

Abstract
Tumor mutational burden (TMB) is an emerging predictive marker of response to immune checkpoint inhibitor therapies. We evaluated the correlation between clinical indicators and high‐throughput sequencing results and TMB in lung adenocarcinoma patients, with the aim of finding simpler and more economical factors as surrogate markers for TMB.
The medical records, next‐generation sequencing data, and immunohistochemistry results of 340 lung adenocarcinoma patients who were admitted to the First Affiliated Hospital of Zhengzhou University between 2019 and 2020 were collected. The mutated genes were screened for, and the obtained mutated genes were subjected to functional enrichment analysis using R software. A protein–protein interaction (PPI) network was also constructed, and significant modules in the network were identified. Gene Ontology (GO) analyses were performed for the core genes. Univariate and multivariate correlation analyses were performed to judge the correlation between gene mutations and TMB.
Genes with a junction mutation rate >1 were selected to construct PPI network and 13 high‐connection core genes were screened. The results of GO enrichment analysis showed that the biological processes related to mutant core genes mainly included mitotic cell cycle and cell aging. Subsequently, ATM (p = 0.006) and PIK3CA (p = 0.008) mutation positivity were identified by univariate and multivariate correlation analysis, while TP53 (p = 0.003) and EGFR (p = 0.008) mutation negativity were significantly associated with elevated TMB.
The results of this study demonstrate that ATM‐ and PIK3CA‐positive and EGFR‐negative mutation status are strongly associated with high levels of TMB and have the potential to be predictive biomarkers of response to immune checkpoint inhibitors in lung adenocarcinoma patients.
Keywords: immune checkpoint inhibitors, lung adenocarcinoma, tumor mutational burden (TMB)
Core genes mutated in lung adenocarcinoma are involved in the regulation of the cell cycle. Age, gender, smoking history, pathological stage, and lymph node infiltration are associated with TMB in patients with lung adenocarcinoma. Mutations in EGFR, ATM, and PIK3CA may provide new ideas for predicting TMB in lung adenocarcinoma patients.
1. INTRODUCTION
Lung adenocarcinoma (LUAD) is a type of malignant non‐small cell lung cancer originating from the glandular epithelium of the bronchus mucosa, accounting for about 45% of all lung cancer. Its incidence rate and mortality rate show an increasing trend. 1 Conventional therapies for LUAD include surgical resection, radiotherapy, and chemotherapy. In recent years, immunotherapy of cancers including LUAD has attracted increasing attention. 2
Cancer is caused by the accumulation of somatic mutations that lead to the expression of neoantigens, 3 which can normally be used by the immune system to recognize and clear tumor cells from the tumor microenvironment. However, to survive and grow, tumor cells can adopt different strategies to suppress the body's immune system so that it becomes unable to kill tumor cells, thereby surviving all stages of the antitumor immune response. Under normal physiological conditions, immune checkpoints are critical for maintaining self‐tolerance and preventing autoimmunity, and can also protect tissues from damage when the immune system responds to pathogen infection. However, tumors may interact with such signals, thereby promoting immune escape. 4 One of the most promising approaches to activate therapeutic antitumor immunity is the blockade of immune checkpoints, which can induce the proliferation and activation of immune cells and fight against cancer cells. 5
Immune checkpoint inhibitors are currently being widely used to treat cancer. Studies have reported that programmed cell death ligand 1 (PD‐L1) and tumor mutational burden (TMB) are independent predictors of response to immunotherapy in patients with non‐small cell lung cancer (NSCLC), 6 , 7 so both PD‐L1 and TMB are promising as biomarkers for the effectiveness of immune checkpoint inhibitors. The use of PD‐L1 expression as a biomarker has been widely studied. In general, the remission rate of anti‐PD‐1/PD‐L1 therapy in patients with PD‐L1‐positive tumors is significantly higher than that of PD‐L1‐negative patients. 8 However, in the tumor microenvironment, PD‐L1 is expressed in both tumor cells and non‐tumor cells. 9 The detection of the PD‐L1 expression level is only suitable for patients receiving PD‐1/PD‐L1‐blocking therapy, not for other types of immunotherapies. However, TMB can predict the prognosis of many types of tumors after anti‐PD‐1/PD‐L1 immunotherapy. 10 TMB is a new predictor of immunosuppressive response. Most studies on TMB have evaluated the relationship between TMB and immunosuppressive response, but the related clinical features have not been well documented. Therefore, in this study, we evaluated the correlation between TMB and clinicopathological features in patients with LUAD to find more convenient factors as alternative markers of TMB.
2. METHODS
2.1. Sample collection
Medical record data, next‐generation high‐throughput sequencing data (NGS), and immunohistochemical results of 340 LUAD patients who were admitted to the First Affiliated Hospital of Zhengzhou University from 2019 to 2020 and who were definitively pathologically diagnosed by lung puncture were collected. The corresponding specimens were lung puncture tissues. Patient enrollment and genomic studies were conducted in accordance with the Declaration of Helsinki. The study was approved by the ethics committee of the First Affiliated Hospital of Zhengzhou University. All patients provided oral and written informed consent for sample acquisition for research purposes. The inclusion criteria were: (1) immunohistochemical results were examined by pathologists and diagnosed as LUAD; (2) no history of other malignant tumors; and (3) not in receipt of chemotherapy or immunotherapy prior to diagnosis. The exclusion criteria were: (1) combination with other lung diseases; (2) combined disease development with neovascularization, such as diabetes or rheumatoid arthritis; (3) pregnant or lactating patients; and (4) patients with incomplete clinical data. All enrolled samples were examined by pathologists to determine the histological subtype and TNM stage. In total, there were 173 men and 167 women, 155 cases were younger than 65 years old and 185 cases were older than 65 years old. There were 104 patients with a history of smoking and 236 patients that did not smoke. There were 45 cases of stage I disease and 295 cases of stages II–IV disease.
2.2. Sample processing and DNA extraction
Fixation of lung puncture tumor tissue with formalin followed by paraffin embedding (FFPE).Genomic DNA was extracted from each FFPE sample using the GeneRead DNA FFPE Kit (Qiagen).
2.3. NGS‐based gene panel test
For pre‐library preparation, purified genomic DNA was first fragmented into DNA pieces around 200 bp in length using enzymatic method. After end‐repairing, A‐tailing, T‐adaptors were ligated on both ends, followed by universal primer‐mediated amplification. The purified pre‐library was hybridized with a customized biotin probe panel (the 457 genes panel, BerryoncoPan, Berryoncology) 11 to capture target DNA fragments. Oncogenes in the probe library were referenced from Catalog of Somatic Mutations in Cancer, the Cancer Genome Atlas, OncoKB, and the Oncomine database oncogene database. The captured DNA fragments were amplified using universal primers and the products were purified to obtain the final library. Paired‐end multiplex samples were sequenced using the NovaSeq 6000 sequencing platform (Illumina). Each sample was sequenced to a depth of around 2000×. The resulting sequences were trimmed, low quality sequences were filtered, and variant calling was performed. The following variant was filtered: non‐synonymous SNPs, indels, and spliced mutations. The allele frequency of variant (cutoff value ≥3%) was defined as somatic variants. Variant allele frequencies (cutoff value ≥1%) and at least 20 high quality reads were defined as cancer hot spots.
2.4. Protein interaction network and module analysis
Protein–protein interaction (PPI) networks were constructed using the STRING database. 12 Subsequently, significant modules in the PPI network were identified using the Cytoscape MCODE plug‐in, with the parameters set as follows: degree cutoff = 2, node score cutoff = 0.2, and K‐core = 2. Subsequently, highly connected genes were taken as core genes in the significant module.
2.5. The calculation of TMB
The tumor mutation burden (TMB) was determined by the number of all the non‐synonymous mutation and indel variants per magabase of coding regions. The 457 gene panels cover the coding region of 1,141,951 bp. Hence, TMB was calculated with the number of all the non‐synonymous mutations and indel variants/1.14 Mb.
2.6. Enrichment analysis of mutant genes
Filtering of mutation characteristics and screening of mutated genes were done according to the previous studies. 11 The R package Cluster Profiler in R software was used for Gene Ontology (GO) enrichment analysis to study the biological significance of core mutation genes. 13 The GO database includes three levels, cellular component, molecular function, and biological process. Here, p < 0.05 was considered statistically significant.
2.7. Statistical analysis
SPSS 21.0 statistical software was used for data analysis. The chi‐squared test was used to analyze the TMB level and clinicopathological parameters of LUAD patients. The Mann–Whitney U test was used to compare TMB and gene mutation. Multivariate linear regression analysis was used for correlation analysis. p < 0.05 was considered statistically significant.
3. RESULTS
3.1. Gene mutation frequency in LUAD
Gene mutation were analyzed by Illumina sequencing platform and target probe capture technology. The panel of this platform allows the simultaneous evaluation of the mutation status of 457 tumor‐associated genes (BerryoncoPan, Berryoncology). We screened genes with a mutation frequency >1, and the mutation frequencies are shown in Figure 1.
FIGURE 1.
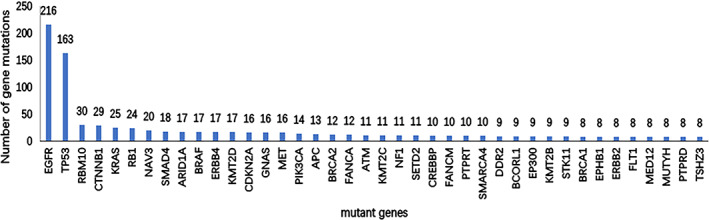
Mutated genes and frequencies in 340 LUAD patients. Genes with the highest mutation frequency are shown here
3.2. PPI network construction and analysis
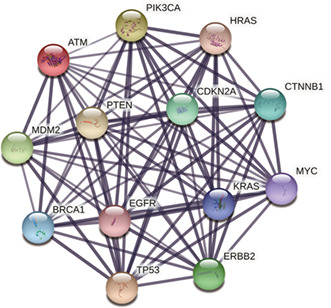
After acquiring data on the differential expression gene interactions using the online database STRING, the PPI network diagram was completed using the MCODE plug‐in in Cytoscape software, and significant modules were screened out by taking an MCODE score value >30 (Figure 2). Subsequently, PPI data were ranked by degree to select 13 genes with high connectivity: TP53, ATM, PTEN, BRCA1, HRAS, KRAS, PIK3CA, MYC, CDKN2A, CTNNB1, MDM2, EGFR, and ERBB2 (Table 1), which were the core genes.
FIGURE 2.
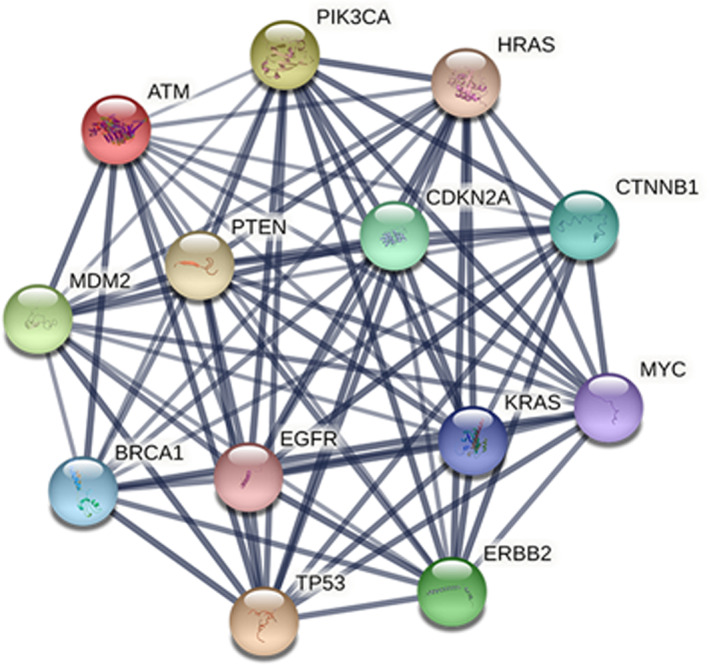
PPI network of 13 core genes
TABLE 1.
Connectivity and mutation rate of core mutated genes in the PPI network
| Genes | Degree | Mutation rate |
|---|---|---|
| TP53 | 64 | 47.9% |
| ATM | 58 | 3.2% |
| PTEN | 57 | 2.1% |
| BRCA1 | 56 | 2.4% |
| HRAS | 55 | 0.6% |
| KRAS | 55 | 7.4% |
| PIK3CA | 55 | 4.1% |
| MYC | 54 | 0.3% |
| CDKN2A | 52 | 4.7% |
| CTNNB1 | 51 | 8.5% |
| MDM2 | 51 | 1.1% |
| EGFR | 50 | 63.5% |
| ERBB2 | 50 | 2.4% |
3.3. Gene set enrichment analysis
To better understand the biological functions of the core mutation genes, GO enrichment analyses were performed. The GO enrichment analysis results showed that the biological processes associated with the mutated genes mainly included negative regulation of mitotic cell cycle, cell cycle arrest, cell aging, and regulation of DNA metabolic process. The molecular functions mainly included 1‐phosphatidylinositol‐3‐kinase activity, phosphatidylinositol 3‐kinase activity, phosphatidylinositol kinase activity, protein phosphatase binding, and ubiquitin protein ligase binding. In addition, these mutated genes were also closely associated with cellular components such as basal plasma membrane, basolateral plasma membrane, basal part of cell, extrinsic component of membrane, membrane raft, membrane microdomain, apical plasma membrane, and membrane region (Figure 3).
FIGURE 3.
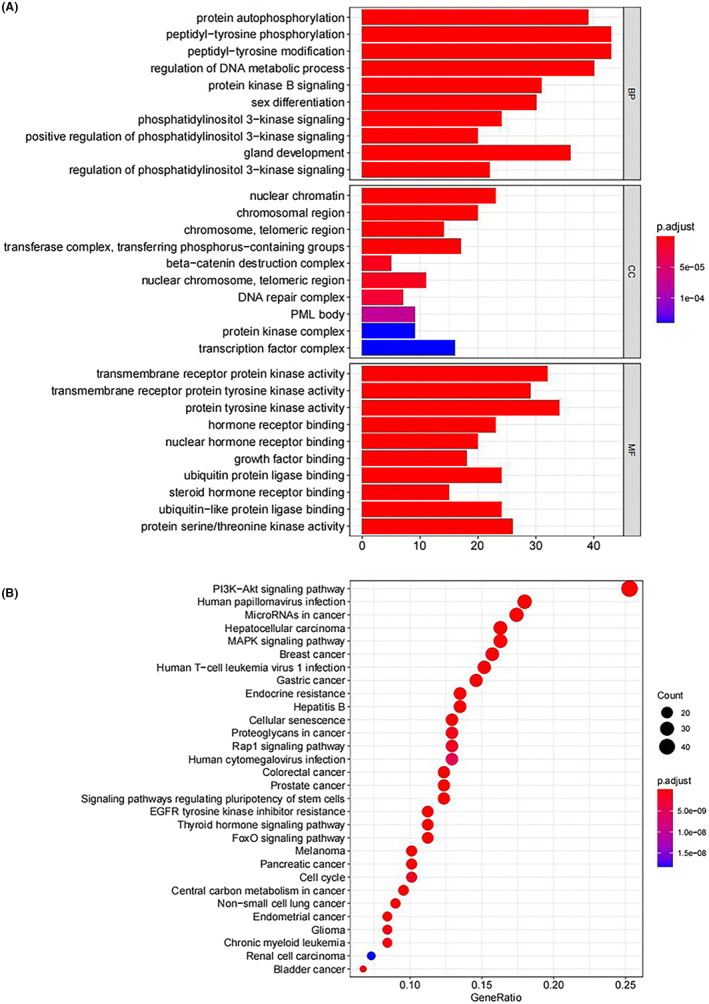
GO enrichment histogram. The abscissa is the number of core genes enriched in the GO analysis, the ordinate is the enriched GO, and the color represents the statistical significance
3.4. Relationship between TMB levels and clinicopathological characteristics
Table 2 shows the percentage of patients with high TMB among the 340 LUAD patients. The threshold for high TMB was set to 10 based on previous studies. 11 It was higher in patients younger than 65 years (65/105 vs. 90/235, χ 2 = 16.305, p < 0.001), male patients (72/105 vs. 101/235, χ 2 = 19.020, p < 0.001), smokers (51/105 vs. 53/235, χ 2 = 21.139, p < 0.001), and patients with stage II–IV (92/105 vs 173/235, χ 2 = 5.708, p = 0.006), and N1‐3 (77/105 vs. 134/235, χ 2 = 10.081, p = 0.002).
TABLE 2.
Relationship between TMB and clinicopathological characteristics
| Clinicopathological characteristics | number | TMB level | χ 2 | p | |
|---|---|---|---|---|---|
| Low TMB | High TMB | ||||
| Age | 235 | 105 | |||
| ≥65 | 185 | 145 | 40 | 16.305 | <0.001 |
| <65 | 155 | 90 | 65 | ||
| Gender | |||||
| Male | 173 | 101 | 72 | 19.020 | <0.001 |
| Female | 167 | 134 | 33 | ||
| Smoking | |||||
| Yes | 104 | 53 | 51 | 21.139 | <0.001 |
| No | 236 | 182 | 54 | ||
| Stage | |||||
| I | 45 | 38 | 7 | 5.708 | 0.011 |
| II–IV | 295 | 197 | 98 | ||
| T | |||||
| T1‐2 | 206 | 148 | 58 | 1.821 | 0.110 |
| T3‐4 | 134 | 87 | 47 | ||
| N | |||||
| N0 | 116 | 93 | 23 | 10.081 | 0.001 |
| N1‐3 | 224 | 142 | 82 | ||
| M | |||||
| M0 | 132 | 95 | 37 | 0.822 | 0.216 |
| M1 | 208 | 140 | 68 | ||
3.5. Relationship between core genes and TMB
Univariate correlation test results indicated that of the core genes, ATM (p = 0.006) and PIK3CA (p = 0.008) mutation positivity were significantly associated with elevated TMB, while TP53 (p = 0.003) and EGFR (p = 0.008) mutation negativity were significantly associated with elevated TMB (Table 3).
TABLE 3.
Relationship between TMB and core genes
| N | Mean | 95%CI | p | |
|---|---|---|---|---|
| TP53 | ||||
| + | 163 | 5.1775 | 4.6550–5.7038 | 0.003 |
| − | 177 | 5.3329 | 4.4239–6.2437 | |
| ATM | ||||
| + | 11 | 10.1909 | 5.8400–14.8874 | 0.006 |
| − | 329 | 5.0935 | 4.5462–5.6248 | |
| PTEN | ||||
| + | 7 | 6.2557 | 3.7528–8.6342 | 0.168 |
| − | 333 | 5.2375 | 4.6827–5.7790 | |
| BRCA1 | ||||
| + | 8 | 7.7738 | 4.3800–11.6352 | 0.08 |
| − | 332 | 5.1987 | 4.6397–5.7482 | |
| HRAS | ||||
| + | 2 | 11.8250 | 8.7600–14.8900 | 0.054 |
| − | 338 | 5.2196 | 4.6696–5.7453 | |
| KRAS | ||||
| + | 25 | 7.3920 | 4.8762–10.1858 | 0.119 |
| − | 315 | 5.0891 | 4.5425–5.6402 | |
| PIK3CA | ||||
| + | 14 | 9.1800 | 5.5717–13.9133 | 0.008 |
| − | 326 | 5.0900 | 4.5554–5.6064 | |
| MYC | ||||
| + | 1 | 4.3800 | 4.3800–4.3800 | 0.865 |
| − | 339 | 5.2610 | 4.7306–5.8124 | |
| CDKN2A | ||||
| + | 16 | 7.6625 | 5.8387–9.3706 | 0.002 |
| − | 324 | 5.1397 | 4.5742–5.6958 | |
| CTNNB1 | ||||
| + | 29 | 6.4021 | 4.9457–7.9788 | 0.005 |
| − | 311 | 5.1518 | 4.5875–5.7167 | |
| MDM2 | ||||
| + | 4 | 10.2900 | 3.500–25.4000 | 0.130 |
| − | 336 | 5.1985 | 4.6432–5.7111 | |
| EGFR | ||||
| + | 216 | 4.3901 | 3.8777–4.8967 | 0.014 |
| − | 124 | 6.7710 | 5.6243–8.0017 | |
| ERBB2 | ||||
| + | 8 | 4.5998 | 2.6300–5.8167 | 0.901 |
| − | 332 | 5.2743 | 4.7019–5.8167 | |
3.6. Relationship between TMB core genes, clinicopathological characteristics, and TMB
Multiple linear regression was used to predict TMB according to the clinicopathological parameters and core mutation genes. As shown in Table 4, the regression model had statistical significance (F = 6.013, p < 0.001, adjusted R 2 = 0.168). The influence of all four independent variables included in the model on TMB was statistically significant (p < 0.05). The results are shown in Table 4. We deduced the equation for predicting TMB:
TABLE 4.
Relationship between TMB core genes, clinicopathological characteristics, and TMB
| n | R | p | |
|---|---|---|---|
| Age | 340 | 0.577 | 0.287 |
| Gender | 340 | 1.396 | 0.044 |
| Smoking | 340 | 1.315 | 0.089 |
| Stage | 340 | 0.499 | 0.454 |
| N | 340 | −0.707 | 0.448 |
| EGFR | 340 | −1.443 | 0.013 |
| TP53 | 340 | −0.134 | 0.803 |
| ATM | 340 | 3.250 | 0.034 |
| PIK3CA | 340 | 4.102 | 0.002 |
| CDKN2A | 340 | 1.951 | 0.121 |
| CTNNB1 | 340 | 1.298 | 0.172 |
TMB = 4.059 + 1.396 × (male: 1, female: 0) − 1.443 × (EGFR+: 1, EGFR−: 0) + 3.250 × (ATM+: 1, ATM−: 0) + 4.102 × (PIK3CA+: 1, PIK3CA−: 0).
4. DISCUSSION
The results showed that a high level of TMB was strongly correlated with ATM and PIK3CA mutation positivity and EGFR mutation negativity. The ATM gene is a pathogenic mutation gene on chromosome 11q22.3, which is one of the frequently mutated genes in cancer. ATM protein is a member of the PI3K‐like protein kinase family, which is involved in genome stability, cell response to DNA damage, and cell cycle control. 14 The main function of ATM protein is to participate in the regulation of the cell cycle and recognize and repair DNA damage.
PI3K is a lipid kinase that regulates important signaling pathways in tumorigenesis, including cell proliferation, adhesion, survival, and motility. 15 , 16 PI3K consists of the p85 regulatory sub‐unit and p110 catalytic sub‐unit. The latter is encoded by three genes, PIK3CA, PIK3CB, and PIK3CD. PIK3CA mutation is the most common gene mutation in cancer and is an independent risk factor for affecting the overall survival (OS) and progression‐free survival (PFS) in NSCLC patients. 4 , 17
In contrast to patients with EGFR mutations who are insensitive to PD‐1/L1 inhibitors (HR = 1.09, p = 0.51), EGFR wild‐type patients were shown to benefit from PD‐1/L1 inhibitors (HR = 0.73, p < 0.00001). The same study provided evidence of a correlation between EGFR gene mutations and a noninflammatory tumor microenvironment with immune tolerance and weak immunogenicity, contributing to the poor response of NSCLCs to PD‐1 blockade, and found that EGFR mutation (EGFR‐mut) was negatively correlated with TMB. 18 Other studies have also demonstrated an inverse correlation between TMB and prognosis with targeted therapies in EGFR‐mut lung cancer patients exhibiting poor responses to immunotherapy. 19 , 20
Studies have reported that smoking is significantly associated with high TMB levels. 21 The TMB level was significantly higher in men than in women. 22 A significant inverse correlation was found between age and TMB level in TP53‐mut LUAD patients. 23 This is consistent with the results of the present study. TMB can predict the prognosis of many types of tumors after anti‐PD‐1/PD‐L1 immunotherapy. 10 Patients with high TMB have longer OS, 10 PFS, 21 and objective response rate (ORR) 24 after receiving immunotherapy compared with those with low TMB. Studies that counted the data of durable clinical benefit (DCB) and TMB in patients who received immunotherapy concluded that TMB was significantly higher in patients who experienced DCB than those who did not. 21
Tumors with a large number of non‐synonymous somatic mutations identified by whole‐exome sequencing (WES) are more likely to respond to checkpoint blocking immunotherapy. Theoretically, these tumors will have a higher diversity of new antigens, and when the inhibition of PD‐1/PD‐L1 is blocked, the immune response can be triggered. 25 , 26 Melanoma and NSCLC showed the strongest response to PD‐1/PD‐L1 blockade, both of which had higher TMB. 27 As assessed by WES, a high burden of non‐synonymous mutations was associated with ORR, long‐term clinical benefit, and PFS in patients receiving anti‐PD‐1/PD‐L1 treatment. However, WES is expensive and time‐consuming, so it is not a routine clinical examination. 28 Targeted panel sequences focusing on cancer‐related genes are also available. Targeted panel sequencing analysis can provide an alternative marker for TMB and may be easier to introduce into clinical application. 29 In addition, the cost of using WES to determine TMB is about 10 times that of using cancer hot spot panel sequencing to determine EGFR and ATM status. Therefore, a simpler and more economical method is needed to estimate TMB and predict the response to checkpoint blockade. It may be a useful alternative to predict TMB by analyzing specific gene changes, such as EGFR.
Because TMB is considered a biomarker of response to immune checkpoint inhibitors, analysis of ATM, PIK3CA, and EGFR mutations may provide a rapid and simple way to predict TMB. Although the correlation of these parameters with TMB in this predictive model is not high enough, biomarkers involving the combination of multiple factors may become more important in the future. Further studies are needed to confirm our results and assess the value of ATM, PIK3CA, and EGFR mutations as predictive biomarkers of response to immune checkpoint inhibitors in patients with NSCLC.
CONFLICT OF INTEREST
Yin Wang and Jian Cao are employees of Berry Oncology.
AUTHOR CONTRIBUTIONS
Kai Ma, Yin Wang, and Fengxiang Huang designed the experiments. Jiaqi Tang, Jiaojiao Lou, and Panfeng Sun collected data. Qilong Wang, Ruiping Qiao and Panfeng Sun analyzed the data. Kai Ma, Jiming Si, and Fengxiang Huang conducted statistical analysis. Kai Ma, Yan Kang, and Jian Cao wrote the manuscript. Lijun Miao revised the manuscript.
ETHICS APPROVAL
Patient enrollment and genomic studies were conducted in accordance with the Declaration of Helsinki. The study was approved by the ethics committee of the First Affiliated Hospital of Zhengzhou University.
PATIENT CONSENT STATEMENT
Clinical information was collected with the informed consent of each patient.
ACKNOWLEDGMENTS
The authors thank Catherine Perfect, MA (Cantab), from Liwen Bianji (Edanz) (www.liwenbianji.cn/), for editing the English text of a draft of this manuscript.
Ma K, Huang F, Wang Y, et al. Relationship between tumor mutational burden, gene mutation status, and clinical characteristics in 340 cases of lung adenocarcinoma. Cancer Med. 2022;11:4389‐4397. doi: 10.1002/cam4.4781
Funding informationThis work was funded by the Joint construction project of medical science and technology in Henan Province (LHGJ20190206), the Key Research Projects of Ordinary Institution of Higher Education in Henan Province (20A320058) and the Youth Innovation Fund of First Affiliated Hospital of Zhengzhou University (YNQN2017170).
Kai Ma, Fengxiang Huang and Yin Wang contributed equally in this study.
DATA AVAILABILITY STATEMENT
The data used to support the findings of this study are available from the corresponding author upon request.
REFERENCES
- 1. Bade BC, Dela Cruz CS. Lung cancer 2020: epidemiology, etiology, and prevention. Clin Chest Med. 2020;41:1‐24. [DOI] [PubMed] [Google Scholar]
- 2. Kleczko EK, Kwak JW, Schenk EL, Nemenoff RA. Targeting the complement pathway as a therapeutic strategy in lung cancer. Front Immunol. 2019;10:954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125:3413‐3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beck JT, Ismail A, Tolomeo C. Targeting the phosphatidylinositol 3‐kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway: an emerging treatment strategy for squamous cell lung carcinoma. Cancer Treat Rev. 2014;40:980‐989. [DOI] [PubMed] [Google Scholar]
- 5. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33:1974‐1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093‐2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carbone DP, Reck M, Paz‐Ares L, et al. First‐line nivolumab in stage IV or recurrent non‐small‐cell lung cancer. N Engl J Med. 2017;376:2415‐2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patel SP, Kurzrock R. PD‐L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847‐856. [DOI] [PubMed] [Google Scholar]
- 9. Santarpia M, Karachaliou N. Tumor immune microenvironment characterization and response to anti‐PD‐1 therapy. Cancer Biol Med. 2015;12:74‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598‐2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng Y, Zhang Y, Yuan Y, et al. The comprehensive analyses of genomic variations and assessment of TMB and PD‐L1 expression in Chinese lung adenosquamous carcinoma. Front Genet. 2021;11:609405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447‐D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen L, Zhang YH, Wang S, Zhang Y, Huang T, Cai YD. Prediction and analysis of essential genes using the enrichments of gene ontology and KEGG pathways. PLoS One. 2017;12(9):e0184129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uziel T, Savitsky K, Platzer M, et al. Genomic organization of the ATM gene. Genomics. 1996;33:317‐320. [DOI] [PubMed] [Google Scholar]
- 15. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3‐kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606‐619. [DOI] [PubMed] [Google Scholar]
- 16. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2009;28:1075‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Y, Wang Y, Li J, Li J, Che G. Clinical significance of PIK3CA gene in non‐small‐cell lung cancer: a systematic review and meta‐analysis. Biomed Res Int. 2020;3608241:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dong ZY. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD‐1 blockade in non‐small cell lung cancer. Onco Targets Ther. 2017;6:e1356145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Offin M, Rizvi H, Tenet M, et al. Tumor mutation burden and efficacy of EGFR‐tyrosine kinase inhibitors in patients with EGFR‐mutant lung cancers. Clin Cancer Res. 2019;25:1063‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee CK, Man J, Lord S, et al. Checkpoint inhibitors in metastatic EGFR‐mutated non‐small cell lung cancer – a meta‐analysis. J Thorac Oncol. 2017;12:403‐407. [DOI] [PubMed] [Google Scholar]
- 21. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science. 2015;348:124‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiao D, Pan H, Li F, Wu K, Zhang X, He J. Analysis of ultra‐deep targeted sequencing reveals mutation burden is associated with gender and clinical outcome in lung adenocarcinoma. Oncotarget. 2016;7:22857‐22864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang W, Flemington EK, Zhang K. Mutant TP53 disrupts age‐related accumulation patterns of somatic mutations in multiple cancer types. Cancer Genet. 2016;209:376‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hellmann MD, Nathanson T, Rizvi H, et al. Genomic features of response to combination immunotherapy in patients with advanced non‐small‐cell lung cancer. Cancer Cell. 2018;33:843‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med. 2014;371:2189‐2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Le DT UJN, Wang H, Bartlett BR, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372:2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD‐1 blockade. Cancer Immunol Res. 2016;4:959‐967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Campesato LF, Barroso‐Sousa R, Jimenez L, et al. Comprehensive cancer‐gene panels can be used to estimate mutational load and predict clinical benefit to PD‐1 blockade in clinical practice. Oncotarget. 2015;6:34221‐34227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.