

Abstract
This review “teaches” researchers how to make their lackluster proteomics data look really impressive, by applying an inappropriate but pervasive strategy that selects features in a biased manner. The strategy is demonstrated and used to build a classification model with an accuracy of 92% and AUC of 0.98, while relying completely on random numbers for the dataset. This “lesson” in data processing is not to be practiced by anyone; on the contrary, it is meant to be a cautionary tale showing that very unreliable results are obtained when a biomarker panel is generated first, using all the available data, then tested by cross-validation. Data scientists describe the error committed in this scenario as having test data leak into the feature selection step, and it is currently a common mistake in proteomics biomarker studies that rely on machine learning. After the demonstration, advice is provided about how machine learning methods can be applied to proteomics data sets without generating artificially inflated accuracies.
Keywords: proteomics, biomarker, machine learning, feature selection, overfitting, AUC, classification, xgboost, validation
Graphical Abstract
Introduction:
Machine learning is entering many aspects of daily life: from Siri to spam filters, our days are becoming influenced by, and better because of, these computational tools. Not surprisingly, proteomics researchers are realizing the benefits of incorporating these methods into their workflows. The problem, though, is that if the approaches are implemented incorrectly, the results are not credible. To demonstrate how biased the reported accuracies can be after making just one common mistake, I committed a single error in feature selection (on purpose) while performing a machine learning study on simulated proteomics biomarker data using a set of completely random numbers. I was able to predict the disease state of my samples with >90% accuracy in a cross-validation experiment. (Random numbers should always produce a very poorly predictive model.) The outcome of this demonstration should not be remarkable to anyone who understands appropriate implementation of feature selection and cross-validation, but surprisingly, the majority of proteomics papers that use machine learning in biomarker discovery studies make the same mistake I commit here. Why are so many people making this mistake? Likely, most researchers have no idea how much this error can impact their results. So a demonstration is provided.
Amazing Machine Learning Results from Random Numbers
Figure 1 shows several analyses of a group of 40 samples, using a small (50 feature), medium (500 feature) and large (10,000 feature) dataset; a detailed explanation of all the experiments and a code example are provided in Supplemental Materials. Each feature in the data sets could be a different protein quantified from plasma in a quantitative proteomics experiment. In reality, the dataset was generated from 400,000 random numbers, installed into a 40 by 10,000 matrix. In the first demonstration experiment, the “best protein” (best feature) of the small, medium, or large data set is identified, by calculating the area under the receiver operator characteristic (ROC) curve, or the AUC, for each simulated protein. See the leftmost bars in Figure 1. Here, one can see that the more features in the data set, the better the AUC can be. The best feature in the biggest data set is able to discriminate all the samples with an AUC of 0.85. (A perfectly discriminating feature generates an AUC of 1.0, while one that is no better than random chance, such as assigning disease state by flipping a coin, would generate an AUC of 0.5.) This finding, that testing more features will naturally increase the likelihood of identifying highly discriminative ones, is a well-known phenomenon that has been studied by statisticians.1–3 While this principle should be understood by proteomics researchers, many researchers still do not understand what they can and cannot do with the highly discriminating features that were identified in datasets with limited numbers of samples. The next demonstration experiments show how using only the highly discriminating features can lead to highly biased accuracy estimates, when only the selected features are used to predict disease state.
Figure 1.
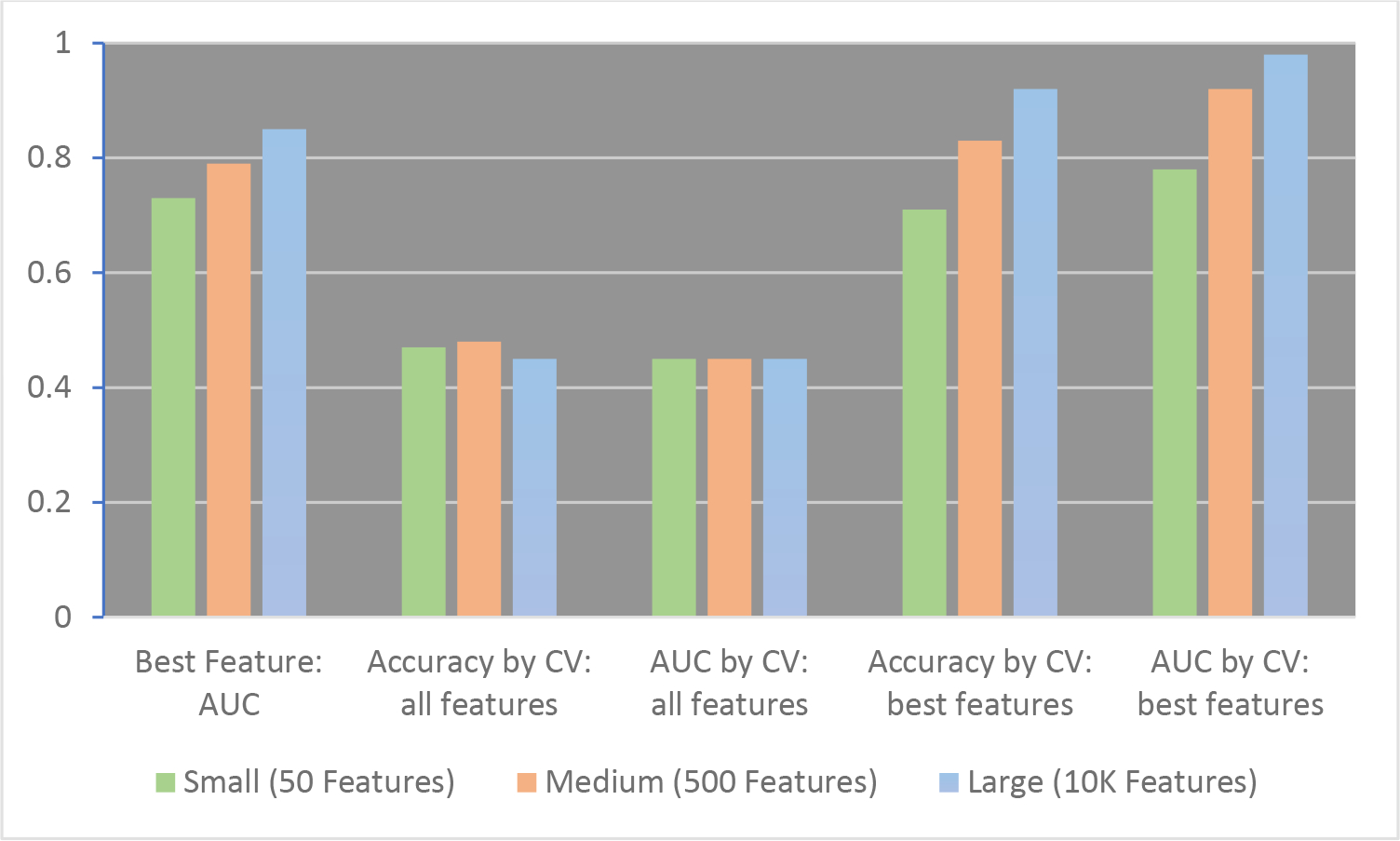
Assigning Disease State Using Radom Numbers: Accuracy and AUC Under Different Conditions. Left to right: The maximum AUC for a single feature in the small (green), medium (orange) and large (blue) data sets is shown (Best Feature AUC). Accuracy by CV and AUC by CV: When all features are used and a classification model is tested by cross-validation (CV), (second and third sets of bars), the accuracy and AUC of the model are ~50%, which is expected for a dataset of random numbers. Fourth and fifth sets of bars: When only the best features are used, both the Accuracy by CV (cross-validation) and the AUC by CV are inappropriately high. All values are generated by averaging results from 50 data sets of random numbers; full details provided in Supplemental Materials.
The most common type of machine learning studies in proteomics, supervised classification, uses multiple features to classify samples into two (or more) groups by first developing a model on known data and then applying it to data with an unknown group assignment. There are several different types of classifiers that can be used, and more detailed background information can be found in references 4 and 5. One critical aspect of supervised classification is that the model’s accuracy needs to be determined by test data, and not the data used to train the model. How is this best accomplished? Machine learning experts contend that the model should first be developed and optimized with one set of data (or a fraction of the original data set), then the accuracy of the model should be reported using only a second set of independent test data (or a fraction of the original data that had not been part of the machine learning optimization).1,2,4 Unfortunately, almost all proteomics experiments suffer from too-few samples to implement this approach, and the field has generally accepted cross-validation as a reasonable alternative, provided the original data set is not too small. (For example, fourteen samples has been reported to be too small.3) Genomics researchers faced with this same problem, of not having enough data for a test set, have implemented the same solution, of using cross-validation to estimate accuracy.6 In cross-validation, a subset of the samples is used to build the model, and the model’s accuracy is found by analyzing the remaining samples. Different combinations of training and test data can be generated so that each sample can be leveraged as a test sample. Reference 4 provides more detail.
Figure 1 also shows the accuracy and AUC generated using leave-one-out cross-validation and XGBoost (extreme gradient boosting) as the classifier, either using all the features in the model (second and third sets of bars), or using only the best features in the model (fourth and fifth sets of bars). In leave-one-out cross-validation, all the samples except the one being assigned are used to build the model; then the left-out sample is assigned; and this process is repeated for every sample in the data set. The Figure shows that this cross-validation approach can be an appropriate strategy to implement in machine learning studies of proteomics data, even for small numbers of samples, because applying it introduces virtually no bias, if it is applied appropriately (with no up-front feature selection). It does not matter if the number of features in the data set are 50, 500, or 10,000; using an XGBoost classifier and cross-validation, only a poorly predictive model is generated. While many different classifiers can be used in this kind of experiment, essentially the same result would have occurred with any of them. This outcome is what is supposed to happen during cross-validation. If there is no underlying trend in the data, the machine learning workflow should not manufacture one.
Unfortunately, “manufactured accuracy” can be, and often is, generated in proteomics experiments that implement machine learning by making a common mistake in the data science steps. If, instead of doing classification on the entire set of features, one had instead only used the features whose abundance changed the most between the two groups, while still applying the same rigorous-seeming cross-validation strategy, an entirely different result occurs. The fourth and fifth sets of bars in Figure 1 show what happens under those circumstances: Even though the same classifier (XGBoost) and strategy (leave-one-out cross-validation) were used, the model is now able to correctly “assign” the samples to the arbitrary classes that they were originally designated to, with up to 92% accuracy for the largest data set. The AUC is an astonishing 0.98 for this dataset. These ultra-high values demonstrate the severity of the problem caused by feature selection on the entire dataset: the model overfits the data, and these accuracies would never be obtainable on a fresh set of samples. (Full experimental details and a code example are in Supplemental Data.)
What matters more: the data used to select the features or the method of feature selection? The problem is using all the data – leaking the test data into the training set – not the feature selection method, which, in this case, was using the features with highest information gain. (This approach was chosen because it is easy to implement using the XGBoost algorithm, and it is becoming prevalent in the literature.) Other feature selection strategies would have led to the same results, inflated accuracies, had all the data been used to select the features. In fact, a similar demonstration of this principle was provided in 2003 to genomics researchers: random numbers were used to generate a highly “accurate” and predictive model, after test data were included in the feature selection step.6 In that case, XGBoost did not yet exist, and a completely different approach for feature selection and classification, using a linear regression model, was employed, but the same outcome as shown here prevailed; the arbitrary class assignments were very accurately predicted.6 A further example is described in reference 7, where SVM (Support Vector Machine) was the classifier and a recursive feature selection algorithm was used: again, starting with random numbers and arbitrary class assignments, then using all the samples for feature selection, a highly predictive model was built, even though the researchers were using random numbers in the data set. The error rate for the model was 2.5%, showing severe overfitting.7 A 50% error rate, which would be expected for random numbers, was obtained when the experiment was set up properly, by leaving the test data out of the feature selection step.7 When researchers use all their data, including their test data, to select the features up front, they are biasing the outcome of the study to find a difference in the samples, even if one does not actually exist.
It is easy to understand why proteomics researchers would fall into this trap. First, their sample sets are often too small to cut out a reasonable-sized test set. But, more important than that, the idea of down-selecting the proteins resonates so well with common sense: Of course most of the proteins in the data set are not going to change due to the biological condition of interest, so it might seem that it would be better to leave most of them out. Also, the end-goal of these kinds of experiments is typically to find a limited biomarker panel that can indicate the disease state; so again, logic may lead someone to want to use all the data for feature selection to identify the few proteins they want to keep in the panel, then to apply a rigorous test method like cross-validation to assess the panel’s accuracy. It sounds like a great idea; except it is not. By leveraging all the data into the feature selection step, the entire experiment becomes flawed, and one can easily generate a model that produces any arbitrary assignments desired with “92% accuracy”, just as is demonstrated in Figure 1.
Improving data science standards
The feature selection mistake described above is a well-known problem infiltrating machine learning studies; it is formally called “leaking test data into the training set.” In a recent metanalysis of over 100 machine learning publications focusing on the gut microbiome, 80% of the studies inappropriately included their test data in the feature selection step of the workflow.8 The problem is so prevalent, researchers are probably learning to do this, simply by following the example of others. And when the pervasiveness of this mistake is coupled with the enormously inflated accuracies the mistake introduces, as shown in Figure 1, this mistake could very likely be the single most problematic machine learning implementation error in proteomics today.
Journal editors must become judicious about preventing the publication of manuscripts where there is a leakage of test data into the machine learning workflow. Feature selection is a legitimate machine learning practice, when its accuracy is measured using data that is independent of the selection process. See references 7 and 9 for examples. However, when the same data to test the strategy are the ones that were used for feature selection in the first place, that is not scientifically justified, and it leads to enormous accuracy inflation, as shown in Figure 1.
The community needs to agree on acceptable alternatives to this problematic practice. Simply requiring additional data held out as a test set for every study is not feasible in certain circumstances. Some sample sets are too small and too hard to get to split into a training group and test group; examples include studies that require amniotic fluid, CSF, brain samples, samples assessing rare diseases, or samples from a racial subgroup. All those are important sample sets. Yet impossible-to-get sample sets cannot justify non-rigorous science. One route forward in these studies is to leave out the machine learning entirely and focus on other aspects of the work. Alternatively, editors and reviewers might refer authors to example studies where researchers still managed to leverage data effectively to offer validation for their findings, even though they were working with very precious samples.10,11 A third possibility would be that journals simply invoke a policy that states authors cannot do feature selection using the test data and let authors decide how to proceed. Many options are available for authors who study small data sets and want to publish on the outcomes while accurately reflecting what can be inferred from the limited data, but those options are not typically the routes chosen now.
My best suggestion to journal editors, in dealing with researchers who do not have enough samples for a test set because they are studying something rare or hard to get, is to take a middle-ground position: tell the authors to limit the types of machine learning done prior to cross-validation (i.e. no feature selection) and to show how much of an improvement is made versus randomly assigned samples undergoing the same workflow (a permutation study; see below). This opinion is not universally endorsed: statistical purists would argue that a separate test set, beyond cross-validation, is always necessary for a truly meaningful evaluation of accuracy.1,2 Cross-validation can provide reasonably accurate results under the right conditions though, as demonstrated above, and this proposed approach is a much better route forward than what is being done now: it allows users to publish on modestly-sized sample sets, provided the machine learning models are not optimized in ways that limit the utility of cross-validation: To keep the bias at a minimum, the full feature set should be used, and common hyperparameters should be used without optimization. That is what I did in the first cross-validation experiment, where the models showed no predictive accuracy. This type of experiment, machine learning without first reducing the feature set, would demonstrate whether the sample type and methods are useful for discriminating the classes studied (health vs disease, for example). My group used this approach on small proteomics data sets with samples originating from brain and CSF; as expected, Alzheimer’s Disease was easily discriminated using data from the brain samples, even when using the full set of >12,000 protein features; CSF also showed utility for discriminating the disease state, albeit at lower accuracies than the brain samples.12 These types of machine learning studies, on data sets with modest sample numbers but lots of features, offer preliminary justification for doing future studies with larger sample sets, where a biomarker panel could be generated using valid feature selection methods and accuracy assessments based on a true test set.
Editors and reviewers could also suggest that a permutation study be done.13 To do this, the original samples are randomly assigned to either the “disease” or “control” groups, and the samples are reclassified based on their new (and meaningless) group assignments. The permutation can be done a bunch of times, with the average accuracy reported. This experiment allows readers to assess the extent to which the accuracy reported is related to the biological condition versus overfitting due to the machine learning strategy. And the beauty of it is that it requires no extra data, so it is a perfect strategy for small sample sets.
Concluding Remarks
Researchers will do better science, and be better reviewers and editors, if they see which data analysis strategies provide reasonable results, even on small sample sets, and which ones cause highly inflated accuracies. Leaking test data into the feature selection step, is a significant problem, and our community should stop doing this. We can reward rigorous studies and stop publishing reports of “biomarker panels” that claim to offer ultra-high predictability, when the same results could have been generated with sets of random numbers.
Supplementary Material
Acknowledgements:
This work was supported by NIH grants R35GM130354 and RF1AG072760 (to HD).
Footnotes
Supporting Information: Additional experimental details to generate the demonstration data in Figure 1; a code example is also provided.
References:
- 1.Broadhurst DI; Kell DB. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2,171–196. [Google Scholar]
- 2.Ransohoff DF. Rules of evidence for cancer molecular-marker discovery and validation. Nature Reviews Cancer 2004, 4, 309–314. [DOI] [PubMed] [Google Scholar]
- 3.Dakna M; Harris K; Kalousis A; Carpentier S; Kolch W; Schanstra JP; Haubitz M; Vlahou A; Mischak H; Girolami M Addressing the Challenge of Defining Valid Proteomic Biomarkers and Classifiers. BMC Bioinformatics, 2010, 11, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chicco D Ten quick tips for machine learning in computational biology. Biodata Mining 2017, 10, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liebal UW; Phan ANT; Sudhakar M; Raman K; Blank LM. Machine Learning Applications for Mass Spectrometry-Based Metabolomics. Metabolites, 2020, 10, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simon R; Radmacher MD; Dobbin K; McShane LM. Pittfalls in the Use of DNA Microarray Data for Diagnostic and Prognostic Classification. J. Natl. Cancer Inst. 2003, 95, 14–18. [DOI] [PubMed] [Google Scholar]
- 7.Zhang XG; Lu X; Shi Q; Xu XQ; Leung HCE; Harris LN; D Iglehart J; Miron A; Liu JS; Wong WH. Recursive SVM feature selection and sample classification for mass-spectrometry and microarray data. BMC Bioinformatics, 2006,7,197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quinn TP. Stool Studies Don’t Pass the Sniff Test: A Systematic Review of Human Gut Microbiome Research Suggests Widespread Misuse of Machine Learning. arXiv [q-bio.GN], July 8, 2021, 2107.03611. https://arxiv.org/abs/2107.03611 (accessed Feb 26, 2022). [Google Scholar]
- 9.Liu QZ; Sung AH; Qiao MY; Chen ZX; Yang JY; Yang MQ; Huang XD; Deng YP. Comparison of feature selection and classification for MALDI-MS data. BMC Genomics. 2009, 10, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein O; Kanter F; Kulbe H; Jank P; Denkert C; Nebrich G; Schmitt WD; Wu ZY; Kunze CA; Sehouli J; Darb-Esfahani S; Braicu I; Lellmann J; Thiele H; Taube ET. MALDI-Imaging for Classification of Epithelial Ovarian Cancer Histotypes from a Tissue Microarray Using Machine Learning Methods. Proteom. Clin. App., 2019, 13(1), 1700181. [DOI] [PubMed] [Google Scholar]
- 11.Desaire H; Stepler KE; Robinson RAS. Exposing the Brain Proteomic Signatures of Alzheimer’s Disease in Diverse Racial Groups: Leveraging Multiple Datasets and Machine Learning. J. Proteome Resch. 2022. 21, 4, 1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hua D; Desaire H Improved Discrimination of Disease States Using Proteomics Data with the Updated Aristotle Classifier. J. Proteome Resch. 2021, 20(5) 2823–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Good P Permutation, Parametric, and Bootstrap Tests of Hypotheses. Third Ed. Springer; New York, NY, 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.