

Abstract
Thromboembolic conditions are a leading cause of death worldwide, and deep vein thrombosis (DVT), or occlusive venous clot formation, is a critical and rising problem that contributes to damage of vital organs, long-term complications, and life-threatening conditions such as pulmonary embolism. Early diagnosis and treatment are correlated to better prognosis. However, current technologies in these areas, such as ultrasonography for diagnostics and anticoagulants for treatment, are limited in terms of their accuracy and therapeutic windows. In this work, we investigated targeting myeloid related protein 14 (MRP-14, also known as S100A9) using plant virus-based nanoparticle carriers as a means to achieve tissue specificity aiding prognosis and therapeutic intervention. We used a combinatorial peptide library screen to identify peptide ligands that bind MRP-14. Candidates were selected and formulated as nanoparticles by using cowpea mosaic virus (CPMV) and tobacco mosaic virus (TMV). Intravascular delivery of our MRP-14-targeted nanoparticles in a murine model of DVT resulted in enhanced accumulation in the thrombi and reduced thrombus size, suggesting application of nanoparticles for molecular targeting of MRP-14 could be a promising direction for improving DVT diagnostics, therapeutics, and therefore prognosis.
Graphical Abstract

INTRODUCTION
Cardiovascular disease is one of the leading causes of death in developed countries. It is estimated that as many as 900000 people each year in the US alone (1–2 per 1000) are affected by venous thromboembolism (VTE), mostly presenting as deep venous thrombosis (DVT) and pulmonary embolism (PE). Estimates suggest that 60000–100000 Americans die of VTE each year. About one-third of the survivors have recurrence within 10 years. In addition, 50% of surviving patients will have post-thrombotic syndrome, a chronic consequence involving symptoms that include swelling, pain, discoloration, and scaling in the affected limb.1 US healthcare costs are staggering and carry a high burden for treatment of VTE patients; it is estimated that $10 billion US dollars are spent each year accounting for treatment and management of VTE.1
There is a clear need in two areas for improving the treatment and prognosis of VTE: (i) accurate clinical diagnosis of VTE, before instituting an intervention, and (ii) targeted therapies to reduce safety issues associated with contemporary thrombolytic drugs. Diagnosis of DVT includes medical imaging and blood tests to assess D-dimer blood levels; however, increased levels are only indicative of DVT.2,3 The established medical imaging modalities for DVT/PE diagnosis are X-ray venography and ultrasonography, with ultrasonography having largely replaced X-ray venography for the diagnosis of DVT due to its wide availability in hospitals and the elimination of radiation exposure of the patient and staff.4 Nevertheless, ultrasonography also has drawbacks; for example, it is not applicable for nonocclusive thrombi.5 In addition, accurate diagnosis is hampered by false negative results.6–8 Both imaging approaches, X-ray venography and ultrasonography, reflect changes in the venous anatomy and enable detection of potential blockages, luminal narrowing, and/or intima-media thickening, but these modalities do not provide information about the molecular composition of the thrombus and therefore do not provide accurate diagnosis and prognosis. Once VTE is identified, on the therapeutics side, many thrombolytic drugs are not targeted and fail due to significant safety issues, particularly bleeding risks, which limit dosage and a therapeutic window.9
Molecularly targeted nanotechnology approaches harness potential to overcome these challenges. There is emerging experimental evidence distinguishing the molecular and cellular mechanisms of hemostasis and thrombosis,10 therefore providing an opportunity for more specifically tailored approaches in DVT and PE for imaging and therapy. Nanoparticles can be engineered to display ligands, such as antibodies and their fragments, peptides, or aptamers, which are designed to interact with specific molecular targets to enable tissue-specific accumulation of contrast agents and/or therapeutics. Cardiovascular nanotechnology is an emerging discipline, and several review articles discuss recent developments and deployment of nanoparticles as (i) contrast agents to aid risk stratification of atherosclerosis and thrombosis, (ii) tools to aid cardiovascular surgeries, and (iii) drug delivery agents to target thrombolytics and mitigate systemic side effects; the reader is referred to the referenced articles.11–15 Many different classes of nanomaterials are undergoing development for applications in medicine; these include lipid, polymeric, metal, and protein-based formulations.16 In our work, we focus on nanoparticles derived from plant viruses: tobacco mosaic virus (TMV, a 300 × 18 nm2 hollow nanotube) and cowpea mosaic virus (CPMV, a 30 nm sized icosahedron or sphere-like nanoparticle). These plant viral nanoparticles (VNPs) offer several unique features. For example, methods to produce these particles through molecular farming in plants (tobacco cowpea for TMV and CPMV) are simple and high-yielding, with 100 mg of pure VNP typically obtained from 100 g of leaf tissue.17,18 Even though VNPs are biologics, the capsids formed by plant viruses are robust and thus do not require cold chain storage or handling. This also means that they are stable to undergo a series of chemical reactions to functionalize the proteinaceous nanoparticles with therapeutics, imaging moieties, and/or targeting ligands.19 Lastly, plant VNPs are noninfectious toward mammals and demonstrate good biocompatibility.20 The materials properties and applications of plant VNPs have been reviewed by us21,22 and others.23,24
Here, we studied TMV and CPMV as platform technologies to design nanoscale materials targeting DVT. This is following previous work demonstrating the applicability of these proteinaceous carriers toward cardiovascular disease. To delineate early stage disease, i.e., atherosclerotic plaques, we described the application of plant VNPs carrying chelated Gd(DOTA) and/or near-infrared dyes to target vascular cell adhesion molecule-1 (VCAM-1) and myeloid related protein 14 (MRP-14, also known as S100A9) by magnetic resonance imaging25 and optical imaging.26 For treatment of late-stage disease, i.e., thrombosis, we utilized streptokinase or tissue plasminogen activator (tPA)-loaded thrombolytic VNPs to treat thrombosis27,28 as well as explored deploying VNPs targeted to fibrin.29
In this work, we set out to contribute new resources enabling noninvasive imaging or targeted therapy of DVT by identifying and validating novel peptide candidates against a key player in this disease context: MRP-14, a targetable regulator of inflammation. MRP-14 is part of the S100 family composed of small proteins (~9–14 kDa) containing two Ca2+-binding domains. MRP-14 and MRP-8 form homodimers and heterodimers, although the MRP-8/14 heterodimer is the dominant extracellular form in humans.30,31 MRP-8/14 complexes are also present in mouse cells, and extensive biochemical characterization has confirmed that mouse MRP-14 is functionally equivalent to its human counterpart.32 Expression of MRP-14 has also been implicated in acute inflammation in rabbits.33 MRP-8/14 broadly regulates vascular inflammation and promotes leukocyte recruitment in models of atherosclerosis, restenosis, and vasculitis.34 MRP-8/14 binding to toll-like receptor (TLR)-4 on leukocytes is known to produce proinflammatory effects that can be attenuated through antibody binding to MRP-14.35 More recently, MRP-14 was identified as a highly expressed mediator of thrombosis. While deficiency of MRP-14 is protective of thrombosis, it has no effect on multiple parameters of hemostasis, including tail bleeding time, activated partial thromboplastin time, thrombin generation, or platelet adhesion/spreading on the von Willebrand factor or collagen.36 Therefore, MRP-14 is a targetable regulator of vascular inflammation and mediator of thrombosis but not hemostasis (i.e., reduced bleeding risk).34,35,37 For these reasons, MRP-14 is a suitable molecular target for the development of next-generation cardiovascular nanomedicine. Here, we identified novel peptide candidates with specificity for MRP-14. We demonstrate the conjugation of select MRP-14 binders to TMV and CPMV and homing of these targeted nanoparticle formulations to DVT in a mouse model.
MATERIALS AND METHODS
Phage Display.
Ph.D.-7 and Ph.D.-C7C phage display peptide libraries (New England Biolabs) were used to identify peptides specific for MRP-14 according to manufacturer’s recommendations. Briefly, three rounds of solution-phase panning were performed. In the first round, phages from the library (1011 pfu/mL) were incubated with His-tagged MRP-14 (2 pmol, Novus Biologicals) in 200 μL of TBST (0.1% (v/v) Tween 20) for 20 min at room temperature. The solution was then mixed for 15 min with 50 μL of HisPur Ni-nitrilotriacetic acid (NTA) resin slurry (Pierce) that had been blocked with bovine serum albumin (BSA). After washing 10 times with TBST, bound phages were eluted with 1 mL of 0.2 M glycine-HCl, pH 2.2, with 1 mg/mL BSA that was quickly neutralized with 150 μL of 1 M Tris-HCl, pH 9.1. Phage amplification was performed with ER2738 E. coli, and subsequent rounds of panning were performed with the amplified phages following the procedure above, except with TBST containing 0.5% (v/v) Tween 20 and an additional negative selection step where the phages were first incubated with blocked resin for 15 min before transferring the supernatant for mixing with MRP-14. After the third round of panning, random phages specific for MRP-14 were selected by using blue-white screening, their DNA was isolated, and their sequences were determined by using the −96 gIII sequencing primer (Eurofins Genomics).
Phage Enzyme-Linked Immunosorbent Assay (ELISA).
Phage specificity for MRP-14 was verified by performing an ELISA. Polysorp 96-well plates (Nunc) were coated with 50 μL of 50 μg/mL MRP-14 in 0.1 M NaHCO3, pH 8.6, in a humidified chamber overnight at 4 °C. The coated wells and additional wells for phage dilutions were blocked with 5 mg/mL BSA in 0.1 M NaHCO3, pH 8.6, for 1 h at 4 °C. After washing six times with TBST (0.5% Tween 20), eight serial 4-fold dilutions from 1012 phages/mL down to 6 × 107 phages/mL were performed in blocked wells, and then 100 μL of the phages was added to MRP-14-coated wells and incubated for 1 h at room temperature. The wells were washed six times with TBST, and then 100 μL of horseradish peroxidase (HRP)-conjugated anti-M13 monoclonal antibody (GE Healthcare) diluted 1:5000 in 2% dry milk in PBS was added to the wells. Another washing step with TBST six times was carried out. Finally, the HRP activity was detected by using 100 μL of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS) colorimetric substrate solution (Life Technologies) at room temperature for 30 min. The absorbance was measured at 410 nm by using a Tecan Infinite 200 plate reader.
Surface Plasmon Resonance (SPR) Characterization.
Short-listed MRP-14 targeting peptides (MTPs) were synthesized with a GGGS spacer and C-terminal amidation (Genscript), and their binding affinities were characterized by SPR using a Biacore T100 (GE Healthcare). PBST (0.05% Tween 20), pH 7.4, containing 50 μM ethylenediaminetetraacetic acid (EDTA) was used as the running buffer. NTA sensor chips (GE Healthcare) were charged with 0.5 mM NiCl2 solution for 2 min at a flow rate of 10 μL/min, and then 10 μg/mL His-tagged MRP-14 was bound for 2 min at 10 μL/min. For testing the affinity to MRP-8/14, untagged MRP-8 (Novus Biologicals) was premixed in a 1:1 ratio with His-tagged MRP-14 for 3 h in the presence of 0.1 mM CaCl2. Peptides at concentrations ranging from 6.25 to 100 μM diluted in running buffer were then injected at 30 μL/min for 1 min association followed by 1 min dissociation. The affinity was calculated by fitting a saturation binding curve to the equilibrium response versus peptide concentration graph.
Plant VNP Preparations and Bioconjugation.
CPMV and TMV were propagated in black-eyed pea No. 5 or Australian tobacco plants by using mechanical inoculation and purification methods as we previously described.17,18 While denoted as TMV, we specify that a mutant of TMV was used: T158K,38 which presents 2130 reactive lysine residues on the external surface, therefore streamlining surface functionalization.
Fluorescent iCy5-TMV was produced as follows: TMV (10 mL at 1 mg/mL) was prepared in 10 mM HEPES (pH 7.4) and mixed with 100 mol equiv of propargylamine per a coat protein (CP). Then, 50 mol equiv of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC, Sigma-Aldrich) was added twice (0 and 6 h) to activate carboxyl groups in TMV for spontaneous reaction with propargylamine (50 equiv per CP, Sigma-Aldrich). The reaction was allowed to proceed for 12–16 h at room temperature. Resultant TMV-iAlk was purified by centrifugation at 160000g for 3 h and resuspended in 2 mL of 10 mM phosphate buffer (KP). Then 2 equiv of sulfo Cy5-azide (Lumiprobe) molecules per CP was added to the TMV solution in the presence of 2 mM aminoguanidine, 2 mM ascorbic acid, and 1 mM CuSO4 (Sigma-Aldrich) and reacted on ice for 30 min. Excess Cy5 dyes were removed by ultracentrifugation as detailed above. iCy5-TMV was resuspended in 10 mM KP at a concentration of 2 mg/mL and stored at 4 °C in the dark until further use.
S100A9-targeted TMV was produced by conjugation of iCy5-TMV with MTP2 and MTP-6 peptides as follows: For MTP2 conjugation, iCy5-TMV was first modified with a bivalent cross-linker (NHS-PEG8-Mal, SM(PEG)8, Thermo Fisher) by using 1 mol equiv per CP, and the reaction was allowed to proceed for 2 h at room temperature. Excess linker was removed by ultracentrifugation. The MTP2 peptide (0.5 equiv per CP) terminated by a Cys side chain was added to the intermediate nanoparticles (iCy5-TMV-Mal) and incubated for 2 h at room temperature on an orbital mixer. Excess peptide was removed by using Zeba spin desalting columns (Thermo Fisher) followed by dialysis against 10 mM KP (pH 7.2) overnight. Recovered iCy5-TMV-MTP2 was kept in 10 mM KP at a concentration of 2 mg/mL and stored at 4 °C in the dark until further use. To conjugate the MTP6 peptides, MTP6 was synthesized with a terminal azide with an intervening GGGS and poly(ethylene glycol) (PEG) spacer (three monomers) (MTP6-N3, JPT); a click chemistry protocol was used to obtain iCy5-TMV-MTP6: iCy5-TMV was modified with NHS-PEG2k-alkyne (NanoCS) by using 10 mol equiv per CP and incubation overnight at room temperature. Excess linkers were removed by ultracentrifugation over 40% sucrose cushion. The pellet was resuspended in 10 mM KP (pH 7.2). MTP6 was then attached to TMV-eAlk by using 2 equiv of MTP6-N3 per CP in a 2 mg/mL solution of TMV in 10 mM KP in the presence of 2 mM aminoguanidine, 2 mM ascorbic acid sodium salt, and 1 mM CuSO4 on ice for 30 min. To chelate the copper and stop the reaction, 5 mM EDTA was added for 5 min. iCy5-TMV-MTP6 nanoparticles were dialyzed against 10 mM KP overnight to remove excess peptides; the product was stored at 4 °C in the dark until further use.
CPMV-Cy5-MTP2 was synthesized as follows: An equimolar ratio (2000 equiv per CPMV) of sulfo-Cy5-NHS (Lumiprobe) and NHS-PEG8-Mal (SM(PEG)8, Thermo Fisher) was added to a 1 mL CPMV (2 mg/mL) solution and incubated for 2 h at room temperature. Excess Cy5 dye and linker were removed by ultracentrifugation (160000g for 3 h). The intermediate CPMV-Cy5-Mal nanoparticles were resuspended in 1 mL of 10 mM KP (pH 7.2). The Cys side chain terminated MTP2 was reacted by using a 2000 equiv per CPMV; the reaction was allowed to proceed for 2 h at room temperature. Excess peptides were removed via dialysis overnight 4 °C; the CPMV-Cy5-MTP2 particles were stored in KP buffer at 4 °C in the dark until further use.
Nanoparticle Characterization.
The resulting CPMV- and TMV-based nanoparticles were characterized by using UV/vis spectroscopy, SDS-PAGE, and transmission electron microscopy (TEM). The concentrations of TMV, CPMV, and Cy5 were determined by the Beer–Lambert law using an extinction coefficient (ε260nm) of 3 mg−1 mL cm−1 and a molecular weight of 39.4 MDa for TMV, an ε260nm of 8.1 mg–1 mL cm−1 and a molecular weight of 5.6 MDa for CPMV, and an ε646nm of 271000 M−1 cm−1 for Cy5. SDS-PAGE was performed with 10 μg of TMV or CPMV samples that were denatured at 100 °C for 5 min. The samples were run on a 4–12% NuPAGE gel (Invitrogen) in 1X MOPS running buffer at 200 V for 50 min. The gel was stained with Coomassie Blue and photographed with an AlphaImager system (Protein Simple). Lane analysis was performed using ImageJ software (http://imagej.nih.gov/ij). For TEM, samples were diluted to a concentration of 0.1 mg/mL in deionized water and applied to Formvar carbon-coated grids (Electron Microscopy Sciences) for 5 min. After excess liquid was blotted away, the grids were rinsed briefly with deionized water and then negatively stained with 2% uranyl acetate solution for 2 min. The samples were imaged at 200 kV with a Zeiss Libra 200FE microscope.
Binding Affinity of MRP-14 Targeted TMV.
We coated 100 μL of hS100A9 or mS100A9 (2 μg/mL, R&D Systems) in 10 mM KP buffer onto the wells of a 96-well plate overnight at 4 °C. After being washed three times with PBS, the plates were blocked with 1% (w/v) bovine serum albumin (BSA, Sigma-Aldrich) in PBS for 1 h. MRP-14-targeted and control TMV (0–5 μg/mL) in 10 mM KP with 1% (w/v) BSA were applied and incubated for 2 h followed by three washings with 10 mM KP. Detection was performed with a polyclonal rabbit α-TMV antibody (100 μL, 0.2 μg/mL in 10 mM PBS containing 1% (w/v) BSA; for 2 h; Pacific Immunology), followed by four washes with PBS, and addition of an α-rabbit IgG conjugated with horseradish peroxidase (HRP; 100 μL, 1:4000 dilution in PBS with 1% (w/v) BSA, for 1 h; Thermo Fisher), followed by four washes with PBS. Colorimetric detection was then performed by using TMB substrate according to the manufacturer’s protocol. The absorbance at 450 nm was measured by using an Infinite 200 PRO (Tecan). The binding affinity (i.e., dissociation constant, KD) of the S100A9-targeted TMV nanoparticles was determined by nonlinear regression analysis using Sigmaplot software and a single-site binding model.
Animal Studies.
DVT Induction.
All animal procedures were performed by using approved protocols from the Institutional Animal Care and Use Committee at Case Western Reserve University. Three-month-old C57BL/6J male mice, fed with alfalfa-free rodent diets (2016 Teklad global 16% protein) for at least 1 week, were subjected to stasis venous thrombus induction by surgery following previously described methods.39 Briefly, once anesthetized, mice underwent laparotomy, exteriorization of the intestines, and exposure of the infrarenal inferior vena cava (IVC). Intestines were kept moist by saline-soaked sterile gauze throughout the procedure. All visible side branches proximal to the iliac bifurcation were ligated with 7–0 polypropylene sutures. The back branches were left patent. The IVC was then isolated away from the aorta and then ligated immediately distal to the renal veins with 6–0 polypropylene suture. Exteriorized organs were replaced, and the peritoneum was closed by using 6–0 polypropylene in a continuous suture, followed by closure of the skin with a 6–0 nylon suture in a simple interrupted pattern.
Nanoparticle Injection and Imaging.
For imaging experiments, 24 h post DVT induction by surgery, mice were divided into groups randomly, and mice were intravenously injected with various Cy5-labeled nanoparticle formulations (PBS, CPMV-PEG, CPMV-MTP2, TMV-PEG, and TMV-MTP2) at a dose of 100 μg/g body weight via the tail vein. After 3 h of circulation of nanoparticles in vivo, mice were euthanized, and the previously ligated IVC with developed thrombi were harvested. Livers were harvested and imaged as controls to monitor that injections were successful. The collected IVCs with thrombi and livers were fixed in 4% (v/v) paraformaldehyde/PBS solution overnight at 4 °C before transferred into PBS. After scanning, tissues surrounding IVC were removed carefully. The clean IVC with thrombi were then imaged by using a Spectrum and Maestro imaging system to detect the Cy5 fluorescence signal accumulated in the thrombi. The obtained images and radiant efficiency were quantitatively analyzed after background signal subtraction.
Histology and Immunofluorescence.
After imaging, each of thrombosed veins were carefully cut into two segments, and one segment was processed for frozen and another for paraffin sections. For frozen sections, thrombi were embedded in optimal cutting temperature (OCT) media, flash frozen by using dry ice and liquid nitrogen, and stored at −80 °C until sectioning using a cryostat. For paraffin sections, thrombi were fixed in 4% (v/v) paraformaldehyde/PBS solution overnight before processed for embedding in paraffin. We cut 7 μm cryosections for all histological analyses. For immunofluorescence staining, cryosections of thrombosed veins were fixed with chilled acetone at −20 °C and then blocked with 20% (v/v) rabbit serum for 30 min at room temperature. Samples were incubated with goat against mouse-MRP-14 (mouse S100A9 antibody, AF2065, 10 μg/mL) at room temperature for 1 h. Following a wash, biotin-conjugated rabbit antigoat antibody (1.4 μg/mL, catalog 305-065-0003, Jackson ImmunoResearch Laboratories) was applied for 60 min at room temperature. Following another wash, fluorescein-conjugated (DTAF-conjugated) streptavidin (2 μg/mL, catalog 016-010-084, Jackson ImmunoResearch Laboratories) was added to the samples and incubated for 30 min in the dark. DNA was visualized with DAPI containing mounting medium (Southern Biotech) after a third wash. Sections were examined, and images were taken by using a microscope equipped with a fluorescent filter cubes for DAPI, fluorescein, and Cy5 to visualize MRP-14 and Cy5-conjugated CPMV or TMV nanoparticles.
Statistical Analysis.
Statistical analyses were performed using GraphPad Prism and SPSS (v27, IBM) software. Data are presented as mean + STD. Statistical significance between the groups was compared by ordinary one-way ANOVA using Tukey’s HSD post hoc test.
RESULTS AND DISCUSSION
Identification of Peptides Specific for MRP-14.
Peptides with affinity to MRP-14 were selected for through three rounds of solution-phase panning by using both linear and cyclic phage display peptide libraries based on bacteriophage M13 from New England Biolabs. Thirty candidates from the linear and 31 candidates from the cyclic library were sequenced, and a set of MRP-14 targeting peptide (MTP) sequences were identified (Table 1). A common motif between the various sequences is the presence of several histidine residues. The recurrence of the positively charged residue indicates that the MRP-14 binding site is shared between the identified peptides, possibly coinciding with one of its calcium-binding domains.
Table 1.
MRP-14 Binding Peptides
| linear Ph.D.-7 library | cyclic Ph.D.-C7C library | ||||
|---|---|---|---|---|---|
| label | sequence | freq | label | sequence | freq |
| MTP1 | TTFHHHK | 2 | MTP4 | ACKAPAHHHC | 1 |
| MTP2 | ASSHIHH | 26 | MTP5 | ACNSHRHGAC | 15 |
| MTP3 | HNHMADP | 2 | MTP6 | ACNSVHQHHC | 11 |
| MTP7 | ACYGKPEHHC | 1 | |||
| MTP8 | ACPHHPQKHC | 1 | |||
| MTP9 | ACPTGLHHAC | 1 | |||
| MTP10 | ACRPKNNHSC | 1 | |||
To narrow down the peptide library to the ones with highest affinity, an enzyme-linked immunosorbent assay (ELISA) was performed with the isolated phages. ELISA plates were coated with MRP-14 and then incubated with varying concentrations of each phage, ranging from 6 × 107 to 1012 phages/mL. The bound phage was detected by using horseradish peroxidase (HRP)-labeled anti-M13 antibodies followed by addition of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS) substrate (Figure 1A). The specificity for MRP-14 was confirmed by high binding of the phages to MRP-14 but not bovine serum albumin (BSA, negative control, Figure 1B). Additionally, unlabeled M13 did not show affinity for MRP-14, indicating the binding of the phages is conferred by the peptide displayed. Based on the phage ELISA results, six peptides were selected for further analysis: MTP2, MTP3, MTP5, MTP6, MTP9, and MTP10 with sequences ASSHIHH, HNHMADP, ACNSHRHGAC, ACNSVHQHHC, ACPTGLHHAC, and ACRPKNNHSC, respectively.
Figure 1.

Peptide identification and ELISA characterization. (a) Phage ELISA to determine binding to MRP-14 by using 4-fold serial dilutions of phages from a starting concentration of 1012 pfu/mL to a final concentration of 6 × 107 pfu/mL. Absorbance values are after background subtraction. (b) For simplicity, values of background binding to BSA and specific binding to MRP-14 are shown for a single dilution (2.5 × 1011 pfu/mL) to more easily compare the affinity of the various displayed peptides. Data shown is one of two replicates, with no statistically significant difference observed between the high binders (MTP2, MTP3, MTP5, MTP6, MTP9, and MTP10).
To determine the binding affinity of the identified MRP-14 binders, peptides were synthesized with a glycine GGGS spacer at the C-terminus along with C-terminal amidation to best match their structure and charge when displayed on M13. Their binding affinity to MRP-14 was then quantified by using surface plasmon resonance (SPR) with an NTA sensor chip. After charging with Ni2+, His-tagged MRP-14 was immobilized on the chip. Then, binding of peptides with concentrations from 6.25 to 100 μM was evaluated, and their dissociation constant (KD) was determined through saturation binding curve fitting (Figure 2 and Table 2). The peptide with the strongest affinity was MTP6 (ACNSVHQHHC), with the lowest KD of 39.6 μM. While the two strongest binders were cyclic (MTP5 and MTP6), the linear peptide MTP2 showed comparable affinity with a KD of 77.6 μM (Table 2). MTP9 demonstrated distinct binding kinetics with fast initial interactions followed by a slower binding phase, which may point to multiple binding sites. Interestingly, while MTP10 was identified as one of the lead candidates based on ELISA (Figure 1), SPR measurements could not confirm specificity. The difference between the assays is the use of free versus phage-bound peptide for SPR vs ELISA.
Figure 2.

SPR analysis of MRP-14 binders. SPR sensorgram data for peptide binding to immobilized MRP-14 and determination of the dissociation constant using a rectangular hyperbola fit of the saturation binding curve are shown as pairs for all peptides, including a scrambled control and azide-functionalized MTP6. KD values are also presented in Table 2. All data shown are one of two similar experiments, and analysis was performed with Prism software. The scrambled peptide experienced low nonspecific binding, and the data could not be fit. MTP9 had a unique profile compared to the others and was fit by using a kinetic curve rather than an equilibrium affinity binding curve.
Table 2.
Values for Dissociation Constants of MRP-14 Binders from SPR Measurements (Italic Font Denotes Cyclic Peptides)
| label | sequence | KD (μM) |
|---|---|---|
| scrambled | MDPHAHNGGGS | no binding |
| MTP2 | ASSHIHHGGGS | 77.6 |
| MTP3 | HNHMADPGGGS | 116.8 |
| MTP5 | ACNSHRHGACGGGS | 67.6 |
| MTP6 | ACNSVHQHHCGGGS | 39.6 |
| MTP9 | ACPTGLHHACGGGS | 190.9 |
| MTP10 | ACTPKNNHSCGGGS | 191.0 |
Because MRP-14 in vivo predominantly exists as a heterodimer with MRP-8,30,31 binding of MTP6 to MRP-8/14 was also investigated by SPR (Figure 3). MRP-8/14 was formed by mixing equal molar concentrations of MRP-8 with MRP-14, with only MRP-14 having a His tag. Immobilization of MRP-8/14 was performed under the same conditions as for MRP-14, and a response of 3600 response units (RU) was observed, which when compared to a response of around 1800 RU for MRP-14 alone indicated successful dimerization of MRP-8/14. For the association of MTP6 with MRP-8/14, the KD was determined to be 57.6 μM, with similar response curves as observed for MRP-14. Therefore, heterodimer formation does not appear to interfere with binding of the peptide.
Figure 3.

SPR analysis of MTP6 binding to MRP-8/14. SPR sensorgram data for peptide binding to immobilized MRP-8/14 (left) and determination of the dissociation constant using a rectangular hyperbola fit of the saturation binding curve (right).
Overall, the affinity of the identified peptides to MRP-14 is only moderate (for comparison, antibodies have KD values in the low micromolar to nanomolar (10−6 to 10−9) range). One thing to note is that the MRP-14 binding peptides all consist of at least one His residue, and the SPR measurements involve MRP-14 immobilization to the sensor chip using His tags. While the protocol ensures that the His tags are oriented away from the free peptides, it is possible that there were charge-based repellent effects that reduced the measured affinity even though we were still able to observe specific binding. Additionally, we hypothesized that affinity would improve upon multivalent display of the MRP-14 targeting peptides on nanoparticles. We therefore proceeded with a couple of the peptides for further evaluation. In particular, we selected MTP2 and MTP6 as representative linear and cyclic peptides, respectively, based on their KD values. Despite its lower affinity overall, MTP5 may be another interesting candidate to explore for future studies to dissect whether the slower dissociation rate that was observed from the SPR binding kinetics would result in a different downstream effect.
MRP-14-Targeted TMV and CPMV Nanoparticles.
For the nanoparticle carriers, we turned to CPMV and TMV, which were obtained by molecular farming in plants using mechanical inoculation. For all studies, wild-type CPMV was used, while for TMV, we utilized a T158 K38 mutant. The T158 K mutant presents 2130 reactive Lys residues on its external surface, therefore streamlining surface functionalization and providing a good match with native CPMV, which offers 300 addressable Lys side chains.40 Functionalization of the particles was performed by using well-established reactions: NHS-, maleimide-, and carbodiimide-based chemistries as well as Cu(I)-catalyzed azide–alkyne cycloaddition (click reaction).41–44 The reactions for attaching MTP2 and MTP6 peptides to the particles are outlined in Figure 4.
Figure 4.

Bioconjugation of MTP2 and MTP6 peptides to CPMV and TMV. Cys or N3 functional groups were added to the C terminus of the peptides via a GGGS spacer; it should be noted that the MTP6 peptide is a cyclic peptide, and cyclization is achieved through formation of a disulfide bridge between the Cys at positions 2 and 10.
MTP6 peptides were synthesized with a terminal N3, and MTP2 peptides were synthesized with either a terminal Cys or N3. The azide-terminated peptides were then attached via a click reaction to NHS-PEG4-alkyne-modified VNPs, while the thiol-terminated peptides were attached to NHS-PEG8-Mal-modified VNPs. To further enable in vivo tracking, Cy5 fluorescent labeling was also performed. iCy5-TMV was produced via two steps: (1) attachment of alkyne functional groups using carbodiimide chemistry targeting Glu side chains on the solvent-exposed surface or within the central channel of TMV44 and (2) attachment of Cy5-azide molecules via click conjugation.45 On the other hand, Cy5 was conjugated to CPMV by using Cy5 functionalized with NHS-active esters.
Resulting nanoparticles were characterized by using UV/vis spectroscopy to determine the number of Cy5 molecules per particle, SDS-PAGE to confirm covalent attachment of the Cy5 dye and MTP2/6 peptides, and TEM to verify that the fluorescent and targeted VNP formulation remained structurally sound upon undergoing several rounds of bioconjugation and purification. UV/vis spectroscopy confirmed the presence of fluorescent nanoparticles as shown by absorbance in the near-infrared range (with an absorbance peak at 654 nm characteristic for Cy5). By use of the Beer–Lambert law, the number of dyes per VNP was determined: ~110 Cy5 per iCy5-TMV-MTP2 and ~14 Cy5 per CPMV-Cy5-MTP2 (Figure 5A) yielding bright nanoparticle formulations; the dye density was chosen to avoid quenching.46 The differences in dye loading can be explained by the different sizes of the TMV measuring 300 × 18 nm2 vs CPMV, which measures 30 nm in diameter. Covalent attachment of the fluorophore and targeting peptides was also confirmed by using SDS-PAGE (Figure 5B): The TMV coat protein (CP) was detectable at ~19 kDa, and the small (S) and large (L) coat proteins of CPMV were detected at ~24 and 42 kDa, respectively. While the tubular nanostructure formed by TMV consists of 2130 identical copies of a single CP,47 CPMV nanoparticles are formed by 60 copies each of a small (S) and large (L) protein.48 Fluorescence imaging of the Cy5-labeled VNPs in gel was also consistent with the presence of fluorescent CPs. In addition, bands with higher molecular weight are detectable in the MTP2/6 peptide-modified VNP formulations, which results from linker and peptide conjugation to the CPs. Densitometric analysis of the band profiles using ImageJ software indicated about 37% or 19% of the CPs modified with MTP2 or MTP6, respectively, or approximately 790 MTP2 or 400 MTP6 peptides per TMV-MTP2/6 nanoparticle (Figure 5B). Similarly, SDS-PAGE profiles of the CPMV nanoparticles also confirmed covalent labeling with Cy5 dye and MTP2; the band pattern was less obvious compared to the TMV nanoparticles. However, additional molecular weight bands were identified for the S and L protein (as expected, because Lys side are present on both protein subunits40). Densitometry analysis indicated labeling of CPMV with ~60 MTP2 per CPMV. Lastly, TEM revealed that the nanoparticle formulations remained intact during modification; high aspect ratio nanotubes are shown for TMV-MTP2/6, and icosahedral (sphere-like) nanoparticles are detected for CPMV-MTP2 (Figure 5C).
Figure 5.
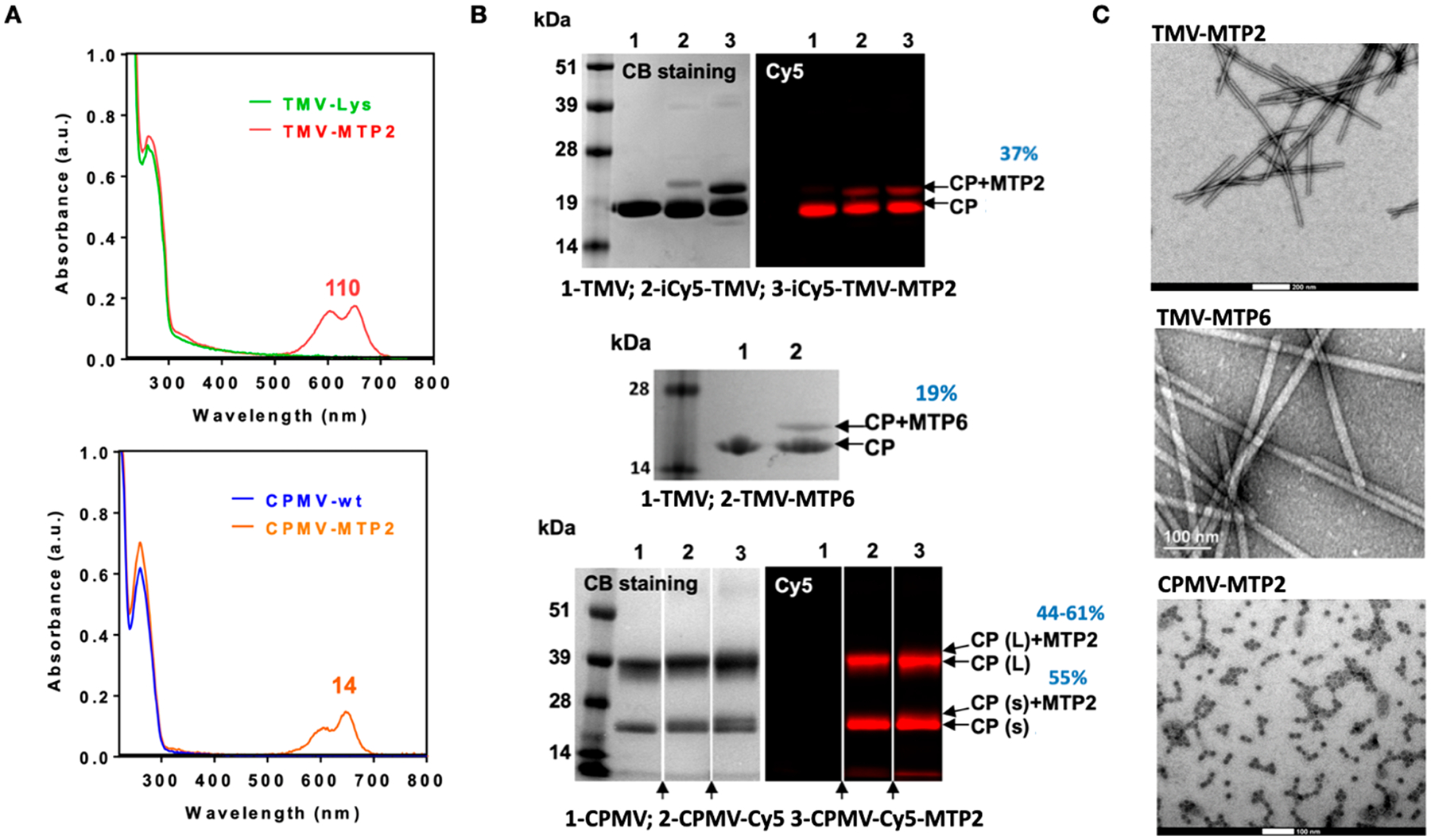
Characterization of CPMV and TMV-MTP2/6 nanoparticle formulations. (A) Cy5 dye loading on TMV and CPMV was quantified by UV/vis spectroscopy, allowing the number of Cy5 molecules per particle to be calculated by using the Beer–Lambert law and the extinction coefficient of TMV/CPMV and Cy5. (B) SDS PAGE analysis under white light and fluorescence imaging was used to confirm the modification of the TMV and CPMV with Cy5 (as detected by fluorescence imaging on the right) and MTP2/6 as detected by additional protein bands (additional to the CP) marked by arrows as CP-MTP2/6. Arrows at the bottom of the CPMV gel indicate that lanes were omitted (these were samples not relevant to the present paper). (C) TEM images confirmed structural integrity of TMV-MTP2/6 and CPMV-MTP2. The scale bars are 200 nm for TMV-MTP2, 100 nm for TMV-MTP6, and 100 nm for CPMV-MTP2.
To verify that the MRP-14-targeted TMV-MTP2/6 nanoparticles maintained their affinity to MRP-14, we performed an ELISA-based assay to estimate the dissociation constant of TMV-MTP2/6 to mouse and human MRP-14 (this assay was not performed with the CPMV nanoparticles). While MTP peptides were produced by phage display against recombinant human MRP-14, MRP-14 proteins for two species are highly homologous as determined by BLAST. Data indicate high affinity of the nanoparticle preparations with affinities in the low nanomolar range (KD = 2.6–13.1 nM) toward both mouse and human MRP-14 (Table 3). Therefore, the dissociation constant of the nanoparticle formulation is about 4 orders of magnitude lower than what was found for the free peptides (see Table 2), indicating a much higher binding affinity. This higher affinity can be attributed to the cooperative effect from display of multiple peptides on a single carrier (400–800 peptides per TMV; see Figure 5).
Table 3.
Estimation of KD Values of MRP-14-Targeted TMV Nanoparticles
| KD (nM) | |||
|---|---|---|---|
| peptide sequence | human MRP-14 | mouse MRP-14 | |
| TMV-MTP6 | ACNSVHQHHCGGGS-N3 | 11.4 | 2.6 |
| TMV-MTP2 | ASSHIHHGGGS-C | 13.1 | 11.9 |
Of note, TMV-MTP6 had a lower dissociation constant for both human and mouse MRP-14 compared to TMV-MTP2. However, TMV-MTP2 exhibited more consistent behavior between the two forms, while in contrast, the binding affinity of TMV-MTP6 appeared to be ~4 times stronger toward mouse than human MRP-14. Despite the equivalent functionality of mouse and human MRP-14, the two proteins only share 59% sequence homology.32 It is therefore highly likely that this structural difference accounts for the difference observed in binding behavior. To have a greater likelihood of translatability of conclusions derived from the animal model, we progressed with MTP2 for further studies. MTP2 also has the added advantage of being a linear peptide for easier synthesis.
DVT Homing of MRP-14-Targeted TMV and CPMV Nanoparticles.
To investigate the in vivo targeting capability of CPMV and TMV nanoparticles conjugated with MRP-14 binding peptide (MTP2), we employed a murine model of DVT. Three-month-old C57BL/6J male mice were fed with an alfalfa-free rodent diet for at least 1 week to reduce autofluorescence background signal prior to being subjected to deep vein thrombus induction by inferior vena cava (IVC) ligation and partial branch ligation as previously described.39 Twenty-four hours postsurgery induction of DVT, mice were injected via the tail vein with PBS (control) (n = 6 for CPMV, n = 4 for TMV), CPMV-PEG (n = 4), CPMV-MTP2 (n = 6), TMV-PEG (n = 6), or TMV-MTP2 (n = 5). Three hours postinjection, the mice were euthanized to harvest thrombi, followed by ex vivo imaging using Maestro and Spectrum Fluorescence Imaging systems. Livers were harvested and imaged as well to serve as controls to verify successful injection (the nanoparticles were cleared via hepatic filtration). Imaging and subsequent quantitative analysis of the images confirmed preferential homing of MRP-14 targeted nanoparticles to DVT; both CPMV-MTP2 and TMV-MTP2 showed significantly enhanced accumulation in the thrombi, with some background binding of the PEGylated versions (CPMV-PEG and TMV-PEG) also observed (Figure 6A,B). PEGylated nanoparticles were chosen to serve as controls in this instance due to the fact that CPMV and TMV are themselves proteinaceous carriers that account for nonspecific protein binding. Because PEG linkers were used for peptide conjugation, this side-to-side comparison allows for observation of the net change resulting from addition of MTP2.
Figure 6.
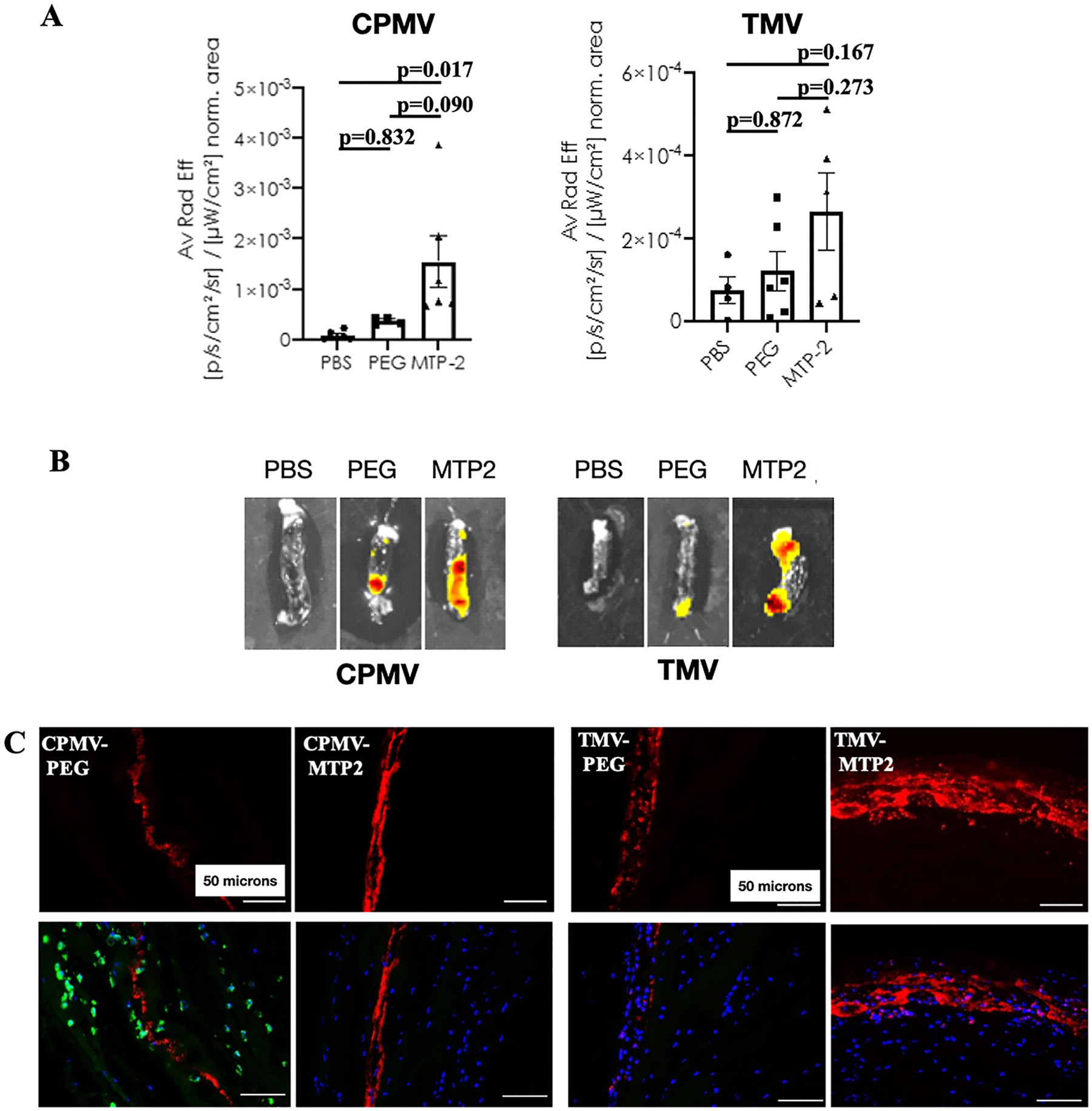
In vivo targeting of deep vein thrombi using CPMV-MTP2 and TMV-MTP2 nanoparticles in the murine model of deep vein thrombosis. (A) Quantification of Cy5 signal from CPMV-MTP2 and TMV-MTP2 in thrombi. Data were normalized to thrombus area of each individual mouse, and each dot represents one individual thrombus from one mouse. Statistical analysis was performed by using one-way ANOVA. Data normalized to thrombus weight is consistent with that normalized to area. (B) Corresponding representative ex vivo fluorescence images of deep vein thrombi harvested 3 h postintravenous administration of the nanoparticles. (C) Immunofluorescence of cryosectioned thrombi showing the Cy5 signals of nontargeted (CPMV-PEG and TMV-PEG) and MRP-14 targeted nanoparticles (CPMV-MTP2 and TMV-MTP2) in deep vein thrombi. CPMV-PEG sections were also stained with MRP-14 (green). Red: nanoparticles; blue: nuclei; green: MRP-14. Scar bar: 50 μm.
Consistent with the results of scanning of the whole thrombus, analysis of nanoparticles in microsections of thrombi also revealed a stronger accumulation of MRP-14-targeted nanoparticles (TMV-MTP2 and CPMV-MTP2) compared to the nontargeted nanoparticles (TMV-PEG and CPMV-PEG) (Figure 6C). Overall, the data were consistent comparing CPMV and TMV in terms of observed targeting benefits.
To gain further insight into the localization and distribution of the MRP-14-targeted TMV or CPMV within the thrombi, we pursued immunofluorescence imaging of cryosectioned IVC thrombi. It is important to note that MRP-14 has both intracellular and extracellular forms, and it is expressed and secreted by both neutrophils and platelets.39 To distinguish between these two forms, 7 μm thick cryosections of venous thrombi were stained for MRP-14, and MRP-14 targeted Cy5-labeled CPMV or TMV nanoparticles were visualized directly without staining based on the Cy5 label. The MRP-14-targeted TMV or CPMV nanoparticles predominantly accumulated in areas close to the edge of the thrombi, and the majority of the nanoparticles did not penetrate into the center of the thrombi (Figure 7A). This may be explained by the fact that the nanoparticles were injected 24 h after thrombi induction and formation; the dense thrombi cannot be penetrated deeply by the nanoparticles within the 3 h window (thrombi were harvested 3 h post-nanoparticle injection). The fact that the nanoparticles were not apparent at the intima of the thrombi, i.e., at the surface, but accumulated right inside the edge of the thrombi may indicate thrombus growth rather than nanoparticle penetration. The sharp layer of nanoparticle signal may indicate that the nanoparticles accumulated at the intima of the thrombi at the time of injection, but the thrombi continued to grow within the 3 h window post-nanoparticle administration (Figure 7B). The major components of thrombi include platelets, red blood cells, fibrin mesh, and neutrophils. While neutrophil-expressed MRP-14 was confirmed (Figure 7C), under these experimental conditions, we did not observe a direct overlapping of Cy5 signal with intracellular form of MRP-14 that were expressed by the leukocytes/neutrophils (Figure 7D), perhaps due to the short time period of nanoparticle circulation in vivo, which was not long enough for the nanoparticles to be internalized by the cells/neutrophils. The enhanced Cy5 signals of CPMV-MTP2 and TMV-MTP2 particles were predominantly due to the binding of MTP2-nanoparticles to extracellular pools of MRP-14 and circulating MRP-14 (Figure 7D,E).
Figure 7.

Immunofluorescence (IF) images of sectioned deep vein thrombi in inferior vena cava. (A) Cross sections of whole thrombus showing bright-field image, nuclei (blue), and nanoparticles (red). (B) IF images of sections showing nanoparticles (red), MRP-14 (green), and nuclei (blue). (C) MRP-14 (red) expression by neutrophils (Ly6G-green) in DVT. (D) MRP-14 (green) and nanoparticles (red) in sections of thrombi. (E) Merged images of thrombi from mice injected with PBS, CPMV-PEG, CPMV-MTP2, TMV-PEG, and TMV-MTP2. The thrombi were stained for MRP-14 and DAPI for nuclei. Scale bars: 250 μm in (A), 50 μm in (B), 20 μm in (C, D), and 50 μm in (E). Arrows indicate the edge of the thrombus.
Data showed that both CPMV-MTP2 and TMV-MTP2 nanoparticles targeting MRP-14 accumulated to a higher degree at sites of DVT compared to nontargeted CPMV-PEG and TMV-PEG control nanoparticles. Side-by-side comparisons of the Cy5 signals from the fluorescent TMV and CPMV nanoparticles in DVT sections revealed that TMV nanoparticles showed a stronger Cy5 signal and accumulated more in DVT. However, TMV also generated higher background and larger variations than CPMV nanoparticles did. Therefore, the difference between TMV-PEG and TMV-MTP2 did not reach significance due to the relative small sample size and the variation. This data may suggest that the smaller CPMV-MTP2 nanoparticles may give more consistent results.
In addition to size, other potential reasons for the differences observed between CPMV and TMV are their distinct shapes as well as differences in surface presentation. Our previous works have described that the higher aspect ratio TMV nanoparticle could be advantageous for cardiovascular disease applications due to experiencing lateral torque and having a higher likelihood of margination to the vessel wall compared to spherical nanoparticles, which are more likely to remain in laminar flow.25,29,49 Because the shape of the thrombus would affect the flow profile and would be highly variable across each mouse, this may account for the lower consistency observed for TMV. Another factor to consider is the potentially different levels and distribution of surface presentation of MTP2 on the VNPs, which could influence the binding affinity. Given that the rod-shaped TMV particle has a higher surface area to volume ratio compared to CPMV, it is expected that would result in higher peptide presentation and thus higher binding. Further exploration is required to tease out the various contributions of these various factors.
Interestingly, we also noticed reduced thrombus weights for the MRP-14 targeted CPMV nanoparticle group compared to CPMV-PEG or PBS group, with CPMV-MTP2 thrombi measuring 10.3 ± 12.8 mg (n = 7) vs CPMV-PEG: 12.7 ± 10.6 mg (n = 7) vs PBS: 20.1 ± 4.9 mg (n = 4), which is consistent with the observed thrombus areas in these groups (CPMV-MTP2:6.6 ± 8.4 mm2; CPMV: 8.6 ± 7.0 mm2; PBS: 13.1 ± 3.7 mm2) (Figure 8A). Thrombus weight and area were reduced by 50% for the CPMV-MTP2 treatment group. Similarly, we also observed reduced thrombus weight and area for the TMV vs PBS groups, but there was no difference between TMV-PEG and TMV-MTP2 groups (TMV-MTP2: 13.7 ± 8.9 mg, n = 7; TMV-PEG: 12.1 ± 6.2 mg, n = 7; PBS: 20.1 ± 7.0 mg, n = 6), which is in agreement with thrombus areas (TMV-MTP2: 8.5 ± 6.7 mm2, n = 7; TMV-PEG: 8.3 ± 4.3 mm2, n = 7; PBS: 14 ± 6.0 mm2, n = 6); with TMV a 35% reduction in thrombus weight and area was observed (Figure 8B). Regardless of the reduced size and high frequency of no thrombus formation in nanoparticle injected groups, due to the relative small sample size and the large variations of thrombus size in this DVT model,50,51 the difference of the thrombus weights does not reach significant levels based on the statistical analyses we performed. Future study with enlarged sample size would provide more clarification. It is known that secreted, platelet-derived MRP-14 directly regulates thrombosis, with CD36 identified as the platelet receptor required for MRP-14 action.36 Because our nanoparticles are primarily interacting with extracellular MRP-14, one potential avenue for thrombus disruption is through this pathway by preventing MRP-14 binding to platelets. It is also possible that innate immune cell activation may play a role; however, the nanoparticles were only allowed to circulate for 3 h. Future longitudinal studies are required to delineate the true mechanism.
Figure 8.

Thrombus weight and area from deep vein thrombi in inferior vena cava. (A) DVT weight and area from PBS, CPMV-PEG, and CPMV-MTP2 treatment groups and (B) corresponding data for TMV treatment groups. Data are presented as mean + STD. Statistical significance between the groups was compared by ANOVA using Tukey’s HSD post hoc test.
In the future, several avenues could be pursued, including delving deeper into the mechanisms of nanoparticle accumulation and the subsequent decrease in thrombus size, pairing this with our previous work using these nanoparticles for delivery of thrombolytics to potentially achieve enhanced therapeutic effect,27,28 and fine-tuning the specificity of the nanoparticle-based delivery systems through improving the targeting of MRP-14. While we chose to explore MTP2-conjugated nanoparticles as a proof-of-concept for in vivo targeting of MRP-14 for DVT, other peptides could also be further explored, such as MTP5, which was mentioned earlier in the text as warranting further investigation given its lower dissociation rate.
CONCLUSIONS
To tackle the challenge of achieving more accurate diagnosis and treatment of DVT, we investigated molecular targeting of MRP-14 using plant virus-based nanoparticle platforms. To this end, peptides specific for MRP-14 were successfully identified by using phage display techniques, and we observed that multivalent display of the MTP2 peptide sequence ASSHIHHGGGS-C on CPMV and TMV nanoparticles resulted in enhanced binding affinity for surgically induced DVT in an in vivo mouse model. In addition to enhanced targeting, some therapeutic effect was also observed from these intravenously delivered nanoparticles in terms of both the size and weight of the thrombi that were harvested. Therefore, these nanoparticles demonstrate a favorable profile that could be used for the development of next-generation diagnostics and safer therapeutic interventions, with the goal of reducing disease burden and improving long-term outcomes.
ACKNOWLEDGMENTS
This work was funded in part by a grant from the National Institutes of Health R01 HL137674 to N.F.S. and fellowship F31 HL129703 to A.M.W. M.S. was supported in part by CBI training program NIH T32GM135142.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biomac.1c00303
The authors declare no competing financial interest.
Contributor Information
Jooneon Park, Department of NanoEngineering, University of California, San Diego, La Jolla, California 92093, United States.
Amy M. Wen, Department of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States
Huiyun Gao, Department of Medicine, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Matthew D. Shin, Department of NanoEngineering, University of California, San Diego, La Jolla, California 92093, United States
Daniel I. Simon, Department of Medicine, Case Western Reserve University, Cleveland, Ohio 44106, United States
Yunmei Wang, Department of Medicine, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Nicole F. Steinmetz, Department of NanoEngineering, Department of Bioengineering, Department of Radiology, Center for Nano-ImmunoEngineering, Moores Cancer Center, and Institute for Materials Discovery and Design, University of California, San Diego, La Jolla, California 92093, United States.
REFERENCES
- (1).Beckman MG; Hooper WC; Critchley SE; Ortel TL Venous thromboembolism: a public health concern. Am. J. Prev. Med 2010, 38 (4), S495–S501. [DOI] [PubMed] [Google Scholar]
- (2).Pulivarthi S; Gurram MK Effectiveness of d-dimer as a screening test for venous thromboembolism: an update. N Am. J. Med. Sci 2014, 6 (10), 491–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Crippa L; D’Angelo SV; Tomassini L; Rizzi B; D’Alessandro G; D’Angelo A The utility and cost-effectiveness of D-dimer measurements in the diagnosis of deep vein thrombosis. Haematologica 1997, 82 (4), 446–51. [PubMed] [Google Scholar]
- (4).Wheeler HB; Anderson FA Jr. Diagnostic methods for deep vein thrombosis. Pathophysiol. Haemostasis Thromb 2004, 25 (1–2), 6–26. [DOI] [PubMed] [Google Scholar]
- (5).Fraser JD; Anderson DR Deep venous thrombosis: recent advances and optimal investigation with US. Radiology 1999, 211 (1), 9–24. [DOI] [PubMed] [Google Scholar]
- (6).Screaton NJ; Gillard JH; Berman LH; Kemp PM Duplicated superficial femoral veins: a source of error in the sonographic investigation of deep vein thrombosis. Radiology 1998, 206 (2), 397–401. [DOI] [PubMed] [Google Scholar]
- (7).Taillefer R Radiolabeled peptides in the detection of deep venous thrombosis. Semin. Nucl. Med 2001, 31 (2), 102–23. [DOI] [PubMed] [Google Scholar]
- (8).Cronan JJ Venous thromboembolic disease: the role of US. Radiology 1993, 186 (3), 619–30. [DOI] [PubMed] [Google Scholar]
- (9).National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med 1995, 333 (24), 1581–1588. [DOI] [PubMed] [Google Scholar]
- (10).Sachs UJ; Nieswandt B In vivo thrombus formation in murine models. Circ. Res 2007, 100 (7), 979–91. [DOI] [PubMed] [Google Scholar]
- (11).Shetty RC Benefits Of Nanotechnology In Cardiovascular Surgery—A Review Of Potential Applications. US Cardiology 2006, 3, 95–96. [Google Scholar]
- (12).Sen Gupta A Bio-inspired nanomedicine strategies for artificial blood components. Wiley Interdiscip Rev. Nanomed Nanobiotechnol 2017, 9 (6), e1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Li T; Liang W; Xiao X; Qian Y Nanotechnology, an alternative with promising prospects and advantages for the treatment of cardiovascular diseases. Int. J. Nanomed 2018, 13, 7349–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Karimi M; Zare H; Bakhshian Nik A; Yazdani N; Hamrang M; Mohamed E; Sahandi Zangabad P; Moosavi Basri SM; Bakhtiari L; Hamblin MR Nanotechnology in diagnosis and treatment of coronary artery disease. Nanomedicine (London, U. K.) 2016, 11 (5), 513–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Deng Y; Zhang X; Shen H; He Q; Wu Z; Liao W; Yuan M Application of the Nano-Drug Delivery System in Treatment of Cardiovascular Diseases. Front. Bioeng. Biotechnol 2020, 7, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chariou PL; Ortega-Rivera OA; Steinmetz NF Nanocarriers for the Delivery of Medical, Veterinary, and Agricultural Active Ingredients. ACS Nano 2020, 14 (3), 2678–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Leong HS; Steinmetz NF; Ablack A; Destito G; Zijlstra A; Stuhlmann H; Manchester M; Lewis JD Intravital imaging of embryonic and tumor neovasculature using viral nanoparticles. Nat. Protoc 2010, 5 (8), 1406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bruckman MA; Steinmetz NF Chemical modification of the inner and outer surfaces of Tobacco Mosaic Virus (TMV). Methods Mol. Biol 2014, 1108, 173–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Chen Z; Li N; Li S; Dharmarwardana M; Schlimme A; Gassensmith JJ Viral chemistry: the chemical functionalization of viral architectures to create new technology. Wiley Interdiscip Rev. Nanomed Nanobiotechnol 2016, 8 (4), 512–34. [DOI] [PubMed] [Google Scholar]
- (20).Manchester M; Singh P Virus-based nanoparticles (VNPs): platform technologies for diagnostic imaging. Adv. Drug Delivery Rev 2006, 58 (14), 1505–22. [DOI] [PubMed] [Google Scholar]
- (21).Wen AM; Steinmetz NF Design of virus-based nanomaterials for medicine, biotechnology, and energy. Chem. Soc. Rev 2016, 45, 4074–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shukla S; Hu H; Cai H; Chan SK; Boone CE; Beiss V; Chariou PL; Steinmetz NF Plant Viruses and Bacteriophage-Based Reagents for Diagnosis and Therapy. Annu. Rev. Virol 2020, 7 (1), 559–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sokullu E; Soleymani Abyaneh H; Gauthier MA Plant/Bacterial Virus-Based Drug Discovery, Drug Delivery, and Therapeutics. Pharmaceutics 2019, 11, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Abrahamian P; Hammond RW; Hammond J Plant Virus-Derived Vectors: Applications in Agricultural and Medical Biotechnology. Annu. Rev. Virol 2020, 7 (1), 513–535. [DOI] [PubMed] [Google Scholar]
- (25).Bruckman MA; Jiang K; Simpson EJ; Randolph LN; Luyt LG; Yu X; Steinmetz NF Dual-modal magnetic resonance and fluorescence imaging of atherosclerotic plaques in vivo using VCAM-1 targeted tobacco mosaic virus. Nano Lett. 2014, 14 (3), 1551–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Park J; Gao H; Wang Y; Hu H; Simon DI; Steinmetz NF S100A9-targeted tobacco mosaic virus nanoparticles exhibit high specificity toward atherosclerotic lesions in ApoE(–/–) mice. J. Mater. Chem. B 2019, 7 (11), 1842–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pitek AS; Park J; Wang Y; Gao H; Hu H; Simon DI; Steinmetz NF Delivery of thrombolytic therapy using rod-shaped plant viral nanoparticles decreases the risk of hemorrhage. Nanoscale 2018, 10 (35), 16547–16555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pitek AS; Wang Y; Gulati S; Gao H; Stewart PL; Simon DI; Steinmetz NF Elongated Plant Virus-Based Nanoparticles for Enhanced Delivery of Thrombolytic Therapies. Mol. Pharmaceutics 2017, 14 (11), 3815–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wen AM; Wang Y; Jiang K; Hsu GC; Gao H; Lee KL; Yang AC; Yu X; Simon DI; Steinmetz NF Shaping bio-inspired nanotechnologies to target thrombosis for dual optical-magnetic resonance imaging. J. Mater. Chem. B 2015, 3 (29), 6037–6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Edgeworth J; Gorman M; Bennett R; Freemont P; Hogg N Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J. Biol. Chem 1991, 266 (12), 7706–13. [PubMed] [Google Scholar]
- (31).Hunter MJ; Chazin WJ High level expression and dimer characterization of the S100 EF-hand proteins, migration inhibitory factor-related proteins 8 and 14. J. Biol. Chem 1998, 273 (20), 12427–35. [DOI] [PubMed] [Google Scholar]
- (32).Nacken W; Sopalla C; Propper C; Sorg C; Kerkhoff C Biochemical characterization of the murine S100A9 (MRP14) protein suggests that it is functionally equivalent to its human counterpart despite its low degree of sequence homology. Eur. J. Biochem 2000, 267 (2), 560–5. [DOI] [PubMed] [Google Scholar]
- (33).Mori S; Goto K; Goto F; Murakami K; Ohkawara S; Yoshlnaga M Dynamic changes in mRNA expression of neutrophils during the course of acute inflammation in rabbits. Int. Immunol 1994, 6 (1), 149–56. [DOI] [PubMed] [Google Scholar]
- (34).Croce K; Gao H; Wang Y; Mooroka T; Sakuma M; Shi C; Sukhova GK; Packard RR; Hogg N; Libby P; Simon DI Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation 2009, 120 (5), 427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Maiseyeu A; Badgeley MA; Kampfrath T; Mihai G; Deiuliis JA; Liu C; Sun Q; Parthasarathy S; Simon DI; Croce K; Rajagopalan S In vivo targeting of inflammation-associated myeloid-related protein 8/14 via gadolinium immunonanoparticles. Arterioscler., Thromb., Vasc. Biol 2012, 32 (4), 962–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wang Y; Fang C; Gao H; Bilodeau ML; Zhang Z; Croce K; Liu S; Morooka T; Sakuma M; Nakajima K; Yoneda S; Shi C; Zidar D; Andre P; Stephens G; Silverstein RL; Hogg N; Schmaier AH; Simon DI Platelet-derived S100 family member myeloid-related protein-14 regulates thrombosis. J. Clin. Invest 2014, 124 (5), 2160–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Healy AM; Pickard MD; Pradhan AD; Wang Y; Chen Z; Croce K; Sakuma M; Shi C; Zago AC; Garasic J; Damokosh AI; Dowie TL; Poisson L; Lillie J; Libby P; Ridker PM; Simon DI Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation 2006, 113 (19), 2278–84. [DOI] [PubMed] [Google Scholar]
- (38).Geiger FC; Eber FJ; Eiben S; Mueller A; Jeske H; Spatz JP; Wege C TMV nanorods with programmed longitudinal domains of differently addressable coat proteins. Nanoscale 2013, 5 (9), 3808–16. [DOI] [PubMed] [Google Scholar]
- (39).Wang Y; Gao H; Kessinger CW; Schmaier A; Jaffer FA; Simon DI Myeloid-related protein-14 regulates deep vein thrombosis. JCI Insight 2017, 2 (11), No. e91356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Chatterji A; Ochoa W; Paine M; Ratna BR; Johnson JE; Lin T New addresses on an addressable virus nanoblock: uniquely reactive Lys residues on cowpea mosaic virus. Chem. Biol 2004, 11 (6), 855–63. [DOI] [PubMed] [Google Scholar]
- (41).Steinmetz NF; Evans DJ; Lomonossoff GP Chemical introduction of reactive thiols into a viral nanoscaffold: a method that avoids virus aggregation. ChemBioChem 2007, 8 (10), 1131–6. [DOI] [PubMed] [Google Scholar]
- (42).Steinmetz NF; Ablack AL; Hickey JL; Ablack J; Manocha B; Mymryk JS; Luyt LG; Lewis JD Intravital imaging of human prostate cancer using viral nanoparticles targeted to gastrin-releasing Peptide receptors. Small 2011, 7 (12), 1664–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Shukla S; Myers JT; Woods SE; Gong X; Czapar AE; Commandeur U; Huang AY; Levine AD; Steinmetz NF Plant viral nanoparticles-based HER2 vaccine: Immune response influenced by differential transport, localization and cellular interactions of particulate carriers. Biomaterials 2017, 121, 15–27. [DOI] [PubMed] [Google Scholar]
- (44).Schlick TL; Ding Z; Kovacs EW; Francis MB Dual-surface modification of the tobacco mosaic virus. J. Am. Chem. Soc 2005, 127, 3718–3723. [DOI] [PubMed] [Google Scholar]
- (45).Presolski SI; Hong VP; Finn MG Copper-Catalyzed Azide-Alkyne Click Chemistry for Bioconjugation. Curr. Protoc Chem. Biol 2011, 3 (4), 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wen AM; Infusino M; De Luca A; Kernan DL; Czapar AE; Strangi G; Steinmetz NF Interface of physics and biology: engineering virus-based nanoparticles for biophotonics. Bioconjugate Chem. 2015, 26 (1), 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Namba K; Stubbs G Structure of tobacco mosaic virus at 3.6 A resolution: implications for assembly. Science 1986, 231 (4744), 1401–6. [DOI] [PubMed] [Google Scholar]
- (48).Lin T; Chen Z; Usha R; Stauffacher CV; Dai JB; Schmidt T; Johnson JE The refined crystal structure of cowpea mosaic virus at 2.8 A resolution. Virology 1999, 265 (1), 20–34. [DOI] [PubMed] [Google Scholar]
- (49).Decuzzi P; Lee S; Bhushan B; Ferrari M A theoretical model for the margination of particles within blood vessels. Ann. Biomed. Eng 2005, 33 (2), 179–90. [DOI] [PubMed] [Google Scholar]
- (50).Diaz JA; Obi AT; Myers DD Jr.; Wrobleski SK; Henke PK; Mackman N; Wakefield TW Critical review of mouse models of venous thrombosis. Arterioscler., Thromb., Vasc. Biol 2012, 32 (3), 556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Schönfelder T; Jäckel S; Wenzel P Mouse models of deep vein thrombosis. Gefasschirurgie. 2017, 22 (S1), 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]