

Abstract

We describe our efforts toward the total synthesis of the natural product elisabethin A. The first route was guided by the proposed biosynthesis, assembling the 6,6-ring system before forming the five-membered ring including the quaternary carbon. The second approach includes a high yielding cyclization under Mitsunobu conditions as a key step. It allowed the preparation of an unusual and highly functionalized bicyclic 6,5-spiro compound. Both routes share a common advanced precursor obtained from an “underdeveloped” Claisen rearrangement of an aryl dienyl ether.
Introduction
Elisabethin A (1) was discovered in 1998 as the first member of the novel elisabethane family.1 It was isolated from the Caribbean gorgonian Pseudopterogorgia elisabethae harvested in the sea near San Andrés Island, Colombia (Figure 1). From approximately 1 kg of dried animal, 25 mg of elisabethin A (1) was isolated as the crystalline material. 2D-NMR analysis and X-ray crystallography allowed the determination of the relative configuration, whereas the absolute configuration remains unclear. This diterpene with the molecular formula C20H28O3 consists of a tricyclic cis-trans-fused 5,6,6-ring system. It bears six contiguous stereocenters, namely, quaternary all-carbon spiro centers C-1, C-2, C-3, C-6, C-7, and C-9. An additional feature is the highly oxidized and fully substituted enedione moiety.
Figure 1.
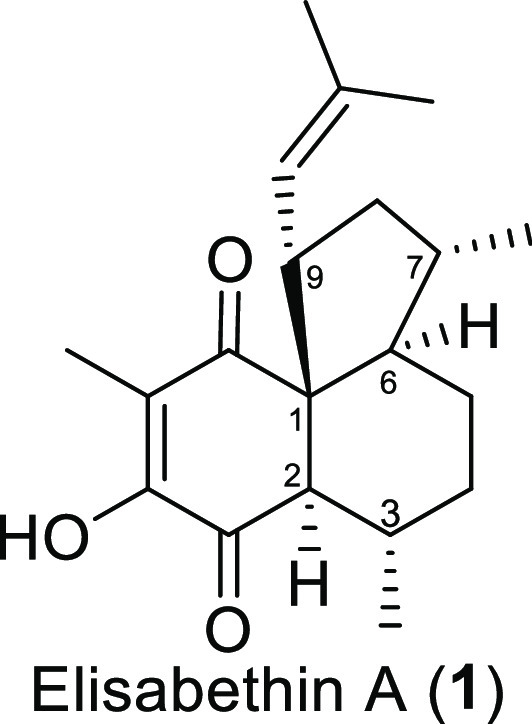
Structure of elisabethin A.
From the same marine organism, various novel metabolites were isolated, namely, elisabethin B (2), elisabethin C (3), elisabethin D (4), and its corresponding acetate (5) (Figure 2).1,2
Figure 2.

Members of the elisabethane family.
Additionally, P. elisabethae was the source for elisapterosin B (6) and tetracyclic colombiasin A (7).2,3 Their novel carbon framework paired with the high density of stereochemical information has made these two molecules attractive targets for total synthesis during the past two decades.4−7
It is speculated that the two latter natural products could be synthesized from elisabethin A (1) as their common precursor (Figure 3). Elisapterosin B (6) might be prepared via C10–C15 bond formation, whereas colombiasin A (7) could be accessible via C12 oxidation and subsequent connection to C2. Despite this potential, none of the published total syntheses relied on this elisabethin A precursor strategy. In 2003, Rawal et al. demonstrated the feasibility of such a possible interconversion in their intended (ent)-elisabethin A (1) synthesis.8
Figure 3.

Precursor role of elisabethin A (1).
While being met with the unsolvable issue of C-2 epimerization (elisabethin A numbering throughout), the synthesis was rerouted to access (ent)-elisapterosin B. This allowed the confirmation of the stereochemical assignment of key intermediates prepared during this approach. In 2003, Mulzer reported the successful completion of the total synthesis of elisabethin A (1). Interestingly, Rawal and Mulzer shared a common intermediate despite being the opposite enantiomer. In Rawal et al.’s case, the C-2 epimerization was reported to be infeasible, whereas Mulzer and Heckrodt claimed a successful transformation of their antipode. This discrepancy and the fact that the spectra of the synthesized and natural materials were not superimposable soon gave rise to considerable doubt with respect to the stereochemical assignment of their final compound.9,10 In 2014, Mulzer et al. reported a second approach to elisabethin A (1) but was not able to prove their claims made in 2003.11
Herein, we wish to report two different approaches toward the total synthesis of elisabethin A (1). Both were running in parallel with the idea to push forward with the most promising approach to completion.
Results and Discussion
First Approach
Our first approach was strongly guided by preliminary results.12,13 Accordingly, target molecule 1 could be traced back to bicyclic ester 8 (Scheme 1). Introduction of the C-7 methyl group was planned to be achievable by conjugate addition to the corresponding α,β-unsaturated ester. Further simplification by means of functional group manipulation would lead to compound 9. Although our first-generation synthesis of the precursor for bicyclic intermediate 9 was successful, the lack of selectivity prompted us to investigate a new strategy to prepare this material. The idea was to perform desymmetrization in the course of RCM reaction of 10, either under a substrate control or by using chiral catalysis.14,15
Scheme 1. Retrosynthetic Analysis of Elisabethin A (1).

Further disconnection in this second-generation approach allowed the identification of two building blocks, chiral alcohol 12 and phenol 11, which was derived from compound 13.12 The first issue to overcome in this approach was whether compound 11 could be synthesized with reasonable effort.
The synthesis commenced with a large-scale preparation of orthogonally protected hydroquinone 13 from commercially available vanillin (see Experimental Section). Selective cleavage of the MOM group16 followed by merging with (E)-5-bromopenta-1,3-diene17 under basic conditions gave rise to dienyl-ether 15 in 97% yield (Scheme 2).18 The following two-step protecting group manipulation produced compound 16 in 90% yield over two steps. This exchange demands some comments. First, the preparation of compound 13 from vanillin was not feasible with OTES protection. Second, TIPS protection of the upper phenolic OH was found to be critical in the later course of the synthesis (see below). Therefore, the lower OTIPS in compound 15 needed to be exchanged to an orthogonal protecting group that could be easily cleaved in the presence of the upper TIPS group.
Scheme 2. Synthesis of Compound 10.

Reagents and conditions: (a) ZnBr2, EtSH, DCM, −32 to −5 °C, 2.5 h, 89%; (b) n-BuLi, (E)-5-bromopenta-1,3-diene, 1.4:1 THF/DMF, −35 °C to rt, 15 h, 97%; (c) TBAF, THF, rt, 2 min, 95%; (d) TESCl, imidazole, DCM, rt, 20 min, 95%; (e) EuFOD, PhMe, 110 °C, 4 h, 84%; (f) n-BuLi, TIPSOTf, THF, −85 to −45 °C, 1.5 h, 92%; (g) TFA, 4:1 THF/H2O, rt, 30 min, 93%; (h) PBu3, ADDP, 12, PhMe, 0 °C to rt, 2 h, 69%; (i) EuFOD, o-xylene, 120 °C, 18 h, 55% (+31% 11).
With compound 16 at hand, the stage was set for the first Claisen rearrangement. After intensive investigations, we found that 5 mol % EuFOD in hot toluene triggered a clean rearrangement of 16, giving the desired phenol 17 as a single product in 84% yield. This protocol demonstrated a considerable improvement compared to the current state-of-the-art literature,19−21 allowing the selective preparation of the desired compound in the multigram scale.
Protection of 17 as a TIPS ether (n-BuLi and TIPSOTf) followed by selective cleavage of the TES group afforded phenol 11 in 86% yield over two steps. The next stage called for a substrate-controlled C–O/C-C chirality transfer to install a tertiary stereogenic center in the key intermediate 10. Accordingly, phenol 11 was coupled with alcohol 12 under Mitsunobu conditions (Bu3P and ADDP) and the resultant allyl ether 19 was subjected to a second EuFOD-catalyzed Claisen rearrangement to give phenol 10 in 55% yield.22,23 This allowed us to proceed with the second key transformation–diastereosective construction of the cyclohexene moiety.
Subjection of phenol 10 to Grubbs II smoothly triggered a relay-RCM furnishing compound 20 in 92% yield (Scheme 3).24 We were delighted to find that product formation proceeded with a full substrate control, affording the desired compound as a single diastereomer. TIPS protection proved to be of utmost importance in this step, as other phenol protecting groups led to significantly lower diastereoselectivity.25 The stereochemical assignment was achieved through NOESY experiments and was later confirmed by X-ray diffraction at the stage of the TIPS-protected product 9 (CCDC no. 2161382). According to the retrosynthetic plan, the next task was C-elongation to introduce an α,β-unsaturated ester moiety. This was accomplished by Ru-catalyzed cross-metathesis, and the desired compound 21 was obtained in 51% yield. With this material at hand, the stage was set for the introduction of a C-7 stereocenter. The treatment of compound 21 with TMSCl and MeMgBr in the presence of a catalytic amount of CuCl cleanly formed the corresponding 1,4-addition product as a single diastereomer in quantitative yield.26 Unfortunately, X-ray crystallography revealed undesired C-7 configuration, forming (7epi)-8 (CCDC no. 2161383). All attempts to overwrite the substrate control using a chiral catalyst proved unsuccessful.27−29 Notably, other cuprate conditions lead to no conversion.30−33
Scheme 3. Synthesis of Compound (7epi)-8.

Reagents and conditions: (a) Grubbs II, DCM, 40 °C, 2 h, 92%; (b) n-BuLi, TIPSCl, 1:1 THF/DMF, −80 to −35 °C, 1.5 h, 97%; (c) GH-II, methyl acrylate, PhMe, 80 °C, 25 h, 51% (+41% 9); (d) CuCl, TMSCl, MeMgBr, THF, −42 to −22 °C, 1 h, quant.
The observed stereochemical outcome of the 1,4-addition suggested that the reaction did not proceed via21A but conformer 21B (A1,3 > A1,2 strain) (Figure 4, aromatic substitution omitted for clarity). Hence, the bulky C-14 and C-17 OTIPS groups are successfully blocking the backside of the molecule and the metalorganic nucleophile can only approach the molecule from the front side (blue arrow) to give an undesired diastereomer. In attempts to overcome this issue, we decided to prepare ester 8 by conjugate reduction of (Z)-22 with the expectation that in this case, A1,3 strain in 22A would be dominant and secure 22B as a reactive conformer.
Figure 4.

Possible explanation for the observed diastereoselectivity.
We anticipated that ester 22 could be derived from compound 9 by minor functional group manipulation (Scheme 4).
Scheme 4. Retrosynthetic Analysis of Ester 8.

Accordingly, compound 9 was subjected to standard Wacker oxidation conditions and ketone 23 was obtained in 92% yield (Scheme 5). Unfortunately, C-elongation under HWE or Peterson olefination conditions proved unsuccessful and only the starting material was isolated.34−40
Scheme 5. Synthesis of Compound 24.

Reagents and conditions: (a) PdCl2(MeCN)2, BQ, 9:3:0.75 DMAC/DCE/H2O, 36 °C, 5 h, 92%; (b) [Ir(cod)(PCy3)(py)]PF6, H2, DCM, rt, 1 h, 99%.
The same lack of reactivity was observed with ketone 24. Faced with this early setback, the plan was adapted and compound 22 was prepared in a two-step sequence. In the first step, compound 21 was subjected to conjugate addition under standard conditions (CuCl, TMSCl, and MeMgBr) followed by addition of PhSeCl to give the corresponding α-phenyl selenide.41 Selective oxidation then triggered selenoxide elimination, forming a set of separable esters, compound (Z)-22 and (E)-22 (Scheme 6).42
Scheme 6. Synthesis of Compound 22.

Reagents and conditions: (a) CuCl, TMSCl, MeMgBr, THF, −50 to −20 °C, 45 min, then PhSeCl; (b) H2O2, pyridine, CHCl3, rt, 30 min, 12% (Z)-22, 20% (E)-22 (over two steps).
Unfortunately, all attempts to reduce esters (Z)-22 and (E)-22 into the desired C-7 methyl ester 8 were unsuccessful.43−50 Inspired by our second approach (see below), in our next attempt to create a C-7 chiral center, we planned to perform conjugated addition of methyl cuprate to lactone 27 (Scheme 7), a reaction that was successful for a similar substrate.
Scheme 7. Synthesis of Compound 27.

Reagents and conditions: (a) TBAF, AcOH, THF, 0 °C to rt, 2 h, 40% (+54% starting material); (b) NaH, acryloyl chloride, THF, −23 to −20 °C, 1.5 h, 83%; (c) GH-II, PhMe, 80 °C, 21 h, 98%.
The sequence commenced with selective deprotection (1:2 TBAF/AcOH) of compound 9 to give phenol 25 in 40% yield (Scheme 7).51 The reaction was stopped short before 50% conversion, and the remaining starting material was recycled. The treatment of 25 with acryloyl chloride provided 26, which was subjected to RCM to form lactone 27 in excellent yield. To our great disappointment, the desired 1,4-addition proved to be infeasible, refusing access to lactone 28. One explanation for the observed lack of reactivity could be that the α,β-unsaturated lactone is out of conjugation. This theory was underpinned by 1H-NMR evidence. The chemical shifts of the α- and β-protons (α: 5.76 ppm; β: 6.51 ppm) suggest a low degree of polarization compared to similar α,β-unsaturated systems, e.g., compound (rac)-35 (α: 5.90 ppm; β: 6.85 ppm). These unsuccessful attempts for direct introduction of C-7 methyl with desired configuration compelled a change in our strategy.
Second Approach
For our second approach, elisabethin A (1) was simplified by scission of the C-2–C-3 bond as well as the C-4–C-5 bond (Scheme 8). The first connection should be accomplished by a Claisen rearrangement, and the second should be accomplished via RCM. Carving out the second six-membered ring allowed the identification of enantiopure spiro compound 29. The said advanced intermediate would be assembled from a (9S)-alcohol-derivative 30 by Pd-catalyzed allylic alkylation.52 Compound 30 could be traced back to chiral ether 31, which in turn should be derived from lactone 32. Finally, enantioenriched lactone 32 could be accessible from ester 33via the Claisen rearrangement, followed by enantioselective RCM and 1,4-addition. The known compound 15 was chosen as a starting material for this approach.
Scheme 8. Second-Generation Retrosynthetic Analysis of Elisabethin A (1).

Guided by the retrosynthetic plan, the known ether 15 was elaborated into phenol 34 by EuFOD catalysis (Scheme 9). Next, phenol 34 was converted into compound 33, which was directly subjected to RCM in the presence of a chiral Mo catalyst.53 Unfortunately, all our attempts to perform this enantioselective cyclization failed and only the starting material was isolated.54,55 Contrastingly, the same reaction in the presence of an HG-II catalyst afforded racemic material (rac)-35. At this point, we decided to go ahead with racemic lactone and to explore the feasibility of the intended five-membered ring spiro cyclization.
Scheme 9. Synthesis of Compound 38.

Reagents and conditions: (a) EuFOD, PhMe, 110 °C, 4 h, 97%; (b) acryloyl chloride, Et3N, DCM, 0 °C to rt, 2 h, 93%; (c) GH-II, PhMe, 80 °C, 15 h; (d) CuCl, LiCl, TMSCl, MeMgBr, THF, −40 °C, 15 min, 42% (over two steps); (e) AlCl3, Me2S, DCM, rt, 30 min, 89%; (f) MOMCl, LiHMDS, NaI, THF, −35 to −26 °C, 25 min, 94%; (g) TBAF, THF, rt, 3 min, 77%; (h) PBu3, ADDP, 12, PhMe, rt, 2 h, 36% (+36% 38′).
Accordingly, organo cuprate addition produced racemic lactone (rac)-36 in 42% yield over two steps. The trans relationship of the newly introduced C-7 methyl and C-6 vinyl substituents was confirmed by X-ray diffraction (CCDC no. 2161384). To elude late-stage difficulties, the methoxy protection needed to be exchanged. Thus, a two-step protocol was employed, commencing with methoxy cleavage (AlCl3 and Me2S) and followed by MOM installation, which gave rise to lactone (rac)-32.56 Subsequently, fluoride-mediated TIPS removal revealed phenol (rac)-37, setting the stage for the Mitsunobu etherification. The merging of compound (rac)-37 and chiral alcohol 12 furnished ether 38 and its C-6/C-7 epimer 38′ (1:1, 72% total yield) accompanied by minor amounts of their corresponding SN2′ products (not shown).
Preliminary experiments in our group suggested that MOM was also not a suitable protecting group for late-stage deprotection. Therefore, it was exchanged again in a two-step sequence. Selective cleavage removed the acetal protecting group of 38, leaving the ether residue unchanged (Scheme 10).57 To our delight, X-ray diffraction of this material confirmed the depicted relative stereochemistry (see Experimental Section, compound S7, and Supporting Information, CCDC no. 2161381). In the second step, TES was installed and the obtained silyl ether 31 was directly converted into the corresponding Weinreb amide. Phenol protection by means of TBS ether led to the formation of compound 39.
Scheme 10. Synthesis of Compound 39.

Reagents and conditions: (a) MgBr2·Et2O, EtSH, DCM, 4 °C to rt, 1.5 h, 95%; (b) TESOTf, 2,6-lutidine, DCM, −85 °C to rt, 25 min; (c) MeONHMe·HCl, AlMe3, PhMe, −20 to −15 °C, 45 min, 93% (over two steps); (d) TBSOTf, 2,6-lutidine, DCM, −92 °C to rt, 83% (+11% 31).
The retrosynthetic analysis depicted in Scheme 8 suggested that (9S)-alcohol was required for the following spiro cyclization. However, to investigate this crucial transformation in detail, it was decided to prepare the corresponding (9R)-alcohol as well. Therefore, two slightly different sequences from compound 39 were conducted in parallel (Scheme 11). First, vinyl Grignard addition gave rise to the corresponding ketone and the following reduction with DIBAL produced (9S)-allylic alcohol 40 in 70% isolated yield over two steps and dr = 11:1.58 The stereochemistry of (9S)-allylic alcohol 40 was unambiguously confirmed by X-ray diffraction (see the Supporting Information). Conversion of compound 40 into the corresponding methyl carbonate proceeded uneventfully. Subsequent acid-mediated TES removal gave rise to phenol 30 in virtually quantitative yield over two steps. To access the corresponding (9R)-allyl carbonate 30′, compound 39 was subjected to DIBAL reduction. The obtained aldehyde was immediately treated with vinyl Grignard to give (9R)-allylic alcohol 40′ as a major epimer in 45% yield over two steps (dr = 2:1). The following two-step sequence was identical to the preparation of phenol 30. Carbonate formation and subsequent TES deprotection transformed compound 40′ into cyclization precursor 30′ in 92% yield. With both epimers 30 and 30′ at hand, the stage was set to investigate the cyclization event.
Scheme 11. Synthesis of Compound Cyclization Precursors 30 and 30′.

Reagents and conditions: (a) CH2=CHMgBr, THF, 0 °C to rt, 1 h; (b) DIBAL, DCM, −90 °C, 15 min, 70% (two steps, dr = 11:1); (c) ClC(O)OMe, pyridine, DCM, −20 to −5 °C, 2 h; (d) TFA, 4:1 THF/H2O, rt, 1.5 h, 99% (two steps); (e) DIBAL, PhMe, −91 °C, 2 min, 98%; (f) CH2=CHMgBr, THF, 4 °C to rt, 20 min, 45% (dr = 2:1); (g) ClC(O)OMe, pyridine, DCM, −20 to 10 °C, 2 h; (h) TFA, 4:1 THF/H2O, rt, 2 h and 50 min, 92% (two steps).
Subjecting phenol 30 to Pd catalysis triggered the desired spiro formation (Scheme 12).52 The product was obtained in 21% yield after column chromatography. Gratifyingly, NOESY experiments confirmed the expected stereochemical outcome of the reaction. To our surprise, HMBC experiments suggested compound 29a to be the reaction product instead of the expected material 29 (see the Supporting Information for details). To gain more insight into this transformation, phenol 30′ was subjected to the same reaction conditions. Surprisingly, compound 29a was isolated in 18% yield as the major product. The observed product distribution could be explained by an equilibrium eroding the initial stereo information before the cyclization event took place.59 Notably, the Pd-catalyzed cyclizations could only be carried out in DCM and CD2Cl2 respectively and all attempts of improving yield and selectivity by means of solvent change (THF, MeCN, PhMe, DMF or 1,4-dioxane) were fruitless.60 Additionally, application of various monodentate ligands (P(2-furyl)3, P(OEt)3, P(OPh)3, AsPh3) or Trost’s chiral bidentate ANDEN ligand led to a complete cessation of the reaction.52,61 In consideration of this, we choose to pursue an alternative process for the construction of the spirocenter. Inspired by literature, we intended to conduct this reaction in a SN2-like manner under Mitsunobu conditions.62 We had good reason to be confident as initial results suggested the feasibility of this approach.
Scheme 12. Synthesis of Compound 29a.

Reagents and conditions: (a) Pd(dba)2, PPh3, DCM, rt, 0.5 h, 21% from 30; 18% from 30′.
In parallel to the synthetic efforts depicted in Scheme 11, we investigated the inversion of the C-9 stereocenter of compound 41 by Mitsunobu reaction (Scheme 13). Therefore, (9S)-allylic alcohol 40 was treated with standard conditions to obtain ester 41 in 63% yield, together with 19% of 41 lacking TES protection (not shown). Additionally, traces of a spiro-compound could be identified, conceivably formed by acid-mediated TES cleavage and following cyclization. The synthesis continued with ester hydrolysis and simultaneous cleavage of TES-liberating phenol 42.
Scheme 13. Synthesis of Compound 42.

Reagents and conditions: (a) p-NO2C6H4CO2H, PPh3, DIAD, PhMe, rt, 40 min, 63%; (b) K2CO3, 2.5:1 EtOH/iPrOH, rt, 48 h, 76%.
With the desired precursor 42 at hand, we were ready to test the envisioned spiro formation. To our great delight, treatment with PPh3 and DIAD in dry toluene at 0 °C cleanly formed compound 43 (Scheme 14). Interestingly, this transformation proceeded with the cleavage of TBS protection. This facile method for the construction of spiro [4,5] ketones was found superior to the Pd catalysis approach with respect to yield and selectivity. To the best of our knowledge, this represents the first example of a spiro [4,5] ketone formation under Mitsunobu conditions. The generality of this method is subject to current investigations within our group. With this satisfying result at hand, we turned our attention toward the remaining six-membered ring of target molecule 1. Hence, next on the agenda was the Claisen rearrangement of allyl-vinyl ether 43 to compound 44. Unfortunately, all attempts to conduct this transformation proved fruitless and the desired material could not be obtained.63−66 This final dead end marked the end of our efforts, and this approach was abandoned.
Scheme 14. Synthesis of Compound 43.

Reagents and conditions: (a) PPh3, DIAD, PhMe, 0 °C, 2 h, 94%.
Conclusions
The synthetic challenges presented led to the improvement and discovery of underdeveloped and unknown reactions. The aryl-dienyl ether Claisen rearrangement proved to be highly reliable as well as high yielding and was successfully applied in both approaches. Although the final goal of preparing the desired target compound 1 was denied to us, our efforts allowed the synthesis of several more advanced intermediates. During our first approach, we achieved the synthesis of compounds 21 and (7epi)-8 in 17 and 18 linear steps with average respective yields per step of 86 and 87%, respectively. The main feature of the second approach was the spiro cyclization, which could be achieved in two different ways. The corresponding material 29a—accessible via Pd catalysis—was prepared in 24 linear steps (average 81% per steps). The unprecedented formation of a spiro [4,5] ketone under Mitsunobu conditions allowed the preparation of compound 43 over 24 linear steps in 1.6% overall yield. The lessons we have learnt during this endeavor will be considered when developing the next-generation approach toward the completion of the total synthesis.
Experimental Section
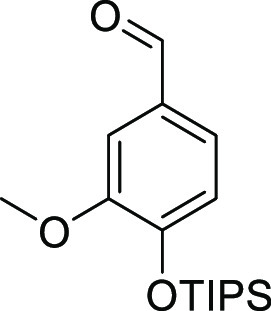
3-Methoxy-4-((triisopropylsilyl)oxy)benzaldehyde (S1)
A 500 mL Schlenk flask was charged with commercially available vanillin (40 g, 263 mmol, 1.00 equiv) in 200 mL of DMF/THF, and DIPEA (89.5 mL, 67.96 g, 526 mmol, 2 equiv) was added, causing a color change from pale yellow to strong orange. After addition of TIPSCl (61.9 mL, 55.76 g, 289 mmol, 1.1 equiv), the color changed to turbid yellow with a white precipitate forming. After 1 h, TLC (petroleum ether/ethyl acetate, 4:1) confirmed full conversion. The reaction was quenched by addition of solid NH4Cl, and the solvents were evaporated under reduced pressure. The residue was taken up in Et2O, filtered, and concentrated in vacuo. It was dried on high vacuum overnight, and the desired product was obtained as a red oil in quantitative yield (81.1 g, 263 mmol). The material was used in the next reaction without further purification. 1H-NMR (400 MHz, CDCl3): δ = 9.83 (s, 1H), 7.38 (d, J = 1.9 Hz, 1H), 7.34 (dd, J = 8.0, 2.0 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 3.85 (s, 3H), 1.32–1.21 (m, 3H), 1.08 (d, J = 7.5 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 191.1, 151.9, 151.7, 130.7, 126.3, 120.2, 110.1, 55.5, 17.9, 13.0; IR [ATR]: ν = 2944, 2892, 2867, 2733, 1696, 1684, 1592, 1506, 1464, 1422, 1390, 1286, 1265, 1238, 1193, 1152, 1124, 1071, 1034, 1016, 997, 961, 938, 896, 882, 819, 781, 729, 705 cm–1; HRMS (ESI): exact mass calculated for C17H29O3Si [(M + H)+], 309.1880; found, 309.1865.
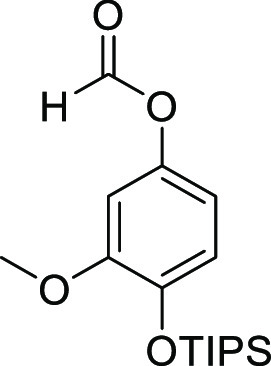
3-Methoxy-4-((triisopropylsilyl)oxy)phenyl Formate (S2)
A 500 mL Schlenk flask was charged with compound S1 (40.0 g, 129.6 mmol, 1 equiv) in 250 mL of CHCl3. To the strong red solution was added m-CPBA (75%, 44.8 g, 194 mmol, 1.5 equiv) in six portions over 75 min. The flask was heated to reflux overnight, and TLC (toluene) on the next day confirmed full conversion. The reaction mixture was transferred into a 1000 mL Erlenmeyer flask and neutralized with 200 mL of saturated NaHCO3 solution. Layers were separated, and the organic layer was washed with NaHCO3 solution, followed by 20 mL of H2O and 20 mL of brine. The dark red solution was dried over MgSO4 and concentrated in vacuo. The product was obtained as a dark red oil and directly subjected to following ester hydrolysis without further purification. 1H-NMR (400 MHz, CDCl3): δ = 8.28 (s, 1H), 6.85 (d, J = 8.6 Hz, 1H), 6.64 (d, J = 2.8 Hz, 1H), 6.58 (dd, J = 8.6, 2.8 Hz, 1H), 3.79 (s, 3H), 1.30–1.18 (m, 3H), 1.09 (d, J = 7.3 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 159.8, 151.5, 144.0, 143.9, 120.3, 112.5, 105.7, 55.7, 18.0, 13.0; IR [ATR]: ν = 2944, 2892, 2867, 1765, 1741, 1601, 1506, 1464, 1450, 1416, 1384, 1314, 1278, 1263, 1227, 1188, 1155, 1102, 1035, 1016, 997, 961, 882, 787, 711 cm–1; HRMS (ESI): exact mass calculated for C16H28O3Si [(M – CO)], 296.1808; found, 296.1806.
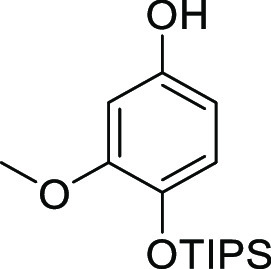
3-Methoxy-4-((triisopropylsilyl)oxy)phenol (S3)
Compound S2 (42 g, 129 mmol, 1.00 equiv) was dissolved in 250 mL of MeOH, and K2CO3 (1.45 g, 26 mmol, 0.2 equiv) was added, causing an instant color change from red to black. After 15 min, TLC (petroleum ether/ethyl acetate, 6:1) showed full conversion and the reaction was quenched with solid NH4Cl. It was filtered and concentrated in vacuo. The desired product was obtained as a brown solid in 92% yield (35.5 g, 118.7 mmol) over two steps. The material was used in the next reaction without further purification. 1H-NMR (400 MHz, CDCl3): δ = 6.71 (d, J = 8.5 Hz, 1H), 6.40 (d, J = 2.9 Hz, 1H), 6.24 (dd, J = 8.5, 2.9 Hz, 1H), 3.74 (s, 3H), 1.27–1.16 (m, 3H), 1.08 (d, J = 7.3 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 151.5, 150.3, 139.4, 120.5, 106.4, 100.9, 55.5, 18.0, 12.9; IR [ATR]: ν = 3411, 2942, 2890, 2865, 2051, 1980, 1602, 1513, 1461, 1450, 1381, 1353, 1293, 1281, 1230, 1214, 1200, 1164, 1112, 1073, 1061, 1025, 1016, 993, 951, 932, 902, 881, 806, 732, 716 cm–1; HRMS (ESI): exact mass calculated for C16H29O3Si [(M + H)+], 297.1880; found, 297.1880.
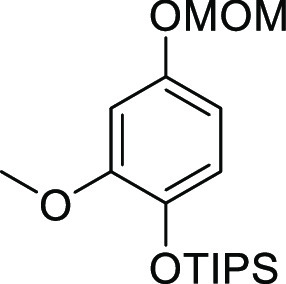
Triisopropyl(2-methoxy-4-(methoxymethoxy)phenoxy)silane (S4)
A 1000 mL three-necked flask was charged with compound S3 (34.6 g, 117 mmol, 1 equiv) dissolved in 750 mL of dry DCM. DIPEA (40 mL, 233 mmol, 2 equiv) was added, and the flask was equipped with a reflux condenser and argon balloon before MOMCl (16.8 mL, 222 mmol, 1.9 equiv) was added dropwise. After full addition, the reaction mixture was heated to reflux for 4 h. After TLC (petroleum ether/ethyl acetate, 6:1) confirmed full conversion, the reaction was quenched by addition of solid NaHCO3 and H2O. Layers were separated, the aqueous layer was extracted three times with DCM, and the combined organic layer was washed with H2O (4×), followed by brine. The dark red solution was dried over MgSO4 and concentrated in vacuo. The desired product was obtained as a dark oil in 97% yield (38.5 g, 113.5 mmol). The product was used in the next reaction without further purification. 1H-NMR (400 MHz, CDCl3): δ = 6.77 (d, J = 8.7 Hz, 1H), 6.58 (d, J = 2.8 Hz, 1H), 6.48 (dd, J = 8.7, 2.8 Hz, 1H), 5.10 (s, 2H), 3.78 (s, 3H), 3.48 (s, 3H), 1.28–1.18 (m, 3H), 1.09 (d, J = 7.3 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 152.1, 151.4, 140.6, 120.3, 107.3, 102.3, 95.4, 56.0, 55.5, 18.0, 12.9; IR [ATR]: ν = 2944, 2892, 2866, 1591, 1508, 1464, 1451, 1418, 1403, 1384, 1367, 1305, 1268, 1227, 1189, 1151, 1127, 1111, 1077, 1014, 998, 937, 921, 882, 836, 805, 731 cm–1; HRMS (ESI): exact mass calculated for C18H33O4Si [(M + H)+], 341.2143; found, 341.2107.
Triisopropyl(2-methoxy-4-(methoxymethoxy)-3-methylphenoxy)silane (13)
A 1000 mL Schlenk flask was charged with compound S4 (39.8 g, 117 mmol, 1 equiv) in 400 mL of dry THF. It was cooled to −50 °C, and n-BuLi (1.6 M, 88 mL, 140 mmol, 1.2 equiv) was added dropwise over 20 min. During addition, the color changed from bright red to strong dark red. Note that if an excess of n-BuLi was used in this transformation, double methylation (C-15 and C-1) was observed. The reaction mixture was allowed to warm to −10 °C over 1.5 h at which point D2O quenching confirmed full lithiation. It was cooled to −20 °C, and MeI (15 mL, 234 mmol, 2 equiv) was added dropwise over 5 min. The reaction was allowed to warm to room temperature overnight, upon which TLC (petroleum ether/ethyl acetate, 10:1) confirmed full conversion. The mixture was quenched by addition of solid NH4Cl and filtered. The residue solution was washed with H2O and brine, dried over MgSO4, and concentrated in vacuo. The desired product was obtained as a yellow oil in quantitative yield (41.4 g, 117 mmol). The product was used in the next reaction without further purification. 1H-NMR (400 MHz, CDCl3): δ = 6.67 (d, J = 8.9 Hz, 1H), 6.63 (d, J = 9.5 Hz, 1H), 5.11 (s, 2H), 3.77 (s, 3H), 3.49 (s, 3H), 2.17 (s, 3H), 1.33–1.19 (m, 3H), 1.10 (d, J = 7.4 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 152.1, 151.4, 140.6, 120.3, 107.3, 102.3, 95.4, 56.0, 55.5, 18.0, 12.9; IR [ATR]: ν = 2944, 2894, 2867, 1591, 1484, 1464, 1418, 1404, 1390, 1297, 1274, 1248, 1222, 1186, 1153, 1101, 1065, 1038, 1013, 991, 922, 881, 869, 824, 804, 761, 734 cm–1; HRMS (ESI): exact mass calculated for C19H33O4Si [M+], 353.2143; found, 353.2141.
3-Methoxy-2-methyl-4-((triisopropylsilyl)oxy)phenol (14)
A 250 mL three-necked flask was charged with compound 13 (20.25 g, 57 mmol, 1 equiv) in 60 mL of dry DCM, and ZnBr2 (12.9 g, 57 mmol, 1 equiv) was added, causing a color change from red to brown. The mixture was cooled to −32 °C, and EtSH (21.1 mL, 286 mmol, 5 equiv) was added quickly in one portion. The reaction was allowed to warm to −5 °C over 2.5 h, upon which TLC (petroleum ether/ethyl acetate, 10:1) confirmed full conversion. Note that with higher reaction temperature or fewer equivalents of EtSH, significant amounts of undesired side products 14′ were obtained. The reaction was quenched by addition of 20 mL of saturated NaHCO3 solution, allowed to warm to room temperature, and stirred for an additional 1.5 h. Layers were separated, and the aqueous layer was extracted three times with DCM. The combined organic layer was washed with H2O (2×) and brine, dried over MgSO4, and concentrated in vacuo. The crude oil was purified by flash chromatography (petroleum ether/ethyl acetate, 10:1), and the pure product was obtained as a yellow oil in 89% yield (15.7 g, 50.7 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.60 (d, J = 8.7 Hz, 1H), 6.41 (d, J = 8.6 Hz, 1H), 4.42 (bs, 1H), 3.78 (s, 3H), 2.17 (s, 3H), 1.34–1.19 (m, 3H), 1.10 (d, J = 7.4 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 149.7, 148.3, 143.2, 118.9, 117.3, 110.0, 60.3, 18.1, 12.9, 9.2; IR [ATR]: ν = 3341, 2967, 2936, 2863, 2846, 1598, 1496, 1460, 1422, 1381, 1367, 1304, 1262, 1211, 1174, 1156, 1075, 1034, 995, 919, 908, 891, 885, 821, 799, 758, 709 cm–1; HRMS (ESI): exact mass calculated for C17H31O3Si [(M + H)+], 311.2037; found, 311.2050.
(E)-Triisopropyl(2-methoxy-3-methyl-4-(penta-2,4-dien-1-yloxy)phenoxy)silane (15)
A 500 mL three-necked flask was charged with compound 14 (5.0 g, 16 mmol, 1 equiv) in 100 mL of dry THF. The pale-yellow solution was cooled to −35 °C, and n-BuLi (1.6 M, 10 mL, 16 mmol, 1 equiv) was added dropwise over 5 min. After 15 min, 5-bromopenta-1,3-diene (3.53 g, 18 mmol, 1.1 equiv, 73% in pentane) dissolved in 70 mL of dry DMF was added dropwise over 30 min. The reaction was allowed to warm to room temperature overnight, upon which TLC (petroleum ether/ethyl acetate, 12:1) confirmed full conversion. It was quenched by addition of solid NH4Cl, filtered, and concentrated in vacuo. The oily residue was taken up in ethyl acetate and little H2O. Layers were separated, and the aqueous layer was extracted three times with ethyl acetate. The combined organic layer was washed with H2O and brine, dried over MgSO4, and concentrated in vacuo. The crude oil was purified by flash chromatography (petroleum ether/ethyl acetate, 10:1), and the pure product was obtained as a yellow oil in 97% yield (5.9 g, 15.5 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.65 (d, J = 8.8, 1H), 6.45 (d, J = 8.9 Hz, 1H), 6.42–6.32 (m, 2H), 5.98–5.87 (m, 1H), 5.31–5.18 (m, 1H), 5.18–5.07 (m, 1H), 4.52–4.49 (m, 2H), 3.78 (s, 3H), 2.18 (s, 3H), 1.33–1.22 (m, 3H), 1.11 (d, J = 7.4 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 151.5, 149.9, 143.6, 136.4, 133.0, 129.3, 121.8, 117.9, 116.7, 107.3, 69.0, 60.2, 18.1, 12.9, 9.4; IR [ATR]: ν = 2944, 2892, 2866, 1605, 1483, 1463, 1417, 1383, 1368, 1274, 1253, 1220, 1185, 1164, 1103, 1037, 999, 951, 904, 881, 822, 793, 762, 733 cm–1; HRMS (ESI): exact mass calculated for C22H37O3Si [(M + H)+], 377.2506; found, 377.2494.
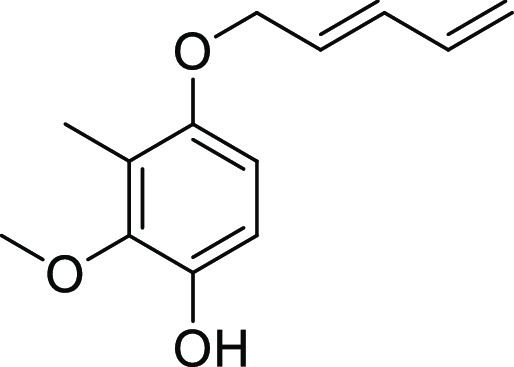
(E)-2-Methoxy-3-methyl-4-(penta-2,4-dien-1-yloxy)phenol (S5)
A 250 mL round-bottom flask was charged with compound 15 (4.22 g, 11.21 mmol, 1 equiv) in 75 mL of dry THF, and TBAF (1.0 M in THF, 11.8 mL, 11.8 mmol, 1.05 equiv) was added. After 2 min, TLC (petroleum ether/ethyl acetate, 10:1) confirmed full conversion. The reaction was quenched by addition of saturated NH4Cl solution, and the aqueous layer was extracted twice with ethyl acetate. The combined organic layer was washed twice with NH4Cl and H2O, followed by brine. It was dried over MgSO4 and concentrated in vacuo. The crude red oil was dried over high vacuum at 40 °C overnight. The received brown solid was purified by flashed chromatography over silica (gradient: petroleum ether/ethyl acetate, 5:1–3:1–1:1). The desired product was obtained as a pale brown solid in 95% yield (2.35 g, 10.67 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.73 (d, J = 8.8, 1H), 6.54 (d, J = 8.8 Hz, 1H), 6.45–6.32 (m, 2H), 5.98–5.84 (m, 1H), 5.35–5.19 (m, 2H), 5.19–5.08 (m, 1H), 4.54–4.48 (m, 2H), 3.78 (s, 3H), 2.21 (s, 3H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 151.0, 146.1, 143.3, 136.4, 133.2, 129.2, 120.7, 118.0, 111.8, 108.7, 69.3, 61.0, 9.6; HRMS (ESI): exact mass calculated for C13H17O3 [(M + H)+], 221.1172; found, 221.1176.
(E)-Triethyl(2-methoxy-3-methyl-4-(penta-2,4-dien-1-yloxy)phenoxy)silane (16)
A 100 mL round-bottom flask was charged with compound S5 (2.33 g, 10.58 mmol, 1 equiv) and imidazole (1.51 g, 22.21 mmol, 2.1 equiv) in 35 mL of dry DCM. TESCl (2.66 mL, 15.87 mmol, 1.5 equiv) was added dropwise to the orange solution. An immediate color change to yellow was observed accompanied by precipitation. TLC (petroleum ether/ethyl acetate, 5:1) after 20 min confirmed full conversion, and the reaction was quenched by addition of solid NaHCO3. It was filtered over silica with DCM and concentrated in vacuo. It was additionally dried on high vacuum overnight, and the desired product was obtained as an orange oil in 95% yield (3.35 g, 10.01 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.63 (d, J = 8.8 Hz, 1H), 6.46 (d, J = 8.8 Hz, 1H), 6.42–6.33 (m, 2H), 5.99–5.86 (m, 1H), 5.31–5.18 (m, 1H), 5.18–5.09 (m, 1H), 4.52–4.48 (m, 2H), 3.77 (s, 3H), 2.17 (s, 3H), 1.00 (t, J = 8.4, 9H), 0.74 (q, J = 8.3 Hz, 6H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 151.8, 150.1, 143.0, 136.4, 133.0, 129.3, 121.6, 117.9, 117.2, 107.4, 69.0, 60.1, 9.4, 6.8, 5.2; HRMS (ESI): exact mass calculated for C19H31O3Si [(M + H)+], 335.2037; found, 335.2038.
3-Methoxy-2-methyl-6-(penta-1,4-dien-3-yl)-4-((triethylsilyl)oxy)phenol (17)
The starting material was azeotropically dried three times with toluene prior to use. A 1000 mL Schlenk flask was charged with compound 16 (9.0 g, 26.9 mmol, 1 equiv) in 270 mL of dry toluene. The pale-yellow solution was schlenked 10–15 times, then EuFOD (660 mg, 0.63 mmol, 2.3 mol %) was added in one portion, and the mixture was schlenked again 10–15 times. The flask was placed in a 110 °C oil bath and heated for 2.5 h. After TLC (petroleum ether/ethyl acetate, 10:1) confirmed full conversion, the reaction was allowed to cool to room temperature. It was quenched by addition of H2O, diluted with toluene, and stirred for 15 h. Layers were separated, and the organic layer was washed with H2O (5×) followed by brine. It was dried over MgSO4, filtered, and concentrated. The crude brown material was flashed over silica, and the desired product was collected as a yellow oil in 84% yield (7.56 g, 22.60 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.49 (s, 1H), 6.04 (ddd, J = 17.3, 10.3, 6.2 Hz, 2H), 5.24 (dt, J = 10.3, 1.5 Hz, 2H), 5.14 (dt, J = 17.3, 1.6 Hz, 2H), 4.75 (broad s, 1H), 4.21–4.12 (m, 1H), 3.76 (s, 3H), 2.15 (s, 3H), 0.99 (t, J = 7.9 Hz, 9H), 0.72 (q, J = 7.9 Hz, 6H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 148.7, 146.6, 142.3, 138.7, 136.4, 133.1, 129.3, 122.2, 118.1, 116.5, 60.1, 47.7, 9.4, 6.8, 5.2; HRMS (ESI): exact mass calculated for C19H31O3Si [(M + H)+], 335.2037; found, 335.2024.
Triethyl(2-methoxy-3-methyl-5-(penta-1,4-dien-3-yl)-4-((triisopropylsilyl)oxy)phenoxy)silane (18)
A 500 mL Schlenk flask was charged with compound 17 (7.56 g, 22.60 mmol, 1 equiv) in 230 mL of dry THF, and the yellow mix was cooled to −85 °C. After 5 min, n-BuLi (1.60 M, 17.0 mL, 27.12 mmol, 1.20 equiv) was added dropwise over 11 min. A change in color was observed from yellow over brown to dark red after full addition. The mixture was stirred at −85 °C for 30 min before TIPSOTf (6.68 mL, 24.86 mmol, 1.10 equiv) was added dropwise over 30 min. The reaction was allowed to slowly warm up, and after 1.5 h at −45 °C, TLC (petroleum ether/ethyl acetate, 20:1) confirmed full conversion. It was quenched with saturated NaHCO3 solution, diluted with toluene, and removed from cooling. The biphasic mixture was stirred for 16 h, and then the aqueous layer was extracted twice with ethyl acetate. The combined organic layer was washed with H2O and brine and dried over MgSO4. Solvents were removed in vacuo, and the crude residue was filtered over silica. The desired product was obtained as a colorless oil in 92% yield (10.19 g, 20.76 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.48 (s, 1H), 5.93 (ddd, J = 17.3, 10.3, 5.8 Hz, 2H), 5.12 (dt, J = 10.3, 1.6 Hz, 2H), 5.00 (dt, J = 17.3, 1.7 Hz, 2H), 4.46–4.40 (m, 1H), 3.75 (s, 3H), 2.18 (s, 3H), 1.38–1.23 (m, 3H), 1.11 (d, J = 7.5 Hz, 18H), 0.97 (t, J = 7.9 Hz, 9H), 0.71 (q, J = 8.3 Hz, 6H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 148.3, 147.1, 142.6, 140.2, 127.1, 122.8, 118.3, 115.3, 59.9, 44.7, 18.3, 14.5, 11.5, 6.8, 5.2; HRMS (ESI): exact mass calculated for C28H49O3Si2 [(M – H)+], 489.3215; found, 489.3182.
2-Methoxy-3-methyl-5-(penta-1,4-dien-3-yl)-4-((triisopropylsilyl)oxy)phenol (11)
A 1000 mL round-bottom flask was charged with compound 18 (10.19 g, 20.76 mmol, 1 equiv) in 280 mL of THF, and TFA (10% in H2O, 71.0 mL, 62.28 mmol, 3 equiv) was added. After 30 min, TLC (petroleum ether/ethyl acetate, 20:1) confirmed full conversion. The reaction was quenched by addition of saturated NaHCO3 solution to pH = 7. The aqueous layer was extracted twice with ethyl acetate, and the combined organic layer was washed with H2O and brine. It was dried over MgSO4, filtered, and concentrated in vacuo. The crude material was filtered over silica, the solvent was removed in vacuo, and the pure product was dried on high vacuum overnight. The desired product was obtained as orange solids in 93% yield (7.28 g, 19.33 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.59 (d, J = 0.7 Hz, 1H), 5.94 (ddd, J = 17.3, 10.3, 6.0 Hz, 2H), 5.24–5.19 (m, 1H), 5.12 (ddd, J = 10.3, 1.6 Hz, 2H), 5.01 (ddd, J = 17.3, 1.7 Hz, 2H), 4.47–4.41 (m, 1H), 3.74 (s, 3H), 2.22 (d, J = 0.6 Hz, 3H), 1.36–1.25 (m, 4H), 1.11 (d, J = 7.4 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 146.2, 144.3, 142.9, 140.0, 128.5, 121.8, 115.5, 112.5, 60.8, 44.9, 18.3, 14.5, 11.6; HRMS (ESI): exact mass calculated for C22H37O3Si [(M + H)+], 377.2506; found, 377.2490.
(S,E)-(4-((1-(Allyloxy)pent-3-en-2-yl)oxy)-3-methoxy-2-methyl-6-(penta-1,4-dien-3-yl)phenoxy)triisopropylsilane (19)
A 250 mL Schlenk flask was charged with compound 18 (1.82 g, 4.83 mmol, 1 equiv) and chiral alcohol 12 (824 mg, 5.80 mmol, 1.2 equiv) in 170 mL of dry toluene. The mixture was cooled in an ice bath, and PBu3 (93%, 1.67 mL, 6.28 mmol, 1.3 equiv) was added. After 5 min, ADDP (1.46 g, 5.80 mmol, 1.2 equiv) dissolved in 25 mL of dry toluene was added, causing a color change from yellow to brown. After 10 min, precipitation was observed. TLC (petroleum ether/ethyl acetate, 5:1) after 2 h confirmed full consumption of the starting material. Et2O was added to further precipitate OPBu3. The mixture was stirred for a few minutes and then flashed with pure Et2O over silica topped with Celite. The solvent was removed in vacuo, and the crude material was purified by column chromatography. The desired product was obtained as a pale-yellow oil in 69% yield (1.68 g, 3.35 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.55 (s, 1H), 5.99–5.83 (m, 3H), 5.65 (dqd, J = 15.5, 6.5, 0.8 Hz, 1H), 5.46 (ddq, J = 15.4, 7.6, 1.6 Hz, 1H), 5.33–5.23 (m, 1H), 5.21–5.14 (m, 1H), 5.13–5.06 (m, 2H), 5.05–4.90 (m, 2H), 4.69–4.60 (m, 1H), 4.46–4.40 (m, 1H), 4.11–4.04 (m, 2H), 3.79 (s, 3H), 3.69 (dd, J = 10.4, 6.4 Hz, 1H), 3.59 (dd, J = 10.4, 4.4 Hz, 1H), 2.17 (s, 3H), 1.67–1.60 (m, 3H), 1.34–1.24 (m, 3H), 1.09 (d, J = 7.4 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 148.2, 147.4, 144.8, 140.2, 140.2, 134.9, 130.7, 128.2, 126.6, 122.7, 117.1, 116.7, 115.3, 115.3, 79.9, 73.0, 72.5, 60.4, 44.7, 18.3, 17.9, 14.5, 11.4; HRMS (ESI): exact mass calculated for C30H48O4SiNa [(M + Na)+], 523.3214; found, 523.3214; [α]D20 = +19.72 (c 1.40, CH2Cl2).
(S,E)-2-(5-(Allyloxy)pent-3-en-2-yl)-6-methoxy-5-methyl-3-(penta-1,4-dien-3-yl)-4-((triisopropylsilyl)oxy)phenol (10)
The starting material was azeotropically dried three times with toluene prior to use. A Schlenk tube was charged with compound 19 (0.90 g, 1.80 mmol, 1 equiv) in 18 mL of dry o-xylene. The solution was schlenked 10 times, then EuFOD (93 mg, 90 μmol, 5 mol %) was added, and the mix was schlenked again 10 times. The pale-yellow solution was placed in a 120 °C oil bath and heated for 8 h. Then, another portion of EuFOD (70 mg, 68 μmol, 3.8 mol %) was added and the reaction was heated again to 120 °C for 10 h. After TLC (DCM) confirmed full conversion, the reaction was cooled to room temperature and the solvent was removed in vacuo. The crude mixture was purified by column chromatography (DCM), and the desired product was obtained as a yellow oil in 55% yield (494 mg, 0.99 mmol). Phenol 11 was collected in 31% yield (211 mg, 0.56 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.17–6.03 (m, 3H), 5.98–5.83 (m, 1H), 5.57–5.46 (m, 1H), 5.40 (s, 1H), 5.24 (dq, J = 17.2, 1.7 Hz, 1H), 5.18–5.11 (m, 3H), 5.12–4.97 (m, 2H), 4.89–4.80 (m, 1H), 4.01–3.89 (m, 4H), 3.80–3.68 (m, 4H), 2.19 (s, 3H), 1.35 (d, J = 7.0 Hz, 3H), 1.31–1.21 (m, 3H), 1.10 (d, J = 7.4 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 146.5, 145.2, 142.8, 140.7, 140.4, 138.3, 135.1, 128.1, 127.0, 125.3, 119.2, 117.0, 115.6, 115.0, 71.1, 70.6, 60.8, 43.3, 37.1, 30.0, 18.7, 18.4, 14.4, 11.8; HRMS (ESI): exact mass calculated for C30H48O4SiNa [(M + Na)+], 523.3214; found, 523.3214; [α]D20 = −8.69 (c 0.70, CH2Cl2).
(5S,8S)-2-Methoxy-3,8-dimethyl-4-((triisopropylsilyl)oxy)-5-vinyl-5,8-dihydronaphthalen-1-ol (20)
The starting material was azeotropically dried with toluene prior to use. A 250 mL Schlenk flask was charged with compound 10 (663 mg, 1.32 mmol, 1 equiv) in 130 mL of dry DCM. Two freeze–thaw cycles were carried out to degas the colorless solution before a Grubbs second-generation catalyst (70 mg, 66 μmol, 5 mol %) was added. The reaction was heated to reflux in a water bath, and after 2 h, TLC (petroleum ether/ethyl acetate, 10:1) confirmed full conversion. The flask was removed from heating and stirred on air for 30 min. The mixture was filtered over silica, and the desired product was obtained as slightly greenish solids in 92% yield (489 mg, 1.21 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.11 (ddd, J = 17.4, 10.3, 5.4 Hz, 1H), 5.84 (ddd, J = 10.2, 3.5, 1.6 Hz, 1H), 5.74 (ddd, J = 10.2, 4.2, 1.8 Hz, 1H), 5.43 (s, 1H), 4.91 (ddd, J = 10.3, 1.5 Hz, 1H), 4.85 (ddd, J = 17.4, 1.6 Hz, 1H), 4.20–4.15 (m, 1H), 3.73 (s, 3H), 3.57–3.48 (m, 1H), 2.20 (s, 3H), 1.34–1.24 (m, 6H), 1.13–1.04 (m, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 145.9, 143.8, 141.8, 141.0, 130.6, 124.1, 123.8, 119.1, 113.0, 60.8, 38.5, 29.9, 23.3, 18.2, 14.6, 11.1; HRMS (ESI): exact mass calculated for C24H37O3Si [(M – H)+], 401.2506; found, 401.2507; [α]D20 = +192.84 (c 1.65, CH2Cl2).
(((5S,8S)-2-Methoxy-3,8-dimethyl-5-vinyl-5,8-dihydronaphthalene-1,4-diyl)bis(oxy))bis(triisopropylsilane) (9)
A 50 mL Schlenk flask was charged with compound 20 (489 mg, 1.21 mmol, 1 equiv) in 10 mL of dry THF and cooled to −80 °C. After 5 min, n-BuLi (2.50 M, 0.59 mL, 1.46 mmol, 1.2 equiv) was added dropwise over 8 min. A change in color was observed from greenish to orange. The reaction was kept at −80 °C, and after 20 min, 10 mL of dry DMF was added dropwise over 9 min, causing a slight color change to stronger yellow. Then, TIPSCl (0.39 mL, 1.82 mmol, 1.5 equiv) was added dropwise and the reaction was allowed to slowly warm up. After 1 h and 40 min, TLC (petroleum ether/ethyl acetate, 20:1) at −35 °C confirmed full conversion. The reaction was quenched by addition of saturated NH4Cl solution, and the aqueous layer was extracted three times with Et2O. The combined organic layer was washed three times with H2O followed by brine. It was dried over MgSO4, filtered, and concentrated in vacuo. The crude material was flashed over silica, and the desired product was collected as greenish white solids in 97% yield (657 mg, 1.18 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.01 (ddd, J = 17.3, 10.2, 5.7 Hz, 1H), 5.80 (ddd, J = 10.1, 3.7, 1.6 Hz, 1H), 5.70 (ddd, J = 10.1, 4.2, 1.7 Hz, 1H), 4.86 (ddd, J = 10.2, 1.4 Hz, 1H), 4.74 (ddd, J = 17.3, 1.5 Hz, 1H), 4.20–4.14 (m, 1H), 3.63 (s, 3H), 3.55–3.47 (m, 1H), 2.18 (s, 3H), 1.37–1.25 (m, 6H), 1.23 (d, J = 6.8 Hz, 3H), 1.13–1.00 (m, 36H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 148.4, 147.1, 142.2, 141.7, 130.1, 129.6, 124.4, 123.2, 120.2, 112.7, 60.5, 39.0, 31.0, 24.6, 18.3, 18.3, 18.2, 18.2, 18.2, 14.6, 14.1, 11.3; HRMS (ESI): exact mass calculated for C33H59O3Si2 [(M + H)+], 559.3997; found, 559.3996; [α]D20 = +152.91 (c 0.95, CH2Cl2).
Methyl (E)-3-((1R,4S)-6-Methoxy-4,7-dimethyl-5,8-bis((triisopropylsilyl)oxy)-1,4-dihydronaphthalen-1-yl)acrylate (21)
The starting material was azeotropically dried three times with toluene prior to use. A 25 mL Schlenk flask was charged with compound 9 (400 mg, 0.716 mmol, 1 equiv) in 4 mL of dry degassed toluene. Freshly distilled methyl acrylate (0.39 mL, 4.29 mmol, 6 equiv) was added, and the flask was placed in an 80 °C oil bath. A Hoveyda–Grubbs second-generation catalyst (23 mg, 37 μmol, 5.1 mol %) dissolved in 2 mL of dry degassed toluene was added via a syringe pump over 10 h. After complete addition, the reaction mixture was heated for another 15 h. The next day, the flask was removed from heating and allowed to cool to room temperature. The reaction mixture was directly flashed over silica, and the desired product was collected as a pale greenish oil in 51% yield (223 mg, 0.361 mmol) accompanied by 41% (162 mg, 0.290 mmol) starting material. 1H-NMR (400 MHz, CDCl3): δ = 7.11 (dd, J = 15.8, 6.0 Hz, 1H), 5.86 (ddd, J = 10.1, 3.8, 1.8 Hz, 1H), 5.65 (ddd, J = 10.1, 4.1, 1.7 Hz, 1H), 5.48 (dd, J = 15.7, 1.4 Hz, 1H), 4.35–4.28 (m, 1H), 3.65 (s, 3H), 3.63 (s, 3H), 3.56–3.47 (m, 1H), 2.17 (s, 3H), 1.36–1.25 (m, 6H), 1.23 (d, J = 6.7 Hz, 3H), 1.13–1.03 (m, 36H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 167.8, 152.2, 149.0, 147.2, 141.8, 131.6, 129.7, 122.4, 121.0, 120.5, 119.3, 60.5, 51.4, 38.2, 30.9, 24.4, 18.3, 18.2, 18.1, 14.6, 14.1, 11.3; HRMS (ESI): exact mass calculated for C33H57O3Si2 [(M – C2H3O2)+], 557.3841; found, 557.3812; [α]D20 = +213.59 (c 1.0, CH2Cl2).
Methyl (R)-3-((1S,4S)-6-Methoxy-4,7-dimethyl-5,8-bis((triisopropylsilyl)oxy)-1,4-dihydronaphthalen-1-yl)butanoate ((7epi)-8)
A 25 mL Schlenk flask was charged with CuCI (37 mg, 0.38 mmol, 0.75 equiv), and compound 21 (310 mg, 0.50 mmol, 1equiv) in 8 mL of dry THF was added. The pale-yellow solution was cooled to −42 °C, and TMSCl (140 μL, 1.11 mmol, 2.2 equiv) was added. After 5 min, MeMgBr (3.0 M in Et2O, 0.67 mL, 2.01 mmol, 4 equiv) was added dropwise. The color changed from yellow to brown over green to red and was orange yellow after full addition. The reaction was allowed to slowly warm to −22 °C over 1 h at which point TLC (petroleum ether/ethyl acetate, 20:1) confirmed full conversion. It was quenched by addition of saturated NH4Cl solution and saturated NaHCO3 solution and stirred for 1 h and 30 min. The aqueous layer was extracted three times with ethyl acetate, and the combined organic layer was washed three times with H2O, followed by brine. It was dried over MgSO4 and concentrated in vacuo. The desired product was obtained as yellow solids in virtually quantitative yield (317 mg, 0.50 mmol). 1H-NMR (400 MHz, CDCl3): δ = 5.85 (ddd, J = 10.4, 3.6, 1.5 Hz, 1H), 5.71 (ddd, J = 10.4, 4.3, 1.8 Hz, 1H), 3.64–3.60 (m, 4H), 3.53 (s, 3H), 3.49–3.42 (m, 1H), 2.59 (m, 1H), 2.17 (s, 3H), 1.76 (dd, J = 15.5, 11.6 Hz, 1H), 1.65 (dd, J = 15.5, 3.5 Hz, 1H), 1.36–1.24 (m, 6H), 1.20 (d, J = 6.7 Hz, 3H), 1.13–1.02 (m, 36H). 13C{1H}-NMR (101 MHz, CDCl3): δ = 147.6, 148.5, 147.0, 141.3, 132.6, 130.6, 123.6, 121.0, 120.7, 60.5, 51.2, 40.9, 35.9, 34.2, 31.4, 24.5, 18.4, 18.3, 18.2, 18.1, 18.1, 14.6, 14.0, 11.5; HRMS (ESI): exact mass calculated for C35H61O5Si2 [(M – CH3)−], 617.4063; found, 617.4056; [α]D20 = +345.84 (c 0.42, CH2Cl2).
1-((1S,4S)-6-Methoxy-4,7-dimethyl-5,8-bis((triisopropylsilyl)oxy)-1,4-dihydronaphthalen-1-yl)ethan-1-one (23)
A 25 mL round-bottom flask was charged with compound 9 (152 mg, 0.271 mmol, 1 equiv) in 9 mL of dimethylacetamide, 3 mL of 1,2-dichlorethane, and 0.75 mL of H2O. Bis(acetonitrile)dichloropalladium(II) (25 mg, 95 μmol, 35 mol %) and 1,4-benzoquinone (323 mg, 2.99 mmol, 11 equiv) were added, and the mix was heated to 36 °C. After 5 h, TLC (petroleum ether/ethyl acetate, 20:1) confirmed full conversion. The mixture was diluted with Et2O, and the organic layer was washed with NaHCO3 (5×; at which point the aqueous layer lost all dark brown coloration) followed by H2O and brine. It was dried over MgSO4, filtered, and concentrated in vacuo. The crude material was flashed over silica, and the desired product was obtained as a pale pink oil in 92% yield (144 mg, 0.251 mmol). 1H-NMR (600 MHz, CDCl3): δ = 5.96 (ddd, J = 10.0, 4.3, 2.6 Hz, 1H), 5.55 (ddd, J = 10.0, 3.8, 1.4 Hz, 1H), 4.30–4.26 (m, 1H), 3.64 (s, 3H), 3.62–3.57 (m, 1H), 2.18 (s, 3H), 1.63 (s, 3H), 1.38–1.29 (m, 6H), 1.24 (d, J = 7.1 Hz, 3H), 1.12–0.99 (m, 36H); 13C{1H}-NMR (151 MHz, CDCl3): δ = 208.8, 149.5, 148.0, 141.8, 133.5, 129.9, 121.4, 120.5, 119.8, 60.5, 52.6, 30.8, 25.1, 24.4, 18.3, 18.2, 18.1, 14.5, 14.1, 11.3; [α]D20 = +57.62 (c 1.74, CH2Cl2).
1-((1S,4S)-6-Methoxy-4,7-dimethyl-5,8-bis((triisopropylsilyl)oxy)-1,2,3,4-tetrahydronaphthalen-1-yl)ethan-1-one (24)
A 50 mL Schlenk flask was charged with compound 23 (144 mg, 0.251 mmol, 1 equiv) in 15 mL of dry degassed DCM, and Crabtree’s catalyst (14 mg, 18 μmol, 7 mol %) was added. The flask was evacuated and backfilled eight times with H2, causing a color change from orange to yellow. The H2 valve was closed, and the reaction was stirred at room temperature. After 1 h, TLC (petroleum ether/ethyl acetate, 50:1) confirmed full conversion. The yellow solution was filtered through silica, and solvents were evaporated. The desired product was obtained as a colorless oil in 99% yield (143 mg, 0.248 mmol). 1H-NMR (400 MHz, CDCl3): δ = 3.72 (dd, J = 6.3, 2.0 Hz, 1H), 3.64 (s, 3H), 3.23–3.15 (m, 1H), 2.18 (m, 4H), 2.03–1.92 (m, 1H), 1.89 (s, 3H), 1.64–1.49 (m, 2H), 1.41–1.27 (m, 6H), 1.28–1.22 (m, 1H), 1.16 (d, J = 6.9 Hz, 3H), 1.13–1.00 (m, 36H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 213.2, 149.0, 148.0, 142.0, 132.6, 121.7, 119.9, 60.6, 47.9, 29.6, 28.1, 26.7, 22.0, 21.6, 18.3, 18.2, 18.0, 18.0, 14.5, 14.1, 11.4; HRMS (ESI): exact mass calculated for C33H61O4Si2 [(M + H)+], 577.4108; found, 577.4105; [α]D20 = +17.9 (c 0.80, CH2Cl2).
Methyl (Z)-3-((1R,4S)-6-Methoxy-4,7-dimethyl-5,8-bis((triisopropylsilyl)oxy)-1,4-dihydronaphthalen-1-yl)but-2-enoate (Z-22) and Methyl (E)-3-((1R,4S)-6-Methoxy-4,7-dimethyl-5,8-bis((triisopropylsilyl)oxy)-1,4-dihydronaphthalen-1-yl)but-2-enoate (E-22)
A 10 mL Schlenk flask was charged with CuCl (9.0 mg, 91 μmol, 0.5 equiv), and compound 21 (112 mg, 0.182 mmol, 1 equiv)/TMSCl (51 μL, 0.399 mmol, 2.2 equiv) in 1.5 mL of dry THF was added. The yellow solution with reddish copper salt was cooled to −50 °C, and after 5 min, MeMgBr (3.0 M, 0.30 mL, 0.91 mmol, 5 equiv) was added dropwise over 3 min. A color change was observed, from strong dark red after the first drop over green and pale green to orange. After full addition, the reaction mixture was yellow. The reaction mixture was allowed to slowly warm up to −20 °C, and after 45 min, TLC (petroleum ether/ethyl acetate, 20:1) confirmed full conversion. A small sample was taken for TLC reference, and then PhSeCl (80 mg, 0.417 mmol, 2.3 equiv) dissolved in 0.5 mL of dry THF was added, causing a color change to strong green. TLC (petroleum ether/ethyl acetate, 20:1) moments after addition already confirmed full conversion. The reaction was quenched by addition of saturated NH4Cl solution and NaHCO3 solution. The mix was stirred for 1 h before layers were separated. The aqueous layer was extracted three times with ethyl acetate. The combined organic layer was washed with H2O and brine and dried over MgSO4. Solvents were removed in vacuo, and the crude material was subjected to column chromatography. The desired product was collected as a yellow oil in 70% yield (100 mg, 0.127 mmol) as an inseparable 1:1 mixture of diastereomers. The material was directly subjected to the next reaction. A 50 mL round-bottom flask was charged with selenide (112 mg, 0.142 mmol, 1 equiv) in 2 mL of CHCl3, and pyridine (23 μL, 0.284 mmol, 2 equiv) was added followed by H2O2 (35% in H2O, 86 μL, 0.995 mmol, 7 equiv). The yellow solution was stirred at room temperature for 30 min, at which point TLC (petroleum ether/diethyl ether, 24:1) confirmed full conversion. The reaction was quenched by addition of saturated Na2S2O3 solution, and the aqueous layer was extracted three times with DCM. The combined organic layer was washed with saturated NaHCO3 solution and H2O. It was dried over MgSO4 and concentrated in vacuo. The crude material was purified by column chromatography, and two products were collected. A Z-isomer (Z-22) (eluting first) was obtained in 17% yield (17 mg, 27 μmol), and an E-isomer (E-22) (eluting second) was obtained in 29% yield (30 mg, 48 μmol). Z-isomer (Z-22): 1H-NMR (600 MHz, CDCl3): δ = 5.81 (ddd, J = 10.0, 4.1, 2.3 Hz, 1H), 5.70–5.65 (m, 2H), 5.47 (dq, J = 4.1, 2.2, 1.7 Hz, 1H), 3.70 (s, 3H), 3.63 (s, 3H), 3.56–3.50 (m, 1H), 2.17 (s, 3H), 1.36–1.24 (m, 9H), 1.22 (d, J = 6.7 Hz, 3H), 1.11–1.04 (m, 27H), 1.00–0.94 (m, 9H); 13C{1H}-NMR (151 MHz, CDCl3): δ = 166.9, 165.4, 148.7, 147.9, 141.3, 130.8, 130.4, 124.8, 122.2, 120.0, 114.8, 60.5, 50.8, 39.4, 30.7, 24.9, 22.0, 18.4, 18.2, 18.1, 18.1, 14.4, 14.0, 11.5; E-isomer (E-22): 1H-NMR (600 MHz, CDCl3): δ = 5.83 (ddd, J = 10.1, 3.9, 2.0 Hz, 1H), 5.58–5.57 (m, 1H), 5.55 (ddd, J = 10.0, 4.3, 1.7 Hz, 1H), 4.27 (ddt, J = 4.1, 2.8, 1.4 Hz, 1H), 3.63 (s, 3H), 3.63 (s, 3H), 3.56–3.53 (m, 1iH), 2.15 (s, 3H), 1.76 (d, J = 1.3 Hz, 3H), 1.37–1.27 (m, 6H), 1.24 (d, J = 6.7 Hz, 3H), 1.12–1.00 (m, 36H); 13C{1H}-NMR (151 MHz, CDCl3): δ = 167.8, 162.9, 149.1, 147.8, 141.5, 131.7, 130.6, 124.1, 120.8, 120.5, 115.7, 60.5, 50.8, 46.4, 31.1, 24.6, 18.4, 18.3, 18.1, 15.9, 14.7, 14.0, 11.4; HRMS (ESI): exact mass calculated for C35H61O5Si2 [(M–CH2 + H)+], 617.4052; found, 617.4033; [α]D20 = +155.91 (c 1.35, CH2Cl2).
(5S,8S)-3-Methoxy-2,5-dimethyl-4-((triisopropylsilyl)oxy)-8-vinyl-5,8-dihydronaphthalen-1-ol (25)
Acetic acid (219.66 mg, 3.66 mmol) was added to a solution of compound 9 (838 mg, 1.50 mmol) in THF (20.0 mL), and the mixture was cooled to 0 °C. After addition of TBAF (1.0 M in THF, 1.50 mL, 1.50 mmol) over a period of 10 min, the solution turned yellow and was stirred at rt for 1 h and 50 min until TLC showed formation of the hydroquinone. The reaction was stopped by adding 5.0 mL of water and 100 mL of ethyl acetate. The organic phase was washed with water and brine. The solvents were removed in vacuo. The crude mixture was separated by column chromatography, giving 40% (338 mg, 605 μmol) of the desired product and 54% (324 mg, 807 μmol) starting material. 1H-NMR (400 MHz, CDCl3): δ = 5.86 (ddd, J = 10.0, 4.3, 2.3 Hz, 1H), 5.70 (ddd, J = 17.4, 9.6, 9.4 Hz, 1H), 5.52 (ddd, J = 10.1, 3.4, 1.4 Hz, 1H), 5.35 (d, J = 17.0 Hz, 1H), 5.22 (dd, J = 9.3, 1.9 Hz, 1H), 5.21 (s, 1H), 3.99–3.92 (1H, m), 3.67 (s, 3H), 3.59–3.49 (m, 1H), 2.15 (s, 3H), 1.18–1.28 (m, 3H), 1.26 (d, J = 6.8 Hz, 3H), 1.11–1.04 (m, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 149.2, 146.9, 142.5, 140.9, 130.8, 129.1124.5, 117.3, 116.8116.1, 60.8, 40.8, 30.5, 23.9, 18.3, 18.2, 14.0, 9.3. [α]D20 = +123.184° (c 0.90, CH2Cl2).
(5S,8S)-3-Methoxy-2,5-dimethyl-4-((triisopropylsilyl)oxy)-8-vinyl-5,8-dihydronaphthalen-1-yl Acrylate (26)
A slurry of NaH (55% in mineral oil, 18 mg, 411 μmol) in THF (3.0 mL) was cooled to −23 °C. Then, compound 25 (83 mg, 205 μmol) in THF (1.0 mL) was added slowly over 20 min and the mixture was stirred for 6 min. Then, acryloyl chloride (37 mg, 411 μmol, 33 μL) was added dropwise over 5 min. The solution became brighter. After 1 h and 10 min at −20 °C, TLC confirmed full conversion of the starting material. The reaction mixture was quenched by adding a spatula of NaHCO3. Then, the solution was diluted with ethyl and water and the phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic phases were washed with water and brine. The solution was dried over Na2SO4, and the solvents evaporated, giving 95 mg of crude material. The crude material was purified over a plug of silica, and the desired product was isolated as a colorless oil in 83% yield (78 mg, 171 μmol). 1H-NMR (600 MHz, CDCl3): δ = 6.57 (broad d, J = 18.2 Hz, 1H), 6.29 (broad dd, J = 17.1, 9.7 Hz, 1H), 5.98 (broad d, J = 7.6 Hz, 1H), 5.77 (ddd, J = 10.1, 3.9, 1.9 Hz, 1H), 5.71–5.53 (broad m, 1H), 5.50 (ddd, J = 10.2, 3.6, 1.6 Hz, 2H), 4.91 (broad d, J = 16.9 Hz, 2H), 3.92–3.57 (m, 1H), 3.69 (s, 3H), 3.57–3.50 (m, 1H), 2.02 (broad s, 3H), 1.33 (m, J = 7.5 Hz, 3H), 1.28 (d, J = 7.3 Hz, 3H), 1.09 (d, J = 7.6 Hz, 9H), 1.06 (d, J = 7.6 Hz, 9H); 13C{1H}-NMR (151 MHz, CDCl3): δ = 164.1, 148.7, 148.6, 145.3, 141.9, 141.6, 132.6, 130.1, 129.6, 128.0, 125.3, 124.2, 122.6, 113.2, 60.9, 40.3, 30.5, 23.8, 18.2, 18.1, 14.0, 10.0; [α]D20 = +3.746° (c 0.37, CH2Cl2).
(4aR,7S)-9-Methoxy-7,10-dimethyl-8-((triisopropylsilyl)oxy)-4a,7-dihydro-2H-naphtho[1,8-bc]oxepin-2-one (27)
A solution of compound 26 (225 mg, 0.497 mmol) in degassed (freeze–pump–thaw, three cycles) toluene (30 mL) was heated to 80 °C. Then, a solution of Hoveyda–Grubbs second-generation catalyst (6 mg, 11 μmol) in toluene (0.5 mL) was added dropwise over 3.5 h to the starting material (syringe pump). After complete addition, the reaction was stirred for 15 h at 80 °C. Then, another portion of the catalyst (1.5 mg) was added, and the solution was stirred for an additional 3 h. The reaction was quenched by bubbling air through the solution for 40 min. Then, toluene was evaporated to obtain 210 mg of the crude product, which was purified via chromatography using toluene and diethyl ether as eluents (very fast filtration to avoid decomposition and isomerization). The desired product was obtained in 98% yield (198 mg, 462 μmol). 1H-NMR (400 MHz, CDCl3): δ = 6.51 (dd, J = 4.5 Hz, 10.9 Hz, 1H), 6.51 (dd, J = 4.5 Hz, 10.9 Hz, 1H), 5.95 (ddd, J = 2.2 Hz, 4.2 Hz, 10.2 Hz, 1H), 5.80 (ddd, J = 1.1 Hz, 3.5 Hz, 10.1 Hz, 1H), 5.76 (dd, J = 2.4 Hz, 10.9 Hz, 1H), 4.38–4.32 (m, 1H), 3.67 (s, 3H), 3.56–3.48 (m, 1H), 2.26 (s, 3H), 1.38–1.27 (m, 3H), 1.26 (d, J = 6.6 Hz, 3H), 1.07 (d, J = 7.5 Hz, 9H), 1.04 (d, J = 7.5 Hz, 9H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 164.1, 154.3, 148.6, 144.6, 143.7, 131.6, 128.4, 124.8, 122.7, 122.6, 119.4, 60.9, 34.5, 29.8, 23.6, 18.2, 18.1, 14.0, 10.3.
3-Methoxy-2-methyl-6-(penta-1,4-dien-3-yl)-4-((triisopropylsilyl)oxy)phenol (34)
A 100 mL Schlenk flask was charged with compound 15 (1.77 g, 4.7 mmol, 1 equiv) and azeotropically dried three times with toluene prior to use. Then, 40 mL of dry toluene was added, and the pale-yellow solution was degassed by applying the Schlenk technique. Subsequently, EuFOD (150 mg, 3 mol %) was added in one portion and the reaction mixture was degassed again. The flask was placed in a 110 °C oil bath and heated for 4 h, upon which TLC (petroleum ether/ethyl acetate, 10:1) confirmed full conversion. The reaction mixture was transferred into a separation funnel, washed with H2O (5×), followed by brine (5×), and dried over MgSO4. It was concentrated under reduced pressure, and the crude oil was purified by flash chromatography. Pure phenol was obtained as a yellow oil in 97% yield (1.72 g, 4.56 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.52 (s, 1H), 6.04 (ddd, J = 17.3, 10.3, 6.2 Hz, 2H), 5.24 (dt, J = 10.3, 1.5 Hz, 2H), 5.14 (dt, J = 17.3, 1.6 Hz, 2H), 4.75 (s, 1H), 4.22–4.14 (m, 1H), 3.77 (s, 4H), 2.18–2.12 (m, 3H), 1.29–1.19 (m, 3H), 1.10 (d, J = 7.4 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 148.4, 146.3, 142.9, 138.7, 122.1, 119.7, 117.6, 116.4, 60.3, 47.6, 18.1, 12.9, 9.4; IR [ATR]: ν = 3511, 2945, 2867, 1635, 1482, 1438, 1390, 1343, 1231, 1119, 1056, 997, 917, 881, 811, 760, 733 cm–1; HRMS (ESI): exact mass calculated for C22H37O3Si [(M + H)+], 377.2506; found, 377.2509.
3-Methoxy-2-methyl-6-(penta-1,4-dien-3-yl)-4-((triisopropylsilyl)oxy)phenyl acrylate (33)
A 250 mL Schlenk flask was charged with compound 34 (5.59 g, 15 mmol, 1 equiv) in 100 mL of dry DCM. The clear yellow solution was cooled in an ice bath, and Et3N (6.2 mL, 45 mmol, 3 equiv) was added, causing a color change to orange. Dropwise addition of acryloyl chloride (1.45 mL, 18 mmol, 1.2 equiv) caused a color change to strong orange. After 2 h, TLC (petroleum ether/ethyl acetate, 10:1) confirmed full consumption of the starting material. The reaction was quenched by addition of saturated NH4Cl solution, and layers were separated. The aqueous layer was extracted three times with DCM. The combined organic layer was washed with H2O and brine. It was dried over MgSO4 and concentrated in vacuo. The crude orange syrup was flashed over silica (petroleum ether/ethyl acetate, 10:1). The pure product was obtained as a colorless to pale yellow oil in 93% yield (5.94 g, 14 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.63–6.57 (m, 2H), 6.33 (dd, J = 17.3, 10.4 Hz, 1H), 6.01 (dd, J = 10.4, 1.3 Hz, 1H), 5.89 (ddd, J = 17.2, 10.3, 6.2 Hz, 2H), 5.12 (dt, J = 10.3, 1.5 Hz, 2H), 5.01 (dt, J = 17.3, 1.6 Hz, 2H), 4.06–4.00 (m, 1H), 3.79 (s, 3H), 2.04 (d, J = 0.5 Hz, 3H), 1.34–1.18 (m, 3H), 1.10 (d, J = 7.3 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 164.2, 148.0, 147.1, 140.9, 138.9, 132.6, 129.4, 127.8, 125.6, 117.3, 115.9, 60.3, 46.3, 18.1, 12.9, 10.2; IR [ATR]: ν = 2945, 2868, 1746, 1634, 1587, 1482, 1436, 1402, 1341, 1292, 1231, 1203, 1148, 1112, 1061, 1016, 997, 917, 905, 881, 803, 771, 734 cm–1; HRMS (ESI): exact mass calculated for C25H39O4Si [(M + H)+], 431.2612; found, 431.2600.
8-Methoxy-9-methyl-7-((triisopropylsilyl)oxy)-5-vinylbenzo[b]oxepin-2(5H)-one ((rac)-35)
Compound 33 (1.53 g, 3.55 mmol, 1 equiv) was azeotropically dried three times with toluene prior to use. It was dissolved in 360 mL of dry toluene, and two freeze–thaw cycles were carried out. Subsequently, a Grubbs–Hoveyda second-generation catalyst (67 mg, 0.11 mmol, 3 mol %) was added and the yellow mixture was heated to 80 °C for 15 h. The next day, TLC (petroleum ether/ethyl acetate, 10:1) confirmed full conversion. The reaction was removed from heating and allowed to cool to room temperature. Then, the flask was opened and stirred on air for approximately 1 h. The mixture was quickly flashed over a plug of silica to remove catalyst residues. Note that fast purification is necessary to prevent isomerization of the α,β-double bond into conjugation with the aromatic system and the methylene group. The desired product was subjected to the next reaction without further purification. 1H-NMR (400 MHz, CDCl3): δ = 6.85 (dd, J = 11.1, 6.8 Hz, 1H), 6.51 (s, 1H), 6.03 (ddd, J = 17.2, 10.3, 7.0 Hz, 1H), 5.90 (dd, J = 11.0, 1.1 Hz, 1H), 5.30 (dt, J = 10.2, 1.1 Hz, 1H), 5.23 (dt, J = 17.2, 1.2 Hz, 1H), 4.20–4.14 (m, 1H), 3.76 (s, 3H), 2.27 (s, 3H), 1.30–1.20 (m, 3H), 1.10 (d, J = 7.0 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 163.8, 149.8, 148.5, 146.5, 143.2, 134.6, 129.7, 125.6, 120.7, 117.9, 115.5, 60.4, 44.7, 18.0, 12.9, 10.3; IR [ATR]: ν = 2944, 2867, 1730, 1589, 1482, 1436, 1383, 1347, 1248, 1202, 1168, 1110, 1061, 1000, 919, 881, 808, 732 cm–1; HRMS (ESI): exact mass calculated for C23H35O4Si [(M + H)+], 403.2299; found, 403.2283.
8-Methoxy-4,9-dimethyl-7-((triisopropylsilyl)oxy)-5-vinyl-4,5-dihydrobenzo[b]oxepin-2(3H)-one ((rac)-36)
A 100 mL Schlenk flask was charged with CuCl (35 mg, 0.39 mmol, 5 mol %) and LiCl (30 mg, 0.68 mmol, 10 mol %), and compound (rac)-35 (2.81 g, 6.98 mmol, 1 equiv) in 46 mL of dry THF was added followed by dropwise addition of TMSCl (0.98 mL, 7.68 mmol, 1.1 equiv). The orange mixture was cooled to −40 °C, and after 20 min, MeMgBr (3.0 M in ether, 2.79 mL, 8.38 mmol, 1.2 equiv) was added dropwise over 5 min. After 10 min, TLC (petroleum ether/ethyl acetate, 10:1) confirmed full conversion. The reaction was quenched by addition of saturated NaHCO3 solution and saturated NH4Cl solution. The blue solution was allowed to warm to room temperature and stirred for 15 min. The aqueous layer was extracted three times with ethyl acetate, and the combined organic layer was washed with H2O and brine. It was dried over MgSO4, filtered, and concentrated in vacuo. The crude orange oil was subjected to column chromatography (petroleum ether/ethyl acetate, 25:1), and the desired product was obtained as a yellow oil in 42% yield (1.24 g, 2.96 mmol) over two steps. The material crystallized in the freezer. 1H-NMR (400 MHz, CDCl3): δ = 6.58 (s, 1H), 5.92 (ddd, J = 17.2, 10.3, 7.9 Hz, 1H), 5.30 (dt, J = 10.4, 1.1 Hz, 1H), 5.17 (dt, J = 17.2, 1.3 Hz, 1H), 3.78 (s, 3H), 3.19–3.10 (m, 1H), 2.59 (dd, J = 11.9, 7.2 Hz, 1H), 2.30–2.12 (m, 6H), 1.32–1.20 (m, 3H), 1.17 (d, J = 6.7 Hz, 3H), 1.13–1.05 (m, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 171.0, 148.3, 146.4, 143.5, 137.2, 127.8, 123.6, 118.4, 116.2, 60.4, 49.1, 38.4, 38.2, 19.8, 18.0, 12.9, 10.0; IR [ATR]: ν = 2945, 2867, 1764, 1587, 1481, 1438, 1384, 1346, 1290, 1239, 1208, 1171, 1131, 1114, 1061, 1027, 999, 943, 917, 882, 858, 818, 797, 785, 762, 731 cm–1; HRMS (ESI): exact mass calculated for C24H39O4Si [(M + H)+], 419.2612; found, 419.2635.
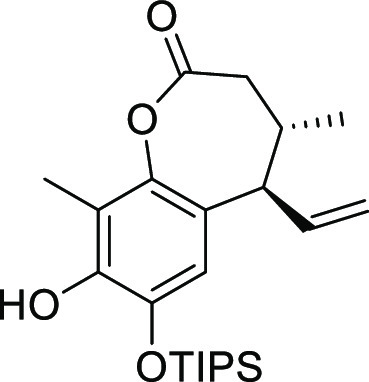
8-Hydroxy-4,9-dimethyl-7-((triisopropylsilyl)oxy)-5-vinyl-4,5-dihydrobenzo[b]oxepin-2(3H)-one ((rac)-S6)
A 25 mL Schlenk flask was charged with AlCl3 (283 mg, 2.12 mmol, 3.21 equiv) in 4 mL of DCM. To the mixture was added dimethyl sulfide (0.15 mL, 1.98 mmol, 3 equiv), causing the solid AlCl3 to dissolve. After 20 min, compound (rac)-36 (277 mg, 0.66 mmol, 1 equiv) dissolved in 4 mL of dry DCM was added dropwise. After 30 min, TLC (petroleum ether/ethyl acetate, 12:1) confirmed full conversion. The reaction was quenched with saturated NaHCO3 solution and carefully brought to pH = 7. The color changed from strong dark brown to orange. The aqueous layer was extracted three times with DCM. The combined organic layer was washed with H2O and brine. It was dried over MgSO4, filtered, and concentrated in vacuo. The crude oil was flashed over a plug of silica, and the desired product was obtained as a red oil in 89% yield (238 mg, 0.59 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.55 (s, 1H), 5.91 (ddd, J = 17.2, 10.4, 7.8 Hz, 1H), 5.69 (s, 1H), 5.30 (dt, J = 10.4, 1.2 Hz, 1H), 5.16 (dt, J = 17.3, 1.3 Hz, 1H), 3.14 (dd, J = 9.3, 7.7 Hz, 1H), 2.59 (dd, J = 11.9, 7.3 Hz, 1H), 2.29–2.11 (m, 5H), 1.37–1.23 (m, 3H), 1.16 (d, J = 6.7 Hz, 3H), 1.11 (dd, J = 7.4, 3.1 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 171.1, 144.6, 143.9, 139.6, 137.4, 123.0, 118.2, 115.8, 113.1, 48.9, 38.6, 38.2, 19.8, 18.0, 12.8, 9.5; IR [ATR]: ν = 3531, 2944, 2867, 1762, 1611, 1488, 1462, 1440, 1383, 1351, 1316, 1245, 1203, 1171, 1131, 1113, 1053, 1025, 999, 946, 919, 881, 856, 803, 787, 743, 683, 647 cm–1; HRMS (ESI): exact mass calculated for C23H37O4Si [(M + H)+], 405.2456; found, 405.2476.
8-(Methoxymethoxy)-4,9-dimethyl-7-((triisopropylsilyl)oxy)-5-vinyl-4,5-dihydrobenzo[b]oxepin-2(3H)-one ((rac)-32)
A 50 mL Schlenk flask was charged with NaI (163 mg, 1.09 mmol, 20 mol %) and compound (rac)-S6 (2.20 g, 5.44 mmol, 1 equiv) dissolved in 25 mL of dry THF. The red solution was cooled to −35 °C, and LiHMDS (1.0 M, 6.0 mL, 6.0 mmol, 1.1 equiv) was added followed by MOMCl (0.58 mL, 7.61 mmol, 1.4 equiv). The reaction was allowed to slowly warm up. After 25 min at −26 °C, TLC (petroleum ether/ethyl acetate, 10:1) confirmed full conversion. The reaction was quenched with saturated NaHCO3 solution and allowed to warm to room temperature. The aqueous layer was extracted four times with Et2O. The combined organic layer was washed with little H2O and brine. It was dried over MgSO4, filtered, and concentrated in vacuo. The crude orange-red oil was flashed over silica (25 g; petroleum ether/ethyl acetate, 20:1), and the desired product was collected as an orange oil in 94% yield (2.30 g, 5.13 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.58 (s, 1H), 5.91 (ddd, J = 17.2, 10.4, 8.0 Hz, 1H), 5.31 (dt, J = 10.5, 1.1 Hz, 1H), 5.20–5.08 (m, 3H), 3.57 (s, 3H), 3.19–3.10 (m, 1H), 2.60 (dd, J = 11.9, 7.2 Hz, 1H), 2.24 (s, 3H), 2.22 (m, 2H), 1.29–1.20 (m, 3H), 1.17 (d, J = 6.7 Hz, 3H), 1.08 (dd, J = 7.4, 3.0 Hz, 18H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 170.9, 145.8, 145.7, 143.4, 137.1, 128.0, 124.1, 118.5, 115.9, 99.1, 57.7, 49.0, 38.4, 38.2, 19.8, 18.0, 13.0, 10.7; IR [ATR]: ν = 2944, 2868, 1754, 1645, 1589, 1482, 1416, 1383, 1342, 1235, 1203, 1162, 1130, 1057, 964, 923, 881, 854 cm–1; HRMS (ESI): exact mass calculated for C25H41O5Si [(M + H)+], 449.2718; found, 449.2738.
7-Hydroxy-8-(methoxymethoxy)-4,9-dimethyl-5-vinyl-4,5-dihydrobenzo[b]oxepin-2(3H)-one ((rac)-37)
A 50 mL round-bottom flask was charged with compound (rac)-32 (2.30 g, 5.13 mmol, 1 equiv) dissolved in 20 mL of dry THF. To the orange solution was added TBAF (1.0 M in THF, 5.38 mL, 5.38 mmol, 1.05 equiv), causing a color change to black. After 3 min, TLC (petroleum ether/ethyl acetate, 5:1) confirmed full conversion. The reaction was quenched by addition of saturated NH4Cl solution, and the aqueous layer was extracted three times with Et2O. The combined organic layer was washed with small portions of H2O (12×) until the aqueous layer became clear and as good as colorless. The orange organic phase was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The crude material was flashed over a plug of silica (gradient: petroleum ether/ethyl acetate, 5:1, to pure ethyl acetate). The purified material was dried on high vacuum overnight, and the desired product was obtained as a yellow-orange oil in 77% yield (1.16 g, 3.97 mmol). 1H-NMR (400 MHz, CDCl3): δ = 7.45–7.02 (broad s, 1H), 6.69 (s, 1H), 5.95 (ddd, J = 17.1, 10.3, 8.2 Hz, 1H), 5.32–5.25 (m, 1H), 5.21–5.12 (m, 1H), 5.03 (d, J = 6.1 Hz, 1H), 4.98 (d, J = 6.1 Hz, 1H), 3.63 (s, 3H), 3.14 (t, J = 8.8 Hz, 1H), 2.58 (dd, J = 11.9, 7.2 Hz, 1H), 2.27–2.12 (m, 5H), 1.16 (d, J = 6.7 Hz, 3H). 13C{1H}-NMR (101 MHz, CDCl3): δ = 171.0, 146.2, 143.4, 142.5, 137.0, 129.6, 122.6, 118.6, 112.8, 99.9, 57.4, 49.3, 38.2, 38.0, 19.8, 10.3; IR [ATR]: ν = 3355, 2977, 2871, 1757, 1642, 1593, 1482, 1437, 1345, 1291, 1242, 1200, 1170, 1157, 1132, 1108, 1048, 966, 913, 856, 803, 730, 647 cm–1; HRMS (ESI): exact mass calculated for C16H21O5 [(M + H)+], 293.1384; found, 293.1384.
(4S,5R)-7-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-8-(methoxymethoxy)-4,9-dimethyl-5-vinyl-4,5-dihydrobenzo[b]oxepin-2(3H)-one (38) and (4R,5S)-7-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-8-(methoxymethoxy)-4,9-dimethyl-5-vinyl-4,5-dihydrobenzo[b]oxepin-2(3H)-one (38′)
A 100 mL Schlenk flask was charged with compound (rac)-37 (539 mg, 1.84 mmol, 1 equiv) and alcohol 12 (367 mg, 2.58 mmol, 1.4 equiv) in 12 mL of dry toluene. The mixture was cooled to 0 °C, and PBu3 (0.50 mL, 2.03 mmol, 1.1 equiv) was added. After 5 min, ADDP (512 mg, 2.03 mmol, 1.1 equiv) dissolved in 8 mL of dry toluene was added. Immediate solidification was observed. After 2 h, TLC (petroleum ether/ethyl acetate, 5:1) confirmed full conversion. The reaction was diluted with Et2O, causing OPBu3 to precipitate. The mixture was filtered over silica gel and Celite. It was concentrated in vacuo, and the oily residue was subjected to column chromatography (petroleum ether/ethyl acetate, 13:1). The desired product 38 was obtained as a colorless to pale yellow oil in 36% yield (278 mg, 0.667 mmol). Diastereomer 38′ was collected as a pale-yellow oil in 36% yield (277 mg, 0.665 mmol). Desired 38: 1H-NMR (400 MHz, CDCl3): δ = 6.65 (s, 1H), 5.97–5.82 (m, 2H), 5.81–5.67 (m, 1H), 5.48 (ddq, J = 15.4, 7.2, 1.6 Hz, 1H), 5.32–5.22 (m, 2H), 5.20–5.08 (m, 4H), 4.66 (td, J = 7.0, 4.0 Hz, 1H), 4.02 (dt, J = 5.6, 1.5 Hz, 2H), 3.65 (dd, J = 10.4, 6.9 Hz, 1H), 3.59–3.53 (m, 5H), 3.14 (t, J = 8.7 Hz, 1H), 2.59 (dd, J = 12.0, 7.2 Hz, 1H), 2.22 (s, 4H), 2.17–2.11 (m, 1H), 1.68 (ddd, J = 6.5, 1.7, 0.7 Hz, 3H), 1.15 (d, J = 6.7 Hz, 3H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 170.8, 147.8, 145.2, 143.7, 137.1, 134.6, 130.5, 127.7, 127.6, 123.7, 118.4, 117.2, 113.4, 99.2, 79.9, 73.0, 72.4, 57.6, 49.3, 38.3, 38.1, 19.8, 18.0, 10.5; IR [ATR]: ν = 2979, 2932, 2870, 1761, 1673, 1644, 1591, 1481, 1435, 1396, 1380, 1346, 1290, 1231, 1157, 1131, 1108, 1062, 1028, 964, 920, 863, 800, 757, 738, 707, 672, 576, 508 cm–1; HRMS (ESI): exact mass calculated for C24H33O6 [(M + H)+], 417.2272; found, 417.2278; [α]D20 = +60.1 (c 0.90, CH2Cl2); Diastereomer 38′: 1H-NMR (400 MHz, CDCl3): δ = 6.66 (s, 1H), 5.97–5.84 (m, 2H), 5.75 (dtd, J = 15.6, 6.5, 1.0 Hz, 1H), 5.44 (ddq, J = 15.5, 7.2, 1.6 Hz, 1H), 5.33–5.23 (m, 2H), 5.23–5.09 (m, 4H), 4.76–4.67 (m, 1H), 4.09–4.01 (m, 2H), 3.67 (dd, J = 10.4, 7.0 Hz, 1H), 3.63–3.55 (m, 4H), 3.20–3.11 (m, 1H), 2.59 (dd, J = 11.9, 7.1 Hz, 1H), 2.28–2.11 (m, 5H), 1.67 (dd, J = 6.6, 0.9 Hz, 3H), 1.16 (d, J = 6.6 Hz, 3H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 170.9, 147.7, 144.8, 143.5, 137.2, 134.7, 130.9, 127.6, 127.4, 123.7, 118.4, 117.3, 112.7, 99.1, 79.5, 73.0, 72.5, 57.6, 49.2, 38.4, 38.1, 19.8, 17.9, 10.5; IR [ATR]: ν = 2870, 1759, 1643, 1592, 1482, 1434, 1379, 1346, 1231, 1157, 1130, 1109, 1062, 964, 919, 863, 787, 731, 669, 647 cm–1; HRMS (ESI): exact mass calculated for C24H33O6 [(M + H)+], 417.2272; found, 417.2243; [α]D = −37.4 (c 1.75, CH2Cl2).
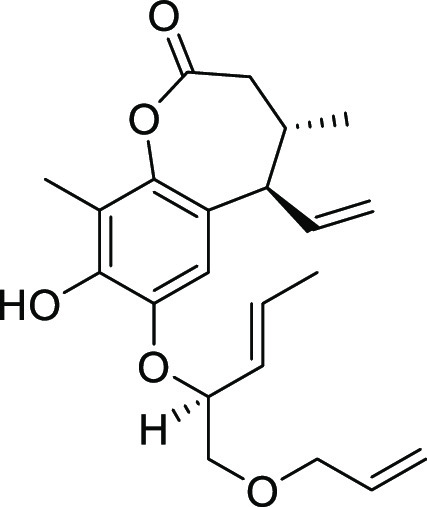
(4S,5R)-7-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-8-hydroxy-4,9-dimethyl-5-vinyl-4,5-dihydrobenzo[b]oxepin-2(3H)-one (S7)
A 50 mL Schlenk flask was charged with MgBr2·Et2O (315 mg, 1.22 mmol, 1.07 equiv) and compound 38 (475 mg, 1.14 mmol, 1 equiv) in 15 mL of dry DCM. The mixture was cooled to 4 °C, and after 10 min, EtSH (0.18 mL, 2.39 mmol, 2.1 equiv) was added dropwise. After 30 min, the cooling bath was removed, and the reaction was stirred at room temperature. TLC (petroleum ether/ethyl acetate, 4:1) after an additional hour confirmed full conversion. The reaction was quenched by addition of saturated NaHCO3 solution, and layers were separated. The aqueous layer was extracted three times with DCM, and the combined organic layer was washed with H2O. It was dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was flashed over a plug of silica, and the desired product was obtained as an orange solid in 95% yield (404 mg, 1.08 mmol). 1H-NMR (400 MHz, CDCl3): δ = 7.65 (d, J = 1.1 Hz, 1H), 6.65 (s, 1H), 6.03–5.85 (m, 2H), 5.79–5.66 (m, 1H), 5.52 (ddq, J = 15.3, 7.8, 1.6 Hz, 1H), 5.34 (dq, J = 17.2, 1.5 Hz, 1H), 5.26 (m, 2H), 5.15 (dt, J = 17.2, 1.3 Hz, 1H), 4.20–4.12 (m, 3H), 3.65 (dd, J = 10.2, 9.3 Hz, 1H), 3.56–3.48 (m, 1H), 3.11 (t, J = 8.9 Hz, 1H), 2.59 (dd, J = 11.9, 7.3 Hz, 1H), 2.22–2.11 (m, 6H), 1.73 (dd, J = 6.5, 1.6 Hz, 3H), 1.15 (d, J = 6.6 Hz, 3H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 171.0, 147.2, 146.0, 142.5, 137.4, 133.8, 131.9, 126.6, 122.7, 118.4, 118.1, 117.2, 116.7, 85.0, 72.6, 72.5, 48.9, 38.6, 38.1, 19.7, 18.0, 9.5; IR [ATR]: ν = 3297, 2976, 2932, 2870, 1746, 1667, 1639, 1609, 1482, 1445, 1419, 1375, 1345, 1317, 1243, 1226, 1196, 1175, 1144, 1131, 1108, 1090, 1056, 1024, 999, 972, 937, 919, 881, 867, 841, 801, 743, 680, 622 cm–1; HRMS (ESI): exact mass calculated for C22H29O5 [(M + H)+], 373.2010; found, 373.2016; [α]D20 = +107.1 (c 1.0, CH2Cl2).
(4S,5R)-7-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-4,9-dimethyl-8-((triethylsilyl)oxy)-5-vinyl-4,5-dihydrobenzo[b]oxepin-2(3H)-one (31)
A 50 mL Schlenk flask was charged with compound S7 (325 mg, 0.87 mmol, 1 equiv) in 5 mL of dry DCM. The yellow solution was cooled to −85 °C, and 2,6-lutidine (0.30 mL, 2.62 mmol, 3 equiv) was added followed by TESOTf (0.30 mL, 1.31 mmol, 1.5 equiv). After 10 min, the reaction mix was removed from the cooling bath and stirred at room temperature. TLC (petroleum ether/ethyl acetate, 5:1) after 15 min confirmed full conversion. The reaction was quenched with saturated NaHCO3 solution, and the aqueous layer was extracted five times with DCM. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The material was used in the next reaction without further purification. 1H-NMR (400 MHz, CDCl3): δ = 6.58 (s, 1H), 5.98–5.84 (m, 2H), 5.82–5.72 (m, 1H), 5.48 (ddd, J = 15.6, 7.0, 1.6, 1H), 5.31–5.22 (m, 2H), 5.20–5.11 (m, 2H), 4.74 (m, 1H), 4.02 (d, J = 5.5, 2H), 3.68 (dd, J = 10.0, 6.4, 1H), 3.56 (dd, J = 10.1, 4.7, 1H), 3.14 (t, J = 8.6, 1H), 2.54 (dd, J = 11.9, 7.2, 1H), 2.23–2.08 (m, 6H), 2.13 (s, 3H), 1.70 (dd, J = 6.6, 0.8, 3H), 1.15 (d, J = 6.64, 3H), 0.96 (t, J = 8.0, 9H), 0.80–0.72 (m, 6H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 171.2, 146.2, 143.3, 137.4, 134.6, 130.3, 128.2, 124.3, 121.3, 118.2, 117.3, 110.7, 78.5, 49.2, 38.6, 38.2, 19.8, 18.0, 10.7, 6.9, 5.6; IR [ATR]: ν = 2955, 2911, 2875, 1762, 1644, 1587, 1482, 1438, 1416, 1379, 1345, 1230, 1171, 1130, 1114, 1063, 1003, 966, 923, 884, 841, 801 cm–1; HRMS (ESI): exact mass calculated for C28H43O5Si [(M + H)+], 487.2874; found, 487.2875; [α]D20 = +70.7 (c 0.83, CH2Cl2).
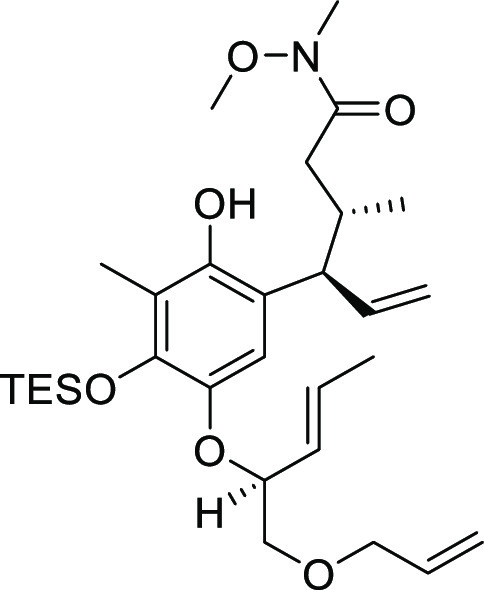
(3S,4R)-4-(5-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-2-hydroxy-3-methyl-4-((triethylsilyl)oxy)phenyl)-N-methoxy-N,3-dimethylhex-5-enamide (S8)
A 25 mL Schlenk flask was charged with N,O-dimethyl hydroxylamine hydrochloride (411 mg, 4.21 mmol, 5 equiv) dissolved in 5 mL of dry toluene, and it was cooled to −20 °C. To the clear colorless solution was added AlMe3 (2.0 M, 2.11 mL, 4.21 mmol, 5 equiv) dropwise, and the mixture was stirred for 50 min. Then, compound 31 (410 mg, 0.84 mmol, 1 equiv) dissolved in 7 mL of dry toluene was added dropwise at −20 °C. After 45 min, TLC (petroleum ether/ethyl acetate, 4:1) at −15 °C confirmed full conversion. The reaction was quenched with saturated NH4Cl and saturated Seignette’s salt solution. It was diluted with ethyl acetate and stirred at room temperature for 2.5 h (until the aqueous layer was clear). Layers were separated, and the aqueous layer was extracted five times with ethyl acetate. The combined organic layer was washed with H2O, dried over MgSO4, and concentrated in vacuo. The desired product was obtained as a yellow amorphous solid in 93% yield (443 mg, 0.809 mmol) over two steps. 1H-NMR (400 MHz, CDCl3): δ = 8.08 (s, 1H), 6.49 (s, 1H), 6.08 (dt, J = 16.8, 9.9 Hz, 1H), 5.98–5.84 (m, 1H), 5.78–5.65 (m, 1H), 5.47 (ddd, J = 15.4, 7.2, 1.7 Hz, 1H), 5.32–5.22 (m, 1H), 5.21–5.13 (m, 2H), 5.08 (dd, J = 16.9, 2.1 Hz, 1H), 4.71–4.62 (m, 1H), 4.08–3.98 (m, 2H), 3.72–3.64 (m, 4H), 3.60–3.50 (m, 2H), 3.24 (s, 3H), 2.58 (dd, J = 17.5, 10.5 Hz, 1H), 2.30–2.10 (m, 5H), 1.69–1.61 (m, 3H), 0.95 (t, J = 7.8 Hz, 9H), 0.87 (d, J = 7.0 Hz, 3H), 0.75 (q, J = 8.6, 8.2 Hz, 6H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 174.9, 147.0, 143.3, 141.4, 135.8, 134.9, 129.8, 129.1, 120.8, 118.2, 117.1, 111.9, 78.5, 72.9, 72.5, 61.3, 46.1, 35.6, 33.5, 32.3, 29.8, 18.0, 14.7, 10.4, 7.0, 5.6; HRMS (ESI): exact mass calculated for C30H50NO6Si [(M + H)+], 548.3402; found, 548.3408; [α]D20 = +22.2 (c 1.05, CH2Cl2).
(3S,4R)-4-(5-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)-3-methyl-4-((triethylsilyl)oxy)phenyl)-N-methoxy-N,3-dimethylhex-5-enamide (39)
A 50 mL Schlenk flask was charged with compound S8 (446 mg, 0.814 mmol, 1 equiv) in 6 mL of dry DCM. It was cooled to −92 °C, and 2,6-lutidine (0.38 mL, 3.26 mmol, 4 equiv) was added followed by TBSOTf (0.26 mL, 1.22 mmol, 1.5 equiv). The reaction was allowed to warm to room temperature upon which TLC (petroleum ether/ethyl acetate, 4:1) confirmed full conversion. The reaction was quenched by addition of saturated NaHCO3 solution, and the aqueous layer was extracted three times with DCM. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The crude mixture was subjected to column chromatography, and the desired product was obtained in 83% yield (445 mg, 0.672 mmol) accompanied by compound 31 in 11% yield (43 mg, 0.088 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.54 (s, 1H), 6.04–5.83 (m, 2H), 5.80–5.66 (m, 1H), 5.44 (ddq, J = 15.4, 7.3, 1.5 Hz, 1H), 5.32–5.21 (m, 1H), 5.21–5.02 (m, 3H), 4.74–4.66 (m, 1H), 4.07–3.96 (m, 2H), 3.67 (dd, J = 10.0, 6.4 Hz, 1H), 3.54 (dd, J = 10.0, 4.9 Hz, 1H), 3.50–3.46 (m, 4H), 3.08 (s, 3H), 2.23–2.03 (m, 6H), 1.65 (dd, J = 6.5, 1.6 Hz, 3H), 1.03 (s, 9H), 1.00–0.90 (m, 12H), 0.74 (q, J = 7.3 Hz, 6H), 0.13 (s, 6H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 174.2 (HMBC), 145.5, 143.5, 142.8, 140.0, 134.9, 130.3, 128.4, 125.3, 121.1, 117.2, 115.7, 111.8, 78.0, 72.9, 72.4, 61.0, 47.2, 26.4, 18.8, 18.1, 18.0, 12.8, 7.0, 5.6, −2.8, −3.0; IR [ATR]: ν = 2955, 2933, 2875, 1669, 1591, 1482, 1415, 1381, 1333, 1302, 1239, 1128, 1077, 1004, 936, 903, 838, 822, 803, 778, 739, 692 cm–1; HRMS (ESI): exact mass calculated for C30H50NO6Si [(M–C6H15Si + H)+], 548.3402; found, 548.3400; [α]D20 = +33.4 (c 1.0, CH2Cl2).
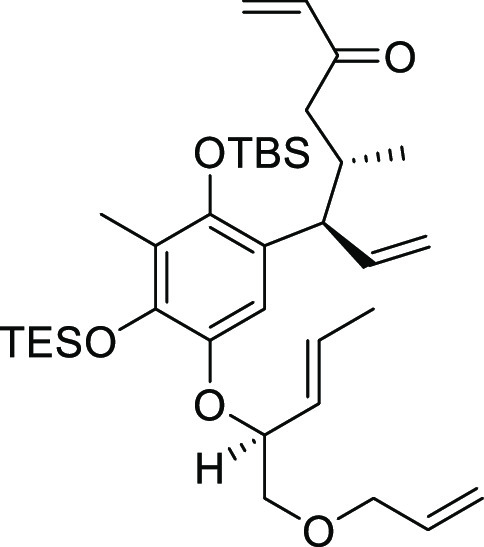
(5S,6R)-6-(5-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)-3-methyl-4-((triethylsilyl)oxy)phenyl)-5-methylocta-1,7-dien-3-one (S9)
A 25 mL Schlenk flask was charged with compound 39 (251 mg, 0.379 mmol, 1 equiv) dissolved in 5 mL of dry THF. The colorless mixture was cooled to 0 °C, and vinyl magnesium bromide (1.0 M, 0.80 mL, 0.80 mmol, 2.1 equiv) was added dropwise. After 1 h, TLC (petroleum ether/ethyl acetate, 5:1) confirmed full conversion. The reaction was quenched by addition of saturated NH4Cl solution, and the aqueous layer was extracted three times with ethyl acetate. The combined organic layer was washed with H2O and brine and dried over MgSO4. The crude material was flashed over a short plug of silica, and the desired product was collected as a pale-yellow oil in 95% yield (227 mg, 0.36 mmol). 1H-NMR (400 MHz, CDCl3): δ = 6.50 (s, 1H), 6.16 (dd, J = 17.7, 10.7 Hz, 1H), 6.04–5.84 (m, 3H), 5.77–5.60 (m, 2H), 5.50–5.39 (m, 1H), 5.27 (dq, J = 17.2, 1.7 Hz, 1H), 5.20–5.02 (m, 3H), 4.75–4.65 (m, 1H), 4.07–3.95 (m, 2H), 3.67 (dd, J = 9.9, 6.6 Hz, 1H), 3.59–3.45 (m, 2H), 2.39–2.28 (m, 1H), 2.24–2.01 (m, 5H), 1.71–1.57 (m, 3H), 1.03 (s, 9H), 0.94 (t, J = 7.9 Hz, 9H), 0.88 (d, J = 6.0 Hz, 3H), 0.75 (q, J = 7.5 Hz, 6H), 0.13 (s, 6H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 201.0, 145.5, 143.6, 142.8, 139.7, 136.9, 134.8, 130.1, 128.4, 127.7, 125.1, 121.3, 117.2, 116.0, 111.4, 77.9, 73.0, 72.4, 47.1, 44.6, 35.0, 26.4, 18.8, 18.0, 18.0, 12.8, 7.0, 5.6, −2.8, −3.0.
(3S,5S,6R)-6-(5-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)-3-methyl-4-((triethylsilyl)oxy)phenyl)-5-methylocta-1,7-dien-3-ol (40)
A 25 mL Schlenk flask was charged with compound S9 (162 mg, 0.258 mmol, 1 equiv) dissolved in 4 mL of dry DCM. The colorless solution was cooled to −90 °C and stirred for 5 min. Then, DIBAL (1.0 M in hexanes, 0.31 mL, 0.31 mmol, 1.2 equiv) was added dropwise. After 15 min, TLC (petroleum ether/ethyl acetate, 6:1) confirmed full conversion. The reaction was quenched by addition of saturated NH4Cl solution and saturated Seignette’s salt solution. The mixture was stirred for 1 h and 20 min before layers were separated. The aqueous layer was extracted three times with DCM, and the combined organic layer was dried over MgSO4. It was concentrated in vacuo, and the crude material was subjected to column chromatography. Pure 40 (eluting first) was obtained in 74% yield (120 mg, 0.19 mmol) accompanied by 7% 12 mg, 0.019 mmol) 40′ (eluting second). Allyl alcohol 40: 1H-NMR (400 MHz, CDCl3): δ = 6.50 (s, 1zfH), 6.05–5.84 (m, 2H), 5.83–5.66 (m, 2H), 5.48 (ddq, J = 15.5, 6.9, 1.6 Hz, 1H), 5.27 (dq, J = 17.3, 1.6 Hz, 1H), 5.21–4.96 (m, 5H), 4.75–4.66 (m, 1H), 4.09–3.99 (m, 3H), 3.68 (dd, J = 10.0, 6.5 Hz, 1H), 3.59–3.49 (m, 2H), 2.08 (s, 4H), 1.96–1.84 (m, 1H), 1.66 (dd, J = 6.4, 1.6 Hz, 3H), 1.34 (ddd, J = 13.3, 10.0, 2.9 Hz, 1H), 1.14 (ddd, J = 14.1, 10.6, 3.8 Hz, 1H), 1.02 (s, 9H), 0.95 (t, J = 7.9 Hz, 9H), 0.88 (d, J = 6.7 Hz, 3H), 0.75 (q, J = 7.7 Hz, 6H), 0.12 (d, J = 4.2 Hz, 6H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 145.6, 143.3, 142.7, 142.2, 139.9, 134.9, 129.9, 128.6, 125.6, 121.3, 117.2, 115.8, 113.6, 112.0, 77.9, 73.0, 72.4, 70.9, 47.4, 42.2, 34.0, 26.5, 18.9, 18.0, 16.7, 12.8, 7.0, 5.6, −2.8, −2.9; [α]D20 = +35.2 (c 1.0, CH2Cl2).
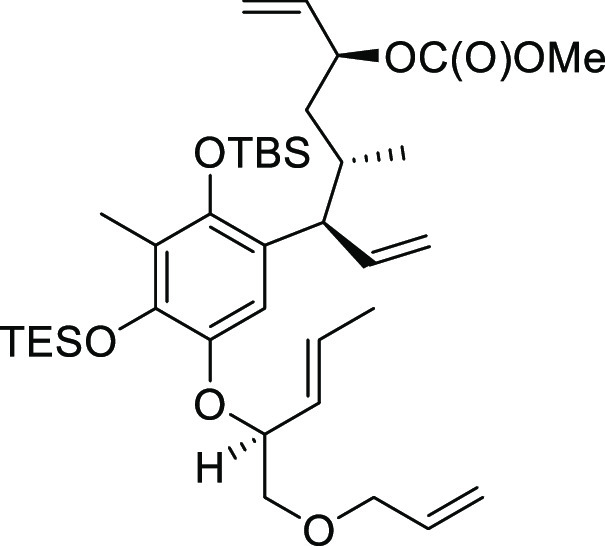
(3S,5S,6R)-6-(5-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)-3-methyl-4-((triethylsilyl)oxy)phenyl)-5-methylocta-1,7-dien-3-yl methyl carbonate (S10)
A 25 mL round-bottom flask was charged with compound 40 (59 mg, 93 μmol, 1 equiv) in 2 mL of dry DCM. The clear colorless solution was cooled to −20 °C, and pyridine (26 μL, 0.327 mmol, 3.5 equiv) was added followed by methyl chloroformate (25 μL, 0.327 mmol, 3.5 equiv). The solution turned orange, and the formation of a precipitate was observed. After 2 h, TLC (petroleum ether/ethyl acetate, 10:1) at −5 °C showed full conversion. The reaction was quenched by addition of brine, and the aqueous layer was extracted twice with DCM. The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude mixture was flashed over a short plug of silica, and the desired product was obtained as a colorless oil in 99% yield (64 mg, 93 μmol). 1H-NMR (400 MHz, CDCl3): δ = 6.49 (s, 1H), 6.03–5.84 (m, 2H), 5.71 (dddd, J = 16.9, 12.3, 8.7, 6.2 Hz, 2H), 5.47 (ddq, J = 15.4, 7.1, 1.6 Hz, 1H), 5.27 (dq, J = 17.3, 1.7 Hz, 1H), 5.23–5.00 (m, 6H), 4.76–4.67 (m, 1H), 4.08–3.99 (m, 2H), 3.74 (s, 3H), 3.68 (dd, J = 10.0, 6.5 Hz, 1H), 3.59–3.49 (m, 2H), 2.07 (s, 3H), 1.88–1.74 (m, 1H), 1.70–1.56 (m, 4H), 1.16 (ddd, J = 14.2, 10.9, 3.0 Hz, 1H), 1.01 (s, 9H), 0.95 (t, J = 8.3 Hz, 9H), 0.86 (d, J = 6.5 Hz, 3H), 0.75 (q, J = 7.7 Hz, 6H), 0.13 (s, 3H), 0.11 (s, 3H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 155.6, 145.6, 143.2, 142.8, 139.6, 137.0, 134.9, 129.9, 128.5, 125.1, 121.2, 117.2, 116.5, 116.0, 112.2, 78.0, 77.1, 73.0, 72.4, 54.6, 47.1, 39.0, 33.5, 26.5, 18.8, 18.0, 16.3, 12.8, 7.0, 5.6, −2.9; HRMS (ESI): exact mass calculated for C36H62O5Si2Na [(M–C2H2O2 + Na)+], 653.4028; found, 653.4031; [α]D20 = +35.7 (c 1.1, CH2Cl2).
(3S,5S,6R)-6-(5-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)-4-hydroxy-3-methylphenyl)-5-methylocta-1,7-dien-3-yl methyl carbonate (30)
A 25 mL round-bottom flask was charged with compound S10 (64 mg, 93 μmol, 1 equiv) in 4 mL of THF and 0.3 mL of H2O. TFA (10% in H2O, 0.7 mL, 0.93 mmol, 10 equiv) was added at room temperature, and the mixture was stirred for 1.5 h. After TLC (petroleum ether/ethyl acetate, 10:1) showed full conversion, the reaction was quenched by addition of saturated NaHCO3 solution. The aqueous layer was extracted three times with ethyl acetate, and the combined organic layer was washed with H2O and brine. It was dried over MgSO4, filtered, and concentrated in vacuo. The crude material was flashed over a plug of silica, and the desired product was obtained as a colorless oil in virtually quantitative yield (53 mg, 92 μmol). 1H-NMR (400 MHz, CDCl3): δ = 6.55 (s, 1H), 6.03–5.84 (m, 2H), 5.77–5.64 (m, 2H), 5.51 (ddq, J = 15.3, 7.7, 1.6 Hz, 1H), 5.37–4.97 (m, 7H), 4.25–4.16 (m, 1H), 4.16–4.11 (m, 2H), 3.74 (s, 3H), 3.63 (dd, J = 10.1, 8.9 Hz, 1H), 3.58–3.49 (m, J = 4.7 Hz, 2H), 2.10 (s, 3H), 1.87–1.77 (m, 1H), 1.75–1.61 (m, 4H), 1.22–1.14 (m, 1H), 1.03 (s, 9H), 0.89 (d, J = 6.6 Hz, 3H), 0.16 (s, 3H), 0.15 (s, 3H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 155.6, 148.2, 146.1, 140.2, 139.5, 137.0, 134.1, 131.3, 127.0, 123.2, 118.2, 117.9, 116.6, 116.1, 115.5, 84.1, 77.2, 72.7, 72.7, 54.6, 46.9, 38.9, 33.5, 26.4, 18.8, 18.0, 16.7, 11.6, −2.6, −2.8; HRMS (ESI): exact mass calculated for C32H51O7Si [(M + H)+], 575.3399; found, 575.3369.
(1R,2S,4S,5R)-9-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-6-((tert-butyldimethylsilyl)oxy)-2,7-dimethyl-1,4-divinylspiro[4.5]deca-6,9-dien-8-one (29a)
A 10 mL Schlenk flask was charged with PPh3 (11.4 mg, 44 μmol, 50 mol %) and Pd(dba)2 (10.0 mg, 17 μmol, 20 mol %), and compound 30 (50 mg, 87 μmol, 1 equiv) dissolved in 2 mL of dry DCM was added. The yellow reaction mixture was stirred for 30 min, at which point TLC (petroleum ether/ethyl acetate, 10:1) confirmed full conversion. The solvent was removed in vacuo, and the residue was purified by column chromatography. The desired product was obtained as a colorless oil in 21% yield (9 mg, 18 μmol). 1H-NMR (600 MHz, CDCl3): δ = 5.90 (ddt, J = 17.3, 10.4, 5.7 Hz, 1H), 5.75 (dqd, J = 15.6, 6.5, 1.0 Hz, 1H), 5.60 (ddd, J = 17.0, 10.3, 8.7 Hz, 1H), 5.52 (ddd, J = 17.0, 10.1, 8.4 Hz, 1H), 5.44 (ddq, J = 15.6, 7.0, 1.6 Hz, 1H), 5.27 (dq, J = 17.2, 1.6 Hz, 1H), 5.19 (dq, J = 10.4, 1.4 Hz, 1H), 5.14 (s, 1H), 4.93 (ddd, J = 10.3, 2.0, 0.7 Hz, 1H), 4.89 (ddd, J = 17.0, 2.0, 1.0 Hz, 1H), 4.81 (ddd, J = 16.9, 2.1, 1.0 Hz, 1H), 4.77 (ddd, J = 10.1, 2.1, 0.7 Hz, 1H), 4.59–4.54 (m, 1H), 4.04–3.97 (m, 2H), 3.64 (dd, J = 10.1, 6.4 Hz, 1H), 3.51 (dd, J = 10.1, 4.7 Hz, 1H), 2.82–2.76 (m, 1H), 2.53 (ddd, J = 11.5, 8.4, 6.0 Hz, 1H), 1.94 (ddt, J = 12.2, 10.6, 6.1 Hz, 1H), 1.84 (dt, J = 11.9, 6.0 Hz, 1H), 1.75–1.73 (m, 7H), 1.06 (d, J = 6.4 Hz, 3H), 0.96 (s, 9H), 0.21 (s, 3H), 0.19 (s, 3H); 13C{1H}-NMR (151 MHz, CDCl3): δ = 203.5, 159.2, 144.3, 138.5, 138.2, 134.6, 130.3, 127.8, 118.4, 117.5, 115.7, 114.7, 113.1, 72.5, 72.3, 63.3, 62.0, 58.0, 39.8, 39.2, 25.9, 18.9, 18.1, 17.8, 9.5, −3.6, −3.7; HRMS (ESI): exact mass calculated for C30H47O4Si [(M + H)+], 499.3238; found, 499.3207.
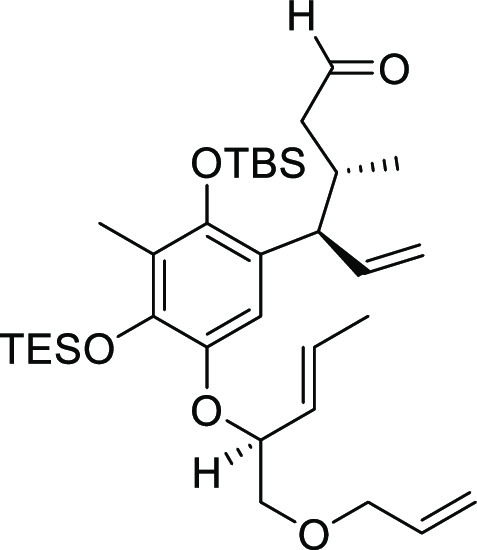
(3S,4R)-4-(5-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)-3-methyl-4-((triethylsilyl)oxy)phenyl)-3-methylhex-5-enal (S11)
A 25 mL Schlenk flask was charged with compound 39 (310 mg, 0.468 mmol, 1 equiv) in 5 mL of dry toluene. The pale-yellow solution was cooled to −91 °C, and DIBAL (1.0 M, 0.56 mL, 0.56 mmol, 1.2 equiv) was added dropwise. After 2 min, TLC (petroleum ether/ethyl acetate, 4:1) showed full conversion. The reaction was quenched with saturated Seignette’s salt solution, and the flask was removed from the cooling bath. The mix was allowed to warm to room temperature and stirred for another 1.5 h. The aqueous layer was extracted four times with Et2O, and the combined organic layer was washed with H2O and brine. It was dried over MgSO4, filtered, and concentrated in vacuo. The crude material was flashed over a short plug of silica, and the desired product was obtained as a very pale-yellow oil in 98% yield (278 mg, 0.461 mmol). 1H-NMR (400 MHz, CDCl3): δ = 9.47 (dd, J = 3.0, 1.2 Hz, 1H), 6.48 (s, 1H), 6.03–5.85 (m, 2H), 5.78–5.65 (m, 1H), 5.44 (ddq, J = 15.5, 7.0, 1.5 Hz, 1H), 5.32–5.22 (m, 1H), 5.22–5.11 (m, 2H), 5.11–5.03 (m, 1H), 4.71 (q, J = 6.3 Hz, 1H), 4.09–3.97 (m, 2H), 3.68 (dd, J = 10.0, 6.6 Hz, 1H), 3.59–3.46 (m, 2H), 2.25–1.97 (m, 6H), 1.70–1.61 (m, 3H), 1.02 (s, 9H), 0.99–0.89 (m, 12H), 0.80–0.69 (m, 6H), 0.12 (s, 6H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 202.9, 145.5, 143.5, 143.0, 139.2, 134.8, 130.1, 128.5, 124.7, 121.5, 117.2, 116.4, 111.5, 77.9, 73.0, 72.5, 49.0, 46.7, 33.5, 26.4, 18.8, 18.2, 18.0, 12.8, 7.0, 5.6, −2.9, −3.0; IR [ATR]: ν = 2954, 2931.04, 2875, 1726, 1590, 1482, 1417, 1378, 1360, 1301, 1239, 1128, 1078, 1004, 964, 937, 894, 838, 822, 802, 778, 739, 691 cm–1; HRMS (ESI): exact mass calculated for C34H58O5Si2Na [(M + Na)+], 625.3715; found, 625.3719; [α]D20 = +53.4 (c 1.0, CH2Cl2).
(3R,5S,6R)-6-(5-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)-3-methyl-4-((triethylsilyl)oxy)phenyl)-5-methylocta-1,7-dien-3-ol (40′)
A 25 mL Schlenk flask was charged with compound S11 (258 mg, 0.427 mmol, 1 equiv) in 5 mL of dry THF. The colorless solution was cooled by an ice bath, and vinyl magnesium bromide (1.0 M, 0.51 mL, 0.51 mmol, 1.2 equiv) was added dropwise over 5 min. TLC (petroleum ether/ethyl acetate, 10:1) after 20 min showed full conversion. The reaction was quenched by addition of saturated NH4Cl solution, and the aqueous layer was extracted four times with Et2O. The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude residue was purified by column chromatography. Allyl alcohol 40 (eluted first) was obtained in 24% yield (65 mg, 0.103 mmol), and allyl alcohol 40′ (eluted second) was obtained in 46% yield (124 mg, 0.197 mmol). Allyl alcohol 40′: 1H-NMR (400 MHz, CDCl3): δ = 6.49 (s, 1H), 6.04–5.84 (m, 2H), 5.80–5.57 (m, 2H), 5.54–5.43 (m, 1H), 5.27 (dq, J = 17.2, 1.7 Hz, 1H), 5.21–4.98 (m, 5H), 4.75–4.63 (m, 1H), 4.08–3.98 (m, 2H), 3.95–3.88 (m, 1H), 3.68 (dd, J = 10.0, 6.5 Hz, 1H), 3.58–3.49 (m, 2H), 2.07 (s, 3H), 1.71–1.56 (m, 4H), 1.41 (ddd, J = 13.3, 7.8, 3.9 Hz, 1H), 1.32–1.25 (m, 1H), 1.02 (s, 9H), 0.95 (t, J = 7.9 Hz, 9H), 0.87 (d, J = 6.7 Hz, 3H), 0.75 (q, J = 7.8 Hz, 6H), 0.13 (s, 3H), 0.11 (s, 3H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 145.4, 143.3, 142.7, 141.3, 139.6, 134.8, 129.8, 128.8, 125.8, 121.3, 117.2, 116.0, 114.9, 111.9, 77.9, 73.0, 72.4, 72.4, 47.3, 42.2, 34.9, 26.5, 18.8, 18.0, 17.4, 12.9, 7.0, 5.6, −2.9, −2.9; HRMS (ESI): exact mass calculated for C30H48O5SiNa [(M–C6H15Si + Na)+], 539.3163; found, 539.3187; [α]D20 = +38.6 (c 1.0, CH2Cl2).
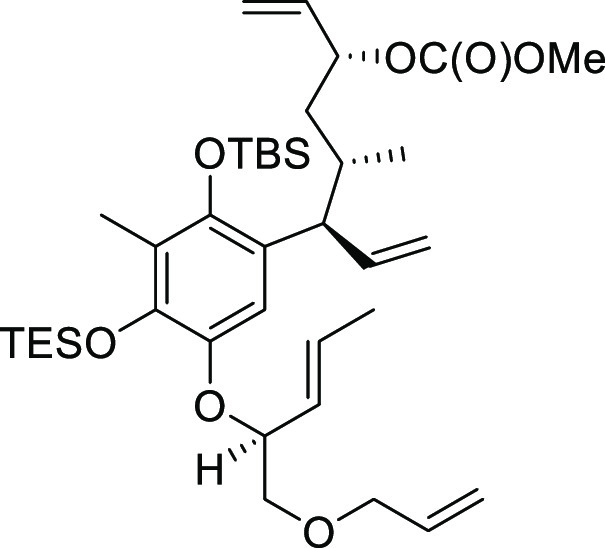
(3R,5S,6R)-6-(5-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)-3-methyl-4-((triethylsilyl)oxy)phenyl)-5-methylocta-1,7-dien-3-yl methyl carbonate (S12)
A 10 mL round-bottom flask was charged with compound 40′ (63 mg, 0.10 mmol, 1 equiv) in 1.5 mL of dry DCM. The clear colorless solution was cooled to −20 °C, and pyridine (28 μL, 0.35 mmol, 3.5 equiv) was added followed by methyl chloroformate (27 μL, 0.35 mmol, 3.5 equiv). The solution turned orange, and the formation of a precipitate was observed. After 2 h, TLC (petroleum ether/ethyl acetate, 10:1) at 10 °C showed full conversion. The reaction was quenched by addition of brine, and the aqueous layer was extracted three times with DCM. The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude mixture was flashed over a short plug of silica, and the desired product was obtained as a colorless oil in 94% yield (65 mg, 94 μmol). 1H-NMR (400 MHz, CDCl3): δ = 6.46 (s, 1H), 6.01–5.83 (m, 2H), 5.79–5.66 (m, 1H), 5.63–5.42 (m, 2H), 5.31–5.00 (m, 6H), 4.98–4.90 (m, 1H), 4.73–4.64 (m, 1H), 4.07–3.96 (m, 2H), 3.73 (s, 3H), 3.67 (dd, J = 10.0, 6.5 Hz, 1H), 3.59–3.48 (m, 2H), 2.07 (s, 3H), 1.66 (dd, J = 6.4, 1.6 Hz, 3H), 1.62–1.37 (m, 3H), 1.01 (s, 9H), 0.95 (t, J = 7.9 Hz, 9H), 0.85 (d, J = 6.5 Hz, 3H), 0.75 (q, J = 8.5, 8.0 Hz, 6H), 0.12 (s, 3H), 0.11 (s, 3H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 155.2, 145.5, 143.3, 142.8, 139.1, 135.8, 134.8, 129.7, 128.8, 125.3, 121.3, 118.6, 117.2, 116.2, 111.8, 79.1, 78.0, 73.0, 72.5, 54.6, 47.5, 38.8, 34.4, 26.5, 18.8, 18.0, 16.4, 12.9, 7.0, 5.6, −2.8, −2.9; HRMS (ESI): exact mass calculated for C38H65O7Si2 [(M + H)+], 689.4263; found, 689.4244; [α]D20 = +29.2 (c 1.0, CH2Cl2).
(3R,5S,6R)-6-(5-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)-4-hydroxy-3-methylphenyl)-5-methylocta-1,7-dien-3-yl methyl carbonate (30′)
A 25 mL round-bottom flask was charged with compound S12 (94 mg, 0.136 mmol, 1 equiv) in 6 mL of THF and 0.5 mL of H2O. TFA (10% in H2O, 1.0 mL, 1.36 mmol, 10 equiv) was added at room temperature, and the mixture was stirred for 2 h and 50 min. After TLC (petroleum ether/ethyl acetate, 10:1) showed full conversion, the reaction was quenched by addition of saturated NaHCO3 solution. The aqueous layer was extracted three times with ethyl acetate, and the combined organic layer was washed with H2O and brine. It was dried over MgSO4, filtered, and concentrated in vacuo. The crude material was flashed over a plug of silica, and the desired product was obtained as a colorless oil in 98% yield (77 mg, 0.134 mmol). 1H-NMR (400 MHz, CD2Cl2): δ = 7.26 (s, 1H), 6.54 (s, 1H), 6.04–5.84 (m, 2H), 5.80–5.67 (m, 1H), 5.66–5.47 (m, 2H), 5.39–5.29 (m, 3H), 5.29–5.12 (m, 1H), 5.07 (ddd, J = 10.4, 2.0, 1.0 Hz, 1H), 5.04–4.97 (m, 1H), 4.91–4.83 (m, 1H), 4.21–4.11 (m, 3H), 3.69 (s, 3H), 3.62 (dd, J = 10.2, 9.1 Hz, 1H), 3.58–3.45 (m, 2H), 2.08 (s, 3H), 1.73 (dd, J = 6.5, 1.6 Hz, 3H), 1.64–1.49 (m, 2H), 1.39 (ddd, J = 13.4, 10.6, 5.4 Hz, 1H), 1.04 (s, 9H), 0.90 (d, J = 6.6 Hz, 3H), 0.17 (s, 3H), 0.15 (s, 3H); 13C{1H}-NMR (101 MHz, CD2Cl2): δ = 155.3, 146.5, 140.4, 140.1, 136.3, 134.5, 131.4, 127.4, 123.7, 118.3, 118.0, 117.9, 116.3, 115.7, 84.6, 79.2, 73.1, 72.8, 54.8, 47.6, 39.0, 34.6, 26.5, 19.0, 18.0, 17.2, 11.7, −2.5, −2.8; HRMS (ESI): exact mass calculated for C32H51O7Si [(M + H)+], 575.3399; found, 575.3372.
(3R,5S,6R)-6-(5-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)-3-methyl-4-((triethylsilyl)oxy)phenyl)-5-methylocta-1,7-dien-3-yl 4-nitrobenzoate (41)
A 10 mL Schlenk flask was charged with compound 40 (50 mg, 79 μmol, 1 equiv) dissolved in 2 mL of dry toluene, and PPh3 (88 μL, 0.367 mmol, 4.5 equiv) was added. After 5 min, p-nitrobenzoic acid (112 mg, 0.555 mmol, 7 equiv) was added and the mixture was stirred for 5 min before DIAD (46 mg, 0.277 mmol, 3.5 equiv) dissolved in 0.9 mL of dry toluene was added. After 40 min, TLC (petroleum ether/ethyl acetate, 5:1) confirmed full conversion and the reaction was quenched by addition of saturated NaHCO3 solution. The aqueous layer was extracted twice with ethyl acetate, and the combined organic layer was washed with NaHCO3 followed by H2O and brine. It was dried over MgSO4 and concentrated in vacuo. The crude material was purified by column chromatography, and the desired product was obtained in 63% yield (39 mg, 50 μmol). 1H-NMR (400 MHz, CDCl3): δ = 8.32–8.22 (m, 2H), 8.20–8.12 (m, 2H), 6.49 (s, 1H), 6.06–5.84 (m, 2H), 5.80–5.60 (m, 2H), 5.49 (ddq, J = 15.5, 6.9, 1.6 Hz, 1H), 5.44–5.35 (m, 1H), 5.32–5.28 (m, 1H), 5.26–5.24 (m, 1H), 5.22–5.02 (m, 4H), 4.73–4.66 (m, 1H), 4.06–4.00 (m, 2H), 3.67 (ddd, J = 12.0, 10.0, 6.3 Hz, 1H), 3.59–3.52 (m, 2H), 2.08 (s, 3H), 1.70–1.60 (m, 4H), 1.55–1.39 (m, 1H), 1.01 (s, 9H), 0.98–0.89 (m, 12H), 0.76 (q, J = 8.0 Hz, 6H), 0.13 (s, 3H), 0.12 (s, 3H); 13C{1H}-NMR (101 MHz, CDCl3): δ = 163.9, 150.6, 145.5, 143.4, 142.8, 139.1, 136.3, 135.7, 134.8, 130.8, 129.6, 128.8, 125.3, 123.6, 121.3, 118.6, 117.2, 116.3, 111.8, 78.0, 76.7, 73.0, 72.5, 47.6, 38.8, 34.7, 26.5, 18.8, 18.0, 16.6, 12.9, 7.0, 5.6, −2.8, −2.8; [α]D20 = +13.2 (c 0.95, CH2Cl2).
6-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-3-((tert-butyldimethylsilyl)oxy)-4-((3R,4S,6R)-6-hydroxy-4-methylocta-1,7-dien-3-yl)-2-methylphenol (42)
A 25 mL round-bottom flask was charged with compound 41 (20 mg, 26 μmol, 1 equiv) in 3.5 mL of EtOH/iPrOH (2.5:1), and K2CO3 (35 mg, 0.256 mmol, 10 equiv) was added. The colorless mixture was stirred for 2 days at which point TLC (petroleum ether/ethyl acetate, 5:1) confirmed full conversion. The reaction was quenched by addition of solid NH4Cl, and solvents were evaporated. The residue was taken up in ethyl acetate and little H2O. The aqueous layer was extracted three times with ethyl acetate, and the combined organic layer was washed with H2O and Brine. It was dried over MgSO4 and concentrated in vacuo. The crude material was subjected to column chromatography and desired product was collected in 76% yield (10 mg, 19 μmol). 1H-NMR (600 MHz, CDCl3): δ = 7.33 (s, 1H), 6.55 (s, 1H), 6.00–5.87 (m, 2H), 5.74–5.62 (m, 2H), 5.51 (ddq, J = 15.4, 7.8, 1.6 Hz, 1H), 5.34 (dq, J = 17.3, 1.6 Hz, 1H), 5.25 (dq, J = 10.3, 1.3 Hz, 1H), 5.11 (dt, J = 17.1, 1.3 Hz, 1H), 5.07–5.03 (m, 2H), 5.00 (dt, J = 17.3, 1.5 Hz, 1H), 4.20–4.09 (m, 3H), 3.97–3.90 (m, 1H), 3.62 (dd, J = 10.1, 9.2 Hz, 1H), 3.55–3.49 (m, 2H), 2.10 (s, 3H), 1.73 (dd, J = 6.5, 1.7 Hz, 3H), 1.65–1.59 (m, 1H), 1.44 (ddd, J = 13.4, 8.3, 3.6 Hz, 1H), 1.33–1.27 (m, 1H), 1.03 (s, 9H), 0.90 (d, J = 6.6 Hz, 3H), 0.16 (s, 3H), 0.15 (s, 3H); 13C{1H}-NMR (151 MHz, CDCl3): δ = 148.1, 146.1, 141.2, 140.2, 139.6, 134.0, 131.3, 127.0, 123.8, 118.3, 118.0, 116.2, 115.5, 115.3, 84.3, 72.7, 72.7, 47.1, 41.9, 34.7, 26.5, 18.9, 18.0, 17.6, 11.6, −2.6, −2.8; [α]D20 = +61.6 (c 0.5, CH2Cl2).
(1R,2S,4S,5R)-9-(((S,E)-1-(Allyloxy)pent-3-en-2-yl)oxy)-8-hydroxy-2,7-dimethyl-1,4-divinylspiro[4.5]deca-7,9-dien-6-one (43)
A 10 mL Schlenk was charged with compound 42 (10 mg, 19 μmol, 1 equiv) in 0.5 mL of dry toluene. The colorless solution was cooled in an ice bath, PBu3 (22 μL, 87 μmol, 4.5 equiv) was added, and the mixture was stirred for 15 min. Then, DIAD (27 mg, 0.135 mmol, 7 equiv) dissolved in 0.3 mL of dry toluene was added dropwise and the mixture was allowed to slowly warm up. After 2 h, TLC (petroleum ether/ethyl acetate, 5:1) confirmed full conversion. Volatiles were removed in vacuo, and the crude residue was subjected to column chromatography. The desired product was collected in 94% yield (7 mg, 18 μmol). 1H-NMR (600 MHz, CDCl3): δ = 8.18 (s, 1H), 5.92 (ddt, J = 17.3, 10.4, 5.8 Hz, 1H), 5.80–5.74 (m, 1H), 5.58 (s, 1H), 5.58–5.51 (m, 2H), 5.43 (ddq, J = 15.3, 8.1, 1.7 Hz, 1H), 5.32 (dq, J = 17.2, 1.5 Hz, 1H), 5.26 (dq, J = 10.4, 1.3 Hz, 1H), 4.93–4.91 (m, 1H), 4.90 (ddd, J = 11.7, 1.9, 0.8 Hz, 1H), 4.84 (ddd, J = 17.0, 2.0, 1.1 Hz, 1H), 4.79 (ddd, J = 10.1, 2.0, 0.8 Hz, 1H), 4.17–4.13 (m, 1H), 4.11 (ddt, J = 5.7, 4.2, 1.4 Hz, 2H), 3.61 (dd, J = 10.2, 9.1 Hz, 1H), 3.52 (dd, J = 10.2, 3.0 Hz, 1H), 2.83 (dd, J = 10.3, 9.0 Hz, 1H), 2.61–2.55 (m, 1H), 1.95–1.88 (m, 1H), 1.87–1.78 (m, 2H), 1.77 (d, J = 1.6 Hz, 3H), 1.76 (s, 3H), 1.05 (d, J = 6.4 Hz, 3H); 13C{1H}-NMR (151 MHz, CDCl3): δ = 201.8, 159.6, 142.4, 138.3, 137.8, 133.7, 132.8, 125.9, 124.3, 118.6, 115.8, 115.1, 111.4, 83.1, 72.7, 72.2, 63.6, 62.2, 56.7, 40.1, 39.3, 18.0, 17.6, 7.7; HRMS (ESI): exact mass calculated for C24H33O4 [(M + H)+], 385.2373; found, 385.2379.
Acknowledgments
We thank David Schachamayr M.Sc. and Daniel Stankic for the large-scale preparation of precursor materials. Additionally, we would like to express our gratitude to Dr. Hametner and Dr. Stanetty for NMR 600 MHz measurements as well as Dr. Nicolas Kratena for valuable input during this project. We thank Christopher Willmott B.Sc. for his assistance in writing this manuscript. The X-ray Centre of TU Wien is acknowledged for providing access to the X-ray diffractometer.
Supporting crystallographic data for this paper are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service at https://www.ccdc.cam.ac.uk/structures/?.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.2c01914.
General information, NMR spectra (1H and 13C) of all prepared compounds, and discussion with respect to compounds 29 and 29a (PDF)
Author Present Address
§ Boehringer Ingelheim RCV GmbH & Co KG, Dr-Boehringer-Gasse 5-11, Vienna 1120, Austria (D.S.)
Author Present Address
∥ Infineon Technologies Austria AG, Siemensstraße 2, Villach 9500, Austria (K.S.).
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was funded by the Austrian Science Fund (FWF, project number P25556-N28). We thank the TU Wien for funding Open Access.
The authors declare no competing financial interest.
Supplementary Material
References
- Rodríguez A. D.; González E.; Huang S. D. Unusual Terpenes with Novel Carbon Skeletons from the West Indian Sea Whip Pseudopterogorgia elisabethae (Octocorallia). J. Org. Chem. 1998, 63, 7083–7091. 10.1021/jo981385v. [DOI] [PubMed] [Google Scholar]
- Rodríguez A. D.; Ramírez C.; Rodríguez I. I.; Barnes C. L. Novel Terpenoids from the West Indian Sea Whip Pseudopterogorgia Elisabethae (Bayer). Elisapterosins A and B: Rearranged Diterpenes Possessing an Unprecedented Cagelike Framework. J. Org. Chem. 2000, 65, 1390–1398. 10.1021/jo9914869. [DOI] [PubMed] [Google Scholar]
- Rodríguez A. D.; Ramírez C. A Marine Diterpene with a Novel Tetracyclic Framework from the West Indian Gorgonian Octocoral Pseudopterogorgia elisabethae. Org. Lett. 2000, 2, 507–510. 10.1021/ol991362i. [DOI] [PubMed] [Google Scholar]
- Kim A. I.; Rychnovsky S. D. Unified Strategy for the Synthesis of (−)-Elisapterosin B and (−)-Colombiasin A. Angew. Chem., Int. Ed. 2003, 42, 1267–1270. 10.1002/anie.200390325. [DOI] [PubMed] [Google Scholar]
- Boezio A. A.; Jarvo E. R.; Lawrence B. M.; Jacobsen E. N. Efficient Total Syntheses of (−)-Colombiasin A and (−)-Elisapterosin B: Application of the Cr-Catalyzed Asymmetric Quinone Diels–Alder Reaction. Angew. Chem., Int. Ed. 2005, 44, 6046–6050. 10.1002/anie.200502178. [DOI] [PubMed] [Google Scholar]
- Harrowven D. C.; Pascoe D. D.; Demurtas D.; Bourne H. O. Total Synthesis of (−)-Colombiasin A and (−)-Elisapterosin B. Angew. Chem., Int. Ed. 2005, 44, 1221–1222. 10.1002/anie.200462268. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Vassilikogiannakis G.; Mägerlein W.; Kranich R. Total Synthesis of Colombiasin A. Angew. Chem., Int. Ed. 2001, 40, 2482–2486. . [DOI] [PubMed] [Google Scholar]
- Waizumi N.; Stankovic A. R.; Rawal V. H. A General Strategy to Elisabethane Diterpenes: Stereocontrolled Synthesis of Elisapterosin B via Oxidative Cyclization of an Elisabethin Precursor. J. Am. Chem. Soc. 2003, 125, 13022–13023. 10.1021/ja035898h. [DOI] [PubMed] [Google Scholar]
- Heckrodt T. J.; Mulzer J. Total Synthesis of Elisabethin A: Intramolecular Diels–Alder Reaction under Biomimetic Conditions. J. Am. Chem. Soc. 2003, 125, 4680–4681. 10.1021/ja034397t. [DOI] [PubMed] [Google Scholar]
- Zanoni G.; Franzini M. Elisabethin A: A Marine Diterpenoid Yet To Surrender to Total Synthesis. Angew. Chem., Int. Ed. 2004, 4837–4841. 10.1002/anie.200460570. [DOI] [PubMed] [Google Scholar]
- Preindl J.; Leitner C.; Baldauf S.; Mulzer J. A Short Access to the Skeleton of Elisabethin A and Formal Syntheses of Elisapterosin B and Colombiasin A. Org. Lett. 2014, 16, 4276–4279. 10.1021/ol501998y. [DOI] [PubMed] [Google Scholar]
- Steiner S.; Gaertner P.; Enev V. S. Synthesis of a Putative Advanced Intermediate En Route to Elisabethin A. Tetrahedron 2016, 72, 4536–4542. 10.1016/j.tet.2016.06.022. [DOI] [Google Scholar]
- Kaiser M.; Gaertner P.; Enev V. S. Studies towards the Enantioselective Synthesis of an Advanced Intermediate of Elisabethin A. Monatsh. Chem. 2017, 148, 49–56. 10.1007/s00706-016-1858-8. [DOI] [Google Scholar]
- Fukuda Y.; Shindo M.; Shishido K. Total Synthesis of (−)-Aspidospermine via Diastereoselective Ring-Closing Olefin Metathesis. Org. Lett. 2003, 5, 749–751. 10.1021/ol034020s. [DOI] [PubMed] [Google Scholar]
- Fukuda Y.; Sasaki H.; Shindo M.; Shishido K. Diastereoselective Ring-Closing Metathesis for the Construction of a Quaternary Carbon Stereogenic Center. Tetrahedron Lett. 2002, 43, 2047–2049. 10.1016/S0040-4039(02)00162-4. [DOI] [Google Scholar]
- Han J. H.; Kwon Y. E.; Sohn J.-H.; Ryu D. H. A Facile Method for the Rapid and Selective Deprotection of Methoxymethyl (MOM) Ethers. Tetrahedron 2010, 66, 1673–1677. 10.1016/j.tet.2010.01.007. [DOI] [Google Scholar]
- Bosque I.; Bagdatli E.; Foubelo F.; Gonzalez-Gomez J. C. Regio- and Stereoselective Aminopentadienylation of Carbonyl Compounds. J. Org. Chem. 2014, 79, 1796–1804. 10.1021/jo402854z. [DOI] [PubMed] [Google Scholar]
- The choice of TIPS as phenol protection proofed to be crucial in this step. Other silyl-groups such as TBS were found to be labile and susceptible to intermolecular attack of the generated phenolate anion. This then led to a scrambling of the silyl group and the dienyl-ether residue. Hence, significantly diminished yields due to side reactions and difficult product isolation were observed.
- Fráter G.; Schmid H. Thermische Umwandlung von Phenyl-penta-2,4-dienyläthern in 4-[Penta-2,4-dienyl]-phenole als Beispiel für [5,5]-sigmatropische Umlagerungen. Vorläufige Mitteilung. HELV. CHIM. ACTA 1968, 51, 190–193. 10.1002/hlca.19680510120. [DOI] [Google Scholar]
- Fráter G.; Schmid H. Thermische Umwandlung von Penta-2, 4-dienyl-phenyläthern in 4-(Penta-2, 4-dienyl)-phenole; [5s, 5s]-sigmatropische Umlagerungen. HELV. CHIM. ACTA 1970, 53, 269–290. 10.1002/hlca.19700530208. [DOI] [Google Scholar]
- Schmid E.; Fráter G.; Hansen H.-J.; Schmid H. Eine neue 2-Allylphenol-Cumaran-Umlagerung. HELV. CHIM. ACTA 1972, 55, 1625–1674. 10.1002/hlca.19720550526. [DOI] [Google Scholar]
- Kanematsu M.; Soga K.; Manabe Y.; Morimoto S.; Yoshida M.; Shishido K. Efficient and Enantioselective Total Syntheses of Heliannuols A and K. Tetrahedron 2011, 67, 4758–4766. 10.1016/j.tet.2011.05.034. [DOI] [Google Scholar]
- Additionally, compound 11, (31%) was isolated as a side product derived from ether-cleavage, which was recycled to improve the yield of 10 to 67% b.r.s.m.
- Hoye T. R.; Jeffrey C. S.; Tennakoon M. A.; Wang J.; Zhao H. Relay Ring-Closing Metathesis (RRCM): A Strategy for Directing Metal Movement Throughout Olefin Metathesis Sequences. J. Am. Chem. Soc. 2004, 126, 10210–10211. 10.1021/ja046385t. [DOI] [PubMed] [Google Scholar]
- TES protection delivered a ratio of approx. 5:1 favoring the desired diastereomer whereat MOM failed to exhibit any selectivity providing the bicyclic product as 1:1 mixture of diastereomers.
- Booker-Milburn K. I.; Barker A. Fe(Ill) Mediated Oxidative Radical Cyclisation of Cyclopropanone Acetals. Tetrahedron 1988, 54, 15321–15344. 10.1016/S0040-4020(98)00958-2. [DOI] [Google Scholar]
- López F.; Harutyunyan S. R.; Minnaard A. J.; Feringa B. L. Copper-Catalyzed Enantioselective Conjugate Addition of Grignard Reagents to Acyclic Enones. J. Am. Chem. Soc. 2004, 126, 12784–12785. 10.1021/ja046632t. [DOI] [PubMed] [Google Scholar]
- Wang S.-Y.; Ji S.-J.; Loh T.-P. Cu(I) Tol-BINAP-Catalyzed Enantioselective Michael Reactions of Grignard Reagents and Unsaturated Esters. J. Am. Chem. Soc. 2007, 129, 276–277. 10.1021/ja0666046. [DOI] [PubMed] [Google Scholar]
- Wang S.-Y.; Lum T.-K.; Ji S.-J.; Loh T.-P. Highly Efficient Copper(I) Iodide-Tolyl-BINAP-Catalyzed Asymmetric Conjugate Addition of Methylmagnesium Bromide to α,β-Unsaturated Esters. Adv. Synth. Catal. 2008, 350, 673–677. 10.1002/adsc.200700567. [DOI] [Google Scholar]
- Bertz S. H.; Chopra A.; Eriksson M.; Ogle C. A.; Seagle P. Re-Evaluation of Organocuprate Reactivity: Logarithmic Reactivity Profiles for Iodo- versus Cyano-Gilman Reagents in the Reactions of Organocuprates with 2-Cyclohexenone and Iodocyclohexane. Chem. – Eur. J. 1999, 5, 2680–2691. . [DOI] [Google Scholar]
- Yang J.; Dudley G. B. Conjugate Addition of Organocopper Reagents in Dichloromethane to α,β-Unsaturated Esters. Tetrahedron Lett. 2007, 48, 7887–7889. 10.1016/j.tetlet.2007.08.105. [DOI] [Google Scholar]
- Moreira R.; Taylor S. D. Highly Efficient and Enantioselective Syntheses of (2S,3R)-3-Alkyl- and Alkenylglutamates from Fmoc-Protected Garner’s Aldehyde. Amino Acids 2020, 52, 987–998. 10.1007/s00726-020-02868-7. [DOI] [PubMed] [Google Scholar]
- Benoit-Marquie F.; Csákÿ A. G.; Esteban G.; Martínez M. E.; Plumet J. An expeditious entry to the hydrophenalene ring system of pseudopterosins. Tetrahedron Lett. 2000, 41, 3355–3358. 10.1016/S0040-4039(00)00413-5. [DOI] [Google Scholar]
- McDevitt J. P.; Lansbury P. T. Glycosamino Acids: New Building Blocks for Combinatorial Synthesis. J. Am. Chem. Soc. 1996, 118, 3818–3828. 10.1021/ja9525622. [DOI] [Google Scholar]
- Kaneko Y.; Kiyotsuka Y.; Acharya H. P.; Kobayashi Y. Construction of a Quaternary Carbon at the Carbonyl Carbon of the Cyclohexane Ring. Chem. Commun. 2010, 46, 5482–5484. 10.1039/c0cc00653j. [DOI] [PubMed] [Google Scholar]
- Tiseni P. S.; Peters R. Catalytic Asymmetric Formation of δ-Lactones from Unsaturated Acyl Halides. Chem. – Eur. J. 2010, 16, 2503–2517. 10.1002/chem.200902896. [DOI] [PubMed] [Google Scholar]
- Reichl K. D.; Dunn N. L.; Fastuca N. J.; Radosevich A. T. Biphilic Organophosphorus Catalysis: Regioselective Reductive Transposition of Allylic Bromides via PIII/PV Redox Cycling. J. Am. Chem. Soc. 2015, 137, 5292–5295. 10.1021/jacs.5b01899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husstedt W.; Thrasher J.; Haufe G. Synthesis of 1-Pentafluorosulfanyl-1,3-Dienes. Synlett 2011, 12, 1683–1686. 10.1055/s-0030-1260809. [DOI] [Google Scholar]
- Brandt D.; Dittoo A.; Bellosta V.; Cossy J. Synthetic Approach to Wortmannilactone C. Org. Lett. 2015, 17, 816–819. 10.1021/ol5036112. [DOI] [PubMed] [Google Scholar]
- Das D.; Khan H.; Chakraborty T. Ti(III)-Mediated Radical-Induced Approach to a Bicyclic δ-Lactone with a Bridgehead β-Hydroxy Group. Synthesis 2018, 50, 3006–3014. 10.1055/s-0037-1609586. [DOI] [Google Scholar]
- Yao H.; Song L.; Liu Y.; Tong R. Cascade Michael Addition/Cycloketalization of Cyclic 1,3-Dicarbonyl Compounds: Important Role of the Tethered Alcohol of α,β Unsaturated Carbonyl Compounds on Reaction Rate and Regioselectivity. J. Org. Chem. 2014, 79, 8774–8785. 10.1021/jo501604e. [DOI] [PubMed] [Google Scholar]
- Lebarillier L.; Outurquin F.; Paulmier C. Preparation and Oxidation of α-Phenylselanyl Esters. Tetrahedron 2000, 56, 7483–7493. 10.1016/S0040-4020(00)00647-5. [DOI] [Google Scholar]
- Kocienski P. J.; Pontiroli A.; Qun L. Enantiospecific Syntheses of Pseudopterosin Aglycones. Part 2. Synthesis of Pseudopterosin K–L Aglycone and Pseudopterosin A–F Aglycone via a B BA BAC Annulation Strategy. J. Chem. Soc., Perkin Trans. 1 2001, 1, 2356–2366. 10.1039/B102961B. [DOI] [Google Scholar]
- Lomba L.; Afarinkia K.; Vinader V. A New Route to Tricyclane Sesquiterpenoids: Total Synthesis of α-Ekasantalic Acid. Org. Biomol. Chem. 2019, 17, 4456–4459. 10.1039/C9OB00630C. [DOI] [PubMed] [Google Scholar]
- Nunn K.; Mosset P.; Gree R.; Saalfrank R. W. Conjugate Reduction Vicinal to Butadiene Tricarbonyl Iron Complexes. Application to the Synthesis of (±)-6,7-Dihydro-LTB4 Methyl Esterr. J. Org. Chem. 1992, 57, 3359–3364. 10.1021/jo00038a026. [DOI] [Google Scholar]
- Raji Reddy C.; Mohammed S. Z. Synthetic Studies toward (±)-Furanocembranoid 1: Construction of the Acyclic Carbon Framework. ACS Omega 2018, 3, 15628–15634. 10.1021/acsomega.8b02328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N.; Ou J.; Miesch M.; Chiu P. Conjugate Reduction and Reductive Aldol Cyclization of α,β-Unsaturated Thioesters Catalyzed by (BDP)CuH. Org. Biomol. Chem. 2011, 9, 6143–6147. 10.1039/c1ob05352c. [DOI] [PubMed] [Google Scholar]
- Pitsinos E. N.; Mavridis I.; Tzouma E.; Vidali V. P. Enantioselective Synthesis of Cassane-Type Furanoditerpenoids: Total Synthesis of Sucutiniranes C and D. Eur. J. Org. Chem. 2020, 30, 4730–4742. 10.1002/ejoc.202000724. [DOI] [Google Scholar]
- Li J.-Q.; Quan X.; Andersson P. G. Highly Enantioselective Iridium-Catalyzed Hydrogenation of α,β-Unsaturated Esters. Chem. – Eur. J. 2012, 18, 10609–10616. 10.1002/chem.201200907. [DOI] [PubMed] [Google Scholar]
- Srikrishna A.; Ravi G. A Stereoselective Total Synthesis of (−)-Seychellene. Tetrahedron 2008, 64, 2565–2571. 10.1016/j.tet.2008.01.035. [DOI] [Google Scholar]
- Chen H. Y.; Kim S.; Wu J. Y.; Birzin E. T.; Chan W.; Yang Y. T.; Dahllund J.; DiNinno F.; Rohrer S. P.; Schaeffer J. M.; Hammond M. L. Estrogen Receptor Ligands. Part 3: The SAR of Dihydrobenzoxathiin SERMs. Bioorg. Med. Chem. Lett. 2004, 14, 2551–2554. 10.1016/j.bmcl.2004.02.084. [DOI] [PubMed] [Google Scholar]
- Nemoto T.; Ishige Y.; Yoshida M.; Kohno Y.; Kanematsu M.; Hamada Y. Novel Method for Synthesizing Spiro[4.5] Cyclohexadienones through a Pd-Catalyzed Intramolecular Ipso-Friedel-Crafts Allylic Alkylation of Phenols. Org. Lett. 2010, 12, 5020–5023. 10.1021/ol102190s. [DOI] [PubMed] [Google Scholar]
- Reader P. W.; Pfukwa R.; Jokonya S.; Arnott G. E.; Klumperman B. Synthesis of α,ω-Heterotelechelic PVP for Bioconjugation, via a One-Pot Orthogonal End-Group Modification Procedure. Polym. Chem. 2016, 7, 6450–6456. 10.1039/C6PY01296E. [DOI] [Google Scholar]
- Ondi L.; Nagy G. M.; Czirok J. B.; Bucsai A.; Frater G. E. From Box to Bench: Air-Stable Molybdenum Catalyst Tablets for Everyday Use in Olefin Metathesis. Org. Process Res. Dev. 2016, 20, 1709–1716. 10.1021/acs.oprd.6b00161. [DOI] [Google Scholar]
- Sattely E. S.; Cortez G. A.; Moebius D. C.; Schrock R. R.; Hoveyda A. H. Enantioselective Synthesis of Cyclic Amides and Amines through Mo-Catalyzed Asymmetric Ring-Closing Metathesis. J. Am. Chem. Soc. 2005, 127, 8526–8533. 10.1021/ja051330s. [DOI] [PubMed] [Google Scholar]
- Kue Lee S.; Kang S.; Tokita M.; Watanabe J. Formation of a Homochiral Antiferroelectric Ground State in Asymmetric Bent-Shaped Molecules. Liq. Cryst. 2010, 37, 593–598. 10.1080/02678291003710482. [DOI] [Google Scholar]
- Kim S.; Park J. H.; Lee S. Conversion of Acetals into Monothioacetals, α-Alkoxyazides and α-Alkoxyalkyl Thioacetates with Magnesium Bromide. Tetrahedron Lett. 1989, 30, 6697–6700. 10.1016/S0040-4039(00)70654-X. [DOI] [Google Scholar]
- various other chiral methods (CBS reduction, transfer hydrogenation applying Noyori’s catalyst) failed. Also, other achiral hydride reagents (NaBH4, LiAlH4, Selectrides, LTBA) delivered the desired product in inferior yield and selectivity.
- Trost B. M.; Bunt R. C.; Lemoine R. C.; Calkins T. L. Dynamic Kinetic Asymmetric Transformation of Diene Monoepoxides: A Practical Asymmetric Synthesis of Vinylglycinol, Vigabatrin, and Ethambutol. J. Am. Chem. Soc. 2000, 122, 5968–5976. 10.1021/ja000547d. [DOI] [Google Scholar]
- Zhao Z.-L.; Xu Q.-L.; Gu Q.; Wu X.-Y.; You S.-L. Enantioselective Synthesis of 4-Substituted Tetrahydroisoquinolines via Palladium-Catalyzed Intramolecular Friedel–Crafts Type Allylic Alkylation of Phenols. Org. Biomol. Chem. 2015, 13, 3086–3092. 10.1039/C4OB02574A. [DOI] [PubMed] [Google Scholar]
- Xiao Q.; Jackson J. J.; Basak A.; Bowler J. M.; Miller B. G.; Zakarian A. Enantioselective Synthesis of Tatanans A–C and Reinvestigation of Their Glucokinaseactivating Properties. Nat. Chem. 2013, 5, 410–416. 10.1038/nchem.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M. A.; Simmons E. M.; Wei C. S.; Park H.; Eastgate M. D. An Enantioselective Total Synthesis of (+)-Duocarmycin SA. J. Org. Chem. 2018, 83, 3928–3940. 10.1021/acs.joc.8b00285. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Schroeder G. M. Cyclic 1,2-Diketones as Building Blocks for Asymmetric Synthesis of Cycloalkenones. J. Am. Chem. Soc. 2000, 122, 3785–3786. 10.1021/ja000274m. [DOI] [Google Scholar]
- Ramadhar T. R.; Kawakami J.; Lough A. J.; Batey R. A. Stereocontrolled Synthesis of Contiguous C(sp3)-C(Aryl) Bonds by Lanthanide(III)-Catalyzed Domino Aryl-Claisen [3,3]-Sigmatropic Rearrangements. Org. Lett. 2010, 12, 4446–4449. 10.1021/ol1018147. [DOI] [PubMed] [Google Scholar]
- van der Baan J. L.; Bickelhaupt F. Palladium(II)-Catalyzed Claisen Rearrangement of Allyl Vinyl Ethers. Tetrahedron Lett. 1986, 27, 6267–6270. 10.1016/S0040-4039(00)85449-0. [DOI] [Google Scholar]
- Bonifazi E. L.; Ríos-Luci C.; León L. G.; Burton G.; Padrón J. M.; Misico R. I. Antiproliferative Activity of Synthetic Naphthoquinones Related to Lapachol. First Synthesis of 5-Hydroxylapachol. Bioorg. Med. Chem. Lett. 2010, 18, 2621–2630. 10.1016/j.bmc.2010.02.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.