

Abstract
A green, quick and sensitive spectrofluorimetric technique was investigated and validated for the assay of three different drugs namely, ketoprofen (KPN), paracetamol (PAR), and chlorzoxazone (CLX). The method is based on fluorescence quenching of the fluorescence probe, silver nanoparticles (SNPs). The fluorescence quenching of SNPs may be attributed to the complexation between each of the studied drugs with SNPs. The fluorescence of SNPs alone or after complexation with the studied drugs were measured at 485 nm (λex 242 nm) without the need to extract the formed complex. Chemical reduction was employed for preparing SNPs, where silver nitrate was reduced by sodium borohydride in deionized water without adding organic stabilizer. SNPs were found soluble in water, had high stability and had a narrow emission band. The studied drugs were found to decrease the fluorescence of SNPs significantly through static quenching according to Stern–Volmer equation. Factors affecting the reaction between the drugs and NPs were carefully examined and optimized. Using the optimum conditions, the difference in the fluorescence intensity of SNPs before and after complexation with the studied drugs was in a good linear relationship with the concentration of the studied drugs, where (R2 = 0.9998, 0.9998 and 0.9991) in the ranges of 0.5–5.0, 0.15–3.0 and 0.5–9.0 μg mL−1 for KPN, PAR and CLX, respectively. Validity of the proposed method was investigated according to ICH recommendations. The proposed technique was also employed for the analysis of each of the three drugs in commercial or laboratory prepared tablets and in spiked human plasma with very good recoveries as well as high level of accuracy and precision. This method was intended to the analysis of the proposed drugs in their single formulation and single drug administration. The suggested technique is considered an eco-friendly method, as it uses water as the safest and least expensive solvent. Moreover, the recommended technique does not involve solvent extraction of the formed complexes. Greenness assessment of the suggested procedure was accomplished by applying the four standard assessment tools. Consequently, the recommended method can be used in the routine quality control analysis of the cited drugs with minimum harmful effect on the environment as well as the individuals.
A green, quick and sensitive spectrofluorimetric technique was investigated and validated for the assay of three different drugs namely, ketoprofen (KPN), paracetamol (PAR), and chlorzoxazone (CLX).
1. Introduction
Ketoprofen (KPN), Fig. 1a, is a derivative of propionic acid and one of the non-steroidal anti-inflammatory drugs (NSAIDs). It is used in musculoskeletal joint disorders, dental pain and in peri-articular disorders. It is also used in menstrual cramps, post-operative pain and as anti-pyretic agent.1 It is official in the United States Pharmacopeia (USP),2 British Pharmacopeia (BP)3 and Japanese Pharmacopeia (JP)4 and assayed by acid–base titration in both USP and JP while in BP by RP-HPLC (reversed phase high performance liquid chromatography).
Fig. 1. Chemical structures of the three drugs.

Different methods were published for the determination of KPN including ion pair chromatography,5 derivative IR (infra-red) spectroscopy,6 HPLC,7 electrochemical method,8 chromatography tandem mass spectrometry9,10 and terbium sensitized luminescence method.11 However, most of these methods have some limitations, such as complicated pre-treatment, wasting of time and possible pollution of the environment. Compared to these methods, the proposed fluorescence determination of KPN is either more sensitive, simpler, faster or safer.
Recently, several research papers about fluorescence determination of KPN have been published. These reported methods include: (a) a near infrared fluorescence imprinted sensor based on polyethyleneimine passivated copper-doped CdS quantum dots and zinc oxide nanorods,12 (b) hybrid nano assemblies using different ligands that were synthesized using various thiourea derivatives13 (c) a mercaptopropionic acid-capped CdTe quantum dots,14 and (d) carbon dot core and molecularly imprinted polymer shell as sensor.15 The suggested method has the advantages of being simpler and greener than the methods cited above ref. 12–15.
Paracetamol (PAR); Fig. 1b, is a para-aminophenol derivative which has analgesic, antipyretic effect and weak anti-inflammatory activity. Paracetamol is administered for relieving mild and moderate pain and as antipyretic for short-term treatment. Usually, PAR is the chosen analgesic or antipyretic, mainly in the old patients in addition to patients who cannot take salicylates or other NSAIDs such as asthmatic patients, patients who are previously diagnosed with peptic ulcer and children. It is also the first choice of painkiller for pregnant or breastfeeding women. It is official in the USP,2 BP3 and JP4 and it was assayed by RP HPLC in both USP and BP and via spectrophotometry in JP.
Different methods including spectroscopic,16–21 chromatographic22,23 and electrochemical24,25 methods were reported for the analysis of PAR.
The reported spectrofluorimetric method16 was based on measuring the fluorescence quenching of quinine sulphate by chloride ions which were produced from oxidation reaction of PAR with potassium chlorate in acidic medium. Other reported spectrofluorimetric method for the determination of paracetamol via oxidation is presented using different oxidizing reagents as sodium hypochlorite,18 cerium(iv)19,20 and N-bromosuccinimide (NBS) to produce a highly fluorescent product.21
Chlorzoxazone (CLX); Fig. 1c, is a skeletal muscle relaxant that act centrally producing sedative properties. It acts primarily by affecting the spinal cord and subcortical areas of the brain that causes inhibition of the muscle spasm. It is considered as an adjunct therapy in relieving the painful muscle spasm coupled with musculoskeletal conditions. Chlorzoxazone is also given with analgesics in compound preparations.1 It is official in USP,2 where it was assayed by spectrophotometry.
Various techniques were stated for CLX analysis including HPLC,26,27 gas chromatography–mass spectrometry,28 spectrophotometry,29 voltammetry,30–32 fluorimetry.33 The reported fluorimetric method33 depends on measuring the native fluorescence of CLX in chloroform at 310 nm after excitation at 286 nm. Using chloroform as solvent decreases the greenness of the reported method.
Recently, the usage of luminescent metal NPs made a revolution in the analysis of drugs either in their raw form or in the pharmaceutical preparations. This is because of what NPs possess of unique and exceptional properties. Nanoparticles means the usage of nanoscale size of metals (1–100 nm).34 SNPs are noble metal nanoparticles that are widely used owing to their astonishing advantages. SNPs are characterized by their elevated chemical and thermal stability, electrical conductivity, catalytic and optical activity,35–38 in addition to surface enhanced Raman scattering for the determination of drugs.39,40 SNPs have been used either in drug delivery or analysis in both pharmaceutical dosage forms and biological fluids. Many drugs were assayed because some of them can cause quenching of SNPs fluorescence. SNPs can also cause fluorescence enhancement of some other drugs.41
In this work, preparation of SNPs was carried out in aqueous solution (deionized water) through chemical reduction of silver nitrate using sodium borohydride as a reducing agent without the addition of organic stabilizers.42,43 The prepared SNPs were used for the development of an accurate, time-saving and reliable technique for the analysis of KPN, PAR and CLX in their pharmaceutical formulations and in biological matrix (spiked human plasma). The proposed method can be used efficiently in quality control of the three drugs.
2. Laboratory investigations
2.1. Materials
All the used reagents were of analytical grade. Authentic samples of KPN, PAR and CLX with certified purities of 99.99, 100.02 and 100.01%, respectively were kindly provided by Pharco Pharmaceutical Co., Alexandria, Eva-Pharma Co., Cairo and Misr pharma Co. Cairo, Egypt, respectively. Silver nitrate with purity of 99.8% and sodium-borohydride with purity of 98% were bought from Sigma-Aldrich (Germany). Pharmaceutical preparations of (Ketofan® ampules, Bi-alcofan® tablets, Ketofan® capsules and Panadol advance® tablets) were bought from a local pharmacy in Egypt. Prepared tablets of CLX, contains 375 mg of CLX and inactive ingredients of lactose, starch, fructose, talc and glucose, were used because of the unavailability of a pharmaceutical preparation that contain CLX alone in the Egyptian market. Human plasma samples were provided by Mansoura University Hospitals (Mansoura, Egypt) and kept frozen until use after gentle thawing at room temperature. The analysis of spiked human plasma was performed after the approval of the ethical committee of the Faculty of Pharmacy, Mansoura University under code number 2022-186. Water used throughout the experiments was deionized (DI). Lactose and starch were brought from laboratory chemicals company, Cairo, Egypt, while fructose, talc and glucose were brought from El Nasr Pharmaceutical Chemicals Company, Cairo, Egypt.
2.2. Apparatus
- Cary Eclipse fluorescence spectrometer was employed for all the spectrofluorimetric measurements. The device was equipped with Xenon flash lamp. Smoothing factor of 19 was applied during the method, the voltage was adjusted at 650 V.
- pH-meter: consort, P-901, Belgium.
- Magnetic stirrer: product of Daihan Scientific Co, limited, S. Korea.
- Spectrophotometer: UV/vis. Shimadzu double-beam 1601 (Kyoto, Japan).
- Transmission electron microscope (TEM), JEOL, JSM -2100 (Tokyo, Japan). Sample was loaded on 200 carbon coated mesh and examined at 200 KV.
- Vortex mixer (IVM-300P, Taiwan).
- Centrifuge (2-16P, SIGMA, Germany).
2.3. Preparation of standard solutions
Standard solutions of KPN, PAR or CLX (200.0 μg mL−1) were prepared by dissolving 10.0 mg of each raw powder separately in 50 mL of ethanol. The stock solutions were diluted using the same solvent to get the suitable working solutions as required.
2.4. SNPs preparation
Colloidal aqueous solution of SNPs was prepared through chemical reduction of silver nitrate using sodium borohydride.42,43 10 mL of a precursor 1 × 10−2 M AgNO3 were added drop by drop to 60 mL of the fresh, ice-cold 2 × 10−2 M NaBH4 solution, in an ice bath, in a ratio of 1 : 6, respectively in conjunction with vigorous and continuous stirring using magnetic stirrer until yellow solution was obtained. Stirring was stopped just after the completion of addition of silver nitrate solution (not more than 2 min). The color of SNPs was brownish-dark-yellow. The prepared NPs were aged for 24 h in cool and dark place, in amber bottle in refrigerator, before the first usage. They were mono-dispersible and stable at five degrees Celsius for 3 weeks. The prepared SNPs were used as the fluorimetric probe for the analysis of the three studied drugs.
2.5. General procedures
2.5.1. Calibration graphs construction
1.0 mL of SNPs (1.4 × 10−4 M) was added to a set of 10 mL volumetric flasks followed by aliquots from the drugs' standard solutions covering the concentration ranges mentioned in Table 1. After that, the volume was made up to the full volume using DI water. Mixing of the flasks contents was followed by measuring the quenching fluorescence intensities (ΔF) at 485 nm after excitation at 242 nm. The blank was treated in the same way. The corresponding regression equations and calibration graphs were obtained through plots between ΔF and each drug concentrations in μg mL−1.
Analytical parameters and assay results for determination of the KPN, PAR and CLX by SNPs.
| Drug | Linear rangea | Slope | Intercept | r | % Found ±S.D.b | LOD, μg mL−1 | LOQ, μg mL−1 |
|---|---|---|---|---|---|---|---|
| KPN | 0.5–5.0 | 24.815 | 97.111 | 0.9995 | 99.98 ± 0.678 | 0.127 | 0.386 |
| PAR | 0.15–3.0 | 82.899 | 109.137 | 0.9998 | 100.02 ± 0.885 | 0.045 | 0.136 |
| CLX | 0.5–9.0 | 24.732 | 139.870 | 0.9997 | 99.95 ± 0.844 | 0.164 | 0.497 |
The concentration in μg mL−1.
Average of three determinations.
2.5.2. Analysis of the studied drugs in pharmaceutical and prepared dosage forms
For ampoules: two mLs of the mixed contents of five Ketofan® ampoules, each contain 100 mg KPN, were dissolved in ethanol to reach the mark of 50 mL volumetric flask. An accurate volume of the previously prepared solution, 2.5 mL, was moved to another 50 mL volumetric flask followed by ethanol addition to the mark to prepare 100 μg mL−1 of KPN. Then, sequential dilutions were performed as required to achieve the working range of KPN as showed in Table 1. Finally, the previously explained method in “2.5.1” was followed.
For tablets and capsules: ten Bi-alcofan® tablets (150 mg), ten Ketofan® capsules (50 mg) or ten Panadol advance® tablets (500 mg) were finely grounded and mixed well. Accurately measured amount of each powder was transferred into 100 mL measuring flask. 60 mL ethanol were added to each flask followed by sonication for 30 min, then ethanol was added to reach the flask mark. After filtration was carried out to remove insoluble excipients, sequential concentrations were prepared as required to achieve the working ranges of KPN or PAR as shown in Table 1. Finally, the previously explained method in “2.5.1” was followed.
For the prepared CLX tablets: 325 mg of the CLX were mixed with 20 mg of talc powder and 15 mg of maize starch, lactose, glucose and fructose to form one tablet. An accurately weighed amount of the prepared tablet equivalent to 10 mg of CLX was dissolved in 60 mL of ethanol in 100 mL volumetric flask. The volumetric flask and its contents were subjected to sonication for 30 min and completed to the required volume with ethanol, then subjected to filtration. Different concentrations inside the working range of CLX were prepared and the previously explained method in “2.5.1” was followed. The content of each dosage form was calculated using the corresponding regression equation.
2.5.3. Analysis of the studied drugs in spiked human plasma
Aliquots (1 mL) of human plasma was transferred using micropipette into a group of centrifuge tubes (15 mL). Aliquots of the standard solution of each drug within the concentration range were added to the tubes, then mixed using vortex for 10 s, and the volume was increased to 5 mL by methanol. Centrifugation was carried out for the tubes at 5800 rpm for half an hour. The clear supernatant was withdrawn and filtered by syringe filter, 0.45 μm porosity, then one mL aliquot of the filtered supernatants was transferred to a series of 10 mL measuring flasks, 1 mL of SNPs was added, and the flasks were completed to the mark with DI water to obtain final concentration covering the linearity range of each drug and within the mean plasma peak concentration. Blank was performed simultaneously and completed as explained before in “2.5.1”.
3. Results and discussion
Preparation of SNPs were carried out through the reduction of AgNO3 as a precursor by sodium borohydride as a reducing agent without using organic stabilizers, as mentioned in previously reported methods.42,43 The prepared NPs were aged for 24 h in the refrigerator before the first usage and stored in the dark to avoid the daylight effect. They were found to have maximum UV absorption at 400 nm (Fig. 2) and strong fluorescence at 485 nm after excitation at 242 nm (Fig. 3a). Ethanolic solutions of KPN or PAR exhibit no native fluorescence, while an ethanolic solution of CLX exhibits native fluorescence at 280/309 nm, as demonstrated in Fig. S1.† The addition of these drugs to SNPs was found to cause quenching of the emission band of the SNPs (Fig. 3b) due to the formation of non-fluorescent complexes.
Fig. 2. Absorbance spectrum of the prepared silver NPs (1.4 × 10−4 M).

Fig. 3. (a) FL excitation and emission spectra of blank and SNPs at 242/485 nm, where (a and a*) are spectra for blank and (b and b*) are for SNPs. (b) FL emission spectra of (a) SNPs and (b, c, d) in the presence of KPN, CLX and PAR 2 μg mL−1 each, respectively at 484 nm.

3.1. UV spectral characteristics and transmission electron microscope images
The UV spectrum of the prepared SNPs was recorded and the obtained spectrum was found to have a narrow absorption peak near 400 nm (Fig. 2) which is a characteristic of SNPs. The presence of a narrow absorption peak proves the SNPs mono-dispersity.44,45
TEM image was also used to know the size and the dispersity of the prepared colloidal SNPs solution. Fig. 4 shows TEM image of SNPs which were found to be spherical in shape, dark colored and monodispersed along with nanoscale average size of 8–9 nm. It was found that the prepared NPs were smaller than NPs prepared by Darya et al.42 and Alothman et al.43 which had an average size of 23 and 18 nm, respectively.
Fig. 4. TEM image of silver NPs.

3.2. Optimization of experimental conditions
3.2.1. Study the effect of (NPs) concentration
The SNPs concentration (calculated based on the final concentration of AgNO3) was found to affect the ΔF of the studied system. Fig. 5 showed that the FL intensity increased and reached the maximum value when the NPs concentration was 1.4 × 10−4 M. That concentration was recommended to be used.
Fig. 5. The influence of SNPs concentration (5 × 10−5 M to 2.5 × 10−4 M) on the ΔF using 1.0 μg mL−1 PAR (as an example).

3.2.2. Effect of pH
The effect of pH on the complex formation and the quenching behavior was studied using Britton–Robinson buffer covering pH range of 3–10. It was found that the pH has no positive effect on increasing the quenching behavior (ΔF) of the studied drugs, as presented in Fig. S2,† so the buffer usage was avoided throughout the study.
3.2.3. Effect of contact time
The time of contact between 1.0 mL of SNPs and each drug solution was studied by monitoring the ΔF as a function of time using various time intervals starting from 1 to 30 min. The interaction between each of the three drugs and SNPs was found to be completed immediately and was stable up to more than 30 min (Fig. S3†).
3.3. Spectral properties of SNPs
Fig. 3a shows the emission spectra of SNPs which have strong emission band at 485 nm after excitation at 242 nm. Upon the addition of each of the three studied drugs separately to SNPs, it was noticed that the FL intensity of SNPs emission spectrum was quenched without any shift (Fig. 3b).
3.4. Reaction mechanism
3.4.1. Quenching mechanism
The fluorescence quenching may originate from a diversity of mechanisms such as inner filter effect (IFE), fluorescence energy transfer (FRET), dynamic quenching, or static quenching.46 The excitation spectrum of SNPs and the UV-absorption spectra of KPN, PAR and CLX showed some overlap (Fig. 6a), thus primary IFE might occur. The primary inner filter effect was checked for the three drugs using eqn (1).
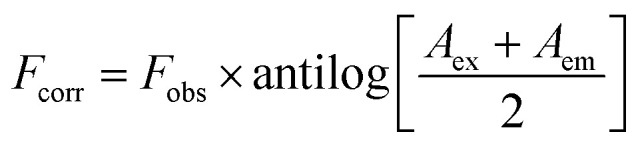 |
1 |
where Fobs is the observed fluorescence intensity, Fcorr is the corrected fluorescence intensity after omitting IFE from Fobs, while Aex and Aem are the absorbance values of the quencher at the excitation and emission wavelengths of the fluorophore, respectively. Then the suppressed efficiency (% E) was calculated for both corrected and observed fluorescence according to eqn (2):
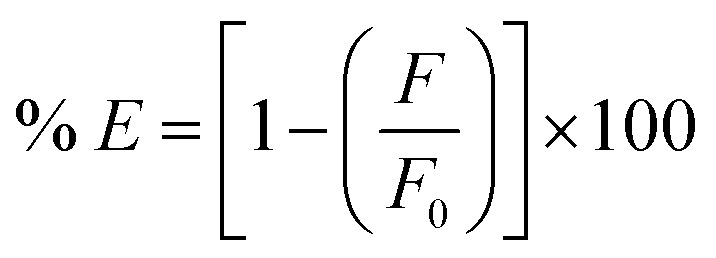 |
2 |
Fig. 6. (a) A co-plot showing the UV-absorption spectra of KPN, PAR and CLX and the excitation fluorescence spectra of SNPs showing slight overlap. (b) Suppressed efficiency of observed and corrected fluorescence of SNPs after addition of different concentrations of KPN and PAR. (c) Fluorescence quenching Stern–Volmer plot of SNPs with increasing concentration KPN, PAR and CLX at different temperatures.

Plots of % E of the observed and corrected fluorescence of KPN, PAR and CLX versus their molar concentration (Fig. 6b) inferring that the IFE participated in the fluorescence quenching by about 11%, 20% and 5% of the total suppressed efficiency for KPN, PAR and CLX, respectively. In order to further explore the main cause of the quenching mechanisms, Stern–Volmer equation47 was employed at three different temperatures to investigate the quenching reaction mechanism between the SNPs and the quenchers. A Stern–Volmer plot was obtained by plotting (F0/F) as a function of drug concentration (quencher) [Q] at three different temperatures, where; F0 is the emission intensity of SNPs without presence of quencher drug and F is the emission intensities of SNPs in presence of different concentration of quencher drug. The produced graph will have an intercept of about 1.0 and a slope called the Stern–Volmer constant (KSV). Table 2 contains the intercepts and Stern–Volmer constants at three different temperatures. The table shows that the quenching constant KSV decreased upon increasing the temperature, as shown in Fig. 6c, indicating a static quenching.46 So, it was concluded that the quenching mechanism was due to a combination of primary inner filter effect and static quenching.
Stern–Volmer parameters of the quencher-SNPs.
| Parameter | Stern–Volmer equation (Q, mol) | Correlation coefficient r2 | K SV (L moL−1) |
|---|---|---|---|
| KPN | |||
| Room temperature | F 0/F = 1.082 + 1.334 × 104[Q] | 0.9819 | 1.334 × 104 |
| 303 | F 0/F = 1.044 + 1.124 × 104[Q] | 0.9901 | 1.124 × 104 |
| 313 | F 0/F = 1.016 + 0.968 × 104[Q] | 0.9636 | 0.968 × 104 |
| PAR | |||
| Room temperature | F 0/F = 0.994 + 3.36 × 104[Q] | 0.9961 | 3.36 × 104 |
| 303 | F 0/F = 1.086 + 3.27 × 104[Q] | 0.9985 | 3.27 × 104 |
| 313 | F 0/F = 0.981 + 2.98 × 104[Q] | 0.9989 | 2.98 × 104 |
| CLX | |||
| Room temperature | F 0/F = 1.084 + 1.075 × 104[Q] | 0.9881 | 1.075 × 104 |
| 303 | F 0/F = 0.989 + 0.994 × 104[Q] | 0.9926 | 0.994 × 104 |
| 313 | F 0/F = 1.093 + 0.926 × 104[Q] | 0.995 | 0.926 × 104 |
3.4.2. Binding sites number, rate constant (K) and free energy change (ΔG°) calculation for reaction of SNPs and PAR
The binding constant K and the binding sites (n) between PAR and SNPs was calculated by utilizing the eqn (3):48
| log((F − F0)/F) = log k + n log[D] | 3 |
where; [D] is the molar concentration of PAR.
Estimating the number of binding sites (n) between PAR and SNPs was approximately one, as represented by the slope value (0.95). The binding constant (K was 1.92 × 104), as the intercept (log K) was 4.2845.
Gibb's free energy (ΔG°) was calculated by utilizing K value through eqn (4):
| Gibb's free energy (ΔG°, kJ mol−1) = −RT ln K = −23.54 kJ mol−1 | 4 |
where; R is the universal gas constant (8.314 J K−1 mol−1), T is the temperature in Kelvin, A negative value for ΔG indicates a spontaneous process.49
3.5. Suggested technique validation
The validity of the suggested technique was evaluated according to ICH Q2R1 suggestions.50
3.5.1. Linearity, range and quantitation & detection limits
Using the previously explained experimental circumstances, standard calibration graphs were constructed by plotting the difference in fluorescence intensities (ΔF) against concentration in μg mL−1. Linear lines with excellent correlation coefficients were obtained. The linear concentration ranges for KPN, PAR and CLX were summarized in Table 1 along with the statistical parameters. Table S1† showed the linearity ranges of the reported and proposed methods for KPN. The detection (LOD) and quantitation (LOQ) limits are shown in Table 1. LOD and LOQ were calculated using the following ICH equation (3.3 × Sa/b) (10 × Sa/b), respectively,50 where Sa represents the standard deviation of intercept and b is the slope of the calibration graph.
3.5.2. Accuracy and precision
The suggested technique's accuracy was confirmed by employing the proposed procedure for the determination of each of the three drugs at different concentrations levels, within the concentration range of each drug, each in three replicates. The results are shown in Table S2† and indicated good accuracy of the recommended technique due to the closeness of the percent found and the true value (100) as well as the low values of the standard deviations.
The two levels precision were applied using the recommended technique to investigate its precision. The results are shown in Table 3 and reveal a very good inter-day and intra-day precision.
Assessment of intra-day and inter-day precision of the proposed method.
| Drug | % Recovery* ± RSDa | |
|---|---|---|
| Intra-day | Inter-day | |
| KPN (3.0 μg mL−1) | 100.26 ± 1.27 | 99.79 ± 1.65 |
| PAR (1.0 μg mL−1) | 99.92 ± 0.96 | 100.16 ± 1.79 |
| CLX (5.0 μg mL−1) | 99.86 ± 1.03 | 100.65 ± 2.18 |
Average of three determinations, RSD is relative standard deviation.
3.5.3. Robustness
The assessment of robustness was carried out by investigating the effect of slightly changing the volume of SNPs (1.0 mL ± 0.02) and excitation wavelength (242 nm ± 2) on the fluorescence intensity. Small % RSD values were produced proving that the recovery was not affected (Table 4).
Evaluation of the suggested technique robustness.
| Parameter | % Recovery | % RSD |
|---|---|---|
| Volume of SNPs, (1.0 mL ± 0.02) | ||
| 0.98 | 99.61 ± 1.26 | 1.26 |
| 1.0 | 100.02 ± 0.76 | 0.76 |
| 1.02 | 100.33 ± 1.53 | 1.53 |
| Excitation wavelength, (242 nm ± 2) | ||
| 240 | 99.07 ± 1.80 | 1.82 |
| 242 | 100.03 ± 0.58 | 0.58 |
| 244 | 98.53 ± 2.08 | 2.09 |
3.5.4. Selectivity and interferences effect
The additives in the dosage forms can be a source of interference. So, interferences from excipients were studied using the recommended technique through the determination of the studied drugs in commercial dosage forms including capsules, tablets and ampules for KPN and tablets for PAR as well as the prepared tablets of CLX. The high recovery of the drugs indicates high selectivity of the proposed method (Table 5). As CLX was not found alone in dosage form in Egyptian local market, interferences from most common excipients such as glucose, lactose, fructose, talc and starch were studied. Five flasks each containing 5.0 μg mL−1 CLX, plus one excipient at a time with a concentration of 20 μg mL−1 51 were examined adopting the recommended approach. No significant interferences were found from the most common additives on the recovery of the studied drug, as presented in Table 6.
Analysis of the studied drugs in their pharmaceutical formulations using the proposed method.
| Dosage forms | Drug conc. | % Found ±S.D. | t test (2.78)a | F test (19.0)a | |
|---|---|---|---|---|---|
| Proposed method | Official methods [2, 3] | ||||
| Ketofan® ampoules | 100 mg ketoprofen/2 mL | 99.95 ± 0.805 | 99.69 ± 1.65 | 0.24 | 4.20 |
| Bi-alcofan® tablets | 100 mg ketoprofen/tablet | 100.15 ± 1.98 | 100.26 ± 1.48 | 0.08 | 1.97 |
| Ketofan® capsules | 50 mg ketoprofen/capsule | 100.32 ± 1.88 | 99.73 ± 1.55 | 0.42 | 1.47 |
| Panadol advance® tablets | 500 mg paracetamol/tablet | 99.98 ± 2.17 | 100.17 ± 0.79 | 0.025 | 7.52 |
| CLX prepared tablets | 325 mg chlorzoxazone/tablet | 100.38 ± 1.43 | 100.04 ± 1.66 | 0.23 | 2.23 |
The figures between parentheses are the tabulated t and F values at P = 0.05.50
Influence of existence of commonly used additives on the determination of 5.0 μg mL−1 CLX, as representative example.
| Additives 20 μg mL−1 | % Recovery |
|---|---|
| Glucose | 100.93 ± 1.82 |
| Fructose | 98.41 ± 1.37 |
| Lactose | 98.48 ± 0.96 |
| Starch | 100.33 ± 1.19 |
| Talc | 99.21 ± 0.69 |
The interference from some commonly co-administrated and/or co-formulated drugs that may be found in human plasma was studied. The tolerance limits of these compounds, namely, phenylephrine hydrochloride, tizanidine, venlafaxine, diacerein, diclofenac sodium, mefenamic acid, aceclofenac and ibuprofen were calculated. The obtained results of the tolerance limits reveal the acceptable selectivity of the proposed method (Table 7).
Effect of some related drugs on the fluorometric determination of 3 μg mL−1 KPN, 1.5 μg mL−1 PAR and 5 μg mL−1 CLX.
| Drug | Reason for interference | Drug tolerance limit (μg mL−1) |
|---|---|---|
| Phenylephrine hydrochloride | - It is used to relieve nasal discomfort caused by colds, allergies, and hay fever | 173 for KPN 234 for PAR |
| -Commonly co-administered with PAR or KPN | ||
| Tizanidine | - Muscle relaxant | 112 for KPN 164 for PAR |
| - Commonly co-administered with PAR or KPN | ||
| Venlafaxine | - An antidepressant medication | 89 for KPN 120 for PAR 125 for CLX |
| - Could be administered with any of the studied drugs | ||
| Diacerein | - Used to treat joint diseases such as osteoarthritis | 32 for KPN 40 for PAR 48 for CLX |
| - Could be administered with any of the studied drugs | ||
| Diclofenac sodium | Analgesic which is commonly prescribed with CLX | 50 for CLX |
| Mefenamic acid | Non-steroidal anti-inflammatory drug which is commonly prescribed with CLX | 7 for CLX |
| Aceclofenac | Non-steroidal anti-inflammatory drug which is commonly prescribed with CLX | 140 for CLX |
| Ibuprofen | Analgesic which is commonly prescribed with CLX | 240 for CLX |
4. Applications
4.1. Pharmaceutical and prepared dosage forms
The recommended technique was applied successfully for determining the studied drugs in commercial dosage forms including capsules, tablets and ampules as well as the prepared tablets of CLX. The results were compared statistically with those of the official methods: BP3 for KPN and USP2 for PAR and CLX using both Student's t-test and variance ratio F-test, as illustrated in Table 5. There was a perfect harmonization between the results of both proposed and official methods, which indicated high levels of accuracy and precision.
4.2. Spiked human plasma
The high sensitivity of the suggested technique made it eligible to be employed successfully in determining the studied drugs in spiked human plasma. The mean peak plasma concentrations were found to be 10 mg L−1, 10–20 mg L−1 and 36.3 mg L−1 for KPN, PAR and CLX, respectively.52 The results shown in Table 8 reveals good percentage recoveries, which proved that the developed method could be effectively involved to quantify the three drugs separately in spiked human plasma.
Analysis of the studied drugs in the spiked human plasma.
| Spiked human plasma | KPN | PAR | CLX | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Conc. taken (μg mL−1) | Conc. found (μg mL−1) | % Recovery | Conc. taken (μg mL−1) | Conc. found (μg mL−1) | % Recovery | Conc. taken (μg mL−1) | Conc. found (μg mL−1) | % Recovery | |
| Spiked human plasma | 1.0 | 0.991 | 99.10 | 0.5 | 0.486 | 97.20 | 2.0 | 1.979 | 98.95 |
| 2.0 | 2.036 | 101.80 | 1.0 | 0.901 | 90.10 | 3.0 | 3.015 | 100.50 | |
| 3.0 | 2.927 | 97.57 | 1.5 | 1.571 | 104.73 | 4.0 | 4.048 | 101.20 | |
| 4.0 | 4.085 | 102.13 | 2.0 | 2.129 | 106.45 | 5.0 | 4.939 | 98.78 | |
| 5.0 | 4.964 | 99.28 | 3.0 | 2.914 | 97.13 | 6.0 | 6.018 | 100.30 | |
| Mean | 99.78 | 99.12 | 99.95 | ||||||
| ±S.D. | 1.94 | 6.59 | 1.04 | ||||||
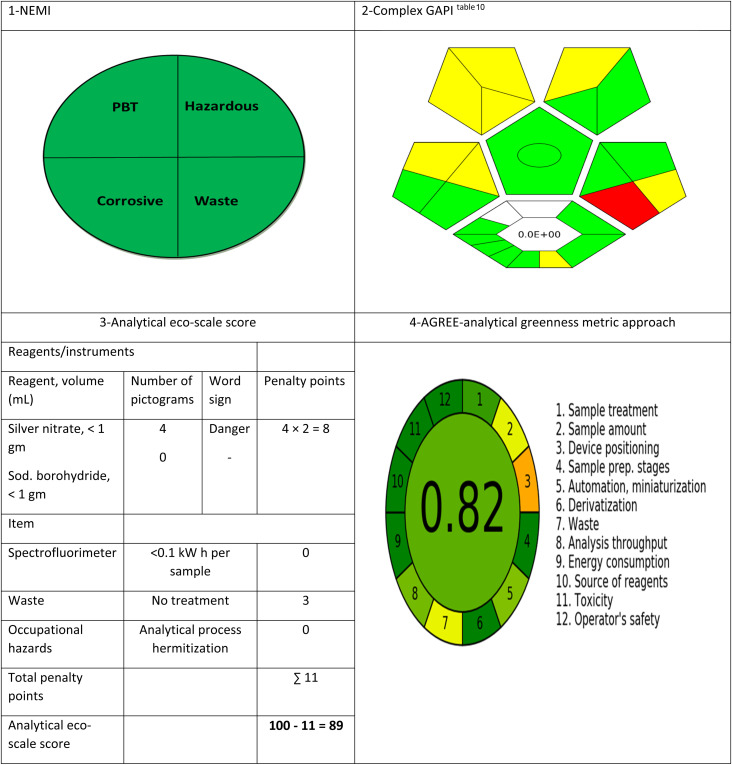
4.3. Greenness assessment
The suggested technique has been evaluated regarding its greenness using the four main assessment tools. First, National Environmental Methods Index (NEMI)53 was applied on the proposed method. The proposed approach fulfilled the four criteria of the greenness profile, as the solvents used during the analysis (water and ethanol) are not classified as either PBT or hazardous by the EPA's Toxic Release Inventory.54,55 The pH of the diluting solvent (water) is 6.5 which is not corrosive and the waste generation is less than 50 g per sample.
The second applied tool is green analytical procedure index (GAPI)56 which is a good semi-quantitative tool for laboratory practice, as it gives information on the whole process including 15 parameters, beginning with sampling until the final determination. Complexed GAPI was presented by Justyna Płotka-Wasylka and Wojciech Wojnowski who provided a very helpful software to automatically generate the complexed GAPI figure depending on the inputs of each method.57 Complex GAPI pictogram for the proposed method is presented in Tables 9 and 10, which ensures its greenness. Third, analytical eco-scale58 was applied to the suggested technique, as shown in Table 9 with a score of 89 which referred to an excellent green methodology (the closer the score to 100, the greener the method). Finally, AGREE-analytical greenness metric software (AGREE),59 created by Francisco Pena-Pereira et al., was applied for assessing the suggested technique. It depends on assessing twelve parameters of green analytical aspects. Those parameters were colored with colors ranging from dark green to orange, as represented in Table 9. The resulted score was 0.82 indicating the greenness of the method, as the closer the score to 1.0, the greener the method.
Assessment of the greenness of the suggested technique by four standard green analytical chemistry metric tools.
|
Green Analytical Procedure Index (GAPI) parameters for the proposed method.
| Categorya | Description |
|---|---|
| Sample preparation | |
| Collection (1) | Off-line |
| Preservation (2) | None |
| Transport (3) | None |
| Storage (4) | Normal condition |
| Type of method: direct or indirect (5) | Direct |
| Scale of extraction (6) | Micro-extraction |
| Solvents/reagents used (7) | Green solvents/reagents used |
| Additional treatments (8) | Simple treatments (clean up, solvent removal, etc.) |
| Reagent and solvents | |
| Amount (9) | <10 mL |
| Health hazard (10) | AgNO3 = 3 Sod. borohydride = 3 |
| Safety hazard (11) | AgNO3 = 0 Sod. borohydride = 1 |
| Instrumentation | |
| Energy (12) | ≤1.0 kW h per sample |
| Occupational hazard (13) | Hermetic sealing of the analytical process |
| Waste (14) | 1–10 mL |
| Waste treatment (15) | No treatment |
| Quantificationb | Yes |
The numbers between parentheses are the numbers in the GAPI pictogram (on the right).
The circle inside the central pentagram represent the quantification property of the method.
5. Conclusion
This suggested method discussed the possibility of using simply prepared SNPs as fluorimetric probe for the analysis of three drugs (KPN, PAR and CLX) by FL quenching technique. The suggested technique is characterized by being green, straightforward, fast and sensitive. It is neither time-consuming nor requires any sophisticated pretreatment. The suggested method is also considered environmentally friendly, as water – which is the greenest solvent – is the main solvent used during the analysis while volatile solvents are excluded. To ensure the greenness of the suggested method, four assessment tools were used. Furthermore, the suggested technique was validated and applied to determine the studied drugs in pharmaceutical dosage forms, prepared tablets and human plasma. This method was intended to the analysis of the proposed drugs in their single formulation and single drug administration. From economic and environmental point of view, this technique was found to have a great potential for the analysis of the studied drugs separately either in pharmaceutical dosage forms or spiked human plasma with high levels of accuracy and precision.
Ethical statement
This article does not include any studies on human participants or animals carried out by any of the authors.
Conflicts of interest
The authors declare that they have no conflict of interest.
Supplementary Material
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra04725j
References
- Brayfield A., Martindale : the complete drug reference, Pharmaceutical Press, London, 2017 [Google Scholar]
- The United States Pharmacopeia, (USP 40), The National Formulary (NF 35), Online version., [01.06.2022], available from, 2017, https://www.uspnf.com
- The British Pharmacopoeia, Electronic version [CD-ROM], 2013
- The Japanese Pharmacopoeia (17th edition), Ministry of Health, Labour and Welfare, 2016 [Google Scholar]
- Muhammad N. Li W. Subhani Q. Wang F. Zhao Y. G. Zhu Y. New J. Chem. 2017;41(17):9321–9329. doi: 10.1039/C7NJ01757J. [DOI] [Google Scholar]
- Azeez A. M. Fakhre N. A. Egypt. J. Chem. 2021;65(1):1–2. [Google Scholar]
- Zheng X. Zhou Y. Liu P. Zhongguo Yaofang. 2017;28(18):2579–2581. [Google Scholar]
- Cebula Z. Niedziałkowski P. Ossowski T. J. Appl. Biosci. 2018;1(3):7–8. [Google Scholar]
- Eichhold T. H. Bailey R. E. Tanguay S. L. Hoke S. H. J. Mass Spectrom. 2000;35(4):504–511. doi: 10.1002/(SICI)1096-9888(200004)35:4<504::AID-JMS958>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Azzouz A. Jurado-Sánchez B. Souhail B. Ballesteros E. J. Agric. Food Chem. 2011;59(9):5125–5132. doi: 10.1021/jf200364w. [DOI] [PubMed] [Google Scholar]
- Al-Kindy S. M. Al-Harasi Z. Suliman F. E. O. Al-Hamadi A. Pillay A. Terbium sensitized luminescence for the determination of ketoprofen in pharmaceutical formulations. J. Fluoresc. 2009;19(2):249–255. doi: 10.1007/s10895-008-0410-6. [DOI] [PubMed] [Google Scholar]
- Chen S. Zhou S. Fu J. Tang S. Wu X. Zhao P. Zhang Z. Anal. Methods. 2021;13(25):2836–2846. doi: 10.1039/D1AY00555C. [DOI] [PubMed] [Google Scholar]
- Saini A. Kaur M. Kuwar A. Kaur N. Singh N. Mol. Syst. Des. Eng. 2020;5(8):1428–1436. doi: 10.1039/D0ME00065E. [DOI] [Google Scholar]
- Molina-García L. Santos J. L. Ruiz-Medina A. Llorent-Martínez E. J. J. Food Drug Anal. 2013;21(4):426–431. doi: 10.1016/j.jfda.2013.09.007. [DOI] [Google Scholar]
- Bhogal S. Kaur K. Maheshwari S. Malik A. K. J. Fluoresc. 2019;29(1):145–154. doi: 10.1007/s10895-018-2322-4. [DOI] [PubMed] [Google Scholar]
- Perera G. W. de Costa M. D. R Mahanama K. J. Photochem. Photobiol., A. 2019;368:248–253. doi: 10.1016/j.jphotochem.2018.09.039. [DOI] [Google Scholar]
- Zhang J. Zhang X. S. Guang Pu Xue Yu Guang Pu Fen Xi. 2002;22(4):665–666. [PubMed] [Google Scholar]
- Vilchez J. L. Blanc R. Avidad R. Navalón A. J. Pharm. Biomed. Anal. 1995;13(9):1119–1125. doi: 10.1016/0731-7085(95)01537-u. doi: 10.1016/0731-7085(95)01537-u. [DOI] [PubMed] [Google Scholar]
- Hossein T. Yahya H. Asian J. Biochem. Pharm. Res. 2011;2(1):684–689. [Google Scholar]
- Jamaluddin A. M. Pak. J. Anal. Environ. Chem. 2019;20(1):17–31. doi: 10.21743/pjaec/2019.06.03. [DOI] [Google Scholar]
- Abdel-Wadood H. M. Mohamed N. A. Mohamed F. A. J. AOAC Int. 2005;88(6):1626–1630. doi: 10.1093/jaoac/88.6.1626. [DOI] [PubMed] [Google Scholar]
- Abdelwahab N. S. Abdelrahman M. M. Boshra J. M. Taha A. A. Biomed. Chromatogr. 2019;33(9):4598. doi: 10.1002/bmc.4598. [DOI] [PubMed] [Google Scholar]
- Mohamed M. A. J. AOAC Int. 2022;105(3):703–716. doi: 10.1093/jaoacint/qsac002. [DOI] [PubMed] [Google Scholar]
- Gürsoy S. Kuralay F. Colloids Surf., A. 2022;633:127846. doi: 10.1016/j.colsurfa.2021.127846. [DOI] [Google Scholar]
- Sripirom J. Sim W. C. Khunkaewla P. Suginta W. Schulte A. Anal. Chem. 2018;90(17):10105–10110. doi: 10.1021/acs.analchem.8b01135. [DOI] [PubMed] [Google Scholar]
- Rajnarayana K. Mada S. R. Vidyasagar J. Kishore P. Krishna D. R. Pharmazie. 2002;57(12):811–813. [PubMed] [Google Scholar]
- Mbinze J. K. Lebrun P. Debrus B. Dispas A. Kalenda N. Mbay J. M. Schofield T. Boulanger B. Rozet E. Hubert P. Marini R. D. J. Chromatogr. A. 2012;1263:113–124. doi: 10.1016/j.chroma.2012.09.038. [DOI] [PubMed] [Google Scholar]
- Eap C. B. Schnyder C. Savary L. J. Chromatogr. B: Biomed. Sci. Appl. 1998;705(1):139–144. doi: 10.1016/S0378-4347(97)00501-X. [DOI] [PubMed] [Google Scholar]
- Sastry C. S. Chintalapati R. Sastry B. S. Lakshmi C. S. Anal. Lett. 2000;33(12):2501–2513. doi: 10.1080/00032710008543205. [DOI] [Google Scholar]
- Zayed S. I. Issa Y. M. Acta Chim. Slov. 2020;67(4):1053–1060. doi: 10.17344/acsi.2020.5731. [DOI] [PubMed] [Google Scholar]
- Abbar J. C. Nandibewoor S. T. Ind. Eng. Chem. Res. 2012;51(1):111–118. doi: 10.1021/ie2021812. [DOI] [Google Scholar]
- Abbar J. Nandibewoor S. Crit. Rev. Anal. Chem. 2012;42(3):272–281. doi: 10.1080/10408347.2012.680336. [DOI] [Google Scholar]
- Stewart J. T. Chan C. W. J. Pharm. Sci. 1979;68(7):910–912. doi: 10.1002/jps.2600680735. [DOI] [PubMed] [Google Scholar]
- Alanazi F. K. Radwan A. A. Alsarra I. A. Saudi Pharm. J. 2010;18(4):179–193. doi: 10.1016/j.jsps.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi Y. Toshima N. J. Mol. Catal. A: Chem. 1999;141(1–3):187–192. doi: 10.1016/S1381-1169(98)00262-3. [DOI] [Google Scholar]
- Tada H. Teranishi K. Inubushi Y. I. Ito S. Langmuir. 2000;16(7):3304–3309. doi: 10.1021/la991315z. [DOI] [Google Scholar]
- Sclafani A. Mozzanega M. N. PichatJ P. Photochem. Photobiol. 1991;59(2):181–189. doi: 10.1016/1010-6030(91)87006-H. [DOI] [Google Scholar]
- Alaqad K. Saleh T. A. J. Environ. Anal. Toxicol. 2016;6(4):525–2161. [Google Scholar]
- Shirtcliffe N. Nickel U. Schneider S. J. Colloid Interface Sci. 1999;211(1):122–129. doi: 10.1006/jcis.1998.5980. [DOI] [PubMed] [Google Scholar]
- Bright R. M. Musick M. D. Natan M. J. Langmuir. 1998;14(20):5695–5701. doi: 10.1021/la980138j. [DOI] [Google Scholar]
- Jouyban A. Rahimpour E. Talanta. 2020;217:121071. doi: 10.1016/j.talanta.2020.121071. [DOI] [PubMed] [Google Scholar]
- Radziuk D. Shchukin D. G. Skirtach A. Möhwald H. Sukhorukov G. Langmuir. 2007;23(8):4612–4617. doi: 10.1021/la063420w. [DOI] [PubMed] [Google Scholar]
- Alothman Z. A. Bukhari N. Haider S. Wabaidur S. M. Alwarthan A. A. Arabian J. Chem. 2010;3(4):251–255. doi: 10.1016/j.arabjc.2010.06.008. [DOI] [Google Scholar]
- Malinsky M. D. Kelly K. L. Schatz G. C. Van Duyne R. P. J. Am. Chem. Soc. 2001;123(7):1471–1482. doi: 10.1021/ja003312a. [DOI] [Google Scholar]
- Riboh J. C. Haes A. J. McFarland A. D. Ranjit Yonzon C. Van Duyne R. P. J. Phys. Chem. B. 2003;107(8):1772–1780. doi: 10.1021/jp022130v. [DOI] [Google Scholar]
- Lakowicz J. R., Principles of fluorescence spectroscopy, Springer US, Boston, MA, 2006 [Google Scholar]
- Xu J. and Wang Z., Fluorimetric Analysis Method, Science Press, Beijing, 3rd edn, 2006 [Google Scholar]
- Bi S. Ding L. Tian Y. Song D. Zhou X. Liu X. Zhang H. J. Mol. Struct. 2004;703(1–3):37–45. doi: 10.1016/j.molstruc.2004.05.026. [DOI] [Google Scholar]
- Paul F., Klaus T., Richard L., Stephen F. and William R., Chemistry 2e, Rice University, 2019, OpenStax [Google Scholar]
- ICH Harmonised Tripartite Guidelines, Validation of analytical procedures: text and methodology Q2 (R1), https://www.ich.org/products/guidelines/quality/article/quality-guidelines.html, accessed, March, 2022
- Derayeaa S. M. Omara M. A. Hammada M. A. Hassanb Y. F. J. Appl. Pharm. Sci. 2017;7(2):016–024. [Google Scholar]
- Moffat A. C. , Osselton M. D. , Widdop B. and Watts J., Clarke's Analysis of Drugs and Poisons, Pharmaceutical Press, London, fourth edn, 2011 [Google Scholar]
- Keith L. H. Gron L. U. Young J. L. Chem. Rev. 2007;107(6):2695–2708. doi: 10.1021/cr068359e. [DOI] [PubMed] [Google Scholar]
- de La Guardia M. Ruzicka J. Analyst. 1995;120(2):17N. doi: 10.1039/AN995200017N. [DOI] [Google Scholar]
- de La Guardia M. Khalaf K. D. Carbonell V. Morales-Rubio A. Anal. Chim. Acta. 1995;308(1–3):462–468. doi: 10.1016/0003-2670(94)00625-V. [DOI] [Google Scholar]
- Płotka-Wasylka J. Talanta. 2018;181:204–209. doi: 10.1016/j.talanta.2018.01.013. [DOI] [PubMed] [Google Scholar]
- Pena-Pereira F. Wojnowski W. Tobiszewski M. Anal. Chem. 2020;92(14):10076–10082. doi: 10.1021/acs.analchem.0c01887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gałuszka A. Migaszewski Z. M. Konieczka P. Namieśnik J. TrAC, Trends Anal. Chem. 2012;37:61–72. doi: 10.1016/j.trac.2012.03.013. [DOI] [Google Scholar]
- Miller J. N. and Miller J. C., Statistics and Chemometrics for Analytical Chemistry, Prentic Hall, Hariow, England, fifth edn, 2005 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.