

SUMMARY
The cyclic AMP pathway promotes melanocyte differentiation by activating CREB and the cAMP-regulated transcription co-activators 1–3 (CRTC1–3). Differentiation is dysregulated in melanomas, although the contributions of CRTC proteins is unclear. We report a selective differentiation impairment in CRTC3 KO melanocytes and melanoma cells, due to downregulation of oculo-cutaneous albinism II (OCA2) and block of melanosome maturation. CRTC3 stimulates OCA2 expression by binding to CREB on a conserved enhancer, a regulatory site for pigmentation and melanoma risk. CRTC3 is uniquely activated by ERK1/2-mediated phosphorylation at Ser391 and by low levels of cAMP. Phosphorylation at Ser391 is constitutively elevated in human melanoma cells with hyperactivated ERK1/2 signaling; knockout of CRTC3 in this setting impairs anchorage-independent growth, migration, and invasiveness, whereas CRTC3 overexpression supports cell survival in response to the mitogen-activated protein kinase (MAPK) inhibitor vemurafenib. As melanomas expressing gain-of-function mutations in CRTC3 are associated with reduced survival, our results suggest that CRTC3 inhibition may provide therapeutic benefit in this setting.
In brief
MAPK/ERK and cAMP pathways influence differentiation of melanocytes and melanoma progression. Ostojić et al. show that the CREB coactivator CRTC3 is activated by both pathways, leading to increased expression of genes regulating melanin production and cell migration. Decreases in CRTC3 activity result in hypopigmentation and impairment of melanoma oncogenesis.
Graphical Abstract
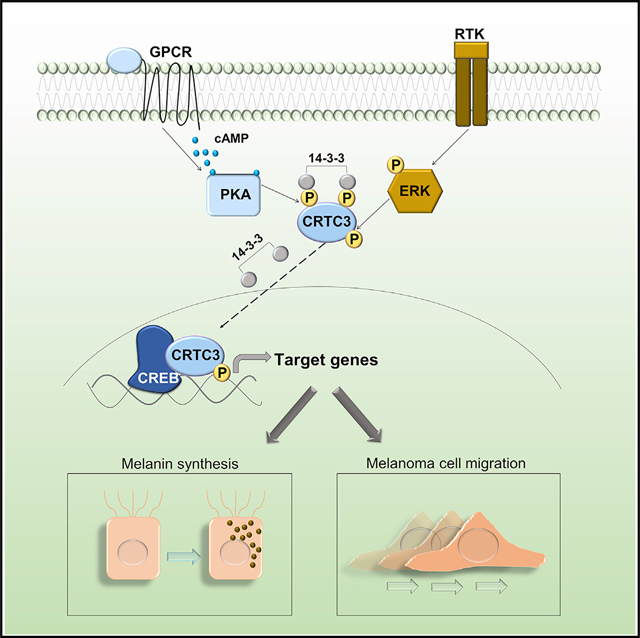
INTRODUCTION
Increased melanoma risk in humans is correlated with pigmentation variants, light skin, and poor tanning ability, although the molecular mechanisms are not fully understood (Lin and Fisher, 2007; Scherer and Kumar, 2010). Regulators of melanocyte homeostasis are frequently involved in melanoma development, as these tumors subvert core signaling pathways present in normal melanocytes during their progression (Shannan et al., 2016; Tsoi et al., 2018; Vandamme and Berx, 2014).
Elevations in intracellular cyclic AMP (cAMP) upregulate melanin synthesis, DNA repair, and survival pathways in melanocytes (Wolf Horrell et al., 2016). cAMP signaling is required for both basal (constitutive) and stimulus-induced pigmentation, depending in part on the intracellular cAMP levels (Cui et al., 2007; Sánchez-Más et al., 2004); they reflect the relative activities of cAMP-generating adenylyl cyclases and hydrolyzing phosphodiesterases (PDEs). Although the underlying mechanisms are unclear, aberrant cAMP signaling has been reported in a variety of human cancers; indeed, this second messenger can have both positive and negative impacts on oncogenesis (Fajardo et al., 2014).
cAMP stimulates the protein kinase A (PKA)-mediated phosphorylation of CREB leading to the expression of target genes, including MITF, the master melanocyte regulator, and a melanoma oncogene (Bertolotto et al., 1998; Buscà and Ballotti, 2000; Levy et al., 2006). In addition to stimulating the expression of genes that promote growth and survival, MITF also regulates the expression of melanogenic enzymes such as tyrosinase (TYR) and dopachrome tautomerase (DCT) (Wellbrock and Arozarena, 2015).
Superimposed on the effects of cAMP, the mitogen-activated protein kinase (MAPK) pathway also modulates melanocyte proliferation and differentiation, in part by regulating MITF activity (Hemesath et al., 1998; Price et al., 1998; Wu et al., 2000). MAPK and cAMP pathways show a nonlinear interaction that can be synergistic or antagonistic, depending on the signaling environment (Buscà et al., 2000; Dumaz et al., 2006; Herraiz et al., 2011; Niwano et al., 2018).
The MAPK pathway is commonly hyperactivated in melanomas, due to mutations in RAS and BRAF that lead to downstream induction of the Ser/Thr kinases ERK1/2 (Wellbrock and Arozarena, 2016). BRAF inhibitors are effective in the treatment of melanomas, but rates of acquired treatment resistance and relapse are also high (Kozar et al., 2019), pointing to the involvement of other signaling pathways that can circumvent BRAF inhibition. Within this group, the CREB pathway has been reported to confer resistance to BRAF blockers (Johannessen et al., 2013).
cAMP-regulated transcription co-activators 1–3 (CRTC1–3) function as effectors of cAMP signaling. In the basal state, salt-inducible kinases (SIKs) sequester CRTCs in the cytoplasm by their phosphorylation at 14-3-3 binding sites. Increases in cAMP promote PKA-mediated phosphorylation and inhibition of the SIKs, leading to CRTC dephosphorylation, nuclear migration, and recruitment to CREB binding sites (Altarejos and Montminy, 2011). In keeping with their considerable sequence homology, CRTCs have been found to exert overlapping effects on CREB target gene expression; yet, knockout (KO) studies also reveal distinct phenotypes for each family member (Altarejos et al., 2008; Blanchet et al., 2015; MacKenzie et al., 2013; Song et al., 2010). In melanocytes, CRTCs have been proposed as targets for the treatment of hyperpigmentary disorders by virtue of their ability to mediate effects of CREB on MITF expression (Bang et al., 2017; Kim et al., 2019, 2020; Yun et al., 2019).
Here, we show that deletion of the CRTC3 but not CRTC1 or CRTC2 genes affects pigmentation and melanocyte fitness. In line with overlapping effects of CRTC family members, MITF expression is only marginally affected in CRTC3 KO animals. Melanosome maturation is defective in CRTC3 mutant melanocytes, reflecting decreases in the expression of the melanosomal transporter oculo-cutaneous albinism II (OCA2), a key regulator of pigmentation and a melanoma susceptibility gene. CREB and CRTC3 were found to stimulate OCA2 expression by binding to cAMP responsive elements (CREs) on a conserved distal enhancer. The specificity of the CRTC3 phenotype reflects in part the ability of this co-activator to integrate signals from both cAMP and MAPK/ERK pathways. By profiling transcriptomes of CRTC3-depleted murine and human melanoma cells, we found that CRTC3 regulates genes involved in cAMP signaling, pigmentation, and cell motility. CRTC3 activity is upregulated in subsets of human melanomas, where it is associated with reduced survival. Our results suggest that CRTC3 has a therapeutic benefit in the treatment of pigmentary disorders, cutaneous melanoma, and potentially other conditions characterized by dysregulated cAMP/MAPK crosstalk.
RESULTS
Absence of CRTC3 in mice impairs melanocyte differentiation
We noticed that mice with a KO of CRTC3 but not CRTC1 or CRTC2 had decreased fur pigmentation, which was readily observable at the onset of pigmentation (3- to 5-day-old pups) and quantified as 65% of wild type (WT) (Figure 1A; Figure S1A). A pigmentation defect was present in CRTC3 KO animals of both sexes and was not progressive or related to premature graying. Knowing the importance of the CREB pathway in regulating MITF expression and melanogenesis, we considered that CRTC3 expression in skin could exceed those of CRTC1 and CRTC2. However, this was not the case in whole skin or primary melanocytes (Figures 1B and 1C; Figures S1B and S1C). CRTC3 KO skin did not show defects in protein accumulation of MITF or several of its target genes (TYR, DCT, and PMEL), likely reflecting compensation by CRTC1 and CRTC2 (Figures 1B and 1C). To identify changes that might explain the CRTC3 KO phenotype, we performed RNA sequencing (RNA-seq) on WT and CRTC3 KO neonatal skins. Consistent with protein accumulation data, mRNA levels for MITF and major melanogenic enzymes were not significantly altered in the KO (Table S1). Transporters were among the most significant downregulated gene categories in this analysis; indeed, three of them were melanocyte specific (Figures 1D, 1E, and 1F). Although melanocytes account for less than 10% of cells in the skin, the melanocyte-specific gene OCA2 scored as the most significantly downregulated gene in CRTC3 KOs (Table S1). OCA2 encodes a transmembrane anion transporter that regulates melanosomal pH and that is essential for the activity of melanogenic enzymes, in particular TYR (Bellono et al., 2014; Park et al., 2015). Alterations in OCA2 underlie natural pigmentation variants, albinism, and melanoma risk in humans (Jannot et al., 2005; Sturm et al., 2008; Duffy et al., 2010; Donnelly et al., 2012; Hawkes et al., 2013).
Figure 1. Hypopigmentation in CRTC3 knockout mice.
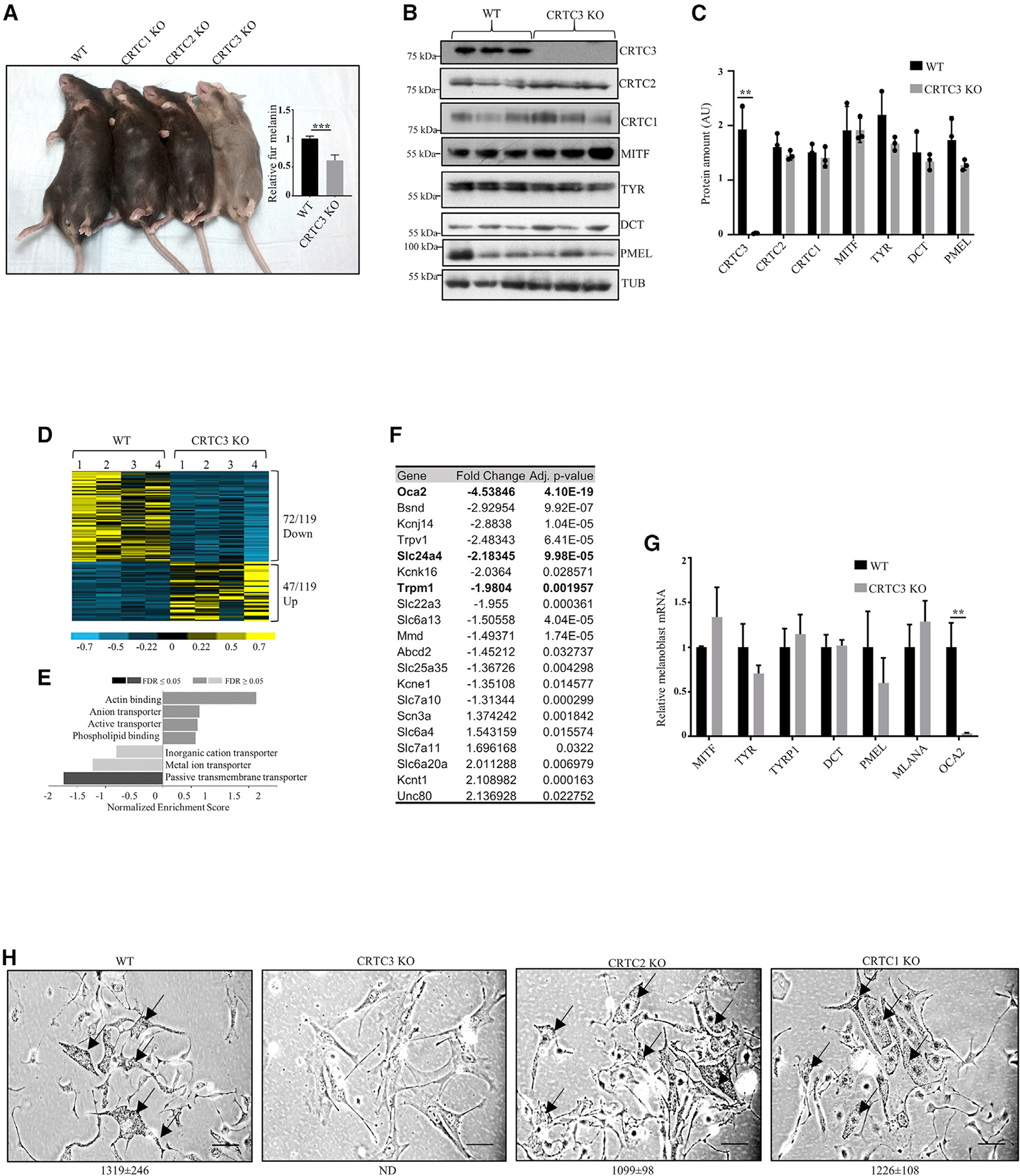
(A) Fur color comparison between mice with individual knockouts of CRTC family members and quantification of melanin content in dorsal hair from WT and CRTC3 KO mice (N = 3 per group, 5 months old). Significance determined by Welch’s t test.
(B) Western blots from whole skin of WT and CRTC3 KO mice (post-natal day 2 [P2]) showing expression of CRTCs, melanogenic enzymes, and structural proteins.
(C) Quantification of protein levels from (B). Significance determined by Welch’s t test.
(D) Heatmap of significant differentially expressed genes in RNA sequencing (RNA-seq) experiments of WT and CRTC3 KO whole skins (P2, N = 4 per group).
(E) Clustering of significant differentially expressed genes in RNA-seq experiments.
(F) List of transporters from RNA-seq experiments shown in (E). Melanocyte-specific transporters are in bold.
(G) qRT-PCR data from sorted melanoblasts of WT and CRTC3 KO mice (P2, N = 4–5 per group). Significance determined by Welch’s t test.
(H) Primary melanocytes isolated from skins of WT, CRTC1 KO, CRTC2 KO, and CRTC3 KO mice (P2), cultured for 2 weeks in differentiation media containing 200 pM cholera toxin and 200 nM TPA. Arrows point to intracellular melanin granules that are absent from CRTC3 culture. Bar, 50 μm. Quantification of melanin granules in cells is shown under each panel (N = 10 cells/genotype). Granules were not detected in CRTC3 (ND); differences between WT, CRTC1 KO, and CRTC2 KO are not significant, as determined by Welch’s t test.
We used DCT promoter-green fluorescent protein (GFP) reporter, to evaluate neonatal melanoblast populations in skins of WT and CRTC3 KO animals (Steel et al., 1992; Zaidi et al., 2011). No significant differences in the percentage of GFP-positive melanoblasts were seen between the two genotypes (Figure S1D). Similar to whole skin, OCA2 mRNA amounts were also reduced in CRTC3 KO melanoblasts, whereas MITF or other core melanogenic enzymes were unaffected (Figure 1G).
We evaluated the differentiation of WT and CRTC3 KO melanoblasts after exposure to cholera toxin (CTX) and phorbol ester 12-o-tetradecanoylphorbol-13-acetate (TPA), activators of cAMP and MAPK cascades. CTX and TPA induced melanin synthesis in WT, CRTC1 KO, and CRTC2 KO melanoblasts, but not in CRTC3 KO (Figure 1H). Rather, surviving CRTC3 KO cells expressed fibroblast markers, but not melanocyte markers (Figure S1E). We considered that ex vivo culture may exacerbate the phenotype of CRTC3 KO mouse melanocytes due to increases in reactive oxygen species (Guyonneau et al., 2004). Supplementing the primary culture with catalase, an antioxidant cocktail, or co-culturing with feeder keratinocytes did not rescue differentiation of CRTC3 KO melanocytes, however. These results indicate that the selective loss of CRTC3, but not CRTC1 or CRTC2, impairs melanocyte differentiation.
CRTC3 promotes pigmentation by regulating the expression of OCA2
To further characterize the role of CRTC3 in pigmentation, we used mouse melanoma cell line B16F1, which produces melanin after exposure to cAMP agonists (Bennett, 1989). CRISPR or RNAi-mediated depletion of CRTC3 reduced B16F1 cell pigmentation, which was rescued by CRTC3 re-expression (Figure 2A; Figures S1F, S1G, and S1H). Despite profound pigmentation defects, CRTC3 KO cells expressed comparable amounts of MITF and TYR proteins as control cells (CTRL); they were able to accumulate both proteins efficiently following Forskolin (FSK) exposure (Figure S1I). TYR desoxy phenyl alanine (DOPA) oxidase activity was decreased in CRTC3-depleted cells, by in-gel and whole-lysate analyses; re-expression of CRTC3 restored TYR activity (Figures 2B, 2C, and 2D). TYR activity depends on the correct maturation of melanosomes (Watabe et al., 2004), prompting us to assess melanosome populations in CTRL and CRTC3 KO cells. Using transmission electron microscopy (TEM), we noted that maturation of melanosomes was severely impaired in CRTC3 KO cells, as evidenced by a high number of early-stage (I and II) versus melanized late-stage (II and IV) melanosomes (Figure 2E; Figure S1J). These results indicate that depletion of CRTC3 reduces pigmentation by altering an essential step in melanosome maturation, which is necessary for TYR enzyme activity.
Figure 2. Loss of CRTC3 impairs OCA2 expression and melanosome maturation.
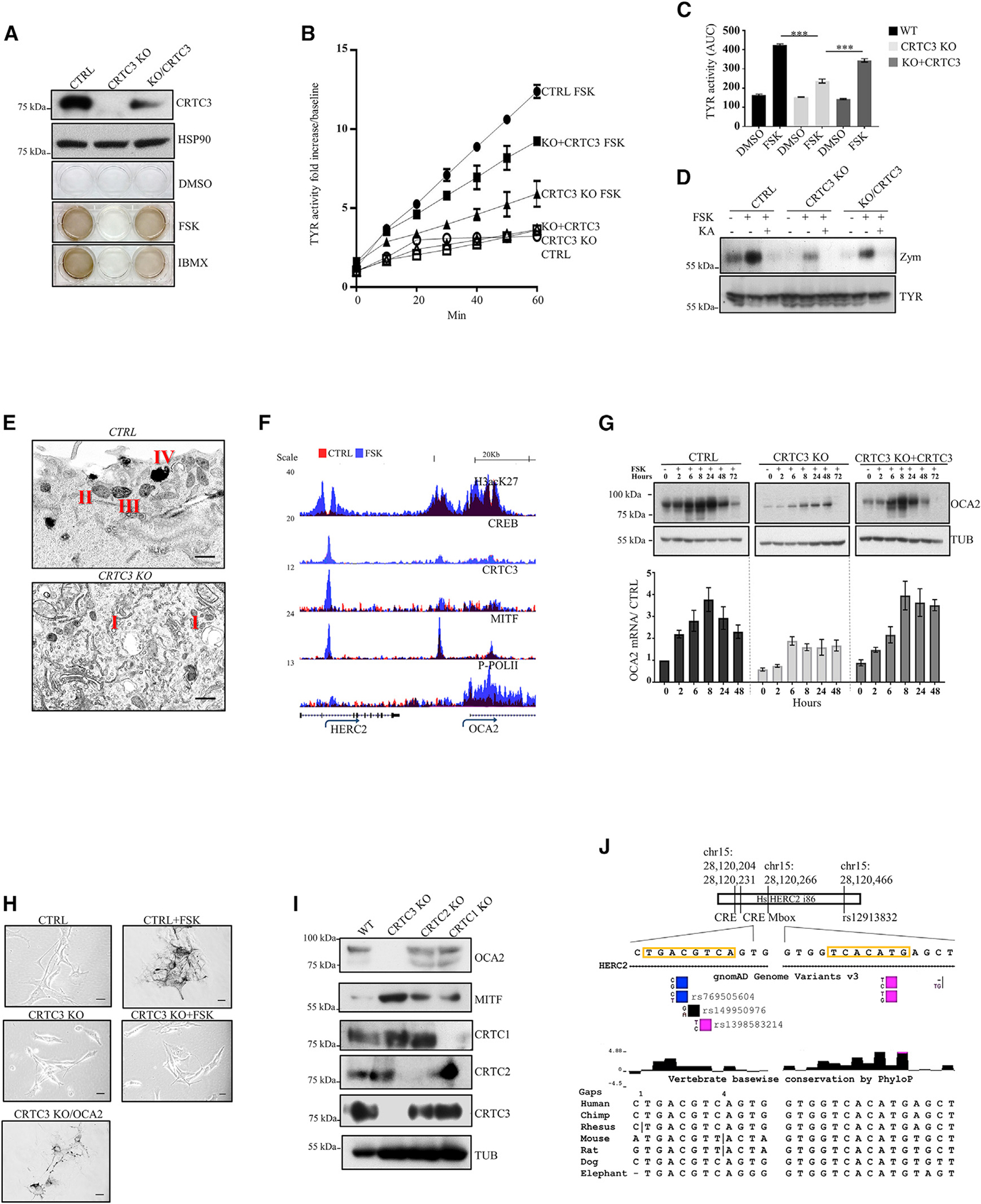
(A) Representative western blot and melanogenesis assay in CTRL, CRTC3 KO, and CRTC3-rescued B16F1 cells following differentiation stimulus with 5 μM FSK or 100 μM 3-isobutyl-1-methylxanthine (IBMX) for 60 h.
(B) Tyrosinase activity in whole lysates of B16F1 cells with indicated genotypes and treatments (5 μM FSK, 48 h; n = 3).
(C) Quantification of tyrosinase activity from (B). Significance determined by one-way ANOVA and Tukey multiple comparisons tests.
(D) Representative tyrosinase in-gel activity (zymography) and western blot showing TYR protein accumulation levels in B16F1 cells of indicated genotypes. Cells were treated with 5 μM FSK or 500 μM TYR inhibitor kojic acid for 48 h.
(E) Representative TEM image of melanosome maturation stages in CTRL and CRTC3 KO B16F1 cells after 48 h of 5 μM FSK stimulation. Observed melanosome maturation stages are indicated with Roman lettering. Bar, 400 nm.
(F) Browser plot of genomic region containing OCA2 enhancer showing occupancy of acetylated histone H3K27, CREB, CRTC3, MITF, and phospho-POLII in B16F1 cells treated with vehicle or 5 μM FSK for 1 h. Scale indicates normalized tag enrichment.
(G) Time course for OCA2 mRNA and protein induction upon 5 μM FSK treatment, assayed through qRT-PCR and western blotting. n = 3.
(H) Baseline rescue of melanogenesis in CRTC3 KO B16F1 cells transiently transfected with OCA2. CTRL and CRTC3 KO cells were treated with 5 μM FSK for 60 h to induce melanin synthesis. Bar, 25 μm.
(I) Representative western blot of adult dorsal skin from mice of indicated CRTC genotypes showing accumulation of OCA2 and MITF.
(J) Schematic representation of the conserved region of intron 86 of the HERC2 gene, containing the SNP rs12913832, which is relevant for pigmentation and melanoma risk in humans. Positions and conservation of the full CRE site and the nearest Mbox site are indicated. Validated SNP variants in the CRE site are shown and colored to indicate allelic prevalence.
To identify direct targets of CRTC3 that contribute to melanin production, we performed chromatin immunoprecipitation sequencing (ChIP-seq) and RNA-seq studies in B16F1 cells. Exposure to FSK triggered CRTC3 recruitment to regulatory regions for genes involved in pigmentation, vesicular trafficking, and MAPK signaling. CRTC3 was primarily recruited to CREB binding sites and in close proximity to MITF-bound M-box sites (Figures S1K and S1L). Pointing to compensation between CRTC family members, KO of CRTC3 decreased the expression of canonical CREB targets and MITF only modestly (Tables S2 and S3). Downregulated genes were clustered in processes involved in substrate interaction and pigmentation; most of these genes were efficiently rescued by re-expression of CRTC3 (Figures S1M and S1N).
OCA2 was prominent within the subset of CRTC3-regulated genes (Table S3). Exposure to FSK simulated binding of CRTC3, CREB, and MITF to a conserved OCA2 enhancer located in intron 86 of the upstream HERC2 gene (Figure 2F; Visser et al., 2012, 2014). Concurrently, exposure to FSK increased occupancy of phosphorylated polymerase II over the OCA2 gene body (Figure 2F). OCA2 mRNA amounts were increased after 2 h of FSK stimulation, followed by increases in protein amounts as determined by immunoblotting with a validated OCA2 antiserum (Figure 2G; Figures S2A–S2D). Induction of OCA2 was severely blunted in CRTC3 KO and recovered with CRTC3 re-expression (Figure 2G). The effects of CRTC3 on OCA2 expression appear to account for reduced pigmentation because overexpression of OCA2 in CRTC3 KO cells rescued melanin production even under basal conditions (Figure 2H). Having seen the selective loss of pigmentation in CRTC3 KO mice, we wondered if the effects on OCA2 were also CRTC3-specific. We generated a CRISPR KO of CRTC1 and assessed its melanin production and OCA2 status in B16F1 cells. Depletion of CRTC3 impacted both basal and FSK-induced OCA2 amounts, whereas CRTC1 KO reduced FSK-induced OCA2 protein amounts only modestly. Extracellular melanin output was largely unchanged in CRTC1 KO, indicating that CRTC1 is dispensable for efficient melanin production in this setting (Figure S2E). OCA2 protein amounts are substantially reduced in CRTC3 KO skin compared to those in the WT as well as CRTC1 and CRTC2 KOs by immunoblot assay (Figure 2I). OCA2 expression in melanocytes is primarily regulated by a distal enhancer in intron 86 of the HERC2 gene. Within this enhancer, we noticed two CREB binding sites that are positioned in the conserved region of intron 86, which is also the site of long-distance chromatin loop formation with the OCA2 proximal promoter (Sturm et al., 2008; Visser et al., 2012; Figure 2J). The CRE site contains validated low-frequency SNP variants, of which all are predicted to decrease CREB binding based on previously reported fluorescence anisotropy assays of physiologically relevant CRE motifs (Luo et al., 2012; Figure 2J).
cAMP and ERK1/2 regulate CRTC3 activity
We hypothesized that CRTC3 may be selectively activated in melanocytes by signals other than cAMP or by low levels of cAMP that are otherwise insufficient to activate CRTC1 and CRTC2. ERK1/2-mediated phosphorylation of CRTC3 at Ser391 increases its interaction with protein phosphatase 2 (PP2A), leading to the dephosphorylation of CRTC3 at inhibitory 14-3-3 binding sites (Sonntag et al., 2019). Exposure of B16F1 cells to several ERK1/2 activators (SCF, HGF, and TPA) promoted the phosphorylation of CRTC3 at Ser391 (Figure 3A). In line with previous observations, Ser391-phosphorylated CRTC3 was predominantly nuclear localized (Figure S3A). Correspondingly, a loss of CRTC3 blocked melanin production, and re-expression of CRTC3 rescued these effects, but the overexpression of CRTC2 did not. Notably, overexpression of the hybrid CRTC2 protein containing the PP2A binding domain of CRTC3 (Sonntag et al., 2019) was competent to rescue melanin synthesis (Figure 3b). Mutation of Ser391 to Ala reduced CRTC3 effects on melanin production relative to WT CRTC3 (Figure S3B).
Figure 3. Selective induction of CRTC3 in response to ERK1/2 activation.
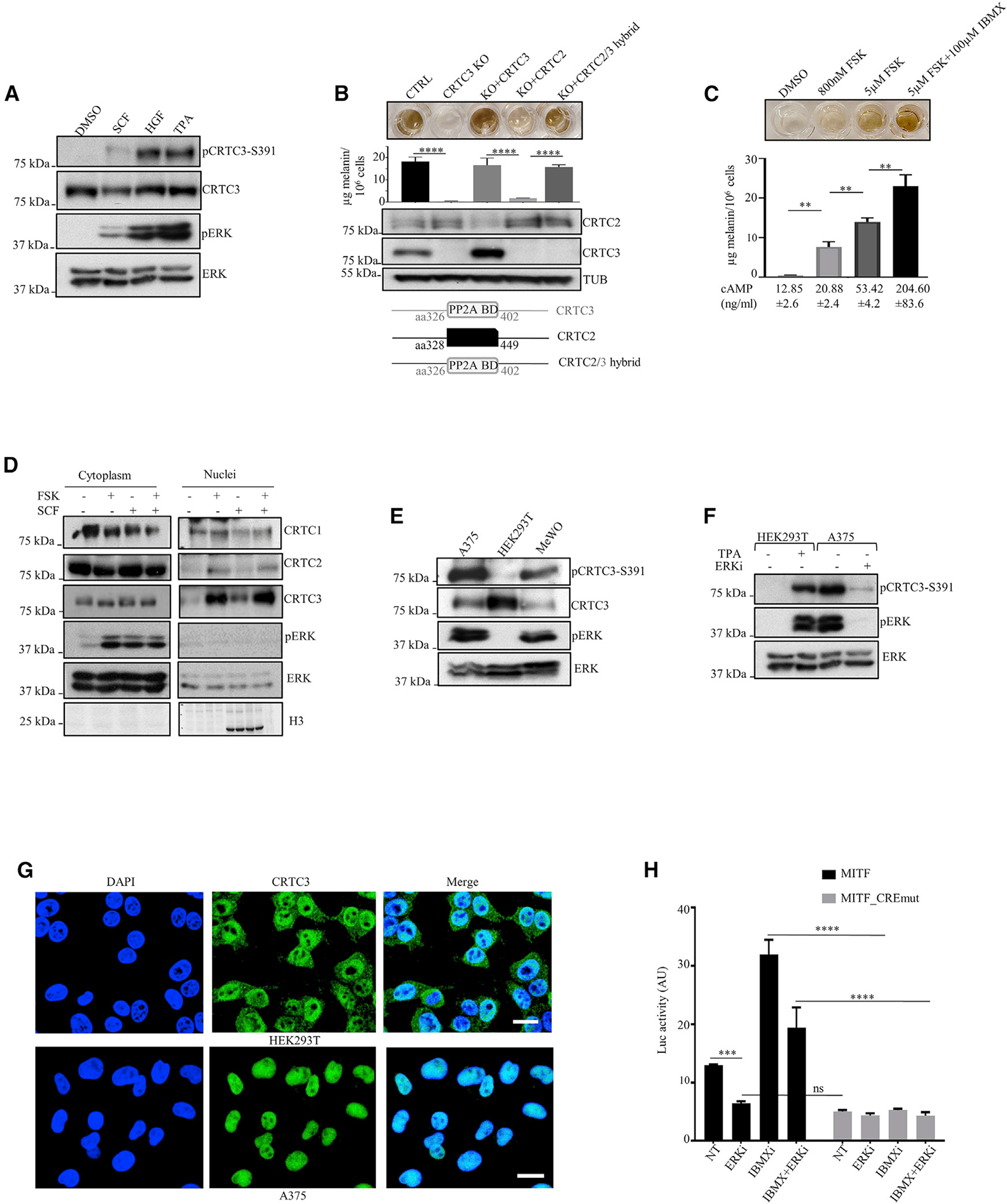
(A) Representative western blot showing phosphorylation of CRTC3 at Ser391 upon stimulation of ERK1/2 in B16F1 cells (SCF, stem cell factor [30 ng/ml]; HGF, hepatocyte growth factor [20 ng/ml]; TPA [200nM]).
(B) Rescue of melanogenesis in CRTC3 KO B16F1 cells after transfection with indicated constructs. A scheme of the CRTC2/3 hybrid protein shows the PP2A binding domain (PP2A BD) of CRTC3 (amino acids [aa] 326–402) replacing the corresponding sequence in CRTC2 (aa 328–449). For PP2A BD sequence details, see Figure S4H. Cells were treated with 5 μM FSK for 60 h, 48 h after transfection. Melanin quantification is shown n = 3) and significance determined by one-way ANOVA and Tukey multiple comparisons tests.
(C) Melanin production related to cAMP content in B16F1 cells stimulated with indicated compounds for 60 h. (n= 3).
(D) Western blot of a representative sub-cellular fractionation experiment after treatment with 800 nM FSK or 30 ng/ml SCF for 20 min.
(E and F) Representative western blots of indicated human cell lines in the basal state and treatment with 200 nM ERK1/2 inhibitor SCH772984 for 8 h.
(G) Immunofluorescence of HEK293T and A375 cells stained with hCRTC3(414–432) antibody and 4′,6-diamidino-2-phenylindole (DAPI). Bar, 20 μM.
(H) Luciferase reporter assay in A375 cells transfected with MITF proximal promoter construct (N = 3). Treatments and mutations of regulatory elements are indicated. Cells were pretreated with 200 nM ERK1/2 inhibitor SCH772984 for 6 h before adding 100 μM IBMX for 5 h. Significance was determined by two-way ANOVA and Šidák’s multiple comparisons tests.
To test whether CRTC3 is activated at lower cAMP levels than other CRTCs, we exposed B16F1 cells to a range of FSK concentrations; at 800 nM, FSK modestly increased intracellular cAMP levels and melanin synthesis (Figure 3C). Low FSK (800 nM) increased nuclear amounts of CRTC3 to a greater extent than CRTC1 or CRTC2 (Figure 3D; Figure S3C). Low FSK treatment also stimulated ERK1/2 activation to a similar degree as treatment with SCF, which promoted nuclear translocation of CRTC3, but not CRTC1 or CRTC2 (Figure 3D). Collectively, these results indicate that CRTC3 has increased sensitivity to both cAMP and ERK1/2 signals.
CRTC3 contributes to oncogenic properties of transformed melanocytes by modulating intracellular cAMP
MAPK hyperactivation is found in over 90% of human melanomas (Cancer Genome Atlas Network, 2015; Wellbrock and Arozarena, 2016), prompting us to test whether corresponding increases in ERK1/2 activity upregulate CRTC3. Amounts of phosphorylated CRTC3-Ser391 correlated with the extent of ERK1/2 activity in several human cell lines. HEK293T cells had low basal ERK1/2 activity and correspondingly low CRTC3 phospho-Ser391; by contrast, A375 and MeWO cells had high basal ERK1/2 activity and increased levels of phospho-Ser391 (Figures 3E and 3F).
A375 human melanoma cells express an oncogenic gain-of-function BRAF (V600E) mutation and low levels of cAMP signaling due to increased expression of phosphodiesterases (Delyon et al., 2017), which provided us with a window to evaluate selective effects of high ERK1/2 signaling on CRTC3 activity. CRTC3 was predominantly nuclear localized in A375 but not HEK293T cells, consistent with their relative ERK1/2 and CRTC3-Ser391 phosphorylation profiles (Figure 3G). Treatment of A375 cells with an ERK1/2 inhibitor decreased CRTC3-pSer391 amounts and correspondingly reduced CREB activity over the MITF promoter (Figures 3F and 3H).
We tested the regulatory contribution of CRTC3 to oncogenic properties of melanoma cells by generating CRISPR-derived CRTC3 KO A375 cells (Figure S3D). A loss of CRTC3 had no effect on proliferation, but it impaired anchorage-independent growth in both A375 and B16F1 melanoma cells (Figures S3E, S3F, S3G, and S3H). Anchorage-independent growth was also reduced in cells expressing a phosphorylation-defective CRTC3-Ser391Ala mutant relative to WT CRTC3 (Figure S3I).
To determine transcriptional effects of CRTC3 in a human melanoma model, we performed RNA-seq on CTRL and CRTC3 KO A375 cells. Loss of CRTC3 downregulated genes involved in cAMP signaling, cell migration, and chemotaxis (Figure 4A; Table S4). A number of canonical CREB targets were significantly downregulated in the KO cells, and among these were the phosphodiesterases PDE4B and PDE4D (Figures 4A; Table S4). PDE4 has been found to promote melanoma progression by reducing inhibitory effects of cAMP on activation of the MAPK pathway (Dumaz et al., 2006; Delyon et al., 2017; Marquette et al., 2011; Watanabe et al., 2012). Based on its ability to stimulate PDE4 expression, we reasoned that the loss of CRTC3 should increase intracellular cAMP. Supporting this idea, CRTC3 KO cells had higher basal concentrations of cAMP, higher PKA activation, and a greater response to intermediate concentrations of FSK (Figure 4B; Figure S3J).
Figure 4. CRTC3 activity is associated with increased tumorigenic potential and decreased survival.
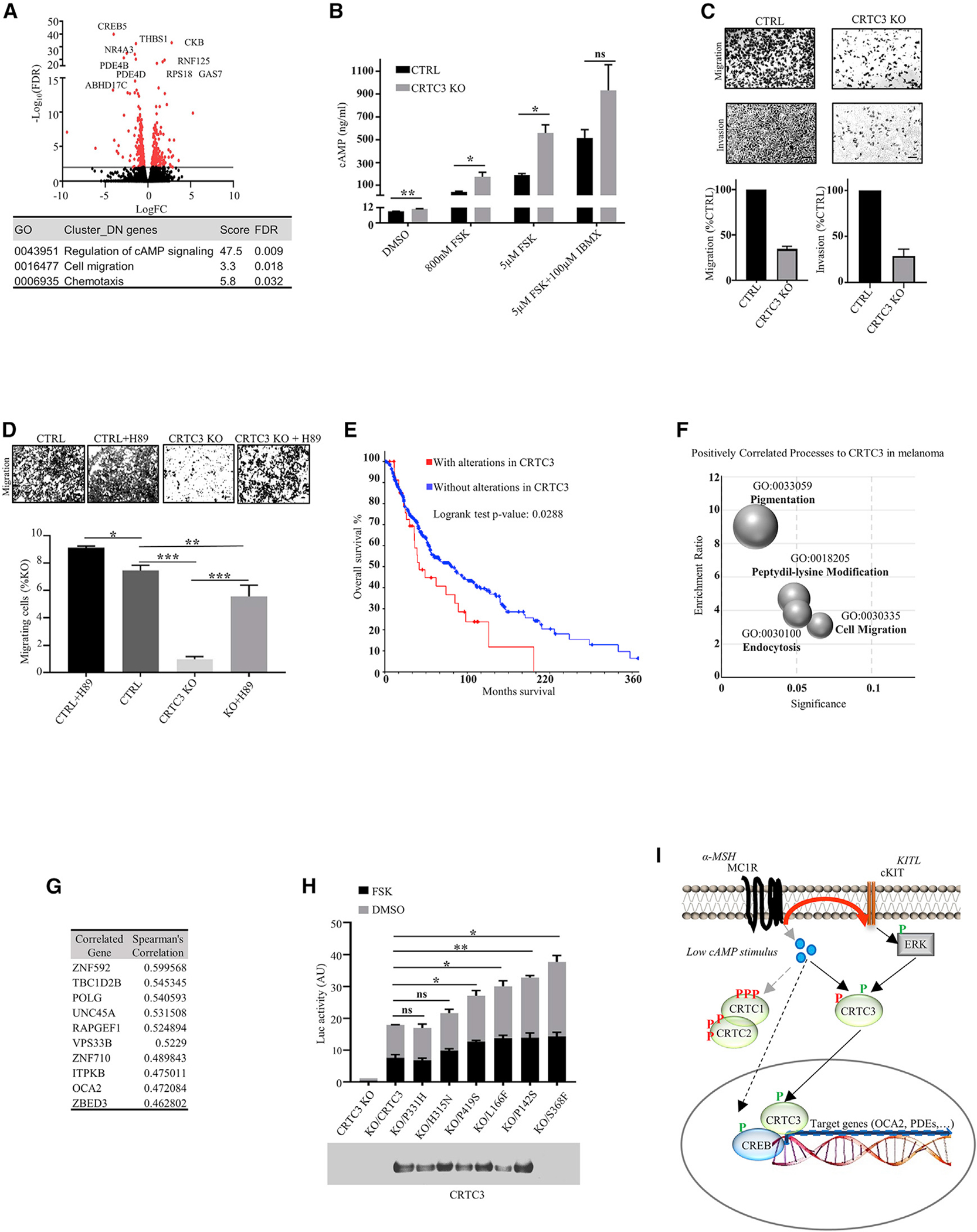
(A) Volcano plot of RNA-seq data showing differentially expressed genes between CTRL and CRTC3 KO A375 cells (n = 2/genotype). Significance determined as adjusted p ≤ 0.05 and Log2 fold changes of ≤−1 or ≥1. Table shows significant cluster enrichment of downregulated genes, listed in Table S4.
(B) Quantification of cellular cAMP in CTRL and CRTC3 KO A375 cells treated with indicated compounds for 15 min (N = 3). Significance determined by Welch’s t test.
(C) Representative images showing migration (24 h) and invasion (72 h) of CTRL and CRTC3 KO A375 cells (N = 3). 2% fetal bovine serum (FBS) was used as a chemotactic agent. Bar, 100μm.
(D) Representative images and quantification of migration (24 h) of CRTC3 KO A375 cells treated with DMSO or PKA inhibitor H89 (10 μM). Bar, 50 μm. Significance was determined by one-way ANOVA and Tukey’s multiple comparisons tests (n = 3).
(E) Kaplan-Meyer graph of the overall survival of cutaneous melanoma patients from TCGA, Firehose Legacy with and without alterations in CRTC3 (N = 367). Survival was compared by log rank tests. Graph and statistical analyses were obtained from https://cbioportal.org. All samples containing copy number alteration data were used for the analysis (N = 367/479), and expression cutoff Z score was set to 1.5. Source data are from GDAC Firehose (https://gdac.broadinstitute.org).
(F) CRTC3 correlation analysis in human melanoma patients from TCGA, Firehose Legacy (N = 367). Bubble chart shows top positively enriched processes distributed by enrichment ratio and significance.
(G) Top 10 genes positively correlated genes with CRTC3 from the cohort in (F).
(H) Luciferase reporter assay in B16F1 cells transfected with EVX-luc-2xCRE reporter and WT or mutated CRTC3 constructs. Cells were treated with 5 μM FSK for 6 h, 48 h after transfection. Assayed patient mutations are indicated and shown on sequence alignment Figure S4H. The experiment was run five different times overall. Data represent two replicas run in parallel, with each assayed in technical duplicates and normalized by expression of transfected CRTC3. Both replicas had a comparable expression of CRTC3 constructs, of which one is shown in the western blot insert. Differences in expression of the reporter constructs were compared to WT CRTC3, and significance was determined by Welch’s t test.
(I) Model for joint regulation of CRTC3 by cAMP and ERK1/2 in low cAMP state.
Increases in cAMP signaling have been shown to inhibit cancer motility, depending on the signaling context (Howe, 2004). Treatment of A375 cells with cAMP agonists inhibited cell migration in a dose-dependent manner; these effects were reversed following exposure to the PKA inhibitor H89 (Figure S4A and S4B). Consistent with this observation, migration and invasiveness were reduced in CRTC3 KO cells; the addition of H89 also rescued migration in this setting (Figures 4C and 4D).
Small molecule inhibitors of BRAF constitute effective treatment options for patients with BRAF mutant melanomas. Upregulation of the CREB pathway has been found to confer resistance to BRAF inhibition (Johannessen et al., 2013), prompting us to evaluate the potential role of CRTC3 in modulating the treatment response of A375 cells to BRAF inhibition by vemurafenib (PLX4032). Exposure to PLX4032 decreased cell viability comparably in CTRL and CRTC3 KO A375 cells. Notably, transient overexpression of CRTC3 increased viability and target gene expression in cells exposed to PLX4032 but not to cisplatin, a DNA cross-linking compound (Figures S4C, S4D, and S4E).
Spontaneous CRTC3 mutations in human melanomas modulate its activity
To determine if CRTC3 activity is altered in human melanoma, we performed CRTC3 profiling on a cohort of 367 melanoma patients from the Cancer Genome Atlas (TCGA) database (Firehose legacy dataset, see legend of Figure 4E). Alterations of CRTC3 were present in 23% of samples, of which most increased its expression; they were associated with reduced patient survival (Figure 4E; Figure S4F). We evaluated the transcriptional signature of melanomas with alterations in CRTC3. To this end, we performed a co-expression analysis in the TCGA patient cohort and compared gene groups showing high unique positive correlation with each CRTC family member (Table S5). In line with our molecular data, transcript signatures related to CRTC3 showed enrichment in pigmentation and cell migration (Figures 4F and 4G; Figure S4G).
Based on public data (Gonzalez-Perez et al., 2013; Tate et al., 2019), we compiled a list of spontaneous CRTC3 mutations in melanoma and noticed that 20% of them occur in or near regulatory 14-3-3 binding sites (Figure S4H; Table S6). Phosphorylation of conserved serines within SIK consensus motifs (LXBS/TXSXXXL) sequesters the CRTC family in the cytoplasm by 14-3-3 binding; disruption of these sites would be expected to increase CRTC activity (Altarejos and Montminy, 2011; Sonntag et al., 2017). Based on their location on the CRTC3 protein sequence, we chose six patient mutations for further analysis in a CRE-luciferase reporter assay. Three of these mutations map to 14-3-3 binding sites (L166F, P331H, and S368F), one flanks a CRTC3 sumoylation site (Hendriks et al., 2017) (P142S), and two are outside discrete domains (H315N and P419S). Almost all mutant CRTC3 proteins stimulated reporter activity to a greater extent than WT, suggesting that CRTC3 is indeed upregulated in subsets of melanoma tumors (Figure 4H). Cells carrying mutant CRTC3 proteins also show upregulation of target genes compared to WT CRTC3 (Figures S4I and S4J).
DISCUSSION
CRTC family members share extensive sequence homology within their CREB-binding, regulatory, and trans-activation domains. Correspondingly, the loss of any one family member can be readily compensated by other family members, as we observed for MITF expression in primary melanoblasts and B16F1 cells. By contrast, OCA2 was significantly downregulated following CRTC3 KO, accounting for the deficiencies in melanin synthesis without corresponding reductions in protein amounts of melanogenic enzymes. We found that the induction of OCA2 is mediated in part by recruitment of CREB/CRTC to CREB binding sites within a conserved enhancer. KO of CRTC3 reduced basal expression of OCA2, leading to lower net induction of pigmentation even in the presence of high cAMP. Consistent with its substantial baseline activity, CRTC3 is the only CRTC family member that modulates constitutive pigmentation in mice.
In contrast with other CRTCs, CRTC3 also appears to stimulate CRE-dependent transcription in response to growth factors that activate ERK1/2. Phosphorylation of Ser391 primes CRTC3 for subsequent activation by modest increases in cAMP that are sufficient to dephosphorylate and fully activate CRTC3, but not CRTC1 or CRTC2. cAMP and ERK1/2 pathways function cooperatively in this setting, explaining in part the higher baseline activity of CRTC3 in melanocytes and melanoma cells. In contrast with the indiscriminate activation of all CRTCs in response to high cAMP levels, ERK1/2 and cAMP activate CRTC3 cooperatively under low cAMP signaling conditions to promote CREB-target gene expression (Figure S4K; Figure 4I). In melanocytes and melanoma cells, several communication points between cAMP and MAPK/ERK cascades have been described, including transactivation of c-Kit by MC1R, activation of ERK1/2 by cAMP, inhibition of CRAF by PKA, and regulation of MITF (Buscà et al., 2000; Dumaz et al., 2006; Herraiz et al., 2011; Niwano et al., 2018). Our results suggest that CRTC3 functions as another communication point between these two pathways, thereby increasing specificity in signal transduction.
BRAF inhibitors represent a mainstay for the treatment of melanoma; reducing resistance to these inhibitors appears critical for improving patient survival. KO of CRTC3 in a BRAF mutant melanoma model increased intracellular concentrations of cAMP as well as PKA activation and impaired chemotactic migration, invasion, and anchorage-free growth. Conversely, upregulation of CRTC3 was associated with improved cell viability during vemurafenib treatment.
Remarkably, spontaneous CRTC3 point mutants identified in a subset of human melanomas map to regulatory regions in the protein that normally sequester this co-activator in the cytoplasm. Our data suggest that the increased activity of CRTC3 in this setting contributes to melanoma progression by increasing the expression of CREB target genes involved in motility, invasiveness, or viability. The increased activity of these CRTC3 mutant proteins in melanomas may promote the constitutive upregulation of CREB target genes while avoiding potential inhibitory effects of PKA activation on tumor growth or metastasis. Future studies should provide further insight into this process.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead contact, Marc Montminy (Montminy@salk.edu).
Materials availability
For the availability of mOCA2 antibody (PBL #7431) please contact Marc Montminy (Montminy@salk.edu).
Data and code availability
Sequencing datasets are available under GEO accession number GSE154117.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal studies
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Salk Institute. Mice were housed in colony cages with a 12 hr light/12 hr dark cycle in a temperature-controlled environment. Age-matched experimental animals of both genders were used; age range was 2–10 months for adult animals, post-natal day two for pups. C57BL6 were purchased from Jackson Laboratories. Knock-outs of CRTC1, CRTC2 and CRTC3 were previously described (Altarejos et al., 2008; Song et al., 2010; Wang et al., 2010). iDCT-GFP mice were obtained from NCI Mouse Repository.
Primary melanoblast isolation and melanocyte culture
Primary cells were isolated from whole epidermises of 2 days old WT, CRTC1 KO, CRTC2 KO and CRTC3 KO pups and cultured in RPMI media containing TPA (P1585, Sigma) and cholera toxin (C8052, Sigma), following previously described detailed protocol (Godwin et al., 2014). For skin melanoblast quantification, WT and CRTC3 KO mice were crossed with mice carrying iDCT:GFP transgene system. Pregnant females were given 1mg/ml doxycycline in water and epidermal cell suspensions were isolated from 2 days old pups (WT N = 5, KO N = 4). GFP positive cells were counted through fluorescence-activated cell sorting (Becton Dickinson Influx cytometer). To account for differences in total amounts of cells isolated from each animal, quantification was expressed as percentage of GFP positive cells per epidermis. In experiments where XB2 feeder keratinocytes were used, cells were obtained from Wellcome Trust Functional Genomics Cell Bank and prepared using mitomycin c treatment (M4287, Sigma), as previously described (Godwin et al., 2014). In some experiments, catalase (LS001896, Worthington Biochemical) and an antioxidant supplement (A1345, Sigma) were added to the primary cultures in the attempt to improve survival and differentiation of CRTC3 KO melanoblasts.
Cell line culture
B16F1, A375, MeWO and HEK293T cells were obtained from the American Type Culture Collection. Cells were tested for mycoplasma contamination with MycoAlert kit (LT07-418, Lonza) before use in experiments. A375, MeWO and HEK293T cells were consistently cultured in DMEM media (GIBCO®, high glucose), supplemented with 10% fetal bovine serum (FBS, Gemini Bio-Products) in 5% CO2 at 37°C. Two different culture modes were tested for B16F1 in order to preserve their melanogenic potential. These were cultures in RPMI 1640 medium (GIBCO®, high glucose) without phenol red supplemented with 10% FBS in 10% CO2 or DMEM without phenol red supplemented with 10% FBS in 5% CO2. In both cases cells were plated sparsely (1×104) for regular maintenance. For induction of melanogenesis, cells were grown to 65% confluence, making sure to avoid cell clumping, and treated with compounds that stimulate melanin synthesis (Forskolin (FSK, F6886; Sigma) or IBMX (I5879, Sigma)). Cells grown in RPMI 1640 were switched to DMEM before stimulation. Maintenance plates were regularly tested (every 3 passages) to confirm the ability of cells to produce melanin. All cells were used for experiments between passages 4 and 12, after which they were discarded and a new low-passage batch thawed for the next experimental round. Under these conditions, there was no difference in melanin output of B16F1 cells between the two culture modes, so we used consistent DMEM for most shown results.
Silencing of CRTC3 in B16F1 cells was performed by transfecting cells with pSilencer 2.1-U6 puro plasmid (AM5762, Applied Biosystems) carrying CRTC3 shRNA constructs. 5′ phosphorylated double stranded CRTC3 oligonucleotides were cloned into BamHI and HindIII sites of the pSilencer 2.1-U6 puro plasmid, according to the manufacturer’s protocol. 0.8×106 cells were transfected with 1.5μg plasmid DNA using Lipofectamine 2000 (11668019, Invitrogen). Selection with 2μg/ml puromycin (P8833, Sigma) was started 48h post transfection and maintained for at least 7 days before testing bulk cell populations for successful CRTC3 knock-down and melanin induction. Empty vector was used as a control.
METHOD DETAILS
Sequences of working constructs:
Sh1: 5′GATCCGCTTCAGCAACTGCGCCTTTTCAAGAGAAAGGCGCAGTTGCTGAAGTTTTTTGGAAA 3′
Sh3:5′GATCCGAAGCTCCTCTGGTCTCCATTCAAGAGATGGAGACCAGAGGAGCTTCTTTTTTGGAAA 3′
Sh4: 5′GATCCGCACATCAAGGTTTCAGCATTCAAGAGATGCTGAAACCTTGATGTGCTTTTTTGGAAA 3′
For CRISPR-Cas9 knockout of CRTC1 and CRTC3, B16F1 cells were separately transfected with two different guide RNAs (gRNAs) per gene, cloned into pSpCas9(BB)-2A-GFP (PX458) (Addgene plasmid # 48138) (Ran et al., 2013). CTRL cells were transfected with the empty vector. Single clones were selected by fluorescence-activated cell sorting (Becton Dickinson Influx cytometer) after 48 hours. The gRNAs target exons 1 and 3 of CRTC1 and exons 1 and 8 of CRTC3:
mC1g1: 5′ CACCGTGACAGGGGTCGACGGTGCG 3′ ex3
mC1g2: 5′ CACCGTCACCCGCGCGGCCCGCGTC 3′ ex1
mC3g1: 5′ CACCGCATCAAGCCGATAATGTTCG 3′ ex1
mC3g2: 5′ CACCGAGCCACTGCCTAAACACCTG 3′ ex8
Rescue experiments were performed by transfecting clonal B16F1 CRTC3 KO cells with pSelect-puro-mcs plasmid (psetp_mcs, Invivogen) carrying full length CRTC3, CRTC2, OCA2, CRTC3-Ser391A or CRTC2/3 hybrid constructs (CRTC2328–449 exchanged for CRTC3326–402, as previously described; Sonntag et al., 2019), cloned into SalI and NheI sites. Transfected cells were selected with 2μg/ml puromycin for at least 7 days before initial testing. Empty vector was used to select for CTRL cells. Sequences of oligonucleotides used for cloning:
CRTC2 F: 5′ GATGGTCGACATGGCGACGTCAGGGGCGAACGGGCCGGGTTCC 3′
CRTC2 R: 5′ CATGCTAGCTCACTGTAGCCGATCACTACGGAATGAGTCCTC 3′
CRTC3 F: 5′ GATGGTCGACATGGCCGCCTCGCCCGGTTCGGGCAGCGCCAAC 3′
CRTC3 R: 5′ CATGCTAGCTCATAGCCGGTCAGCTCGAAACGTCTCCTCCAC 3′
OCA2 F: 5′ GATGGTCGACATGCGCCTAGAGAACAAAGACATCAGGC 3′
OCA2 R: 5′ CATGCTAGCTTAATTCCATCCCACCACAATGTGAGCAATCAGG 3′
Site directed mutagenesis of CRTC3-Ser391 to Ala residue was performed from pSelect-puro-mcs-CRTC3 construct with oligonucleotides:
S391A_F: 5′ GGCGCAGGCAGCCTCCAGTCGCCCCTCTCACGCTCTCTCCTGG 3′
S391A_R: 5′ CCAGGAGAGAGCGTGAGAGGGGCGACTGGAGGCTGCCTGCGCC 3′
Q5 high fidelity polymerase (M0492S, New England Biolabs) was used for the PCR reaction (95°C 3min, 95°C 30sec, 55°C 1min, 72°C for 7min, 18 cycles, 72°C for 7min final extension). Template plasmid was digested with DpnI for 2h at 37°C before amplification in Top10 competent cells (Invitrogen). Presence of S391A mutation was confirmed by sequencing.
CRTC3 knock-out and single cell selection in A375 cells was performed with the same CRISPR protocol used for B16F1 cells. The gRNAs targeted exons 1 and 11 of human CRTC3:
hC3g1: 5′ CACCGTGAGCGGCCCGTCTCGGCGT 3′ ex11
hC3g2: 5′ CACCGCGCGCTGCACACGCAGAGAC 3′ ex1
Melanin quantification
1.5 μg of dorsal hair was plucked from 8 weeks old WT and CRTC3 KO animals and solubilized in 1ml of 1M NaOH at 85°C for 4h under agitation. Samples were centrifuged at 12000 rpm for 5 minutes and the absorbance of supernatants was read at 475nm in the Synergy-H1 microplate reader (BioTek). Absorbance values were compared to the standard curve of synthetic melanin (155343, MP Biomedicals) dissolved in 1M NaOH. The standard curve was in a linear range of the experimental values.
For extracellular melanin quantification, B16F1 cells were plated in 6-well dishes at the density of 0.5×106 cells/well in phenol red-free media and treated with FSK (F6886, Sigma) or IBMX (I5879, Sigma) for 60h, 24h after seeding. Final concentrations of small molecules used for treatment are indicated in figure legends. Melanin in the collected medium was determined by comparison of experimental absorbance values at 405nm with synthetic melanin standard curve diluted in the same media.
Transmission electron microscopy (TEM)
B16F1 cells were fixed in a solution of 2% paraformaldehyde, 2.5% glutaraldehyde, and 2mM CaCl2 in 0.1M sodium cacodylate buffer (pH 7.4) for 2 h at room temperature, post-fixed in 1% osmium tetroxide for 40 and 1.5% potassium ferrocyanide in sodium cacodylate buffer for 1 hour at 4°C in the dark, stained in 1% aqueous uranyl acetate at 4°C in the dark, dehydrated in ethanol graded series, and embedded in Eponate12 resin (Ted Pella). Ultra-thin sections of 70 nm were obtained using a diamond knife (Diatome) in an ultramicrotome (Leica EM UC7) and placed on copper grids (300 mesh). Sections were imaged on Zeiss TEM Model Libra 120, operated at 120 kV and captured as 2048 × 2040 pixel tiffs using a Zemas camera system.
Tyrosinase activity
B16F1 cells were plated in 6-well dishes at a density of 0.5×106 cells/well and treated with FSK for 48h, 24h after seeding. Cells were lysed in 100 μL of 50 mM sodium phosphate buffer (pH 6.8) containing 1% Triton X-100 and 0.1 mM phenylmethylsulfonyl fluoride (PMSF, Thermo Fisher) and frozen at −80°C for 30 min. Thawed cellular extracts were centrifuged at 12,000 g for 30 min at 4°C. For whole lysate activity, supernatants (80 μl) and 20 μL of 3,4-Dihydroxy-L-phenylalanine (L-DOPA, 2 mg/ml freshly dissolved in 50mM sodium phosphate buffer) were placed in a 96-well plate, and the absorbance at 492 nm was read every 10 min for 1 h at 37°C using the Synergy-H1 plate reader. For in-gel activity, protein concentration of cell lysates was determined by micro BCA assay (Pierce), equal protein amounts were mixed with Laemmli sample buffer without β-mercaptoethanol and incubated at 37°C for 15 min with slight agitation. Proteins were resolved on 8% sodium-dodecyl-sulfate (SDS)-acrylamide gel by electrophoresis. Gels were equilibrated in 50 mM sodium phosphate buffer (pH 6.8) for 1h before adding 1mg/ml L-DOPA. Further incubation was monitored until clear colorimetric detection of tyrosinase activity. L-DOPA was obtained from Tocris Bioscience (3788).
OCA2 antibody production
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Salk Institute. Female New Zealand white rabbits (10–12 weeks old; I.F.P.S. Inc., Norco, California, USA) were used for mOCA2 antibody production as previously described (Sonntag et al., 2019). OCA2 synthetic peptide (Cys27 mOCA2(2–27)-NH2) was synthesized by RS Synthesis (Louisville, KY) and conjugated to maleimide activated Keyhole Limpet Hemocyanin (KLH) per manufacturer’s instructions (77610, Thermo Fisher). Antisera with highest titers against the synthetic peptides were tested for the ability to recognize endogenous OCA2 in B16F1 cell line and primary mouse melanocytes. Rabbit PBL #7431 anti-mOCA2 was purified using Cys27 mOCA2(2–27)-NH2 covalently attached to Sulfolink agarose (20401, Thermo Fisher). Validation of the purified antibody is available in Figures S2A–S2D.
Soft agar, migration and invasion assays
Soft agar assays were carried out as previously described (Borowicz et al., 2014). Briefly, 6-well dishes were coated with 0.5% noble agar in DMEM media and cells were seeded at 3×104 cells/well in DMEM containing 0.3% noble agar and 10% FBS. Cells were allowed to form colonies for 28 days and were subsequently imaged on Zeiss VivaTome microscope. Diameters of imaged colonies were measured using ImageJ. For comparison between B16F1 KO cells carrying full length CRTC3 or CRTC3-S391A constructs, cells were subjected to initial 7 day selection with 2μg/ml puromycin and tested for comparable expression of CRTC3. Puromycin was also added to noble agar layers in these experiments. For migration and invasion assays, A375 cells were grown in serum-free DMEM media for 24h prior to seeding into 24-well permeable 8μM pore PET transwell inserts, with or without Matrigel coating (Corning 353097 and 354480, respectively). Inserts and companion plates were equilibrated in serum-free media at 37°C in 5% CO2 for at least 2h before starting the experiments. 3–7×104 cells in serum-free media were added to the transwell inserts and 2% FBS was used as chemotactic agent in the bottom well. For certain experiments, cAMP elevating compounds were mixed with cells prior to seeding with concentrations indicated in Figures and Legends. Cells were allowed to migrate for 24h or invade for 72h. Transwell membranes containing migrating/invading cells were fixed in 3.7% paraformaldehyde for 2 min at RT, permeabilized in methanol for 15 min at RT and stained with a solution of 0.2% crystal violet/20%methanol for 20 min in the dark. Non-migrating cells were removed from the inside of membranes by swabbing with cotton tips before imaging on Zeiss VivaTome microscope. Cells were counted with ImageJ.
cAMP measurement
B16F1 or A375 cells were plated at 0.5×106 cells/well in 6-well dishes and treated with cAMP inducing compounds for 15min, 24h after seeding. Different compounds and concentrations are indicated in Figures and Legends. Cellular cAMP levels were measured using an ELISA kit (581001, Cayman Chemical Company) according to manufacturer’s instructions.
Immunofluorescence
HEK293T and A375 cells were plated in Poly-D-Lysine coated glass bottom dishes (P35GC-0-10-C, MatTek Corporation). 24h post seeding, cells were fixed with 4% paraformaldehyde. After incubation with the primary hCRTC3(414–432) (PBL #7019) antibody, microscopy samples were incubated with secondary antibodies conjugated with Alexa Fluor®-568 (goat anti-rabbit). Counterstaining with DAPI (14285, Cayman Chemical Company) was performed before image acquisition (LSM 710, Zeiss).
Real time qRT-PCR analysis
RNA was extracted from cultured cells, sorted primary melanoblasts or whole skin with TRIzol (15596026, Invitrogen)-chloroform. Skin samples were weighted, frozen in liquid nitrogen and equal amounts of frozen tissue were ground with mortar and pestle before extracting RNAs in TRIzol. RNA extracts were further purified and DNase treated by using RNAeasy QIAGEN columns (DNase 79254, QIAGEN). cDNA was synthesized from 400 ng (melanoblasts) or 1μg (cell lines and tissues) input RNA using Transcriptor first-strand cDNA synthesis kit (04897030001, Roche) according to the manufacturer’s instructions. Quantitative PCR was performed using LightCycler 480 SYBR green I master mix (04887352001, Roche) in a LightCycler 480 II (Roche). Relative mRNA levels were calculated using the 2-ΔΔCt method, normalized to L32. Primer sequences:
RPL32: F 5′ TCTGGTGAAGCCCAAGATCG 3′ R: 5ˊ CTCTGGGTTTCCGCCAGTT 3′
MITF: F 5′ GACTAAGTGGTCTGCGGTGT 3′ R: 5′ CTGGTAGTGACTGTATTCTA 3′
TYR: F 5′ TGGACAAAGACGACTACCACA 3′ R: 5′TTTCAGTCCCACTCTGTTTCC 3′
TYRP1: F: 5′ GGCATCAGGGGAAAAGCAGA 3′ R: 5′ GCTCAGATGAAAATACAGCAGTACC 3′
DCT: F: 5′ AACAGACACCAGACCCTGGA 3′ R: 5′ AAGTTTCCTGTGCATTTGCATGT 3′
PMEL: F: 5′ ACTGCCAGCTGGTTCTACAC 3′ R: 5′ CACCGTCTTGACCAGGAACA 3′
MLANA: F: 5′ GTGTTCCTCGGGGAAGGTGT 3′ R: 5′ CAGCAGTGACATAGGAGCGT 3′
OCA2: F: 5′ ATAGTGAGCAGGGAGGCTGT 3′ R: 5′ ACTGATGGGCCAGCAAAAGA 3′
COL11A1: F: 5′ CGATGGATTCCCGTTCGAGT 3′ R: 5′ GAGGCCTCGGTGGACATTAG 3′
S100A4: F: 5′ CCTCTCTCTTGGTCTGGTCTC 3′ R: 5′ GTCACCCTCTTTGCCTGAGT 3′
Proliferation and viability assays
Cells were seeded in white 96-well plates with clear bottom (6005181, PerkinElmer) at 3500 cells/well and cell proliferation over 3–5 day periods was determined by measuring luminescence with CellTiter-Glo® assay kit (G7570, Promega), according to manufacturer’s instructions. For experiments measuring A375 cell viability after CRTC3 overexpression and treatment with inhibitors, 1×106 cells (grown in 6-well dishes) were subjected to nucleofection with Amaxa® Cell Line Nucleofector® kit V (VCA-1003, Lonza), following the manufacturer’s protocol. Total of 2μg pSelect-puro-CRTC3 or empty vector were used per well. Cells were treated with 5μM vemurafenib (PLX4032, RG7204, S1267, Selleckchem) or 20μM cisplatin (S1166, Selleckchem) for 72h, 24h post nucleofection. Prior to experiments, cells were tested for sensitivity to vemurafenib and cisplatin (inhibitor range 0–20μM) and IC50 determined to be 0.0829μM and 6.99μM, respectively.
Sub-cellular fractionation and western blotting
Cells were grown to 65% confluency on 10cm dishes, treated with compounds indicated and fractionated using Calbiochem® fractionation kit (539790), according to the manufacturer’s instructions. For some fractionation experiments a previously described protocol (Andrews and Faller, 1991) was used with some modifications. Briefly, cells were treated with indicated compounds for 20min, washed and scraped in cold phosphate-buffered saline (PBS), resuspended in buffer A (10mM HEPES-KOH pH 7.4, 1.5mM MgCl2, 10mM KCl, 0.2mM PMSF, protease inhibitor cocktail (P9599, Sigma), phosphatase inhibitors 2 and 3 (P5726 and P0044, Sigma), 0.1% NP-40) and allowed to swell on ice for 10min. Cell swelling was monitored through light microscope visualization. Cells were spun and supernatant used as cytosolic fraction. Nuclear proteins were extracted in high salt buffer C (20mM HEPES-KOH pH 7.9, 25% glycerol, 420mM NaCl, 1.5mM MgCl2, 0.2mM EDTA, 0.2mM PMSF, protease inhibitor cocktail, phosphatase inhibitors 2 and 3) for 20min on ice. Compounds used for cell treatments in these experiments were SCF (S9915, Sigma), HGF (H9661, Sigma), FSK (F6886; Sigma) and IBMX (I5879, Sigma). Whole cell or tissue extracts were prepared by collecting the samples in KB (Killer buffer, 2M urea, 4% sucrose, 5% SDS, 1mM EDTA, protease inhibitor cocktail, phosphatase inhibitors 2 and 3) and then passing through Qiashredder columns (79656, QIAGEN). Tissue samples were weighted, frozen in liquid nitrogen and ground using mortar and pestle before lysis. Protein concentrations were determined with Pierce micro BCA protein assay kit (23235, Thermo Fisher). Samples were resolved by SDS-polyacrylamide gel electrophoresis and transferred onto PVDF membranes (IPVH00010, Millipore). Membranes were blocked in 5% milk before adding antibodies of interest. HyGlo HRP detection kit (E2500, Denville) was used to visualize proteins.
Luciferase assays
MITF proximal promoter was cloned into SacI and HindIII sites of the PGL4 promoter vector (E6651, Promega). EVX_2xCRE promoter cloned into PGL4 (Sonntag et al., 2017) was used to measure transcriptional activity of CRTC3 mutations found in melanoma patients. Mutations in CRE sites and CRTC3 were produced via site-directed mutagenesis. All constructs were sequenced to confirm the presence of mutations. Cells were seeded in 24-well dishes at 0.03×106 cells/well and transfected with reporter constructs 24h post seeding, using Lipofectamine 2000 (11668019, Invitrogen) according to the manufacturer’s instructions. Total of 400ng of DNA was added in each well. In the case of CRTC3 melanoma mutations, EVX and pSelect_CRTC3 constructs were co-transfected at 200ng DNA each. Expression of CRTC3 constructs was visualized via western blotting and was used to normalize activity. Media was changed 24h after transfection. Various treatments that the cells were subjected to 48h after transfection are indicated in respective Figures and Legends. Cells were lysed in luciferase extraction buffer (25mM Gly-Gly, 15mM MgSO4, 4mM EGTA, 1mM DTT, 1% Triton X-100). 50 μL of extract was added to 50μl of assay buffer (25mM Gly-Gly, 15mM MgSO4, 4mM EGTA, 15mM K2HPO4, pH 7.8, 2mM DTT, 2.5mM ATP) and 50μl of 0.1mM d-luciferin K+ salt before measuring luminescence in a GloMax® multi microplate reader (Promega).
Oligonucleotides used for cloning:
MITFprom_F: 5′CATGAGCTCAGACTCGGGTGCAAGATGAAG 3′
MITFprom_R: 5′GTGAAGCTTAGCAAGGTTTCAGGCAGCCCC 3′
CRTC3 F: 5′ GATGGTCGACATGGCCGCCTCGCCCGGTTCGGGCAGCGCCAAC 3′
CRTC3 R: 5′ CATGCTAGCTCATAGCCGGTCAGCTCGAAACGTCTCCTCCAC 3′
Oligonucleotides used for site-directed mutagenesis:
MITFprom_CRE_F: 5′TATCTATGAAAAAAAGCATGAAATCAAGCCAGCAGGGAAACTGATATC 3′
CRTC3-P142S_F: 5′ GAGCTGGCCACGGCAACAGTCTCCTTGGAAAGAAGAGAAGCA 3′
CRTC3-L166F_F: 5′ GGACCAATTCTGATTCTGCTTTTCACACGAGTGCTCTGAGCACC 3′
CRTC3-H315N_F: 5′ CAACCTTCCAGCTGCCATGACTAACCTGGGGATAAGAACCTCCTC 3′
CRTC3-P331H_F: 5′ CTCCAAAGTTCTCGAAGTAACCATTCCATCCAAGCCACACTCAGT 3′
CRTC3-S368F_F: 5′ CACCCCTCCCTCCGGCTCTTCTTCCTTAGCAACCCGTCTCTTTCC 3′
CRTC3-P419S_F: 5′ GACCAGCCCACTGAACCCGTATTCTGCCTCCCAGATGGTGACCTCA 3′
Chromatin immunoprecipitation and sequencing (ChIP-seq)
ChIP was performed as previously described (Van de Velde et al., 2019). Briefly, cells were grown to 80% confluency in 150mm dishes (one per IP), treated with DMSO or 5μM FSK for 30min, fixed in 1% formaldehyde for 10min and quenched with 125mM glycine for 5min. Cells were washed and scraped in ice-cold phosphate-buffered saline (PBS) and then resuspended in buffer LB3 (10mM Tris-HCl (pH 8.0), 100mM NaCl, 1mM EDTA, 0.5mM EGTA, 0.1% Na-deoxycholate, 0.5% N-laurylsarcosine, protease inhibitor cocktail). Resuspended cells were mixed with 100mg glass beads and sonicated (Virtis Virsonic 100 sonicator) at power 9 for 9 pulses (20sec on, 1min off on ice). Sonication testing was performed before every ChIP experiment to determine the optimal shearing conditions. After sonication, 1% final Triton X-100 was added and the extract clarified by centrifugation at 17,000 × g for 5 min. Thirty microliters of protein A-agarose beads (20333, Thermo Scientific) were prepared for IP by washing in LB3 buffer, then washing twice with PBS/0.5% bovine serum albumin (BSA) and incubating with 5μg antibody in PBS/0.5% BSA for 3h at 4°C with rotation. For ChIP, beads were washed once in PBS/0.5% BSA and 500 μL of clarified extract was added to antibody-coupled beads and incubated overnight at 4°C with rotation.
Beads were washed 3 times in 500 μL wash buffer 1 (20mM Tris-HCl pH 7.4, 150mM NaCl, 2mM EDTA, 0.1% SDS, 1% Triton X-100) and three times in wash buffer 2 (20mM Tris-HCl (pH 7.4), 250mM LiCl, 1mM EDTA, 1% Triton X-100, 0.7% Na-deoxycholate). Elution was then performed by incubating beads in 50 μL elution buffer 1 (Tris-EDTA, 1% SDS) for 15min at 50°C and 45 μL elution buffer 2 (TE, 1% SDS, 300 mM NaCl) for 15min at 50°C with shaking. Both elutions were combined and cross-links reversed overnight at 65°C with shaking. Samples were incubated with RNase A for 1h at 37°C and proteinase K for 1h at 50°C. ChIP DNA was purified using Agencourt AMPure XP beads (A63881, Beckman Coulter) and DNA amounts determined by Qubit® 2.0 fluorometer (Invitrogen). All samples had more than 0.5ng/μl DNA, which was considered as the cutoff to proceed to library preparation. Libraries were prepared with NebNext® ChIP seq reagents for Illumina (E6240 and E7335, New England Biolabs), following the manufacturer’s protocol. High-throughput sequencing was performed on the HiSeq 2500 system (Illumina) at a run configuration of single read 50 bases. Image analysis and base calling were done with Illumina CASAVA-1.8.2.on HiSeq 2500 system and sequenced reads were quality-tested using FASTQC.
ChIP-seq analysis
Reads were aligned to the mouse (mm10) genomes using STAR (Dobin et al., 2013). Tag directories of uniquely mapped reads were generated with HOMER (Heinz et al., 2010) and peaks were called and annotated using HOMER (findPeaks in “factor” mode for CREB, CRTC2, CRTC3, POLII and MITF and in “histone” mode for H3AcK27; annotatePeaks.pl, default parameters). Motif enrichment near ChIP-seq peaks was performed with HOMER using findMotifsGenome.pl. Overlapping peaks were visualized by generating BigWig files with HOMER (makeMultiWigHub.pl, default parameters) and uploading the hubs in the UCSC Genome Browser (http://genome.ucsc.edu). Enrichment analysis was preformed using WebGestalt (Liao et al., 2019).
DNA binding motif screen in intron 86 of H.sapiens HERC2 gene
HOMER annotatePeaks.pl was used to scan for CRE and Mbox motifs in intron 86 of the HERC2 gene in the hg38 human genome. Motifs of interest were visualized in the UCSC genome browser and overlapped with variants from gnomAD (Collins et al., 2020), PhyloP (Pollard et al., 2010) conservation tracks.
RNA-sequencing and analysis
RNAs were prepared as indicated in RT-qPCR protocol. The quality of the isolated total RNA was assessed using the Tape Station 4200 and RNA-seq libraries were prepared using the TruSeq stranded mRNA Sample Preparation Kit v2 (RS-122-2001) according to Illumina protocols. RNA-seq libraries were multiplexed, normalized and pooled for sequencing. High-throughput sequencing was performed on the HiSeq 2500 system (Illumina) at a run configuration of single read 50 bases. Image analysis and base calling were done with Illumina CASAVA-1.8.2.on HiSeq 2500 system and sequenced reads were quality-tested using FASTQC. FASTQ files were aligned to the mouse mm10 or human hg19 genome builds using STAR (Dobin et al., 2013). Independent biological replicates (2 for CTRL and CRTC3 KO B16F1 and A375 cell line samples, 1 for CRTC3 rescue sample in B16F1 cells, 4 for WT and CRTC3 KO skin samples) were used for differential expression analysis with HOMER (analyzeRepeats.pl with option –raw; getDiffExpression.pl using DESeq2; Love et al., 2014). Expression was compared between vehicle and FSK treatment or CTRL and KO genomes and differentially expressed genes were defined as having Log2 fold change of ≥ 1 or ≤ −1 (1.3 and −1.3 for whole skin samples) and an adjusted p value of ≤ 0.05. Rescued genes were defined with a Log2 fold change of ≥ 0.58 or ≤ −0.58 with respect to their relative KO values. Enrichment analysis was preformed using WebGestalt (Liao et al., 2019). Heatmaps were generated with Cluster 3.0 (Eisen et al., 1998) and Java TreeView (version 1.1.6r4) (Saldanha, 2004).
Antibodies
Rabbit anti-CREB serum (244, in-house), rabbit anti-CRTC1 (C71D11, 2587, CST), rabbit anti-CRTC2 serum (6865, in-house), rabbit anti-CRTC3 (C35G4, 2720, CST), rabbit anti-CRTC3-pSer391 (PBL #7408, in-house; Sonntag et al., 2019), rabbit-anti-hCRTC3 (PBL #7019, in-house; Sonntag et al., 2019), rabbit anti-mOCA2 (PBL #7431, in-house, this work), rabbit anti-H3AcK27 (ab4729, Abcam), Anti-RNA polymerase II (CTD repeat YSPTSPS (phospho S2) antibody - ChIP Grade (ab5095)), mouse anti-Tubulin (05-829; EMD Millipore), mouse anti-MITF (clone C5, MAB3747-1 Millipore; MITF ChIP grade ab12039 Abcam), rabbit anti-TYR (ab61284, Abcam), rabbit anti-DCT (ab74073, Abcam), rabbit anti-MLANA (NBP254568H, Novus), rabbit anti-PMEL (ab137078, Abcam) rabbit anti-ERK1/2 (4695, CST), rabbit anti-pERK1/2 (9101, CST), mouse anti-HSP90α/β (SC-13119, Santa Cruz Biotechnology), rabbit anti-Histone H3 (9715, CST).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using the GraphPad Prism software (9.0.1 (151)). Statistical tests run for each experiment can be found in the corresponding Figure Legends. N represents the number of biological replicates (number of animals, number of biologically independent cell experiments or number of melanoma patient samples from TCGA database). Bar graphs represent means ± SEM. Determination of significance for RNA-sequencing experiments is detailed in the corresponding STAR Methods section. For all other experiments, a p value of ≤% 0.05 was considered significant (* < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001). Graphical presentations were generated with GraphPad Prism software (9.0.1(152)).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| anti-CRTC3 | CST | C35G4, 2720; RRID:AB_2083845 |
| anti-CRTC3-pSer391 | Sonntag et al., 2019 | PBL #7019; https://doi.org/10.1016/j.isci.2018.12.012 |
| anti-mOCA2 | This work | PBL #7431 |
| anti-ERK1/2 | CST | 4695; RRID:AB_390779 |
| anti-pERK1/2 | CST | 9101; RRID:AB_331646 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Forskolin | Sigma | F6886 |
| IBMX | Sigma | I5879 |
| Stem cell factor (SCF) | Sigma | S9915 |
| TPA | Sigma | P1585 |
| Cholera toxin | Sigma | C8052 |
| Vemurafenib | Selleckchem | S1267 |
| Cisplatin | Selleckchem | S1166 |
|
| ||
| Critical commercial assays | ||
|
| ||
| cAMP ELISA kit | Cayman Chemicals | 581001 |
| Amaxa® Cell Line Nucleofector® kit V | Lonza | VCA-1003 |
| Sub-cellular fractionation kit | Calbiochem | 539790 |
| Transwell migration assay | Corning | 354480 |
|
| ||
| Deposited data | ||
|
| ||
| Sequencing datasets | This work | GSE154117 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| A-375 human melanoma line | ATCC | CRL-1619 |
| B16-F1 | ATCC | CRL-6323 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57/BL6_CRTC3 | Song et al., 2010 | https://doi.org/10.1038/nature09564 |
| Mouse: iDCT-GFP | NCI Mouse Repository | FVB.Cg-Tg(Dct-rtTA2S*M2)#Mrln Tg(tetO-HIST1H2BJ/GFP)47Efu/Nci |
|
| ||
| Oligonucleotides | ||
|
| ||
| 5′ CACCGCATCAAGCCGATAATGTTCG 3′ | This work | mC3g1 |
| 5′ CACCGAGCCACTGCCTAAACACCTG 3′ | This work | mC3g2 |
| 5′GGCGCAGGCAGCCTCCAGTC GCCCCTCTCACGCTCTCTCCTG 3′ | This work | S391A_F |
| 5′ CACCGTGAGCGGCCCGTCTCG GCGT 3′ | This work | hC3g1 |
| 5′ CACCGCGCGCTGCACACGCAGAGAC 3′ | This work | hC3g2 |
|
| ||
| Software and algorithms | ||
|
| ||
| HOMER software | Heinz et al., 2010 | https://doi.org/10.1016/j.molcel.2010.05.004 |
Highlights.
CRTC3 knockout mice have decreased fur pigmentation
Melanosome maturation is defective in CRTC3 knockout melanocytes
CREB and CRTC3 promote OCA2 expression in melanocytes
Increased CRTC3 expression/activity in melanomas correlates with reduced survival
ACKNOWLEDGMENTS
This work was supported by NIH grant R01 DK083834, the Leona M. and Harry B. Helmsley Charitable Trust, the Clayton Foundation for Medical Research, and the Salkexcellerator Fund. We acknowledge the core facilities of the Salk Institute: Flow Cytometry Core Facility with funding from NIH-NCI CCSG grant P30 014195 and Shared Instrumentation Grant S10-OD023689 (Aria Fusion cell sorter), the Waitt Advanced Biophotonics Core Facility with funding from NIH-NCI CCSG grant P30 014195, and the Waitt Foundation and the NGS Core Facility of the Salk Institute with funding from NIH-NCI CCSG grant P30 014195, the Chapman Foundation, and the Leona M. and Harry B. Helmsley Charitable Trust.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109136.
REFERENCES
- Altarejos JY, and Montminy M (2011). CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 12, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarejos JY, Goebel N, Conkright MD, Inoue H, Xie J, Arias CM, Sawchenko PE, and Montminy M (2008). The Creb1 coactivator Crtc1 is required for energy balance and fertility. Nat. Med. 14, 1112–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews NC, and Faller DV (1991). A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 19, 2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang S, Won KH, Moon H-R, Yoo H, Hong A, Song Y, and Chang SE (2017). Novel regulation of melanogenesis by adiponectin via the AMPK/CRTC pathway. Pigment Cell Melanoma Res. 30, 553–557. [DOI] [PubMed] [Google Scholar]
- Bellono NW, Escobar IE, Lefkovith AJ, Marks MS, and Oancea E (2014). An intracellular anion channel critical for pigmentation. eLife 3, e04543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DC (1989). Mechanisms of differentiation in melanoma cells and melanocytes. Environ. Health Perspect. 80, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotto C, Abbe P, Hemesath TJ, Bille K, Fisher DE, Ortonne J-P, and Ballotti R (1998). Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. J. Cell Biol. 142, 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet E, Van de Velde S, Matsumura S, Hao E, LeLay J, Kaestner K, and Montminy M (2015). Feedback inhibition of CREB signaling promotes beta cell dysfunction in insulin resistance. Cell Rep. 10, 1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowicz S, Van Scoyk M, Avasarala S, Karuppusamy Rathinam MK, Tauler J, Bikkavilli RK, and Winn RA (2014). The soft agar colony formation assay. J. Vis. Exp., e51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscà R, and Ballotti R (2000). Cyclic AMP a key messenger in the regulation of skin pigmentation. Pigment Cell Res. 13, 60–69. [DOI] [PubMed] [Google Scholar]
- Buscà R, Abbe P, Mantoux F, Aberdam E, Peyssonnaux C, Eychène A, Ortonne J-P, and Ballotti R (2000). Ras mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 19, 2900–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2015). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, Khera AV, Lowther C, Gauthier LD, Wang H, et al. ; Genome Aggregation Database Production Team; Genome Aggregation Database Consortium (2020). A structural variation reference for medical and population genetics. Nature 581, 444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui R, Widlund HR, Feige E, Lin JY, Wilensky DL, Igras VE, D’Orazio J, Fung CY, Schanbacher CF, Granter SR, and Fisher DE (2007). Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell 128, 853–864. [DOI] [PubMed] [Google Scholar]
- Delyon J, Servy A, Laugier F, André J, Ortonne N, Battistella M, Mourah S, Bensussan A, Lebbé C, and Dumaz N (2017). PDE4D promotes FAK-mediated cell invasion in BRAF-mutated melanoma. Oncogene 36, 3252–3262. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly MP, Paschou P, Grigorenko E, Gurwitz D, Barta C, Lu R-B, Zhukova OV, Kim J-J, Siniscalco M, New M, et al. (2012). A global view of the OCA2-HERC2 region and pigmentation. Hum. Genet. 131, 683–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy DL, Zhao ZZ, Sturm RA, Hayward NK, Martin NG, and Montgomery GW (2010). Multiple pigmentation gene polymorphisms account for a substantial proportion of risk of cutaneous malignant melanoma. J. Invest. Dermatol. 130, 520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumaz N, Hayward R, Martin J, Ogilvie L, Hedley D, Curtin JA, Bastian BC, Springer C, and Marais R (2006). In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 66, 9483–9491. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, and Botstein D (1998). Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo AM, Piazza GA, and Tinsley HN (2014). The role of cyclic nucleotide signaling pathways in cancer: targets for prevention and treatment. Cancers (Basel) 6, 436–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godwin LS, Castle JT, Kohli JS, Goff PS, Cairney CJ, Keith WN, Sviderskaya EV, and Bennett DC (2014). Isolation, culture, and transfection of melanocytes. Curr. Protoc. Cell Biol. 63, 1.8.1–1.8.20. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder MP, Jene-Sanz A, Santos A, and Lopez-Bigas N (2013). IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 10, 1081–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyonneau L, Murisier F, Rossier A, Moulin A, and Beermann F (2004). Melanocytes and pigmentation are affected in dopachrome tautomerase knockout mice. Mol. Cell. Biol. 24, 3396–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes JE, Cassidy PB, Manga P, Boissy RE, Goldgar D, Cannon-Albright L, Florell SR, and Leachman SA (2013). Report of a novel OCA2 gene mutation and an investigation of OCA2 variants on melanoma risk in a familial melanoma pedigree. J. Dermatol. Sci. 69, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemesath TJ, Price ER, Takemoto C, Badalian T, and Fisher DE (1998). MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature 391, 298–301. [DOI] [PubMed] [Google Scholar]
- Hendriks IA, Lyon D, Young C, Jensen LJ, Vertegaal ACO, and Nielsen ML (2017). Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 24, 325–336. [DOI] [PubMed] [Google Scholar]
- Herraiz C, Journé F, Abdel-Malek Z, Ghanem G, Jiménez-Cervantes C, and García-Borrón JC (2011). Signaling from the human melanocortin 1 receptor to ERK1 and ERK2 mitogen-activated protein kinases involves transactivation of cKIT. Mol. Endocrinol. 25, 138–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe AK (2004). Regulation of actin-based cell migration by cAMP/PKA. Biochim. Biophys. Acta 1692, 159–174. [DOI] [PubMed] [Google Scholar]
- Jannot A-S, Meziani R, Bertrand G, Gérard B, Descamps V, Archimbaud A, Picard C, Ollivaud L, Basset-Seguin N, Kerob D, et al. (2005). Allele variations in the OCA2 gene (pink-eyed-dilution locus) are associated with genetic susceptibility to melanoma. Eur. J. Hum. Genet. 13, 913–920. [DOI] [PubMed] [Google Scholar]
- Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK, Narayan R, Flaherty KT, Wargo JA, Root DE, and Garraway LA (2013). A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 504, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y-H, Kim D, Hong AR, Kim J-H, Yoo H, Kim J, Kim I, Kang S-W, Chang SE, and Song Y (2019). Therapeutic Potential of Rottlerin for Skin Hyperpigmentary Disorders by Inhibiting the Transcriptional Activity of CREB-Regulated Transcription Coactivators. J. Invest. Dermatol. 139, 2359–2367.e2. [DOI] [PubMed] [Google Scholar]
- Kim J-H, Hong AR, Kim Y-H, Yoo H, Kang S-W, Chang SE, and Song Y (2020). JNK suppresses melanogenesis by interfering with CREB-regulated transcription coactivator 3-dependent MITF expression. Theranostics 10, 4017–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozar I, Margue C, Rothengatter S, Haan C, and Kreis S (2019). Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta Rev. Cancer 1871, 313–322. [DOI] [PubMed] [Google Scholar]
- Levy C, Khaled M, and Fisher DE (2006). MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 12, 406–414. [DOI] [PubMed] [Google Scholar]
- Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, and Fisher DE (2007). Melanocyte biology and skin pigmentation. Nature 445, 843–850. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Q, Viste K, Urday-Zaa JC, Senthil Kumar G, Tsai W-W, Talai A, Mayo KE, Montminy M, and Radhakrishnan I (2012). Mechanism of CREB recognition and coactivation by the CREB-regulated transcriptional coactivator CRTC2. Proc. Natl. Acad. Sci. USA 109, 20865–20870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie KF, Clark K, Naqvi S, McGuire VA, Nöehren G, Kristariyanto Y, van den Bosch M, Mudaliar M, McCarthy PC, Pattison MJ, et al. (2013). PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J. Immunol. 190, 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquette A, André J, Bagot M, Bensussan A, and Dumaz N (2011). ERK and PDE4 cooperate to induce RAF isoform switching in melanoma. Nat. Struct. Mol. Biol. 18, 584–591. [DOI] [PubMed] [Google Scholar]
- Niwano T, Terazawa S, Nakajima H, and Imokawa G (2018). The stem cell factor-stimulated melanogenesis in human melanocytes can be abrogated by interrupting the phosphorylation of MSK1: evidence for involvement of the p38/MSK1/CREB/MITF axis. Arch. Dermatol. Res. 310, 187–196. [DOI] [PubMed] [Google Scholar]
- Park S, Morya VK, Nguyen DH, Singh BK, Lee H-B, and Kim E-K (2015). Unrevealing the role of P-protein on melanosome biology and structure, using siRNA-mediated down regulation of OCA2. Mol. Cell. Biochem. 403, 61–71. [DOI] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, and Siepel A (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price ER, Ding H-F, Badalian T, Bhattacharya S, Takemoto C, Yao T-P, Hemesath TJ, and Fisher DE (1998). Lineage-specific signaling in melanocytes. C-kit stimulation recruits p300/CBP to microphthalmia. J. Biol. Chem. 273, 17983–17986. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ (2004). Java Treeview—extensible visualization of microarray data. Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Sánchez-Más J, Hahmann C, Gerritsen I, García-Borrón JC, and Jiménez-Cervantes C (2004). Agonist-independent, high constitutive activity of the human melanocortin 1 receptor. Pigment Cell Res. 17, 386–395. [DOI] [PubMed] [Google Scholar]
- Scherer D, and Kumar R (2010). Genetics of pigmentation in skin cancer—a review. Mutat. Res. 705, 141–153. [DOI] [PubMed] [Google Scholar]
- Shannan B, Perego M, Somasundaram R, and Herlyn M (2016). Heterogeneity in Melanoma. Cancer Treat. Res. 167, 1–15. [DOI] [PubMed] [Google Scholar]
- Song Y, Altarejos J, Goodarzi MO, Inoue H, Guo X, Berdeaux R, Kim J-H, Goode J, Igata M, Paz JC, et al. ; CHARGE Consortium; GIANT Consortium (2010). CRTC3 links catecholamine signalling to energy balance. Nature 468, 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag T, Moresco JJ, Vaughan JM, Matsumura S, Yates JR III, and Montminy M (2017). Analysis of a cAMP regulated coactivator family reveals an alternative phosphorylation motif for AMPK family members. PLoS One 12, e0173013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag T, Ostojić J, Vaughan JM, Moresco JJ, Yoon Y-S, Yates JR III, and Montminy M (2019). Mitogenic Signals Stimulate the CREB Coactivator CRTC3 through PP2A Recruitment. iScience 11, 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel KP, Davidson DR, and Jackson IJ (1992). TRP-2/DT, a new early melanoblast marker, shows that steel growth factor (c-kit ligand) is a survival factor. Development 115, 1111–1119. [DOI] [PubMed] [Google Scholar]
- Sturm RA, Duffy DL, Zhao ZZ, Leite FPN, Stark MS, Hayward NK, Martin NG, and Montgomery GW (2008). A single SNP in an evolutionary conserved region within intron 86 of the HERC2 gene determines human blue-brown eye color. Am. J. Hum. Genet. 82, 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 47, D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell 33, 890–904.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Velde S, Wiater E, Tran M, Hwang Y, Cole PA, and Montminy M (2019). CREB Promotes Beta Cell Gene Expression by Targeting Its Coactivators to Tissue-Specific Enhancers. Mol. Cell. Biol. 39, e00200–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandamme N, and Berx G (2014). Melanoma cells revive an embryonic transcriptional network to dictate phenotypic heterogeneity. Front. Oncol. 4, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser M, Kayser M, and Palstra R-J (2012). HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome Res. 22, 446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser M, Kayser M, Grosveld F, and Palstra R-J (2014). Genetic variation in regulatory DNA elements: the case of OCA2 transcriptional regulation. Pigment Cell Melanoma Res. 27, 169–177. [DOI] [PubMed] [Google Scholar]
- Wang Y, Inoue H, Ravnskjaer K, Viste K, Miller N, Liu Y, Hedrick S, Vera L, and Montminy M (2010). Targeted disruption of the CREB coactivator Crtc2 increases insulin sensitivity. Proc. Natl. Acad. Sci. USA 107, 3087–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe H, Valencia JC, Yasumoto K, Kushimoto T, Ando H, Muller J, Vieira WD, Mizoguchi M, Appella E, and Hearing VJ (2004). Regulation of tyrosinase processing and trafficking by organellar pH and by proteasome activity. J. Biol. Chem. 279, 7971–7981. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Murata T, Shimizu K, Morita H, Inui M, and Tagawa T (2012). Phosphodiesterase 4 regulates the migration of B16-F10 melanoma cells. Exp. Ther. Med. 4, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellbrock C, and Arozarena I (2015). Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res. 28, 390–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellbrock C, and Arozarena I (2016). The Complexity of the ERK/MAP-Kinase Pathway and the Treatment of Melanoma Skin Cancer. Front. Cell Dev. Biol. 4, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Horrell EM, Boulanger MC, and D’Orazio JA (2016). Melanocortin 1 Receptor: Structure, Function, and Regulation. Front. Genet. 7, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Hemesath TJ, Takemoto CM, Horstmann MA, Wells AG, Price ER, Fisher DZ, and Fisher DE (2000). c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes Dev. 14, 301–312. [PMC free article] [PubMed] [Google Scholar]
- Yun C-Y, Hong SD, Lee YH, Lee J, Jung D-E, Kim GH, Kim S-H, Jung J-K, Kim KH, Lee H, et al. (2019). Nuclear Entry of CRTC1 as Druggable Target of Acquired Pigmentary Disorder. Theranostics 9, 646–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi MR, Hornyak TJ, and Merlino G (2011). A genetically engineered mouse model with inducible GFP expression in melanocytes. Pigment Cell Melanoma Res. 24, 393–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing datasets are available under GEO accession number GSE154117.