

Familial dilated cardiomyopathy (DCM) is an inherited DCM defined as having ≥2 first-degree relatives with DCM in a patient with idiopathic DCM and is characterized by variable expression and incomplete penetrance leading to heterogeneity in presentation.1 With increasing disease recognition and widespread availability of genetic testing, it is important to characterize the clinical outcomes and prognosis among familial DCM patients with stage D heart failure requiring a mechanical circulatory support device (MCSD).2 We examined the nationwide INTERMACS (Interagency Registry for Mechanical Assisted Circulatory Devices) database to examine the clinical characteristics and outcomes of familial DCM patients who received an MCSD.
Anonymized study data are publicly available at the NHLBI BioLINCC repository. The University of Alabama at Birmingham Institutional Review Board provided ethical oversight for this project. DCM patients receiving an MCSD between June 2005 and December 2017 were included and stratified into familial DCM, ischemic DCM, idiopathic DCM, and DCM due to other causes based on the diagnoses recorded in INTERMACS. The primary outcome of interest was death, and the secondary outcome was heart transplant. Early (≤3 months from the day of device implantation) and late (>3 months from the day of device implantation) adverse event frequency was also assessed. Baseline characteristics were compared using descriptive statistics with continuous data compared using Wilcoxon rank-sum test and categorical data compared using the χ2 test. Multivariable-adjusted Cox proportional hazard model, using ischemic DCM as the reference group, accounting for age, sex, race, body mass index (BMI), INTERMACS profile, device type, device strategy, education level, implant year, and work-for-income was used to estimate the risk of death. The Fine-Gray subdistribution model was used to account for heart transplant (in the model for death) and death (in the model for heart transplantation) as competing risks. The evolution of MCSD over time may influence study outcomes and therefore time stratified analyses were performed. The population was stratified into early-era (2006-2012) and late-era (2013-2017) of MCSD implantation. A two-sided p-value of < 0.05 was considered statistically significant.
Among 19,928 patients diagnosed with DCM, 8,622 (43.3%), 7,091 (35.7%), 568 (2.9%), and 3,647 (18.3%) had ischemic DCM, idiopathic DCM, familial DCM, and DCM due to other causes, respectively. The age at implantation was lowest among familial DCM patients [46 (34, 56) years] compared with other DCM etiologies (p<0.001). Familial DCM patients receiving an MCSD included 70.4% males, with a median BMI of 27.9 (23.6, 33.1) kg/m2, and 85.1% with an INTERMACS profile 1-3. The baseline characteristics were similar across all DCM etiologies. In the familial DCM group, bridge to transplant was the most common device strategy (78.4%). Among familial DCM patients, 45.6% were listed, and 32.8% were eligible for a heart transplant. Compared with other DCM etiology, the familial DCM group had the lowest early and late adverse events for bleeding, device thrombosis, infection, and respiratory failure compared with the other groups (p<0.05 for all). The cumulative incidence of death at 4 years was 45.9%, 35.5%, 24.5%, and 35.5% in the ischemic DCM, idiopathic DCM, familial DCM, and DCM due to other causes groups, respectively. The cumulative incidence of heart transplantation at 4 years was 31.0%, 41.2%, 60.0%, and 41.7% in the ischemic DCM, idiopathic DCM, familial DCM, and DCM due to other causes groups, respectively. Over a median follow-up of 0.95 (0.36, 2.04) years, familial DCM patients had the lowest risk of death (HRadj:0.68 [95%CI:0.55-0.83]) compared with patients with other DCM etiology, accounting for heart transplant as a competing risk event (Figure 1). The familial DCM patients were more likely to receive a heart transplant earlier than the other DCM etiological groups receiving an MCSD (HRadj:1.47 [95%CI:1.28-1.69]). Similar results were noted in population stratification based on the year of implantation.
Figure 1. Clinical Outcomes Among Familial Dilated Cardiomyopathy Patients Receiving a Mechanical Circulatory Support Device.
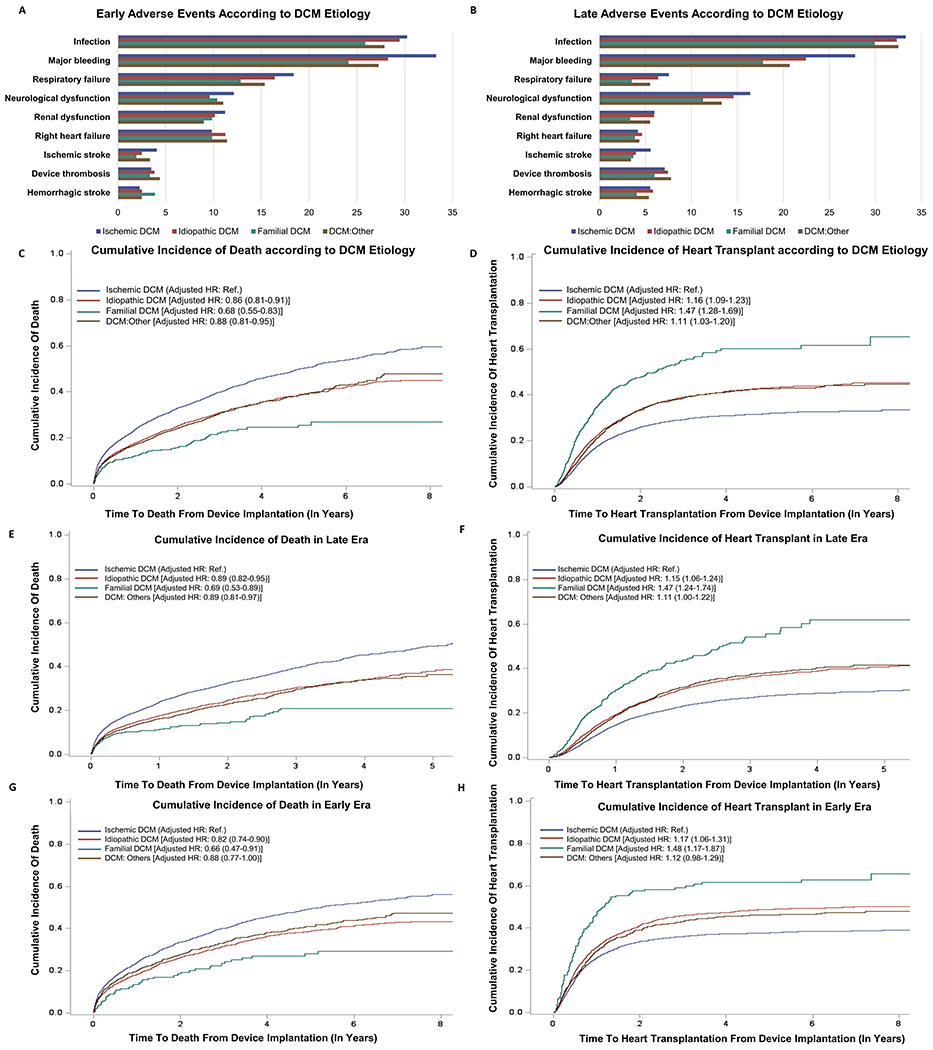
Part A and B: These bar graphs depict the percentage of early and late adverse events stratified by DCM etiology. The blue, red, green, and brown bar depicts ischemic DCM, idiopathic DCM, familial DCM, and DCM due to other causes, respectively.
Part C: Kaplan-Meier curves for the risk of death post MCSD implantation stratified by DCM etiology, taking heart transplantation as a censoring event. The blue, red, green, and brown curve depicts ischemic DCM, idiopathic DCM, familial DCM, and DCM due to other causes, respectively.
Part D: Kaplan-Meier curves for the cumulative incidence of heart transplantation stratified by DCM etiology. The blue, red, green, and brown curve depicts ischemic DCM, idiopathic DCM, familial DCM, and DCM due to other causes, respectively.
Part E: Kaplan-Meier curves for the cumulative incidence of death in the late era stratified by DCM etiology. The blue, red, green, and brown curve depicts ischemic DCM, idiopathic DCM, familial DCM, and DCM due to other causes, respectively.
Part F: Kaplan-Meier curves for the cumulative incidence of heart transplantation in the late era stratified by DCM etiology. The blue, red, green, and brown curve depicts ischemic DCM, idiopathic DCM, familial DCM, and DCM due to other causes, respectively.
Part G: Kaplan-Meier curves for the cumulative incidence of death in the early era stratified by DCM etiology. The blue, red, green, and brown curve depicts ischemic DCM, idiopathic DCM, familial DCM, and DCM due to other causes, respectively.
Part H: Kaplan-Meier curves for the cumulative incidence of heart transplantation in the early era stratified by DCM etiology. The blue, red, green, and brown curve depicts ischemic DCM, idiopathic DCM, familial DCM, and DCM due to other causes, respectively.
In this study of ~20,000 DCM patients receiving an MCSD, familial DCM patients were noted to be younger at the time of implantation and had fewer post-implantation adverse events, lower risk of death, and a higher likelihood of receiving a heart transplant compared with patients with other forms of DCM. Compared with idiopathic DCM, pediatric familial DCM patients have been previously noted to have a similar incidence of heart transplant or pre-transplant death.3 Prior data indicates that familial DCM patients listed in the United Network for Organ Sharing (UNOS) registry are younger and have a higher total artificial heart use compared with patients of other DCM etiologies.4 DCM prevalence in first-degree relatives of familial DCM patients is estimated to be 30% by 80 years of age, and a large proportion of these individuals may be eligible for an MCSD based on the disease stage.5 The current study findings may facilitate discussion about clinical prognosis and outcomes among familial DCM patients and first-degree family members identified by cascade phenotypic and genotypic screening in cardiovascular and genetics centers across the country. Disease identification and treatment in the early disease course and access to MCSD in the advanced disease course may improve survival among familial DCM patients. This study is limited by the unavailability of genotypic data among study participants. In summary, familial DCM patients receiving an MCSD have a relatively lower risk of death and a higher likelihood of receiving a heart transplant compared with MCSD patients with other DCM etiologies.
Funding Sources:
Dr. Pankaj Arora is supported by the National Heart, Lung, And Blood Institute of the NIH awards R01HL160982, R01HL163852, R01HL163081, and K23HL146887, and by the Doris Duke Charitable Foundation COVID-19 Fund to Retain Clinician Scientists (Grant #2021255); University of Alabama at Birmingham COVID-19 Caregiving Affected Research Early-Career Scientist Retention Program (CARES at UAB).
Footnotes
Conflicts of Interest: None.
References
- 1.Burkett EL and Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. Journal of the American College of Cardiology. 2005;45:969–981. [DOI] [PubMed] [Google Scholar]
- 2.Michels VV, Driscoll DJ, Miller FA, Olson TM, Atkinson EJ, Olswold CL, Schaid DJ. Progression of familial and non-familial dilated cardiomyopathy: long term follow up. Heart. 2003;89:757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khayata M, Al-Kindi SG, Oliveira GH. Contemporary characteristics and outcomes of adults with familial dilated cardiomyopathy listed for heart transplantation. World Journal of Cardiology. 2019;11:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rusconi P, Wilkinson JD, Sleeper LA, Lu M, Cox GF, Towbin JA, Colan SD, Webber SA, Canter CE, Ware SM, et al. Differences in presentation and outcomes between children with familial dilated cardiomyopathy and children with idiopathic dilated cardiomyopathy: a report from the Pediatric Cardiomyopathy Registry Study Group. Circulation: Heart Failure. 2017;10:e002637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huggins GS, Kinnamon DD, Haas GJ, Jordan E, Hofmeyer M, Kransdorf E, Ewald GA, Morris AA, Owens A, Lowes B, et al. Prevalence and cumulative risk of familial idiopathic dilated cardiomyopathy. JAMA. 2022;327:454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]