

Abstract
Trimethylamine N-oxide (TMAO) is a gut microbiota metabolite derived from trimethylamine-containing nutrient precursors such as choline, L-carnitine, and betaine, which are rich in many vegetables, fruits, nuts, dairy products, and meats. An increasing number of clinical studies have demonstrated a strong relationship between elevated plasma TMAO levels and adverse cardiovascular events. It is commonly agreed that TMAO acts as both an independent risk factor and a prognostic index for patients with cardiovascular disease. Although most animal (mainly rodent) data support the clinical findings, the mechanisms by which TMAO modulates the cardiovascular system are still not well understood. In this context, we provide an overview of the potential mechanisms underlying TMAO-induced cardiovascular disease at the cellular and molecular levels, with a focus on atherosclerosis. We also address the direct effects of TMAO on cardiomyocytes (a new and under-researched area) and finally propose TMAO as a potential biomarker and/or therapeutic target for diagnosis and treatment of patients with cardiovascular disease.
Keywords: Metabolites, Trimethylamine N-oxide, Atherosclerosis, Cardiomyopathy, Cardiomyocytes, Signalling pathway, Mechanism
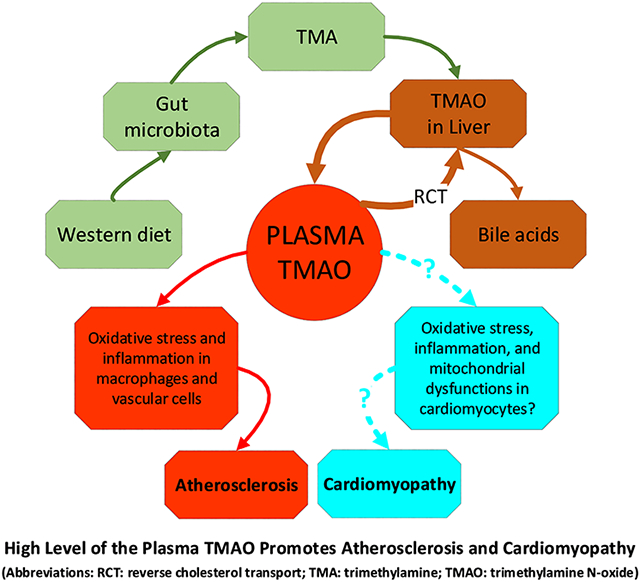
Graphical Abstract
INTRODUCTION
Despite remarkable progress in drug treatment and interventional therapy in recent years, cardiovascular diseases (CVDs), such as coronary artery disease, heart failure, dilated cardiomyopathy, peripheral artery disease, and stroke, still remain the leading cause of both morbidity and mortality worldwide [1]. Meanwhile, the increasing incidence of metabolic disorders (e.g. obesity and Type 2 diabetes) and an aging population further escalate the prevalence of CVDs [2].
Over the past 10 years, substantial research has focussed on gut microbiota-host interactions (e.g. with the brain, liver, kidneys and heart) [3-7]. Emerging evidence demonstrated that the gut microbiota plays a crucial role in the development of metabolic-associated disorders, including CVDs [5,7-9]. Gut microbiota can directly and indirectly affect cardiovascular outcomes through modulation of gut barrier integrity, host immunity, and gut microbiota-derived metabolites [7,9-11]. Because of technical advances in the field of metabolomics, human plasma metabolites were analysed leading to the identification of several disease-relevant gut microbiota metabolites such as trimethylamine-N-oxide (TMAO) [3,12].
TMAO, as an intermediate metabolite, directly links gut microbial activities to the pathogenesis of various metabolic disorders [3,12]. In the case of CVDs, the majority of published work, especially review articles, addresses the broad clinical impacts and outcomes of TMAO on cardiology and vascular pathobiology, as well as interactions among the gut microbiota, gut microbial metabolism, immune responses, lipid metabolism, and the cardiovascular system [6,7,9,13,14]. Currently, the central topic of research in the field is related to how gut-microbiota-associated TMAO mediates atherosclerotic progression [15]. However, whether TMAO has direct acute and/or chronic effects on cardiomyocytes is not well studied and review articles reporting on TMAO and cardiomyocytes seem not to have been published.
In this mini-review, we focus on the current progress of how TMAO contributes to the pathogenesis of atherosclerosis (i.e. endothelial cells) and cardiomyopathy (i.e. cardiomyocytes) at the cellular and molecular levels, while the impact of TMAO on the cardiovascular system has been described [16-18].
TMAO ORIGIN AND METABOLISM
Trimethylamine (TMA), a precursor of TMAO, is plentiful in deep-sea fish and milk [19] and it can be metabolically produced in the gut and vagina by endogenous microbiota [20]. In the gut of mammals, L-carnitine, choline or betaine from foods (e.g. red meat, eggs and fish) are enzymatically converted into TMA by TMA lyase from gut bacteria [e.g. a class of bacteria from the Phylum Firmicutes] [21]. TMA is then absorbed into the bloodstream via intestinal epithelial cells before being transported into the liver, where it is further processed into TMAO by the liver enzyme flavin-dependent mono oxygenase 3 (FMO3) [22]. Over 95% of TMA that is oxidized into TMAO by FMO3 in the liver is then rapidly cleared into the urine [23]. On the other hand, a lack of, or diminished, FMO3 activity leads to the accumulation of TMA in the blood, resulting in a disease called “fish odor syndrome” (also known as trimethylaminuria) [24]. Patients with FMO3 mutation(s) show a reduction of FMO3 expression, which is accompanied with excessive excretion of TMA in sweat, urine, and breath, causing patients to present with the odor of rotting fish [24]. Therefore, changes to any components along the TMAO metabolic pathway (from gut microbiota, food composition, the liver FMO3 expression/activity, to kidney function) could affect plasma TMAO levels and the potential for subsequent complications, such as atherosclerosis [17].
PATHOGENIC MECHANISMS OF TMAO-INDUCED CVDS
The correlation between TMAO and CVDs was first reported in patients with CVD in 2011 [12]. In this study, Wang et al. analysed the metabolic profiles of plasma collected from patients with CVD and age-gender-matched healthy controls using Mass Spectrometric techniques. Of the over 2000 analytes screened, TMAO was found to be highly correlated with CVD and could thus be proposed as a biomarker for CVD [12]. In a study with a germ-free animal model, the same group found that the level of TMAO in the bloodstream was highly dependent on dietary choline uptake and the existence of gut microbiota [12]. Importantly, supplementation of choline or TMAO in the diet significantly promoted atherosclerosis in a mouse model of atherosclerosis in a gut-microbiota dependent manner, demonstrating a direct correlation between the gut microbiota-associated TMAO and vascular pathogenesis [12]. Since the initial discovery of TMAO, multiple clinical studies (see Table 1) have been conducted and the overall results support the conclusion that an increased plasma level of TMAO is directly proportional to the risk of a cardiovascular event and to the prognosis of a patient with CVD [25-30].
Table 1.
In vivo clinical studies of the role of TMAO on CVDs.
| Authors | Study Design | Study Subjects | Conclusion |
|---|---|---|---|
| Tang, WH et al. (2013)[25] | Prospective | 4007 patients undergoing elective coronary angiography | TMAO is an independent risk factor of major adverse cardiovascular events. |
| Tang, WH et al. (2014)[26] | Prospective cohort | 720 patients with stable HF | HF patients showed high TMAO levels and elevated TMAO indicates a higher long-term mortality risk |
| Troseid, M et al. (2015)[27] | Prospective | 155 patients with CHF;100 patients with stable CAD | TMAO levels were increased in HF patients and associated with disease severity and survival |
| Haissman, JM et al. (2015)[28] | Cross section Cohort/nested case control | 105 asymptomatic HIV-infected persons/55 HIV-infected persons with confirmed first-time MI & 182 HIV infected controls | TMAO is elevated in silent ischaemia but is not associated with first-time MI in HIV patients |
| Mafune, A et al. (2016)[29] | Cross-section | 227 patients who underwent cardiovascular surgery | The number of infarcted coronary arteries in patients undergoing cardiovascular surgery are related to the serum TMAO level |
| Suzuki, T et al. (2017)[30] | Prospective | 1079 acute MI patients | TMAO is an independent prognostic factor for patients hospitalized due to acute MI |
Abbreviations: CAD: coronary artery disease; CHF: congestive heart failure; CM: cardiomyocytes; CVDs: cardiovascular diseases; HF: heart failure; HIV: human immunodeficiency virus; MI: myocardial infarction; TMAO: Trimethylamine N-oxide.
TMAO-associated Atherosclerosis
Atherosclerosis is a vascular (often referred to arterial) disease manifested as a narrowing of the lumen and a thick, hardening of the vessel wall caused by inflammatory plaques comprised of cholesterol, fatty cells, and inflammatory cells [31]. Although endothelial dysfunction, lipid deposits, migration of inflammatory cells, formation of foam cells, and degradation of the extracellular matrix are all believed to be involved in atherogenesis, the details about this mechanism, especially regarding TMAO-associated pathways, are not completely understood [32].
As a recently-discovered biomarker and mediator of metabolic disorders, TMAO has been found to play an important role in mediating atherogenesis based on human clinical observations and pre-clinical animal studies. Five proatherosclerotic mechanisms that are potentially modulated by TMAO have been proposed (Fig.1):
Fig. (1).

Potential metabolic and signalling pathways underlying TMAO-induced atherosclerotic coronary artery and cardiomyopathy. The proposed mechanisms are mostly extracted from studies of macrophages, ECs and VSMCs at both in vitro and in vivo levels, while it is largely unknown how TMAO directly modulates EPCs and cardiomyocytes.
1) The first pathway describes that TMAO-induced atherosclerosis is caused by enhanced migration of macrophages and the formation of foam cells in plaque [4,32]). Park et al. found that both mRNA and protein expression of CD36 and scavenger receptor A (SR-A), two cell surface receptors on macrophages that are involved in atherogenesis, increased in mice supplemented with either choline or TMAO in a microbiota-dependent manner [33]. Additionally, the formation of cholesterol-laden macrophage foam cells, one of the earliest cellular markers of atherosclerosis, was also significantly escalated in mice fed with a choline supplemented diet [12, 34]. Other studies found that TMAO could induce cellular stress in murine macrophages (J774A cell line) by increasing expression of the stress-promoted heat shock protein 70kDa alpha (HSP70) and heat shock protein 90kDa beta member 1 (HSP90B1), in a time- and dose-dependent manner, suggesting that TMAO may promote foam cell formation via activation of macrophages [35].
2) The second potential pathway involves modulation of cholesterol metabolism, by inhibiting the reverse cholesterol transport (RCT) system and reducing biliary excretion of cholesterol [36]. The RCT is a process involving blood transporting cholesterol from peripheral tissues back to the liver for biliary excretion into the gastrointestinal system, thus maintaining homeostasis of blood cholesterol [37]. However, how TMAO affects RCT transportation remains largely unknown. Koeth et al. found that ApoE−/− mice fed with a diet containing TMAO showed a 35% decrease in RCT compared with controls [36]. On the other hand, an in vitro study of macrophages loaded with cholesterol did not show significant changes of expression in low-density-lipoprotein (LDL) receptor and cholesterol synthesis enzymes under a physiological concentration of TMAO [36], implying that TMAO-induced atherogenesis may target other components in the RCT system or that TMAO at the physiological level is unable to alter RCT transportation capacity in healthy individuals [38].
Furthermore, TMAO could increase blood cholesterol levels by reducing biliary excretion [39]. Previous studies showed that downregulation of the cytochrome P450 family 7, subfamily a member 1 (Cyp7a1, a gene coding the enzyme “cholesterol 7α-hydroxylase” that catalyses the initial step in cholesterol catabolism and bile acid synthesis) leads to decreased bile acid synthesis and bile acid secretion, thus, reducing excretion of excess cholesterol and facilitating atherosclerosis [40, 41]. Ding et al. reported that administration of TMAO to ApoE−/− mice accelerated aortic lesion formation and decreased bile acid synthesis in the liver through down-regulation of hepatic Cyp7a1 gene expression [42, 43]. Another key bile acid synthetic enzyme, cytochrome P450 family 27, subfamily a polypeptide 1 (Cyp27a1) was also significantly decreased in the liver tissue of mice supplemented with TMAO [36], suggesting that both Cyp7a1 and Cyp27a1 genes may be involved in TMAO-promoted atherogenesis. However, exactly how TMAO modulates RCT is not fully understood [39].
3) The third pathway proposes that TMAO-associated atherosclerosis is due to dysfunctional endothelial cells under chronic inflammation and oxidative stress, especially in the early stage of atherosclerosis [31,44]. This hypothesis is supported by in vitro and in vivo studies. Seldin et al. found that atherosclerosis-prone LDLR−/− mice fed with a choline diet for 3 weeks, or after acute injection of TMAO at the physiological level, developed a significant phenotype of vascular inflammation in vivo [45]. The in vitro experiments by the same group using human aortic endothelial cells (HAECs) and human vascular smooth muscle cells (VSMCs) indicated that TMAO-induced inflammation responses in HAECs and VSMCs were mediated by mitogen-activated protein kinase (MAPK) and nuclear factor-ƙB (NF-κB) signalling. In addition, TMAO was also found to enhance recruitment of leukocytes to endothelial cells, which is likely triggered by a G-protein-coupled receptor (GPCR) [45]. Moreover, another study revealed that TMAO activates NF-κB in human umbilical vein endothelial cells (HUVECs) and leads to increased expression of vascular cell adhesion molecule-1 (VCAM-1). TMAO treatment significantly inhibited the proliferation and migration of HUVECs but stimulated monocyte adherence on HUVECs, implying that the self-repair capacity of HUVECs was impaired by TMAO [46]. Finally, protein kinase C (PKC) activity, which plays a crucial role in mediating endothelial dysfunction [47], was found to significantly increase in a dose dependent manner after TMAO treatment, suggesting that the activation of PCK may partially participate in TMAO-induced endothelial dysfunction [46].
4) The fourth likely pathway presumes that TMAO could induce atherosclerosis by activating the nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome in endothelial cells. Activation of NLRP3 triggers the sirtuin-3 (SIRT3) > superoxide dismutase 2 (SOD2) > mitochondrial reactive oxygen species (mtROS) signalling pathway, leading to ROS production [48,49]. Oxidative stress is known to cause cell damage and has been implicated in various diseases, including atherosclerosis [50]. Under normal conditions, activation of the NLRP3 inflammasome triggers inflammatory responses and production of pro-inflammatory cytokines while activations of SIRT3 and SOD2 have the opposite effects by reducing oxidative stress-induced cellular damage [48,49].
Studies have found that TMAO probably facilitates atherosclerosis by simultaneously affecting all 3 critical components in the pathways described above. Although the mechanism(s) is/are not completely known, TMAO increases both ROS and mtROS generation, which then activates the NLRP3 inflammasome, leading to the production of pro-inflammatory cytokines [48,49,51]. Meanwhile, by inhibiting both SIRT3 and SOD2 activity, TMAO increases ROS production, thus, exacerbating the pro-inflammatory effects of the NLRP3 inflammasome [48,49,51]. The finding that the over-expression of SIRT3 protects endothelial cells from TMAO-induced dysfunction support the above hypothesis [49,52].
Several other TMAO > ROS-associated pathways have also been proposed [51,53,54]. By increasing oxidative stress in endothelial cells, TMAO impairs endothelial nitric-oxide synthase (eNOS) and the bioavailability of nitric oxide (NO), eventually leading to endothelial dysfunction [53]. TMAO can also induce inflammation and endothelial dysfunction through activating ROS/TXNIP/NLRP3 inflammasome signalling in HUVECs [51]. Furthermore, by downregulating SIRT1 expression, TMAO facilitated oxidative stress via stimulating the p53 > p21 > Rb pathway, which, in turn, led to endothelial cell senescence and endothelial dysfunction [54]. Collectively, these findings demonstrate that oxidative stress and ROS are involved in TMAO-associated atherosclerosis.
5) The fifth, but possibly not the last, signalling pathway that mediates TMAO-related cardiovascular disorders operates by suppressing proliferation and differentiation of endothelial progenitor cell (EPC) [55]. Blood-circulating EPCs, which originate from the bone marrow, are recognized for repairing or regenerating the damaged endothelium following vascular injury [56]. Studies showed that a decreased number of EPCs were associated with endothelial dysfunction and atherosclerosis [57,58]. An investigation of 81 patients with stable angina reported that the level of plasma TMAO is proportional to the level of plasma inflammatory markers (e.g. high-sensitivity C-reactive protein, hsCRP), but inversely proportional to the level of circulating EPCs [55]. In vitro studies using cultured human EPCs have found that high-dose TMAO treatments (200 and 500 μM for 24 h) significantly increase levels of pro-inflammatory factors (e.g. interleukin-6 “IL-6”, CRP and TNF-α) and ROS generation, but decrease NO production and impaired capillary Tube formation [55], indicating that TMAO modulates numerous signalling molecules in both differentiated somatic cells and stem cells.
TMAO-associated cardiomyopathy
As described above, existing studies of TMAO-associated cardiovascular events have mainly focused on vascular cells (i.e. endothelial and vascular smooth muscle cells), while the direct effects of TMAO on cardiomyocytes are largely disregarded. To date, only a few studies that investigated TMAO’s impact on isolated cardiomyocytes or excised heart were published (see Table 2).
Table 2.
In vitro and ex vivo effects of TMAO on CMs.
| Authors | Targets | Experiment types |
Cell types | Phases | TMAO concentrations and exposure duration |
|---|---|---|---|---|---|
| Li, T et al. (2018)[53] | Hypertrophy & fibrosis | In vitro | Isolated rat CMs | Acute | 5 μM for 18 h |
| Makrecka-Kuka, M et al. (2017)[60] | Mitochondria | In vitro and in vivo | Isolated mouse CMs | Acute & chronic | 20 μM for 1 h or 8 weeks |
| Savi, M et al. (2018)[59] | Contraction | In vitro | Isolated rat ventricular CMs | Acute | 20 μM for 2 h |
| Oakley, C et al. (2020)[61] | Contraction | Ex vivo and in vitro | Excised mouse hearts, human heart tissue, and E18 rat CMs | Acute | 300 and 3000 μM for 30 min |
Abbreviations: CMs: cardiomyocytes; E18 rat: E18 Sprague Dawley rat; TMAO: trimethylamine N-oxide.
Savi et al. found that TMAO treatment (20 μM for 2 h) impairs cardiomyocyte contractility and intracellular Ca2+ cycling of isolated rat cardiomyocytes, independent of TMAO concentration. TMAO also increases the number of mitochondria and promotes glycogen accumulation and lipofuscin-like pigmentation deposits in cardiomyocytes [59]. A similar study using isolated mouse cardiomyocytes revealed that both acute (1h in isolated cells) and chronic (8 week administration in mice) exposure to 20 μM TMAO disrupted oxidative cycling of pyruvate and fatty acids in cardiac mitochondria, leading to disturbances of energy metabolism in the heart [60].
Interestingly, an ex vivo study using Langendorff-perfused mouse heart found that high concentrations of TMAO (300 and 3000 μM for 30 min) increases the force and rate of cardiac contractility, which, in turn, could lead to left ventricular hypertrophy, cardiac remodelling, and eventually heart failure [61]. TMAO-associated cardiac hypertrophy was also observed in a rat cardiac hypertrophy model induced by transverse aortic constriction (TAC). In this study, the blood levels of TMAO in TAC groups were significantly increased 6 weeks after surgery compared with control animals without TAC. In isolated cardiomyocytes, TMAO treatment (5 μM for 18 h) increased cardiomyocyte size and upregulated hypertrophic markers (e.g. β-myosin heavy chain) [62].
In a Western diet-induced (rich in choline, a TMA precursor) mouse obese model, the associated cardiac inflammation and fibrosis, as well as the elevated concentration of plasma TMAO, could be significantly reduced or prevented by administrating TMA inhibitor (3,3-Dimethyl-1-butanol “DMB”), further confirming TMAO-associated cardiomyopathy.
Taken together, these findings demonstrate that pathological concentrations of TMAO can promote dysfunction of both vascular cells and cardiomyocytes via multiple signalling pathways, eventually leading to the development of atherosclerosis (coronary artery disease, peripheral artery diseases), cardiomyopathy (hypertrophy, fibrosis), and heart failure. A more complete understanding of the mechanisms underlying TMAO-associated CVDs will facilitate new drug discovery and treatments for patients with CVD.
Potential therapeutic targets of the TMAO metabolic pathway for CVD therapy
The strongly positive correlation between the plasma level of TMAO and CVD risks/outcomes reported in clinical studies suggests that modulations of the TMAO metabolic pathway (via drug or gene editing) may lead to new therapeutic approaches to prevent or treat CVDs that are promoted by TMAO [18, 27]. It is likely that any major component along the TMAO metabolic pathway could be a potential therapeutic target for treatment of TMAO-induced disorders [63]. For example, the plasma level of TMAO can be controlled by reduced intake of carnitine-/choline/betaine-rich foods [16]. Clinical and animal studies have shown that a high-fat or Western diet is highly related with a significantly increased plasma TMAO level, while a vegetarian diet supplemented with pistachios decreased TMA production and led to reduced TMAO levels [63]. This is in accordance with research showing that the Mediterranean diet (plant-based foods) is associated with better cardiovascular health outcomes [64].
Modification of the gut microbiota composition and their synthesis pathways could be another way to control the plasma level of TMAO [5]. Use of probiotics, such as Lactobacillus and Bifidobacterium, has shown beneficial effects on atherosclerosis [65]. Interestingly, a mouse study found that probiotics (Bifidobacterium animal subsp., Lactis F1-3-2) reduced plasma TMAO levels and improved lipid metabolism [66], but human clinical trials failed to show a correlation between the plasma level of TMAO and probiotic supplementation [13,67]. This dissociation may be due to short-term probiotic administration (4 weeks [67] vs 3 months [13]), the complexity of the gut environment in each individual, or diverse inclusion criteria. On the other hand, suppression of microbiota enzymes could also be used therapeutically. For example, inhibition of the gut microbiota CntA/B and Cut C/D enzymes stopped conversion of choline and carnitine to TMA [26], while inhibition of gut microbiota TMA lyase with DMB (a choline analogue) significantly decreased the plasma level of TMAO in both mice and rats [68].
Other components along the TMAO metabolic pathway can also be therapeutic targets but come with potential limitations. For example, down-regulating FMO3 activity and expression in the liver could, for example, significantly decrease the plasma level of TMAO [69]; however, FMO3 is important for morphine, propranolol, and tyramine metabolism [4] and inhibition of FMO3 could lead to excessive production of TMA, causing "fish odor syndrome".
Taken together, the current findings from both human clinical trials and pre-clinical animal studies indicate that multiple potential targets along the TMAO metabolic pathway can be modulated to decrease the plasma level of TMAO, including, but not limited to, intake of a Western diet, inhibition of gut microbiota composition and enzymes, and possible interventions of critical signaling components in the cells and tissues shown in Fig. 1. Further investigation using both animal models and patients are needed to fully understand the mechanisms underlying TMAO-associated CVDs and the development of drugs to effectively treat CVD without significant side effects [70].
CONCLUSIONS
Existing studies based on clinical trials, in vivo animal studies, ex vivo organ perfusion, and in vitro cell cultures, have demonstrated that multiple distinct pathways strongly link TMAO to cardiovascular diseases, especially atherosclerosis and cardiomyopathy (a new and under-researched area). TMAO appears to chronically modulate the cardiovascular system via altering metabolic, inflammatory, ROS signalling, and cellular apoptosis. By targeting endothelial cells, vascular smooth muscle cells, cardiomyocytes, and inflammatory macrophages/foam cells, TMAO promotes atherogenesis and cardiomyopathy. Although significant advances have recently been made in our understanding of these signalling pathways, the detailed mechanisms underlying TMAO-associated pathogenesis largely remain unanswered. The role of TMAO in CVD risk and the subsequent evaluation of TMA inhibitors support the concept that TMAO could potentially serve as a biomarker for CVDs, while the TMAO metabolic pathway may become potential therapeutic targets to treat CVDs.
ACKNOWLEDGEMENT
We thank Dr. Janet Webster and Ms Annika Nelson for providing a review of the manuscript. This work was supported by an NIH grant (1R15HL140528-01 for JQH), a One-Health seed grant (PJ6SPVHJ for JQH) from the College of Veterinary Medicine at Virginia Tech and the Edward Via College of Osteopathic Medicine, the Interdisciplinary Graduate Education Program in Regenerative Medicine (IGEP-RM, for YJZ), and the IRC Seed Grant (#178391 for JQH) from the College of Veterinary Medicine at Virginia Tech. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations:
- BA
Bile acids
- Cyp7a1/27a1
cytochrome P450 family 7, subfamily a member 1/ cytochrome P450 family 27, subfamily a polypeptide 1
- CD36
cluster of differentiation 36 (also known as platelet glycoprotein 4, fatty acid translocase, scavenger receptor class B member 3, and glycoproteins 88, IIIb, or IV)
- ECs
endothelial cells
- eNOS
endothelial nitric oxide synthase
- EPCs
endothelial progenitor cells
- FMO3
flavin-containing monooxygenase 3
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor kappa B
- NLRP3
NOD-, LRR- and pyrin domain-containing protein 3
- NO
nitric oxide
- p21/53
tumour protein 21/53
- Rb
retinoblastoma protein
- ROS
reactive oxygen species
- RCT
reverse cholesterol transport
- SIRT1/3
sirtuin 1/3
- SR-A1
scavenger receptors type 1
- SOD2
superoxide dismutase 2
- VSMCs
vascular smooth muscle cells
- TMA
trimethylamine
- TMAO
trimethylamine N-oxide
- TXNIP
thioredoxin interacting protein
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- [1].Benjamin EJ, Muntner P, Alonso A, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019;139:e56–e528. [DOI] [PubMed] [Google Scholar]
- [2].Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation 2011;123:933–44. [DOI] [PubMed] [Google Scholar]
- [3].Dumas ME, Kinross J, Nicholson JK. Metabolic phenotyping and systems biology approaches to understanding metabolic syndrome and fatty liver disease. Gastroenterology 2014;146:46–62. [DOI] [PubMed] [Google Scholar]
- [4].Zeisel SH, Warrier M. Trimethylamine N-Oxide, the Microbiome, and Heart and Kidney Disease. Annu Rev Nutr 2017;37:157–81. [DOI] [PubMed] [Google Scholar]
- [5].Anbazhagan AN, Priyamvada S, Priyadarshini M. Gut Microbiota in Vascular Disease: Therapeutic Target? Curr Vasc Pharmacol 2017;15:291–5. [DOI] [PubMed] [Google Scholar]
- [6].Nam HS. Gut Microbiota and Ischemic Stroke: The Role of Trimethylamine N-Oxide. J Stroke 2019;21:151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hosseinkhani F, Heinken A, Thiele I, et al. The contribution of gut bacterial metabolites in the human immune signaling pathway of non-communicable diseases. Gut Microbes 2021;13:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ussher JR, Lopaschuk GD, Arduini A. Gut microbiota metabolism of L-carnitine and cardiovascular risk. Atherosclerosis 2013;231:456–61. [DOI] [PubMed] [Google Scholar]
- [9].Battson ML, Lee DM, Weir TL, et al. The gut microbiota as a novel regulator of cardiovascular function and disease. J Nutr Biochem 2018;56:1–15. [DOI] [PubMed] [Google Scholar]
- [10].Burcelin R, Serino M, Chabo C, et al. Gut microbiota and diabetes: from pathogenesis to therapeutic perspective. Acta Diabetol 2011;48:257–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhou W, Cheng Y, Zhu P, et al. Implication of Gut Microbiota in Cardiovascular Diseases. Oxid Med Cell Longev 2020;2020:5394096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011;472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Borges NA, Stenvinkel P, Bergman P, et al. Effects of Probiotic Supplementation on Trimethylamine-N-Oxide Plasma Levels in Hemodialysis Patients: a Pilot Study. Probiotics Antimicrob Proteins 2019;11:648–54. [DOI] [PubMed] [Google Scholar]
- [14].Jin M, Qian Z, Yin J, et al. The role of intestinal microbiota in cardiovascular disease. J Cell Mol Med 2019;23:2343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhao X, Oduro PK, Tong W, et al. Therapeutic potential of natural products against atherosclerosis: Targeting on gut microbiota. Pharmacol Res 2021;163:105362. [DOI] [PubMed] [Google Scholar]
- [16].Thomas MS, Fernandez ML. Trimethylamine N-Oxide (TMAO), Diet and Cardiovascular Disease. Curr Atheroscler Rep 2021;23:12. [DOI] [PubMed] [Google Scholar]
- [17].Gatarek P, Kaluzna-Czaplinska J. Trimethylamine N-oxide (TMAO) in human health. EXCLI J 2021;20:301–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Papandreou C, More M, Bellamine A. Trimethylamine N-Oxide in Relation to Cardiometabolic Health-Cause or Effect? Nutrients 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Miller NB, Beigelman A, Utterson E, et al. Transient massive trimethylaminuria associated with food protein-induced enterocolitis syndrome. JIMD Rep 2014;12:11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brugere JF, Borrel G, Gaci N, et al. Archaebiotics: proposed therapeutic use of archaea to prevent trimethylaminuria and cardiovascular disease. Gut Microbes 2014;5:5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Spencer MD, Hamp TJ, Reid RW, et al. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011;140:976–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Krueger SK, Williams DE. Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol Ther 2005;106:357–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fennema D, Phillips IR, Shephard EA. Trimethylamine and Trimethylamine N-Oxide, a Flavin-Containing Monooxygenase 3 (FMO3)-Mediated Host-Microbiome Metabolic Axis Implicated in Health and Disease. Drug Metab Dispos 2016;44:1839–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rehman HU. Fish odour syndrome. Postgrad Med J 1999;75:451–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tang WH, Wang Z, Levison BS, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 2013;368:1575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tang WH, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J Clin Invest 2014;124:4204–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Troseid M, Ueland T, Hov JR, et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med 2015;277:717–26. [DOI] [PubMed] [Google Scholar]
- [28].Haissman JM, Knudsen A, Hoel H, et al. Microbiota-Dependent Marker TMAO Is Elevated in Silent Ischemia but Is Not Associated With First-Time Myocardial Infarction in HIV Infection. Acquir Immune Defic Syndr 2015;71:130–6. [DOI] [PubMed] [Google Scholar]
- [29].Mafune A, Iwamoto T, Tsutsumi Y, et al. Associations among serum trimethylamine-N-oxide (TMAO) levels, kidney function and infarcted coronary artery number in patients undergoing cardiovascular surgery: a cross-sectional study. Clin Exp Nephrol 2016;20:731–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Suzuki T, Heaney LM, Jones DJ, et al. Trimethylamine N-oxide and Risk Stratification after Acute Myocardial Infarction. Clin Chem 2017;63:420–8. [DOI] [PubMed] [Google Scholar]
- [31].Tabas I, Garcia-Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol 2015;209:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wolf D, Ley K. Immunity and Inflammation in Atherosclerosis. Circ Res 2019;124:315–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med 2014;46:e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Febbraio M, Podrez EA, Smith JD, et al. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest 2000;105:1049–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mohammadi A, Vahabzadeh Z, Jamalzadeh S, et al. Trimethylamine-N-oxide, as a risk factor for atherosclerosis, induces stress in J774A.1 murine macrophages. Adv Med Sci 2018;63:57–63. [DOI] [PubMed] [Google Scholar]
- [36].Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 2013;19:576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ohashi R, Mu H, Wang X, et al. Reverse cholesterol transport and cholesterol efflux in atherosclerosis. QJM 2005;98:845–56. [DOI] [PubMed] [Google Scholar]
- [38].Spann NJ, Garmire LX, McDonald JG, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 2012;151:138–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen ML, Yi L, Zhang Y, et al. Resveratrol Attenuates Trimethylamine-N-Oxide (TMAO)-Induced Atherosclerosis by Regulating TMAO Synthesis and Bile Acid Metabolism via Remodeling of the Gut Microbiota. mBio 2016;7:e02210–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Charach G, Rabinovich A, Argov O, et al. The role of bile Acid excretion in atherosclerotic coronary artery disease. Int J Vasc Med 2012;2012:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lu Y, Feskens EJ, Boer JM, et al. The potential influence of genetic variants in genes along bile acid and bile metabolic pathway on blood cholesterol levels in the population. Atherosclerosis 2010;210:14–27. [DOI] [PubMed] [Google Scholar]
- [42].Miyake JH, Duong-Polk XT, Taylor JM, et al. Transgenic expression of cholesterol-7-alpha-hydroxylase prevents atherosclerosis in C57BL/6J mice. Arterioscler Thromb Vasc Biol 2002;22:121–6. [DOI] [PubMed] [Google Scholar]
- [43].Ding L, Chang M, Guo Y, et al. Trimethylamine-N-oxide (TMAO)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis 2018;17:286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004;109:III27–32. [DOI] [PubMed] [Google Scholar]
- [45].Seldin MM, Meng Y, Qi H, et al. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-kappaB. J Am Heart Assoc 2016;5:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ma G, Pan B, Chen Y, et al. Trimethylamine N-oxide in atherogenesis: impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci Rep 2017;37:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Durpes MC, Morin C, Paquin-Veillet J, et al. PKC-beta activation inhibits IL-18-binding protein causing endothelial dysfunction and diabetic atherosclerosis. Cardiovasc Res 2015;106:303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhou X, Chen M, Zeng X, et al. Resveratrol regulates mitochondrial reactive oxygen species homeostasis through Sirt3 signaling pathway in human vascular endothelial cells. Cell Death Dis 2014;5:e1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chen ML, Zhu XH, Ran L, et al. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J Am Heart Assoc 2017;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012;24:981–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sun X, Jiao X, Ma Y, et al. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem Biophys Res Commun 2016;481:63–70. [DOI] [PubMed] [Google Scholar]
- [52].Tao R, Coleman MC, Pennington JD, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 2010;40:893–04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li T, Chen Y, Gua C, et al. Elevated Circulating Trimethylamine N-Oxide Levels Contribute to Endothelial Dysfunction in Aged Rats through Vascular Inflammation and Oxidative Stress. Front Physiol 2017;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ke Y, Li D, Zhao M, et al. Gut flora-dependent metabolite Trimethylamine-N-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radic Biol Med 2018;116:88–00. [DOI] [PubMed] [Google Scholar]
- [55].Chou RH, Chen CY, Chen IC, et al. Trimethylamine N-Oxide, Circulating Endothelial Progenitor Cells, and Endothelial Function in Patients with Stable Angina. Sci Rep 2019;9:4249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Toya T, Ozcan I, Corban MT, et al. Compositional change of gut microbiome and osteocalcin expressing endothelial progenitor cells in patients with coronary artery disease. PLoS One 2021;16:e0249187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Michowitz Y, Goldstein E, Wexler D, et al. Circulating endothelial progenitor cells and clinical outcome in patients with congestive heart failure. Heart 2007;93:1046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Rauscher FM, Goldschmidt-Clermont PJ, Davis BH, et al. Aging, progenitor cell exhaustion, and atherosclerosis. Circulation 2003;108:457–63. [DOI] [PubMed] [Google Scholar]
- [59].Savi M, Bocchi L, Bresciani L, et al. Trimethylamine-N-Oxide (TMAO)-Induced Impairment of Cardiomyocyte Function and the Protective Role of Urolithin B-Glucuronide. Molecules 2018;23:549–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Makrecka-Kuka M, Volska K, Antone U, et al. Trimethylamine N-oxide impairs pyruvate and fatty acid oxidation in cardiac mitochondria. Toxicol Lett. 2017;267:32–8. [DOI] [PubMed] [Google Scholar]
- [61].Oakley CI, Vallejo JA, Wang D, et al. Trimethylamine-N-oxide acutely increases cardiac muscle contractility. Am J Physiol Heart Circ Physiol 2020;318:H1272–H82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Li Z, Wu Z, Yan J, et al. Gut microbe-derived metabolite trimethylamine N-oxide induces cardiac hypertrophy and fibrosis. Lab Invest 2019;99:346–57. [DOI] [PubMed] [Google Scholar]
- [63].Janeiro MH, Ramirez MJ, Milagro FI, et al. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Martinez-Gonzalez MA, Gea A, Ruiz-Canela M. The Mediterranean Diet and Cardiovascular Health. Circ Res 2019;124:779–98. [DOI] [PubMed] [Google Scholar]
- [65].O'Morain VL, Ramji DP. The Potential of Probiotics in the Prevention and Treatment of Atherosclerosis. Mol Nutr Food Res 2020;64:e1900797. [DOI] [PubMed] [Google Scholar]
- [66].Liang X, Zhang Z, Lv Y, et al. Reduction of intestinal trimethylamine by probiotics ameliorated lipid metabolic disorders associated with atherosclerosis. Nutrition 2020;79–80:110941. [DOI] [PubMed] [Google Scholar]
- [67].Chen S, Jiang PP, Yu D, et al. Effects of probiotic supplementation on serum trimethylamine-N-oxide level and gut microbiota composition in young males: a double-blinded randomized controlled trial. Eur J Nutr 2021;60:747–58. [DOI] [PubMed] [Google Scholar]
- [68].Wang Z, Roberts AB, Buffa JA, et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015;163:1585–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bennett BJ, de Aguiar Vallim TQ, Wang Z, et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab 2013;17:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sethi NJ, Safi S, Korang SK, et al. Antibiotics for secondary prevention of coronary heart disease. Cochrane Database Syst Rev 2021;2:CD003610. [DOI] [PMC free article] [PubMed] [Google Scholar]