

Abstract
As the targets of chimeric antigen receptor (CAR)-T cells expand to a variety of cancers, autoimmune diseases, viral infections, and fibrosis, there is an increasing demand on identifying new antigens and designing new CARs that can be effectively activated. However, the rational selection of antigens and design of CARs are limited by a lack of knowledge regarding the molecular mechanism by which CARs are activated by antigens. Here, we present data supporting a “size-exclusion” model explaining how antigen signals are transmitted across the plasma membrane to activate the intracellular domains of CARs. In this model, antigen engagement with CAR results in a narrow intermembrane space that physically excludes CD45, a bulky phosphatase, out of the CAR zone, thus favoring CAR phosphorylation by kinases, which further triggers downstream pathways leading to T cell activation. Aligned with this model, increasing the size of CAR extracellular domains diminished CAR T activation both in vitro and in a mouse lymphoma model; membrane-proximal epitopes activated CAR-Ts better than membrane-distal epitopes. Moreover, increasing the size of CD45 by antibody conjugation enhanced the activation of CARs that recognize membrane-distal epitopes. Consistently, CAR-Ts expressing CD45RABC, the larger isoform, were activated to a higher level than those expressing a smaller isoform CD45RO. Together, our work revealed that CAR-T activation depends on the size difference between the CAR-antigen pair and CD45; the size of CAR, antigen, and CD45 can be targets for tuning CAR-T activation.
One Sentence Summary:
The size difference between CAR-antigen pair and CD45 regulates CAR-T activation.
INTRODUCTION
The chimeric antigen receptor (CAR) emerges as a powerful platform to reprogram immune cell function. T cells armed with CARs show remarkable efficacy for the treatment of refractory or relapsed B cell malignancies (1–3). Excitingly, the application of CAR-T therapy is expanding towards treating solid tumors, autoimmune diseases, virus infection, and cardiac and hepatic fibrosis (4–9). As the number of CAR-T targets keeps increasing, there is a growing demand on identifying new antigens and designing corresponding CARs that can be effectively activated. However, the rational selection of antigens and design of CARs are limited by a lack of knowledge on the molecular mechanism underlying antigen-dependent CAR activation.
CAR is a synthetic receptor with modular properties. A CAR can be viewed as a “Lego” receptor in which its extracellular, transmembrane, and intracellular domains are “borrowed” and recombined from other receptors or designed de novo. This modular feature enables the flexibility of CAR in recognizing a variety of target cells and in triggering multiple signaling pathways (10). Meanwhile, it also raises the question as to how different antigens trigger activation of the same intracellular domains. Textbooks usually describe a specific communication, using EGFR as an example, between the extracellular and intracellular domain that underlies receptor activation in responses to ligand binding (11). However, in the case of CAR, the activation of the cytosolic domain is no longer necessarily coupled to a specific ligand-binding domain because the ligand-binding domain can be replaced with a variety of single-chain antibodies or even synthetic binding modules including FKBP-FRB or complementary DNAs (12, 13). These facts challenge the traditional view of how receptors are activated by ligands and raise the critical question of the underlying principles that govern the activation of modular receptors like CAR.
Spatial reorganization of proteins at the cell-cell interface serves as an important principle in understanding receptor activation (14, 15). This is illustrated by the “kinetic segregation” model, in which TCR-pMHC interactions exclude a bulky phosphatase CD45 away from the TCR, leading to TCR phosphorylation by the kinase Lck (12, 16–20). A similar size-dependent spatial segregation is found in the phagocytic receptor (21, 22), immunoglobulin E receptor (23), and Notch receptor signaling (24). In vitro reconstitution experiments demonstrate that the size difference between receptor-ligand pair and CD45 (or an unligated molecule) is sufficient to cause their spatial segregation in the synapse (25, 26). Interestingly, CD45 is also excluded, at least partially, from the CAR-antigen interface (12, 27), but it has not been rigorously tested how CAR size affects CD45 exclusion. Several studies show that CAR activation is affected by a spacer or hinge inserted between the transmembrane domain and the single chain variable fragment (scFv). These works provide rich information on how to design better CARs for specific antigens. However, the reported results are controversial: the spacer either enhances (28–30), reduces (31), or has no effect (32) on CAR-T activation. Resolving these conflicting results requires a comprehensive characterization of individual signaling steps along the CAR pathway and an evaluation of the contribution of each key player (CAR, antigen, CD45) in CAR-T activation.
In this manuscript, we comprehensively investigated how the sizes of CAR, antigen, and CD45 affect CAR-T activation. We aimed to test a “size-exclusion” hypothesis that the binding of antigen to CAR induces a narrowed intermembrane space between T cells and cancer cells. Consequently, CD45 is excluded out of the “CAR zone” because of its large extracellular domain, leading to a net phosphorylation of CAR, which initiates downstream signaling leading to CAR-T activation. Supporting this hypothesis, our data showed that CAR-T activation was closely related to CD45 exclusion from the CAR-antigen interacting zone, both of which were inversely proportional to the length of the CAR extracellular domain; CAR-T with a longer extracellular domain displayed a reduced anti-tumor effect in a mouse lymphoma model; membrane-proximal epitopes activated CAR-T better than membrane-distal epitopes. Moreover, we implemented chemical and genetic approaches to increase the size of CD45 extracellular domain, which resulted in an enhancement of CAR-T activity against membrane-distal epitopes. Together, these efforts highlighted physical size, in addition to antigen binding affinity, as a critical factor in determining CAR-T activation.
RESULTS
CAR-T activation decreases as the length of CAR extracellular domain increases
As a first step to test the size-exclusion hypothesis for CAR activation (Fig. 1A), we investigated how the length of the extracellular domain of CAR affected CAR signaling. We chose the CD19 CAR as a model, as this is the most widely used CAR in both research and clinics. This CAR was composed of an scFv recognizing CD19 (FMC63), a stalk and transmembrane domain from CD8α, and a cytosolic part containing tandem signaling domains from CD28, 4-1 BB, and CD3ζ. A GFP tag was additionally included at the C-terminus for visualizing CAR expression. The size of the CAR-CD19 pair in the extracellular space was estimated to be around 12 nm (Alphafold2 prediction or (33)), which was smaller than the size of the extracellular domain of CD45 (22–28 nm for the RO isoform (34, 35)). To determine if CAR-T activation depended on the length difference between the CAR-CD19 pair and CD45, we increased the length of the CAR extracellular domain by inserting single or multiple immunoglobulin domains between the scFv and stalk of CAR (Fig. 1B). These immunoglobulin domains were derived from carcinoembryonic antigen CEACAM5 (CEA), and named N, N-A1, N-A1-B1, and N-A1-B1-A2, which had an estimated length of 4, 8, 12, or 16 nm, respectively (36). These domains are successfully implemented to quantitatively increase cell surface protein size (22). The above CAR variants were introduced into Jurkat T cells by lentiviral transduction. All the CAR variants showed a similar cell surface expression level as determined by flow cytometry (Fig. 1C and S1A). To exclude the possibility that inserting immunoglobulin domains abolished CAR’s binding to cancer cells, we incubated CAR-T cells with Raji B cells expressing CD19. The cell-cell conjugation efficiency was similar across all CAR variants, suggesting that these CARs can still bind CD19 normally (Fig. 1D). In contrast, CD45 exclusion from the CAR synapse was gradually reduced as the length of CAR increased (Fig. 1E–F). Consistent with the biochemical activity of CD45 as a phosphatase, phosphorylation of ITAMs on CAR, as determined by flow cytometry, was gradually reduced as the CD45 exclusion was decreased (Fig. 1G and S1B). Flow cytometry further showed that ERK phosphorylation, which was triggered following ITAM phosphorylation (37), displayed a corresponding reduction (Fig. 1H and S1C). We further investigated long-term signaling outputs which involve transcription induction. Both CD69 expression and IL-2 release displayed a CAR size-dependent reduction. Intriguingly, a significant drop was revealed between the CEA-N and CEA-N-A1 CAR (Fig. S1D–F), suggesting a threshold to activate these two signaling outputs in Jurkat T cells. Together, these data supported a size-exclusion model in which CD19 binds to CAR, excludes CD45, enables a net phosphorylation of CAR, which triggers downstream signaling leading to T cell activation.
Fig. 1. The length of CAR extracellular domain regulated CD45 exclusion and CAR signaling in Jurkat T cells.
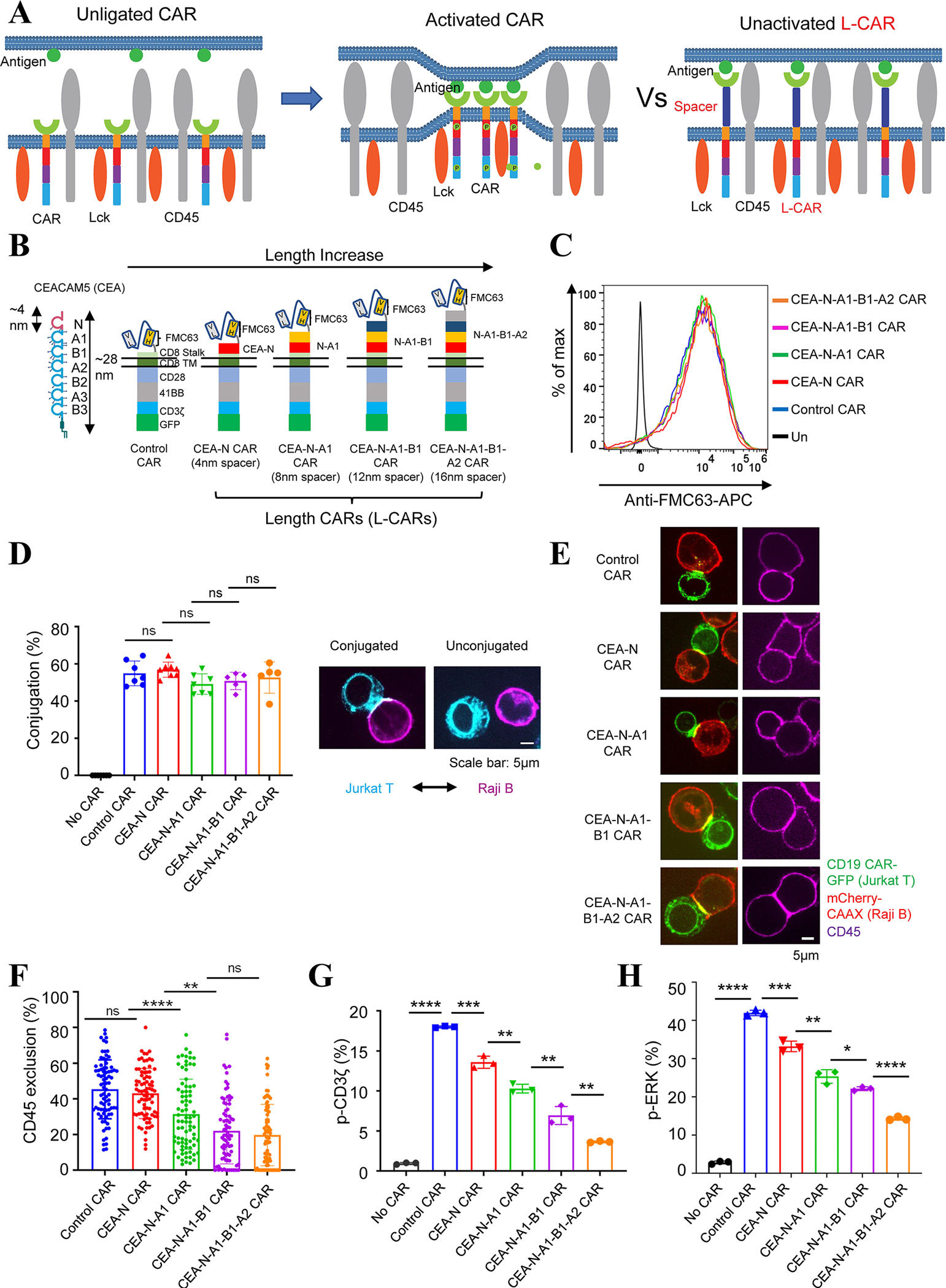
(A) Schematic representation of the ‘size exclusion’ hypothesis explaining antigen-dependent CAR activation. LEFT: In resting cells, the phosphorylation of CAR by the kinase Lck is antagonized by the phosphatase CD45. MIDDLE: The binding of antigen to CAR creates a narrowed intermembrane space (synapse), from which CD45 is excluded, leading to the phosphorylation of CAR. RIGHT: The binding of antigen to a CAR with a longer extracellular domain (L-CAR) does not exclude CD45. Consequently, CAR is not activated (phosphorylated).
(B) Generation of CD19 CARs with increasing lengths (L-CARs) by inserting tandem immunoglobulin domains of CEACAM5 (CEA) into the extracellular part of the CAR. Each immunoglobulin domain of CEA is around 4 nm long.
(C) CAR variants expression on the surface of Jurkat T cells, as determined by staining with an anti-FMC63 (CD19 scFv) antibody.
(D) Effects of inserted Ig domains on CAR’s binding to antigen. Jurkat T cells expressing CAR-GFP and Raji B cells expressing mCherry-CAAX (E:T=1:1) were co-cultured for 30 mins and the cell-cell conjugation was imaged by confocal microscopy. LEFT: Quantification of conjugation percentage (%), which was calculated by dividing the number of CAR T cells that bound to Raji B cells by the total number of CAR T cells. Each dot indicates one independent experiment. Data were presented as mean with SD (Unpaired T test, p>0.05 (ns)). N=5–7 co-culture experiments. RIGHT: representative images of conjugated cells versus unconjugated cells.
(E) Effects of CAR length on the exclusion of CD45 from the CAR synapse. Jurkat T cells expressing CAR-GFP and Raji B cells expressing mCherry-CAAX (E:T =1:1) were co-cultured for 30 mins, fixed, and stained with an anti-CD45 antibody conjugated with APC. Images were acquired by confocal microscopy. The exclusion percentage = (1 − ICD45 in car zone/ICD45out car zone) *100%. Data were presented as mean with SD (Mann-Whitney U test, p>0.05 (ns), p<0.01 (**), p<0.0001 (****)). N=80 synapses. Representative images were shown.
(F) Phosphorylation of the CD3ζ domain in Jurkat T cells expressing CAR variants. pCD3ζ in GFP+ CAR Jurkat T cells was detected by flow cytometry 5 min after co-culture with Raji B cells (E:T =1:1). Data were presented as mean with SD (Unpaired T test, p<0.0001 (****), p<0.001 (***), p<0.01 (**)). N=3 co-culture experiments.
(G) ERK activation in Jurkat T cells expressing CAR variants. pERK in GFP+ CAR Jurkat T cells was detected by flow cytometry 5 min after co-culture with Raji B cells (E:T =1:1). Data were presented as mean with SD (Unpaired T test, p<0.0001 (****), p<0.001 (***), p<0.01 (**), p<0.05 (*)). N=3 co-culture experiments.
Next, we verified the size-dependent activation in human primary T cells. Pan T cells were purified from peripheral blood mononuclear cells from healthy donors and infected with lentiviruses of CAR variants (Fig. 2A and S2A). These CAR-Ts were co-cultured with Raji B cells and displayed a similar cell-cell conjugation percentage (Fig. 2B). Meanwhile, the level of CD45 exclusion from the CAR synapse decreased as the CAR length increased (Fig. 2C–D). Consistently, the ERK phosphorylation (Fig. S2B), cell proliferation (Fig. 2E), cytotoxicity (Fig. 2F), production of IFNγ (Fig. 2G) and TNFα (Fig. 2H) in CAR-Ts were gradually compromised as the length of CAR extracellular domain increased. Because T cell activation is linked to exhaustion, we further determined the expression of exhaustion markers PD1, TIM3, and LAG3, after coculturing CAR-T with Raji B cells for 7 days. Consistent with the results evaluating activation markers, the expression of all the three exhaustion markers displayed a size-dependent reduction (Fig. S3). Together, these data suggested that the length of the CAR extracellular domain is critical for the activation of primary CAR T cells.
Fig. 2. The length of CAR extracellular domain regulated CAR-triggered human primary T cell activation.

(A) Human T cells were purified from PMBC from anonymous healthy donors, expanded, and engineered with lentivirus to express CD19 CARs. CAR variants were expressed in a similar level on the surface of primary T cells, as determined by an anti-FMC63 (CD19 scFv) antibody.
(B) Antigen binding capacity after insertion of Ig domains into the CAR. Human T cells expressing CAR-GFP and Raji B cells expressing mCherry-CAAX (E:T =1:1) were co-cultured for 30 mins and the cell conjugation was imaged by confocal microscopy. Conjugation percentage (%) was calculated by dividing the number of CAR T cells that bound to Raji B cells by the total number of CAR T cells. Data were presented as mean with SD (Unpaired T test, p>0.05 (ns)). N=3 co-culture experiments.
(C) Effects of increasing CAR length on the exclusion of CD45 from the CAR synapse. Human T cells expressing CAR-GFP and Raji B cells expressing mCherry-CAAX (E:T =1:1) were co-cultured for 30 mins, fixed, and stained with an anti-CD45 antibody conjugated with APC. Images were acquired by confocal microscopy. Data were presented as mean with SD (Mann-Whitney U test, p>0.05 (ns), p<0.05 (*), p<0.01 (**), p<0.0001 (****)). N=33~34 synapses. Representative images were shown.
(D) Proliferative capacity of CAR-T cells after exposure to Raji B cells (E:T =1:1). The human CD8+ CAR-T cell number upon Raji B cell stimulation was calculated with counting beads by flow cytometry at indicated time points. Data were presented as mean with SD (Unpaired T test, p<0.0001 (****)). N=3 co-culture experiments.
(E) The tumor cell-killing capacity of CAR-T. Human CD8+ CAR T cells were co-cultured with Raji B-LUC-mCherry cells (E:T =5:1) for 24 hours. The Raji B cells remained were quantified by luciferase-generated fluorescence as detected by microplate photoreaders. Data were presented as mean with SD (Unpaired T test, p>0.05 (ns), p<0.05 (*), p<0.01 (**), p<0.0001 (****)). N=3 co-culture experiments.
(F-G) The co-cultured medium from (E) was used for the measurement of IFNγ and TNFα by ELISA. Data were presented as mean with SD (Unpaired T test, p>0.05 (ns), p<0.05 (*), p<0.01 (**), p<0.001 (***)). N=3 co-culture experiments.
To determine how the CAR length affected the anti-tumor efficacy in vivo, we exploited a lymphoma xenograft model in immune-deficient NSG mice (Fig. 3A). Raji B cells were injected subcutaneously into the mice. Seven days after, the mice received one dose of CAR-Ts with different extracellular domain lengths (Fig. S4A). The tumor size was monitored for up to 6 weeks, and blood was drawn for determining CAR-T number and exhaustion on Day 7, 14, and 21. We found that CAR-Ts inhibited tumor growth in a length-dependent manner (Fig. 3B and S4B). Mice received the control CAR-Ts survived longer compared to those that received CEA-N-A1 or CEA-N-A1-B1-A2 CAR-Ts (Fig. 3C). Flow cytometry analysis of circulating CAR T cells showed no significant difference in cell number among the control CAR T, CEA-N-A1 CAR T, and CEA-N-A1-B1-A2 CAR T (Fig 3D and Data File S1). However, consistent with our in vitro data (Fig. S3), the control CAR-Ts displayed a higher exhaustion percentage as compared to the CAR-Ts with long extracellular domains, as indicated by the exhaustion marker PD1, TIM3, and LAG3 (Fig. 3E and S4C). Together, these data suggested that increasing the CD19 CAR length reduced its anti-tumor effect, which is consistent with the “size-exclusion” model (Fig. 1A).
Fig. 3. Length-dependent anti-tumor activity of CAR-T in vivo.
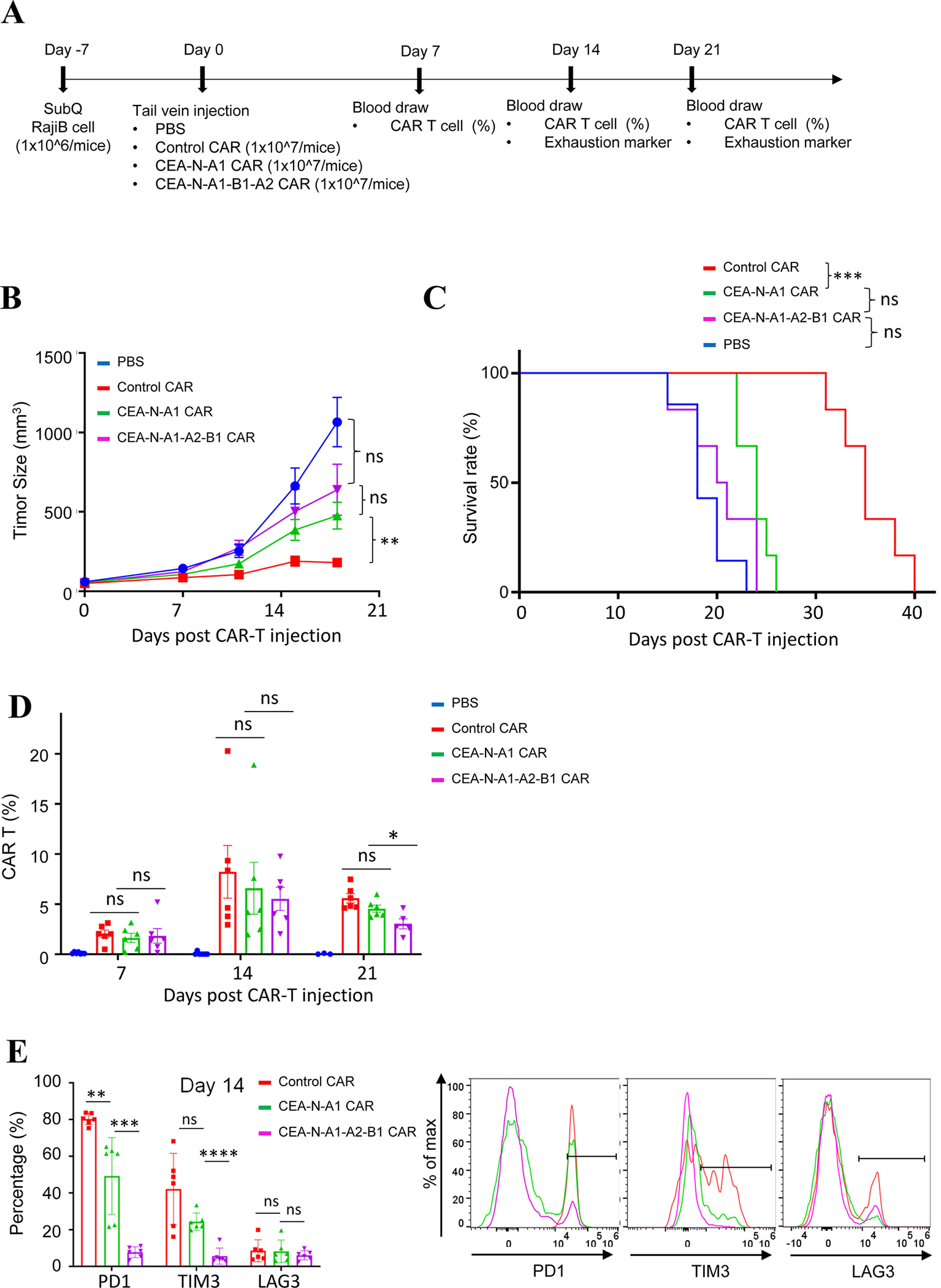
(A) Experimental settings of CAR-T mediated killing of xenografted tumors.
(B) Tumor growth monitored in NSG mice inoculated with Raji B cells and treated with PBS, control CAR T, CEA-N-A1 CAR T, or CEA-N-A1-B1-A2 CAR T cells. Data were presented as mean with SD (Unpaired T test was performed on data of day 18; P >0.05 (ns), P < 0.01 (**)). N = 6–7 mice.
(C) Mice survival with CAR-T treatment. Log-rank (Mantel-Cox) test was performed on data at day 28; P>0.05 (ns); P < 0.001 (***). N = 6–7 mice.
(D) CAR T percentage in circulation at day 7, 14, 21 post injection was determined via flow cytometry. Data were presented as mean with SD (Unpaired T test; P>0.05 (ns)). N= 6–7 mice.
(E) Flow cytometry analysis of exhaustion marker PD1, TIM3, LAG3 on CAR T cells at day 14. Data were presented as mean with SD (Unpaired T test; P>0.05 (ns), P < 0.01 (**), P < 0.001 (***), P < 0.0001 (****)). N=6–7 mice.
Membrane-proximal epitopes stimulate CAR-T better than membrane-distal epitopes
According to the size-exclusion model (Fig. 1A), a shorter intermembrane distance created by the CAR-antigen pair induces stronger exclusion of CD45 and thus higher activation of the CAR. Therefore, this model predicts that a small-sized antigen, or an antigen epitope located proximal to the cancer cell membrane, will trigger higher activation of CAR than an epitope located distal to the membrane (Fig. 4A). This is supported by previous studies showing that proximal epitopes activate CAR better than distal epitopes (38–40). However, because different epitope-scFv pairs are compared in some of these studies, the difference in CAR activation could result from a difference in antigen affinities, configuration, or binding partners (40). To rigorously test the positional effect of epitopes on CD45 exclusion and CAR-T activation, we followed a strategy in which CAR-Ts are compared between the same epitope on a wild-type versus truncated antigen (38); we engineered the epitope position on CD22, a large-sized antigen on the B cell surface (41). CD22 contains multiple immunoglobulin domains. One of the scFvs for CD22, RFB4, recognizes membrane-distal epitopes on Domain 3 (~20 nm from the membrane) whereas the other scFv m971 recognizes membrane-proximal epitopes on Domain 7 (~2 nm from the membrane) (42, 43) (Fig. 4B). The wild-type CD22 activated Jurkat T cells expressing m971 CAR better than those expressing RFB4 CAR (Fig. S5), which is consistent with published results (39). In order to compare CAR-Ts targeting the same epitope, we constructed a shortened version of CD22 of which Domain 5, 6, and 7 were deleted (CD22 short, Fig. 4B). The full-length (FL) or short extracellular domain of CD22, together with a transmembrane domain and an mCherry tag, was ectopically expressed in a leukemia line KU812 that did not naturally express CD22 (Fig. 4C). KU812 cells expressing the full-length or short CD22 displayed a similar conjugation efficiency to primary T cells expressing RFB4 CAR (Fig. 4D&E), suggesting that the truncation of CD22 did not alter its binding capacity to RFB4. In contrast, the exclusion of CD45 from the CAR synapse was significantly increased as CD22 was shortened (Fig. 4F). Accordingly, CAR-T activation was increased when encountering shortened CD22 as determined by ERK phosphorylation, cytotoxicity against KU812 cells, and production of TNFα (Fig. 4G–I, Data File S1), although the production of IFNγ were comparable between the full-length and short CD22-triggered stimulation (Fig. 4J). This could be explained by a potentially different threshold in activating IFNγ as compared to TNFα. Similar to primary T cells, Jurkat T cells expressing RFB4 CAR were better activated by the short CD22 as compared to the full-length CD22 (Fig. S6). As a control, T cells expressing m971 CAR, which binds membrane-proximal epitopes in Domain 7 (Fig. 4B), was only activated by KU812 expressing the full-length but not the short CD22 (Fig. S6). Together, these data supported our hypothesis that membrane-proximal epitopes on CD22 activate CAR-Ts better than membrane-distal epitopes.
Fig. 4. Effects of epitope position on human primary CAR-T targeting CD22.

(A) Schematics of CAR activation by membrane-proximal versus membrane-distal epitopes. LEFT: CARs bind membrane proximal epitopes to create a narrow intermembrane space that excludes phosphatase CD45 and induces CAR activation. Right: CARs recognize membrane-distal epitopes, which cannot form a close-contact zone to exclude CD45 effectively.
(B) Schematics of the binding sites of two scFvs, m971 and RFB4, that recognize CD22. CD22 contains seven extracellular immunoglobulin domains. RFB4 recognizes distal epitopes on Domain 3 whereas m971 recognizes proximal epitopes on Domain 7. A short version of CD22 was constructed by removing Domain 5–7 so that the epitopes on Domain 3 become membrane proximal. An mCherry tag replaced the intracellular part of both the full-length and short CD22.
(C) Expression of the full-length or short extracellular domain of CD22 in KU812 cells, as detected by western blot.
(D) Human primary T cells were purified from the peripheral blood of anonymous healthy donors, expanded, and engineered with lentivirus to express RFB4 CARs. CAR expression was evaluated by flow cytometry.
(E) Conjugation of RFB4 CAR-T cells with KU812 cells expressing the full-length (FL) or short CD22. Human primary T cells expressing RFB4 CAR were co-cultured with KU812 cells expressing FL or short CD22 extracellular domain (E:T =1:1) for 30 min before being imaged by confocal microscopy. Data were presented as mean with SD (Unpaired T test, p>0.05(ns)). N=3 co-culture experiments.
(F) Effects of epitope position on CD45 exclusion from the RFB4 CAR synapses. Primary T cells expressing RFB4 CAR were co-cultured with KU812 cells expressing the FL or short CD22 (E:T =1:1) for 30 min, followed by staining with an anti-CD45 antibody. Imaging was acquired by confocal microscopy. Data were presented as mean with SD (Mann-Whitney U test, p<0.01 (**)). N=50 synapses.
(G) ERK phosphorylation in RFB4 CAR-T cells engaged with the full-length or short CD22. Primary RFB4 CAR-T cells were cocultured with KU812 cells (E:T =1:1) for 10 minutes. ERK phosphorylation was assessed by flow cytometry. Data were presented as mean with SD (Unpaired T test, p<0.01 (**), p<0.05 (*)). N=3 co-culture experiments.
(H) Cytotoxicity of RFB4 CAR-T cells to cells expressing the full-length or short CD22. Primary RFB4 CAR T cells were co-cultured with FL or short CD22 cells (E:T =5:1) for 24 hours, followed by flow cytometry analysis. Data were presented as mean with SD (Unpaired T test, p<0.05 (*), p<0.01 (**)). N=3 co-culture experiments.
(I-J) TNFα and IFNγ production in RFB4 CAR-T cells. The co-cultured medium from (H) was used for the measurement of IFNγ and TNFα by ELISA. Data were presented as mean with SD (Unpaired T test, p>0.05 (ns), p<0.05 (*), p<0.01 (**), p<0.0001 (****)). N=3 co-culture experiments.
To generalize the finding on positional effect of epitope in activating CAR-Ts, we investigated another large-sized CAR antigen CEA (CEACAM5) which is expressed on the surface of multiple solid tumors (44). The scFv MFE23 recognizes a membrane-distal epitope (~24 nm from the membrane) that is located on the N-terminal domain and A1 domain of CEA (36). A truncated CEA was made by deleting the A2, B2, and A3 domain so that the MFE23 epitope was placed 12 nm closer to the membrane based on structural prediction (36) (Fig. 5A). The full-length and shortened CEA was ectopically expressed in HeLa cells that do not normally express CEA (Fig. 5B). Primary T cells expressing MFE23 CAR (Fig. 5C) were incubated with HeLa cells expressing the full-length or short CEA. MFE23 CAR-T displayed a similar cell-cell conjugation efficiency to HeLa cells expressing the full-length versus short CEA (Fig. 5D). Meanwhile, short CEA, as compared to the full-length CEA, induced higher CD45 exclusion from the CAR synapse, and higher ERK activation, killing capacity, and secretion of IFNγ and TNFα in MFE23 CAR-T cells (Fig. 5E–I, Data File S1). Consistent with the result on primary T cells, we found that short CEA activated Jurkat MFE23 CAR T better than the full-length CEA did (Fig. S7), supporting the conclusion that membrane-proximal epitopes activated CAR better than membrane-distal epitopes. These results highlighted the importance of physical position of epitopes in affecting CAR activation and provided a criterion for selecting new antigens for CAR development.
Fig. 5. Effects of epitope position on human primary CAR-T targeting CEA.
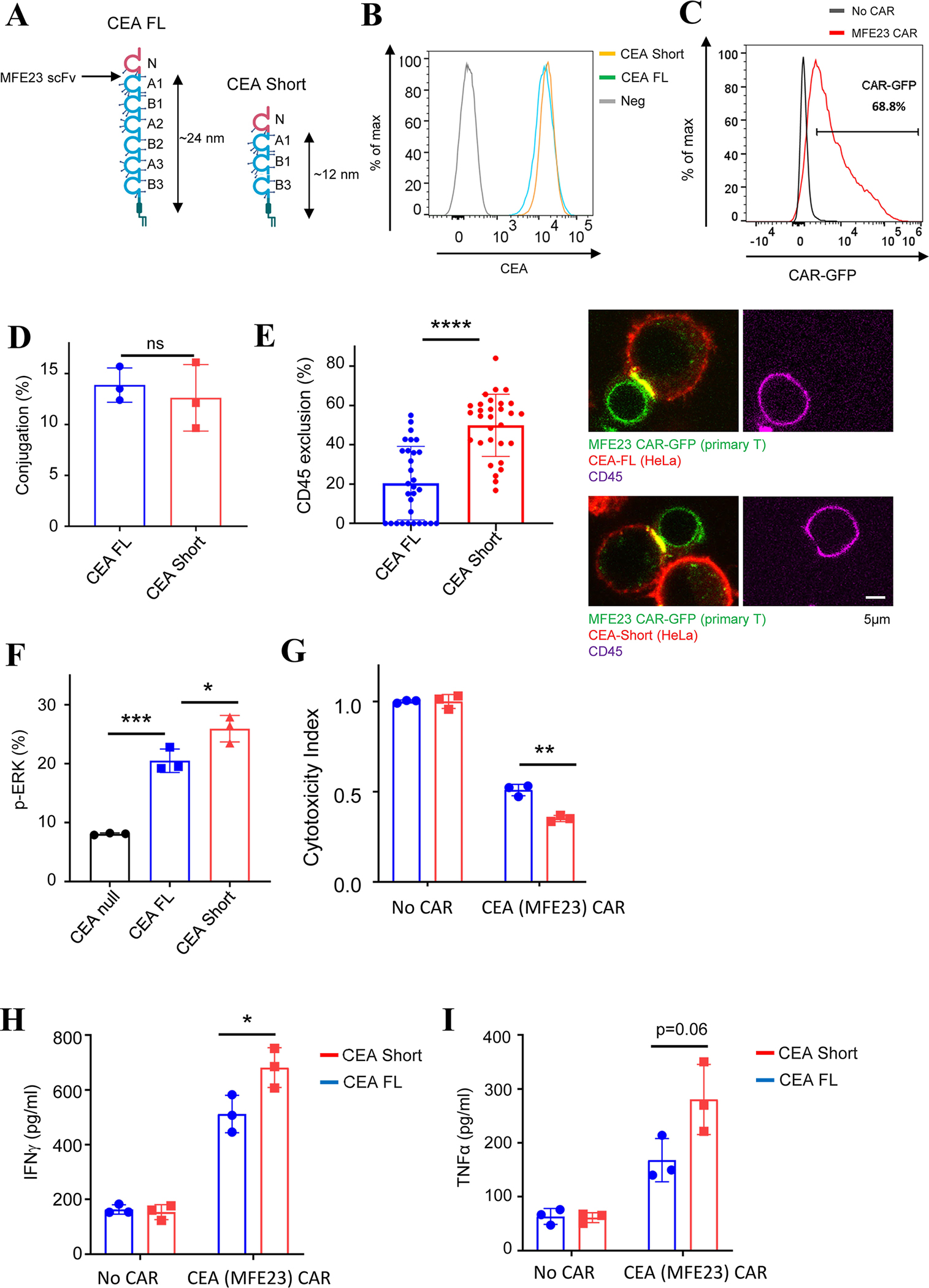
(A) The full-length (FL) CEA contains multiple immunoglobulin domains. scFv MFE23 recognize epitopes between the N and A1 domain. A short CEA was constructed by deleting domain A2, B2, and A3 so that the epitope for MFE23 was positioned closer to the membrane.
(B) Comparing the expression level of the FL and short CEA in HeLa cells, as determined by flow cytometry.
(C) Primary T cells expressing MFE23 CAR that recognizes CEA. CAR expression was evaluated by flow cytometry.
(D) Binding of primary MEF23 CAR-T cells to HeLa cells expressing the FL or short CEA. Primary T cells expressing MFE23 CAR were co-cultured with HeLa cells expressing the FL or short CEA (E:T =1:1) for 30 min before being imaged by confocal microscopy. Data were presented as mean with SD (Unpaired T test, p>0.05 (ns)). N=3 co-culture experiments.
(E) Effects of epitope position on CD45 exclusion from the MFE23 CAR synapses. Primary T cells expressing MFE23 CAR were co-cultured with HeLa cells expressing the FL or short CEA (E:T =1:1) for 30 min, followed by staining with an anti-CD45 antibody. Imaging was acquired by confocal microscopy. Data were presented as mean with SD (Mann-Whitney U test, p<0.0001 (****)). N=30 synapses.
(F) ERK phosphorylation in MFE23 CAR-T cells engaged with the full-length or short CEA. Primary MFE23 CAR-T cells were cocultured with HeLa cells (E:T =1:1) for 10 minutes. pERK was assessed by flow cytometry. Data were presented as mean with SD (Unpaired T test, p<0.001 (***), p<0.05 (*)). N=3 co-culture experiments.
(G) Cytotoxicity of MFE23 CAR-T cells to HeLa cells expressing the full-length or short CEA. Primary MFE23 CAR T cells were co-cultured with HeLa cells expressing the FL CEA or short CEA cells (E:T =5:1) for 24 hours, followed by flow cytometry analysis. Data were presented as mean with SD (Unpaired T test, p<0.01 (**)). N=3 co-culture experiments.
(H-I) TNFα and IFNγ production in MFE23 CAR-T cells. The co-cultured medium from (G) was used for the measurement of IFNγ and TNFα by ELISA. Data were presented as mean with SD (Unpaired T test, p>0.05 (ns), p<0.05 (*)). N=3 co-culture experiments.
To evaluate if the above conclusions about CAR size and epitope position were relevant to CAR-Ts used in clinics, we explored protein structures deposited in Protein Data Bank or predicted by alphafold2 (45) to estimate the size of CAR-antigen pair of the six FDA-approved CAR-Ts, of which four target CD19 and two target BCMA. The epitope position of all six CARs was membrane-proximal (~2 nm to the membrane). The estimated size of every CAR-antigen pair (12–15 nm) (Table S1) is significantly smaller than CD45 (22–28 nm). Therefore, the size-exclusion model is also relevant to clinically effective CAR-Ts.
Increasing CD45 size promotes activation of CAR-Ts targeting membrane-distal antigens
Although membrane-proximal epitopes activated CAR better than membrane-distal epitopes (Fig. 4&5), their availability depends on individual protein structures. Moreover, the glycocalyx surrounding cancer cell surface might bury these membrane-proximal epitopes and reduce their accessibility to CAR. Therefore, a solution to increase the activation efficiency of CARs targeting membrane-distal epitopes is still valuable. According to the size-exclusion model (Fig. 1A), the CD45 exclusion is not only determined by the size of CAR-antigen pair, but also the size of CD45. We rationalized that CAR-T activation can be enhanced by increasing the size of the extracellular domain of CD45 (Fig. 6A). As a proof-of-principle experiment, we exploited an anti-CD45 antibody to increase the physical size of the extracellular domain of CD45. To further enhance the size of CD45, the CD45 antibody was biotinylated and crosslinked by streptavidin. An isotype antibody, which does not interact with CD45, was used a control. The crosslinked-CD45 antibody was added to human primary T cells expressing RFB4 CAR that recognizes a membrane-distal epitope on CD22 (Fig. 4B). The CD45 antibody effectively bound CAR-Ts (Fig. 6B) but did not affect cell-cell conjugation efficiency (Fig. 6C). Meanwhile, it increased the exclusion of CD45 from the CAR synapse (Fig. 6D), which was consistent with the prediction from the size-exclusion model (Fig. 6A). Consequently, ERK phosphorylation, cytotoxicity, secretion of IFNγ and TNFα were significantly increased when CAR-Ts were treated with the CD45 antibody as compared to the isotype control (Fig. 6E–H, Data File S1). A similar enhancing effect by the CD45 antibody was revealed on Jurkat RFB4 CAR-Ts (Fig. S8).
Fig. 6. Effects of a CD45 antibody on the activation of human primary CAR-T cells recognizing membrane-distal antigens.
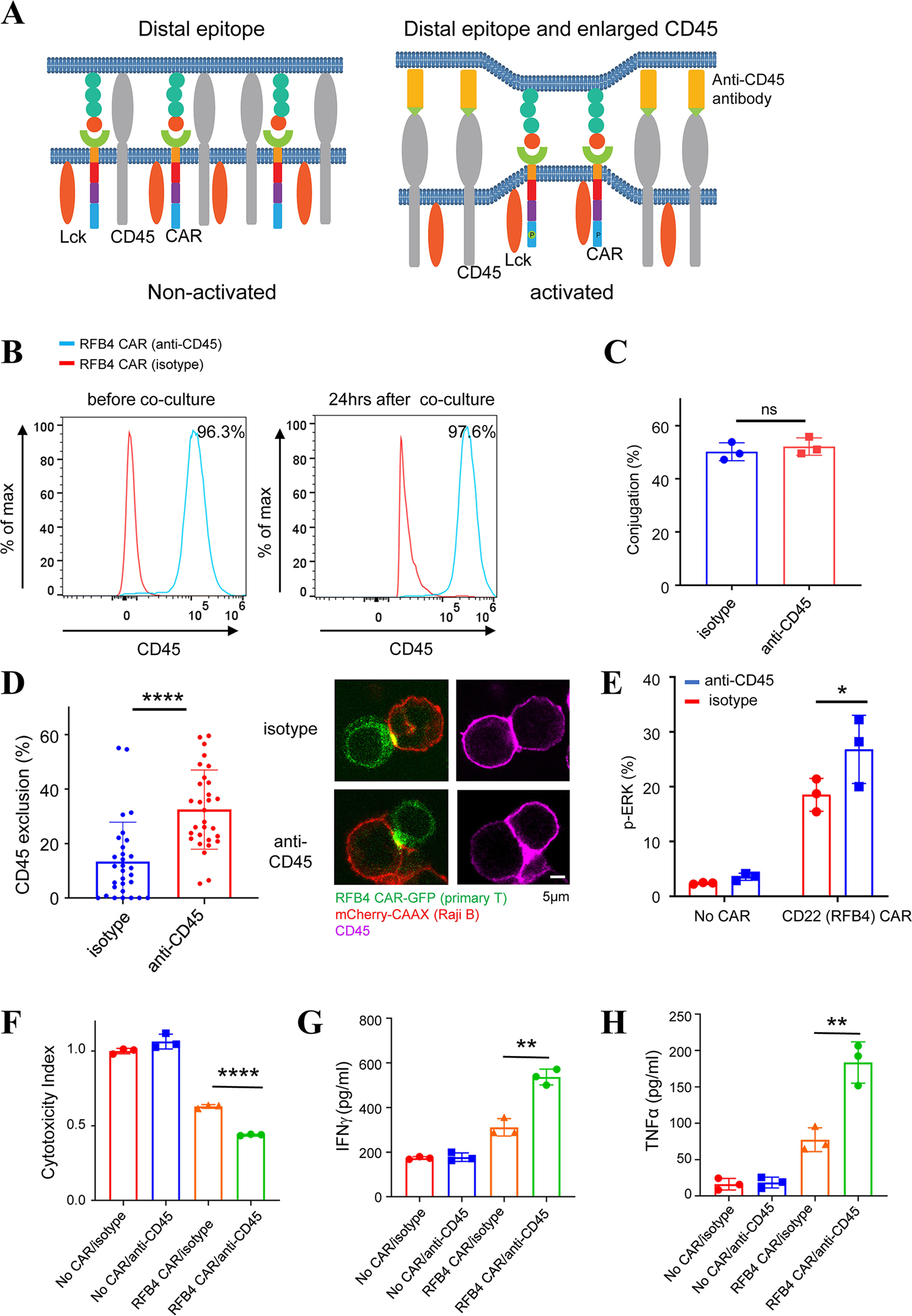
(A) Illustration of a size increase in CD45 by conjugation with an anti-CD45 antibody. LEFT: CARs recognize membrane-distal epitopes, which does not result in sufficient CD45 exclusion and CAR activation. RIGHT: An ani-CD45 antibody increases the size of CD45 and promotes CD45 exclusion from the CAR synapse, which results in CAR activation.
(B) Human primary RFB4 (CD22) CAR-T cells were labeled with a biotinylated anti-CD45 antibody, crosslinked with streptavidin, and co-cultured with Raji B cells. Flow cytometry analysis showed that the CD45 antibody remained bound with CAR-T cells 24 hrs after co-culture with Raji B cells. The isotype antibody served as a negative control.
(C) Effects of the CD45 antibody on the RFB4 CAR-T conjugation with Raji B cells. Primary T cells expressing RFB4 CAR were co-cultured with Raji B cells expressing mCherry-CAAX (E:T =1:1) for 30 min before being imaged by confocal microscopy. Data were presented as mean with SD (Unpaired T test, p>0.05(ns)). N=3 co-culture experiments.
(D) Effects of the CD45 antibody on CD45 exclusion from the CAR synapse. Primary RFB4 CAR-T cells were labeled with CD45 or isotype antibodies, and cocultured with Raji B cells expressing mCherry-CAAX (E:T =1:1) for 30 min, followed by fixation and staining. Images were acquired by confocal microscopy. Data were presented as mean with SD (Mann Whitney U test, p<0.0001 (****). N=60 synapses.
(E) ERK phosphorylation affected by the CD45 antibody. RFB4 CAR-T cells were labeled with CD45 or isotype antibodies and cocultured with Raji B cells (E:T=1:1) for 10 minutes. ERK phosphorylation was assessed by flow cytometry. Data were presented as mean with SD (Unpaired T test, p<0.05 (*)). N=3 co-culture experiments.
(F) Cytotoxicity of RFB4 CAR T cells against Raji B cells in the presence of the CD45 antibody. Primary RFB4 CAR T cells were co-cultured with Raji B-LUC-mCherry cells (E:T =5:1) for 24 hours. The Raji B cells remained were quantified by luciferase-generated fluorescence as detected by a microplate photoreader. Data were presented as mean with SD (Unpaired T test, p<0.0001 (****)). N=3 co-culture experiments.
(G-H) Cytokine production in primary RFB4 CAR-T cells with the CD45 antibody. The co-cultured medium from (F) was used for the measurement of IFNγ and TNFα by ELISA. Data were presented as mean with SD (Unpaired T test, p<0.01 (**)). N=3 co-culture experiments.
In parallel to RFB4 CAR against CD22, we tested if the CD45 antibody promoted MFE23 CAR activation against CEA. Human primary MFE23 CAR-Ts were labeled with the CD45 antibody (Fig. S9A) and cocultured with HeLa cells expressing the full-length CEA which presents membrane-distal epitopes to MFE23. The CD45 antibody did not affect cell-cell conjugation percentage (Fig. S9B) but increased CD45 exclusion from the CAR synapses (Fig. S9C). Consequently, the CD45 antibody enhanced ERK phosphorylation, cytotoxicity, and secretion of IFNγ of primary MFE23 CAR-Ts (Fig. S9D–F). A similar pro-activation effect by the CD45 antibody was observed on Jurkat MFE23 CAR Ts (Fig. S10).
The CD45 antibody could not only increase CD45 size but also cause its dimerization, and previous reports show that dimerization induces a reduction in phosphatase activity(46, 47). We reasoned that if the pro-activation effect of CD45 antibody is attributed to a reduction in phosphatase activity, the antibody is expected to generally enhance CAR-T activation regardless of the CAR type or epitope position; alternatively, if the antibody effect is size-related, it is expected to increase the activation of CARs targeting membrane-distal epitopes more than the CARs targeting membrane-proximal epitopes. To test these possibilities, we determined if the CD45 antibody promoted the activation of Jurkat MFE23 CAR-T against short CEA (Fig. 5A). The CD45 antibody did not promote MFE23 CAR activation against the short CEA (Fig. S10), which was in contrast with its pro-activation effect on the full-length CEA (Fig. S10). These data suggested that the pro-activation effect of CD45 depended on epitope position; CARs targeting a membrane-distal epitope were more likely to be affected by the CD45 antibody than those targeting a membrane-proximal epitope. To strengthen this conclusion, we further tested how the CD45 antibody affected the activation of m971 CAR, which targets a membrane-proximal epitope on CD22 (Fig. 4B). In contrast to the pro-activation effect on RFB4 CAR which targets a membrane-distal epitope (Fig. 6 and S8), the CD45 antibody did not increase the CD69 expression and IL-2 release in Jurkat m971 CAR-Ts. Neither did it affect the cytotoxicity, secretion of IFNγ or TNFα in human primary m971 CAR-Ts (Fig. S11). Together, these data suggested that the CD45 antibody did not generally enhance CAR-T activation; it preferentially promoted the activation of CARs targeting membrane-distal epitopes. This result argued for the possibility that the pro-activation effect of CD45 antibody was attributed to a size increase although we cannot completely exclude the contribution from a dimerization-induced reduction in phosphatase activity.
CD45 has multiple isoforms due to alternative splicing (48) (Fig. 7A). The largest isoform RABC (40–51 nm) is expressed in an early stage of T cell development whereas the smallest isoform RO (22–28 nm) is expressed in a later stage (34, 35, 49, 50). A previous study shows that RABC is more strongly excluded from a TCR-pMHC synapse than RO (26). Therefore, we decided to test how the two isoforms of CD45 affected CAR-T activation. The CD22 (RFB4) CAR was introduced into a CD45-deficient Jurkat T cell line J45.01 (51), in which CD45RO or CD45RABC was ectopically expressed (Fig. 7B). These cells were incubated with Raji B cells expressing CD22. CAR-T cells expressing CD45 RABC were activated to a higher level than those expressing CD45 RO, as determined by CD69 expression and IL-2 release (Fig. 7C–E). Consistent with the results on RFB4 CAR-T, we found that CD45 RABC induced a higher Jurkat MFE23 CAR-T activation than CD45 RO did (Fig. 7F–H). Together, consistent with the size-exclusion model (Fig. 1A), these data suggested that CD45 isoforms with different sizes can influence CAR-T activation. The size of CD45 and isoform expression could be a new target for tunning CAR T activity.
Fig. 7. Regulation of Jurkat CAR-T activation by CD45 isoforms of different sizes.
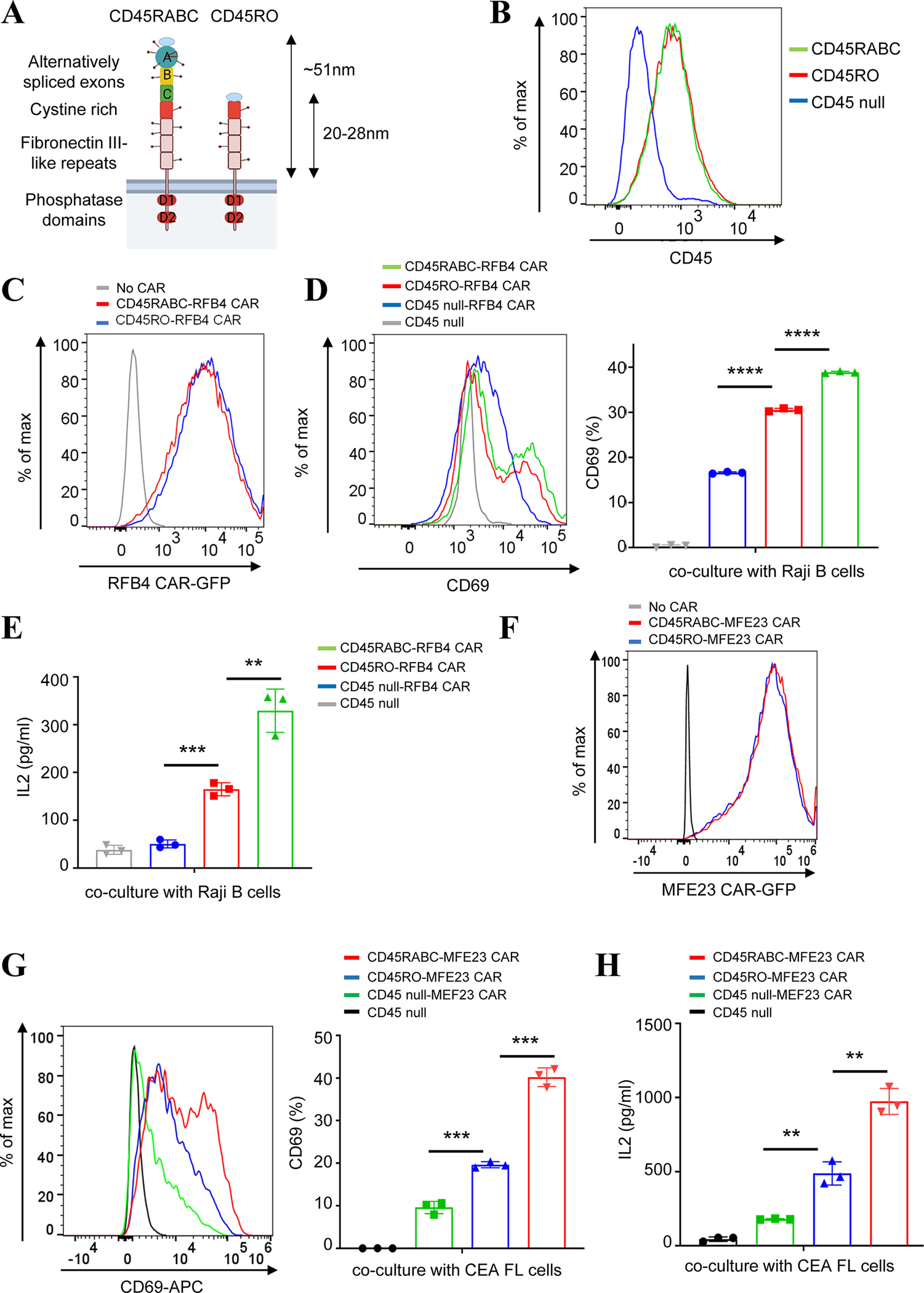
(A) Illustration of CD45 isoforms. CD45RABC is the longest isoform that is expressed in naïve T cells. CD45RO, the shortest splicing isoform, is expressed in activated T cells.
(B) Flow cytometry analysis showed the expression level of CD45RO and RABC when they were reconstituted in a CD45-deficient Jurkat T cell line.
(C) Flow cytometry analysis showed the expression level of RFB4 (CD22) CAR in CD45RO or RABC-expressing Jurkat T cells.
(D-E) Comparing CD45RABC to CD45RO on the activation of RFB4 CAR T against CD22. RFB4 (CD22) CAR Jurkat T cells expressing either the CD45RABC or CD45RO were cocultured with Raji B cells (E:T =1:1) for 24 hrs. CD69 expression was assessed by flow cytometry and IL-2 release was measured by ELISA. Data were presented as mean with SD (Unpaired T test, p<0.01 (**), p<0.001 (***), p<0.0001 (****)). N=3 co-culture experiments.
(F) Flow cytometry analysis showed the expression level of MEF23 (CEA) CAR in CD45RO or RABC-expressing Jurkat T cells.
(G-H) Comparing CD45RABC to CD45RO on the activation of MFE23 CAR T against CEA. MFE23 (CEA) CAR Jurkat T cells expressing either the CD45RABC or CD45RO were cocultured with HeLa cells expressing full-length CEA (E:T=1:1) for 24 hrs. CD69 expression was assessed by flow cytometry and IL-2 release was measured by ELISA. Data were presented as mean with SD (Unpaired T test, p<0.01 (**), p<0.001 (***)). N=3 co-culture experiments.
The large CD45 isoform RABC also enabled us to address the concern whether inserting a long spacer into the CD19 CAR (Fig. 1B) abolished its signaling capability. We reasoned that CD45RABC should rescue, at least partially, the signaling function of the long CD19-CARs (CEA-N-A1-B1-A2) which showed low activity in regular Jurkat T cells that expressed CD45RO (Fig. 1F &1G). This is because that the size-exclusion model predicts that CAR-T activation depends on the size difference between the CAR-antigen pair and CD45. Indeed, we found that expressing CD45RABC could partially rescue the signaling function of long CARs, which were lost in cells expressing CD45RO (Fig. S12). These data suggested CARs with spacers inserted did not lose signaling function per se; their impairment in signaling function depended on CD45 size.
Although in this manuscript we focused on investigating how the interaction between CAR and antigen regulates CD45 distribution, there are other binding pairs in the CAR synapses that can potentially regulate CD45 exclusion, as demonstrated in the TCR synapse (52–55). LFA-1 and LFA-2 are two important integrins that are expressed on the T cell surface that can interact with ICAM-1 and CD58 on the B cell surface, respectively. Using blocking antibodies that inhibit the function of LFA-1 and LFA-2, we tested how these two adhesion molecules affected CD45 exclusion in the synapse formed between human primary CD19 CAR-Ts and Raji B cells. As a result, we did not detect significant effects from either antibody on CD45 exclusion (Fig. S13), suggesting neither LFA1 nor LFA2 plays a critical role in CD45 exclusion from the CAR synapse. Our data are also consistent with a previous report showing that the CAR synapse formation is not dependent on LFA-1 (56). These observations could potentially be explained by the strong interactions between CAR and antigen, the affinity of which is much higher than the pMHC-TCR pair so that the contribution of integrin to CAR synapse becomes less prominent. Meanwhile, we do not exclude the possibilities that other receptor-ligand pair, e.g. PD1-PDL1 or CD2-CD58, could regulate CD45 exclusion.
DISCUSSION
The key to transmembrane receptor activation is to convert extracellular stimuli to intracellular signal transduction. This process is traditionally depicted as a specific communication between the ligand-binding extracellular domain and the intracellular signaling domain, a model that is based on extensive experimental evidence. The success of swapping and recombining parts of cell surface receptors to build functional CARs suggests the need to expand the model of receptor activation beyond this conventional view, and indeed raises the question of the underlying nature of receptor activation. We addressed this fundamental question in receptor activation in the context of CAR. The “size-exclusion” model suggests a critical role of surrounding membrane context in activating signaling receptors. This expands the traditional view of receptor activation which focuses on the conformational change in receptor itself.
Our work also suggested that manipulating CAR size could serve as an effective strategy to tune CAR-T activation. Poor activation will not lead to effective killing whereas robust activation can drive exhaustion and potential tissue toxicity (57, 58). Therefore, an intermediate CAR-T activity is desired which could enable a sustained anti-tumor effect. This is traditionally achieved by either reducing the binding affinity between antigen and scFv (59–64) or selecting intercellular domains with attenuated signaling functions (57, 58, 65). Our results suggested a new way to adjust CAR-T: modifying spacer length to tune CAR activation into a window where a balance between killing, tissue toxicity, and exhaustion can be achieved. Compared to traditional ways, for example, by tuning CAR-antigen affinity, which usually involves a screen of point mutations on specific scFvs, manipulating CAR size by inserting Ig domains or changing the hinge/stalk length is relatively straightforward and can serve as a promising universal strategy to tune the activation of various CARs.
It has been extensively studied how the spacer (stalk) length affects CAR activation. One study shows that CARs with a short spacer of 12 residues can be better activated than CARs with a long spacer of 229 residues (hinge-CH2-CH3 domain from IgG4) (66). Meanwhile, other studies suggest that CARs with the spacer from IgG4 can function reasonably well (67, 68), which leads to the conclusion that space length does not affect CAR-T activation. It is worth noting that the spacer from IgG4 (~7 nm), although longer than other commonly used spacers including CD28 (~6 nm) or CD8α (~5 nm), is still significantly shorter than the long spacer used in our study (~21 nm for the N-A1-B1-A2 domain plus a CD8 stalk) and the size of CD45 (22–28 nm for RO isoform). Therefore, we emphasized that it is important to describe “short” or “long” spacers using numbers and in the context of CD45 size when comparing their functional activities. This would help to resolve some seemingly controversial conclusions in the past about whether spacer length affects CAR-T activation (28–32).
The current study is limited by focusing on a single mechanism explaining CAR-T activation. Although the size-exclusion model is supported by our data, we do not exclude alternative mechanisms that might work in parallel or synergistically to activate CAR-T. For example, in the case of soluble antigens, ligand-induced dimerization has been shown to activate CAR-T (69); antigen binding-induced mechanotransduction could also potentially trigger CAR signaling or tune the threshold (70). Future work will be needed to evaluate how these different mechanisms contribute to CAR-T activation.
Together, our study supported a size-exclusion model in which CAR-antigen interactions exclude the phosphatase CD45 out of the CAR zone, triggering CAR phosphorylation and CAR-T activation. Our data suggested that CAR-T activity can be tuned by manipulating the size of CAR or CD45 and that CAR-T activation was affected by antigen size and epitope position. These highlighted physical size as a key element that needs to be taken into consideration when designing future CAR-Ts.
MATERIALS AND METHODS
Study Design
The objective of this study was to determine how antigen signals were transmitted across the plasma membrane to activate the intracellular signaling downstream of CARs. We used protein engineering, microscopy, and immunology approaches to test a “size-exclusion” mechanism for antigen-dependent CAR-T activation. Unless noted, experiments were performed three or more independent times. Data from multiple experiments were pooled unless noted. Investigators were not blinded to groups. No data were excluded. The details of the reagents including cell lines, plasmids, and antibodies are provided in Table S2.
Cell line culture
Jurkat T (E6.1), J45.01, Raji B, and KU812 cells were grown in RPMI1640 supplemented with 10% FBS, PenStrep-Glutamine, and 10 mM HEPES pH 7.4. HEK293T and HeLa cells were grown in DMEM supplemented with 10% FBS and PenStrep-Glutamine. The resource of cell lines used in this study were specified in Table S2. All cell lines were tested as mycoplasma free.
Cell engineering
HEK293T cells were co-transfected with the pHR plasmids and second-generation lentiviral packaging plasmids pMD2.G and psPAX2 (Addgene plasmid #12259 and #12260) using Genejuice transfection reagent (EMD Millipore, #70967-3). 48 hrs after transfection, cell culture media containing viral particles were harvested, centrifuged, and filtered through 0.45 um pore size filters and mixed with Jurkat T/Raji B/KU812 cells for infection in RPMI media for 72 h. Jurkat T/Raji B/KU812 cells expressing fluorescently (GFP or mCherry) tagged proteins were sorted by flow cytometry. HeLa cells expressing CEA FL or CEA shot were stained with Alexa Fluor® 647 anti-human CD66a/c/e Antibody (Biolegend, #342318), and sorted by flow cytometry.
Generation of primary CAR T cells
T cell isolation and expansion were performed as previously described (65, 71). Briefly, pan or CD8+ T cells were isolated from PBMCs (Zenbio #SER-PBMC-200) using MojoSort™ Human CD3 or CD8 T Cell Isolation Kit (BioLegend, #480022 or #480012) and activated with Human T-Activator CD3/CD28 Dynabeads (ThermoFisher, #11161D). Cells were cultured in RPMI1640 supplemented with 10% FBS, 50 nM 2-mercaptoethanol, 300 U/ml Interleukin-2 (PEPROTECH, # 200-02). 5 days after activation, the cells were infected with concentrated lentivirus (MOI = 20–50). To produce the concentrated virus, HEK293T cells were transfected with pHR plasmids, pMD2.G, and psPAX2 (Addgene plasmid #12259 and #12260) using GeneJuice transfection reagent (EMD Millipore, #70967-3). The cell culture media were collected 48 h after transfection and concentrated 100× using PEG-it Virus Precipitation Solution (System Biosciences, # LV810A-1). 48 hrs after viral transduction, CD8+ T cells were washed with PBS once, Dynabeads were removed, and CAR-T cells were resuspended in a fresh culture medium. Those cells were harvested and used for in vitro assays one or two weeks post initial stimulation with dynabeads.
Cell conjugation and CD45 exclusion
Raji B cells expressing a membrane marker mCherry-CAAX or KU812 cells expressing CD22 FL-mCherry/CD22 Short-mCherry, or HeLa cells expressing CEA FL/CEA Short were cocultured at a 1:1 ratio with primary or Jurkat CAR T cells in RPMI1640 medium supplemented with 20 mM HEPES pH 7.4 for 0.5 h at 37°C. Cells were fixed in 4% paraformaldehyde (Santa Cruz Biotechnology, # 30525-89-4) for 15 min at RT, washed with PBS, and stained with an anti-CD45-APC antibody (BioLegend, # 304012) for 30 min on ice. Then the cells were washed with PBS and resuspended in RPMI1640 medium supplemented with 20 mM HEPES and imaged by confocal microscopy (see details in “Microscopy and image analysis”).
Cytokine production
Raji B cells, KU812 CD22 FL-mCherry/CD22 Short-mCherry cells, or HeLa CEA FL/CEA Short cells were cocultured at a 1:1 or 5:1 ratio with Jurkat CAR T cells or primary CAR T for 24 hrs at 37°C. The supernatant was collected for cytokine measurement using ELISA kits (IL2 ELISA kit, BioLegend #431801; IFNγ ELISA kit, BioLegend #430101; TNFα ELISA kit, BioLegend # 430204) according to the manufacturer’s instruction.
Flow cytometry
To stain cell surface proteins, cells were collected and blocked with an anti-human Fc receptor binding inhibitory antibody in the staining buffer (PBS with 2% FBS and 1 mM EDTA) for 15 minutes at 4°C. Then, cells were incubated with individual antibodies in staining buffer for 30 minutes on ice. The cells were washed twice with staining buffer before sending for flow cytometry analysis. To determine p-CD3ζ or p-ERK, co-cultured cells were collected and fixed with 4% PFA for 5 min, blocked in flow cytometry staining buffer for 30 min on ice, followed by antibody staining with the BD Fixation/Permeabilization Solution Kit. The details of antibodies were provided in Table S2. Data were acquired on LSRII (BD Biosciences) and analyzed with FlowJo software (Treestar).
Cytotoxicity assay
Cytotoxicity was measured by Luciferase assay or flow cytometry, as indicated in the figure legend. Briefly, ffLuc-expressing Raji B cells or KU812 cells were resuspended in RPMI medium supplemented with 10% FBS in 96-well tissue culture plates. CAR-T cells were added at an effector to target ratio of 5:1. After 24 hrs of incubation, cells were collected and washed with PBS. Cell pellets were lysed in a lysis reagent (Promega). The luminescence of lysates was analyzed using a plated spectrophotometer. Spontaneous release control was set up using cancer cells with co-culturing with T cells not expressing CAR. Cytotoxicity index = experimental readout/spontaneous readout. To perform the flow cytometry-based cytotoxicity assay, tumor cells expressing mCherry or pre-stained with antibodies targeting surface antigens (CEA) were cocultured with CAR-T cells for 24 hrs. The cocultures were supplemented with counting beads (Thermofisher) for normalization before flow cytometry analysis. Cytotoxicity index= live tumor cell number CAR-treated group/live tumor cell number No CAR-treated group.
Proliferation assay
CAR-T cells were cocultured with tumor cells at a ratio of 1:1 for 4 or 10 days, stained with anti-CD3 or CD8 antibodies, and quantified using CountBright™ absolute counting beads (ThermoFisher #C36950) on a BD LSRII flow cytometer.
Immunoblotting
Cells were washed twice in cold PBS and lysed in 4x Laemmli Sample Buffer (Bio-Rad #1610747). Cell lysates were heated for 10 min at 95°C. The supernatants were processed for SDS–PAGE and immunoblot analysis. Briefly, the supernatants were loaded onto a 4–20% protein gel (Bio-Rad #4568096) for SDS-PAGE analysis, followed by a transferring onto PVDF membrane (Bio-Rad #1620177). The membrane was blocked with TBST buffer containing 5% nonfat milk for 1 h at room temperature and blotted with indicated primary antibodies overnight at 4°C. The next day, the membrane was further blotted with HRP-conjugated secondary antibody for 1 h at room temperature. Target proteins were detected with a chemiluminescent HRP substrate (Thermo Fisher Scientific #34577) and visualized by a Bio-Rad ChemiDoc imaging system (Bio-Rad). Images were quantified by ImageJ. The details of antibodies were provided in Table S2.
Cell labeling with CD45 antibody
Jurkat or human primary T cells expressing RFB4 CAR or CEA CAR were incubated with biotin anti-human CD45 Antibody or biotin Isotype Ctrl Antibody (See Table S2) on ice for 30 min, washed, and further incubated with Strepavtin-Alex647 for 30 min. The labeled cells were co-cultured with Raji B cells for 24 hours before being sent for measuring CD69 expression by FACS and IFNγ, TNFα, and IL2 secretion by ELISA. Detailed information could be found in “Flow Cytometry” and “Cytokine production”.
Cell labeling with integrin-blocking antibodies
Human primary T cells expressing CD19 CAR were treated with an anti-LFA1, anti-LFA2, or isotype control antibody (see Table S2) on ice for 30 min and washed once with staining buffer (PBS with 2% FBS and 1 mM EDTA). The labeled cells were co-cultured with Raji B cells for analyzing cell-cell conjugation and CD45 exclusion as described in “Cell conjugation and CD45 exclusion”.
Microscopy and Image analysis
Confocal microscopy was performed on a Nikon Ti2-E inverted motorized microscope stand equipped with motorized stage with stagetop Piezo, Yokogawa CSU-X1 spinning disk confocal, Agilent laser combiner with four lines, 405, 488, 561, and 640 nm, scientific CMOS camera Photometrics Prime 95B. Images were acquired using Nikon Elements and analyzed in Fiji (ImageJ). The same brightness and contrast were applied to images within the same panels. Cell conjugation percentage was calculated by dividing the number of tumor cell-associated GFP+ CAR-T cells by the number of total GFP+ CAR T cells. To quantify CD45 exclusion in the synapse, the line scan function was used to measure the intensities of CD45 in the synapse between CAR-T and target Tumor cells. The exclusion percentage = (1 − Intensity of CD45 in CAR zone/Intensity of CD45 out of CAR zone) *100%.
Anti-tumor efficacy of CAR-Ts in a mouse xenograft model
Mice were housed in specific-pathogen-free conditions and cared for in accordance with US National Institutes of Health guidelines, and all procedures were approved by the Rutgers University Animal Care and Use Committee (PROTO202100020). Raji B cells (1×106) were mixed with 50% Matrigel (Matrix Growth Factor Reduced, Corning, CAT# 354230) in 100 μL PBS and injected subcutaneously into the right flank of NSG mice (7–8 weeks old, purchased from The Jackson Laboratory, Strain # 005557). 7 days later, mice were divided into 4 groups randomly: PBS, control CAR T, CEA-N-A1 CAR T, and CEA-N-A1-B1-A2 CAR T. 107 CAR T cells in 100 μL PBS were injected into each mouse through tail vein. Blood was drawn on day 7, 14, and 21 post CAR T injection for flow cytometry analysis of CAR T percentage and the expression of exhaustion markers. Tumor growth was measured with calipers, and size was expressed as one-half of the product of perpendicular length and square width in cubic millimeters (Volume=1/2*L*W*W). For survival tests, mice were euthanized when the tumor size exceeded 1,000 mm3.
Statistical analysis
Student’s T test, Mann–Whitney U test, or Log-rank (Mantel-Cox) test analysis was used to assess significance, with P < 0.05 considered significant. Data were analyzed on GraphPad Prism. The statistical details for each experiment were provided in the associated figure legends.
Supplementary Material
Acknowledgments:
We thank Dr. Daniel Fletcher for sharing plasmids encoding CEA, Dr. Christopher Burd for providing comments on the manuscript, and Dr. Longhui Zeng for discussion and technical assistance.
Funding:
XS was supported by an American Cancer Society Research Scholar Grant, the Charles H. Hood Foundation Child Health Research Awards, the Andrew McDonough B+ Foundation Research Grant, the Gilead Sciences Research Scholars Program in Hematology/Oncology, the Rally Foundation and Bear Necessities Foundation A Collaborative Pediatric Cancer Research Awards Program, the Rally Young Scholar Program, the Yale SPORE in skin cancer DRP Award CA121974, the Yale DeLuca Pilot Award, the NIGMS MIRA (R35) program GM138299, the Human Frontier Science Program Early-Career Research Grant. CH is supported the Duncan and Nancy MacMillan Cancer Immunology and Metabolism Center of Excellence. JC is supported by a Start-up Fund from the Rutgers Cancer Institute of New Jersey (State of NJ appropriation and Cancer Center Support Grant P30CA072720) and the Center of Excellence in Cancer Immunology and Metabolism Seed Grant.
Footnotes
Competing interests:
CH provides consulting for GlaxoSmithKine and PACT Pharma, holds NIH patents and royalties in the field of immunotherapy and cell therapy, and receives research funding from Neogene Therapeutics and T-Cure Bioscience. XS is a co-applicant for a provisional patent on CAR, which is not based on the specific results in this manuscript.
Data and materials availability:
All data are available in the main text or the supplementary materials. Reagents generated from this study will be available upon request. Please contact Xiaolei Su (xiaolei.su@yale.edu).
References and Notes
- 1.Porter DL, Levine BL, Kalos M, Bagg A, June CH, Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365, 725–733 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, Timmerman JM, Holmes H, Jaglowski S, Flinn IW, McSweeney PA, Miklos DB, Pagel JM, Kersten MJ, Milpied N, Fung H, Topp MS, Houot R, Beitinjaneh A, Peng W, Zheng L, Rossi JM, Jain RK, Rao AV, Reagan PM, KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med 382, 1331–1342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH, Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368, 1509–1518 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leibman RS, Richardson MW, Ellebrecht CT, Maldini CR, Glover JA, Secreto AJ, Kulikovskaya I, Lacey SF, Akkina SR, Yi Y, Shaheen F, Wang J, Dufendach KA, Holmes MC, Collman RG, Payne AS, Riley JL, Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog 13, e1006613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hale M, Mesojednik T, Romano Ibarra GS, Sahni J, Bernard A, Sommer K, Scharenberg AM, Rawlings DJ, Wagner TA, Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol Ther 25, 570–579 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G, Milone MC, Payne AS, Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 353, 179–184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elinav E, Waks T, Eshhar Z, Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology 134, 2014–2024 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, Mansilla-Soto J, Boyer JA, Li X, Giavridis T, Kulick A, Houlihan S, Peerschke E, Friedman SL, Ponomarev V, Piersigilli A, Sadelain M, Lowe SW, Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, Wang T, Bedi K, Morley MP, Linares Saldana RA, Bolar NA, McDaid K, Assenmacher CA, Smith CL, Wirth D, June CH, Margulies KB, Jain R, Pure E, Albelda SM, Epstein JA, Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim WA, June CH, The Principles of Engineering Immune Cells to Treat Cancer. Cell 168, 724–740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bessman NJ, Freed DM, Lemmon MA, Putting together structures of epidermal growth factor receptors. Curr Opin Struct Biol 29, 95–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.James JR, Vale RD, Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature 487, 64–69 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor MJ, Husain K, Gartner ZJ, Mayor S, Vale RD, A DNA-Based T Cell Receptor Reveals a Role for Receptor Clustering in Ligand Discrimination. Cell 169, 108–119 e120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belardi B, Son S, Felce JH, Dustin ML, Fletcher DA, Cell-cell interfaces as specialized compartments directing cell function. Nat Rev Mol Cell Biol 21, 750–764 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Dustin ML, Choudhuri K, Signaling and Polarized Communication Across the T Cell Immunological Synapse. Annu Rev Cell Dev Biol 32, 303–325 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Davis SJ, van der Merwe PA, The kinetic-segregation model: TCR triggering and beyond. Nat Immunol 7, 803–809 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Anton van der Merwe P, Davis SJ, Shaw AS, Dustin ML, Cytoskeletal polarization and redistribution of cell-surface molecules during T cell antigen recognition. Semin Immunol 12, 5–21 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Davis SJ, van der Merwe PA, The structure and ligand interactions of CD2: implications for T-cell function. Immunol Today 17, 177–187 (1996). [DOI] [PubMed] [Google Scholar]
- 19.Choudhuri K, Wiseman D, Brown MH, Gould K, van der Merwe PA, T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature 436, 578–582 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Wilhelm KB, Morita S, McAffee DB, Kim S, O’Dair MK, Groves JT, Height, but not binding epitope, affects the potency of synthetic TCR agonists. bioRxiv, 2021.2005.2013.443919 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williamson AP, Vale RD, Spatial control of Draper receptor signaling initiates apoptotic cell engulfment. J Cell Biol 217, 3977–3992 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bakalar MH, Joffe AM, Schmid EM, Son S, Podolski M, Fletcher DA, Size-Dependent Segregation Controls Macrophage Phagocytosis of Antibody-Opsonized Targets. Cell 174, 131–142 e113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Felce JH, Sezgin E, Wane M, Brouwer H, Dustin ML, Eggeling C, Davis SJ, CD45 exclusion- and cross-linking-based receptor signaling together broaden FcepsilonRI reactivity. Sci Signal 11, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwak M, Southard KM, Kim WR, Kim NH, Gopalappa R, An M, Lee HJ, Kang MK, Choi SH, Farlow J, Georgakopoulos A, Robakis NK, Kutys ML, Seo D, Kim HB, Kim YH, Cheon J, Gartner ZJ, Jun Y.-w., Size-dependent protein segregation creates a spatial switch for Notch signaling and function. bioRxiv, (2021). [Google Scholar]
- 25.Schmid EM, Bakalar MH, Choudhuri K, Weichsel J, Ann H, Geissler PL, Dustin ML, Fletcher DA, Size-dependent protein segregation at membrane interfaces. Nat Phys 12, 704–711 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carbone CB, Kern N, Fernandes RA, Hui E, Su X, Garcia KC, Vale RD, In vitro reconstitution of T cell receptor-mediated segregation of the CD45 phosphatase. Proc Natl Acad Sci U S A 114, E9338–E9345 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong R, Libby KA, Blaeschke F, Fuchs W, Marson A, Vale RD, Su X, Rewired signaling network in T cells expressing the chimeric antigen receptor (CAR). EMBO J 39, e104730 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moritz D, Groner B, A spacer region between the single chain antibody- and the CD3 zeta-chain domain of chimeric T cell receptor components is required for efficient ligand binding and signaling activity. Gene Ther 2, 539–546 (1995). [PubMed] [Google Scholar]
- 29.Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, Arif S, Mather SJ, Taylor-Papadimitriou J, Burchell JM, Maher J, Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol 180, 4901–4909 (2008). [DOI] [PubMed] [Google Scholar]
- 30.McComb S, Nguyen T, Shepherd A, Henry KA, Bloemberg D, Marcil A, Maclean S, Gilbert R, Gadoury C, Pon R, Sulea T, Zhu Q, Weeratna RD, Antigenic Sensitivity of Membrane-Proximal Targeting Chimeric Antigen Receptors can be Fine-Tuned through Hinge Truncation. bioRxiv, (2021). [Google Scholar]
- 31.Almasbak H, Walseng E, Kristian A, Myhre MR, Suso EM, Munthe LA, Andersen JT, Wang MY, Kvalheim G, Gaudernack G, Kyte JA, Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther 22, 391–403 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Qin L, Zhao R, Li P, Incorporation of functional elements enhances the antitumor capacity of CAR T cells. Exp Hematol Oncol 6, 28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teplyakov A, Obmolova G, Luo J, Gilliland GL, Crystal structure of B-cell co-receptor CD19 in complex with antibody B43 reveals an unexpected fold. Proteins 86, 495–500 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Chang VT, Fernandes RA, Ganzinger KA, Lee SF, Siebold C, McColl J, Jonsson P, Palayret M, Harlos K, Coles CH, Jones EY, Lui Y, Huang E, Gilbert RJC, Klenerman D, Aricescu AR, Davis SJ, Initiation of T cell signaling by CD45 segregation at ‘close contacts’. Nat Immunol 17, 574–582 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCall MN, Shotton DM, Barclay AN, Expression of soluble isoforms of rat CD45. Analysis by electron microscopy and use in epitope mapping of anti-CD45R monoclonal antibodies. Immunology 76, 310–317 (1992). [PMC free article] [PubMed] [Google Scholar]
- 36.Boehm MK, Perkins SJ, Structural models for carcinoembryonic antigen and its complex with the single-chain Fv antibody molecule MFE23. FEBS Lett 475, 11–16 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Abraham RT, Weiss A, Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol 4, 301–308 (2004). [DOI] [PubMed] [Google Scholar]
- 38.James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, Raubitschek AA, Forman SJ, Press OW, Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol 180, 7028–7038 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ, Barrett DM, Wayne AS, Mackall CL, Orentas RJ, Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 121, 1165–1174 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z, Jiang D, Yang H, He Z, Liu X, Qin W, Li L, Wang C, Li Y, Li H, Xu H, Jin H, Qian Q, Modified CAR T cells targeting membrane-proximal epitope of mesothelin enhances the antitumor function against large solid tumor. Cell Death Dis 10, 476 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ereno-Orbea J, Sicard T, Cui H, Mazhab-Jafari MT, Benlekbir S, Guarne A, Rubinstein JL, Julien JP, Molecular basis of human CD22 function and therapeutic targeting. Nat Commun 8, 764 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiao X, Ho M, Zhu Z, Pastan I, Dimitrov DS, Identification and characterization of fully human anti-CD22 monoclonal antibodies. MAbs 1, 297–303 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ereno-Orbea J, Liu X, Sicard T, Kucharska I, Li W, Borovsky D, Cui H, Feng Y, Dimitrov DS, Julien JP, Structural details of monoclonal antibody m971 recognition of the membrane-proximal domain of CD22. J Biol Chem 297, 100966 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hammarstrom S, The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol 9, 67–81 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Desai DM, Sap J, Schlessinger J, Weiss A, Ligand-mediated negative regulation of a chimeric transmembrane receptor tyrosine phosphatase. Cell 73, 541–554 (1993). [DOI] [PubMed] [Google Scholar]
- 47.Majeti R, Bilwes AM, Noel JP, Hunter T, Weiss A, Dimerization-induced inhibition of receptor protein tyrosine phosphatase function through an inhibitory wedge. Science 279, 88–91 (1998). [DOI] [PubMed] [Google Scholar]
- 48.Hermiston ML, Xu Z, Weiss A, CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol 21, 107–137 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Fukuhara K, Okumura M, Shiono H, Inoue M, Kadota Y, Miyoshi S, Matsuda H, A study on CD45 isoform expression during T-cell development and selection events in the human thymus. Hum Immunol 63, 394–404 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Woollett GR, Williams AF, Shotton DM, Visualisation by low-angle shadowing of the leucocyte-common antigen. A major cell surface glycoprotein of lymphocytes. EMBO J 4, 2827–2830 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koretzky GA, Picus J, Schultz T, Weiss A, Tyrosine phosphatase CD45 is required for T-cell antigen receptor and CD2-mediated activation of a protein tyrosine kinase and interleukin 2 production. Proc Natl Acad Sci U S A 88, 2037–2041 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dustin ML, Modular design of immunological synapses and kinapses. Cold Spring Harb Perspect Biol 1, a002873 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siokis A, Robert PA, Demetriou P, Kvalvaag A, Valvo S, Mayya V, Dustin ML, Meyer-Hermann M, Characterization of mechanisms positioning costimulatory complexes in immune synapses. iScience 24, 103100 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Graf B, Bushnell T, Miller J, LFA-1-mediated T cell costimulation through increased localization of TCR/class II complexes to the central supramolecular activation cluster and exclusion of CD45 from the immunological synapse. J Immunol 179, 1616–1624 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson KG, Bromley SK, Dustin ML, Thomas ML, A supramolecular basis for CD45 tyrosine phosphatase regulation in sustained T cell activation. Proc Natl Acad Sci U S A 97, 10138–10143 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davenport AJ, Cross RS, Watson KA, Liao Y, Shi W, Prince HM, Beavis PA, Trapani JA, Kershaw MH, Ritchie DS, Darcy PK, Neeson PJ, Jenkins MR, Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc Natl Acad Sci U S A 115, E2068–E2076 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, Dobrin A, Cabriolu A, Hamieh M, Sadelain M, Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med 25, 82–88 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, Mackall CL, 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 21, 581–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, Albon SJ, Casanovas-Company J, Castro F, Popova B, Villanueva K, Yeung J, Vetharoy W, Guvenel A, Wawrzyniecka PA, Mekkaoui L, Cheung GW, Pinner D, Chu J, Lucchini G, Silva J, Ciocarlie O, Lazareva A, Inglott S, Gilmour KC, Ahsan G, Ferrari M, Manzoor S, Champion K, Brooks T, Lopes A, Hackshaw A, Farzaneh F, Chiesa R, Rao K, Bonney D, Samarasinghe S, Goulden N, Vora A, Veys P, Hough R, Wynn R, Pule MA, Amrolia PJ, Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med 25, 1408–1414 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, Mi T, Switzer K, Singh H, Huls H, Lee DA, Heimberger AB, Champlin RE, Cooper LJ, Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res 75, 3505–3518 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, Cogdill AP, Li N, Ramones M, Granda B, Zhou L, Loew A, Young RM, June CH, Zhao Y, Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res 75, 3596–3607 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, Martens ACM, Zweegman S, van de Donk N, Groen RWJ, Lokhorst HM, Mutis T, A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol Ther 25, 1946–1958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arcangeli S, Rotiroti MC, Bardelli M, Simonelli L, Magnani CF, Biondi A, Biagi E, Tettamanti S, Varani L, Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol Ther 25, 1933–1945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vander Mause ER, Atanackovic D, Lim CS, Luetkens T, Roadmap to affinity-tuned antibodies for enhanced chimeric antigen receptor T cell function and selectivity. Trends Biotechnol, (2022). [DOI] [PubMed] [Google Scholar]
- 65.Wu W, Zhou Q, Masubuchi T, Shi X, Li H, Xu X, Huang M, Meng L, He X, Zhu H, Gao S, Zhang N, Jing R, Sun J, Wang H, Hui E, Wong CC, Xu C, Multiple Signaling Roles of CD3epsilon and Its Application in CAR-T Cell Therapy. Cell 182, 855–871 e823 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR, The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res 3, 125–135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, Chang WC, Bretzlaff W, Starr R, Priceman S, Ostberg JR, Forman SJ, Brown CE, Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther 23, 757–768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mamonkin M, Rouce RH, Tashiro H, Brenner MK, A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood 126, 983–992 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY, Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol 14, 317–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li R, Ma C, Cai H, Chen W, The CAR T-Cell Mechanoimmunology at a Glance. Adv Sci (Weinh) 7, 2002628 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu L, Bi E, Ma X, Xiong W, Qian J, Ye L, Su P, Wang Q, Xiao L, Yang M, Lu Y, Yi Q, Enhanced CAR-T activity against established tumors by polarizing human T cells to secrete interleukin-9. Nat Commun 11, 5902 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials. Reagents generated from this study will be available upon request. Please contact Xiaolei Su (xiaolei.su@yale.edu).