

Keywords: cachexia, colon cancer, colorectal cancer, inflammation, muscle wasting, ribosomal RNA
Abstract
We investigated the impact of tumor burden on muscle wasting in metastatic (m) and xenograft (x) models of colorectal cancer (CRC). Male Nod SCID γ and CD2F1 mice were injected subcutaneously or intrasplenically with HCT116 or C26 tumor cells, respectively. CRC tumors resulted in significant muscle wasting regardless of tumor type or model, although muscle loss was exacerbated in mHCT116 hosts. The mHCT116 model decreased ribosomal (r)RNA content and rDNA transcription, whereas the mC26 model showed no loss of rRNA and the upregulation of rDNA transcription. The xHCT116 model reduced mTOR, RPS6, and 4E-BP1 phosphorylation, whereas the mHCT116 model had a similar effect on RPS6 and 4E-BP1 without altering mTOR phosphorylation. The C26 models caused a reduction in 4E-BP1 phosphorylation independent of mTOR. Muscle interleukin (IL)-6 mRNA was elevated in all models except xHCT116, and the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) mRNA was induced only in the mC26 model. IL-1β mRNA increased in all groups with greater expression in metastatic relative to the xenograft model regardless of tumor types. Our findings indicate that HCT116 tumor burden results in more drastic muscle wasting and anabolic deficits, whereas C26 tumor burden causes similar muscle wasting but exhibits a divergent proinflammatory phenotype. These results highlight potentially important divergence in the pathogenesis of muscle wasting among preclinical models of CRC and demonstrate that tumor burden plays a role in determining anabolic deficits and the expression of proinflammatory effectors of muscle wasting in a tumor-type-dependent manner.
NEW & NOTEWORTHY We provide evidence demonstrating that colorectal tumor burden plays a role in determining anabolic deficits and the expression of proinflammatory effectors of muscle wasting in a tumor-type-dependent manner.
INTRODUCTION
Colorectal cancer (CRC) is the second deadliest cancer in the United States (1). Over 60% of patients with advanced CRC develop liver metastases, which is a major contributor to the development of cachexia (2). Despite strong evidence demonstrating the occurrence of cachexia in patients with metastatic hepatic tumors (2–4), preclinical studies often obviate the metastatic component (5). This represents a confounding variable that limits the understanding of how tumor burden affects muscle wasting. In the preceding study (6), we reported that different lung tumors (i.e., LP07 vs. LLC) promote muscle wasting via divergent mechanisms. This discrepancy may be a consequence of the tumor burden associated with each model, that is, a twofold difference in tumor-to-body weight ratio resulted in similar muscle wasting. A direct comparison between these two models is limited by specific features, such as the host genetic background and the intrinsic properties of each tumor type (e.g., growth rate and tumor-derived factors). The use of metastatic and xenograft preclinical models of cancer is an ideal approach to investigate the downstream consequences associated with tumor burden. Huot et al. (7, 8) recently demonstrated that metastatic HCT116 or C26 CRC tumors exacerbate the wasting phenotype relative to xenograft implantation of the same tumor type. Based on this important observation, we designed the present study to determine if tumor burden, modeled by injecting CRC cells to develop into metastatic or xenograft tumors, would involve similar or elicit divergent muscle-wasting mechanisms.
One frequent incidence in cancer-induced muscle wasting is a diminished capacity for muscle protein synthesis (9–11). This anabolic deficit is partly due to a decrease in the ribosomal capacity of the muscle (12, 13) consequent to reduced transcription of the ribosomal (r) RNA genes (rDNA). In a model of ovarian cancer, we recently demonstrated that muscle wasting involved a significant reduction in rDNA transcription (14), which together with alterations in signal transduction modulating translational control, may contribute to anabolic deficits that promote muscle wasting. Whether these mechanisms are sensitive to tumor burden or whether they are common to all tumors remains an unanswered question. Another important contributor to muscle wasting in cancer is the presence of inflammatory cytokines (2, 3, 15, 16). Circulating procachectic cytokines such as IL-6 and TNFα can trigger the production of local cytokines (17) via inflammasomes (18). These multiprotein complexes convert cytokine precursors into their mature and active forms (19). For example, NLRP3 mediates changes in local inflammation by processing IL-1β and IL-18 (19) and is particularly relevant to cancer because deletion of Nlrp3 can attenuate inflammation and muscle wasting (20).
In this study, we sought to determine 1) whether tumor burden promotes muscle wasting via common or divergent mechanisms, and 2) whether any potential divergence is tumor-type dependent. We found that tumor burden not only exacerbated muscle wasting but also promoted anabolic deficits and impaired intracellular signaling in a tumor-type-specific manner. The expression of proinflammatory effectors of muscle wasting also displayed tumor burden and tumor-type dependence.
MATERIALS AND METHODS
Cell Culture
Human HCT116 cells were purchased from American Type Culture Collection (No. CRL-247, RRID: CVCL_0291) and cultured in McCoy’s medium supplemented with 10% fetal bovine serum (FBS), 1% glutamine, 1% sodium pyruvate, and 1% penicillin/streptomycin (P/S) in 5% CO2 at 37°C. C26 cells (RRID: CVCL_0240) were obtained from Dr. Donna McCarthy (Ohio State University, Columbus, OH) and then were cultured in DMEM (Thermo Fisher Scientific, Waltham, MA) medium supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% sodium pyruvate (Thermo Fisher Scientific, Waltham, MA) and maintained in a 5% CO2, 37°C humidified incubator. All cells were cultured, passaged, and trypsinized when subconfluent to be prepared for animal injections.
Animals
Animal studies were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine and were in compliance with the National Institutes of Health Guidelines for Use and Care of Laboratory Animals and with the 1964 Declaration of Helsinki and its later amendments. The HCT116 and C26 preclinical models used in this study have been thoroughly characterized and previously reported to undergo significant muscle wasting (7, 8). Briefly, 8-wk-old male NOD scid γ (NSG; NOD-scid/IL2Rgnull) immunodeficient mice (In Vivo Therapeutics Core Facility, IU Simon Cancer Center, Indianapolis, IN) were used for the HCT116 model and were housed in a pathogen-free facility at IUSM’s laboratory animal resource center. Animals were randomized into one of the following experimental models: subcutaneous injection of 3.0 × 106 HCT116 tumor cells in sterile saline (xHCT116, n = 5) or an isovolumetric subcutaneous injection of vehicle (control, n = 5); intrasplenic injection of 1.25 × 105 HCT116 tumor cells in sterile saline (mHCT116, n = 8) or an isovolumetric intrasplenic injection of vehicle (sham, n = 5). Sham and mHCT116 animals were euthanized at day 24 due to an average weight loss of ∼2 g of body weight and a significant decline in activity and hunched over appearance (7). Control and xHCT116 animals were euthanized at day 30. In the C26 model, 8-wk-old CD2F1 male mice (Envigo, Indianapolis, IN) were either injected subcutaneously (xC26; 1.0 × 106) or intrasplenically (mC26; 2.5 × 105) with C26 cells (n = 4–6/group). Nontumor-bearing (control) and sham-operated (Sham) mice were used as controls. Mice bearing C26 tumors were weighed daily and euthanized at day 15 under isoflurane anesthesia upon reaching their predetermined end point (weight loss greater than 10%) as previously described (8). Gastrocnemius muscles were harvested, weighed, snap-frozen in liquid nitrogen, and stored at −80°C for further studies.
RNA Extraction and Quantification, cDNA Synthesis, and qRT-PCR
Total RNA was isolated from gastrocnemius muscles using TRizol (No. 15596018, Invitrogen, Carlsbad, CA) and was subsequently purified using Direct-zol RNA MiniPrep columns (R2052; Zymo Research, Irvine, CA). RNA quantity, purity, and integrity were determined on a CLARIOstar microplate reader in the LVis plate (BMG Labtech, Ortenberg, Germany) followed by agarose gel electrophoresis, respectively. cDNA was synthesized from 1 μg of total RNA using the SuperScript VILO cDNA synthesis kit (No. 11-754-050, Invitrogen, Carlsbad, CA) and subjected to a quantitative real-time polymerase chain reaction (qRT-PCR) using GoTaq qPCR master mix (A6002; Promega, Madison, WI) on a Bio-Rad touch CFX-384 system (Bio-Rad, Hercules, CA). Relative expression levels were obtained by normalizing target genes of interest to GAPDH by the comparative Ct (ΔΔCt) method using the Bio-Rad CFX manager (v. 3.0) software. Primer sequences used in the present study were reported in the preceding study 6).
Western Blotting
Total protein was extracted from the gastrocnemius muscle, lysed in ice-cold RIPA buffer [50 mM Tris·HCl pH 8, 150 mM NaCl, 1% Nonidet P-40 (NP-40), 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1 mM sodium fluoride, and 1 mM sodium orthovanadate] supplemented with one of each Pierce protease and phosphatase inhibitor (×1) mini tablets (A32959; Thermo Fisher Scientific, Waltham, MA). Tissue homogenization was performed using a 7-mm generator probe coupled to an OMNI TH tissue homogenizer (OMNI International, Kennesaw, GA). Homogenates were centrifuged for 20 min at 4°C, and supernatants were transferred to new tubes. Protein concentration was determined using the DC protein assay (No. 500-0113; No. 500-0114, Bio-Rad, Hercules, CA), and the lysate was diluted with lysis buffer before mixing 1:1 with ×2 Laemmli buffer (Bio-Rad) containing 5% β-mercaptoethanol. Samples were boiled at 95°C for 10 min and immediately cooled on ice before being stored at −20°C until further use. Western blotting was performed using standard techniques. Samples containing equal amounts of protein (10 μg) were separated by SDS-PAGE on 15% gel and transferred to PVDF membranes (No. 1620177, Bio-Rad, Hercules, CA) activated in 100% methanol. After transfer, membranes were washed in Tris-buffered saline with 0.1% Tween (TBS-T) and blocked in a protein-containing buffer (TBS-T + 5% milk). Membranes were incubated with primary [mTOR: No. 2983, 1:1,000, RRID: AB_2105622; Phospho-mTOR (Ser2448): No.2971, 1:1,000, RRID: AB_330970; rpS6: No. 2217, 1:2,000, RRID: AB_331355; Phospho-rpS6 (Ser235/236): No. 2211, 1:2,000, RRID: AB_331679; 4E-BP1: No. 9644, 1:2,000, RRID: AB_2097841; Cell Signaling, Danvers, MA] and subsequently detected with secondary (Antirabbit IgG: No. 7074, RRID: AB_2099233, Cell Signaling, Danvers, MA) antibodies diluted in blocking buffer according to the manufacturer’s recommendations. Bands were captured with a Bio-Rad ChemiDoc XRS+ (Bio-Rad, Hercules, CA), and band intensity was quantified with the Image Lab software (Bio-Rad, Hercules, CA). Densitometric analysis of p-mTOR and p-rpS6 was conducted by normalizing the phospho signal to its corresponding pan band. The 4E-BP1 γ band was normalized to the band density of all three 4E-BP1 bands (α, β, and γ). All blots were verified for equal loading by staining the membranes with Ponceau S (P7170; Sigma Aldrich, St. Louis, MO).
Statistical Analysis
Values are reported as means ± SD. To reduce variability, potential outliers were identified by a ROUT test with the maximum false discovery rate set to 1%. Significant differences were determined by two-way ANOVA (tumor type × model) followed by a Tukey’s multiple comparisons test when a significant interaction or main effect was detected. The level of significance was set at P ≤ 0.05. Correlations were calculated using Pearson product-moment correlation coefficients where appropriate. All data were analyzed using GraphPad Prism 8 (GraphPad Prism, RRID: SCR_002798, San Diego, CA).
RESULTS
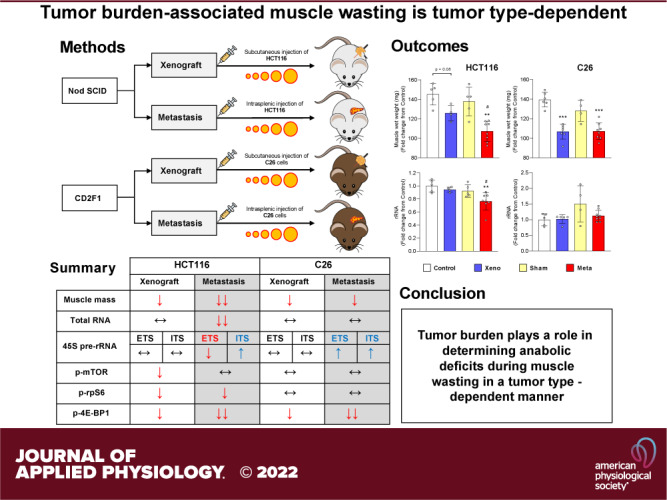
Skeletal Muscle Wasting Is Exacerbated in mHCT116 Compared with xHCT116 but Is Similar in the C26 Models
To define whether tumor burden is tumor-type dependent, we generated metastatic and xenograft models using HCT11 or C26 tumor cells as previously described (7, 8). mHCT116 tumors exacerbated the loss of gastrocnemius wet weight compared with control (−26.2%, P < 0.01) and xHCT116 (P < 0.05), whereas both xC26 and mC26 displayed a similar degree of muscle loss compared with control (xC26: −23.4%, P < 0.001; mC26: −23.2%, P < 0.001; Fig. 1, A and B). HCT116 tumors caused a significant loss in relative muscle mass (xHCT116: −14.2%, P = 0.006; mHCT116: −29.0%, P < 0.001) with a greater decline in the mHCT116 (P = 0.002) despite a shorter disease course (24 days vs. 30 days, respectively; Fig. 1C). The magnitude of muscle wasting did not differ significantly in the C26 models (xC26: −20.4%, P < 0.001; mC26: −17.1%, P < 0.001; Fig. 1D).
Figure 1.

Skeletal muscle wasting is exacerbated in mHCT116 compared with xHCT116 but is similar in the C26 model. Muscle wet weight of gastrocnemius muscle in HCT116 (A) and C26 models (B). Gastrocnemius muscle mass was normalized to the initial body weight for the HCT116 (C) and C26 models (D). Data are expressed as fold change from controls ± SD. A two-way ANOVA followed by a Tukey’s multiple comparisons test was performed for all direct comparisons between groups. Significant difference from control: *P ≤ 0.05, **P < 0.01, and ***P < 0.001; xenograft vs. metastasis: #P < 0.05, ##P < 0.01.
Ribosomal Capacity and rDNA Transcription Are Reduced in mHCT116, but C26 Tumors Display Elevated rDNA Transcription and Maintain rRNA Levels
A reduction in rRNA content was evident in the mHCT116 compared with control (−21.2%, P = 0.005) and xHCT116 (P = 0.05; Fig. 2A), whereas there was no difference in C26 tumor hosts (Fig. 2A). The mHCT116 model showed a reduction in 45S pre-rRNA levels (5′ external transcribed spacer, ETS) relative to control (−40%, P < 0.001) and xHCT116 (P = 0.009; Fig. 2B), whereas mC26 showed upregulation of 45S pre-rRNA ETS compared with control (> twofold, P < 0.001) and xC26 (P = 0.002; Fig. 2B). To assess rDNA transcription elongation, we determined 45S pre-rRNA levels (Internal Transcribed Spacer 1, ITS-1), which was higher in mHCT116 (P = 0.04) relative to xHCT116, but it was not statistically different from the sham control (Fig. 2C). ITS-1 was significantly upregulated in mC26 compared with control (>1.5-fold, P < 0.001) and xC26 (P < 0.001; Fig. 2C). The reduction in rRNA and ETS in mHCT116 strongly correlated with muscle wasting (rRNA: r = 0.859, P < 0.001; ETS: r = 0.699, P < 0.001; Fig. 2, D and E).
Figure 2.
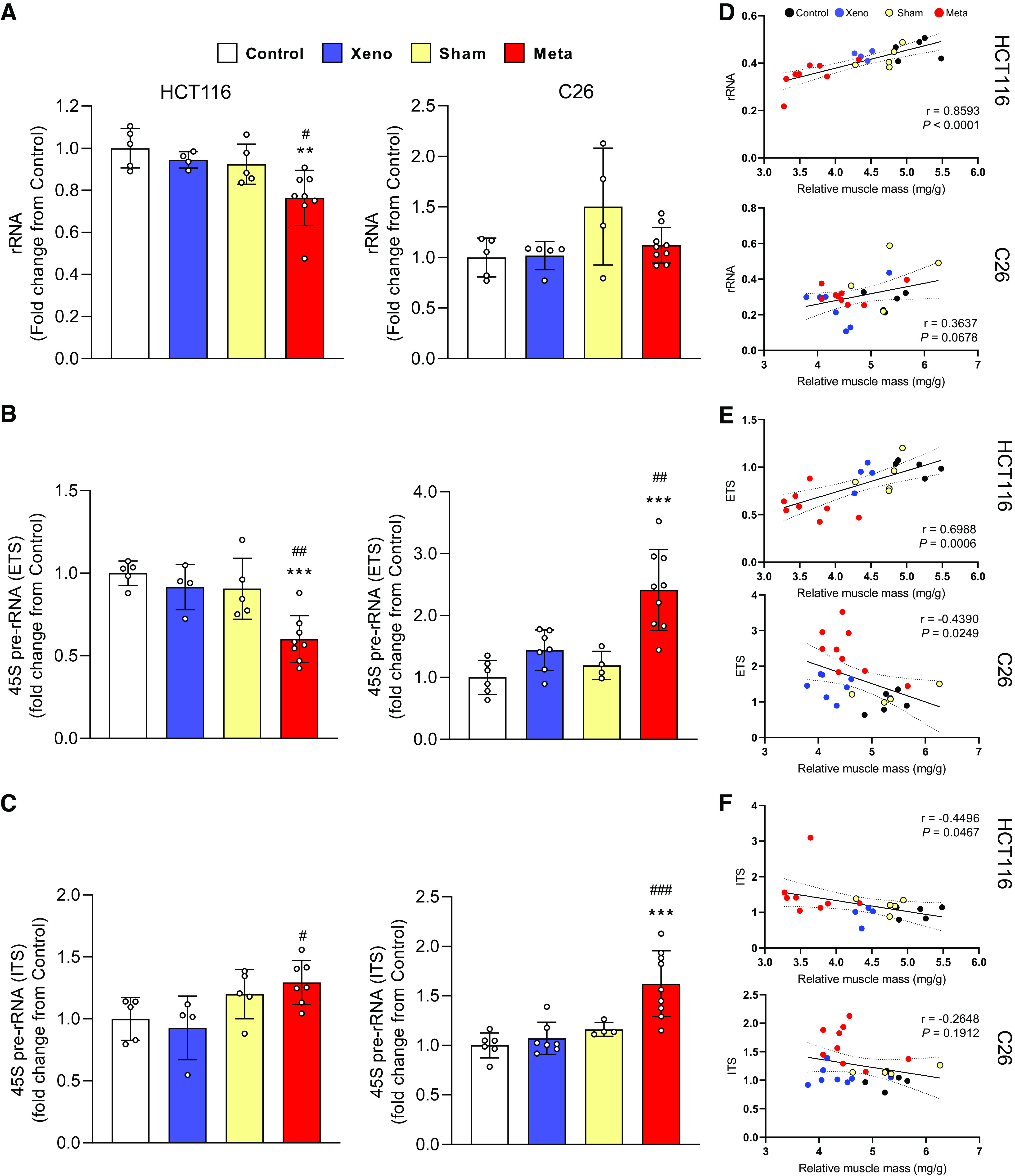
Ribosomal capacity and rDNA transcription are reduced in mHCT116, but C26 tumors display elevated rDNA transcription and maintain rRNA levels. A: total rRNA content was quantified to compare between groups. Expression of 45S pre-rRNA 5′ external transcribed spacer (ETS; B) and internal transcribed spacers (ITS; C) were analyzed by qPCR. D–F: Pearson product-moment correlations between each factor and muscle mass, respectively. Data are expressed as fold change from controls ± SD. A two-way ANOVA followed by a Tukey’s multiple comparisons test was performed for all direct comparisons between groups. Significant difference from control: *P ≤ 0.05, **P < 0.01, and ***P < 0.001; xenograft vs. metastasis: #P < 0.05, ##P < 0.01, and ###P < 0.001.
Intracellular Signaling Phosphorylation in CRC-Induced Muscle Wasting Is Tumor-Type Dependent
To determine if tumor burden exerted divergent alterations in signal transduction, we assessed phosphorylation changes of intracellular signaling proteins involved in translational control. mTOR phosphorylation was reduced in xHCT116 (P = 0.04) but it was not affected in mHCT116. RPS6 phosphorylation was significantly reduced in xHCT116 (P = 0.05) and mHCT116 (P = 0.003). Changes in 4E-BP1 phosphorylation were significantly reduced in both HCT116 tumors, mHCT116 exhibited 73.4% less γ4E-BP1 compared with control (P < 0.001) and xHCT116 (P = 0.067), whereas xHCT116 also experienced a 57.8% reduction in γ4E-BP1 compared with control (P = 0.0022; Fig. 3A). Both mTOR and RPS6 phosphorylation showed no statistical differences in either C26 model. In the xC26 model, γ4E-BP1 declined by 50.9% (P < 0.001), whereas in the mC26 model it was barely detectable (−99.2%, P < 0.001; Fig. 3B). The reduction in RPS6 and 4E-BP1 phosphorylation in the HCT116 model demonstrated a strong correlation with muscle wasting (RPS6: r = 0.439, P = 0.001; 4E-BP1: r = 0.795, P < 0.001; Fig. 3E).
Figure 3.
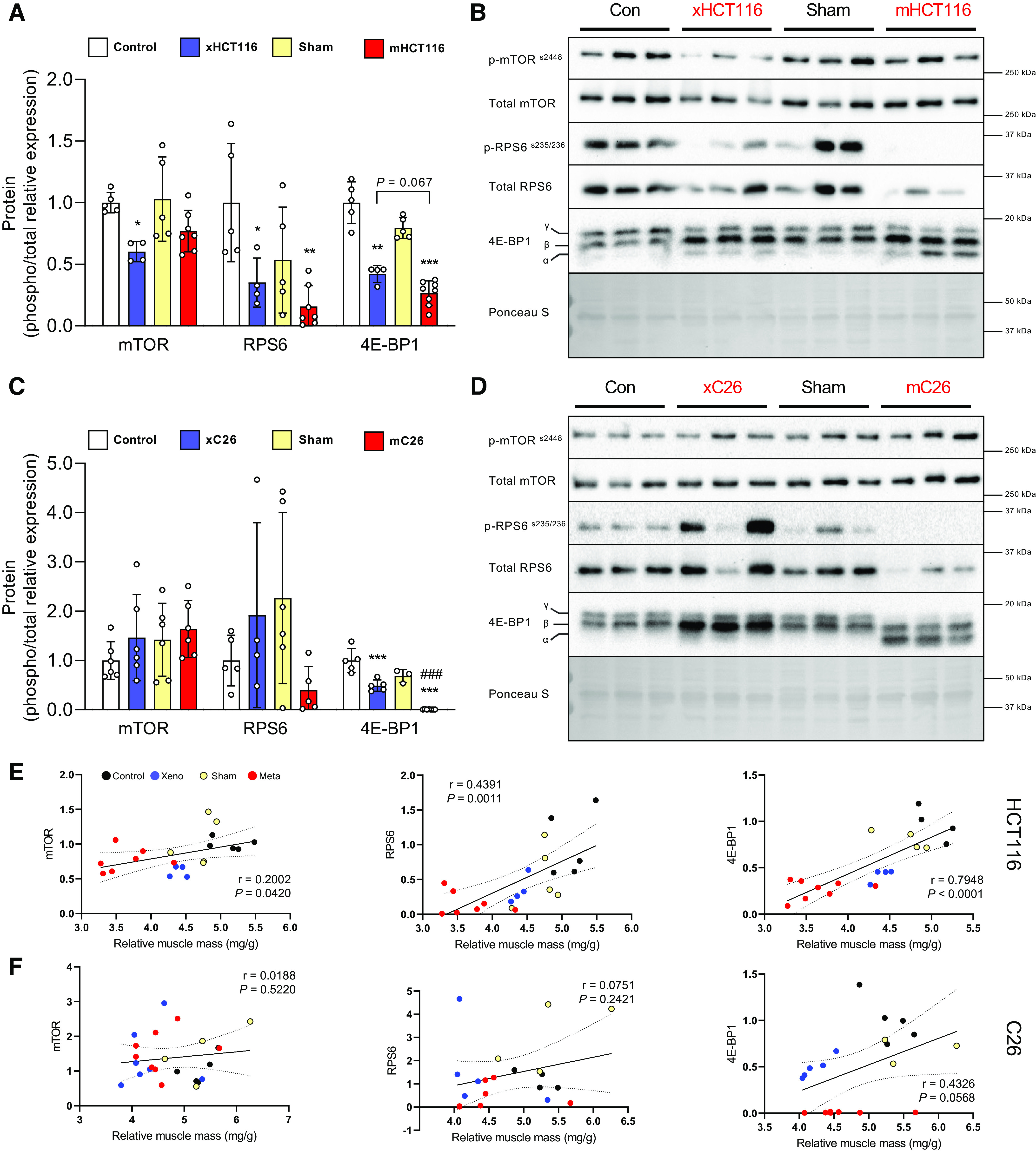
Intracellular signaling phosphorylation in colorectal cancer (CRC)-induced muscle wasting depends on the tumor type. A and C: quantification of Western blots for mTOR, RPS6, and 4E-BP1 phosphorylation in HCT116 and C26 model, respectively. B and D: representative image of Western blots. E and F: Pearson product-moment correlations between each factor and muscle mass, respectively. Data are expressed as fold change from controls ± SD. A two-way ANOVA followed by a Tukey’s multiple comparisons test was performed for all direct comparisons between groups. Significant difference from control: *P ≤ 0.05, **P < 0.01, and ***P < 0.001; xenograft vs. metastasis: #P < 0.05, ##P < 0.01, and ###P < 0.001.
Local IL-6 Expression Is Higher in Both mHCT116 and mC26 but Not TNF-α
IL-6 and TNF-α mRNA levels were determined to assess the expression of muscle-derived proinflammatory effectors of muscle wasting. IL-6 expression was significantly elevated in mHCT116 than control (1.8-fold, P = 0.02) and xHCT116 (P = 0.02; Fig. 4A). C26 tumor hosts, either in the xenograft or metastatic models, showed increased expression of IL-6 (xC26: 3.6-fold, P = 0.02; mC26: 4.1-fold, P = 0.005), but it was not different between xC26 and mC26 (Fig. 4A). IL-6 expression in both CRC models was inversely and significantly correlated with muscle mass (HCT116: r = −0.528, P = 0.018; C26: r = −0.577, P = 0.003; Fig. 4C). We did not detect significant changes in TNF-α expression in any of the tumor models (Fig. 4B) and neither correlated with muscle mass (Fig. 4D).
Figure 4.
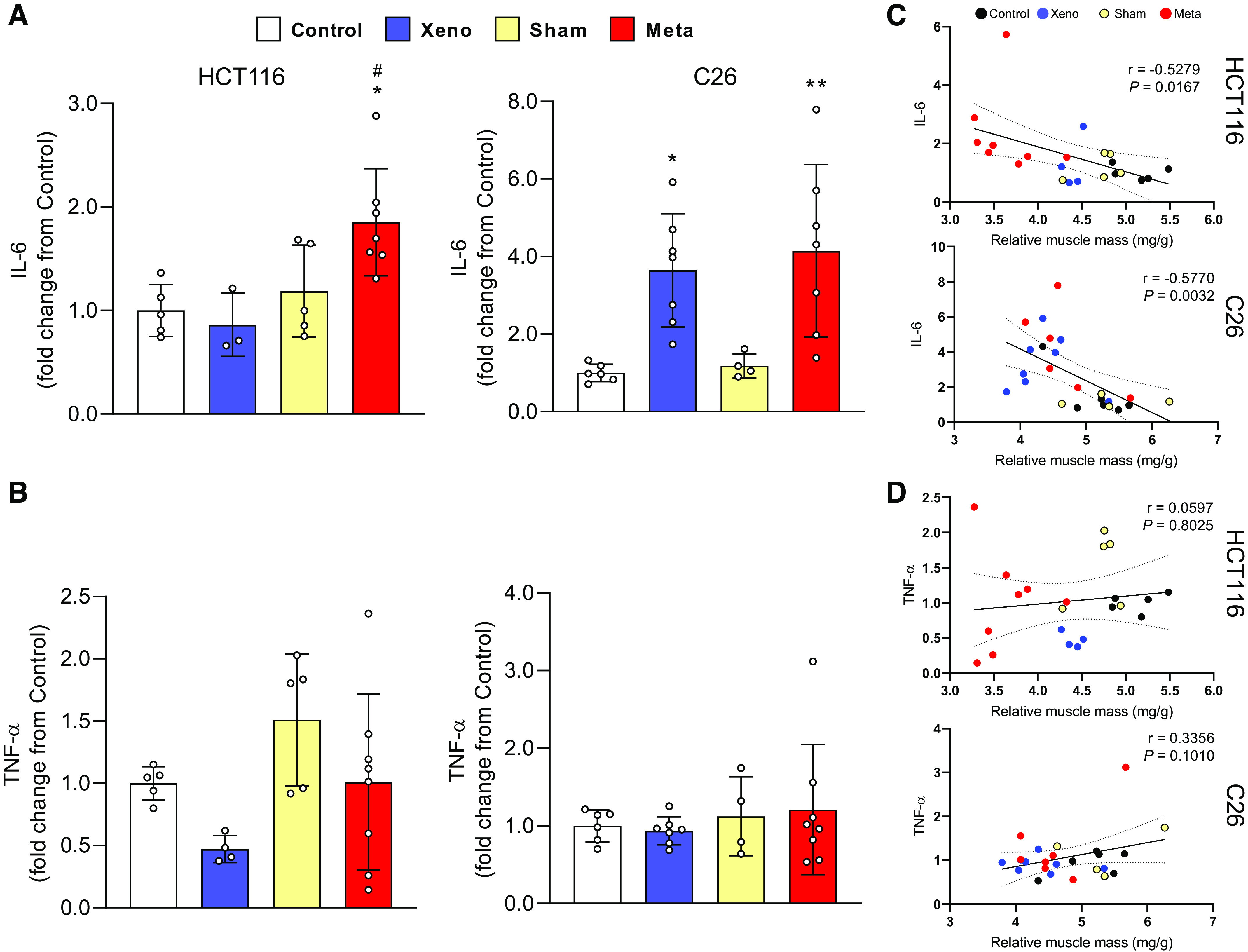
Local interleukin 6 (IL-6) expression is higher in both mHCT116 and mC26 but not TNF-α. Expression of IL-6 (A) and TNF-α mRNA (B) was analyzed by qPCR. C and D: Pearson product-moment correlations between each factor and muscle mass, respectively. Data are expressed as fold change from controls ± SD. A two-way ANOVA followed by a Tukey’s multiple comparisons test was performed for all direct comparisons between groups. Significant difference from control: *P ≤ 0.05, **P < 0.01; xenograft vs. metastasis: #P < 0.05.
Expression of Inflammasome-Related Factors Is Elevated in mC26 but Not in mHCT116 Tumor Mice
NLRP3, IL-1β, and IL-18 mRNA levels were measured to determine the expression of proinflammatory factors at the local muscle level. NLRP3 showed no change across groups in HCT116-injected animals, whereas mC26 showed a significant upregulation of NLRP3 compared with control (fivefold, P < 0.001) and xC26 (P < 0.001; Fig. 5A). IL-1β mRNA increased significantly in both xHCT116 (5.5-fold, P = 0.03) and mHCT116 (10-fold, P < 0.001; Fig. 5B) and was also significantly elevated in both xC26 (2.5-fold, P = 0.003) and mC26 (3.5-fold, P < 0.001). Both metastatic models displayed statistical differences in IL-1β expression compared with the corresponding xenografts (mHCT116, P = 0.02; mC26, P < 0.001), and this increase in IL-1β expression was strongly correlated with the loss of muscle mass in both CRC models (HCT116: r = −0.756, P < 0.001; C26: r = −0.577, P = 0.002; Fig. 5E). IL-18 was not affected by either HCT116 model, however, its expression was significantly elevated in the mC26 (threefold, P = 0.001) and xC26 (P < 0.001) models.
Figure 5.
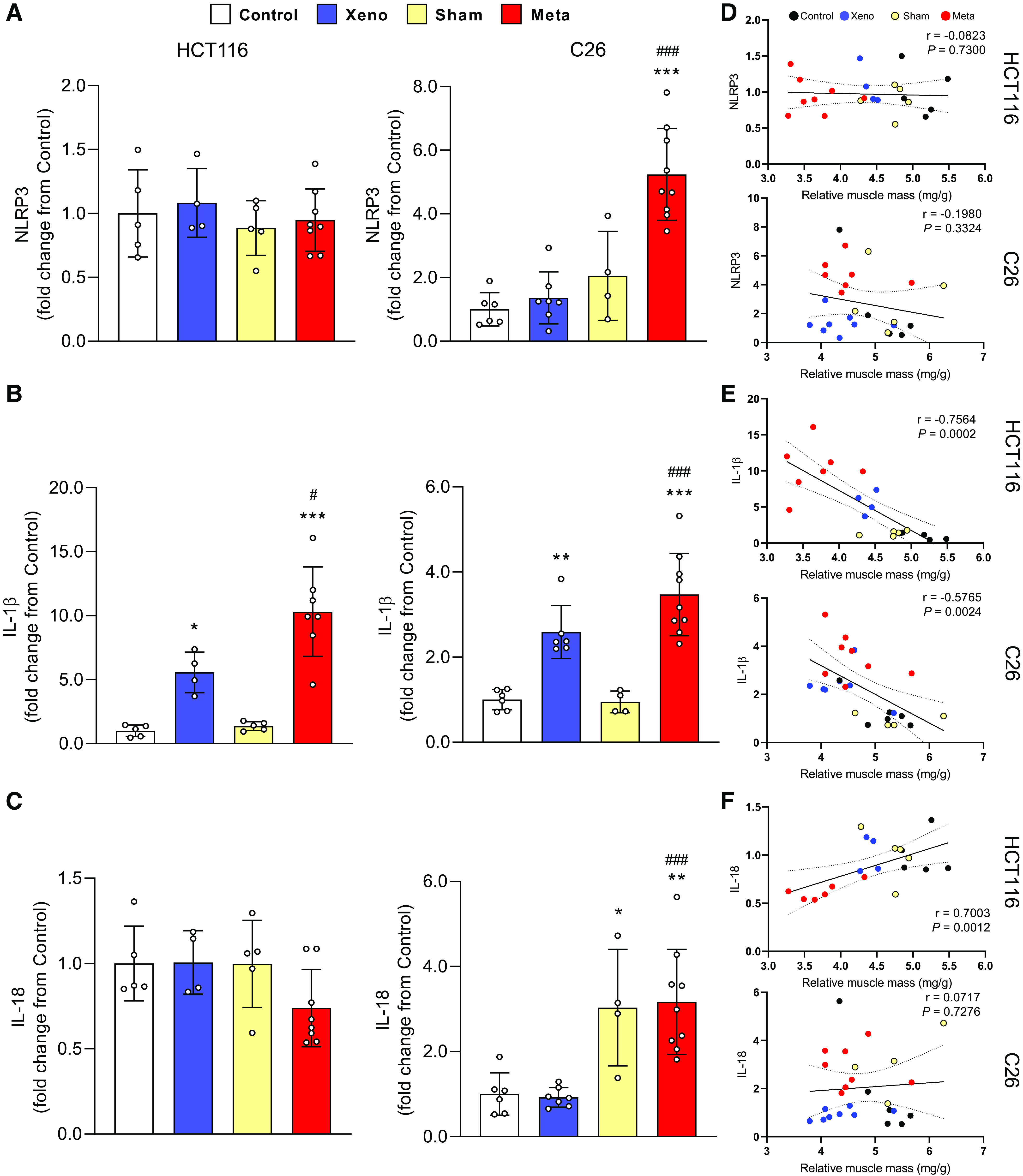
The expression of inflammasome-related factors is elevated in mC26 but not in mHCT116 tumor mice. Expression of NLRP3 (A), IL-1β (B), and IL-18 mRNA (C) was analyzed by qPCR. D–F: Pearson product-moment correlations between each factor and muscle mass, respectively. Data are expressed as fold change from controls ± SD. A two-way ANOVA followed by a Tukey’s multiple comparisons test was performed for all direct comparisons between groups. Significant difference from control: *P ≤ 0.05, **P < 0.01, and ***P < 0.001; xenograft vs. metastasis: #P < 0.05, ##P < 0.01, and ###P < 0.001.
DISCUSSION
We investigated whether metastatic or xenograft CRC tumors (i.e., tumor burden) promoted muscle wasting via common mechanisms. Unlike the clinical population, where over 60% of advanced CRC cases develop metastases (2), preclinical models rely on different tumor types and administration routes that may not always metastasize at the time of sacrifice. Together with the lack of well-characterized clinical specimens, potential differences in these preclinical models limit our understanding of the mechanisms promoting muscle wasting and may hinder the development of effective treatments to prevent the loss of muscle mass in patients with CRC. Here, we report new findings indicating that different tumor models, while producing similar wasting phenotypes, do so via divergent mechanisms in a tumor-type-dependent manner.
Preclinical cancer models are valuable tools for studying the mechanisms underlying muscle wasting. To determine if tumor burden exacerbates muscle wasting via divergent mechanisms, we examined metastatic or xenograft models using two CRC tumor types. In the present study, tumor burden is defined as number of cancer cells, the size of a tumor, the amount of cancer in the body (sometimes referred as tumor load), or the disease severity associated with the tumor. Muscle wasting was more pronounced in the mHCT116 tumor than in the xHCT116 tumor hosts, but the C26 tumors caused a similar degrees of muscle loss regardless of the model. This suggests that metastatic tumors produce a more aggressive wasting phenotype in a tumor-type-dependent manner. Because both metastatic and xenograft models were developed in the same host for each tumor type, we can rule out the possibility that genetic background was a confounding variable in our assessment of tumor burden-induced muscle wasting. This is a frequent concern when comparing different preclinical cancer models and complicates the identification of tumor-specific effects. To further understand whether tumor burden plays a role in anabolic deficits, we investigated changes in ribosomal capacity and rDNA transcription. The level of ribosomes is a major determinant of the muscle’s capacity for protein synthesis (12, 13), and has been previously reported to be reduced in human and rodent models of cancer with muscle wasting (11, 14, 21). In the mHCT116 model, exacerbated muscle wasting involved a larger decrease in rRNA than in the xHCT116 model, whereas rRNA levels were not affected in either C26 model. The changes in muscle mass and ribosomal capacity in the HCT116 models strongly support the interpretation that tumor burden is a major factor in the wasting process. But because there were no differences in the C26 models, we conclude that tumor burden elicits muscle wasting via divergent mechanisms of anabolic deficits in a tumor-type-dependent manner. Our findings in the mHCT116 model are consistent with previous studies showing that rats bearing Yoshida ascites hepatoma exhibited a 10%–14% loss in muscle mass with a 20%–40% reduction in rRNA content (22), and mice bearing MAC16 colon adenocarcinoma experienced ∼60% protein loss with ∼86% lower rRNA levels (23). Thus, muscle wasting in preclinical models with metastatic hepatic compromise appears to involve a significant reduction in rRNA. It is unclear, however, why the C26 tumors did not affect rRNA levels despite the large differences in tumor burden. Following intrasplenic tumor cell injection, mC26 hosts experienced a 21% increase in liver size relative to sham-operated animals, likely due to the 30% increase in hepatic tumor area (8). While his model is more severe in terms of tumor burden than the xC26, the amount of muscle loss was not. Thus, it becomes clear that tumor burden affects muscle loss in a tumor-type-dependent manner. One possible explanation for this discrepancy is that production of C26-specific tumor factors may trigger alternative mechanisms of muscle wasting (e.g., high inflammation and excessive protein degradation). Another possibility is that different muscle groups may be more sensitive than others to the disease burden caused by the tumor. Huot et al. (8) recently reported using the mC26 model that muscle wasting was more pronounced in the quadriceps than in the gastrocnemius and tibialis anterior muscles. This suggests that the interactions between tumor-derived factors and specific muscle groups may represent another layer of complexity in the wasting process, and needs to be considered when studying preclinical cancer models. The reduction in ribosomal capacity in the mHCT116 model could be accounted for by a decrease in ribosomal production as reflected by a significant (∼ 40%) reduction in rDNA transcription at the level of initiation (i.e., decrease in ETS signal). The results from the HCT116 models indicate that rDNA transcription is negatively affected in concordance with tumor burden, but this anabolic deficit is tumor-type dependent because mice bearing the C26 tumors showed no change in rRNA in either the xenograft or metastasis models. Interestingly, the mC26 displayed a significant ∼2.4- and ∼1.6-fold increase in rDNA transcription at the initiation (ETS) and elongation (ITS) levels, respectively. This unexpected finding may explain why the ribosomal capacity was maintained in the mC26 models and suggests the presence of a feedback mechanism that increases rDNA transcription to maintain rRNA levels in unfavorable anabolic setting (e.g., when ribosomal production falls). Our results extend previous findings demonstrating that muscle wasting 11 days following subcutaneous C26 tumor injection did not result in a significant loss of rRNA content (24), and support our interpretation that tumor burden negatively affects the ribosomal capacity of the muscle in a tumor-type-dependent manner.
Discrepancies in intracellular signaling were also dependent on tumor type. The HCT116 tumor model resulted in lower mTOR, RPS6, and 4E-BP1 phosphorylation and was independent of tumor burden. In contrast, mTOR and RPS6 phosphorylation was not affected by either C26 model. This should be interpreted with caution as we encountered large variability, particularly in RPS6 phosphorylation, which appears to be common in the C26 model. 4E-BP1 phosphorylation was lower in both C26 models, but significantly more reduced in mC26, suggesting a more prominent derailment of cap-dependent translation. Although RPS6 was highly variable, the fact that 4E-BP1 was not, indicates that the source of variability was biological and not technical. Alteration in mTOR/RPS6 signaling in the HCT116 models was consistent with other models of CRC-induced muscle wasting such as the Apcmin/+ mice (25), or ES-2 ovarian cancer (14), again indicating that not all CRC tumor types affect anabolic signaling in a similar manner. As cap-dependent initiation is rate-limiting for translation (26), reductions in 4E-BP1 phosphorylation in both models of CRC suggests that translation initiation is targeted independent of tumor type or model, but it is more sensitive to tumor burden with certain tumor types (i.e., mC26). Thus, in addition to reductions in translational (ribosomal) capacity, intracellular signaling may contribute to the anabolic deficits typically described in wasting skeletal muscle in a tumor-type-dependent manner.
Another important cancer-induced contributor to muscle wasting is the elevation of circulating proinflammatory cytokines (27, 28). In addition to their systemic procachectic effects, circulating cytokines stimulate the local expression of proinflammatory effectors of muscle wasting (29, 30). The most obvious discrepancy between the HCT116 and C26 models was the elevation of IL-6 mRNA in all tumor models except in xHCT116. This indicates that IL-6 expression is associated with tumor burden in a tumor-type-specific manner, i.e., IL-6 mRNA was higher in the mHCT116 than in xHCT116, but was not different in either C26 model. Clearly, in terms of IL-6 expression, the C26 tumors caused a more pronounced inflammatory response than the HCT116 tumors. In contrast, TNF-α mRNA did not differ in either tumor type or model. Although TNFα mRNA was highly variable, it appears to be expressed to some degree at this late stage in the wasting process. TNF-α is an acute-phase procachectic factor (31), which may explain the variable expression at this late time point. To further determine alterations in local cytokine production, we quantified changes in NLRP3 mRNA, a component of the NLRP3 inflammasome (19). This multiprotein complex is responsible for the conversion of IL-1β and IL-18 precursors into their mature active forms (19), both of which may stimulate muscle protein turnover by upregulating the expression of the muscle-specific ubiquitin ligases MuRF1 and atrogin-1 (32). NLRP3 mRNA was only elevated in the mC26 model, but the expression of IL-1β mRNA was stimulated by both tumor types with significantly higher expression levels in the metastatic relative to the xenograft model. Thus, while IL-1β expression exhibits dependence on tumor burden irrespective of tumor type, the discordance with NLRP3 expression indicates that NLRP3 and IL-1β are not transcriptionally co-regulated. IL-18 mRNA was not induced by either tumor, but it was higher in both sham and mC26 mice suggesting that the surgical stress associated with this model exerted a more powerful response than the metastatic tumors. IL-18 is considered to be an acute phase response cytokine, and this may explain the lack of changes in either tumor model at this advanced stage. A stated above, a possible limitation of the analysis of cytokines in the present investigation may be related to the late time point studied.
In summary, our findings demonstrate that metastatic models of CRC produce a more robust alteration in ribosomal capacity, intracellular signaling and local proinflammatory profiles than xenograft models in a tumor-type-dependent manner. A better understanding of precise molecular mechanisms underlying muscle wasting in metastatic CRC preclinical models will provide new opportunities to design and implement novel treatments to enhance muscle anabolism and prevent the loss of muscle mass in cancer.
GRANTS
This study was supported by grants from the National Institutes of Health (NIH) Grants AR073385 and AR078430 (to G.A.N.), and the Departments of Surgery and Otolaryngology—Head and Neck Surgery at Indiana University, by grants from the American Cancer Society (Research Scholar Grant 132013-RSG-18-010-01-CCG) and NIH Grant AR079379 (to A.B.). J.H. was supported by an NIH-T32 Institutional Training Grant AR065971. We thank the Simon Comprehensive Cancer Center at Indiana University School of Medicine, funded by the IU Simon Cancer Center Support Grant P30 CA082709, for the use of the In Vivo Therapeutic Core, which provided the NSG mice.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.R.H., A.B., and G.A.N. conceived and designed research; H.-G.K., J.R.H., F.P., and D.J.B. performed experiments; H.-G.K. and D.J.B. analyzed data; H.-G.K., D.J.B., and G.A.N. interpreted results of experiments; H.-G.K. prepared figures; H.-G.K. and G.A.N. drafted manuscript; H.-G.K., A.B., and G.A.N. edited and revised manuscript; H.-G.K., J.R.H., F.P., D.J.B., A.B., and G.A.N. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address for A. Bonetto: Dept. of Pathology, Univ. of Colorado Anschutz Medical Campus, Aurora, CO (andrea.bonetto@cuanschutz.edu).
REFERENCES
- 1. Siegel RL, Miller KD, Sauer AG, Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics. CA: Cancer J Clin 70.3: 145–164, 2020. doi: 10.3322/caac.21601. [DOI] [PubMed] [Google Scholar]
- 2. Loberg RD, Bradley DA, Tomlins SA, Chinnaiyan AM, Pienta KJ. The lethal phenotype of cancer: the molecular basis of death due to malignancy. CA Cancer J Clin 57: 225–241, 2007. [Erratum in CA Cancer J Clin 57: 380, 2007]. doi: 10.3322/canjclin.57.4.225. [DOI] [PubMed] [Google Scholar]
- 3. Consul N, Guo X, Coker C, Lopez-Pintado S, Hibshoosh H, Zhao B, Kalinsky K, Acharyya S. Monitoring metastasis and cachexia in a patient with breast cancer: a case study. Clin Med Insights Oncol 10: 83–94, 2016. doi: 10.4137/CMO-S40479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shiono M, Huang K, Downey RJ, Consul N, Villanueva N, Beck K, Fenn K, Dietz D, Yamaguchi T, Kato S, Divgi C, Kalinsky K, Wei Y, Zhang Y, Borczuk AC, Inoue A, Halmos B, Acharyya S. An analysis of the relationship between metastases and cachexia in lung cancer patients. Cancer Med 5: 2641–2648, 2016. doi: 10.1002/cam4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tomasin R, Martin ACBM, Cominetti MR. Metastasis and cachexia: alongside in clinics, but not so in animal models. J Cachexia Sarcopenia Muscle 10: 1183–1194, 2019. doi: 10.1002/jcsm.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belcher DJ, Guitart M, Hain B, Kim HG, Waning D, Barreiro E, Nader GA. LP07 and LLC pre-clinical models of lung cancer induce divergent anabolic deficits and expression of pro-inflammatory effectors of muscle wasting. J Appl Physiol (1985). doi: 10.1152/japplphysiol.00246.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huot JR, Novinger LJ, Pin F, Bonetto A. HCT116 colorectal liver metastases exacerbate muscle wasting in a mouse model for the study of colorectal cancer cachexia. Dis Model Mech 13: dmm043166, 2020. doi: 10.1242/dmm.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huot JR, Novinger LJ, Pin F, Narasimhan A, Zimmers TA, O’Connell TM, Bonetto A. Formation of colorectal liver metastases induces musculoskeletal and metabolic abnormalities consistent with exacerbated cachexia. JCI Insight 5: e136687, 2020. doi: 10.1172/jci.insight.136687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Svaninger G, Bennegard K, Ekman L, Ternell M, Lundholm K. Lack of evidence for elevated breakdown rate of skeletal muscles in weight-losing, tumor-bearing mice. J Natl Cancer Inst 71: 341–346, 1983. [PubMed] [Google Scholar]
- 10. Emery PW, Lovell L, Rennie MJ. Protein synthesis measured in vivo in muscle and liver of cachectic tumor-bearing mice. Cancer Res 44: 2779–2784, 1984. [PubMed] [Google Scholar]
- 11. Emery PW, Edwards RH, Rennie MJ, Souhami RL, Halliday D. Protein synthesis in muscle measured in vivo in cachectic patients with cancer. Br Med J (Clin Res Ed) 289: 584–586, 1984. doi: 10.1136/bmj.289.6445.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Millward DJ, Garlick PJ, James WPT, Nnanyelugo DO, Ryatt JS. Relationship between protein synthesis and RNA content in skeletal muscle. Nature 241: 204–205, 1973. doi: 10.1038/241204a0. [DOI] [PubMed] [Google Scholar]
- 13. Millward DJ, Garlick PJ, Nnanyelugo DO, Waterlow JC. The relative importance of muscle protein synthesis and breakdown in the regulation of muscle mass. Biochem J 156: 185–188, 1976. doi: 10.1042/bj1560185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim H, Huot JR, Pin F, Guo B, Bonetto A, Nader GA. Reduced rDNA transcription diminishes skeletal muscle ribosomal capacity and protein synthesis in cancer cachexia. FASEB J 35: e21335, 2021. doi: 10.1096/fj.202002257R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lang CH, Frost RA, Nairn AC, MacLean DA, Vary TC. TNF-α impairs heart and skeletal muscle protein synthesis by altering translation initiation. Am J Physiol Endocrinol Metab 282: E336–E347, 2002. doi: 10.1152/ajpendo.00366.2001. [DOI] [PubMed] [Google Scholar]
- 16. White JP, Puppa MJ, Gao S, Sato S, Welle SL, Carson JA. Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am J Physiol Endocrinol Metab 304: E1042–E1052, 2013.doi: 10.1152/ajpendo.00410.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Webster JM, Kempen LJAP, Hardy RS, Langen RCJ. Inflammation and skeletal muscle wasting during cachexia. Front Physiol 11: 597675, 2020. doi: 10.3389/fphys.2020.597675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kalbitz M, Fattahi F, Grailer JJ, Jajou L, Malan EA, Zetoune FS, Huber‐Lang M, Russell MW, Ward PA. Complement‐induced activation of the cardiac NLRP3 inflammasome in sepsis. FASEB J 30: 3997–4006, 2016. doi: 10.1096/fj.201600728R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell 10: 417–426, 2002. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 20. Huang N, Kny M, Riediger F, Busch K, Schmidt S, Luft FC, Slevogt H, Fielitz J. Deletion of Nlrp3 protects from inflammation-induced skeletal muscle atrophy. Intensive Care Med Exp 5: 3–15, 2017. doi: 10.1186/s40635-016-0115-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goodlad GAJ, Clark CM. Response of skeletal muscle RNA polymerases I and II to tumour growth. Biochim Biophys Acta 950: 296–302, 1988. doi: 10.1016/0167-4781(88)90125-X. [DOI] [PubMed] [Google Scholar]
- 22. Baracos VE, DeVivo C, Hoyle DH, Goldberg AL. Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am J Physiol 268: E996–E1006, 1995. doi: 10.1152/ajpendo.1995.268.5.E996. [DOI] [PubMed] [Google Scholar]
- 23. Bhogal AS, Lorite ML, Tisdale MJ. Changes in nucleic acid and protein levels in atrophying skeletal muscle in cancer cachexia. Anticancer Res 26: 4149–4154, 2006. [PubMed] [Google Scholar]
- 24. Lautaoja JH, Lalowski M, Nissinen TA, Hentilä J, Shi Y, Ritvos O, Cheng S, Hulmi JJ. Muscle and serum metabolomes are dysregulated in colon-26 tumor-bearing mice despite amelioration of cachexia with activin receptor type 2B ligand blockade. Am J Physiol Endocrinol Metab 316: E852–E865, 2019. doi: 10.1152/ajpendo.00526.2018. [DOI] [PubMed] [Google Scholar]
- 25. White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, Carson JA. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the ApcMin/+ mouse. PLoS One 6: e24650, 2011. doi: 10.1371/journal.pone.0024650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433: 477–480, 2005. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- 27. Deans C, Wigmore SJ. Systemic inflammation, cachexia and prognosis in patients with cancer. Curr Opin Clin Nutr Metab Care 8: 265–269, 2005. doi: 10.1097/01.mco.0000165004.93707.88. [DOI] [PubMed] [Google Scholar]
- 28. Zimmers TA, Fishel ML, Bonetto A. STAT3 in the systemic inflammation of cancer cachexia. Semin Cell Dev Biol 54: 28–41, 2016. doi: 10.1016/j.semcdb.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frost RA, Nystrom GJ, Lang CH. Lipopolysaccharide regulates proinflammatory cytokine expression in mouse myoblasts and skeletal muscle. Am J Physiol Regul Integr Comp Physiol 283: R698–R709, 2002. doi: 10.1152/ajpregu.00039.2002. [DOI] [PubMed] [Google Scholar]
- 30. Lang CH, Silvis C, Deshpande N, Nystrom G, Frost RA. Endotoxin stimulates in vivo expression of inflammatory cytokines tumor necrosis factor alpha, interleukin-1β,-6, and high-mobility-group protein-1 in skeletal muscle. Shock 19: 538–546, 2003. doi: 10.1097/01.shk.0000055237.25446.80. [DOI] [PubMed] [Google Scholar]
- 31. Patel HJ, Patel BM. TNF-α and cancer cachexia: molecular insights and clinical implications. Life Sci 170: 56–63, 2017. doi: 10.1016/j.lfs.2016.11.033. [DOI] [PubMed] [Google Scholar]
- 32. Li W, Moylan JS, Chambers MA, Smith J, Reid MB. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am J Physiol Cell Physiol 297: C706–C714, 2009. doi: 10.1152/ajpcell.00626.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]