

Abstract
The CAMP reaction is a synergistic lysis of erythrocytes by the interaction of an extracellular protein (CAMP factor) produced by some streptococcal species with the Staphylococcus aureus sphingomyelinase C (beta-toxin). Group A streptococci (GAS [Streptococcus pyogenes]) have been long considered CAMP negative, and this reaction commonly has been used to distinguish GAS from Streptococcus agalactiae. We here provide evidence that GAS possess this gene and produce an extracellular CAMP factor capable of participating in a positive CAMP reaction. The S. pyogenes CAMP factor is specified by a 774-bp open reading frame homologous to the CAMP factor genes from S. agalactiae and Streptococcus uberis. This gene, designated cfa, was isolated on a 1,256-bp fragment and cloned in Escherichia coli. Recombinant clones of E. coli expressing cfa secreted an active CAMP factor. The deduced 28.5-kDa protein encoded by cfa consists of 257 amino acids, with a predicted 28-amino-acid signal peptide. The cfa gene is widely spread among GAS: 82 of 100 clinical GAS isolates produced a positive CAMP reaction. Of the CAMP-negative strains, 17 of the 18 GAS strains contained the cfa gene. Additionally, CAMP activity was detected in streptococci from serogroups C, M, P, R, and U. The cfa gene was cloned and actively expressed in Escherichia coli and gene fusions were made, placing the β-galactosidase gene (lacZ) under control of the cfa promoter. These cfa promoter-lacZ fusions were introduced into S. pyogenes via a bacteriophage-derived site-specific integration vector where they showed that the cfa gene has a strong promoter that may be subject to as-yet-unidentified regulatory factors. The results presented here, along with previous reports, indicate that the CAMP factor gene is fairly widespread among streptococci, being present at least in groups A, B, C, G, M, P, R, and U.
The CAMP reaction, as originally described by Christie et al. (3), is the synergistic lysis of sheep erythrocytes by Staphylococcus aureus sphingomyelinase C (beta-toxin) and CAMP factor (cohemolysin), a secreted 25.3-kDa protein from group B streptococci (GBS [Streptococcus agalactiae]) (26). The CAMP reaction consists of two sequential steps and is performed with erythrocytes whose cell membranes contain at least 45 mol% sphingomyelin (35, 41). The first step is the hydrolysis of membrane sphingomyelin and phospholipids by the action of a sphingomyelinase or phospholipase (5). In the second step, the CAMP factor interacts nonenzymatically with the resulting metastable membrane, commonly by binding of previously released ceramide, leading to cell lysis (2, 5). Besides its role in cohemolysis, the CAMP protein binds in a nonimmune reaction with the Fc part of immunoglobulins G and M, leaving the Fab sites active (17). Although the role of CAMP factor in pathogenesis is unclear, several investigations suggest that the release of this protein during systemic infections could impair the host immune response. For example, rabbits receiving a single intravenous injection of partially purified CAMP protein from GBS rapidly died. In mice, the dose of the same protein had to be 45 times higher in relation to the body mass for killing (32). However, mice that had received prior injections of highly purified CAMP factor from GBS developed fatal septicemia when infected intraperitoneally with sublethal doses of GBS (17). These observations provide support for a function of CAMP protein as a pathogenicity factor.
The CAMP factor genes of S. agalactiae (29), Actinobacillus pleuropneumoniae (10), and Streptococcus uberis (16) have been cloned and expressed in Escherichia coli. The genes from S. agalactiae (cfb) (26) and S. uberis (cfu) (16) encode 28.4- and 28.3-kDa proteins, respectively, with N-terminal signal peptides. The sequence identity of the two CAMP factor proteins is 66.4%. A number of other gram-positive and gram-negative bacteria are known to react positively in the CAMP test, including Rhodococcus equi (9), Pasteurella haemolytica (8), Listeria monocytogenes, Listeria seeligeri (27), Aeromonas sp. (7), certain Vibrio spp. (18), and group G streptococci (34).
Group A streptococci (GAS) have long been considered CAMP negative. Moreover, the CAMP reaction has been commonly used as a diagnostic test for streptococci of serological group B and has been accepted as an important way to differentiate GAS from GBS (4, 14, 24, 40). Several reports have shown that GAS produce a positive CAMP reaction under certain conditions, particularly anaerobic incubation (4, 14), but synergistic hemolysis seen in the CAMP reaction system with GAS was interpreted as a false-positive reaction caused by small amounts of unoxidized streptolysin O (38). Given this long period of acceptance that GAS were CAMP negative, it was a surprise to find in the sequence data of the Streptococcus pyogenes SF370 genome sequencing project at the University of Oklahoma the existence of a 774-bp open reading frame (ORF) with 67% identity to the S. agalactiae cfb gene and with 65% identity to the S. uberis cfu gene.
In this study, we demonstrate that GAS do possess and express the gene for an active CAMP factor. Cloning in E. coli of the reading frame homologous to the hitherto sequenced CAMP factor genes demonstrates that it represents the gene designated cfa for the CAMP factor of GAS. Using transcriptional-translational lacZ fusions, cfa promoter activity was studied in both homologous and heterologous backgrounds. In S. pyogenes SF370, the highest level of activity of the cfa promoter was observed during the early stationary phase of growth.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
E. coli JM109 (44) was used for cloning and expression studies. S. pyogenes SF370 (36) was the source of chromosomal DNA used for cloning of cfa and for electrotransformation with plasmid DNA. Streptococcal strains surveyed for CAMP activity were clinical isolates from the strain collection at the University of Oklahoma. Staphylococcus aureus ATCC 25923 was used as the beta-toxin-producing test strain, and a clinical S. agalactiae isolate (Children’s Hospital, Oklahoma City, Okla.) served as a positive control for the CAMP reaction. E. coli were grown in L broth medium (28), and streptococcal strains were grown in Todd-Hewitt broth (Difco) supplemented with 0.2% yeast extract or on 5% sheep blood agar plates (Fisher Scientific). For solid media, 1.5% Bacto Agar (Difco) was added. Antibiotics were used in the following concentrations: for E. coli, 500 mg of erythromycin per ml and 100 mg of ampicillin per ml; for S. pyogenes, 3 mg of erythromycin per ml. 5-Bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) was used at a concentration of 40 mg/ml to detect β-galactosidase activity.
Computer analysis.
The sequence of the chromosomal CAMP factor gene was identified with BLASTX database searches (1, 13). Multiple sequence alignment of the CAMP factor genes from S. pyogenes, S. agalactiae, and S. uberis was done with CLUSTALX (39). SignalP was used to predict the probable signal peptide (23).
CAMP test.
Strains were tested for CAMP activity in a conventional diffusion test with slight modification. In brief, the test strains, and S. agalactiae as a positive control, were streaked on 5% sheep blood agar plates perpendicular to, but not touching, a streak of the beta-toxin-producing strain S. aureus ATCC 25923. After 12 to 20 h of incubation at 30°C, streptococcal strains were inspected for CAMP hemolysis.
Recombinant DNA techniques and DNA primers.
All protocols were performed as recommended by the manufacturers of the enzymes or by standard methods (28). Chromosomal DNA was isolated from S. pyogenes SF370 by the guanidium thiocyanate method (25). Plasmid DNA was isolated with the QIAprep-spin plasmid kit (Qiagen Inc.). PCRs were performed with Taq polymerase (Fisher Scientific); conditions were 30 cycles of 94°C (1 min), 37°C (1 min), and 72°C (2 min). The following oligonucleotides were used as PCR primers: CAMP1 (5′ GGAAATTAAAGAAATATTGACC), CAMP2 (5′ TCACCTCAAACTCTACAATGG), CAMP3 (5′ CCAAATTAAATCTTAAAAAGG), CAMP4 (5′ GGCGCCGGATCCTGTTTGGGTTTCATATTATTCCTC), CAMP5 (5′ TATGATTTTAGTTCAATTGG), CAMP6 (5′ CCAACCGCATGTGTGATAGCTTT), INT1 (5′ AATCTAAAACAAAAAGCCCCACG), INT2 (5′ AAAAAGGTTATTACAATTCACCA), and M13R (5′ TCACACAGGAAACAGCTATGAC).
Primers CAMP1 and CAMP3 recognize the cfa upstream region, and CAMP2 recognizes the region downstream of the gene. CAMP4 anneals to the 5′ cfa codons and introduces a BamHI site into cfa promoter fragments for in-frame fusions to lacZ (BamHI site and sequence antiparallel to the ATG start codon are underlined; sequence containing the BamHI linker is in italics). Primers CAMP5 and CAMP6 correspond to the cfa regions with the highest identity to cfb from GBS and cfu from S. uberis and were used to amplify an internal cfa fragment. Primer pair INT1 and INT2 allows amplification of the bacteriophage T12 integrase gene and attP site (21). Primer M13R anneals to the ribosome binding site of lacZ present on pGEM-T Easy (Promega) and its derivatives. Sequencing primers used were CAMP1, CAMP2, CAMP3, T7 (5′ AATACGACTCACTATAGGG), SP6 (5′ GATTTAGGTGACACTATAG), and M13 −40 (5′ GTTTTCCCAGTCACGAC). Primers SP6 and T7 served for the sequencing of PCR fragments cloned into pGEM-T Easy. The M13 −40 primer was used to ensure the accuracy of cfa-lacZ reporter fusions. Sequencing was done with an ABI model 377 automated sequencer.
PCR analysis of cfa in CAMP-negative S. pyogenes strains.
To detect the cfa gene in CAMP-negative S. pyogenes strains, DNA from crude cell lysates (15) was used as a template for PCR with primer pair CAMP4 and CAMP5 to amplify an internal 386-bp cfa fragment. For analysis, PCR products were separated on a 1.5% agarose gel.
Cloning and expression of the S. pyogenes cfa gene.
To clone the cfa gene, PCR was performed with S. pyogenes SF370 chromosomal DNA as a template. Fragments generated with primers CAMP2 and CAMP3 (1,256 bp) containing cfa with the complete promoter upstream region including the 3′ end of the adjacent gene (see Fig. 1) were cloned into pGEM-T Easy (Promega) to create pCAMP1 (Plac and Pcfa in opposite orientations) and pCAMP2 (Plac and Pcfa in tandem arrangement). Primers CAMP1 and CAMP2 generated a 992-bp fragment containing cfa with only its core promoter. Cloning of this fragment into pGEM-T Easy was possible only when spontaneous cfa promoter down mutations occurred: pCAMP62 and pCAMP64, both with Plac and cfa in the same orientation, carried a T→C transition mutation in the −10 region and a deletion of 30 nucleotides including the −35 promoter region, respectively (see Fig. 1). In contrast, the 148-bp cfa core promoter PCR fragment generated by primers CAMP1 and CAMP4 with pCAMP2 as the template was cloned into pGEM-T Easy to create pCAMP14. This unstable plasmid has two vector fragments in opposite orientations connected by two PCR fragments in the same orientation. Because of this arrangement, pCAMP14 contained the cfa core promoter and lacZ in the same orientation.
FIG. 1.

(a) Nucleotide sequence of the promoter and upstream regions of cfa from S. pyogenes SF370. The putative −35 and −10 cfa promoter hexanucleotides and ribosomal binding site (RBS) are indicated by underlining, and arrows mark the longest inverted repeats found in the upstream sequence. The region referred to in the text as the “core” promoter is indicated, starting at the beginning of the arrow. The promoter portion containing the −35 region that was deleted on plasmids pCAMP64 and pCAMP13 is indicated (“Δ −35 Region”). (b) Alignment of amino acid sequences of the CAMP proteins from S. pyogenes, S. agalactiae (26), and S. uberis (16). Homologous residues are shaded.
Construction of lacZ reporter fusions.
The 3,072-bp BamHI fragment from pMC1871 (30) containing the 3′ portion of lacZ starting with codon 10 was cloned into plasmid pPRG10 (12) digested with BamHI and BglII. The resulting plasmid, pPRGLAC (6.1 kb), contained the lacZ coding sequence from codon 10 with a BamHI site at its 5′ end, the p15A origin of replication, and the ermAM erythromycin resistance gene selectable both in E. coli and S. pyogenes. Primers CAMP4 and M13R were used to amplify by PCR a 550-bp product from pCAMP2. This product was digested with BamHI and SalI, releasing a 436-bp digestion fragment containing the cfa promoter that was cloned into pPRGLAC to create pCAMP11 (6.5 kb), a vector containing a transcriptional and in-frame translational fusion of cfa to lacZ. The cfa-lacZ fusion plasmids pCAMP12, pCAMP13, and pCAMP16 were created by replacing the cfa portion of pCAMP11 with SalI-BamHI cfa promoter cassettes obtained by amplification of pCAMP62, pCAMP64, and pCAMP14, respectively, with the primer pair M13R and CAMP4. For the construction of cfa-lacZ fusion vectors capable of site-specific integration into the chromosome of S. pyogenes, PCR with primer pair INT1 and INT2 and p7INT, a derivative of pWM139 (20), as a template was performed. The resulting 1,802-bp fragment, containing the functional integrase gene and the phage attachment site from S. pyogenes bacteriophage T12 (21), was cloned into pGEM-T Easy. The integrase gene and attP were released from this construct by digestion with SphI and SalI and cloned into pCAMP11 digested with the same enzymes to create pCAMP17 (8.4 kb [see Fig. 5]). The 442-bp PstI-BamHI cfa promoter fragment of pCAMP17 was replaced by the 430- and 166-bp PstI-BamHI cfa promoter cassettes from pCAMP11 and pCAMP12, respectively, to create integration plasmids pCAMP18 (8.3 kb) and pCAMP19 (8.1 kb). Cloning of the cfa core promoter from pCAMP16 as a PstI-BamHI fragment in pCAMP17 was attempted repeatedly, but no stable constructs were obtained. Plasmids pCAMP18 and pCAMP19 were integrated into the chromosome of S. pyogenes SF370 following electroporation, as previously described (31). The 3′ limit of the cfa portion on all reporter plasmids is cfa codon 4 fused in frame, via a BamHI site, to codon 10 of lacZ.
FIG. 5.

β-Galactosidase activities determined by cfa promoter constructs fused to lacZ in the indicated plasmids in a multicopy plasmid state in E. coli JM109 (top) or in plasmids pCAMP18 (A), pCAMP19 (B), and p7INT (C) present as genomic integrations in S. pyogenes SF370 (bottom). Assays were performed in the logarithmic growth phase (E. coli) or in the early logarithmic (2 h), late logarithmic (4 h), and early stationary (6 h) growth phases (S. pyogenes SF370). Plasmids pPRGLAC and p7INT served as negative controls.
β-Galactosidase assay.
β-Galactosidase was assayed as described previously (11), according to the method of Miller (22). Fresh medium was inoculated with 10−2 and 10−3 volumes of overnight cultures of S. pyogenes and E. coli, respectively. Samples of E. coli were taken in the logarithmic growth phase (optical density at 600 nm <1), and samples of S. pyogenes were taken in the early logarithmic, late logarithmic, and early stationary growth phases, which were experimentally determined by standard methods to occur at about 2, 4, and 6 h of culture incubation. Cells were harvested by centrifugation, resuspended, and made permeable to o-nitrophenyl-β-d-galactopyranoside (ONPG) by adding 20 μl of chloroform per sample and vortex mixing for 10 s. β-Galactosidase activity was determined at a temperature of 28°C by measuring ΔE at 420 nm to detect o-nitrophenyl released from ONPG and was calculated in Miller units, proportional to enzyme activity per cell.
Nucleotide sequence accession number.
The nucleotide sequence of cfa has been submitted to the EMBL/GenBank/DDBJ data banks and assigned accession no. AF079502.
RESULTS
The CAMP factor of GAS: native expression and sequence analysis.
To determine whether the ORF with significant homology to the CAMP factor genes from S. agalactiae (26) and S. uberis (16) found in the course of sequencing the genome of S. pyogenes SF370 (36) actually encoded an active CAMP protein, this M1 strain was tested for CAMP activity in a conventional diffusion test. CAMP activity was detected, especially when incubation of the bacteria was done at 30°C to limit the amount of hemolysis due to streptolysin activity, and the ORF was considered the putative CAMP factor gene of group A streptococci (cfa). Examination of the genome region containing cfa revealed an ORF flanking cfa in the 5′ direction with the same orientation as cfa and encoding a hypothetical protein that is homologous to bacterial amino acid binding proteins (42). Another hypothetical ORF neighboring cfa in the 3′ direction and oriented oppositely to cfa has homology to a number of hypothetical bacterial proteins of unknown function (6). The intergenic regions between the cfa coding sequence and neighboring ORFs in both the 5′ and 3′ directions have lengths of 369 and 368 bp, respectively. Putative cfa promoter −35 and −10 hexanucleotides resembling E. coli ς70 consensus promoter sequences (19) were identified 101 and 78 bp upstream from the cfa start codon. The 264-bp region upstream of position −50 of the putative cfa promoter (cfa promoter upstream region; nucleotide positions 1 to 264 in Fig. 1a) contains inverted repeats of 11 and 10 bp. A potential Shine-Dalgarno sequence is located at an appropriate distance from the ATG start codon of cfa. The coding sequence of cfa consists of 774 nucleotides (35.7% G+C) and encodes a 257-amino-acid protein with a molecular mass of 28.5 kDa and a predicted N-terminal 28-amino-acid signal peptide. The identities of the GAS CAMP factor with the homologous proteins from S. agalactiae (255 amino acids, 28.4 kDa) and S. uberis (256 amino acids, 28.3 kDa) are 59.9 and 54.9%, respectively (Fig. 1b). Mature GAS CAMP factor is predicted to be a 25.5-kDa protein consisting of 229 amino acids.
Distribution of cfa among GAS.
To investigate the general distribution of the cfa gene among GAS, 100 S. pyogenes strains of 60 different M types were surveyed in standard CAMP tests. Of these strains, 82 were CAMP positive. The level of CAMP factor production varied from strain to strain and ranged from a level similar to that produced by most GBS to completely negative (Fig. 2). PCR analysis of the 18 CAMP-negative strains with internal primers CAMP5 and CAMP6, and streptococcal chromosomal DNA as a template, revealed the existence of cfa in 17 of these strains. Only one isolate of S. pyogenes, originating from Canada (M31, T25), did not yield a cfa internal 386-bp fragment. The genes for CAMP proteins seem to be more common among streptococci than previously expected. CAMP tests also were performed with streptococci from serogroups other than A, and cohemolytic activity was found among isolates from streptococcal groups C, M, P, R, and U (not shown).
FIG. 2.

CAMP reactions of different GAS strains. Vertical streak: Sau, S. aureus ATCC 25923. Horizontal streaks: 1, S. agalactiae (positive control); 2, S. pyogenes NZ131; 3, S. pyogenes AS119; 4, S. pyogenes D794; 5, S. pyogenes 3728.
Cloning and expression of cfa in E. coli JM109.
The cfa gene from S. pyogenes SF370 was cloned and expressed in E. coli JM109, resulting in conversion of this recombinant strain to CAMP+ (Fig. 3). The amount of the promoter and flanking upstream region cloned with the coding region had a strong influence upon the level of CAMP expression in E. coli and the stability of the recombinant plasmid. Plasmids pCAMP1 and pCAMP2, which differed only in the orientation of the insert with respect to the vector lacZ promoter, stably carried cfa, its “core” promoter elements (i.e., −10 and −35 regions), and the 5′ upstream region to the end of the neighboring 5′ ORF (Fig. 1a). However, attempted cloning by PCR of the sequence containing only the cfa coding region and its core promoter yielded only 5% of the expected number of recombinant clones, and all clones analyzed carried mutations in the cfa promoter −10 or −35 element. This instability of the native sequence in E. coli is not related to vector copy number (not shown). The native promoter could be cloned by including only the first four codons of the cfa gene (pCAMP14). Two of these plasmids, pCAMP62 (T→C transition in the −10 hexanucleotide [TATGAT to TATGA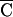 ]) and pCAMP64 (deletion of the −35 promoter region [Fig. 1a]), were chosen for further investigation. On both plasmids, cfa was oriented so that CAMP expression might be under the control of Plac in E. coli. CAMP activity tests performed with recombinant E. coli JM109 strains containing plasmid pCAMP1, pCAMP2, pCAMP62, or pCAMP64 demonstrated production of an active CAMP factor in these strains; however, the constructs differed in the amount of protein released (Fig. 3). The highest level of CAMP activity, comparable to that found with the positive control S. agalactiae, was produced by E. coli JM109 containing pCAMP62, followed by pCAMP64, pCAMP2, and pCAMP1, which secreted the same amount of active CAMP protein as donor strain S. pyogenes SF370 (Fig. 3 and data not shown).
]) and pCAMP64 (deletion of the −35 promoter region [Fig. 1a]), were chosen for further investigation. On both plasmids, cfa was oriented so that CAMP expression might be under the control of Plac in E. coli. CAMP activity tests performed with recombinant E. coli JM109 strains containing plasmid pCAMP1, pCAMP2, pCAMP62, or pCAMP64 demonstrated production of an active CAMP factor in these strains; however, the constructs differed in the amount of protein released (Fig. 3). The highest level of CAMP activity, comparable to that found with the positive control S. agalactiae, was produced by E. coli JM109 containing pCAMP62, followed by pCAMP64, pCAMP2, and pCAMP1, which secreted the same amount of active CAMP protein as donor strain S. pyogenes SF370 (Fig. 3 and data not shown).
FIG. 3.

CAMP reactions of GAS and E. coli containing recombinant cfa. Vertical streak: Sau, S. aureus ATCC 25923. Horizontal streaks: 1, S. agalactiae (positive control); 2, S. pyogenes AS119; 3, E. coli JM109(pCAMP62) (recombinant clone); 4, E. coli JM109(pUC19) (negative control).
cfa promoter activity in E. coli and S. pyogenes.
The different levels of CAMP activity detected among GAS and the importance of the 264-bp cfa promoter 5′ upstream region for the stability of cfa clones in E. coli suggested that expression of cfa might be regulated at the transcriptional level. To investigate cfa expression and to determine the influence of the cfa promoter upstream region on cfa promoter activity, transcriptional-translational reporter fusions were made. These fusions, containing the ribosome binding site and the first four codons of cfa fused in frame with lacZ (from codon 10), carried different cfa promoter constructs on a p15A replicon in E. coli. Plasmid pCAMP11, shown in Fig. 4, has the core promoter and complete cfa 5′ upstream region. Plasmids pCAMP12, pCAMP13, and pCAMP16 are identical to pCAMP11 except they derive their cfa promoter regions from pCAMP62, pCAMP64, and pCAMP14, respectively. The activity of the different cfa promoter lacZ fusion constructs in E. coli JM109 during the logarithmic growth phase was determined by β-galactosidase assays, according to Miller (22). E. coli JM109(pPRGLAC) was used as a negative control.
FIG. 4.

Plasmid pCAMP17 was used for introducing cfa-lacZ reporter fusions into the S. pyogenes genome by site-specific recombination into a unique site on the S. pyogenes chromosome by the action of the bacteriophage T12 integrase gene (int) and attP site (21). The cfa-lacZ fusions were made by replacing the cfa promoter fragment (P) with promoter inserts from pCAMP11 or pCAMP12 to create pCAMP18 and pCAMP19, respectively.
As shown in Fig. 5, the cfa promoter with the native upstream region showed strong activity in E. coli JM109 (pCAMP11, 21,800 Miller units). The deletion of the 264-bp cfa promoter upstream region increased cfa promoter activity 1.9-fold (pCAMP16, 40,700 Miller units). The additional single base pair exchange from TATGAT to TATGAC in the −10 region or the additional deletion of the −35 cfa promoter region resulted in a 300-fold decrease of promoter activity (pCAMP12, 150 Miller units; pCAMP13, 130 Miller units). The E. coli strains with the different constructs grew equally well, and expression of the CAMP–β-galactosidase fusion protein did not noticeably affect cell viability (not shown).
Although the results obtained in E. coli were suggestive, only examination of promoter activity in the native host would provide convincing evidence of possible translational control. Our laboratory had previously identified the integrase gene (int) and bacterial and phage attachment sites (attB and attP, respectively) used by the temperate bacteriophage T12 of S. pyogenes (21), showing that plasmid vectors containing T12 int and attP that were capable of site-specific integration at the unique attB site in the S. pyogenes genome (a serine tRNA gene) could be constructed (20, 21). The phage T12 genomic region containing int and attP was added as a functional module to pCAMP11 to create pCAMP17. The cfa promoter regions from pCAMP11 (core and 5′ upstream) and pCAMP62 (core containing the T→C transition in the −10 region) were cloned into pCAMP17 to create pCAMP18 and pCAMP19, respectively. These cfa-lacZ fusion constructs were introduced into S. pyogenes SF370, where the phage T12 integrase specified by these plasmids mediated site-specific integration of one copy of the plasmid at the attB site (not shown). The resulting recombinant strains were tested for β-galactosidase activity in the early logarithmic (2 h), late logarithmic (4 h), and early stationary (6 h) growth phases. S. pyogenes SF370 containing p7INT, a plasmid containing only the T12 integrative elements and lacZ under the control of its native promoter (20), was used as a negative control. The results of the assays are shown in Fig. 5.
In the homologous host S. pyogenes SF370, the activity of the cfa promoter with the native upstream region increased 10-fold during growth from early logarithmic to early stationary phase (pCAMP18, 39, 133, and 401 Miller units at the specified times). Deletion of the promoter upstream region and the single base pair exchange in the −10 region caused a dramatic decrease in cfa promoter strength. The β-galactosidase activity mediated by this promoter construct was only slightly above the values determined for the negative control (p7INT) and decreased during growth (pCAMP 19, 1.1, 0.9, and 0.7 Miller units).
DISCUSSION
The data presented in this paper demonstrate that, in contrast to the generally accepted view (4, 14, 24, 38, 40), S. pyogenes does possess the gene for an active CAMP factor protein. As shown by CAMP tests and by PCR analysis, the CAMP factor gene (cfa) is widespread among GAS. Of the 100 tested GAS strains, 82 gave a positive CAMP reaction. Among the 18 strains showing a CAMP-negative reaction, all but one contained at least part of the cfa gene, as shown by PCR amplification of a cfa internal fragment. The absence of CAMP factor expression among these 18 GAS strains was not clustered among any particular M serotype.
Cloning and expression of the cfa gene in recombinant E. coli strains confirms the existence of an active CAMP gene in GAS. Heterologous expression of the cfa gene demonstrated that the positive CAMP tests performed with different S. pyogenes strains were due to GAS CAMP protein and not to false-positive reactions, as assumed by Tapsall and Phillips (38). Indeed, the commonly accepted opinion that GAS are CAMP negative may have persisted over the years because of the relatively low level of CAMP activity produced by S. pyogenes. The level of secreted CAMP activity of tested GAS strains was in most cases clearly below the level produced by most GBS. Moreover, the clear zones caused by streptolysin often overlapped with the CAMP factor lysis zones, making clear identification of a positive CAMP reaction difficult (Fig. 4).
The CAMP reaction is temperature dependent between 15 and 30°C (35). Cooling of erythrocytes treated with S. aureus sphingomyelinase C to 4°C causes complete lysis of cells without the action of any CAMP-like protein (33). We performed CAMP tests at 30°C. At this temperature, the rate of CAMP factor induced-lysis is maximal, and erythrocyte membranes modified by sphingomyelinase C remain stable. Reduction of the temperature at which the CAMP tests were performed from 37 to 30°C resulted in sharper lysis zones and better identification of positive CAMP reactions.
The observed differences in CAMP production may be due to regulatory effects or to chromosomal alterations. The cfa promoter upstream region contains two distinct inverted repeats longer than 9 bp, of which the more upstream could act as a transcriptional terminator for the adjacent gene. The failure to clone cfa without this upstream region led us to the construction of transcriptional-translational cfa-lacZ fusions. They were used to study the influence of the native upstream region on cfa promoter activity and to investigate the expression of cfa during growth of the homologous host S. pyogenes SF370.
In E. coli, the presence of the native cfa promoter upstream region reduces cfa promoter activity significantly. The cfa core promoter exhibited an activity of 40,700 Miller units (pCAMP16). Addition of the upstream region decreased cfa promoter activity 1.9-fold (pCAMP11). Because of the low-level activities measured for the mutated core promoters (pCAMP12 and pCAMP13) a read-through from another promoter located on the vector plasmid and a function of the region as a transcriptional terminator can be excluded as a reason for this effect. Since E. coli is not the normal host, this reduction of promoter activity may be due to steric properties of the cloned DNA and not to any specific regulatory molecule. The high-level activity of the cfa core promoter in pCAMP16 is comparable to that determined for the promoter of the streptokinase gene (skc) from S. equisimilis in E. coli (11). The failure to clone the cfa core promoter without the native upstream region in other plasmid constructions on ColE1 and p15A replicons seems to be not due to toxicity of the gene products to the host E. coli, but rather due to plasmid instabilities induced by the activity of this strong promoter. β-Galactosidase produced at high levels is not detrimental to E. coli because of the formation of inclusion bodies (43).
The −35 and −10 cfa promoter sequences were detected by their strong homology to the E. coli ς70 consensus promoter sequences (19). In E. coli, cfa promoter activity was reduced about 300-fold on plasmids pCAMP12 and pCAMP13 carrying cfa promoter down mutations resulting from a single base pair exchange in the putative −10 promoter region and deletion of the putative −35 promoter region, respectively. These dramatic effects confirm the predicted location of the cfa promoter.
When integrated into the S. pyogenes SF370 chromosome, the transcriptional-translational cfa-lacZ fusion with a mutation in the −10 region of the cfa promoter (pCAMP19) released, depending upon growth phase, 35-fold to 570-fold less β-galactosidase activity than the lacZ fusion with the native upstream region (pCAMP18). These results could therefore be considered evidence for the lack of read-through from any promoter located upstream. The level of cfa expression in S. pyogenes SF370::pCAMP18 increased 10-fold during growth from the early logarithmic phase to the early stationary phase. This activity corresponds to about 4 and 40%, respectively, of the activity known for the promoter of the streptokinase gene (skc) in S. equisimilis (11). The results of β-galactosidase assays suggest that GAS CAMP factor is produced at the highest level when a limitation of nutrients occurs. Similarly to these results, Takaisi-Kikuni et al. (37) detected in S. agalactiae the maximal accumulation of CAMP factor in the late logarithmic phase of growth.
Although the function of the CAMP protein as a cohemolytic agent is well characterized, the common distribution of the genes among streptococci and the variable expression levels of CAMP genes in GAS suggest that these factors may fulfill another, more substantial function in physiology or pathogenicity. As demonstrated by Jürgens et al. (17), one additional function of CAMP factor from GBS is the nonspecific binding to immunoglobulins, thus impairing the immune response of the host. Further investigations might better define the potential role of CAMP factor proteins in pathogenesis of streptococci.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants AI19304 and AI38406 from the National Institutes of Health to J.J.F.
REFERENCES
- 1.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 2.Bernheimer A W, Linder R, Avigad L S. Nature and mechanism of action of the CAMP protein of group B streptococci. Infect Immun. 1979;23:838–844. doi: 10.1128/iai.23.3.838-844.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christie R, Atkins N E, Munch-Petersen E. A note on a lytic phenomenon shown by group B streptococci. Aust J Exp Biol. 1944;22:197–200. doi: 10.1038/icb.1945.30. [DOI] [PubMed] [Google Scholar]
- 4.Darling C L. Standardization and evaluation of the CAMP reaction for the prompt, presumptive identification of Streptococcus agalactiae (Lancefield group B) in clinical material. J Clin Microbiol. 1975;1:171–174. doi: 10.1128/jcm.1.2.171-174.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fehrenbach F J, Jürgens D, Rühlmann J, Sterzik B, Özel M. Role of CAMP factor (protein B) for virulence. In: Fehrenbach F J, Alouf J E, Falmagne P, Goebel W, Jeljaszewicz J, Jürgens D, Rappuoli R, editors. Bacterial protein toxins. Stuttgart, Germany: Gustav Fischer Verlag; 1988. pp. 351–357. [Google Scholar]
- 6.Ferson A E, Wray L V J, Fisher S H. Expression of the Bacillus subtilis gabP gene is regulated independently in response to nitrogen and amino acid availability. Mol Microbiol. 1996;22:693–701. doi: 10.1046/j.1365-2958.1996.d01-1720.x. [DOI] [PubMed] [Google Scholar]
- 7.Figura N, Guglielmetti P. Differentiation of mobile and mesophilic Aeromonas strains into species by testing for a CAMP-like factor. J Clin Microbiol. 1987;25:1341–1342. doi: 10.1128/jcm.25.7.1341-1342.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fraser G. The hemolysis of animal erythrocytes by Pasteurella haemolytica produced in conjunction with certain staphylococcal toxins. Res Vet Sci. 1962;3:104–110. [Google Scholar]
- 9.Fraser G. Hemolytic activity of Corynebacterium ovis. Nature (London) 1961;189:246. doi: 10.1038/189246a0. [DOI] [PubMed] [Google Scholar]
- 10.Frey J, Perrin J, Nicolet J. Cloning and expression of a cohemolysin, the CAMP factor of Actinobacillus pleuropneumoniae. Infect Immun. 1989;57:2050–2056. doi: 10.1128/iai.57.7.2050-2056.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gase K, Ellinger T, Malke H. Complex transcriptional control of the streptokinase gene of Streptococcus equisimilis H46A. Mol Gen Genet. 1995;247:749–758. doi: 10.1007/BF00290407. [DOI] [PubMed] [Google Scholar]
- 12.Gase K, Gase A, Schirmer H, Malke H. Cloning, sequencing and functional overexpression of the Streptococcus equisimilis H46A gapC gene encoding a glyceraldehyde-3-phosphate dehydrogenase that also functions as a plasmin(ogen)-binding protein. Purification and biochemical characterization of the protein. Eur J Biochem. 1996;239:42–51. doi: 10.1111/j.1432-1033.1996.0042u.x. [DOI] [PubMed] [Google Scholar]
- 13.Gish W, States D J. Identification of protein coding regions by database similarity search. Nat Genet. 1993;3:266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- 14.Gubash S M. Synergistic hemolysis phenomenon shown by an alpha-toxin-producing Clostridium perfingens and streptococcal CAMP factor in presumptive streptococcal grouping. J Clin Microbiol. 1978;8:480–488. doi: 10.1128/jcm.8.5.480-488.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hynes W L, Ferretti J J, Gilmore M S, Segarra R A. PCR amplification of streptococcal DNA using crude cell lysates. FEMS Microbiol Lett. 1992;73:139–142. doi: 10.1016/0378-1097(92)90597-h. [DOI] [PubMed] [Google Scholar]
- 16.Jiang M, Babiuk L A, Potter A A. Cloning, sequencing and expression of the CAMP factor gene of Streptococcus uberis. Microb Pathog. 1996;20:297–307. doi: 10.1006/mpat.1996.0028. [DOI] [PubMed] [Google Scholar]
- 17.Jürgens D, Sterzik B, Fehrenbach F J. Unspecific binding of group B streptococcal cocytolysin (CAMP factor) to immunoglobulins and its possible role in pathogenicity. J Exp Med. 1987;165:720–732. doi: 10.1084/jem.165.3.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Köhler W. CAMP-like phenomena of vibrios. Zentbl Bakteriol Hyg A. 1988;270:35–40. doi: 10.1016/s0176-6724(88)80139-1. [DOI] [PubMed] [Google Scholar]
- 19.McClure W R. Mechanism and control of transcription initiation in procaryotes. Annu Rev Biochem. 1985;54:171–204. doi: 10.1146/annurev.bi.54.070185.001131. [DOI] [PubMed] [Google Scholar]
- 20.McShan W M, McLaughlin R E, Nordstrand A, Ferretti J J. Vectors containing streptococcal bacteriophage integrases for site-specific gene insertion. 1998. Methods Cell Sci., in press. [Google Scholar]
- 21.McShan W M, Tang Y-F, Ferretti J J. Bacteriophage T12 of Streptococcus pyogenes integrates into the gene encoding a serine tRNA. Mol Microbiol. 1997;23:719–728. doi: 10.1046/j.1365-2958.1997.2591616.x. [DOI] [PubMed] [Google Scholar]
- 22.Miller J H. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Plainview, N.Y: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- 23.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of procaryotic and eucaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 24.Phillips E A, Tapsall J W, Smith D D. Rapid tube CAMP test for identification of Streptococcus agalactiae (Lancefield group B) J Clin Microbiol. 1980;12:135–137. doi: 10.1128/jcm.12.2.135-137.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pitcher D G, Saunders N A, Owen R J. Rapid extraction of bacterial genomic DNA with guanidium thiocyanate. Lett Appl Microbiol. 1989;8:151–156. [Google Scholar]
- 26.Podbielski A, Blankenstein O, Lütticken R. Molecular characterization of the cfb gene encoding group B streptococcal CAMP-factor. Med Microbiol Immunol. 1994;183:239–256. doi: 10.1007/BF00198458. [DOI] [PubMed] [Google Scholar]
- 27.Rocourt J, Grimont P A D. Listeria welshimeri sp. nov. and Listeria seeligeri sp. nov. Int J Syst Bacteriol. 1983;33:866–869. [Google Scholar]
- 28.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 29.Schneewind O, Friedrich K, Lütticken R. Cloning and expression of the CAMP factor of group B streptococci in Escherichia coli. Infect Immun. 1988;56:2174–2179. doi: 10.1128/iai.56.8.2174-2179.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shapira S K, Chou J, Richaud F V, Casadaban M J. New versatile plasmid vectors for expression of hybrid proteins coded by a cloned gene fused to lacZ gene sequences encoding an enzymatically active carboxy-terminal portion of beta-galactosidase. Gene. 1983;25:71–82. doi: 10.1016/0378-1119(83)90169-5. [DOI] [PubMed] [Google Scholar]
- 31.Simon D, Ferretti J J. Electrotransformation of Streptococcus pyogenes with plasmid and linear DNA. FEMS Microbiol Lett. 1991;82:219–224. doi: 10.1016/0378-1097(91)90336-9. [DOI] [PubMed] [Google Scholar]
- 32.Skalka B, Smola J. Lethal effect of CAMP-factor and UBERIS-factor—a new finding about diffusible exosubstances of Streptococcus agalactiae and Streptococcus uberis. Zentbl Bakteriol. 1981;249:190–194. [PubMed] [Google Scholar]
- 33.Smyth C J, Mollby R, Wadstrom T. Phenomenon of hot-cold hemolysis: chelator-induced lysis of sphingomylinase-treated erythrocytes. Infect Immun. 1975;12:1104–1111. doi: 10.1128/iai.12.5.1104-1111.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soedermanto I, Lämmler C. Comparative studies on streptococci of serological group G isolated from various origins. Zentbl Vet Reihe B. 1996;43:513–523. doi: 10.1111/j.1439-0450.1996.tb00349.x. [DOI] [PubMed] [Google Scholar]
- 35.Sterzik B, Fehrenbach F J. Reaction componenets influencing CAMP factor induced lysis. J Gen Microbiol. 1985;131:817–820. doi: 10.1099/00221287-131-4-817. [DOI] [PubMed] [Google Scholar]
- 36.Suvorov A N, Ferretti J J. A physical and genetic map of an M type 1 strain of Streptococcus pyogenes. Dev Biol Stand. 1995;85:215–218. [PubMed] [Google Scholar]
- 37.Takaisi-Kikuni N B, Jürgens D, Wecke J, Fehrenbach F J. Immunochemical localization of CAMP factor (protein B) in Streptococcus agalactiae. Microbios. 1997;89:171–185. [PubMed] [Google Scholar]
- 38.Tapsall J W, Phillips E A. Streptococcus pyogenes streptolysin O as a cause of false-positive CAMP reactions. J Clin Microbiol. 1984;19:534–537. doi: 10.1128/jcm.19.4.534-537.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thompson J D, Higgins D G, Gibson T J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilkinson H W. CAMP-disk test for presumptive identification of group B streptococci. J Clin Microbiol. 1977;6:42–45. doi: 10.1128/jcm.6.1.42-45.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiseman G M, Caird J D. The nature of staphylococcal beta hemolysin. I. Mode of action. Can J Microbiol. 1967;13:369–376. doi: 10.1139/m67-049. [DOI] [PubMed] [Google Scholar]
- 42.Wissenbach U, Keck B, Unden G. Physical map location of the new artPIQMJ genes of Escherichia coli, encoding a periplasmatic arginine transport system. J Bacteriol. 1993;175:3687–3688. doi: 10.1128/jb.175.11.3687-3688.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Worrall D M, Goss N H. The formation of biologically active beta-galactosidase inclusion bodies in Escherichia coli. Aust J Biotechnol. 1989;3:28–32. [PubMed] [Google Scholar]
- 44.Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]