described for K1-70 IgG. CS-17 Fab generation also followed identical steps to K1-70 Fab generation.
Expression and purification of TR1402
The human TSH analogue TR1402 was produced at Trophogen (Rockville), following the procedures for site-directed mutagenesis and expression of recombinant hormones as described previously19,20. In brief, TR1402 was produced in stably transfected Chinese hamster ovary CHO-DG44-TR1402 cells (unauthenticated and tested negative for mycoplasma contamination, American Type Culture Collection, ATCC) and purified by a combination of dye, ion exchange, and gel filtration HPLC.
Preparation of inactive and active-state TSHR complexes
To prepare the TSH, TR1402, and M22-activated TSHR complexes, purified TSHR(ΔCpep)–miniGs399 was incubated with a 2-fold molar excess of purified Gβ1γ2, Nb35, and either native human TSH (National Hormone and Pituitary Program, resuspended from lyophilized power in 50 mM HEPES, pH 7.4 150 mM NaCl), human TSH analogue TR1402 (resuspended from lyophilized power in 50 mM HEPES, pH 7.4 150 mM NaCl), or M22 Fab and incubated overnight at 4 °C. After incubation, the complexed material was purified with anti-protein C antibody Sepharose resin to purify Nb35-bound complex. Protein C Sepharose was washed with 20 column volumes of 50 mM HEPES, 150 mM NaCl, 0.0075% GDN (w/v), 2 mM CaCl2 before elution with 50 mM HEPES, 150 mM NaCl, 0.005% GDN (w/v), 5 mM EDTA, 0.2 mg ml−1 protein C-peptide. The eluted fractions were concentrated with a 100 kDa MWCO Amicon filter, and injected onto a Superdex200 Increase 10/300 GL gel filtration column equilibrated in 50 mM HEPES, 150 mM NaCl, 0.005% GDN (w/v), pH 7.5. Monodisperse fractions were concentrated with a 100 kDa MWCO Amicon filter immediately prior to cryo-EM grid preparation.
For formation of the inactive-state CS-17-bound TSHR(ΔCpep) complex, the CS-17 Fab was incubated at a 2× molar excess with SEC-purified TSHR(ΔCpep) overnight at 4 °C, then concentrated in a 100 kDa MWCO Amicon filter prior to cryo-EM grid preparation prior to size-exclusion chromatography over a Superdex200 Increase 10/300 GL gel filtration column equilibrated in 50 mM HEPES, 150 mM NaCl, 0.005% GDN (w/v), pH 7.5 and 1 μM Org 274179-0. Monodisperse fractions of the resulting complex were concentrated with a 50 kDa MWCO concentrator immediately prior to cryo-EM grid preparation.
Cryo-EM vitrification, data collection and processing
TSH-bound TSHR–Gs complex.
The TSH-bound TSHR–Gs complex was concentrated to 23 μM and 3 μl was applied onto a glow-discharged 300 mesh 1.2/1.3 gold grid covered in a holey gold film (UltrAufoil). After a 30 s hold at 4 °C, excess sample was removed with a blotting time of 3 s and a blotting force of 0 prior to plunge freezing into liquid ethane using a Vitrobot Mark IV (Thermo Fisher). A total of 15,346 super-resolution movies were recorded with a K3 detector (Gatan) on a Titan Krios (Thermo Fisher) microscope operated at 300 keV with a BioQuantum post-column energy filter set to a zero-loss energy selection slit width set of 10 eV. The 66-frame movies were recorded for 2 s at a nominal magnification of 130,000× (physical pixel size of 0.664 Å per pixel) and a defocus range of −0.8 to −2.2 μm for a total dose of 77 e− Å−2. Exposure areas were acquired with automated scripts in a 3 × 3 image shift collection strategy using SerialEM 3.8 (ref. 57).
Super-resolution movies of the TSH–TSHR–Gs complex were motion-corrected, binned to physical pixel size, and dose-fractionated on-the-fly during data collection using UCSF MotionCor2 (ref. 58). Corrected micrographs were imported into cryoSPARC v3.1 (ref. 59) for CTF estimation via the Patch CTF Estimation job type. Templates for particle picking were generated from projections of the same TSHR complex reconstructed from a previous 200 keV imaging session. Particle picking templates were low-pass filtered to 20 Å and used to pick 9,151,778 particles. After picking, estimated CTF fit resolution >5 Å and relative ice thickness outlier measurements were used to remove low-quality micrographs before further processing. 6,185,950 curated particles were extracted in a 512-pixel box and Fourier cropped to 128 pixels before undergoing a round of 3D classification with alignment utilizing one 20 Å low-pass filtered reconstruction and three ‘random’ reconstructions generated from a prematurely truncated ab initio reconstruction job. 2,474,380 particles classified into the TSHR class were extracted in a 512-pixel box and Fourier cropped to 256 pixels for two additional rounds of 3D classification with alignment, utilizing the same class distributions as previously described. From these 2 rounds of 3D classification, and because the TSHR ECD remained poorly resolved, 734,891 particles belonging to the TSHR class were extracted into a 512-pixel box for a new 3D classification workflow. First, these particles were subject to one round of non-uniform refinement60 in cryoSPARC and then classified using two different masking schemes (TSHR 7TM domain or TSHR ECD–TSH complex) using alignment-free 3D classification in RELION v3.1.2 (ref. 61). Particles in qualitatively good-looking classes were re-imported into cryoSPARC for focused refinements. Particles from the TSHR 7TM domain-masked 3D classification were subject to a round of focused refinement with a mask encompassing the TSHR 7TM domain and G protein–Nb35 complex. Particles from the TSHR ECD–TSH complex masked 3D classification were subject to two rounds of focused refinement using the same mask as in RELION, decreasing mask pixel dilation between rounds. For each focused refinement, pose/shift gaussian priors (7° standard deviation of prior (rotation), 4 Å standard deviation of prior (shifts)) were used to limit large deviations from the initially determined poses. Directional FSC curves for each final reconstruction were calculated, and each map was loaded into ChimeraX v1.362 for generation of a composite map via the vop maximum command.
TR1402-bound TSHR–Gs complex.
The human TSH analogue TR1402-bound TSHR–Gs complex was concentrated to 11.6 μM and 3 μL was applied onto a glow-discharged 300 mesh 1.2/1.3 gold grid covered in a holey gold film and plunge frozen into liquid ethane with identical blotting conditions as with the TSH complex. 14,277 super-resolution movies were recorded with a K3 detector on a 300 kV FEI Titan Krios microscope located at the HHMI Janelia Research Campus equipped with a spherical aberration corrector and a post-column BioQuantum energy filter set to a zero-loss energy selection slit width set of 20 eV. Sixty-frame movies were recorded across a defocus range of −0.8 to −2.2 μm at a nominal magnification of 105,000× (physical pixel size of 0.831 Å per pixel) for 4.4 s, resulting in a total exposure dose of 60 e− Å−2.
A total of 14,276 super-resolution movies were motion-corrected, binned to physical pixel size and dose-weighted, post acquisition, using UCSF MotionCor2. Contrast transfer function estimation was performed with the cryoSPARC Patch CTF Estimation job before template-assisted, automated particle picking. Templates were generated from projections of the final native human TSH-bound TSHR–Gs complex. Micrographs for additional data processing were curated as previously described, and the subsequent 7,904,651 particles were extracted in a 416-pixel box and Fourier cropped to 104 pixels. To generate initial models for 3D classification, 200,000 particles were used to generate 3 ab initio reconstructions. The class containing a reconstruction resembling a TR1402-bound complex was used along with three additional reconstructions generated from a truncated ab initio reconstruction job, as previously described. The picked, curated particles were then subject to 2 rounds of 3D classification without alignment. Particles classified into the TR1402-bound class at the end of these 2 rounds were extracted in a 416-pixel box and Fourier cropped to 208 pixels. Particles then underwent one round of 3D classification with alignment, using four TR1402-bound TSHR-Gs complex reconstructions as initial models. The 375,930 particles in the highest resolution reconstruction class were then extracted in a 416-pixel box without additional Fourier cropping. One round of non-uniform refinement was performed to assign accurate initial angles for subsequent alignment-free classification jobs. At this point, based on the native human TSH processing particles results, these particles were split into separate ECD and transmembrane domain–G protein refinement pathways. For the ECD refinement approach, using a mask encompassing only the ECD, hinge region and TR1402 density, particles were locally refined with the same pose/shift gaussian priors as in the native human TSH processing. Then, particle subtraction using a mask of the transmembrane domain–G protein complex was performed. These subtracted particles were subject to a final round of local refinement using the same parameters as previously described.
For the transmembrane domain–G protein classification/refinement workflow, particles from the non-uniform refinement job were imported into RELION and subject to 3D classification without alignment using a mask encompassing only the transmembrane domain density. The 259,702 particles belonging to the highest resolution class from this alignment-free classification run were re-imported into CryoSPARC and subject to another round of non-uniform refinement. A final round of local refinement using a mask encompassing the transmembrane domain–G protein complex was performed, and the subsequent reconstruction was used for generating a composite map with the ECD-TR1402 reconstruction in ChimeraX v1.3 using the vop maximum command. Directional FSC curves were calculated for each independent reconstruction.
M22-bound TSHR–Gs complex.
The M22-bound TSHR-Gs complex was concentrated to 13.8 μM and 3 μl was applied onto a glow-discharged 300 mesh 1.2/1.3 gold grid covered in a holey gold film and plunge frozen into liquid ethane utilizing identical blotting procedures as with the TSH complex. 25,030 super-resolution movies were recorded with the same 300 kV microscope and detector used in the TR1402-bound TSHR–Gs complex collection. Sixty-frame movies were recorded across a defocus range of −0.8 to − 2.0 μm at a nominal magnification of 81,000× (physical pixel size of 0.844 Å per pixel) for 4.4s, resulting in a total exposure dose of 60 e− Å−2.
A total of 25,030 super-resolution movies were motion-corrected, binned to physical pixel size and dose-weighted, post acquisition, using UCSF MotionCor2. Contrast transfer function estimation was performed with the cryoSPARC Patch CTF Estimation job before automated particle picking with a gaussian template. After curating for high-quality micrographs as previously described, 9,078,725 particles were extracted in a 512-pixel box and Fourier cropped to 128 pixels. Due to the size of the dataset, image processing was performed in three cohorts. In the first cohort, 2,978,754 particles were subjected to two rounds of 2D classification followed by ab initio reconstruction of selected particles. Distinct conformations of the TSHR ECD in up and side states were apparent from 2D classification. Particles were selected for one round of 3D classification without alignment irrespective of their ECD conformation, then split into a second separate round 3D classification in up or side classes only. Particles from the best looking and highest resolution side class were extracted in an unbinned 512-pixel box and subject to non-uniform refinement. The resulting reconstruction was of low quality, with uninterpretable 7TM domains that contoured at thresholds similar to the detergent micelle. As a result, particle images belonging to side class were not further processed. Particles classified into a high-quality ECD up class were extracted into a 512-pixel box and subject to non-uniform refinement. The remaining 16,159 micrographs were processed in a similar workflow of multiple rounds of 2D classification followed by 3D classification with alignment. Unbinned particles from each cohort in qualitatively good ECD up classes were subject to independent non-uniform refinement jobs. The three refined sets of particles were then combined and subject to unmasked, alignment-free 3D classification in RELION. All classes exhibited high-quality 7TM domain features. Thus, particles in classes containing distinct ECD–M22 VH/VL density were selected, combined, and re-imported into cryoSPARC for 1 round of focused refinement using masked regions separately encompassing the TSHR ECD–M22 domain or the TSHR transmembrane–G protein complex. Directional FSC curves for each focused refinement reconstruction were calculated, and each map was loaded into ChimeraX for generation of a composite map via the vop maximum command.
Org 274179-0-bound TSHR complex.
The Org 274179-0- bound TSHR(ΔCpep) complex was concentrated to 55 μM and 3 μl was plunge frozen on holey gold grids as previously described for other TSHR samples after a 30 s hold and a 3 s blotting time at a blotting force of 0. Cryo-EM data were collected on a 300 keV Titan Krios microscope equipped with a K3 detector and a BioQuantum post-column energy filter with a slit width of 20 eV. 10,003 super-resolution movies were recorded at a nominal magnification of 105,000x (physical pixel size of 0.86 Å/pixel) for 1.35 s each and fractionated across 50 frames. The defocus range for this collection was −0.8 to −2.4 μm.
10,003 TSHR(ΔCpep)–Org 274179-0 movies were motion-corrected, binned to physical pixel size and dose-weighted, post acquisition, using UCSF MotionCor2. CTF estimation and reference-free ‘blob’ particle autopicking were performed in cryoSPARC. Micrographs were curated following similar thresholds as previously described prior to extraction of 5,922,556 particles in a 288-pixel box that was subsequently Fourier cropped to 72 pixels. To generate a suitable reference for 3D classification, 2 rounds of 2D classification were performed. From 2D classification, the particles belonging to classes that exhibited ‘micelle-like’ density were then subjected for ab initio 3D reconstruction. Then, 3D classification with alignment was performed on the entire set of initially picked particles using one class suggestive of a micelle–ECD reconstruction and three classes generated from prematurely truncated ab initio jobs, as previously described. Particles classified into the micelle–ECD class were subject to two further rounds of the same classification workflow prior to being extracted without Fourier cropping. Finally, the 357,869 remaining particles were subject to non-uniform refinement. The reconstruction quality of the TSHR–Org 274179-0 complex was poor, and was not improved upon further rounds of unbinned 3D classification with, or without, alignment. 2D classification of the particles in the initial non-uniform refinement job revealed clear density for the ECD with more diffuse alignment on the 7TM domain.
CS-17-bound TSHR complex.
3 μL of purified CS-17-bound TSHR(ΔCpep)–Org 274179-0 complex was concentrated to 40.8 μM and similarly plunge frozen into liquid ethane after a 30 s hold, a blotting time of 2 s, and a blotting force of 0. The TSHR(ΔCpep)–Org 274179-0 complex at 55 μM was plunge frozen on holey gold grids (1.2/1.3 UltrAufoil 300 Mesh) with blotting parameters as previously described for other TSHR samples after a 30 s hold and a 3 s blotting time at a blotting force of 0. For data collection, 9,244 super-resolution movies were recorded with identical parameters on the same Titan Krios microscope as described for the TSHR–Org 274179-0 complex.
For data processing, super-resolution movies of the TSHR(ΔCpep)–CS-17 Fab fragment complex were motion-corrected, dose-weighted, and binned on-the-fly with MotionCor2 during data collection, as previously described. CTF estimation, blob particle autopicking, and Fourier cropped particle extraction were all performed in cryoSPARC, after curating for high-quality micrographs. 2,742,080 picked particles were subject to 4 rounds of 2D classification followed by ab initio reconstruction to generate reference maps for 3D classification. Then, all picked particles were subject to 4 rounds of 3D classification with alignment, two rounds at a 4×-Fourier cropped box size of 112 pixels, and two rounds at a 2×-Fourier cropped box size. A total of 114,707 particles were then subject to 3D classification with alignment using four identical input reference volumes. The shifts and poses of the 41,054 particles belonging to the highest resolution reconstruction from this final 3D classification were then refined using the non-uniform refinement job type. In an attempt to improve reconstruction quality in the TSHR(ΔCpep)–CS-17 Fab binding interface, two rounds of focused refinement were performed. The first focused refinement utilized a mask encompassing only the ECD and CS-17 variable domains and was followed by a refinement with the entire TSHR(ΔCpep)–CS-17 complex masked. Both focused refinements used the same rotation/shift prior restrictions as in the TSHR–Gs processing workflow. However, in comparison to the initial non-uniform refinement reconstruction, the focused refinements yielded lower quality 7TM domain reconstructions. As a result, the pre-focused refinement reconstruction half-maps were used for directional FSC calculation and subsequent atomic model building.
Model building and refinement
We first modelled the M22-bound TSHR-Gs complex in a composite cryo-EM map (Extended Data Fig. 10). For TSHR, we started with an AlphaFold63 model of full-length human TSHR, which had high structural agreement with a previously determined X-ray crystal structure of the M22 Fab bound to the TSHR ECD (PDB ID: 3G04 (ref. 44)). After truncating unresolved regions, the 7TM domain in this model was fit into the composite cryo-EM map of M22-bound TSHR-Gs with ChimeraX v1.3 (ref. 62). This template model was rebuilt in Coot64 for rigid-body fitting of the ECD. For the M22 Fab, we used the PDB ID: 3G04 as a starting structure. For the G protein (miniGs, Gβ and Gγ) we used PDB ID: 7LJC65 as a starting template. Finally, for Nb35, we used PDB ID: 3SN6 (ref. 18) as a starting template. For DPPC, restraints were generated using the Prodrg server66 and the aliphatic tails of the lipid were manually docked into cryo-EM density using Coot. Each of these components was individually fit into cryo-EM density with ChimeraX v1.2.5. We subsequently iteratively refined the model with manual refinement in Coot and ISOLDE 1.0b3 (ref. 67) and real space refinement in Phenix68.
To model TSH- and TR1402-bound TSHR, we started with the M22-activated structure described above and fit it into a composite map of the TSH–TSHR–Gs complex using ChimeraX (Extended Data Fig. 2). The fit of the TSHR ECD was further optimized by rigid-body fitting in Coot and Phenix. A key distinction between the M22- and TSH or TR1402- activated structures of TSHR is in the hinge region. For TSH- and TR1402- activated TSHR, we were able to resolve residues 291–302 and 387–396 in the hinge domain. We first built a model for TR1402 bound to the TSHR ECD using AlphaFold2, which revealed a partially ordered hinge region consistent with our cryo-EM structure. This model was fit into the TR1402–TSHR–Gs complex composite map in ChimeraX, and then iteratively refined with manual changes in Coot and ISOLDE and real space refinement in Phenix. As the TSH-bound TSHR complex was of lower resolution in the extracellular region, we used the TR1402 structure as a starting point and iteratively refined TSHR ECD and TSH using the composite map of the TSH–TSHR–Gs complex.
For the CS-17-Org 274179-0-bound TSHR complex, we started with AlphaFold2 predictions for the TSHR and for the CS-17 Fab which were fit into the cryo-EM map with ChimeraX.The TSHR ECD was fit into the cryo-EM density by rigid-body refinement using Coot. Similarly, the constant regions of the CS-17 Fab were fit into the cryo-EM density by rigid-body refinement in Coot. The resulting model was refined in ISOLDE with manual changes in both ISOLDE and Coot followed by real space refinement in Phenix. All maps and models were validated using MolProbity69.
Modelling TSH, M22 and K1-70 binding poses on inactive-state TSHR (as in Fig. 4) was performed in ChimeraX with the matchmaker command to align the active-state ECD residues C29-F405 to the inactive-state structure as a reference model. The same operation was performed for modelling CS-17 onto the active-state TSHR ECD. Structure coordinate files for these models are available in the Supplementary Information.
Identification of lipid in M22-bound TSHR–Gs
Lipids were extracted using a modified version of the Bligh–Dyer method70. In brief, samples of PC(16:0/16:0) (DPPC) for the standard curve were manually shaken for 30 s in a glass vial (VWR) with 1 ml PBS, 1 ml methanol and 2 ml chloroform containing the internal standard Cer(d18:1-d7/18:1). The resulting mixtures were vortexed for 15 s and centrifuged at 2,400g for 6 min to induce phase separation. The organic (bottom) layer was retrieved using a glass pipette, dried under a gentle stream of nitrogen, and reconstituted in 2:1 chloroform:methanol for LC/MS analysis. Targeted lipidomic analysis was performed on a Dionex Ultimate 3000 LC system (Thermo) coupled to a TSQ Quantiva mass spectrometer (Thermo). Data was acquired in positive ionization mode. Solvent A consisted of 95:5 water:methanol, solvent B was 70:25:5 isopropanol:methanol:water. Solvents A and B contained 5 mM ammonium formate with 0.1% formic acid. A XBridge (Waters) C8 column (5 μm, 4.6 mm × 50 mm) was used. The gradient was held at 0% B between 0 and 5 min, raised to 20% B at 5.1 min, increased linearly from 20% to 100% B between 5.1 and 35 min, held at 100% between 35 and 40 min, returned to 0% B at 40.1 min, and held at 0% B until 50 min. Flow rate was 0.1 ml min−1 from 0 to 5 min, 0.3 ml min−1 from 5.1 to 50 min. Mass spectrometry analyses were performed using electrospray ionization in positive ion mode, with spay voltages of 3.5 kV, ion transfer tube temperature of 325 °C, and vaporizer temperature of 200 °C. Sheath, auxiliary, and sweep gases were 40, 10 and 1, respectively. Internal standard Cer(d18:1-d7/18:1) was detected using the following transitions: 571.6>271.2, 571.6>289.3, 571.6>259.3. DPPC was detected using the following transition: 734.6>184.1, 734.6>125.1, 734.6>86.1. Other PCs were detected using the specific parent ion m/z and the same diagnostic fragments as DPPC. Chromatography and peak integration of lipid targets were verified with Skyline71. Peak areas were used in data reporting, data was normalized using internal standards.
Native mass spectrometry of K1-70 fab fragments
Purified Fab fragments were prepared for native mass spectrometry by buffer exchange into 200 mM ammonium acetate with a Zeba spin desalting column. Samples, normalized to 5 μM, were injected directly in a Q Exactive Extended Mass Range mass spectrometer using nanoES electrospray capillaries. The instrument parameters were: capillary voltage 1.1 kV; S-lens RF 100%; quadrupole selection 300–10,000 m/z, collisional activation in the HCD cell 150 °C, trapping gas pressure setting 8; temperature 250 °C, instrument resolution 35,000. Data were analysed with UniDec 4.2.2 (ref. 72).
Mass spectrometry of human TSH
Native human TSH (National Hormone and Pituitary Program) was resuspended in water, reduced with 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Sigma-Aldrich) and alkylated with 10 mM chloroacetamide (Sigma-Aldrich). Protein was digested at 37 °C in 100 mM Triethylammonium bicarbonate buffer, pH 8.5, with trypsin (Promega) for 1 h. Digestion was quenched with formic acid, 5% (v/v) final concentration.
The digested TSH was analysed on a Orbitrap Eclipse mass spectrometer (Thermo). The digest was injected directly onto a 25 cm, 100 μm ID column packed with BEH 1.7 um C18 resin (Waters). The sample was separated at a flow rate of 300 nl min−1 on an Easy nLC 1200 (Thermo). Buffer A and B were 0.1% formic acid in water and 0.1% formic acid in 90% acetonitrile, respectively. A gradient of 1–25% B over 100 min, an increase to 40% B over 20 min, an increase to 90% B over 10 min and held at 90% B for a final 10 min was used for 140 min total run time. Column was re-equilibrated with 15 μl of buffer A prior to the injection of sample. Peptides were eluted directly from the tip of the column and nanosprayed directly into the mass spectrometer by application of 2.5 kV voltage at the back of the column. The Eclipse was operated in a data dependent mode. Full mass spectrometry scans were collected in the Orbitrap at 120 k resolution with a mass range of 350 to 1,800 m/z and an AGC target of 4 × 105. The cycle time was set to 3 s, and within this 3 s the most abundant ions per scan were selected for HCD MS/MS in the Orbitrap with an AGC target of 5 × 104, resolution of 30 k and minimum intensity of 50,000. Maximum fill times were set to 50 ms and 54 ms for MS and MS/MS scans, respectively. Quadrupole isolation at 2 m/z was used, monoisotopic precursor selection was enabled and dynamic exclusion was used with exclusion duration of 60 s. Protein and peptide identification were done with Byonic node (Protein Metrics) within Proteome Discoverer 2.5 (Thermo).
Gs signalling assays
We measured cAMP production to determine ligand-dependent activation of Gs signalling by TSHR. For each TSHR construct (wild-type (WT), ΔC-peptide, Y385F, Y385A, E409A, I486F, A644K, A647K, K262C, N473C, K262C-N483C), a 20-ml suspension culture of Expi293F-TetR cells was co-transfected with a pcDNA3.1 plasmid expressing TSHR and a luciferase-based cAMP biosensor (pGlosensor-22F; Promega). DNA quantities for each construct were titrated and resultant receptor expression levels were determined by flow cytometry using an Alexa647-conjugated anti-M1 Flag antibody. Transfection quantities of all constructs resulting in similar expression levels to wild-type TSHR were selected, and were co-transfected with a fixed 3:1 DNA mass ratio of the receptor construct to the cAMP biosensor construct as used for wild-type TSHR transfections. Cells were collected 24 h post-transfection, resuspended in Expi293 expression media (Gibco) supplemented with 10% DMSO, and gradually frozen to −80 °C in a Mr. Frosty Freezing container for future use. To perform the assay, frozen Expi293F-TetR cells were rapidly thawed in a 37 °C water bath and resuspended in fresh Expi293 expression medium. Cells were diluted to a final concentration of 500,000 cells per ml in Expi293 expression medium plus 2% (v/v) Glosensor assay reagent (Promega) and incubated for 75 min at room temperature with gentle rotation. Expi293F-TetR cells were then plated into a white 384-well plate (Greiner) to a final density of 10,000 cells per well. Separately, a 5× ligand stock plate (13-point half-log dilution series, 3 wells per condition) was made in Hank’s balanced salt solution + 0.1% (w/v) bovine serum albumin. An aliquot from the ligand plate was transferred into new 384-well plates to achieve a final 1× concentration upon addition of Glosensor reagent incubated, co-transfected TSHR:cAMP biosensor cells. Immediately after cell addition, luminescence was measured in 0.1 s intervals for 10 min using a CLARIOstar instrument at an emission wavelength of 580 nm ± 80 nm band-pass (BMG LabTech). The resulting dose response curves from the 5-min read point were fit to a nonlinear regression three-parameter log(agonist) versus response fit in GraphPad Prism 9 (Dotmatics).
Molecular dynamics simulations
Simulation setup.
We performed simulations of four distinct conditions, using the M22-bound active-state TSHR and the CS-17-bound inactive-state TSHR structures as starting points. The four conditions are the following: (A) from active, with M22 bound; (B) from active, with M22 removed; (C) from inactive, with CS-17 bound; (D) from inactive, with CS-17 removed. For each simulation condition, we performed 10 independent 1-μs simulations.
N-acetylglucosamine residues as well as fragments other than TSHR, M22, or CS-17 were removed from the structures. For conditions A and B, the lipid in the transmembrane binding pocket was modelled as DPPC in Maestro v12.8.117 (Schrödinger). Missing loops and amino acid side chains were modelled using Prime in Schrödinger Maestro. We did not model the ~100-residue TSHR hinge region. Neutral acetyl and methylamide groups were added to cap the N and C termini, respectively, of the protein chains. Titratable residues were kept in their dominant protonation state at pH 7, except for residues E506 and D633, which were neutral. Histidine residues were modelled as neutral, with a hydrogen atom bound either to the epsilon nitrogen or the delta nitrogen, depending on which optimized the local hydrogen bonding network upon visual inspection. Dowser73 was used to add water molecules to protein cavities. We used the OPM PPM webserver47 to align the proteins, after which the aligned structures were inserted into a pre-equilibrated palmitoyloleoyl-phosphatidylcholine (POPC) membrane bilayer using Dabble v2.7.9 (ref. 74). Sodium and chloride ions were added to neutralize each system at a concentration of 150 mM.
The final system of condition A comprised 308,127 atoms, including 592 lipid molecules and 70,982 water molecules (initial system dimensions: 150 Å × 150 × 135 Å). The final system of condition B comprised 131,486 atoms, including 246 lipid molecules and 29,762 water molecules (initial system dimensions: 100 Å × 100 Å × 130 Å). The final system of condition C comprised 306,262 atoms, including 576 lipid molecules and 71,057 water molecules (initial system dimensions: 150 Å × 150 Å × 135 Å). The final system of condition D comprised 131,806 atoms, including 245 lipid molecules and 29,907 water molecules (initial system dimensions: 100 Å × 100 Å × 130 Å).
Simulation protocols.
For each simulation, initial atom velocities were assigned randomly and independently. We employed the CHARMM36m force field for protein molecules, the CHARMM36 parameter set for lipid molecules and salt ions, and the associated CHARMM TIP3P model for water75,76. Simulations were run using the AMBER20 software77 under periodic boundary conditions with the Compute Unified Device Architecture (CUDA) version of Particle-Mesh Ewald Molecular Dynamics (PMEMD) on one GPU78.
After energy minimization, the systems were first heated over 12.5 ps from 0 K to 100 K in the NVT ensemble using a Langevin thermostat with harmonic restraints of 10.0 kcal mol−1 Å−2 on the non-hydrogen atoms of the lipids, protein, and ligand. Initial velocities were sampled from a Boltzmann distribution. The systems were then heated to 310 K over 125 ps in the NPT ensemble. Equilibration was performed at 310 K and 1 bar in the NPT ensemble, with harmonic restraints on the protein and ligand non-hydrogen atoms tapered off by 1.0 kcal mol−1 Å−2 starting at 5.0 kcal mol−1 Å−2 in a stepwise manner every 2 ns for 10 ns, and finally by 0.1 kcal mol−1 Å−2 every 2 ns for an additional 18 ns. All restraints were completely removed during production simulation. Production simulations were performed at 310 K and 1 bar in the NPT ensemble using the Langevin thermostat and Monte Carlo barostat. Lengths of bonds to hydrogen atoms were constrained using SHAKE, and the simulations were performed using a timestep of 4.0 fs while using hydrogen mass repartitioning79. Non-bonded interactions were cut off at 9.0 Å, and long-range electrostatic interactions were calculated using the particle-mesh Ewald (PME) method with an Ewald coefficient (β) of approximately 0.3 Å and B-spline interpolation of order 4. The PME grid size was chosen such that the width of a grid cell was approximately 1 Å. Snapshots of each trajectory were saved every 200 ps.
Simulation analysis protocols.
The AmberTools17 CPPTRAJ package80 was used to reimage trajectories at 1 ns per frame. Visual Molecular Dynamics (VMD v1.9.4a12)81 was used for visualization and analysis.
For the analysis of ECD conformation shown in Fig. 4 and Extended Data Fig. 12, we performed a projection analysis as described in Latorraca, N. et al.82. In brief, we monitor ECD conformation by representing the Cα atoms of ECD residues 30–260 as a single vector containing the Cartesian coordinates. After aligning on transmembrane (TM) helices TM1–TM4 of the receptor (Cα atoms of residues 415–441, 448–476, 492–525 and 536–558), we project this vector in a given simulation frame onto the line connecting the experimentally determined inactive ECD conformation (from the CS-17-bound structure reported in this paper) and experimentally determined active ECD conformation (from the M22-bound structure reported in this paper). We then report the position of the projected point on the line, using the convention that the inactive ECD conformation is at 0 and positive values indicate change toward the active conformation. The experimentally determined active ECD conformation is thus assigned a value equal to its r.m.s.d. from the inactive conformation (in this case, 33.5 Å).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Coordinates for TSH-bound TSHR–Gs, TR1402-bound TSHR–Gs, M22-bound TSHR–Gs and CS-17-bound TSHR have been deposited in the PDB under accession codes 7T9I, 7UTZ, 7T9N and 7T9M, respectively. Sharpened and unsharpened cryo-EM density maps for TSH-bound TSHR–Gs (composite), TR1402-bound TSHR–Gs (composite), M22-bound TSHR–Gs (composite), CS-17-bound TSHR, and Org 2274179-0-bound TSHR have been deposited in the Electron Microscopy Data Bank under accession codes EMD-25758, EMD-26795, EMD-25763, EMD-25762 and EMD-27640, respectively. Sharpened maps, unsharpened maps, half-maps and masks for each composite map component (ligand-bound ECD or 7TM–G protein), for TSH-bound TSHR–Gs, TR1402-bound TSHR–Gs and M22-bound TSHR-Gs have been deposited in the Electron Microscopy Data Bank under accession codes EMD-27649 (TSH-bound ECD), EMD-27650 (TSH-bound 7TM–Gs), EMD-27647 (TR1402-bound ECD), EMD-27648 (TR1402 7TM–Gs), EMD-27651 (M22-bound ECD) and EMD-27652 (M22-bound 7TM–Gs). Final particle stacks and .star files for TSH-bound TSHR–Gs, TR1402-bound TSHR–Gs and M22-bound TSHR–Gs and CS-17-bound TSHR-containing particle shift and pose assignments have been uploaded to the Electron Microscopy Public Image Archive under the accession codes EMPIAR-11120. Structure files for OPM-based modelling of inactive and active TSHR are available in the Supplementary Information.
Extended Data
Extended Data Fig. 1 |. Signaling studies for TSHR.

a, b) DNA titrations of TSHR mutants tested in cAMP production assays. Median fluorescence intensity ± SD (n = 3) of anti-FLAG-A647 staining is plotted. Filled in bars represent the DNA concentration used for co-transfection experiments with pGlo cAMP biosensor plasmid. Horizontal red line indicates the mean wild-type (WT) TSHR fluorescence intensity. Flow cytometry experiments were performed in triplicate, with identical gating across all cell lines tested. c) cAMP production of TSHR cysteine mutants comparing the basal level (−) to 100 nM of TSH, M22, or CS-17 for untreated cells (left panel) and cells treated with 500 μM TCEP (right panel). Data points are means ± SD from three biological replicates. d, e) cAMP production curves comparing TSH and M22-mediated activation of WT and C-peptide deleted TSHR constructs. f) cAMP production curves for TSH and TR1402-mediated WT TSHR activation. g) cAMP production curves for TSH-mediated WT and TSHR mutant cell line (Y385F, Y385A) activation. h) cAMP production curves for M22-mediated WT and lipid-displacing TSHR mutant cell line (A644K, A647K) activation. Plotted data points in panels c-g are means of triplicate measurements ± SD from a representative experiment of n = 3 biological replicates.
Extended Data Fig. 2 |. Cryo-EM data processing for TSH-bound TSHR-Gs complex.
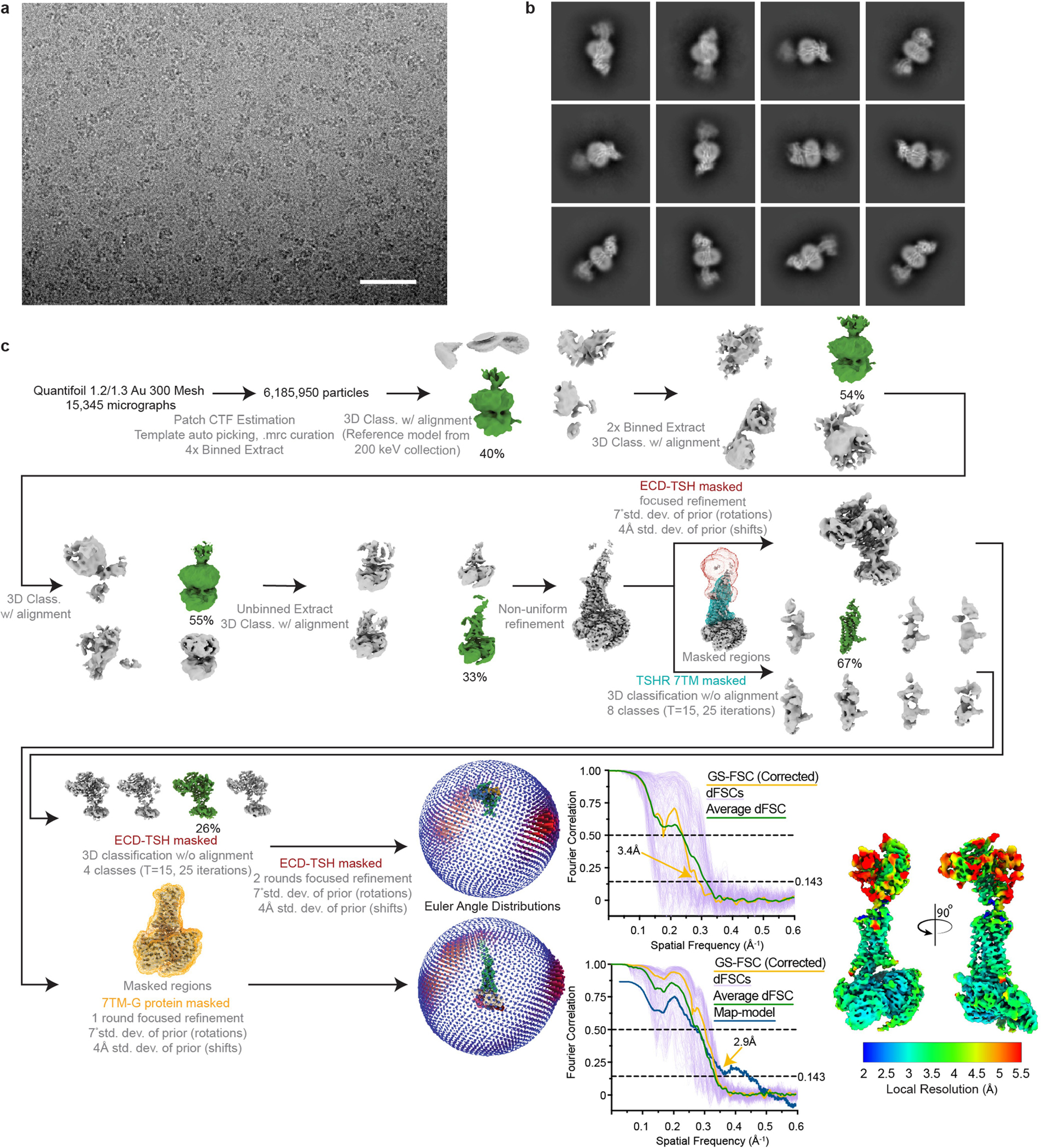
a) Representative image from 15,345 micrographs. Scale bar, 50 nm. b) Selected 2D class averages. c) Processing approach used for reconstruction of TSH-bound TSHR-Gs complex. A local resolution map was calculated from cryoSPARC using masks from indicated local refinement, then visualized with the composite map in the same scale. A viewing distribution plot was generated using scripts from the pyEM software suite83 and visualized in ChimeraX. GS-FSC and Directional FSC (dFSC, shown as purple lines) curves were generated in cryoSPARC and as previously described in Dang, S. et al. Nature 552, 426–429 (2017).
Extended Data Fig. 3 |. Model map correlation coefficients for cryo-EM structures.

Correlation values for resolved residues in each modeled chain are shown. Low values indicate regions that are poorly resolved, e.g. the 7TM domain of TSHR in CS-17-TSHR complex or the constant domains of the Fab fragments for CS-17 and M22.
Extended Data Fig. 4 |. Cryo-EM data processing for TR1402-bound TSHR-Gs complex.

a) Representative image from 14,277 micrographs. Scale bar, 50 nm. b) Selected 2D class averages from final reconstruction. c) Processing approach used for reconstruction of TR1402-bound TSHR-Gs complex. A local resolution map was calculated from cryoSPARC using masks from indicated local refinement, then visualized with the composite map in the same scale. A viewing distribution plot was generated using scripts from the pyEM software suite and visualized in ChimeraX. GS-FSC and dFSC curves were generated in cryoSPARC and as previously described in Dang, S. et al. Nature 552, 426–429 (2017).
Extended Data Fig. 5 |. Glycosylation status of native human TSH.
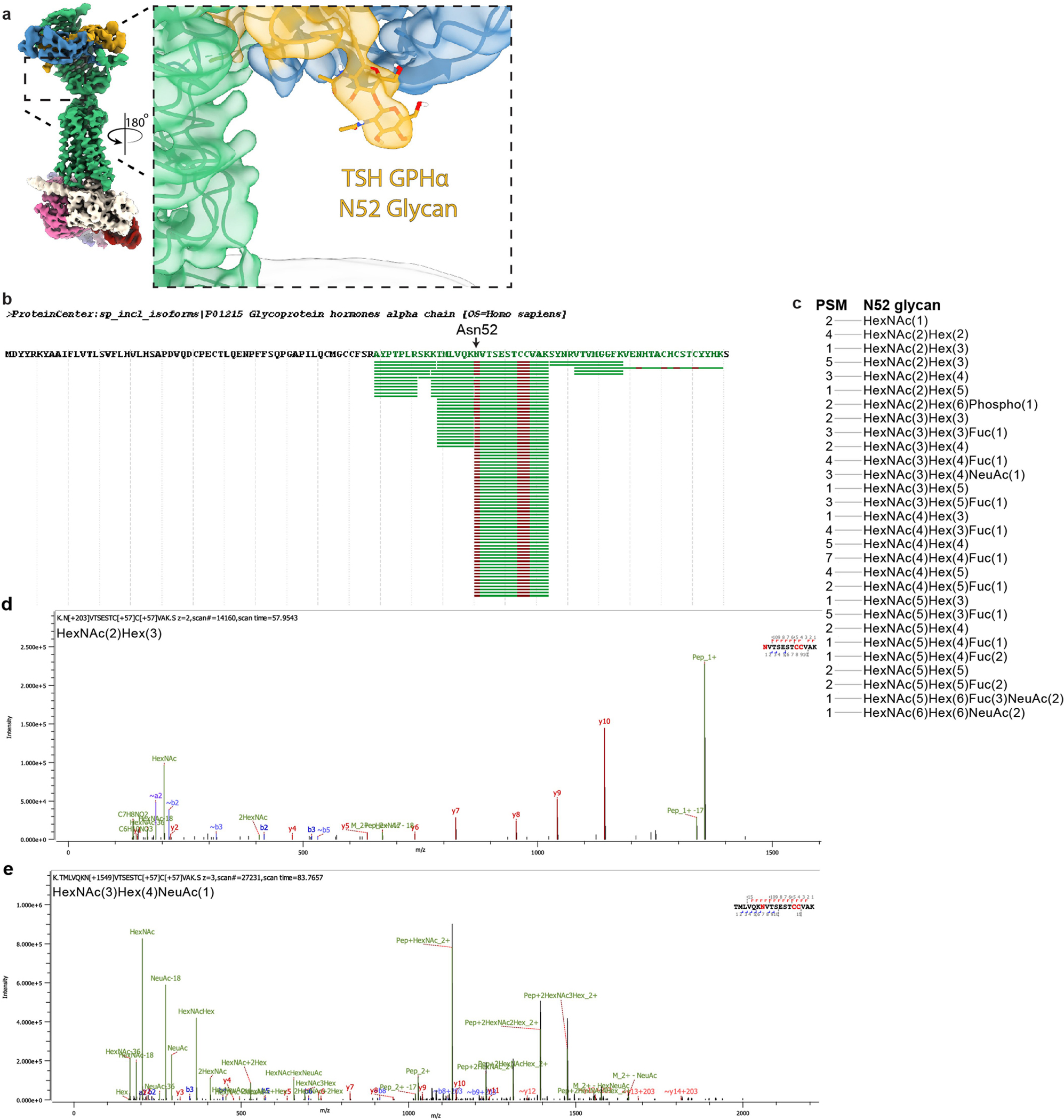
a) Cryo-EM density map of TSH-bound TSHR showing resolved glycan density for Asn52. b) Peptide coverage of Asn52 from native human TSH GPHα chain in mass spectrometry experiments. c) Peptide spectra match (PSM) of glycans detected on Asn52 of GPHα chain. d,e) Representative MS/MS spectra showing Asn52 with various length glycans.
Extended Data Fig. 6 |. Receptor:hormone interaction comparisons across the glycoprotein hormone receptor family.
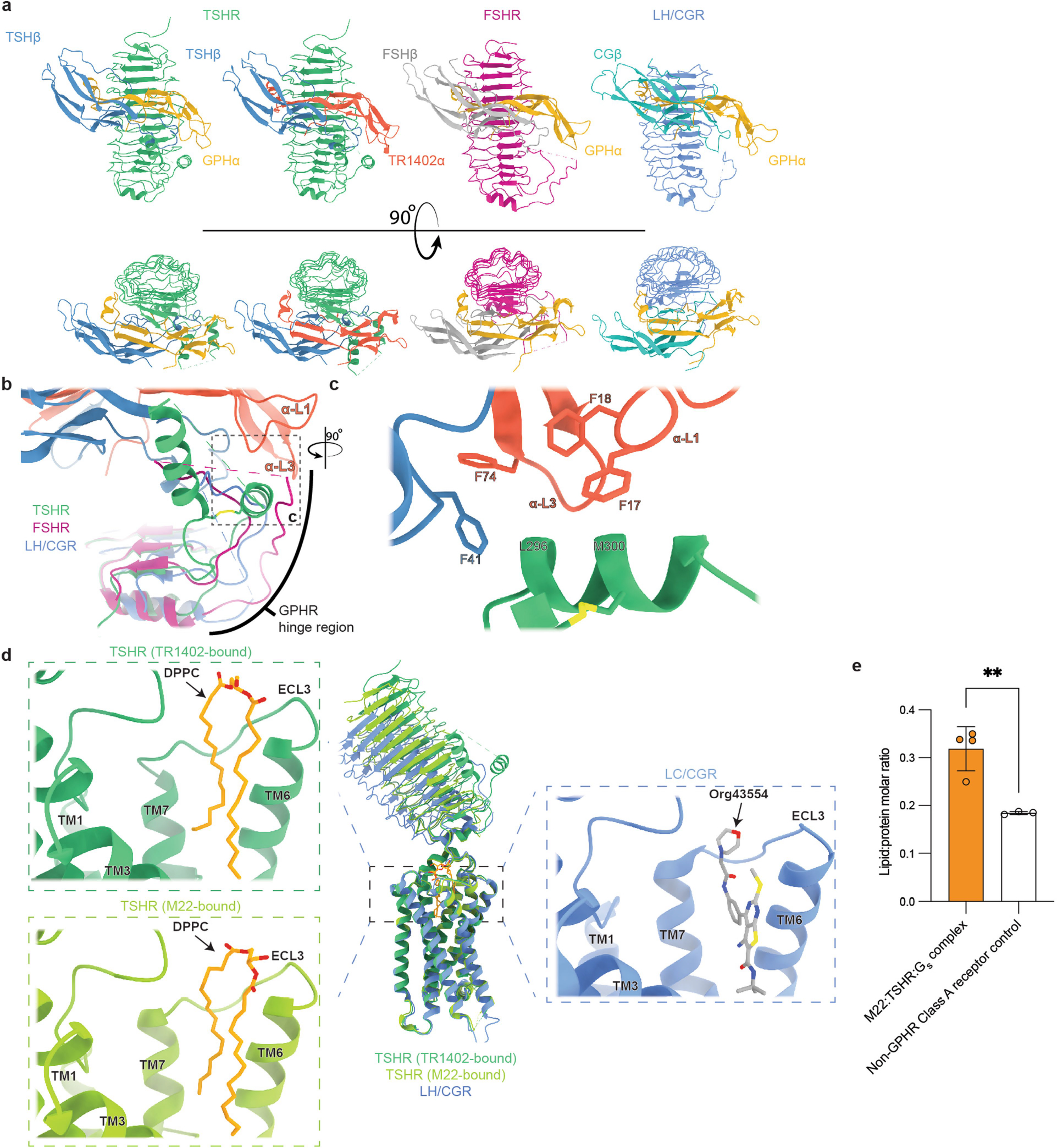
a) Comparison of TSHR, FSHR (PDB: 4AY9), and LH/CGR ECD:hormone (PDB: 7FIH) interactions shows the comparable concave binding-interface and lateral-ECD contacts made by GPHα and receptor-specific β subunits. b) Alignment of the GPHR ECDs reveals the variability in modeled poses of hinge-hormone contacts observed in prior GPHR:hormone structures. c) The N-terminal hinge-hormone disulfide-linked alpha helix presents a hydrophobic face to the α-L1 and α-L3 loops of the TR1402 α-chain and is stabilized by multiple phenylalanine contacts in the GPHα and TSHβ chains. C-terminal hinge-region α helix hidden for visualization. d) Comparison of the TSHR and LHCGR (PDB: 7FIH) transmembrane pockets highlights the similarity in the DPPC and the allosteric agonist Org43554 binding sites. TM4/5 and ECL2 not shown. e) Lipid:protein molar ratio comparison between the M22:TSHR:Gs complex and a non-GPHR Class A receptor control. Data points represent individual measurements from technical replicates (n = 3 for non-GPHR control and n = 4 for M22:TSHR:Gs complex) of the ratio of pmol of lipid DPPC per pmol of protein. **P = 0.0088; Unpaired two-tailed t test was used to calculate statistical differences in lipid:protein molar ratios.
Extended Data Fig. 7 |. Cryo-EM data processing for Org 274179-0 bound TSHR.
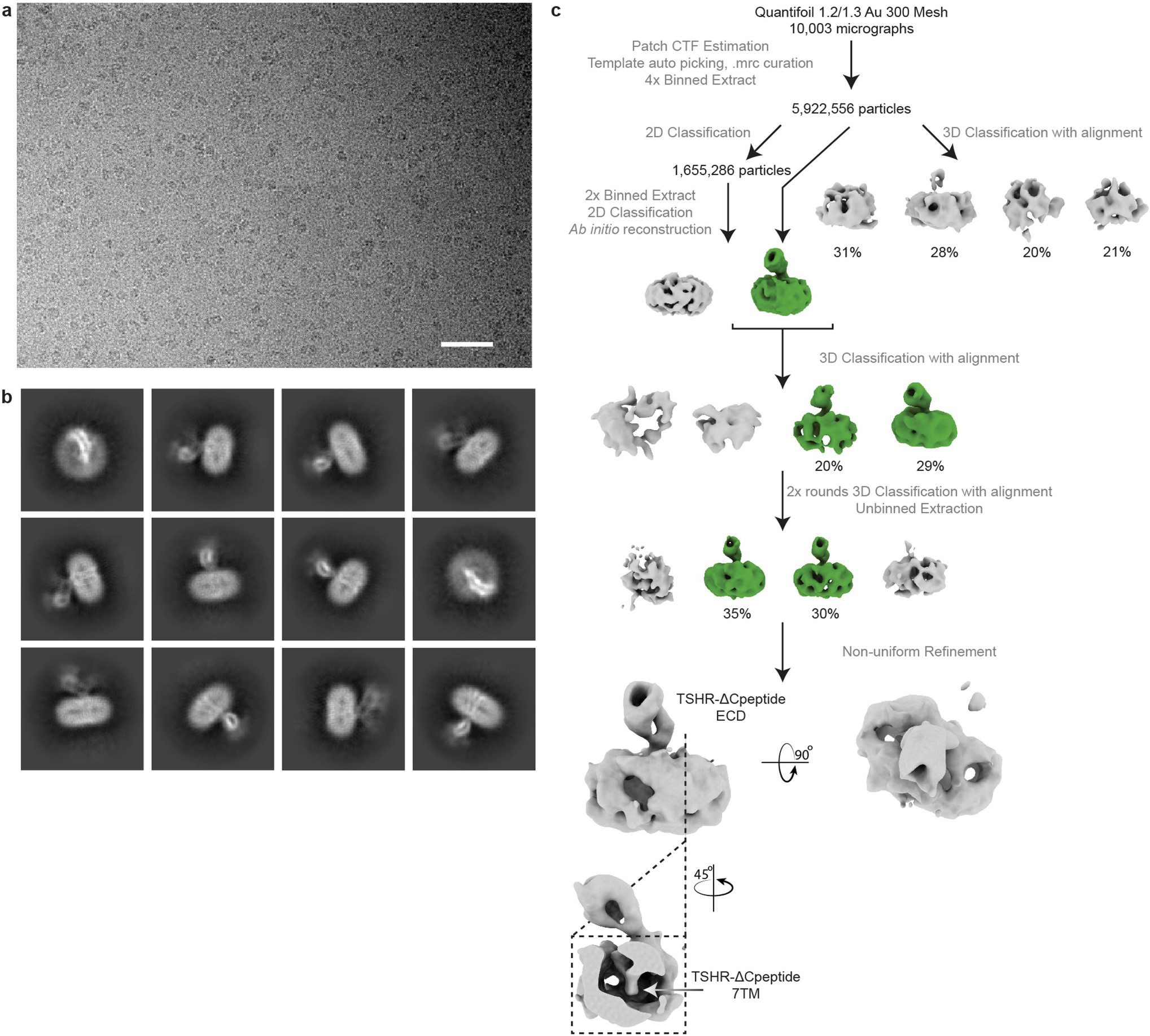
a) Representative image from 10,003 micrographs. Scale bar, 50 nm. b) Selected 2D class averages generated from curated particles. c) Processing approach used for low resolution reconstruction. Despite starting with a similar or larger sized dataset as for other TSHR samples, the TSHR–Org 274179-0 complex did not yield high resolution reconstruction of the 7TM domain. A low-resolution reconstruction of the TSHR ECD was observed. This suggests potential flexibility between the TSHR 7TM domain and the ECD.
Extended Data Fig. 8 |. Cryo EM data processing for the CS-17 bound TSHR:Org 274179-0 complex.
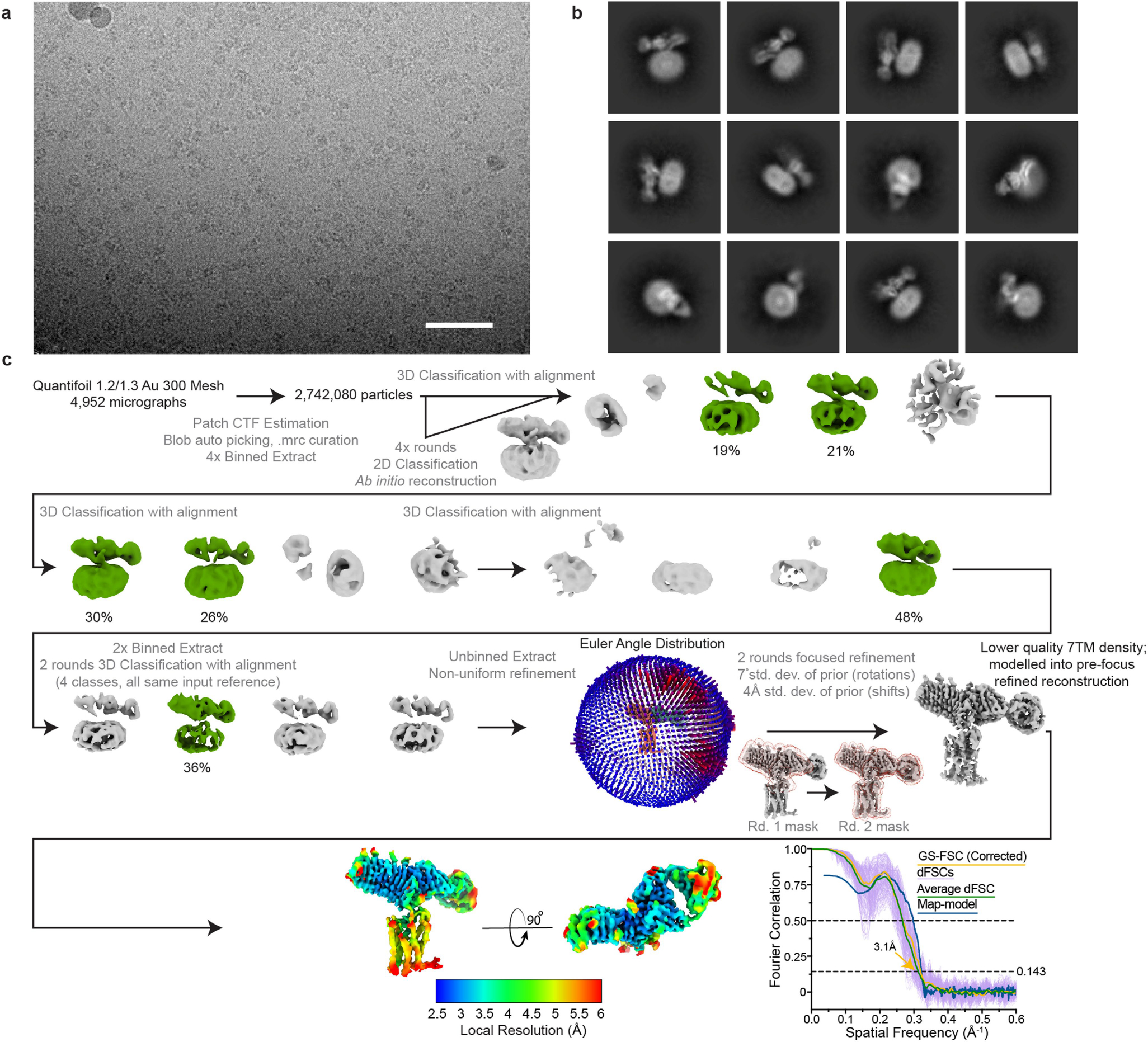
a) Representative image from 4,952 micrographs. Scale bar, 50 nm. b) Selected 2D class averages generated from the final reconstruction. c) Processing approach used for reconstruction of the complex. A viewing distribution plot was generated using scripts from the pyEM software suite and visualized in ChimeraX. Local resolution map generated from non-uniform refinement mask in cryoSPARC. GS-FSC and dFSC curves were generated in cryoSPARC and as previously described in Dang, S. et al. Nature 552, 426–429 (2017).
Extended Data Fig. 9 |. Comparison of Org 274179-0 bound- and CS-17 bound TSHR EM maps.
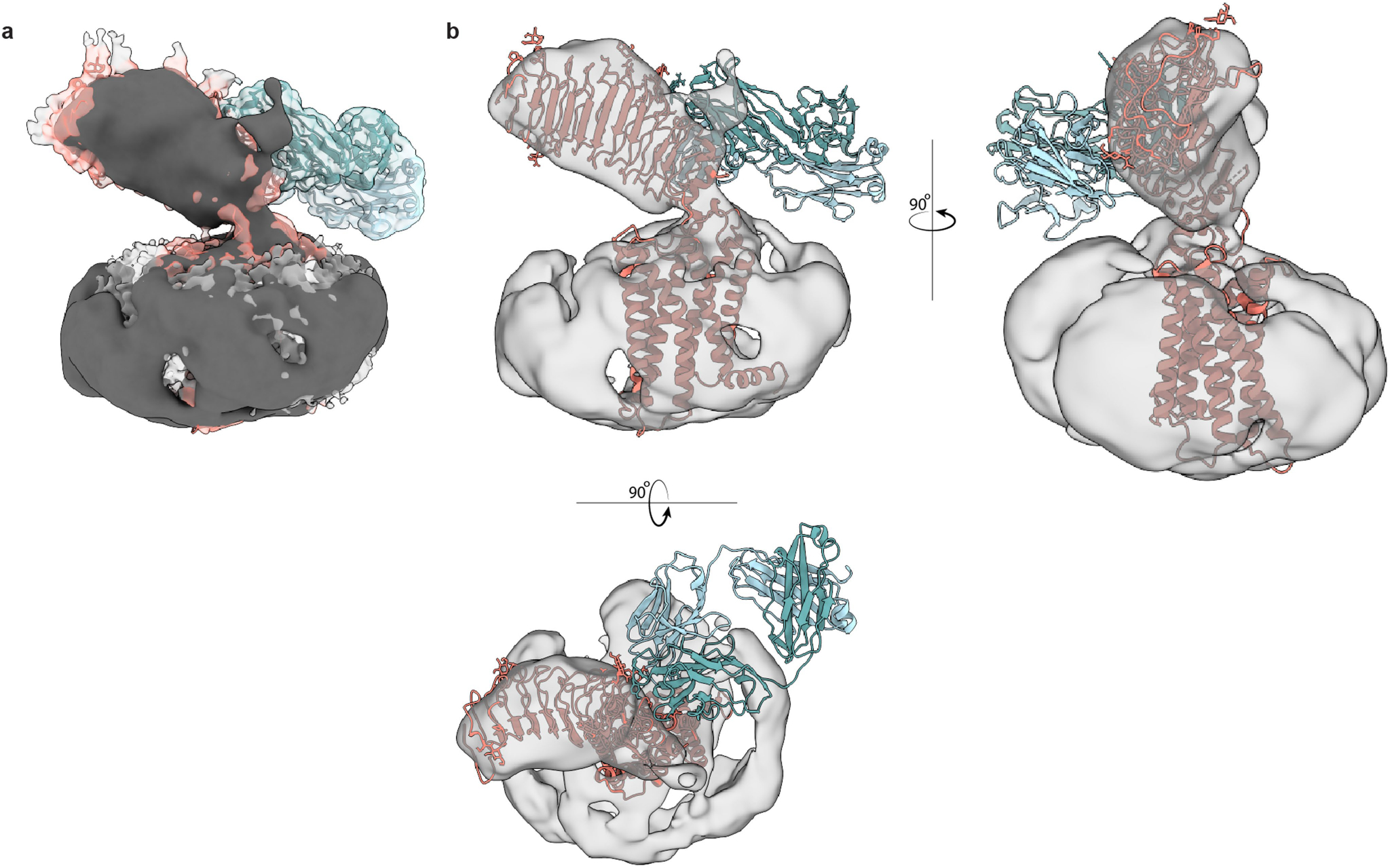
a) Org 274179-0 bound-TSHR EM density map (dark) fit into the CS-17-bound TSHR map (colored by TSHR-CS-17 model). b) CS-17-bound TSHR model fit into Org 274179-0 bound-TSHR map, suggesting that inactive state TSHR ECD orientations are similar between CS-17 and Org 274179-0-bound states.
Extended Data Fig. 10 |. Comparison of TSHR activation with other GPCRs.
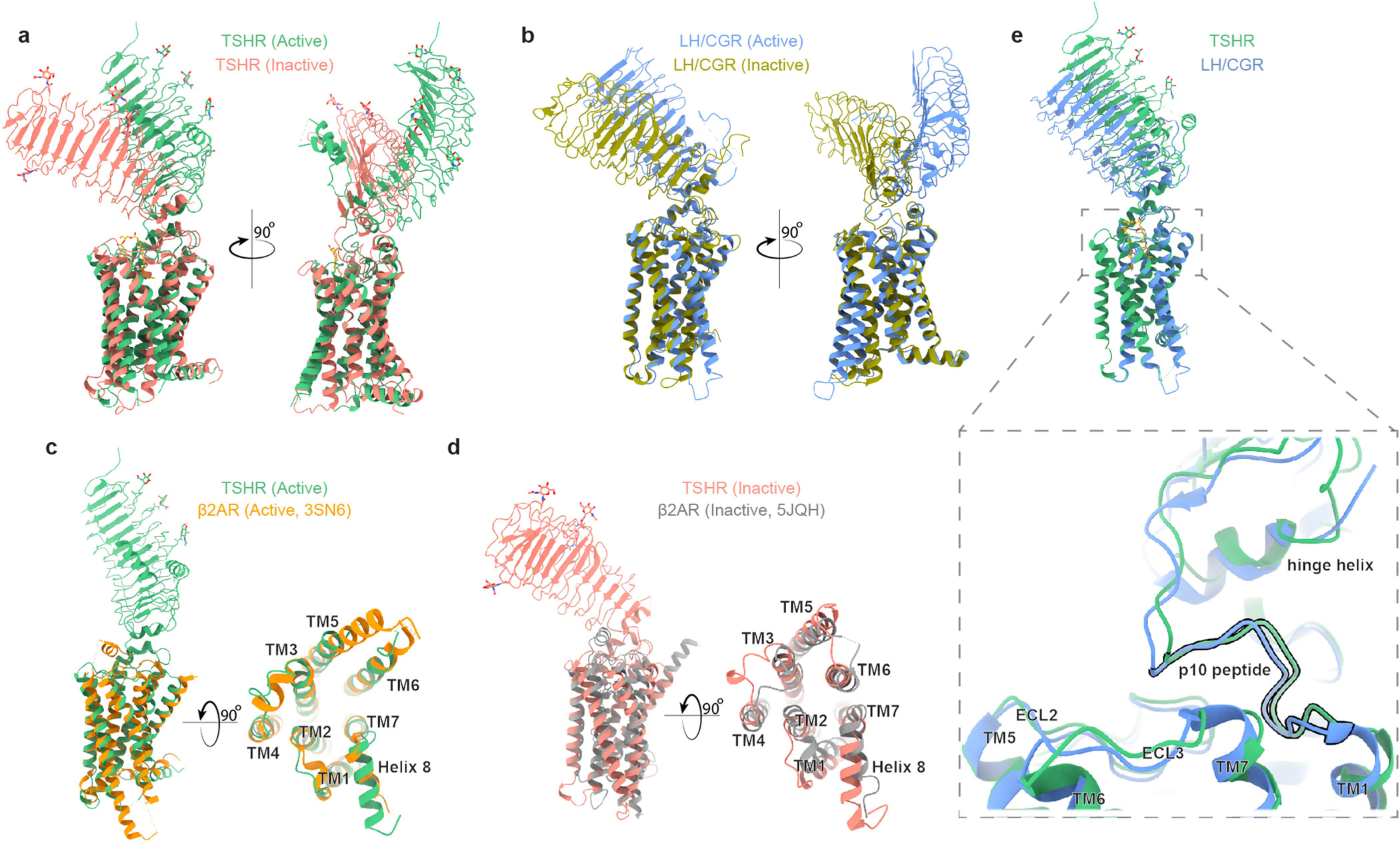
a) Ribbon diagram of TSHR in active and inactive conformations. b) Ribbon diagram of LH/CGR in active (PDB:7FIH) and inactive (PDB:7FIJ) conformations. A similar reorientation of the ECD is shared between TSHR and LH/CGR upon activation. c) Comparison of active TSHR to active β2-adrenoceptor (β2AR, PDB:3SN6). d) Comparison of inactive TSHR to inactive β2AR, PDB:5JQH). e) Comparison of active conformations of TSHR and LH/CGR reveals similar overall structures of the 7TM domain and the p10 peptide.
Extended Data Fig. 11 |. Cryo-EM data processing for M22-bound TSHR-Gs complex.
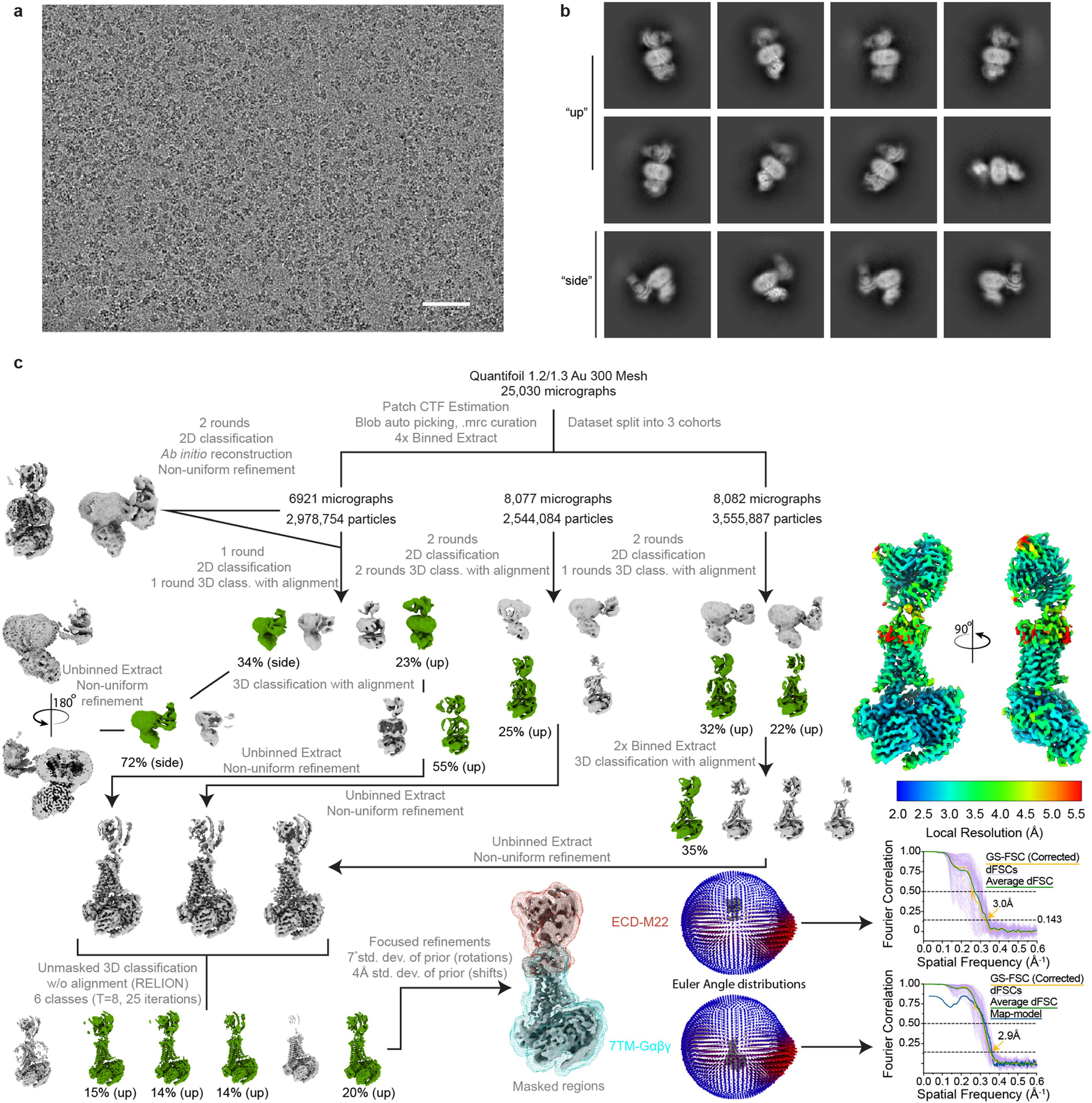
a) Representative image from 25,030 micrographs. Scale bar, 50 nm. b) Selected 2D class averages from final reconstruction. c) Processing approach used for reconstruction of M22-bound TSHR-Gs complex. A local resolution map was calculated from cryoSPARC using masks from indicated local refinement, then visualized with the composite map in the same scale. A viewing distribution plot was generated using scripts from the pyEM software suite and visualized in ChimeraX. GS-FSC and dFSC curves were generated in cryoSPARC and as previously described in Dang, S. et al. Nature 552, 426–j429 (2017).
Extended Data Fig. 12 |. Extent of ECD activation for all individual simulations across four simulation conditions.

Dashed lines indicate the projection metric values (see Methods) in the inactive (0.0 Å) and active (33.5 Å) state cryo-EM structures. Thick traces indicate the moving average smoothed over a 25-ns window, and thin traces represent unsmoothed data. Each column represents a distinct simulation condition: started from the active structure, with M22 bound (brown traces, first column); started from the active structure, with M22 removed (grey traces, second column); started from the inactive structure, with CS-17 bound (blue traces, third column); started from the inactive structure, with CS-17 removed (grey traces, fourth column).
Extended Data Fig. 13 |. Glycosylation of engineered K1-70glyco construct.
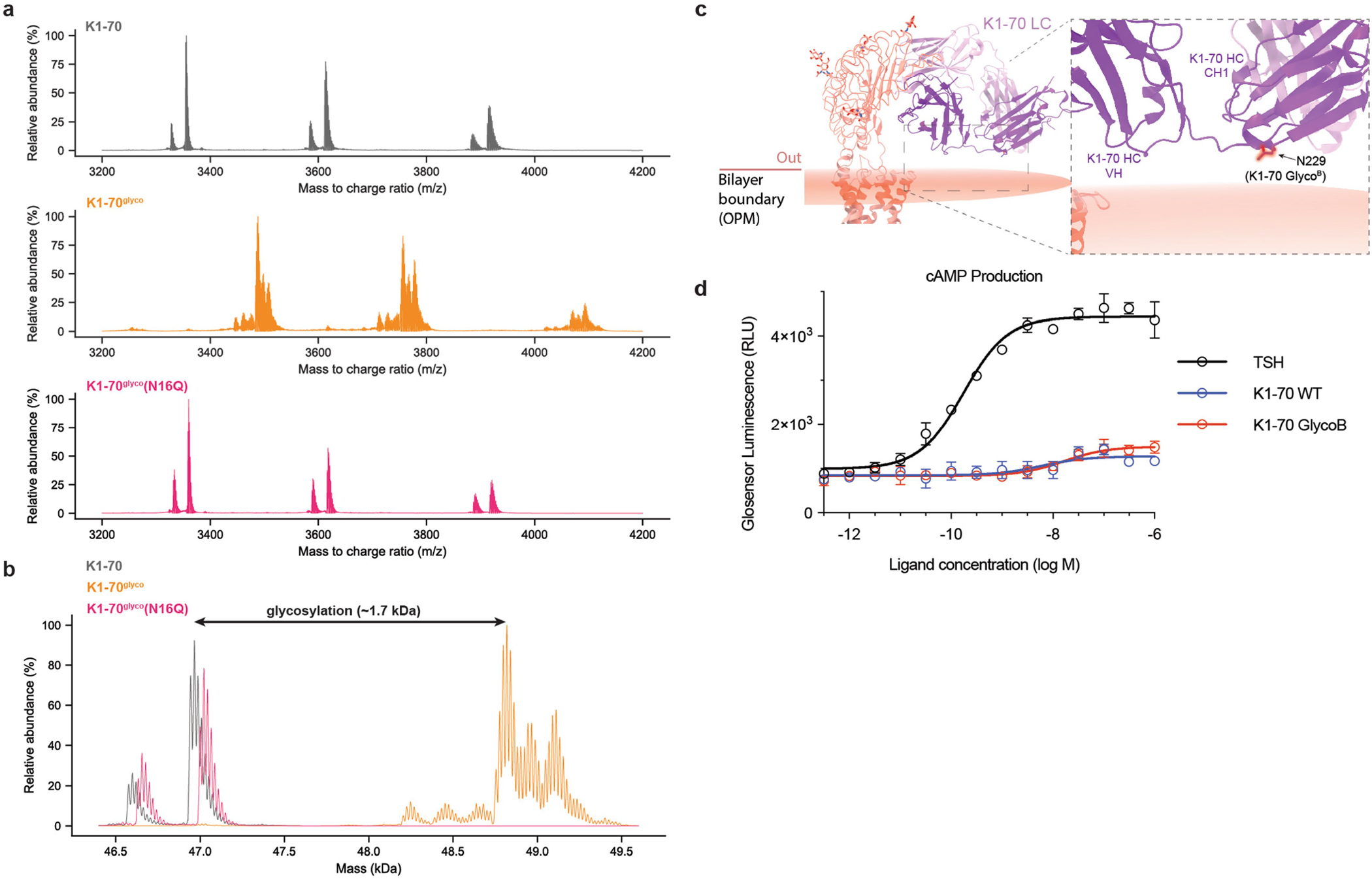
a) Native mass spectrum (nMS) of K1-70, K1-70glyco, and K1-70glyco(N16Q) Fab fragments. b) Accurate mass assignment of Fab fragments. nMS demonstrates ~1.7 kDa increased mass for K1-70glyco and more heterogeneity consistent with N-linked glycosylation. The smaller mass and increased homogeneity of the K1-70glyco(N16Q) construct further supports glycosylation at the engineered N16 position. c) Crystal structure of K1-70 TSHR-ECD complex (PDB: 2XWT) aligned to CS-17 bound TSHR. The membrane-proximal N229 residue (of the “GlycoB” glycosylation motif) is highlighted in red. d) cAMP production comparing K1-70 WT, K1-70 GlycoB and M22 Fab fragment-mediated activation of TSHR. Plotted data points are means of triplicate measurements ± SD from one of three biological replicates.
Extended Data Table 1 |.
Cryo-EM data collection, refinement, and validation statistics
| TSH-bound TSHR-Gs | TR1402-bound TSHR-Gs | Org 2274179-0-bound TSHR | CS-17 Fab/Org 2274179-0-bound TSHR | M22-bound TSHR-Gs | |
|---|---|---|---|---|---|
| Composite/full map | EMDB-25758 | EMDB-26795 | EMDB-27640 | EMDB-25762 | EMDB-25763 |
| ECD-ligand map | EMDB-27649 | EMDB-27647 | EMDB-27651 | ||
| 7TM-Gs map | EMDB-27650 | EMDB-27648 | EMDB-25762 | ||
| RCSB PDB | PDB: 7T9I | PDB: 7UTZ | PDB: 7T9M | PDB: 7T9N | |
|
| |||||
| Data collection and processing | |||||
| Magnification | 130,000 | 105,000 | 105,000 | 130,000 | 81,000 |
| Voltage (kV) | 300 | 300 | 300 | 300 | 300 |
| Electron exposure (e-/Å2) | 77 | 60 | 50 | 77 | 60 |
| Defocus range (μm) | −0.8 to −2.2 | −0.8 to −2.2 | −0.8 to −2.4 | −0.8 to −2.2 | −0.8 to −2.0 |
| Pixel size (Å) (physical) | 0.662 | 0.831 | 0.85 | 0.644 | 0.844 |
| Symmetry imposed | C1 | C1 | C1 | C1 | C1 |
| Initial particle images (no.) | 6,185,950 | 7,940,651 | 5,922,556 | 2,742,080 | 9,078,725 |
| Final particle images (no.) | 60,446 (7TM-Gs) 80,483 (ECD-TSH) |
259,702 (7TM-Gs) 375,930 (ECD-TR1402) |
57,018 | 41,054 | 244,973 |
| Map resolution (Å) (masked) | 2.9 (7TM-G protein) | 2.4 (7TM-G protein) | 6.7 | 3.1 | 2.9 (7TM-G protein) |
| 3.4 (ECD-TSH) | 2.7 (ECD-TSH) | 3.0 (ECD-M22) | |||
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Refinement | |||||
| Initial model used (PDB code) | TR1402-bound structure | AlphaFold (TR1402) M22-bound structure |
AlphaFold (TSHR) AlphaFold (CS-17) |
AlphaFold (TSHR), 7LJC (G protein) 3SN6 (Nb35) 3G04 (M22) |
|
| Model resolution (Å) (unmasked/masked) | 3.9/3.8 | 3.2/3.1 | 3.5/3.3 | 3.2/3.1 | |
| FSC threshold | 0.5 | 0.5 | 0.5 | 0.5 | |
| Model resolution range (Å) | 3.8–50 | 3.5–50 | 3.2–50 | ||
| Map sharpening B factor (Å2) | −102 (ECD), | −100 (ECD), | −91 | −90 (ECD) | |
| −83 (7TM/MiniGαsβγ) | −73 (7TM/MiniGαsβγ) | −80 (7TM/MiniGαsβγ) | |||
| Model composition | |||||
| Non-hydrogen atoms | 11991 | 12057 | 7827 | 13405 | |
| Protein residues | 1532 | 1536 | 995 | 1723 | |
| Ligands | NAG: 7 | NAG:7, DPC:1 | NAG: 5 | NAG: 3, DPC:1 | |
| B factors (Å2) | |||||
| Protein | 104.8 | 79.2 | 93.5 | 89.9 | |
| Ligand | 96.2 | 88.5 | 49.4 | 45.6 | |
| R.m.s. deviations | |||||
| Bond lengths (Å) | 0.005 | 0.004 | 0.006 | 0.006 | |
| Bond angles (°) | 0.916 | 0.678 | 1.221 | 1.013 | |
| Validation | |||||
| MolProbity score | 1.36 | 1.36 | 1.53 | 1.58 | |
| Clashscore | 3.3 | 3.91 | 5 | 5.62 | |
| Poor rotamers (%) | 0.62 | 0 | 0.91 | 0.14 | |
| CaBLAM outliers (%) | 1.34 | 1.34 | 1.63 | 1.32 | |
| Ramachandran plot | |||||
| Favored (%) | 96.42 | 96.96 | 96.05 | 95.99 | |
| Allowed (%) | 3.58 | 3.04 | 3.95 | 4.01 | |
| Disallowed (%) | 0 | 0 | 0 | 0 | |
Supplementary Material
Acknowledgements
We thank R. Yan and D. Toso for help in microscope operation and data collection; C. Bracken for mass spectrometry troubleshooting; and H. Shan for sample preparation for lipid mass spectrometry. This work was supported by National Institutes of Health (NIH) grants DP5OD023048 (A.M.), 1R35GM140847 (Y.C.), P30CA014195, R01GM102491 (A.S.), R01GM127359 (R.O.D.), 5R44CA224376 (M.W.S.) and a Human Frontier Science Program Long-Term Fellowship LT000916/2018-L (C.-M.S.). Cryo-EM equipment at UCSF is partially supported by NIH grants S10OD020054 and S10OD021741. Some of this work was performed at the Stanford-SLAC Cryo-EM Center (S2C2), which is supported by the National Institutes of Health Common Fund Transformative High-Resolution Cryo-Electron Microscopy program (U24 GM129541). This research was, in part, supported by the National Cancer Institute’s National Cryo-EM Facility at the Frederick National Laboratory for Cancer Research under contract HSSN261200800001E. Some of this work was supported by the Mass Spectrometry Core of the Salk Institute with funding from NIH-NCI CCSG: P30CA014195. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. A.M. acknowledges support from the Pew Charitable Trusts, the Esther A. & Joseph Klingenstein Fund, the Searle Scholars Program, the Edward Mallinckrodt Jr Foundation, and the Vallee Foundation. Y.C. is an Investigator of Howard Hughes Medical Institute. A.M. is a Chan Zuckerberg Biohub investigator.
Footnotes
Competing interests A.M. and R.O.D. are consultants for and stockholders in Septerna Inc. Y.C. is a consultant and advisor of Shuimu BioScience Ltd.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-022-05159-1.
Peer review information Nature thanks Marvin Gershengorn, Patrick M. Sexton and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Reprints and permissions information is available at http://www.nature.com/reprints.
References
- 1.Oppenheimer JH & Samuels HH Molecular Basis of Thyroid Hormone Action (Academic Press, 1983). [Google Scholar]
- 2.Vassart G & Dumont JE The thyrotropin receptor and the regulation of thyrocyte function and growth. Endocr. Rev. 13, 596–611 (1992). [DOI] [PubMed] [Google Scholar]
- 3.Smith TJ & Hegedüs L Graves’ disease. N. Engl. J. Med. 375, 1552–1565 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Fekete C & Lechan RM Central regulation of hypothalamic–pituitary–thyroid axis under physiological and pathophysiological conditions. Endocr. Rev. 35, 159–194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laurent E, Mockel J, Van Sande J, Graff I & Dumont JE Dual activation by thyrotropin of the phospholipase C and cyclic AMP cascades in human thyroid. Mol. Cell. Endocrinol. 52, 273–278 (1987). [DOI] [PubMed] [Google Scholar]
- 6.Taylor PN et al. Global epidemiology of hyperthyroidism and hypothyroidism. Nat. Rev. Endocrinol. 14, 301–316 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Zimmermann MB & Boelaert K Iodine deficiency and thyroid disorders. Lancet Diabetes Endocrinol. 3, 286–295 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Mincer DL & Jialal I Hashimoto Thyroiditis. in StatPearls (StatPearls Publishing, 2021); https://www.ncbi.nlm.nih.gov/books/NBK459262/ [PubMed] [Google Scholar]
- 9.Lane LC, Cheetham TD, Perros P & Pearce SHS New therapeutic horizons for Graves’ hyperthyroidism. Endocr. Rev. 41, 873–884 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flack MR, Froehlich J, Bennet AP, Anasti J & Nisula BC Site-directed mutagenesis defines the individual roles of the glycosylation sites on follicle-stimulating hormone. J. Biol. Chem. 269, 14015–14020 (1994). [PubMed] [Google Scholar]
- 11.Matzuk MM, Keene JL & Boime I Site specificity of the chorionic gonadotropin N-linked oligosaccharides in signal transduction. J. Biol. Chem. 264, 2409–2414 (1989). [PubMed] [Google Scholar]
- 12.Grossmann M, Weintraub BD & Szkudlinski MW Novel insights into the molecular mechanisms of human thyrotropin action: structural, physiological, and therapeutic implications for the glycoprotein hormone family. Endocr. Rev. 18, 476–501 (1997). [DOI] [PubMed] [Google Scholar]
- 13.Jiang X et al. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc. Natl Acad. Sci. USA 109, 12491–12496 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duan J et al. Structures of full-length glycoprotein hormone receptor signalling complexes. Nature 598, 688–692 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Chazenbalk GD et al. Evidence that the thyrotropin receptor ectodomain contains not one, but two, cleavage sites. Endocrinology 138, 2893–2899 (1997). [DOI] [PubMed] [Google Scholar]
- 16.Chen C-R, Salazar LM, McLachlan SM & Rapoport B Deleting the redundant TSH receptor C-peptide region permits generation of the conformationally intact extracellular domain by insect cells. Endocrinology 156, 2732–2738 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nehmé R et al. Mini-G proteins: novel tools for studying GPCRs in their active conformation. PLoS ONE 12, e0175642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rasmussen SGF et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature 477, 549–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szkudlinski MW, Teh NG, Grossmann M, Tropea JE & Weintraub BD Engineering human glycoprotein hormone superactive analogues. Nat. Biotechnol. 14, 1257–1263 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Reinfelder J et al. Effects of recombinant human thyroid-stimulating hormone superagonists on thyroidal uptake of 18F-fluorodeoxyglucose and radioiodide. Thyroid 21, 783–792 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Ulloa-Aguirre A, Timossi C, Damián-Matsumura P & Dias JA Role of glycosylation in function of follicle-stimulating hormone. Endocrine 11, 205–215 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Fan QR & Hendrickson WA Structure of human follicle-stimulating hormone in complex with its receptor. Nature 433, 269–277 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Costagliola S et al. Tyrosine sulfation is required for agonist recognition by glycoprotein hormone receptors. EMBO J. 21, 504 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kosugi S, Ban T, Akamizu T & Kohn LD Site-directed mutagenesis of a portion of the extracellular domain of the rat thyrotropin receptor important in autoimmune thyroid disease and nonhomologous with gonadotropin receptors. Relationship of functional and immunogenic domains. J. Biol. Chem. 266, 19413–19418 (1991). [PubMed] [Google Scholar]
- 25.Caltabiano G et al. The specificity of binding of glycoprotein hormones to their receptors. Cell. Mol. Life Sci. 65, 2484–2492 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moyle WR et al. Co-evolution of ligand-receptor pairs. Nature 368, 251–255 (1994). [DOI] [PubMed] [Google Scholar]
- 27.Lapthorn AJ et al. Crystal structure of human chorionic gonadotropin. Nature 369, 455–461 (1994). [DOI] [PubMed] [Google Scholar]
- 28.Wu H, Lustbader JW, Liu Y, Canfield RE & Hendrickson WA Structure of human chorionic gonadotropin at 2.6 Å resolution from MAD analysis of the selenomethionyl protein. Structure 2, 545–558 (1994). [DOI] [PubMed] [Google Scholar]
- 29.Grossmann M et al. Substitution of the seat-belt region of the thyroid-stimulating hormone (TSH) β-subunit with the corresponding regions of choriogonadotropin or follitropin confers luteotropic but not follitropic activity to chimeric TSH. J. Biol. Chem. 272, 15532–15540 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Dias JA, Zhang Y & Liu X Receptor binding and functional properties of chimeric human follitropin prepared by an exchange between a small hydrophilic intercysteine loop of human follitropin and human lutropin. J. Biol. Chem. 269, 25289–25294 (1994). [PubMed] [Google Scholar]
- 31.Vischer HF, Granneman JCM & Bogerd J Opposite contribution of two ligand-selective determinants in the N-terminal hormone-binding exodomain of human gonadotropin receptors. Mol. Endocrinol. 17, 1972–1981 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Chen F, Wang Y & Puett D Role of the invariant aspartic acid 99 of human choriogonadotropin β in receptor binding and biological activity. J. Biol. Chem. 266, 19357–19361 (1991). [PubMed] [Google Scholar]
- 33.Smits G et al. Glycoprotein hormone receptors: determinants in leucine-rich repeats responsible for ligand specificity. EMBO J. 22, 2692–2703 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Koppen CJ et al. Mechanism of action of a nanomolar potent, allosteric antagonist of the thyroid-stimulating hormone receptor. Br. J. Pharmacol. 165, 2314–2324 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Sande J et al. In Chinese hamster ovary K1 cells dog and human thyrotropin receptors activate both the cyclic AMP and the phosphatidylinositol 4,5-bisphosphate cascades in the presence of thyrotropin and the cyclic AMP cascade in its absence. Eur. J. Biochem. 229, 338–343 (1995). [PubMed] [Google Scholar]
- 36.Chen C-R, McLachlan SM & Rapoport B A monoclonal antibody with thyrotropin (TSH) receptor inverse agonist and TSH antagonist activities binds to the receptor hinge region as well as to the leucine-rich domain. Endocrinology 150, 3401–3408 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mueller S, Jaeschke H, Günther R & Paschke R The hinge region: an important receptor component for GPHR function. Trends Endocrinol. Metab. 21, 111–122 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Mizutori Y, Chen C-R, McLachlan SM & Rapoport B The thyrotropin receptor hinge region is not simply a scaffold for the leucine-rich domain but contributes to ligand binding and signal transduction. Mol. Endocrinol. 22, 1171–1182 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brüser A et al. The activation mechanism of glycoprotein hormone receptors with implications in the cause and therapy of endocrine diseases. J. Biol. Chem. 291, 508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schulze A et al. The intramolecular agonist is obligate for activation of glycoprotein hormone receptors. FASEB J. 34, 11243–11256 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Parma J et al. Somatic mutations causing constitutive activity of the thyrotropin receptor are the major cause of hyperfunctioning thyroid adenomas: identification of additional mutations activating both the cyclic adenosine 3′,5′-monophosphate and inositol phosphate-Ca2+ cascades. Mol. Endocrinol. 9, 725–733 (1995). [DOI] [PubMed] [Google Scholar]
- 42.Vlaeminck-Guillem V, Ho S-C, Rodien P, Vassart G & Costagliola S Activation of the cAMP pathway by the TSH receptor involves switching of the ectodomain from a tethered inverse agonist to an agonist. Mol. Endocrinol. 16, 736–746 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Sanders J et al. Human monoclonal thyroid stimulating autoantibody. Lancet 362, 126–128 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Sanders J et al. Crystal structure of the TSH receptor in complex with a thyroid-stimulating autoantibody. Thyroid 17, 395–410 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Evans M et al. Monoclonal autoantibodies to the TSH receptor, one with stimulating activity and one with blocking activity, obtained from the same blood sample. Clin. Endocrinol. 73, 404–412 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Sanders P et al. Crystal structure of the TSH receptor (TSHR) bound to a blocking-type TSHR autoantibody. J. Mol. Endocrinol. 46, 81–99 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Lomize MA, Pogozheva ID, Joo H, Mosberg HI & Lomize AL OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reily C, Stewart TJ, Renfrow MB & Novak J Glycosylation in health and disease. Nat. Rev. Nephrol. 15, 346–366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erbel PJA, Haseley SR, Kamerling JP & Vliegenthart JFG Studies on the relevance of the glycan at Asn-52 of the α-subunit of human chorionic gonadotropin in the αβ dimer. Biochem. J. 364, 485–495 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amr S et al. Activities of deglycosylated thyrotropin at the thyroid membrane receptor-adenylate cyclase system. J. Endocrinol. Invest. 8, 537–541 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Fares FA, Levi F, Reznick AZ & Kraiem Z Engineering a potential antagonist of human thyrotropin and thyroid-stimulating antibody. J. Biol. Chem. 276, 4543–4548 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Feng X, Müller T, Mizrachi D, Fanelli F & Segaloff DL An intracellular loop (IL2) residue confers different basal constitutive activities to the human lutropin receptor and human thyrotropin receptor through structural communication between IL2 and helix 6, via helix 3. Endocrinology 149, 1705–1717 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang M et al. Intrinsic differences in the response of the human lutropin receptor versus the human follitropin receptor to activating mutations. J. Biol. Chem. 282, 25527–25539 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Skiba MA & Kruse AC Autoantibodies as endogenous modulators of GPCR signaling. Trends Pharmacol. Sci. 42, 135–150 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Girdlestone C & Hayward S The DynDom3D webserver for the analysis of domain movements in multimeric proteins. J. Comput. Biol. 23, 21–26 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Sanders J et al. Characteristics of a human monoclonal autoantibody to the thyrotropin receptor: sequence structure and function. Thyroid 14, 560–570 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Mastronarde DN SerialEM: a program for automated tilt series acquisition on Tecnai microscopes using prediction of specimen position. Microsc. Microanal. 9, 1182–1183 (2003). [Google Scholar]
- 58.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Punjani A, Zhang H & Fleet DJ Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020). [DOI] [PubMed] [Google Scholar]
- 61.Scheres SHW A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 415, 406–418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pettersen EF et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Zhuang Y et al. Mechanism of dopamine binding and allosteric modulation of the human D1 dopamine receptor. Cell Res. 31, 593–596 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schüttelkopf AW & van Aalten DMF PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. D 60, 1355–1363 (2004). [DOI] [PubMed] [Google Scholar]
- 67.Croll TI ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D 74, 519–530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bligh EG & Dyer WJ A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 (1959). [DOI] [PubMed] [Google Scholar]
- 71.MacLean B et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marty MT et al. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal. Chem. 87, 4370–4376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang L & Hermans J Hydrophilicity of cavities in proteins. Proteins 24, 433–438 (1996). [DOI] [PubMed] [Google Scholar]
- 74.Betz R Dabble. 10.5281/zenodo.836914 (2017). [DOI] [Google Scholar]
- 75.Huang J et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klauda JB et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Case DA et al. Amber 2021 (Univ. California, San Francisco, 2021). [Google Scholar]
- 78.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S & Walker RC Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888 (2013). [DOI] [PubMed] [Google Scholar]
- 79.Hopkins CW, Le Grand S, Walker RC & Roitberg AE Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11, 1864–1874 (2015). [DOI] [PubMed] [Google Scholar]
- 80.Roe DR & Cheatham TE III . PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013). [DOI] [PubMed] [Google Scholar]
- 81.Humphrey W, Dalke A & Schulten K VMD: visual molecular dynamics. J. Mol. Graph. 14, 27–28 (1996). [DOI] [PubMed] [Google Scholar]
- 82.Latorraca NR et al. How GPCR phosphorylation patterns orchestrate arrestin-mediated signaling. Cell 183, 1813–1825.e18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Asarnow D, Palovcak E & Cheng Y UCSF pyem v0. 5. 10.5281/zenodo.3576630 (2019). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates for TSH-bound TSHR–Gs, TR1402-bound TSHR–Gs, M22-bound TSHR–Gs and CS-17-bound TSHR have been deposited in the PDB under accession codes 7T9I, 7UTZ, 7T9N and 7T9M, respectively. Sharpened and unsharpened cryo-EM density maps for TSH-bound TSHR–Gs (composite), TR1402-bound TSHR–Gs (composite), M22-bound TSHR–Gs (composite), CS-17-bound TSHR, and Org 2274179-0-bound TSHR have been deposited in the Electron Microscopy Data Bank under accession codes EMD-25758, EMD-26795, EMD-25763, EMD-25762 and EMD-27640, respectively. Sharpened maps, unsharpened maps, half-maps and masks for each composite map component (ligand-bound ECD or 7TM–G protein), for TSH-bound TSHR–Gs, TR1402-bound TSHR–Gs and M22-bound TSHR-Gs have been deposited in the Electron Microscopy Data Bank under accession codes EMD-27649 (TSH-bound ECD), EMD-27650 (TSH-bound 7TM–Gs), EMD-27647 (TR1402-bound ECD), EMD-27648 (TR1402 7TM–Gs), EMD-27651 (M22-bound ECD) and EMD-27652 (M22-bound 7TM–Gs). Final particle stacks and .star files for TSH-bound TSHR–Gs, TR1402-bound TSHR–Gs and M22-bound TSHR–Gs and CS-17-bound TSHR-containing particle shift and pose assignments have been uploaded to the Electron Microscopy Public Image Archive under the accession codes EMPIAR-11120. Structure files for OPM-based modelling of inactive and active TSHR are available in the Supplementary Information.