

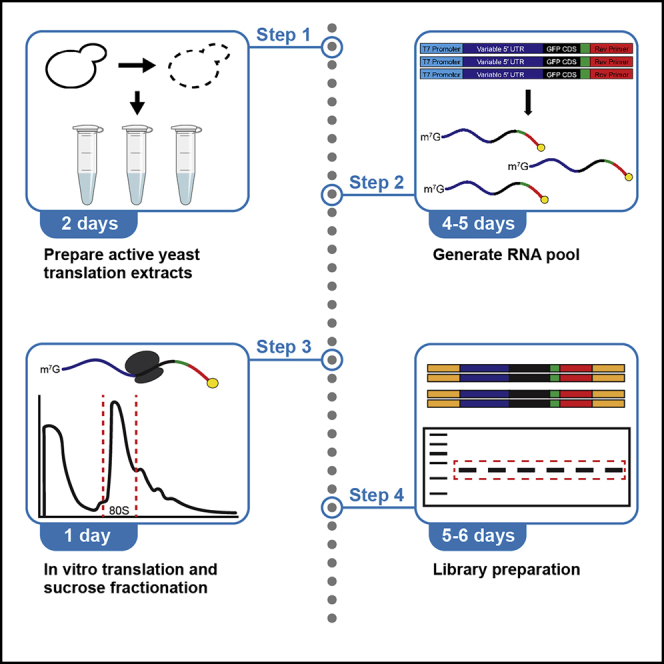
Summary
Direct analysis of ribosome targeting (DART) allows investigators to measure the translation initiation potential of thousands of RNAs in parallel. Here, we describe an optimized protocol for generating active translation extract from S. cerevisiae, followed by in vitro translation, purification of ribosome-bound RNAs, and subsequent library preparation and sequencing. This protocol can be applied to a variety of cell types and will enable high-throughput interrogation of translational determinants.
For complete details on the use and execution of this protocol, please refer to Niederer et al. (2022).1
Subject areas: Sequencing, Molecular Biology, Gene Expression, Systems biology
Graphical abstract
Highlights
-
•
Preparation of translation-competent yeast extracts
-
•
Generation of a methylguanosine-capped and biotinylated RNA pool
-
•
In vitro translation and sucrose fractionation to isolate ribosome-bound RNAs
-
•
Generation of next-generation sequencing libraries to quantify ribosome recruitment
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Direct analysis of ribosome targeting (DART) allows investigators to measure the translation initiation potential of thousands of RNAs in parallel. Here, we describe an optimized protocol for generating active translation extract from S. cerevisiae, followed by in vitro translation, purification of ribosome-bound RNAs, and subsequent library preparation and sequencing. This protocol can be applied to a variety of cell types and will enable high-throughput interrogation of translational determinants.
Before you begin
Preparation of yeast translation extracts
Timing: 2 days
-
1.
Inoculate a starter culture by scooping a single yeast colony from a streak plate into 6 mL of PAD media. Incubate at 30°C on a shaker for 12–14 h.
-
2.Dilute the starter culture 1:20 and measure absorbance at 600 nm (OD600).
-
a.Calculate the required volume of starter culture to inoculate 2 750 mL yeast cultures to achieve a final OD600 of 12–15 in the desired length of time given the wild-type doubling time of approximately 90 min.
-
a.
Note: We generally inoculate 750 mL cultures the night before lysate preparation, targeting the desired OD600 of 12–15 the following morning (approximately 12 h of growth).
CRITICAL: Do not dilute the yeast below an OD600 of 0.004 as the yeast will not recover consistently.
Note: The OD600 values described here should be taken only as general guidelines. Cells should be harvested in late exponential growth phase to yield active lysate. OD600 measurements can vary significantly between instruments and both the doubling time and OD600 at which exponential growth occurs differs between strains. We recommend performing growth curve measurements to determine this empirically before proceeding to lysate preparation.
-
3.Inoculate two 2 L flasks containing 750 mL of YPAD media with the calculated volume of starter culture.
-
a.Incubate at 30°C on a shaker until the absorbance at OD600 reaches approximately 12–15.
-
a.
-
4.
Transfer the cultures to 1 L centrifuge tubes and pellet yeast at 9,000 × g for 5 min at 4°C.
-
5.Discard the supernatant from pelleted yeast and resuspend both pellets in 15 mL of Buffer A + M.
-
a.Weigh an empty 50 mL conical tube (with cap) and record.
-
b.Transfer the yeast suspension to the single weighed 50 mL tube.
-
a.
-
6.
Spin down the yeast at 2,600 × g for 5 min at 4°C.
-
7.
Wash the cells twice more by resuspending in 15 mL of pre-chilled Buffer A + M, then pelleting the yeast by centrifuging at 2,600 × g for 5 min at 4°C.
Note: Remove the supernatant after each wash.
Pause point: The washed cell pellets may be flash frozen in liquid nitrogen and stored at −80°C.
-
8.
Weigh the 50 mL tube containing the yeast pellet and subtract the weight of the empty tube to determine the mass of the cell pellet.
-
9.Transfer a volume of Buffer A + M equal to at least 1.5 times the pellet mass, rounding up to the nearest 10 mL (i.e., 20 mL for a 10 g pellet) to a new 50 mL conical tube.
-
a.Add 1 cOmplete Mini protease inhibitor tablet per 10 mL of Buffer A + M and dissolve.
-
a.
-
10.
Resuspend the cell pellet in a volume 1.5 times the pellet mass (i.e., 15 mL for a 10 g pellet) of Buffer A + M.
-
11.
Add a mass of 0.5 mm zirconia/silica beads, pre-chilled to −20°C, equal to 5 times the mass of the cell pellet to the 50 mL tube.
-
12.Lyse the cells by vortexing for 1 min at maximum speed.
-
a.Repeat this 5 times (for a total of 6 rounds) with 1 min of cooling on ice between vortexing to avoid heating of the solution.
-
a.
-
13.
Pellet the cell debris by centrifuging at 2,600 × g for 5 min at 4°C. Transfer the supernatant to 50 mL Oak Ridge tubes.
-
14.
Place the Oak Ridge tubes in an SS-34 or other compatible rotor and clarify by centrifuging at 26,700 × g for 35 min at 4°C.
-
15.
Transfer the supernatant to new Oak Ridge tubes and centrifuge again at 26,700 × g for 12 min at 4°C.
-
16.
While samples are centrifuging, hydrate a dialysis cassette in a beaker containing 2 L of Buffer A for 2 min at 4°C.
-
17.
Carefully remove the supernatant, avoiding the bottom residual pellet and top lipid layer, and transfer it to a new 15 mL tube on ice.
-
18.
Transfer the clarified extract into a 20 mL syringe.
-
19.
Affix an 18-gauge needle and inject the extract into the hydrated dialysis cassette, carefully avoiding puncturing the cassette membrane.
-
20.Place the cassette into the beaker containing 2 L of Buffer A and dialyze for 45 min at 4°C.
-
a.Replace the buffer with 2 L of fresh Buffer A and continue dialysis for another 45 min at 4°C.
-
a.
-
21.
Collect the dialyzed extract from the cassette and aliquot into pre-chilled 1.5 mL tubes. Flash freeze in liquid nitrogen and store at −80°C.
Preparation of reactivated creatine phosphokinase solution
-
22.
Prepare the creatine phosphokinase resuspension buffer as follows:
| Reagent | Final concentration | Amount |
|---|---|---|
| HEPES KOH pH 7.6 (1 M) | 20 mM | 100 μL |
| DTT (1 M) | 20 mM | 100 μL |
| ddH2O | N/A | 9.8 mL |
| Total | 10 mL |
-
23.
Resuspend 200 mg of creatine phosphokinase powder in 10 mL of creatine phosphokinase resuspension buffer (20 mg / mL creatine phosphokinase).
-
24.
Incubate the solution at 26°C for 30 min to reactivate the creatine phosphokinase.
-
25.
Add 10 mL of 100% glycerol and mix well. The final 10 mg / mL creatine phosphokinase solution can be aliquoted and stored at −20°C for 6 months.
Pool design
Design a pool of DNA oligonucleotide sequences corresponding to the 5′ UTR RNA sequences of interest. The pool used in our original1 study consisted of up to 122 nucleotides of 5′ UTR sequence and at least 24 nucleotides of coding sequence to cover the binding footprint of initiating ribosomes.2 Append the T7 promoter sequence to the 5′ end and a common primer sequence (key resources table) to the 3′ end of the oligos. These sequences provide common handles for PCR amplification, in vitro transcription, and reverse transcription of the oligo pool. A 10-nucleotide unique barcode may also be included at the 3′ end of the oligo prior to the common primer sequence to facilitate assignment of highly similar sequences. A schematic illustrating the oligo design is shown in Figure 1. Note that it is essential to remove any upstream start codons located upstream of the desired initiation codon. This is required to ensure that each RNA molecule can only recruit a single ribosome and to unambiguously assign a ribosome recruitment score to a given initiation context. In our original study,1 upstream ATGs were changed to AGT.
Note: The oligonucleotide pool used in our original study1 contained 12,000 unique 5′ UTR sequences, and we have since performed DART analysis successfully on pools consisting of 24,000 unique sequences.
Figure 1.

Schematic of designed DNA pool oligos
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Cycloheximide | Millipore Sigma | 01810 |
| Phenylmethanesulfonyl fluoride (PMSF) solution 0.1 M | Millipore Sigma | 93482 |
| RNasin Plus Ribonuclease Inhibitor | Promega | N2615 |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530L |
| GlycoBlue Coprecipitant (15 mg/mL) | Thermo Fisher | AM9516 |
| TURBO DNase (2 U/μL) | Thermo Fisher | AM2239 |
| Phenol:chloroform:IAA 25:24:1 pH 6.6 | Thermo Fisher | AM9732 |
| Micrococcal nuclease | NEB | M0247S |
| cOmplete Mini Protease Inhibitor Cocktail Tablets | Millipore Sigma | 11836153001 |
| Creatine phosphokinase | Millipore Sigma | C3755 |
| 100 bp DNA ladder | NEB | N3231L |
| ssDNA ladder (6 Ultramer oligos from 100 to 200 nucleotides in 20 nucleotide steps, combined) | IDT | N/A |
| SYBR Gold (10,000× concentrate in DMSO) | Thermo Fisher | S11494 |
| T4 RNA Ligase 1, high concentration | NEB | M0437M |
| Buffer RLT | Qiagen | 79216 |
| SuperScript III Reverse Transcriptase | Thermo Fisher | 18080-044 |
| Water, nuclease-free | AmericanBio | AB02123-00500 |
| Ethanol 200 Proof | Decon Laboratories | 2701 |
| Bacto Peptone | Thermo Fisher | 211677 |
| Adenine hemisulfate salt | Sigma-Aldrich | A9126 |
| D-(+)-Glucose | Sigma-Aldrich | G8270 |
| HEPES | Sigma-Aldrich | H3375 |
| Potassium acetate | Sigma-Aldrich | 236497 |
| Magnesium acetate | Sigma-Aldrich | M0631 |
| D-Mannitol | Sigma-Aldrich | M9647 |
| Glycerol, anhydrous | J.T. Baker | 2136-03 |
| Bromophenol blue sodium salt | Sigma-Aldrich | B5525 |
| Xylene cyanol FF | Sigma-Aldrich | 335940 |
| Tris hydrochloride | AmericanBio | AB02005-0500 |
| Formamide DI (deionized) | AmericanBio | AB00600 |
| Ethylenediaminetetraacetic acid | Sigma-Aldrich | EDS-1KG |
| Guanidine hydrochloride | Sigma-Aldrich | G3272 |
| MES hydrate | Sigma-Aldrich | M2933 |
| Yeast extract | Thermo Fisher | 212750 |
| Sucrose | Sigma-Aldrich | S9378 |
| Triton X-100 | Sigma-Aldrich | T9284 |
| DL-dithiothreitol | Sigma-Aldrich | D9779 |
| 2-Propanol | J.T. Baker | 9084-05 |
| Phenol solution | Millipore Sigma | P4557 |
| Critical commercial assays | ||
| Vaccinia Capping System | NEB | M2080S |
| Pierce RNA 3′ End Biotinylation Kit | Thermo Fisher | 20160 |
| High Sensitivity DNA Kit | Agilent | 5067-4626 |
| Oligonucleotides | ||
| Primer: T7 forward: GCTAATACGACTCACTATAGGG |
Niederer et al.1 | N/A |
| Primer: RT handle reverse: CCTTGGCACCCGAGAATTCCA |
Niederer et al.1 | N/A |
| Primer: RT primer: GCCTTGGCACCCGAGAATTCC |
Niederer et al.1 | N/A |
| 5′ adaptor: /5Phos/NNNNNNNNNN GATCGTCGGACTGTAGAACTCTGA ACGTG/3SpC3/ |
Niederer et al.1 | N/A |
| Primer: RP1 reverse: AATGATACG GCGACCACCGAGATCTACACGT TCAGAGTTCTACAGTCCGA |
Niederer et al.1 | N/A |
| Primer: barcoded forward: CAAGCAGAAGACGGCATAC GAGATNNNNNNGTGACTGG AGTTCCTTGGCACCCGAGAATTCCA |
Niederer et al.1 | N/A |
| Software and algorithms | ||
| STAR | Dobin et al., 20133 | https://github.com/alexdobin/STAR |
| BBTools Suite | Bushnell B. - sourceforge.net/projects/bbmap/ | https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/bbmap-guide/ |
| Other | ||
| Seton Open-Top Polyclear Centrifuge Tubes 1 × 3.5 in. | Seton Scientific | 7052 |
| Zymo-Spin V Columns | Zymo Research | C1012 |
| Slide-A-Lyzer Dialysis Cassettes, 3.5K MWCO | Thermo Fisher | 66330 |
| MaXtract High Density 15 mL Tubes | Qiagen | 129065 |
| MaXtract High Density 1.5 mL Tubes | Qiagen | 129046 |
| Gradient Master Base Unit | Biocomp | 108 |
| 1″ Magnabase Holder for SW28/32 Rotors | Biocomp | 105-925-I |
| 10 mm Isopycnic (Long) Caps (SW28/32) | Biocomp | 105-525-6 |
| Marker Block for SW28/32 | Biocomp | 105-625 |
| Hydrophilic streptavidin magnetic beads | NEB | S1421S |
| Dynabeads MyOne Silane | Thermo Fisher | 37002D |
| AMPure XP Reagent | Beckman Coulter | A63881 |
| DNA LoBind 1.5 mL Tubes | Eppendorf | 022431021 |
| 0.5 mm dia. zirconia/silica beads | Biospec Products | 11079105z |
| Oak Ridge Centrifuge Tube, PC | Thermo Scientific | 3118-0050 |
| Qubit 4 Fluorometer | Thermo Fisher | Q33238 |
| 1 L (1,000 mL) Polypropylene Bottle 95 × 191 mm | Beckman Coulter | A98814 |
| Avanti J-26 XPI Centrifuge | Beckman Coulter | 393127 |
| J-Lite JLA-8.1000 Fixed-Angle Rotor | Beckman Coulter | 363688 |
| Falcon 50 mL Conical Centrifuge Tubes | Corning | 352070 |
| Sorvall Legend RT+ Centrifuge | Thermo Fisher | 75004377 |
| Sorvall Swing Bucket Rotor | Thermo Fisher | 75006445 |
| Vortex mixer | VWR | 58816-121 |
| SS-34 Fixed Angle Rotor | Thermo Fisher | 28020TS |
| Sorvall RC 6 Plus Centrifuge | Thermo Fisher | 49610 |
| Oligo Pool (custom) | Twist Bioscience | N/A |
| NanoDrop One Microvolume UV-Vis Spectrophotometer | Thermo Fisher | ND-ONE-W |
| Benchrocker 2D Variable Speed Rocker | Benchmark Scientific | BR2000 |
| Compact transilluminator | Clare Chemical Research | DR-46B |
| Single-edge razor blades | Personna | 94-120-2 |
| Analog Rotisserie Tube Rotator | Scilogex | 824220019999 |
| SW 28 Swinging-Bucket Rotor | Beckman Coulter | 342207 |
| SW 28 Buckets | Beckman Coulter | 342217 |
| Optima L-90K Ultracentrifuge | Beckman Coulter | 365672 |
| Precision General Purpose Bath | Thermo Fisher | TSGP10 |
| Thermomixer C | Eppendorf | 5382000023 |
| 2100 Bioanalyzer | Agilent | G2939BA |
Materials and equipment
YPAD
| Reagent | Final concentration | Amount |
|---|---|---|
| Yeast extract | 1% | 7.5 g |
| Peptone | 2% | 15 g |
| Adenine hemisulfate | 0.01% | 75 mg |
| Glucose (40%) | 2% | 37.5 mL |
| ddH2O | N/A | 690.5 mL |
| Total | 750 mL |
Autoclave to sterilize YPAD medium.
Storage conditions: Store at 4°C for 6 months.
Buffer A + M
| Reagent | Final concentration | Amount |
|---|---|---|
| HEPES KOH pH 7.4 (1 M) | 30 mM | 3 mL |
| Potassium acetate (2 M) | 100 mM | 5 mL |
| Magnesium acetate (1 M) | 2 mM | 200 μL |
| DTT (1 M) | 2 mM | 200 μL |
| PMSF (0.1 M) | 0.1 mM | 100 μL |
| Mannitol (17.5%) | 8.5% | 48.5 mL |
| ddH2O | N/A | 43 mL |
| Total | 100 mL |
Add DTT and PMSF immediately before use.
Storage conditions: Buffer without DTT and PMSF may be stored at 4°C for 6 months.
Buffer A
| Reagent | Final concentration | Amount |
|---|---|---|
| HEPES KOH pH 7.4 (1 M) | 30 mM | 60 mL |
| Potassium acetate (2 M) | 100 mM | 100 mL |
| Magnesium acetate (1 M) | 2 mM | 4 mL |
| DTT (1 M) | 2 mM | 4 mL |
| PMSF (0.1 M) | 0.1 mM | 2 mL |
| ddH2O | N/A | 1,830 mL |
| Total | 2 L |
Add DTT and PMSF immediately before use.
Storage conditions: Buffer without DTT and PMSF may be stored at 4°C for 6 months.
DNA loading dye (6×)
| Reagent | Final concentration | Amount |
|---|---|---|
| Glycerol | 30% | 3 mL |
| Bromophenol blue | 0.025% | 2.5 mg |
| Xylene cyanol FF | 0.025% | 2.5 mg |
| ddH2O | N/A | 7 mL |
| Total | 10 mL |
Storage conditions: Store at −20°C indefinitely.
DNA elution buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl (5 M) | 300 mM | 6 mL |
| Tris-HCl pH 8.0 (1 M) | 10 mM | 1 mL |
| ddH2O | N/A | 93 mL |
| Total | 100 mL |
Storage conditions: Store at 25°C for 1 year.
Formamide buffer (2×)
| Reagent | Final concentration | Amount |
|---|---|---|
| Formamide | 95% | 9.5 mL |
| EDTA pH 8.0 (0.5 M) | 5 mM | 100 μL |
| SDS | 0.025% | 2.5 mg |
| Bromophenol blue | 0.025% | 2.5 mg |
| Xylene cyanol FF | 0.025% | 2.5 mg |
| ddH2O | N/A | 400 μL |
| Total | 10 mL |
Storage conditions: Store at −20°C for 6 months.
RNA elution buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Sodium acetate pH 5.3 (3 M) | 300 mM | 1 mL |
| EDTA pH 8.0 (0.5 M) | 1 mM | 20 μL |
| RNasin Plus (40 U / μL) | 100 U / mL | 25 μL |
| ddH2O | N/A | 8.55 mL |
| Total | 10 mL |
Note: Add RNasin Plus immediately before use.
Storage conditions: Buffer without RNasin Plus added may be stored at 25°C for 1 year.
Binding buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Guanidine hydrochloride | 8 M | 38.21 g |
| EDTA pH 8.0 (0.5 M) | 20 mM | 2 mL |
| MES hydrate | 20 mM | 213.25 mg |
| ddH2O | N/A | to 50 mL |
| Total | 50 mL |
Storage conditions: Store covered in foil at 25°C for 6 months.
Translation mix (6×)
| Reagent | Final concentration | Amount |
|---|---|---|
| HEPES KOH pH 7.4 (1 M) | 132 mM | 1,320 μL |
| Potassium glutamate (2.5 M) | 720 mM | 2,880 μL |
| Magnesium glutamate (1 M) | 6 mM | 60 μL |
| ATP (100 mM) | 4.5 mM | 450 μL |
| GTP (100 mM) | 0.6 mM | 60 μL |
| Creatine phosphate | 150 mM | 382.62 mg |
| DTT (1 M) | 10.2 mM | 102 μL |
| ddH2O | N/A | 4,745 μL |
| Total | 10 mL |
Storage conditions: Separate into 200 μL aliquots and store at −20°C for 6 months.
Cycloheximide (10 mg / mL) solution
-
•
Dissolve 50 mg of cycloheximide powder in 5 mL of nuclease-free water.
Storage conditions: Prepare fresh before use.
cOmplete mini solution
-
•
Dissolve one cOmplete mini protease inhibitor tablet in 2 mL of nuclease-free water.
Storage conditions: Prepare fresh before use.
Streptavidin binding buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl (5 M) | 0.5 M | 10 mL |
| Tris-HCl pH 7.5 (1 M) | 20 mM | 2 mL |
| EDTA pH 8.0 (0.5 M) | 1 mM | 200 μL |
| ddH2O | N/A | 87.8 mL |
| Total | 100 mL |
Storage conditions: Store at 25°C for 1 year.
Streptavidin wash buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl pH 7.5 (1 M) | 10 mM | 1 mL |
| EDTA pH 8.0 (0.5 M) | 1 mM | 200 μL |
| ddH2O | N/A | 98.8 mL |
| Total | 100 mL |
Storage conditions: Store at 25°C for 1 year.
Streptavidin elution buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Formamide | 95% | 9.5 mL |
| EDTA pH 8.0 (0.5 M) | 10 mM | 200 μL |
| ddH2O | N/A | 300 μL |
| Total | 10 mL |
Storage conditions: Prepare fresh before use.
Step-by-step method details
PCR amplification of the DNA pool
Amplification of the designed DNA pool provides input material for subsequent in vitro transcription. We recommend optimizing the number of PCR cycles for each DNA pool to maximize yield while minimizing spurious overamplification products.
-
1.
Resuspend the DNA pool to a final concentration of 0.5 ng/μL in 10 mM Tris-HCl, pH 8.0.
-
2.
Prepare an 11× PCR master mix.
| Reagent | Amount (1×) | Amount (11×) |
|---|---|---|
| HF Buffer (5×) | 10 μL | 110 μL |
| T7 forward primer (10 μM) | 1 μL | 11 μL |
| RT handle reverse primer (10 μM) | 1 μL | 11 μL |
| dNTPs (10 mM) | 1 μL | 11 μL |
| DMSO | 1.5 μL | 16.5 μL |
| Oligo pool (0.5 ng/ μL) | 1 μL | 11 μL |
| Phusion polymerase | 0.25 μL | 2.75 μL |
| ddH2O | 34.25 μL | 376.25 μL |
| Total | 50 μL | 550 μL |
-
3.
Aliquot 50 μL of PCR master mix into 11 individual PCR tubes. Place in thermocycler for 12 PCR cycles (or pre-determined optimal cycle number) as below:
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial denaturation | 98°C | 30 s | 1 |
| Denature | 98°C | 10 s | 12 cycles |
| Anneal | 58°C | 30 s | |
| Extend | 72°C | 30 s | |
| Final extension | 72°C | 5 min | 1 |
| Hold | 4°C | Indefinitely | |
-
4.Gel purify the amplified template DNA.
-
a.Cast two 8% non-denaturing TBE polyacrylamide mini gels.4
-
b.Pool the PCR reactions and add 110 μL of 6× DNA loading dye.
-
c.Load the PCR product across all available lanes. Run the samples at 200 V for 50 min in 0.5× TBE.
-
d.Remove the gels from the plates and stain by covering in 15 mL of 0.5× TBE plus 2 μL of SYBR Gold.
-
i.Incubate at 25°C on a rocking platform for 5 min.
-
ii.Visualize under blue light.Note: An example pool PCR optimization gel is shown in Figure 2.CRITICAL: The use of blue light illumination is necessary to prevent DNA damage that can result from UV illumination.
-
i.
-
e.Excise PCR products with a razor blade and place gel slices into two 1.5 mL tubes containing 750 μL of DNA elution buffer.
-
i.Rotate for 12–14 h at 25°C.
-
i.
-
f.Transfer the eluted DNA solution to new 1.5 mL tubes.
-
i.Add 750 μL of isopropanol and 2 μL of Glycoblue to each tube.
-
ii.Vortex the sample and incubate at −20°C for at least 30 min.
-
iii.Centrifuge at 21,000 × g at 4°C for 30 min to pellet DNA.Pause point: The purified DNA template can be stored at −20°C in isopropanol until ready for use.
-
i.
-
g.Wash pellets with 750 μL of 70% ethanol and centrifuge at 21,000 × g at 4°C for 10 min.
-
i.Remove supernatant and air-dry pellets at 25°C for 5 min.
-
ii.Resuspend the precipitated DNA in 8 μL of nuclease-free water.
-
i.
-
a.
Figure 2.

Optimization of pool DNA PCR
The pool DNA template was amplified for the indicated number of cycles and ran on an 8% TBE polyacrylamide gel.
Transcribing, capping, and biotinylating the RNA pool
This step generates the RNA pool to be used in the ribosome recruitment assay. Addition of the 5′ methylguanosine cap is required to emulate physiological cap-dependent translation. 3′ biotinylation ensures efficient RNA recovery after incubation in translation extract and fractionation.
-
5.
Prepare the in vitro transcription reaction by adding the following to the 8 μL of purified DNA template (from 4f above):
| Reagent | Amount |
|---|---|
| Transcription buffer (10×) | 2 μL |
| ATP (75 mM) | 2 μL |
| CTP (75 mM) | 2 μL |
| GTP (75 mM) | 2 μL |
| UTP (75 mM) | 2 μL |
| T7 Enzyme Mix | 2 μL |
| Total | 12 μL |
| Reaction Total | 20 μL |
-
6.
Incubate the reaction at 37°C for 4 h.
-
7.
Add 2 μL of TURBO DNase and incubate at 37°C for 15 min to degrade the template DNA.
-
8.Gel-purify the RNA pool by denaturing polyacrylamide gel electrophoresis.
-
a.Cast an 8% denaturing 1× TBE/7 M urea polyacrylamide mini gel. Pre-run the gel in 0.5× TBE at 200 V for 20 min.
-
b.Add 22 μL of 2× formamide buffer to the RNA and incubate at 70°C for 5 min to denature the RNA. Transfer the mixture to ice to cool.
-
c.Using a syringe, flush the wells of the mini gel with 0.5× TBE to remove urea.
-
d.Load 6 μL of denatured RNA per well. Run the gel at 200 V for 60 min.
-
e.Remove the gels from the plates and stain by covering in 15 mL of 0.5× TBE plus 2 μL of SYBR Gold.
-
i.Incubate at 25°C on a rocking platform for 5 min.
-
ii.Visualize under blue light.Note: An example in vitro transcription product gel is shown in Figure 3.CRITICAL: The use of blue light illumination is necessary to prevent RNA damage that can result from UV illumination.
-
i.
-
f.Excise RNA bands using a razor blade and place gel slices into a 1.5 mL tube containing 750 μL of RNA elution buffer.
-
i.Rotate for 12–14 h at 4°C.
-
i.
-
g.Transfer the eluted RNA solution to a new 1.5 mL tube for precipitation.
-
i.Add 750 μL of isopropanol and 2 μL of Glycoblue.
-
ii.Vortex the sample and incubate at −20°C for at least 30 min. Centrifuge at 21,000 × g at 4°C for 30 min to pellet RNA.Pause point: The purified RNA can be stored at −20°C in isopropanol until ready for use.
-
iii.Wash pellets with 750 μL of 80% ethanol and centrifuge at 21,000 × g at 4°C for 10 min.
-
iv.Remove supernatant and air-dry pellets at 25°C for 5 min.
-
v.Resuspend the precipitated RNA in 50 μL of nuclease-free water.
-
i.
-
h.Transfer 0.5 μL of RNA to a tube containing 9.5 μL of water. Load 2 μL of diluted RNA onto a Nanodrop and measure concentration, targeting approximately 40 micrograms total.
-
a.
-
9.Add 5′ methylguanosine cap as follows:
-
a.Add 2 μL of RNasin Plus to the 50 μL of purified RNA. Heat-denature by incubating at 65°C for 5 min. Transfer denatured RNA to ice for 2 min.
-
b.Prepare the capping reaction using the Vaccinia capping system (NEB).
Reagent Amount Denatured RNA (30–40 μg) 50 μL Capping buffer (10×) 8 μL GTP (10 mM) 12 μL SAM (32 mM) 4 μL Vaccinia capping enzyme 4 μL ddH2O 2 μL Total 80 μL -
c.Incubate the capping reaction at 37°C for 1 h.
-
a.
-
10.Clean-up the capping reaction.CRITICAL: Keep the samples on ice for the duration of the reaction clean-up.
-
a.Add 560 μL of binding buffer and 640 μL of isopropanol to the capping reaction.
-
b.Apply 800 μL (maximum loading capacity) of the RNA solution to a Zymo V column. Centrifuge at 20,000 × g for 1 min at 4°C. Repeat with the remaining 480 μL.
-
c.Wash the column by adding 200 μL of 80% ethanol, then centrifuging at 20,000 × g for 1 min at 4°C.
-
d.Repeat the wash step twice more (for a total of three washes).
-
e.To elute the RNA, add 200 μL of nuclease-free water and spin at 21,000 × g for 2 min at 4°C.
-
f.Precipitate the eluted RNA by adding 22 μL of 3 M sodium acetate, pH 5.3 and 1 mL of 100% ethanol.
-
i.Incubate at −20°C for 30 min. Centrifuge at 21,000 × g at 4°C for 30 min to pellet the RNA.
-
i.
-
g.Wash pellets with 1 mL of 80% ethanol and centrifuge at 21,000 × g at 4°C for 10 min.
-
i.Remove supernatant and air-dry pellets at 25°C for 5 min.
-
ii.Resuspend the precipitated RNA in 6 μL of nuclease-free water.
-
i.
-
h.Transfer 0.5 μL of capped RNA to a tube containing 9.5 μL of water for concentration measurement.
-
i.Load 2 μL of diluted RNA onto a Nanodrop and measure the capped RNA concentration.
-
ii.Determine the molarity of the capped RNA solution using the following approximate molecular weight calculation: Molecular Weight of ssRNA = (number of nucleotides × 320.5) + 159.0.
-
i.
-
a.
-
11.Transfer 50 pmol of capped RNA to a new 0.2 mL tube. Add water to bring the total volume to 6 μL.
-
a.Add 2 μL of DMSO. Heat-denature the RNA by incubating at 85°C for 4 min.
-
b.Prepare the 3′ biotinylation reaction using the Pierce 3′ biotinylation kit as follows:
-
a.
| Reagent | Amount |
|---|---|
| Capped RNA/DMSO solution | 8 μL |
| 10× T4 RNA ligase reaction buffer | 3 μL |
| RNase inhibitor | 1 μL |
| Biotinylated cytidine (bis)phosphate | 1 μL |
| PEG 30% | 15 μL |
| T4 RNA ligase | 2 μL |
| Total | 30 μL |
-
12.
Incubate at 16°C for 12–14 h in a thermocycler.
-
13.Clean-up the biotinylation reaction.
-
a.Add 70 μL of water and transfer reaction to 1.5 mL MaXtract tube.
-
i.Add 100 μL of phenol:chloroform:IAA, 25:24:1, pH 6.6.
-
ii.Vortex briefly and centrifuge at 16,000 × g for 10 min at 4°C to separate the phases.
-
i.
-
b.Carefully transfer the top aqueous layer to a new 1.5 mL tube.
-
i.Add 10 μL of 5 M NaCl, 2 μL of Glycoblue, and 300 μL of 100% ethanol.
-
ii.Incubate at −20°C for at least 30 min to precipitate RNA.
-
iii.Centrifuge at 21,000 × g at 4°C for 30 min to pellet the RNA.
-
i.
-
c.Wash pellets with 1 mL of 80% ethanol and centrifuge at 21,000 × g at 4°C for 10 min. Remove supernatant and repeat for a second wash.
-
d.Remove the supernatant and air-dry pellets at 25°C for 5 min.
-
e.Resuspend the precipitated RNA in 1.2 mL of nuclease-free water.
-
a.
Figure 3.

In vitro transcription of pool RNA
The pool DNA PCR template was transcribed with T7 RNA polymerase and ran on an 8% denaturing TBE polyacrylamide gel. Full-length RNA indicated by the dye-excluded band in the dashed box was excised and purified.
Yeast extract preparation and ribosome recruitment
The yeast translation extract is first treated with micrococcal nuclease to degrade endogenous mRNA, increasing ribosome recruitment to the pool RNA. The ribosome recruitment reactions are carried out in the presence of cycloheximide to ensure that each RNA is only able to recruit a single ribosome that is then trapped on that RNA.
-
14.Degrade endogenous mRNA from the yeast translation extract by treating with micrococcal nuclease (MNase):
-
a.Add 42 μL of 40 mM CaCl2 to 3.5 mL of yeast translation extract.CRITICAL: Supplementing the extract with CaCl2 is essential as MNase is a Ca2+-dependent enzyme.
-
b.Degrade endogenous mRNAs by adding 35 μL of MNase and incubating for 10 min at 25°C on a rotator.
-
c.Deactivate the MNase by adding 63 μL of 0.1 M EGTA and transfer to ice.CRITICAL: The use of EGTA in place of other chelators (i.e., EDTA) is essential. EGTA has a significantly higher affinity for Ca2+ than Mg2+, thus inactivating MNase without significantly perturbing Mg2+-dependent processes.
-
a.
-
15.Prepare the in vitro ribosome recruitment reaction. The amount shown for the 3.2× allows for 3 individual replicate reactions with additional volume to allow for pipetting error.
Reagent Amount (1×) Amount (3.2×) Cycloheximide (10 mg/mL)
(Prepare fresh before use)100 μL 320 μL 6× Translation mix 333.4 μL 1,067 μL RNasin Plus 10 μL 32 μL PMSF (0.1 M) 40 μL 128 μL cOmplete Mini solution (Prepare fresh before use) 66.8 μL 213.8 μL MNase-treated yeast extract 984.8 μL 3151.4 μL Creatine phosphokinase solution (10 mg/mL) 65 μL 208 μL ddH2O 20 μL 60 μL Total 1.62 mL 5.18 mL -
a.Split each reaction in half by transferring 810 μL to 2 1.5 mL tubes.
-
b.Add 190 μL of the capped and biotinylated RNA pool (20 pmol of RNA per tube).
-
c.Incubate the reaction for 30 min on a thermomixer set to 600 RPM and 26°C. Immediately place on ice.
-
d.Transfer 200 μL of each reaction into a new 1.5 mL tube. Snap-freeze these samples in liquid nitrogen and store at −80°C for subsequent input library preparation.
-
a.
Isolation of 80S-RNA complexes and RNA extraction
In this step, RNAs bound to an 80S ribosome are separated from unbound through sucrose gradient fractionation. Cycloheximide is maintained in the sucrose solutions to ensure 80S-RNA complexes are maintained during fractionation. Hot phenol plus SDS RNA extraction is used to ensure complete denaturation of the concentrated ribosomal RNA and proteins.
-
16.
Before you begin, pre-chill SW28 rotor and swing buckets to 4°C. Turn on ultracentrifuge and allow it to reach 4°C.
-
17.Prepare the sucrose gradients. This is done using a Biocomp Gradient Master/Fractionator station and associated parts and accessories.
-
a.Begin by preparing 10% and 50% sucrose solutions.
Reagent 10% sucrose solution 50% sucrose solution HEPES KOH pH 7.4 (1 M) 2.4 mL 2.4 mL Magnesium acetate (1 M) 240 μL 240 μL Potassium acetate (2 M) 6 mL 6 mL Triton X-100 1.2 mL 1.2 mL DTT (1 M) 360 μL 360 μL Cycloheximide (10 mg/mL) (Prepare fresh before use) 1.2 mL 1.2 mL Sucrose (2 M) 17.5 mL 87.6 mL ddH2O 91.1 mL 21 mL Total 120 mL 120 mL -
b.Using the Marker Block, mark the long cap halfway point of Seton Polyclear ultracentrifuge tubes.
-
c.Add the 10% sucrose solution until approximately 1 cm above the mark.
-
d.Draw 60 mL of 50% sucrose solution into a 60 mL syringe.
-
e.Affix a wide-bore pipetting needle to the syringe.
-
f.Depress the plunger until the solution is at the end of the needle, then hold tension to prevent dripping. Insert the needle to the bottom of the tube containing 10% sucrose.
-
g.Slowly dispense the 50% sucrose solution until the interface reaches the marked point. Hold tension on the syringe to prevent dripping and remove from the tube.
-
i.Repeat the underlaying process for the remaining tubes.
-
i.
-
h.Place the long caps on the filled tubes, avoiding bubbles, and remove any excess solution from the top of the cap.
-
i.Place the tubes in the Magnabase holder, then place the holder onto the Gradient Master mixing station.
-
j.Select the pre-installed program for the SW28 10%–50% sucrose gradient (long) and run the gradient preparation.
-
k.Place the prepared gradients in SW28 buckets and chill at 4°C for 30 min.
-
a.
-
18.
Slowly remove the caps and then gently load the 1.8 mL ribosome recruitment reaction onto the top of the gradient.
-
19.
Precisely balance the swing buckets and spin the gradients for 3 h at 27,000 RPM (approximately 97,000 × g) at 4°C.
-
20.
Fractionate the sucrose gradients using a Gradient Master with a fraction collector attached.
Note: An example gradient trace is shown in Figure 4.
-
21.
Collect fraction tubes corresponding to the 80S fraction. Pool the 80S fractions in 15 mL conical tubes. Keep on ice.
Note: In the following RNA extraction steps, the 80S and input samples are processed in parallel. 80S fraction processing steps are denoted by “80S:” while input processing steps are denoted by “Input:”.
-
22.
80S: Pre-spin 15 mL MaXtract tubes for 2 min at 1,500 × g. Input: Pre-spin 1.5 mL MaXtract tubes for 1 min at 12,000 × g.
-
23.
80S: Add 650 μL of chloroform to the tube per 600 μL of pooled fraction volume. Input: Add 220 μL of chloroform to the tubes.
-
24.
80S: Add 650 μL of acid phenol and 40 μL of 20% SDS per 600 μL of pooled fraction volume to tubes containing pooled fractions. Vortex briefly to mix. Input: Add 220 μL of acid phenol and 13.3 μL of 20% SDS to the tubes containing the reserved input samples. Vortex briefly to mix.
-
25.
Incubate 80S and input samples for 10 min in a water bath at 65°C, vortexing every minute.
-
26.
Cool 80S and input samples on ice for 3 min.
-
27.
80S: Transfer the phenol/aqueous mixture to pre-spun 15 mL MaXtract tubes containing chloroform. Mix by inversion. Input: Transfer the phenol/aqueous mixture to pre-spun 1.5 mL MaXtract tubes containing chloroform. Mix by inversion.
-
28.
80S: Centrifuge MaXtract tubes at 4,500 × g for 5 min at 4°C. Input: Centrifuge MaXtract tubes at 12,000 × g for 5 min at 4°C.
-
29.
80S: Transfer the aqueous fractions to new 15 mL tubes. Input: Transfer the aqueous fractions to new 1.5 mL tubes.
-
30.
80S: Add 650 μL of phenol:chloroform:IAA pH 6.6 per 600 μL of aqueous volume. Mix by inversion. Input: Add 220 μL of phenol:chloroform:IAA pH 6.6 to the input samples. Mix by inversion.
-
31.
80S: Centrifuge 15 mL tubes at 4,500 × g for 5 min at 4°C. Input: Centrifuge 1.5 mL tubes at 12,000 × g for 5 min at 4°C.
-
32.
80S: Transfer the aqueous fractions to new 15 mL tubes. Input: Transfer the aqueous fractions to new 1.5 mL tubes.
-
33.
80S: Add 650 μL of chloroform per 600 μL of aqueous volume. Mix by inversion. Input: Add 220 μL of chloroform to the input samples. Mix by inversion.
-
34.
80S: Centrifuge 15 mL tubes at 4,500 × g for 5 min at 4°C. Input: Centrifuge 1.5 mL tubes at 12,000 × g for 5 min at 4°C.
-
35.
80S: Transfer the aqueous fractions to 50 mL Oak Ridge tubes. Input: Transfer the aqueous fractions to new 1.5 mL tubes.
-
36.
80S: Add 1/9th volume of 3 M sodium acetate pH 5.3. Input: Add 22.2 μL of 3 M sodium acetate pH 5.3.
-
37.
80S: Add an equal volume (including sodium acetate volume) of isopropanol to the Oak Ridge tubes. Mix and incubate at −20°C for at least 30 min. Input: Add 250 μL of isopropanol to the input samples. Mix and incubate at −20°C for at least 30 min.
-
38.
80S: Transfer the Oak Ridge tubes to an SS-34 or other compatible rotor and spin at 23,500 × g for 30 min at 4°C to pellet RNA. Input: Pellet the RNA by centrifuging at 21,000 × g for 30 min at 4°C in a benchtop centrifuge.
-
39.
80S: Remove the supernatant. Add 750 μL of 80% ethanol to wash pellet. Input: Add 750 μL of 80% ethanol to wash pellet.
-
40.
80S: Centrifuge at 23,500 × g for 20 min at 4°C. Input: Centrifuge at 21,000 × g for 20 min at 4°C.
-
41.
Air dry the pellets for 5 min on ice. Resuspend 80S and input samples in 500 μL of streptavidin binding buffer and proceed immediately to streptavidin pulldown.
Figure 4.

Sucrose gradient fractionation and 80S isolation
Shown here is a representative A260 trace of the in vitro translation reaction following sucrose gradient ultracentrifugation and fractionation using a Biocomp Gradient Master/Fractionator. Fractions 20–24 corresponding to the 80S monosome peak were collected for subsequent steps.
Isolation of pool RNA
The RNA extracted from the 80S fraction contains a large amount of contaminating ribosomal RNA. In this step, the biotinylated RNA pool is pulled down with streptavidin beads to remove this contamination from subsequent library preparation steps. The reserved RNA pool and input samples are also pulled down to account for potential biases in pull-down efficiency.
-
42.
Briefly vortex the magnetic streptavidin beads to resuspend. Transfer 125 μL of bead slurry per sample to a LoBind 1.5 mL tube.
-
43.
Apply the tubes to a magnetic rack to separate the beads for 30 s. Remove the supernatant. Equilibrate the beads by washing the beads twice with 100 μL of streptavidin binding buffer.
-
44.
Denature the RNA samples by incubating at 65°C for 5 min. Transfer to ice for 3 min.
-
45.
Transfer the RNA samples to the tubes containing the magnetic beads. Vortex briefly to mix. Incubate the samples for 12–14 h at 4°C, rotating.
-
46.
Apply the tubes to the magnetic rack and allow to separate for 2 min. Remove the supernatant. Wash the samples three times with 1 mL of streptavidin wash buffer.
-
47.
To elute, resuspend the beads in 50 μL of streptavidin elution buffer. Incubate at 70°C for 5 min.
-
48.
Apply the tubes to the magnetic rack and transfer the eluted RNA to new 1.5 mL tubes.
-
49.
Add 400 μL of water, 50 μL of 3 M sodium acetate pH 5.3, 2 μL of Glycoblue, and 600 μL of isopropanol. Incubate the samples at −20°C for at least 30 min to precipitate RNA.
-
50.
Centrifuge at 21,000 × g at 4°C for 30 min to pellet the RNA.
-
51.
Wash pellets twice with 1 mL of 80% ethanol and centrifuge at 21,000 × g at 4°C for 10 min.
-
52.
Remove supernatant and air-dry pellets at 25°C for 5 min.
-
53.
Resuspend the RNA in 11 μL of water and proceed immediately to reverse transcription.
Reverse transcription and adaptor ligation
Here, the purified RNA is reverse transcribed into single-stranded cDNA. We gel-purify full-length cDNA to capture only the desired product for subsequent steps. An adaptor containing a common PCR handle and a 10N randomer unique molecular identifier (UMI) sequence for PCR duplicate collapsing is then ligated to the 3′ end of the cDNA. The adaptor ligation reactions are then cleaned up using Silane beads.
-
54.Transfer the resuspended RNA to PCR tubes for RT primer annealing.
-
a.Add 1 μL of 10 μM RT primer to the resuspended RNA.
-
b.Transfer the samples to a thermocycler. Denature the RNA and anneal the RT primer with the following program:
-
a.
| Steps | Temperature | Time |
|---|---|---|
| RNA Denaturation | 65°C | 4 min |
| Annealing | 55°C | 2 min |
| Annealing | 45°C | 2 min |
| Annealing | 42°C | 2 min |
-
55.
Transfer the samples to ice.
-
56.
Prepare the Superscript III reverse transcription master mix as follows:
Note: The amount shown is for one reaction and should be scaled as appropriate.
| Reagent | Amount |
|---|---|
| 5× First-Strand Buffer | 4 μL |
| DTT (0.1 M) | 1 μL |
| dNTPs (10 mM) | 1 μL |
| RNasin Plus | 1 μL |
| Superscript III enzyme | 1 μL |
| Total | 8 μL |
-
57.
Add 8 μL of RT master mix to each tube containing RNA. Incubate at 50°C for 1 h.
-
58.
Add 2 μL of 1 M sodium hydroxide to each sample. Incubate at 98°C for 15 min to degrade the RNA. Neutralize by adding 2 μL of 1 M hydrochloric acid.
-
59.
Add 22 μL of formamide loading buffer and denature the cDNA at 70°C for 5 min.
-
60.
Load the samples onto an 8% denaturing TBE polyacrylamide mini gel.
-
61.
Run the gel at 200 V for 45 min.
-
62.Remove the gel from the plate and stain by covering in 15 mL of 0.5× TBE plus 2 μL of SYBR Gold.
-
a.Incubate at 25°C on a rocking platform for 5 min.
-
b.Visualize under blue light.
-
a.
Note: An example cDNA gel is shown in Figure 5.
-
63.
Excise cDNA synthesis products with a razor blade and place gel slices into two 1.5 mL tubes containing 750 μL of DNA elution buffer.
-
64.
Rotate the samples for 12–14 h at 25°C.
-
65.
Transfer the eluted DNA solution to new 1.5 mL tubes.
-
66.
Add 750 μL of isopropanol and 2 μL of Glycoblue to each tube. Vortex the sample and incubate at −20°C for at least 30 min.
-
67.
Centrifuge at 21,000 × g at 4°C for 30 min to pellet DNA.
-
68.
Wash pellets twice with 1 mL of 80% ethanol and centrifuge at 21,000 × g at 4°C for 10 min. Remove supernatant and air-dry pellets at 25°C for 5 min.
-
69.
Resuspend the cDNA in 5 μL of 10 mM Tris-HCl pH 7.5 and transfer to 1.5 mL LoBind tubes.
-
70.
Add 0.8 μL of 80 μM 5′ adaptor and 1 μL of 100% DMSO.
-
71.
Denature at 75°C for 2 min and place on ice.
-
72.
Prepare the ligation master mix.
Note: The amount shown is for one reaction and should be scaled as appropriate.
| Reagent | Amount |
|---|---|
| 10× T4 RNA Ligase Reaction Buffer | 2 μL |
| ATP (100 mM) | 0.2 μL |
| 50% PEG 8000 | 9 μL |
| T4 RNA Ligase 1, High Concentration | 1 μL |
| ddH2O | 1 μL |
| Total | 13.2 μL |
-
73.
Add 13.2 μL of ligation master mix to the cDNA samples.
-
74.
Mix to homogenize and incubate at 25°C on a thermomixer, shaking at 1000 RPM for 12–14 h.
-
75.Clean up the adaptor ligation reaction with MyOne Silane beads.
-
a.Vortex the bead solution to resuspend. Transfer 10 μL of bead slurry per ligation reaction to a 1.5 mL LoBind tube.
-
b.Apply to a magnetic rack to separate for 30 s and remove the supernatant.
-
c.Equilibrate the beads by washing with 500 μL of Buffer RLT.
-
d.Apply to magnet for 30 s and discard supernatant.
-
e.Resuspend the beads in 60 μL of Buffer RLT per sample.
-
f.Vortex to resuspend the beads and transfer 60 μL of bead slurry to the ligated cDNA.
-
g.Add 60 μL of 100% ethanol, mix by pipetting and incubate at 25°C for 5 min to bind cDNA to the beads.
-
h.Magnetically separate the beads for 1 min and remove the supernatant.
-
i.Resuspend the beads in 1 mL of 75% ethanol and transfer to a new 1.5 mL LoBind tubes.
-
j.Incubate at 25°C for 30 s, separate on the magnet for 30 s, and remove the supernatant.
-
k.Wash twice more with 1 mL of 75% ethanol. Discard the supernatant each time.
-
l.Spin down the samples briefly and remove residual ethanol with a 20 μL pipette. Air dry the beads for 5 min at 25°C.
-
m.Resuspend the beads in 27 μL of 10 mM Tris-HCl pH 7.5.
-
n.Separate the beads on the magnetic rack for 1 min and transfer the eluted cDNA to a new 1.5 mL tube.
-
a.
Figure 5.

Reverse transcription of pool RNA
Pool RNA was reverse transcribed followed by degradation of the template RNA, then ran on an 8% denaturing 1× TBE/7 M urea polyacrylamide gel. This example gel is of the input samples, exhibiting a clear full-length cDNA product.
Diagnostic PCRs, library construction, and quality control
These steps prepare next-generation sequencing libraries from the adaptor-ligated cDNA samples. The PCR primers used provide Illumina adaptor sequences with the reverse primer including a sample-specific 6-nucleotide barcode for sample identification. In the first step, we run small-scale diagnostic PCRs across a range of PCR cycles to identify the optimal cycle number for library construction without producing spurious overamplification products and large numbers of PCR duplicates. With the optimal cycle number identified, we run full-scale library construction PCRs and gel-purify the library products. The libraries are then quantified on Qubit and run on a Bioanalyzer to check library purity.
-
76.
Prepare the diagnostic PCR reactions master mix.
Note: The amount shown is sufficient for 30 diagnostic reactions:
| Reagent | Amount (1×) | Amount (30×) |
|---|---|---|
| HF Buffer (5×) | 3.33 μL | 100 μL |
| RP1 forward primer (10 μM) | 0.83 μL | 25 μL |
| Barcoded reverse primer (10 μM) | 0.83 μL | 25 μL |
| dNTPs (10 mM) | 0.33 μL | 10 μL |
| DMSO | 0.5 μL | 15 μL |
| Phusion polymerase | 0.17 μL | 5 μL |
| ddH2O | 9.67 μL | 290 μL |
| Total | 15.66 μL | 470 μL |
-
77.
Aliquot 15.66 μL of the PCR master mix per PCR tube containing 1 μL of adaptor-ligated cDNA.
-
78.
Run the PCR program as follows:
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial denaturation | 98°C | 30 s | 1 |
| Denature | 98°C | 10 s | Variable |
| Anneal | 58°C | 30 s | |
| Extend | 72°C | 30 s | |
| Final extension | 72°C | 5 min | 1 |
| Hold | 4°C | Indefinitely | |
Note: We generally recommend testing at least 3 cycle numbers ranging from 10 – 20 cycles as a first pass. Libraries requiring more than 20 cycles may be sequenced but are likely to contain large numbers of PCR duplicates that hinder analysis.
-
79.
Run the diagnostic PCRs on 8% non-denaturing TBE polyacrylamide mini gels at 200 V for 45 min in 0.5× TBE.
-
80.Remove the gels from the plates and stain by covering in 15 mL of 0.5× TBE plus 2 μL of SYBR Gold.
-
a.Incubate at 25°C on a rocking platform for 5 min.
-
b.Visualize under blue light.
-
a.
Note: An example diagnostic PCR gel is shown in Figure 6.
-
81.
With the optimal PCR conditions determined, prepare the final PCR reaction master mix.
Note: The amount shown is sufficient for 10 reactions.
| Reagent | Amount (1×) | Amount (10×) |
|---|---|---|
| HF Buffer (5×) | 20 μL | 200 μL |
| RP1 reverse primer (10 μM) | 5 μL | 50 μL |
| Barcoded forward primer (10 μM) | 5 μL | 50 μL |
| dNTPs (10 mM) | 2 μL | 20 μL |
| DMSO | 3 μL | 30 μL |
| Phusion polymerase | 1 μL | 10 μL |
| ddH2O | 58 μL | 580 μL |
| Total | 94 μL | 940 μL |
-
82.
Aliquot 94 μL of the PCR master mix per PCR tube containing 6 μL of adaptor-ligated cDNA.
-
83.
Run the same PCR program used for the diagnostic PCRs for the determined number of cycles.
-
84.
Split each 100 μL PCR reaction into two 50 μL aliquots in PCR tubes.
-
85.
Add 90 μL of Ampure XP bead solution to each tube. Mix thoroughly by pipetting. Incubate at 25°C for 5 min to bind DNA.
-
86.
Place the tubes on a magnetic rack and allow beads to separate for 2 min. Remove the supernatant and discard.
-
87.
Wash the beads twice with 200 μL of 70% ethanol. Remove the ethanol and allow the beads to air dry.
-
88.
Elute the DNA by adding 27 μL of water to the beads, mixing thoroughly. Incubate the sample for 2 min at 25°C.
-
89.
Place the sample back onto the magnetic rack to separate beads for 1 min. Transfer 25 μL of the eluted DNA to a clean 1.5 mL tube.
-
90.
Run the final PCRs on 8% non-denaturing TBE polyacrylamide mini gels at 200 V for 50 min in 0.5× TBE.
-
91.Remove the gel from the plate and stain by covering in 15 mL of 0.5× TBE plus 2 μL of SYBR Gold.
-
a.Incubate at 25°C on a rocking platform for 5 min.
-
b.Visualize under blue light.
-
a.
Note: An example final PCR gel is shown in Figure 7.
-
92.
Excise the library products with a razor blade and place gel slices into two 1.5 mL tubes containing 750 μL of DNA elution buffer.
-
93.
Incubate on a rotator for 12–14 h at 25°C.
-
94.
Transfer eluted DNA solution to new 1.5 mL tubes.
-
95.
Add 750 μL of isopropanol and 2 μL of Glycoblue to each tube. Vortex the sample and incubate at −20°C for at least 30 min.
-
96.
Centrifuge at 21,000 × g at 4°C for 30 min to pellet DNA.
-
97.
Wash pellets twice with 1 mL of 80% ethanol and centrifuge at 21,000 × g at 4°C for 10 min. Remove supernatant and air-dry pellets at 25°C for 5 min.
-
98.
Resuspend the DNA library in 10 μL of water.
-
99.
Run 1 μL of each sample on an Agilent Bioanalyzer using a High Sensitivity DNA chip to determine the library size distribution.
Note: A successful library preparation should yield a single dominant peak of the appropriate size. An example size distribution is shown in Figure 8.
-
100.
Pool the samples into a single multiplexed library at a final concentration of 1–2 nM each.
Note: To obtain sufficient coverage for a 12,000-sequence pool, we used one lane of Illumina HiSeq 2500 sequencer to obtain 150 million 75 bp paired end reads.
Figure 6.

Diagnostic library PCRs
Adapter-ligated cDNA was PCR amplified for the indicated number of cycles and ran on an 8% TBE polyacrylamide gel.
Figure 7.

Final library PCRs
Representative 8% TBE polyacrylamide gel of final PCR products. Library bands indicated by the black dashed box were excised and purified to avoid contamination from primers and undesired PCR products above and below these bands.
Figure 8.

Bioanalyzer quality confirmation of DART libraries
Size-selected libraries analyzed on a Bioanalyzer using a High Sensitivity DNA chip. A sharp peak corresponding to the desired library size indicates a successful library preparation.
Expected outcomes
A successful outcome is shown in Figure 5 (RT gel), 7, and 8. First, we expect to see a clear and defined RT product from the input lane (Figure 5), though we do not always observe signal at this stage for the other samples. Relatedly, we expect to see products of the correct size at the final PCR stage (Figure 7), which should correspond to a high quality and narrow size distribution when visualized on a Bioanalyzer or other similar quality control instrument. Lastly, we expect greater than 75% mapping efficiency to the synthetic genome used in the experiment.
Quantification and statistical analysis
Adapter trimming
This step trims Illumina adapter sequences that may be present in the sequenced read. The adapters.fa file containing Illumina adapter sequences may be obtained from here: https://github.com/BioInfoTools/BBMap/blob/master/resources/adapters.fa.
>bbduk.sh in1=${name} in2=${name2} out1=${name}.trimmed out2=${name2}.trimmed ref=adapters.fa ktrim=r k=23 mink=11 hdist=1 tpe tbo
PCR duplicate collapse
Here, reads arising from PCR duplication are collapsed based on a unique molecular identifier (UMI) present in the 5′ adaptor.
>clumpify.sh in1=${name}.trimmed in2=${name2}.trimmed out1=${name}.collapsed out2=${name2}.collapsed dedupe subs=0
Note: The percentage of PCR duplicates should not exceed 30%. Usable data may still be obtained from libraries containing a higher percentage of duplicates, but this increases the number of wasted sequencing reads.
UMI removal
Next, we remove the UMI sequence from the collapsed reads.
bbduk.sh in1=${name}.collapsed in2=${name2}.collapsed out1=${name1}.clean out2=${name2}.clean ftl=10
Generate a STAR reference genome
Before aligning the processed reads, create a reference genome to align to. The input for this genome generation is a fasta-formatted file containing the oligo sequences minus the T7 promoter, as this is not included in the transcribed RNA sequence. In this example, this input file is referred to as “DARTref.fa”.
>STAR --runMode genomeGenerate --genomeDir STARgenome/ --genomeFastaFiles DARTref.fa --genomeSAindexNbases 9
Aligning the reads with STAR
With the reference genome created and the reads processed, align the reads using STAR.
>STAR –runThreadN 10 –genomeDir STARgenome/ --readFilesIn ${name}.clean ${name2}.clean –outFileNamePrefix ${name} –soloStrand Forward –alignSJoverhangMin 999 –alignIntronMax 1 –alignIntronMin 999
Note: Reads that fail to align should be excluded from further analysis. A successful experiment and analysis should yield > 80% uniquely mapping reads.
Count the reads mapping to each oligo sequence
>pileup.sh in=${name}Aligned.out.sam out=${name}_STAR_coverage.txt
Next, convert the raw read counts to counts per million (CPM) by dividing the number of reads per sequence by the total number of reads in that library (in millions).
Calculate ribosome recruitment scores (RRS)
The CPM values can now be used to calculate the RRS values for each sequence by taking the ratio of normalized 80S reads to normalized initial pool reads as follows:
Limitations
Each extract preparation must be tested to determine the optimal RNA concentration for DART. We generally target high, but non-saturating concentrations as evaluated by luciferase assays. To achieve the desired 20 pmol per reaction this may require scaling up the size of the reaction, and therefore the amount of extract required. Lastly, any RNAs that may co-sediment with the 80S fraction as part of an unrelated large RNP will be identified as ribosome bound.
Troubleshooting
Problem 1
Difficulty with phenol/chloroform extraction or resuspension in step 41.
Potential solution
When reaction volumes are large (greater than 2 mLs) resuspension of the pellet can be challenging, presumably due to the large amount of ribosomal RNA in the samples. In this case an alternative is to forgo the phenol/chloroform extraction and precipitation altogether and instead run the combined fractions onto a desalting column to achieve the desired buffer conditions for streptavidin pulldown.
Problem 2
Low yields or broad size distribution of final library prep in step 99.
Potential solution
When fraction collection takes an extended amount of time the already collected samples may experience some degree of degradation. If this is a concern, fractions can be collected directly into phenol/chloroform solution to preserve the integrity of the samples.
Problem 3
Low yield in final library prep in step 99.
Potential solution
One of the main reasons for low yield is low input RNA followed by consistent loss of RNA throughout the procedure. To minimize loss during handling we extensively utilize GlycoBlue to ensure RNA pellets are not lost. If low yield continues to be an issue it is likely that the reaction volume needs to be scaled up to increase the amount of total input RNA from the beginning of the procedure.
Problem 4
High percentage of PCR duplicates in final library following step 100.
Potential solution
Decrease the number of PCR cycles used to generate the final library.
Problem 5
Smeared cDNA gel in step 62.
Potential solution
This often occurs as a result of incomplete RNA degradation. Increase the concentration of sodium hydroxide used and/or extend the incubation time to fully degrade RNA.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wendy V. Gilbert (wendy.gilbert@yale.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
We thank the lab members for their advice and many discussions. This work was funded by NIH R01GM132358 to W.V.G., NIH 1F31DK129022 to C.J.T.L., as well as NIH 1K99GM135533 and ACS 133477-PF-19-092-01-RMC to R.O.N.
Author contributions
Conceptualization, W.V.G.; investigation and testing, C.J.T.L., R.O.N., and R.N.; writing – original draft, C.J.T.L.; writing – reviewing & editing, C.J.T.L., R.O.N., R.N., and W.V.G.; funding acquisition, R.O.N. and W.V.G.
Declaration of interests
The authors declare no competing interests.
Data and code availability
All datasets associated with the original study1 have been deposited at GEO and the accession numbers are available therein. This study did not generate original code.
References
- 1.Niederer R.O., Rojas-Duran M.F., Zinshteyn B., Gilbert W.V. Direct analysis of ribosome targeting illuminates thousand-fold regulation of translation initiation. Cell Syst. 2022;13:256–264.e3. doi: 10.1016/j.cels.2021.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Archer S.K., Shirokikh N.E., Beilharz T.H., Preiss T. Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature. 2016;535:570–574. doi: 10.1038/nature18647. [DOI] [PubMed] [Google Scholar]
- 3.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGookin R. Electrophoresis of DNA in nondenaturing polyacrylamide gels. Methods Mol. Biol. 1988;4:75–79. doi: 10.1385/0-89603-127-6:75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All datasets associated with the original study1 have been deposited at GEO and the accession numbers are available therein. This study did not generate original code.