

Summary
Small-cell lung cancer (SCLC) methylome is understudied. Here, we comprehensively profile SCLC using cell-free methylated DNA immunoprecipitation followed by sequencing (cfMeDIP-seq). Cell-free DNA (cfDNA) from plasma of 74 patients with SCLC pre-treatment and from 20 non-cancer participants, genomic DNA (gDNA) from peripheral blood leukocytes from the same 74 patients, and 7 accompanying circulating tumor cell-derived xenografts (CDXs) underwent cfMeDIP-seq. Peripheral blood leukocyte methylation (PRIME) subtraction to improve tumor specificity. SCLC cfDNA methylation is distinct from non-cancer but correlates with CDX tumor methylation. PRIME and k-means consensus identified two methylome clusters with prognostic associations that related to axon guidance, neuroactive ligand−receptor interaction, pluripotency of stem cells, and differentially methylated at long noncoding RNA and other repeats features. We comprehensively profiled the SCLC methylome in a large patient cohort and identified methylome clusters with prognostic associations. Our work demonstrates the potential of liquid biopsies in examining SCLC biology encoded in the methylome.
Subject areas: Cancer, Cancer systems biology, Epigenetics, Genomics
Graphical abstract
Highlights
-
•
SCLC methylome can be assessed from cell-free DNA in patient plasma
-
•
PRIME subtraction improves tumor specificity of cell-free methylome
-
•
Two methylation-defined SCLC clusters have prognostic association
-
•
Clusters were differentially hypermethylated at lncRNA and repetitive DNA elements
Cancer; Cancer systems biology; Epigenetics; Genomics
Introduction
Small-cell lung cancer (SCLC) is a highly aggressive subset of lung cancer.1 While often sensitive to first-line therapy, most patients with SCLC develop recurrent disease accompanied by therapeutic resistance.1 While SCLC genomics have been recently characterized,2,3,4,5,6,7 the epigenome has not been as extensively profiled.8,9 Epigenetic mechanisms, specifically DNA methylation, may contribute toward SCLC oncogenesis, recurrence, and resistance,1 through tumorigenesis and epigenetic reprogramming.10
Two key SCLC DNA methylome studies examined patient tissue to identify a methylation-defined differentiation block9 and unique methylation-defined subtypes.8 However, progress in comprehensive methylome profiling of SCLC is impeded by lack of primary tumor tissue. Liquid biopsy using cell-free DNA (cfDNA) presents a solution. Previous cfDNA analysis in SCLC evaluated genetic changes, demonstrating a high tumor mutation burden, an increased mutant allele fraction in plasma cfDNA,11,12 identified druggable targets,12 and prediction of disease relapse.13 These findings demonstrate the potential of liquid biopsy analyses of SCLC tumor biology.
Here, we comprehensively profiled the methylome of plasma cfDNA from patients with SCLC to identify novel putative biomarkers and epigenetic mechanisms of disease. For this, we conducted cell-free methylated DNA immunoprecipitation and high-throughput sequencing (cfMeDIP-seq),14 which has previously been applied to pancreatic,14 non-SCLC,14 renal cell,15,16 glioma,17 head and neck,18 prostate,19 and other cancers. While cfMeDIP-seq has revolutionized the study of cancer biology, especially for cancers that have limited conventional tissue biopsy samples, removing non-cancer from the cancer signal remains a challenge. Many of the studies used cfDNA from healthy non-cancer patients as a reference to filter out non-cancer signal.14,15,17,18 Although a tractable approach, this does not fully account for the non-tumor signal present within the plasma of each cancer patient.
For our study, we developed the peripheral blood leukocyte (PBL) methylation (PRIME) subtraction algorithm to simultaneously use PBLs acquired from the same patient and timepoint as the cfMeDIP-seq analyzed patient blood plasma. We analyzed the methylome of 74 plasma samples of patients with SCLC enriched for tumor-derived signal by PRIME subtraction to evaluate biological, clinical, and prognostic associations. Our findings demonstrated the utility of liquid biopsies to examine SCLC methylome and identified by unsupervised machine learning two methylation-defined clusters of patients with prognostic association.
Results
SCLC cfDNA methylome reflects tumor tissue methylation and is distinct from NCC
We examined the cell-free methylome of 74 patients with SCLC using blood samples collected prior to first-line treatment (Figure 1A). 88% of patients (n = 66) were current or former smokers and 57% had extensive-stage (ES) SCLC (n = 42; Figure 1A) with the remainder limited-stage (LS). We also examined the cell-free methylomes of 20 non-cancer control (NCC) participants. NCC participants were smokers and were similar in age and sex to the SCLC cohort (Figure 1A).The distinct clustering pattern of the NCC methylome profiles compared to SCLC suggests that the tumor methylome is being captured. Although a small fraction of patients with SCLC were self-reported never smokers (8/74), by PCA, this subset of patients were distributed throughout current/former smoker patients with SCLC and did not cluster with NCC methylation. Based on our analysis, the methylome did not correlate with smoking status; however, this current study is underpowered to detect this effect because never smokers (n = 8) are a small proportion in our cohort and there is a possibility of inconsistent self-reporting of smoking status20 (Figure S1). Detailed demographic characteristics can be found in Table S1.
Figure 1.

SCLC cell-free methylome reflects tissue and is distinct from non-cancer
(A) Demographics and clinical characteristics of 74 patients with small-cell lung cancer (SCLC) and non-cancer donors.
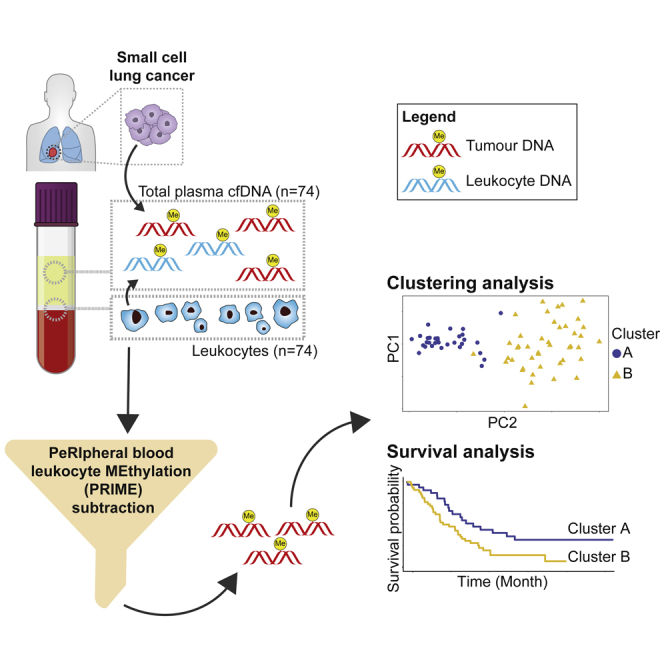
(B) Schematic of overall structure of study and major analyses done.
(C) Principal component analysis of genome-wide methylation profiles of total methylated cell-free DNA (cfDNA) from patients with SCLC (n = 7) and methylated genomic DNA from paired circulating tumor cell-derived xenograft (CDX) models (n = 7).
(D) Principal component analysis of genome-wide methylation profiles of total methylation cfDNA from patients with SCLC (n = 74) and non-cancer donors (n = 20).
(E) Volcano plot of differentially methylated region (DMR) analysis of patients with SCLC (n = 74) and non-cancer donors (n = 20). Each dot corresponds to a 300bp region of the genome. The horizontal line corresponds to p-adjusted = 0.05 and vertical lines correspond to log2 fold-change of +/−2. There are 51,666 hypermethylated DMRs in SCLC and 1,019 in non-cancer donors.
(F) Bar plot of significantly hypermethylated DMRs corresponding to CpG features observed in patients with SCLC (n = 51,666 DMRs) and non-cancer donors (n = 1,019 DMRs).
(G) Permutation analysis of the hypermethylated DMRs observed in SCLC (n = 51,666 DMRs).
To assess whether cfMeDIP-seq can ascertain SCLC tumor tissue methylation, we performed a genome-wide concordance analysis of methylome profiles (n = 8,828,974 300bp windows) of cfDNA of patients with SCLC (n = 7) and gDNA of their respective circulating tumor cell (CTC)-derived xenograft (CDX) tumor tissue (n = 7). Patient cfDNA was highly concordant with CDX methylation by principal component analysis (PCA; Figure 1C) and strongly correlated by correlation analysis of normalized read counts within each window (median r = 0.92, n = 7). Thus, cfMeDIP-seq data appeared representative of tissue-level DNA methylation. Reassuringly, this concordance between cfDNA and tumor tissue methylome has been recently reported in another independent study.21However, as others have reported, patients with SCLC that successfully engraft CDXs have increased CTCs which could increase tumor cfDNA.22 For our patients with SCLC without a corresponding CDX (n = 67), the contribution of non-cancer cfDNA is expected to be higher. Therefore, we assessed if cfMeDIP-seq could distinguish between SCLC and NCC methylation through examining genome-wide cfDNA methylome profiles by PCA (Figure 1D). NCCs were distinct from SCLC suggesting cfMeDIP-seq distinguished non-cancer methylation from cancer. Through differentially methylated region (DMR) analysis between SCLC and NCC, 51,666 and 1,019 significantly hypermethylated DMRs were identified in SCLC and NCC, respectively (p-adj < 0.05; log2FC > 1; Figure 1E). SCLC significant DMRs were enriched in CpG islands and shores relative to NCCs, whereas NCC significant DMRs were enriched in open-sea regions (Figure 1F). Permutation analysis revealed that CpG features were significantly enriched in SCLC cfDNA DMRs and verified that cfMeDIP-seq is tumor specific (Figure 1G).
PRIME removes non-cancer methylation using paired PBLs from same patients with SCLC
Non-tumor methylation signal in the SCLC cfDNA data was examined and quantified using MethylCIBERSORT. This allowed us to approximate the proportion of methylation in plasma cfDNA from non-tumor cells. We found PBLs to be a large contributor to plasma cfDNA methylation (Figures S2–S6).
To increase specificity of the SCLC signal to non-cancer noise ratio, we implemented a novel approach utilizing paired PBL gDNA collected from the same plasma source material of patient with SCLC at identical timepoints (Figure 2A). Comparison of total plasma cfDNA to PBL gDNA by PCA revealed that PBLs exhibited a distinct methylation signal (Figure 2B). Next, we examined the methylome of SCLC total plasma cfDNA alone (Figure 2C), and after applying our novel algorithmic filter, PRIME, to reduce PBL methylation signals (Figure 2D). In brief, we started with whole-genome windows (n = 9,603,445), removed ENCODE-blacklist regions, and then selected windows hypomethylated across PBLs (median beta per window <0.2). Within these PBL-hypomethylated windows, we further selected for windows with a CG-density threshold >= 5 to account for the functionality of the 5-methylcytosine antibody.18 Thus, PRIME filtered out non-tumor noise in the cfDNA, reducing 9,603,445 whole-genome windows to 196,582 SCLC-specific windows. By PCA, PRIME increased the variance explained by methylation in PC1 from 40% (Figures 2C) to 57% (Figure 2D) and decreased the variance in PC2 from 7% (Figure 2C) to 4% (Figure 2D). PRIME also identified two distinct PCA groups (Figure 2D), refined our cfDNA methylation, and increased cfMeDIP-seq SCLC-specificity.
Figure 2.

Filtering out non-cancer methylation signal from plasma cfDNA
(A) Overall schematic outlining our approach to first compare SCLC total cfDNA and SCLC PBL gDNA methylome (2B), SCLC total cfDNA methylome (2C), and PRIME-filtered cfDNA methylome (2D).
(B) Principal component analysis (PCA) of methylation profiles of 74 patients with SCLC comparing total plasma cfDNA methylation to PBL gDNA methylation. For each patient, plasma cfDNA and PBL gDNA are extracted using peripheral blood samples collected prior to starting first-line chemotherapy. Genome-wide methylation profiles are examined in this PCA plot (8,828,974 300bp windows). Red filled circles correspond to cfDNA samples and blue triangles are PBL samples.
(C) PCA of total plasma cfDNA methylation profiles of the 74 patients with SCLC. Genome-wide methylation profiles are examined in this PCA plot (8,828,974 300bp windows). Red filled circles correspond to cfDNA samples.
(D) PCA of PRIME filtered plasma cfDNA methylation profiles of the 74 patients with SCLC. In this PCA, methylation profiles for 196,582 300bp windows are examined.
Identifying methylation-defined prognostic clusters and examining differentially methylated pathways and features
Our goal was to determine what we can understand about underlying SCLC biology using only the methylome. As such, we decided on using a non-supervised approach to determine if there are any intrinsic methylome-defined subgroups in SCLC. To define potential SCLC methylome subgroups, we applied k-means consensus clustering on the PRIME-filtered cfDNA methylome profiles for all 74 patients with SCLC. We determined 2 methylation-defined clusters were the optimal number (Figure S7), which we designated as Clusters A and B (Figure 3A).
Figure 3.

Identifying prognostic clusters and examining differentially methylated pathways and features
(A) Consensus clustering done on PRIME-filtered SCLC methylated cfDNA identified two clusters, A and B.
(B) Volcano plot of differentially methylated region (DMR) analysis between cluster A and B. The horizontal line corresponds to p-adjusted = 0.05 and vertical lines correspond to log2 fold-change of +2 and −1. There are 174 hypermethylated DMRs in cluster A and 9,037 in cluster B.
(C) KEGG pathway analysis of hypermethylated DMRs in cluster A and cluster B.
(D) Kaplan-Meier survival analysis on cluster A and B identified by consensus clustering (p = 0.013).
To examine biologic differences in Clusters A and B, we conducted a DMR analysis (Figure 3B). This analysis identified 174 significantly hypermethylated DMRs in Cluster A (p-adj < 0.5 and log2FC < -1) and 9,037 in Cluster B (p-adj < 0.05 and log2FC > 2). To understand methylation differences in terms of biological pathways, a KEGG pathway analysis was performed on the significantly hypermethylated DMRs (Figure 3C). In Cluster A, 174 significant DMRs corresponded to 137 genes whereas in Cluster B, 9,037 DMRs corresponded to 2,131 genes. Pathways corresponding to axon guidance or phospholipase D signaling pathway, or non-small-cell lung cancer, were enriched in Cluster A (Figure 3C). On the other hand, Cluster B had several pathways like neuroactive ligand−receptor interaction, immune chemokine signaling, and pathways regulating pluripotency of stem cells (Figure 3C).
In order to understand clinical relevance of these intrinsic methylome-defined clusters, we performed an overall survival (OS) analysis on patients in Cluster A and B. Patients in these clusters had significantly different OS (HR = 2.02, p = 0.014; Figure 3D and Table S2) where Cluster B had a median OS of 13 months compared to 21 in Cluster A. With respect to clinical stage, interestingly, Cluster A had a predominance of patients with LS-SCLC (n = 21, 68% of 31 Cluster A patients), whereas Cluster B had a greater proportion of patients with ES-SCLC (n = 32, 74% of 43 Cluster B patients) (Figure S8). Therefore, after adjusting for stage with the smaller sample size per stage, Clusters A and B were no longer significant for OS suggesting these SCLC methylome clusters and associated aggressive biology has some correlation with clinical stage or disease burden. Interestingly, after we stratified stage by cluster (Table S3), Cluster B consistently had worse OS in either stage, which was more apparent in LS-SCLC (Figure S9).
To further examine stage-specific methylome associations, we performed supervised machine learning on an 80:20 training:test split of the 74 samples. We developed a cross-validated elastic-net penalized regression model to classify patients with LS vs. ES. Our model demonstrated a balanced accuracy of 85% (Table S4). After performing DMR analysis of LS vs. ES, KEGG pathway analysis of ES revealed pathways similar to Cluster B (Figure S10). Our dual unsupervised and supervised machine learning approaches of methylome analysis of SCLC samples reveal distinct methylation patterns associated with either aggressive biology (Cluster B; k-means consensus) or worse clinical stage (ES; elastic net), respectively.
Taken together, cfMeDIP-seq may be identifying a more aggressive SCLC biological phenotype that is partially associated with stage but may also have some stage-independent value that the current study is underpowered to detect. However, future larger studies are needed to determine molecular prognostic methylome features that may supplement or refine clinical stage.
Hypermethylation of biological features in methylation-defined prognostic clusters A and B
To interrogate the differences detected by k-means consensus algorithm, we then examined DMRs that associated with Cluster A or Cluster B. Both clusters had similar hypermethylated DMR proportions among all CpG features (Figure 4A). Interestingly, approximately 40% of all significantly hypermethylated DMRs in both Cluster A and B also corresponded to non-protein coding features such as short interspersed nuclear elements (SINEs), long interspersed nuclear elements (LINEs), long terminal repeats (LTRs), retrotransposons, and satellites (Figure 4B). Strikingly, 65% of significantly hypermethylated DMRs in Cluster A corresponded to long noncoding RNA (lncRNA) windows compared to 38% in Cluster B (Figure 4C) suggesting loss of methylation at some lncRNAs may be associated with more aggressive biology. This change in hypermethylation, which was observed for lncRNAs only, suggested some underlying role of lncRNAs in mediating the aggressiveness of SCLC. For most features, a similar proportion of windows (Figures 4A and 4B) were observed suggesting different LINEs, SINEs, LTRs, or CpG features were differentially hypermethylated in Cluster A and B.
Figure 4.

Understanding differentially methylated biological features in Cluster A and B
(A) Bar plots of significantly hypermethylated DMRs observed in Cluster A (n = 174) and Cluster B (n = 9,037) corresponding to CpG features.
(B) Bar plots of significantly hypermethylated DMRs observed in Cluster A (n = 174) and Cluster B (n = 9,037) corresponding to repeats features.
(C) Bar plots of significantly hypermethylated DMRs observed in Cluster A (n = 174) and Cluster B (n = 9,037) corresponding to long noncoding RNA (lncRNA).
(D) Overall schematic of analyzing biological features of interest in PRIME-filtered hypermethylated 300bp windows.
(E) Correlating the number of PRIME-filtered hypermethylated 300bp windows corresponding to specific methylomic features with principal component 1 (PC1) variance.
(F) Principal component analysis (PCA) plot of 300bp PRIME-filtered windows subset to lncRNA (80,236/196,582 windows).
(G) Principal component analysis (PCA) plot of 300bp PRIME-filtered windows subset to SINEs (64,027/196,582 windows).
(H) Principal component analysis (PCA) plot of 300bp PRIME-filtered windows subset to LINEs (59,901/196,582 windows).
To better understand which differentially hypermethylated features are contributing to the segregation of Cluster A versus B, we subset our PRIME-filtered data to various features including protein coding genes (promoters, exons; Figure S11), non-protein coding genes (lncRNA, miRNA; Figure S11), and repeats regions (LINEs, SINEs, LTRs, etc.; Figures 4D and S11). Our goal was to identify specific methylome features that would account for the differences between Cluster A and B. For each feature, we correlated the number of windows associated with such feature with PC1 variance (Figure 4E). We subset the 196,582 PRIME-filtered windows to correspond specifically to promoters (11,702 PRIME-filtered windows), transcription factors (1,626 windows), microRNA (65 windows), lncRNA (80,236 windows), SINEs (64,027 windows), LINEs (59,901 windows), LTRs (25,917 windows), satellites (1,000 windows), clinically actionable SNVs (919 windows), exons (34,042 windows), and CTCF-sites (10,715 windows) (Figure S11). After subsetting for each of the respective features and performing clustering analysis, we observed that three features, lncRNA (Figure 4F), SINEs (Figure 4G), and LINEs (Figure 4H), have a PC1 variance of 58% (Figure 4E) identical to the original clustering (Figure 3A). This suggested that differential hypermethylation of lncRNAs, LINEs, and SINEs may explain the underlying biological difference between Cluster A and B. Our data suggest the possible role of hypermethylation of noncoding elements like lncRNAs and repeat regions like LINEs/SINEs in mediating the prognostic associations identified in Cluster A and B.
Discussion
Our study demonstrated the utility of the cfMeDIP-seq assay in studying the methylome of SCLC and demonstrated that plasma methylation is representative of tissue (Figure 1C) and is enriched in regions that are cancer specific (Figures 1D and 1E).23 We report one of the largest studies to comprehensively examine the DNA methylation of patients with SCLC by whole-genome analysis alongside a contemporaneously published study that demonstrates the value of liquid biopsy assays.21 In contrast to prior tissue-based studies,8,9 our genome-wide approach profiled methylated DNA loci beyond 450K/EPIC array probes and identified hypermethylated noncoding and repeat elements (e.g. LINE, SINE, lncRNA, etc.). In addition, our available clinical annotation with these treatment naive patient blood samples allowed us to correlate tumor methylation with clinical outcomes.
Previous liquid biopsy studies have observed PBL being major contributors to the cfDNA signal.24,25 In our data, we quantified this non-cancer signal using MethylCIBERSORT underscoring the need to filter the non-cancer contribution to plasma cfDNA. Currently, most liquid biopsy studies do not control for PBL cfDNA and this may affect the tumor specificity of the resultant methylome analysis and impact certain application goals. Using PRIME, a bespoke PBL algorithmic to refine SCLC-specific methylome signal using paired PBLs from the same patients’ blood sample, we greatly increased tumor specificity of our resultant cfMeDIP-seq data (Figure 2D). Using our PRIME approach, we opted to do our analysis on 196,582 windows. This approach is a trade-off as we sacrifice sensitivity of other interesting methylated regions; however, we gain specificity of calling and identifying the tumor methylome in plasma. Our MethylCIBERSORT analysis (Figures S2, S3, and S6) identified the presence of immune cell cfDNA in plasma. While some of these immune cells may constitute the tumor microenvironment, the vast majority of immune cell cfDNA is expected to be non-tumor related and it would be difficult to discriminate between these two immune cell populations in plasma. Therefore, to increase tumor specificity in our analysis, we opted to remove methylation from the immune cells by PRIME. Nonetheless, with the recent incorporation of immunotherapy into the SCLC treatment paradigm,26,27,28 the tumor-immune microenvironment is of great interest and future work with tumor tissue and immune infiltrate will be required to more directly interrogate the role of epigenetics in affecting immunotherapy response.
We identified two Clusters, A (better OS) and B (worse OS) by unsupervised machine learning with prognostic association suggests that methylation may be contributing to cancer progression and metastasis. These findings highlight the need to unravel SCLC biology that is mediated by epigenetic mechanisms. Moreover, DMR analysis of these clusters identified noncoding repeat features such as LINEs/SINEs and lncRNA and chromatin architecture binding sites not previously comprehensively characterized in the SCLC methylome. Methylation of these regions may implicate them in epigenetically mediating biological pathways in SCLC and may elucidate novel biologic insights worthy of future investigation. These findings are especially important as they suggest the potential difference in aggressiveness and patient outcomes in LS-SCLC and ES-SCLC may in fact be mediated by a combination of hypermethylation of noncoding and chromatin architecture elements.
Here, we show that cfMeDIP-seq allows interrogation of the SCLC methylome in a non-invasive, comprehensive manner.
For future applications, this liquid biopsy assay can be applied longitudinally to interrogate the SCLC methylome using pre-, on-, and post-treatment samples at a scale and accessibility not possible by invasive tissue collection methods. Therefore, in addition to defining methylome biology at diagnosis, cfMeDIP-seq can also be used to identify treatment-induced changes in SCLC that may be contributing to therapeutic resistance. By unraveling the biology of these potential epigenetic oncogenic and resistance mechanisms, we hope to continually improve the outcomes of our patients with SCLC.
Limitations of the study
A limitation of this current study is the lack of tumor fraction data for the patients with SCLC. However, SCLC has high disease burden and multiple liquid biopsy studies have reported 15%–87%11,12 of plasma cfDNA as being tumor derived. Due to our sample size, we limited our cluster and differential methylation analysis to discovery-based examination; future additional samples for a validation cohort would be necessary to confirm these findings. Valuable future directions would be to examine the tumor fraction content from this cohort of patients with SCLC and to validate these associations by functional experiments and independent cohorts.
STAR★Methods
Key resources table
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| All SCLC patients and non-cancer donors are detailed in Table S1. | Princess Margaret Cancer Centre, University Health Network (UHN) | N/A |
| Circulating tumour cell derived xenograft models | Princess Margaret Cancer Centre, University Health Network (UHN) | N/A |
| Critical commercial assays | ||
| QIAamp Circulating Nucleic Acid Kit | Qiagen | Cat. no. 55114 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat. no. Q32851 |
| λ DNA | Thermo Fisher Scientific | Cat. no. SD0011 |
| QIAquick PCR Purification Kit | Qiagen | Cat. no. 28106 |
| Kapa Hyper Prep Kit | Roche | Cat. no. 07962363001 |
| KAPA HiFi HotStart ReadyMix | Roche | Cat. no. 07958935001 |
| Agencourt AMPure XP beads | Beckman Coulter | Cat. no. A63881 |
| MagMeDIP Kit | Diagenode | Cat. no. C02010021; RRID: AB_442823 |
| DNA methylation control package | Diagenode | Cat. no. C02040012 |
| IPure Kit v2 | Diagenode | Cat. no. C03010015 |
| Sigma-Aldrich DNA Isolation Kit for Mammalian Blood | Roche | Cat. no. 11667327001 |
| Deposited data | ||
| cfMeDIP-seq data (SCLC patients, CDX models, non-cancer donors) | This paper | Zenodo: https://doi.org/10.5281/zenodo.7235989 |
| Software and algorithms | ||
| Original code used in study | This paper | CodeOcean: https://doi.org/10.24433/CO.7544854.v1 |
| R (version 3.6) | https://www.r-project.org/ | |
| RStudio | N/A | https://www.rstudio.com/ |
| R package MeDEStrand (version 0.0.0.9000) | Xu et al.29 | https://github.com/jxu1234/MeDEStrand |
| R package DESeq2 (version 1.30.1) | Love et al.30 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R package annotatr (1.16.0) | Cavalcante and Sartor31 | https://bioconductor.org/packages/release/bioc/html/annotatr.html |
| R package clusterprofiler (version 3.16.1) | Yu et al.32 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| R package ConsensusClusterPlus | Wilkerson and Hayes33 | https://bioconductor.org/packages/release/bioc/html/ConsensusClusterPlus.html |
| R package ggplot2 | https://ggplot2.tidyverse.org/ | |
| SAMtools (version 1.12) | Li et al.34 | https://github.com/samtools/samtools/releases |
| Burrows-Wheeler Alignment tool (BWA; version 0.7.17) | https://github.com/lh3/bwa | |
| Human genome (hg19) | Genome Reference Consortium Human Build36 | genome.ucsc.edu |
| TrimGalore! (version 0.6.5) | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Fastqc | https://github.com/s-andrews/FastQC | |
| UCSC RepeatMasker (version: 2021-09-03) | UCSC | https://genome.ucsc.edu/cgi-bin/hgTrackUi?g=rmsk |
Resource availability
Lead contact
Requests for additional information and resources should be directed to the lead contact, Benjamin H. Lok (Benjamin.Lok@rmp.uhn.ca).
Materials availability
This study did not generate unique reagents.
Experimental model and subject details
Patient recruitment & sample acquisition
All patients and donors provided written informed consent, and all samples were obtained upon approval of the institutional ethics committee and Research Ethics Board from the Princess Margaret Cancer Centre, University Health Network (UHN). SCLC patients and healthy donors were recruited, and peripheral blood was collected.
Method details
cfDNA extraction
Peripheral blood collected in EDTA tubes was first spun down at 4000 × g at 4 degrees Celsius for 10 min. Subsequently, the top plasma layer was transferred to 15mL Falcon tubes and spun again at 16000 × g at 4 degrees Celsius for 10 min. The supernatant was then transferred to 1.5mL Eppendorf tubes and stored at −80 degrees Celsius. For cfDNA extraction, approximately 3mL of the processed plasma was used with the QIAamp Circulating Nucleic Acid Kit (Qiagen, cat. no. 55114) as described in this protocol.35 Concentration of extracted cfDNA was quantified using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, cat. no. Q32851).
Peripheral blood leukocyte (PBL) DNA extraction
Peripheral blood collected in EDTA tubes was first spun down at 4000 × g at 4 degrees Celsius for 10 min. Subsequently, the buffy coat layer was transferred to a 1.5mL Eppendorf and stored at −80 degrees Celsius. Genomic DNA was extracted using the Sigma-Aldrich DNA Isolation Kit for Mammalian Blood (Roche, cat. no. 11667327001) and then sonicated to 150bp using the Covaris M220 Focused-ultrasonicator. Sonicated DNA was then size selected using Agencourt AMPure XP beads (Beckman Coulter, cat. no. A63881) using a 0.8× ratio.
CDX generation
Circulating tumour cells (CTCs) were extracted from one EDTA tube of peripheral blood. The blood was first incubated with 50ul RosetteSepTM (cat. no. 15705) per mL of patient blood for 20 min in a rotator at room temperature. After 20 min, blood was diluted with equal volume of wash buffer (WB; 10% HITES media in HBSS) and layered on top of 15 mL of Ficol-Plaque Plus in a 50 mL SepMateTM tube (cat. no. 85450), followed by centrifugation at 1200g for 10 min. Contents above plastic insert of SepMateTM tube were collected in fresh 50mL tube, 30 mL of WB was added, followed by another centrifugation at 300g for 10 min. The supernatant was discarded, and the pellet was resuspended in 3 mL of 1× StemCell Technologies' RBC lysis buffer (cat. no. 20120) and incubated for 10 min at room temperature. Subsequently, 10 mL of WB was added, followed by centrifugation at 300g for 10 min. Finally, the supernatant was removed, and the pellet was resuspended in 100ul of 50% of Matrigel (cat. no. 354230) in HITES media. CTCs in HITES/Matrigel mixture were injected subcutaneously into flank of 6 –16-week immunocompromised (NSG) mice.
CDX gDNA extraction
CDX tumour tissue gDNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen, cat. no. 69504). Up to 25mg of tumour tissue was used. The extracted gDNA was then sonicated and size-selected in an identical manner as described in the “peripheral blood leukocyte (PBL) DNA extraction” section.
cfMeDIP library preparation
cfMeDIP libraries were made using either extracted cfDNA or processed genomic DNA according to the protocol outlined by Shen/Burgener et al.35 For all samples, 10ng sample DNA as input and 90ng of methylated/unmethylated lambda filler DNA was used.
Next-generation sequencing
cfMeDIP-seq libraries were sequenced on an Illumina NovaSeq 6000 instrument (2 × 100 bp paired-end reads) according to the manufacturer’s recommendations using NovaSeq 6000 SP Reagent Kit v1.5. (Illumina, San Diego, CA, USA). All cfMeDIP-seq libraries were sequenced to 100 million reads.
Quantification and statistical analysis
Processing sequenced reads
Fastq files were processed as follows. First, terminal adaptor sequences were removed using TrimGalore! (version 0.6.5) and aligned to the reference human genome (hg19) using Burrows-Wheeler Alignment tool (BWA; version 0.7.17). Resulting SAM files were then indexed, and duplicate reads removed using SAMtools (version 1.12).
PRIME filter
For all PBL MeDIP libraries, sequenced reads were binned into 300bp windows spanning the entire human genome, excluding ENCODE blacklisted regions. For each 300bp window, beta-value was estimated using the R package MeDEStrand (version 0.0.0.9000). PBL windows with a median beta-value of less than 0.2 (across all PBL samples). Within these windows, CG density per window greater than or equal to five were selected to account for the functionality of the 5-methylcytosine antibody.18 These windows (n = 196,582) are referred to as PRIME-filtered windows.
Differentially hyper-methylated regions (DMR) analysis
DMR analysis was done using the R package DESeq2 (version #1.30.1). For any DMR analysis, samples of interest were divided up into two groups. Sequenced reads were binned into 300bp windows covering the entire human genome. For each window, a normalization factor was applied where read counts for each sample was divided by the mean read count of all samples (Normalization Factor = sample count/Mean of all samples). Bayesian statistical approaches were used to minimize within sample variation and bring extreme values close to the mean, per window. Subsequently, for each window, a general linear model was fitted using the negative binomial distribution.
Principal component analysis (PCA)
PCA was done using the built-in plotPCA() function of DESeq2 on counts data from processed samples. For whole-genome methylation profile analysis, 300bp windows corresponding to ENCODE blacklist regions were removed and the remaining windows (n = 8,828,974) were examined by PCA. For PRIME-filtered methylation profile analysis, counts data was subset to windows corresponding to PRIME-filtered windows (n = 196,582). Subsequently, counts data was transformed by variance stabilizing transformation to produce transformed data on the log2 scale and this was normalized with respect to library size. The transformed counts data were visualized by PCA using plotPCA().
Annotation of genomic regions
The R package annotatr (1.16.0) was used to annotate genomic regions (300bp windows). Specifically, annotatr was used to annotate CpG features, i.e. if a window falls in a CpG island/shelf/shore/open-sea region or gene features (i.e. promoter, 3’/5′ UTR, exon). ENSEMBL release-104 was used to annotate non-coding gene features such as lncRNA. UCSC RepeatMasker (version: 2021-09-03) was used to annotate repeats features such as LINEs, SINEs, LTRs, retrotransposons, and satellites features.
Consensus clustering
To identify methylation-defined biologically relevant subtypes of SCLC, consensus clustering was applied to sequenced read data in the following manner. For each sample, read data was subset to PRIME-filtered windows. The median absolute deviation was calculated for each window, and the top 5000 most deviant windows were selected. Consensus clustering was performed on these top 5000 windows. Consensus clustering was done for k values of 2 to 20, with 1000 resamplings.
Permutation analysis
To calculate significance of CpG feature enrichment, permutation analysis was done. First, the human genome was segmented into 300bp windows spanning the entire genome (except sex chromosomes). 51,666 windows were randomly sampled (because this is the number of hypermethylated DMRs observed in SCLC) and the occurrence of CpG feature (i.e. CpG island) was calculated. This was repeated 1000 times. Subsequently, the 1000× calculated frequencies were converted into Z-scores and a null distribution was determined. The observed CpG feature frequency was converted into a Z-score and statistical analysis was done.
KEGG pathway analysis
The R package clusterprofiler (version 3.16.1) was used to perform pathway analysis on methylated regions. Gene symbols corresponding to significantly hypermethylated 300bp windows were determined using annotatr. Subsequently, clusterprofiler function enrichKEGG() was run on the gene symbols list.
Patient clinical/demographics and Kaplan-Meier survival analysis
The clinical-demographic features were summarized descriptively, using median and IQR and for continuous variables, and counts and percentages for ordinal/categorical variables. p-values were obtained via fisher's exact test for categorical/ordinal variables and Kruskal Wallis H test for continuous variables.
Overall survival (OS) was defined as the time from small-cell lung cancer diagnosis until death from any cause or censored at last follow-up. Kaplan-Meier curves and log-rank tests were used to visualize survival differences between groups. The association between cluster and overall survival stratified by VA stage at diagnosis was explored using Cox proportional-hazard model. Hazard ratios (HRs) were reported with 95% confidence intervals. The proportionality assumption was met via assessing Schoenfeld residuals against time. Statistical analyses were performed using R software (version 3.6).
Acknowledgments
This study was supported by funding from the Ontario Molecular Pathology Research Network of the Ontario Institute for Cancer Research. Research in the B.H. Lok laboratory is supported by the Canada Foundation for Innovation, Cancer Research Society, Canadian Institutes of Health Research, National Institute of Health/National Cancer Institute (U01CA253383), Clinical and Translational Science Center at Weill Cornell Medical Center, MSKCC (UL1TR00457). S.U.H. is supported by the Institute of Medical Science, Canadian Institutes of Health Research, and The Strategic Training in Transdisciplinary Radiation Science for the 21st Century (STARS21). S.V.B. is supported by the Gattuso-Slaight Personalized Cancer Medicine Fund at the Princess Margaret Cancer Centre and the Dr. Mariano Elia Chair in Head & Neck Cancer Research at University Health Network and University of Toronto.
Author contributions
S.U.H.: Conceptualization, Methodology, Software, Investigation, Formal analysis, Data Curation, Writing - Original Draft, Writing - Review & Editing, Visualization. S.S.: Data Curation, Writing – Review & Editing. M.K.A.: Resources, Methodology, Writing - Review & Editing. K.H.: Data Curation. L.J.Z.: Data Curation, Formal analysis, Visualization. D.S.: Data Curation, Writing - Review & Editing. J.J.N.L.: Data Curation. N.M.: Data Curation. D.P.: Project administration. D.C.: Project administration. V.P.: Project administration, Writing – Original Draft, Writing – Review & Editing. M.S.T.: Conceptualization, Funding acquisition, Resources, Supervision, Writing - Review & Editing. M.C.: Conceptualization, Funding acquisition, Resources, Supervision, Writing - Review & Editing. D.d.C.: Conceptualization, Funding acquisition, Resources, Supervision, Writing - Review & Editing. G.L.: Conceptualization, Funding acquisition, Resources, Supervision, Writing – Review & Editing. S.V.B.: Conceptualization, Funding acquisition, Resources, Supervision, Writing - Review & Editing. B.H.L.: Conceptualization, Funding acquisition, Resources, Project administration, Supervision, Writing - Original Draft, Writing - Review & Editing.
Declaration of interests
S.S. reports grants from AstraZeneca, BMS, Janssen, Von Tobel foundation, Fill the Gap, and has served on all institutional advisory board for BMS, AstraZeneca, MSD. N.M. reports personal fees from Takeda Oncology, Pfizer, and Novartis, outside of the submitted work. M.-S.T. reports research grant from Bayer and AstraZeneca, outside the submitted work, and personal fees from AstraZeneca, Amgen, BMS, Daiichi-Sankyo and Lilly, outside the submitted work. D. de Carvalho received research funds from Pfizer and is co-founder and shareholder of Adela Inc. S.V.B. is inventor on patents related to cell-free DNA mutation and methylation analysis technologies that have been licensed to Roche and Adela, respectively, and is co-founder of, has ownership in, and serves in a leadership role at Adela. G.L. reports grants and personal fees from AstraZeneca and Takeda; grants from Boehringer Ingelheim; and personal fees from Hoffman La Roche, Merck, Bristol Myers Squibb, and Pfizer outside the submitted work. B.H.L. reports grants from Pfizer; and grants, personal fees, and non-financial support from AstraZeneca outside the submitted work.
Inclusion and diversity
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure that the study questionnaires were prepared in an inclusive way. We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105487.
Supplemental information
Data and code availability
-
•
All original code has been deposited at Code Ocean and is publicly available as of the date of publication. DOI is listed in the key resources table.
-
•
Deidentified patient and CDX methylation cfMeDIP-seq counts data have been deposited at Zenodo and are publicly available as of the date of publication. DOI is listed in the key resources table.
References
- 1.Rudin C.M., Brambilla E., Faivre-Finn C., Sage J. Small-cell lung cancer. Nat. Rev. Dis. Primers. 2021;7:3. doi: 10.1038/s41572-020-00235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rudin C.M., Durinck S., Stawiski E.W., Poirier J.T., Modrusan Z., Shames D.S., Bergbower E.A., Guan Y., Shin J., Guillory J., et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peifer M., Fernández-Cuesta L., Sos M.L., George J., Seidel D., Kasper L.H., Plenker D., Leenders F., Sun R., Zander T., et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.George J., Lim J.S., Jang S.J., Cun Y., Ozretić L., Kong G., Leenders F., Lu X., Fernández-Cuesta L., Bosco G., et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53. doi: 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagner A.H., Devarakonda S., Skidmore Z.L., Krysiak K., Ramu A., Trani L., Kunisaki J., Masood A., Waqar S.N., Spies N.C., et al. Recurrent WNT pathway alterations are frequent in relapsed small cell lung cancer. Nat. Commun. 2018;9:3787. doi: 10.1038/s41467-018-06162-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dowlati A., Lipka M.B., McColl K., Dabir S., Behtaj M., Kresak A., Miron A., Yang M., Sharma N., Fu P., Wildey G. Clinical correlation of extensive-stage small-cell lung cancer genomics. Ann. Oncol. 2016;27:642–647. doi: 10.1093/annonc/mdw005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tlemsani C., Takahashi N., Pongor L., Rajapakse V.N., Tyagi M., Wen X., Fasaye G.-A., Schmidt K.T., Desai P., Kim C., et al. Whole-exome sequencing reveals germline-mutated small cell lung cancer subtype with favorable response to DNA repair-targeted therapies. Sci. Transl. Med. 2021;13:eabc7488. doi: 10.1126/scitranslmed.abc7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poirier J.T., Gardner E.E., Connis N., Moreira A.L., de Stanchina E., Hann C.L., Rudin C.M. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene. 2015;34:5869–5878. doi: 10.1038/onc.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalari S., Jung M., Kernstine K.H., Takahashi T., Pfeifer G.P. The DNA methylation landscape of small cell lung cancer suggests a differentiation defect of neuroendocrine cells. Oncogene. 2013;32:3559–3568. doi: 10.1038/onc.2012.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kulis M., Esteller M. DNA methylation and cancer. Adv. Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 11.Almodovar K., Iams W.T., Meador C.B., Zhao Z., York S., Horn L., Yan Y., Hernandez J., Chen H., Shyr Y., et al. Longitudinal cell-free DNA analysis in patients with small cell lung cancer reveals dynamic insights into treatment efficacy and disease relapse. J. Thorac. Oncol. 2018;13:112–123. doi: 10.1016/j.jtho.2017.09.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devarakonda S., Sankararaman S., Herzog B.H., Gold K.A., Waqar S.N., Ward J.P., Raymond V.M., Lanman R.B., Chaudhuri A.A., Owonikoko T.K., et al. Circulating tumor DNA profiling in small-cell lung cancer identifies potentially targetable alterations. Clin. Cancer Res. 2019;25:6119–6126. doi: 10.1158/1078-0432.CCR-19-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iams W.T., Kopparapu P.R., Yan Y., Muterspaugh A., Zhao Z., Chen H., Cann C., York S., Horn L., Ancell K., et al. Blood-based surveillance monitoring of circulating tumor DNA from patients with SCLC detects disease relapse and predicts death in patients with limited-stage disease. JTO Clin. Res. Rep. 2020;1 doi: 10.1016/j.jtocrr.2020.100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen S.Y., Singhania R., Fehringer G., Chakravarthy A., Roehrl M.H.A., Chadwick D., Zuzarte P.C., Borgida A., Wang T.T., Li T., et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563:579–583. doi: 10.1038/s41586-018-0703-0. [DOI] [PubMed] [Google Scholar]
- 15.Nuzzo P.V., Berchuck J.E., Korthauer K., Spisak S., Nassar A.H., Abou Alaiwi S., Chakravarthy A., Shen S.Y., Bakouny Z., Boccardo F., et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat. Med. 2020;26:1041–1043. doi: 10.1038/s41591-020-0933-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lasseter K., Nassar A.H., Hamieh L., Berchuck J.E., Nuzzo P.V., Korthauer K., Shinagare A.B., Ogorek B., McKay R., Thorner A.R., et al. Plasma cell-free DNA variant analysis compared with methylated DNA analysis in renal cell carcinoma. Genet. Med. 2020;22:1366–1373. doi: 10.1038/s41436-020-0801-x. [DOI] [PubMed] [Google Scholar]
- 17.Nassiri F., Chakravarthy A., Feng S., Shen S.Y., Nejad R., Zuccato J.A., Voisin M.R., Patil V., Horbinski C., Aldape K., et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat. Med. 2020;26:1044–1047. doi: 10.1038/s41591-020-0932-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burgener J.M., Zou J., Zhao Z., Zheng Y., Shen S.Y., Huang S.H., Keshavarzi S., Xu W., Liu F.-F., Liu G., et al. Tumor-naïve multimodal profiling of circulating tumor DNA in head and neck squamous cell carcinoma. Clin. Cancer Res. 2021;27:4230–4244. doi: 10.1158/1078-0432.CCR-21-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peter M.R., Bilenky M., Isserlin R., Bader G.D., Shen S.Y., De Carvalho D.D., Hansen A.R., Hu P., Fleshner N.E., Joshua A.M., et al. Dynamics of the cell-free DNA methylome of metastatic prostate cancer during androgen-targeting treatment. Epigenomics. 2020;12:1317–1332. doi: 10.2217/epi-2020-0173. [DOI] [PubMed] [Google Scholar]
- 20.Soulakova J.N., Crockett L.J. Consistency and recanting of ever-smoking status reported by self and proxy respondents one year apart. J. Addict. Behav. Ther. Rehabil. 2014;3 doi: 10.4172/2324-9005.1000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chemi F., Pearce S.P., Clipson A., Hill S.M., Conway A.-M., Richardson S.A., Kamieniecka K., Caeser R., White D.J., Mohan S., et al. cfDNA methylome profiling for detection and subtyping of small cell lung cancers. Nat. Cancer. 2022;3:1260–1270. doi: 10.1038/s43018-022-00415-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vickers A.J., Frese K., Galvin M., Carter M., Franklin L., Morris K., Pierce J., Descamps T., Blackhall F., Dive C., Carter L. Brief report on the clinical characteristics of patients whose samples generate small cell lung cancer circulating tumour cell derived explants. Lung Cancer. 2020;150:216–220. doi: 10.1016/j.lungcan.2020.11.002. [DOI] [PubMed] [Google Scholar]
- 23.Yates J., Boeva V. Deciphering the etiology and role in oncogenic transformation of the CpG island methylator phenotype: a pan-cancer analysis. Brief. Bioinform. 2022;23:bbab610. doi: 10.1093/bib/bbab610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Croitoru V.M., Cazacu I.M., Popescu I., Paul D., Dima S.O., Croitoru A.E., Tanase A.D. Clonal hematopoiesis and liquid biopsy in gastrointestinal cancers. Front. Med. 2021;8 doi: 10.3389/fmed.2021.772166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan H.T., Nagayama S., Chin Y.M., Otaki M., Hayashi R., Kiyotani K., Fukunaga Y., Ueno M., Nakamura Y., Low S.-K. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol. Oncol. 2020;14:1719–1730. doi: 10.1002/1878-0261.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudin C.M., Awad M.M., Navarro A., Gottfried M., Peters S., Csőszi T., Cheema P.K., Rodriguez-Abreu D., Wollner M., Yang J.C.-H., et al. Pembrolizumab or placebo plus etoposide and platinum as first-line therapy for extensive-stage small-cell lung cancer: randomized, double-blind, phase III KEYNOTE-604 study. J. Clin. Oncol. 2020;38:2369–2379. doi: 10.1200/JCO.20.00793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horn L., Mansfield A.S., Szczęsna A., Havel L., Krzakowski M., Hochmair M.J., Huemer F., Losonczy G., Johnson M.L., Nishio M., et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N. Engl. J. Med. 2018;379:2220–2229. doi: 10.1056/NEJMoa1809064. [DOI] [PubMed] [Google Scholar]
- 28.Paz-Ares L., Dvorkin M., Chen Y., Reinmuth N., Hotta K., Trukhin D., Statsenko G., Hochmair M.J., Özgüroğlu M., Ji J.H., et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet. 2019;394:1929–1939. doi: 10.1016/S0140-6736(19)32222-6. [DOI] [PubMed] [Google Scholar]
- 29.Xu J., Liu S., Yin P., Bulun S., Dai Y. MeDEStrand: an improved method to infer genome-wide absolute methylation levels from DNA enrichment data. BMC Bioinf. 2018;19:540. doi: 10.1186/s12859-018-2574-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cavalcante R.G., Sartor M.A. annotatr: genomic regions in context. Bioinformatics. 2017;33:2381–2383. doi: 10.1093/bioinformatics/btx183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilkerson M.D., Hayes D.N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup Genome Project data processing subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen S.Y., Burgener J.M., Bratman S.V., De Carvalho D.D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 2019;14:2749–2780. doi: 10.1038/s41596-019-0202-2. [DOI] [PubMed] [Google Scholar]
- 36.Church D.M., Schneider V.A., Graves T., Auger K., Cunningham F., Bouk N., Chen H.-C., Agarwala R., McLaren W.M., Ritchie G.R.S., et al. Modernizing reference genome assemblies. PLoS Biol. 2011 doi: 10.1371/journal.pbio.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All original code has been deposited at Code Ocean and is publicly available as of the date of publication. DOI is listed in the key resources table.
-
•
Deidentified patient and CDX methylation cfMeDIP-seq counts data have been deposited at Zenodo and are publicly available as of the date of publication. DOI is listed in the key resources table.