

Abstract
Reduced dithiolene ligands are bound to high valent Mo centers in the active site of the oxotransferase family of enzymes. Related model complexes have been studied with great insight by Prof. Holm and his colleagues. This study focuses on the other limit of dithiolene chemistry: an investigation of the 2-electron oxidized dithiolene bound to low-valent late transition metal (TM) ions (ZnII, CuI, and CuII). The bonding descriptions of the oxidized dithiolene [N,N-dimethyl piperazine 2,3-dithione (Me2Dt0)] complexes are probed using S K-edge X-ray absorption spectroscopy (XAS) and the results are correlated to density functional theory (DFT) calculations. These experimentally supported calculations are then extended to explain the different geometric structures of the three complexes. The ZnII(Me2Dt0)2 complex has only ligand-ligand repulsion so it is stabilized at the D2d symmetry limit. The CuI(Me2Dt0)2 complex has additional weak backbonding thus distorts somewhat from D2d toward D2h symmetry. The CuII(Me2Dt0)2 complex has a strong σ donor bond that leads to both a large Jahn-Teller stabilization to D2h and an additional covalent contribution to the geometry. The combined strong stabilization results in the square planar, D2h structure. This study quantifies the competition between the ligand-ligand repulsion and the change in electronic structures in determining the final geometric structures of the oxidized dithiolene complexes, and provides quantitative insights into the Jahn-Teller stabilization energy and its origin.
Keywords: oxidized dithiolene, S K-edge X-ray Absorption Spectroscopy (XAS), Density Functional Theory (DFT) calculations, electronic structures, Jahn-Teller distortion, backbonding
Graphical Abstract

Among the bis oxidized dithiolene compounds, the ZnII complexe only has ligand-ligand repulsion thus a D2d geometry. The CuI complex has additional weak backbonding thus distorts from D2d toward D2h. The CuII complex has a strong σ donor bonding that leads to both a large Jahn-Teller stabilization to D2h and an additional covalent contribution to this distortion.
1. Introduction
Prof. Richard H. Holm was a pioneer in the field of Bioinorganic Chemistry.1–4 His group successfully synthesized a wide range of model complexes, including Iron-sulfur and Molybdenum dithiolene compounds, characterized their structures, physical properties, and measured their reactivities.5–14 Our groups collaborated with Prof. Holm and his group for decades, and applied both spectroscopic techniques and density functional theory (DFT) calculations to understand the electronic structures associated with the geometric structures, and how this enables the reactivities of these complexes as related to the metalloenzymes.15–22
For the past decades, one component of our collaboration provided significant insights into the oxo transfer reactions of the Mo enzymes.16–17, 19–20 One of the interesting properties of the dithiolene ligands is their non-innocent behavior.18, 23–24 That is, in a redox reaction, the ligand orbital can be active while the metal orbital shows little change. The two limits are the fully reduced dithiolene and the 2-electron oxidized form. The former complexes have been studied extensively by the Holm group.8–9, 11–12, 15–20, 25
The dithiolene ligands are strong π donors of up to two electrons when bound to high oxidation state transition metal (TM) ions (e.g. MoVI, WVI). This π donor contribution is responsible for their non-innocent behavior. In this study, we investigate two 2-electron oxidized dithiolene ligands bound to low valent late TM ions (ZnII, CuI, and CuII).26 The oxidized dithiolene ligands, N,N-dimethyl piperazine 2,3-dithione (Me2Dt0) and N,N-diisopropyl piperazine 2,3-dithione (iPr2Dt0) were chosen for the stability of their oxidized forms.27–30
The geometric and electronic structures of the fully reduced and 2 electron oxidized forms of the [Me2Dt] free ligand are plotted on the left and right respectively in Figure 1. The three key valence orbitals that are involved in M-L bonds are the in plane σ−, out of plane π+, and out of plane π−, from low to high energies, respectively. The σ− and π+/− refer to the symmetric and anti-symmetric combinations of in and out of plane p orbitals on the S atoms. Upon oxidation, the filled out of plane π+ orbital of the reduced dithiolene is oxidized, as indicated by the dotted line in Figure 1. With respect to geometry, the C(S)=C(S) bond becomes longer and weaker, while the C-S and C-N bonds become shorter and stronger upon oxidation. In addition, the S-C-C-S dihedral angle increases dramatically from 8° to 40° in the DFT calculations. Similar distortions (increase in S-C-C-S dihedral angle) upon oxidation are generally observed in other dithiolene ligands including bdt (benzene-1,2-dithiolate) and mnt (maleonitriledithiolate) (Figure S1). This ligand distortion is responsible for some interesting properties and geometric distortions of their metal complexes30–31, which will be analyzed in later sections.
Figure 1,

The top and side view of the fully reduced dithiolene Me2Dt2- (left) and the 2-electron oxidized dithiolene Me2Dt0 (right). The change in C=C, C-N, C-S bond distances and S-C-C-S dihedral angle are indicated. Their molecular orbital (MO) diagrams are shown in the middle, with the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) separated by the horizontal dashed line. Three key MOs are listed. From bottom to top, they are the in plane σ−, out of plane π+, and out of plane π−, respectively. Note that the HOMO of the dithiolene becomes the LUMO upon oxidation, as indicated by the dotted line.
As indicated above, the reduced dithiolene ligand has been studied with early TM ions in high oxidations states (i.e. MoVI, WVI) with respect to their non-innocent behavior and ligand π to TM dπ charge transfer (LMCT).17, 19–20 Certain oxidized dithiolene ligands, however, can be bound to the late TM ions in low oxidation states where the electronic structure can have contributions from the metal to ligand charge transfer (MLCT).26 The role of this backbonding in the CuI complexes as well as a σ donor bonding in the CuII complexes will be evaluated in this study.
The S K-edge XAS method developed by our group has proven to be a powerful experimental tool to study the electronic structures of the M-S bonds. The S p character in the unoccupied orbitals can be probed directly from the intensity of the Sulfur 1s to the LUMOs pre-edge peak.19–20, 32–35 It is applied here to the study of oxidized dithiolene metal complexes for the first time. Multiple combinations of functionals and basis sets in DFT calculations as well as different data analysis methods are systematically tested in this study. DFT calculations that reasonably correlate with the S K-edge XAS data are employed to understand how the electronic structures impact the geometric structures of the CuII, CuI, and ZnII oxidized dithiolene complexes.
2. Experimental
2.1. Sample preparation
The oxidized dithiolene metal complexes were synthesized as in the literature.28
2.2. S K-edge XAS data collection and analysis
S K-edge XAS data were collected at beam line 4–3 at the Stanford Synchrotron Radiation Lightsource (SSRL). Samples from two independent preparations were measured at different beamtimes to confirm reproducibility. Samples were ground into fine powders and mounted on Kapton tape with a polypropylene front window. Data were processed and analyzed using EXAFSPAK36 and PySpline37. More details can be found in our previous literature.19–20, 33–35
2.3. DFT calculations
DFT calculations were carried out using Gaussian 0938. Combinations of different functionals (B3LYP, BP86, B3PW91, TPSSh) and basis sets (6-311G(d) and TZVP) were tested. Different guesses (initial geometry, charge and spin distributions) were also tested. DFT results were analyzed using QMForge39. For orbital contributions, both C2 population analysis40 and fragment analysis were applied.
3. Results and Analysis
3.1. S K-edge XAS
Normalized S K-edge XAS spectra of the oxidized dithiolene complexes are shown in Figure 2A, and the baseline subtracted pre-edge regions are given in Figure 2B. Quantitative values of the sulfur contributions to the unoccupied orbitals derived from these experimental pre-edge areas are listed in Table 1, last column. From our previous studies, the S% character in the LUMO is proportional to the pre-edge intensity of the S K-edge XAS, and the details about the conversion is obtained are presented in reference23, 32 Note that the pre-edge features have multiple LUMOs and α and β spin contributions, thus the total %S value can exceed 100. Comparison of the experimental pre-edge energies and intensities (Figure 2 and Table 1) give the following observations: 1) The S K pre-edge of the CuII complex is more intense than its CuI counterpart; 2) the S K pre-edge of the CuI complex is more intense than its ZnII counterpart; 3) with respect to varying the ligand, the pre-edges of the complexes with [Me2Dt0] are slightly less intense than those of the same metal ion with the [iPr2Dt0] ligands, and 4) the pre-edge energies with [Me2Dt0] ligands are also systematically lower by ~0.4 eV. These observations are analyzed with the assistance of DFT calculations in the next section.
Figure 2.

A) Normalized S K-edge XAS data of the oxidized dithiolene complexes; B) The baseline subtracted pre-edge region. The integrated intensities are converted to percentage S p characters based on ref23 and are given in the last column of Table 1.
Table 1.
Comparison of the Experimental Data and the DFT Calculated Results
| Compound | M-S (Å) | C-S (Å) | S-S (Å) | C=C (Å) | S-M-S angle | S-C-C-S Dihedral Angle | Dithiolene Angle (Θ) | %S | |
|---|---|---|---|---|---|---|---|---|---|
| CuII(iPr2Dt0)2 | Exp | 2.27 | 1.69 | 3.19 | 1.50 | 89 | 23 | 0 | 142 |
| DFT | 2.33 | 1.71 | 3.27 | 1.51 | 89 | 31 | 5 | 132 | |
| CuII(Me2Dt0)2 | Exp | NA | NA | NA | NA | NA | NA | NA | NA |
| DFT | 2.33 | 1.70 | 3.28 | 1.51 | 89 | 22 | 0 | 138 | |
| CuI(iPr2Dt0)2 | Exp | 2.29 | 1.70 | 3.27 | 1.50 | 91 | 35 | 61 | 107 |
| DFT | 2.34 | 1.69 | 3.31 | 1.51 | 90 | 33 | 74 | 114 | |
| CuI(Me2Dt0)2 | Exp | 2.27 | 1.67 | 3.26 | 1.51 | 91 | 14 | 78 | 115 |
| DFT | 2.33 | 1.69 | 3.30 | 1.51 | 90 | 18 | 72 | 124 | |
| ZnII(iPr2Dt0)2 | Exp | NA | NA | NA | NA | NA | NA | NA | 100 |
| DFT | 2.38 | 1.71 | 3.41 | 1.53 | 91 | 33 | 90 | 94 | |
| ZnII(iPr2Dt0)Cl2 | Exp | 2.36 | 1.68 | 3.36 | 1.53 | 91 | 31 | 87 | 104 |
| DFT | 2.59 | 1.67 | 3.39 | 1.52 | 82 | 40 | 85 | 98 | |
| ZnII(Me2Dt0)Cl2 | Exp | 2.37 | 1.68 | 3.33 | 1.52 | 89 | 19 | 85 | 104 |
| DFT | 2.59 | 1.67 | 3.38 | 1.52 | 81 | 27 | 85 | 91 |
There are some additional features at greater than 2eV above the pre-edge. They are assigned as overlapping contributions of S1s to C-S σ*/ π* and M4s-S3p σ* transitions. Their total %S values could not be experimentally quantified because they overlap with the edge jump.
3.2. DFT calculations
DFT results with different functionals and basis sets are given in Table S1. Their results are reasonably consistent with respect to trends relative to experiments. For a given functional and basis set, the relative change in geometric structure and %S values are nicely reproduced. Here we present the results with B3LYP/6-311G(d) to be consistent with our previous studies.17–20, 33–34, 41 We also tested the effects of different initial guesses on geometry and fragment charge/spin distributions, and all converged to the same geometric and electronic structures.
The crystal structures of several of the oxidized dithiolene complexes were published in ref28. For bis oxidized dithiolene complexes, the DFT optimized structures obtained here are in good agreement with the available crystal structures, including the key bond distances, bond angles, S-C-C-S dihedral angles in the dithiolene ligands, and the relative distortion angles between the two dithiolenes (defined by Θ). For Mixed-ligand compounds ZnII(iPr2Dt0)Cl2 and ZnII(Me2Dt0)Cl2, calculated structures are less consistent with crystal structures. Thus we only present their experimental S K-edge XAS, while the analysis in the rest of this study is focused on the bis oxidized dithiolene complexes. Some of the crystal structures could not be obtained experimentally28 (CuII(Me2Dt0)2 and ZnII(iPr2Dt0)2), thus only their geometry optimized structures are given. Geometric parameters from both the crystal structures and DFT optimizations are listed in Table 1, and the molecular structures are shown in Figure 3 and in the SI. Overall, the oxidized dithiolene S-C-C-S dihedral angles in the metal complexes are systematically smaller compared with those of the free oxidized dithiolene ligands. In comparing these complexes, the largest changes are in the Θ angles between the two dithiolene planes (or the angles between the dithiolene plane and the Cl-Zn-Cl plane in the Zn mixed-ligand compounds). In the ZnII complexes, they are almost perpendicular to each other (Θ=90°, i.e. close to tetrahedral), while in the CuII complexes they are almost co-planar (Θ=0°, i.e. close to square planar), while in the CuI complexes the values are in between, with Θ=~75°.
Figure 3.


The geometry optimized molecular structures (coordinates in SI) and the MO diagrams of the ZnII(Me2Dt0)2 (A), CuI(Me2Dt0)2 (B), and CuII(Me2Dt0)2 (C) complexes. Only the alpha orbitals were plotted for ZnII and CuI, thus the total covalent mixing contributions need to be multiplied by 2.
Figure 3A shows the optimized structure and the MO diagram of the ZnII(Me2Dt0)2 complex. The LUMO orbital (out of plane π+) of each ligand only weakly interacts with each other, leading to two almost degenerate MOs (the + and − combinations) each with ~25% S p character and ~0 Zn d character. Due to the high effective nuclear charge (Zeff) of the Zn2+, the fully occupied Zn 3d orbitals are deep in energy. The highest MO with significant Zn 3d character is calculated at ~7 eV below the HOMO. Fragment analysis also shows no ligand LUMO character in the occupied orbitals (Table S4). There is a weak M4s-S3p σ interaction, but the M4s σ* orbital is ~3eV above the LUMO (Figure S2), thus the pre-edge of the ZnII(Me2Dt0)2 complex mainly reflects the ligand LUMO.
The geometry optimized structure and the MO diagram of the CuI(Me2Dt0)2 complex are shown in Figure 3B. The LUMOs are similar to those in the ZnII(Me2Dt0)2 complex (the + and − combinations of the LUMOs of the free ligands). Their energy splitting is ~ 0.4 eV, which is higher than that in the ZnII complex. This is consistent with the structure of the tilted oxidized dithiolene planes (Θ=78° experimentally, Θ=72° computationally) resulting in some S π p orbital overlap. There is also more S p character in the LUMOs, which is attributed to the decreased charge of the CuI center. In testing a series of closed shell metal ions that have different charges with the oxidized dithiolene ligands with the same geometry, increasing the positive charge on the metal center significantly decreases the S p character in the LUMOs (Table S2). The increasing positive charge attracts electron density thus resulting in more coordinated S character in the occupied orbitals and less S p character in the unoccupied levels. Alternatively, varying the M-S bond distances or the oxidized dithiolene plane angles has little impact (Table S3). Although both CuI and ZnII have d10 configurations, the d manifold for CuI is higher in energy due to its lower Zeff. This results in a small amount of filled Cu d character mixed into the unoccupied ligand based LUMOs, and a small amount of oxidized dithiolene LUMO mixed into the occupied valence orbitals due to backbonding. This backbonding contributes to the tilt of the dithiolene planes (Θ decreases from 90° in Figure 3A to 78° in Figure 3B), and is analyzed in the next section.
The CuII(Me2Dt0)2 complex has a d9 configuration, which requires a spin polarized electronic structure description with an additional unoccupied β LUMO having Cu d character that is σ antibonding with the in-plane σ− orbital from both oxidized dithiolene ligands (Figure 3C, β-LUMO). This additional β-LUMO contributes an additional covalent interaction between the CuII center and the ligands. However, the energy of the additional β-LUMO is close to the ligand out of plane π+ LUMO and thus cannot be separated experimentally. This results in a significant additional amount of S p character in the LUMO of the CuII complex, and is consistent with its broader, more intense S K-pre-edge peak observed experimentally (Figure 2B).23
In all of these compounds, there are a few MOs ~3eV above the LUMO. They are assigned as C-S π* and σ*, as well as a weak M4s-S3p σ* orbitals, as shown in Figure S2. The calculated S p characters in these MOs are relatively small compared to the S p characters in the LUMO.
The [iPr2Dt0] complexes were also evaluated by DFT. In general, the experimental S K-edge XAS pre-edge intensities and their peak energies are consistent with the calculated S p character in their LUMOs and their energy splitting. Compared with the M(Me2Dt0)2 complexes with the same metal ion, the [iPr2Dt0] compounds have both slightly less S p character and higher orbital energies as observed experimentally (Figure 2 and Table 1). The isopropyl (iPr) group is a better electron donor than the methyl (Me) group, leading to more charge density on the S. This results in the higher unoccupied orbital energies and their decreased S p characters.
3.3. Electronic structure contributions to geometric structures
When the geometric structures of the ZnII(Me2Dt0)2, CuI(Me2Dt0)2, and CuII(Me2Dt0)2 complexes are compared, the most obvious difference is in the relative angles Θ between the two oxidized dithiolene planes in each molecule (Figure 3). The ZnII(Me2Dt0)2 complex has almost D2d (tetrahedral) symmetry with the two oxidized dithiolene planes perpendicular (Θ=90°) to each other. This minimizes their overall ligand-ligand repulsion, which has both steric and electrostatic contributions. The CuII(Me2Dt0)2 complex on the other hand, has almost D2h symmetry (square planar) with the two oxidized dithiolenes in the same plane (Θ=0°), because of strong electronic structure contributions. Both the Jahn-Teller driving force and an increase in the covalency of the Cu-S bonds in D2h favor the square planar geometry. The CuI(Me2Dt0)2 complex may be expected to have a similar geometric structure to that of the Zn complex due to its d10 configuration, but both the crystal structure and DFT optimized structure show a distortion away from D2d (Θ=78° experimentally and Θ=72° computationally). This results from a competition between ligand-ligand repulsion and electronic stabilization.
Electronic structure contributions to the geometric structures were analyzed by a series of calculations on the M(Me2Dt0)2 complexes (M = ZnII, CuI, CuII) with systematically varying the Θ angles from 90° (D2d, tetrahedral) to 0° (D2h, square planar), but with all other structural parameters fixed.
In Figure 4A, the total energy vs the Θ angle is plotted for both the ZnII(Me2Dt0)2 (green) and the CuII(Me2Dt0)2 (red) complexes. Because the ZnII complexes have only ligand-ligand repulsion that increases from D2d to D2h and no Θ angular dependence of the M4s-S3p σ bonding (Table S4), the difference of the two (magenta) reflects the angle dependent of the electronic contributions to the total energy, which gives 22 kcal/mol for the electronic energy stabilization in going from D2d to D2h. This large electronic stabilization energy for the CuII(Me2Dt0)2 complex overcomes the ligand-ligand repulsion (12kcal/mol at the D2h limit) and results in its square planar geometry. This has both Jahn-Teller and covalency contributions (Figure 4A, orange line scale on the right gives the β-LUMO covalency) that are discussed below. In Figure 4B, the total energy vs Θ is plotted for the ZnII (green) and CuI (black) complexes. Compared with the CuII(Me2Dt0)2 complex which has significant electronic contributions due to the Jahn-Teller effect and σ donor bonding, the electronic contribution for the CuI(Me2Dt0)2 complex is due to the backbonding. The energy difference of the CuI(Me2Dt0)2 relative to the ZnII(Me2Dt0)2 complex (purple) shows that the maximum backbonding contribution (at Θ=30°, Figure 4B, cyan line scale on the right for amount of backbonding) is worth ~ 3kcal/mol, which leads to the distortion away from the D2d steric limit as observed experimentally. In Figure 4B, the amount of M d backbonding is from the C2 population analysis. A fragment analysis of the ligand LUMO mixed into the valence occupied orbitals gives similar results (Table S4). The backbonding dependence on Θ is discussed below.
Figure 4.
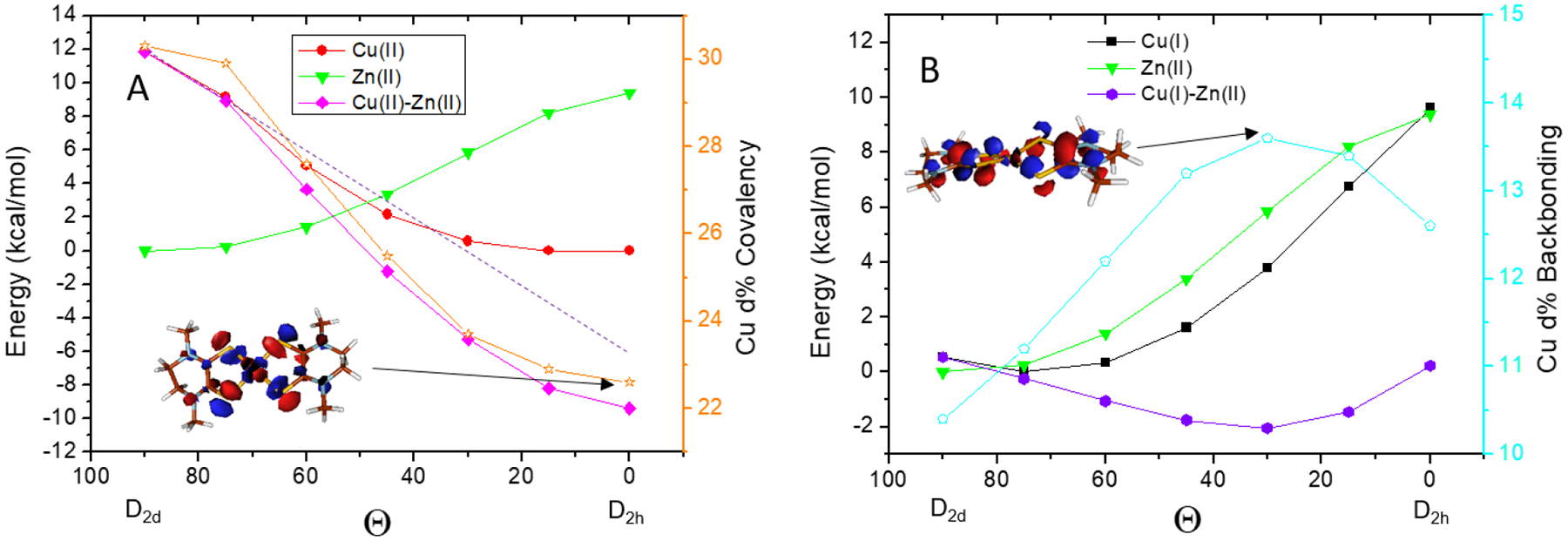
Dependence of total energy on the relative dithiolene angles Θ of (A) ZnII(Me2Dt0)2, CuII(Me2Dt0)2 and (B) ZnII(Me2Dt0)2, CuI(Me2Dt0)2. The magenta curve in (A) reflects the total electronic contribution in CuII(Me2Dt0)2 (from 12 to −10 kcal/mol), which can be further divided into the Jahn-Teller electron-nuclear coupling and the covalency contributions. The covalency (Cu d%) is plotted on the right axis in orange. The MO contour at 0° is given as inset. The purple curve in (B) reflects the contribution from the backbonding in CuI(Me2Dt0)2 (from 1 to −2 kcal/mol). The amount of backbonding is plotted on the right axis in cyan. The MO contour with maximum backbonding in CuI(Me2Dt0)2 (Θ=30°) is given as an inset.
4. Discussion
As presented above, varying the metal ions (CuI, CuII, ZnII) in the bis-oxidized dithiolene complexes results in a change in the relative angle Θ between the two oxidized dithiolene planes. In the ZnII(Me2Dt0)2 complex where there is little electronic stabilization and no angle dependence in Zn 4s mixing in the occupied orbitals, Θ=90° minimizes the ligand-ligand repulsion (Figure 5, green). In the CuII(Me2Dt0)2 complex, there are both a Jahn-Teller stabilization energy and a covalent contribution to the geometry (deriving from the increase in ligand character from ~70% at 0° to 78% at 90°), which gives 22 kcal/mol total electronic stabilization (Figure 4A). This significant stabilization energy overcomes the 12 kcal/mol ligand-ligand repulsion and results in the square planar geometry (Figure 5, red). In the CuI(Me2Dt0)2 complex, there is backbonding which makes a small contribution to the stabilization energy of 3 kcal/mol (Figure 4B). This competes with the ligand-ligand repulsion and distorts the geometry by Θ=15° from the D2d limit (Figure 5, black).
Figure 5.

The final geometry (Θ angle) of the bis oxidized dithiolene complexes are determined by the competition between the ligand-ligand repulsion and electronic contributions. In ZnII(Me2Dt0)2 complex (green), there is only ligand-ligand repulsion favoring the D2d geometry. In the CuI(Me2Dt0)2 complex (black), relatively weak backbonding distorts the geometry away from D2d. In the CuII(Me2Dt0)2 complex (red), significant electronic terms (Jahn-Teller + covalency) distorts to the D2h limit.
The electronic stabilization can be divided into 2 parts. The Jahn-Teller stabilization energy and the change in M-S covalent bonding with changing geometry. In the CuI(Me2Dt0)2 complex, there is no Jahn-Teller contribution as it has a d10 configuration, but there is some backbonding due to the relatively low Zeff of the CuI. Figure 4B shows that the stabilization maximizes (purple) along with the maximum backbonding (cyan); ~3% increase in the Cu d character in the π LUMO of the oxidized dithiolene corresponds to ~ 3kcal/mol in stabilization. Interestingly, the amount of backbonding maximizes at Θ=30°. This is because the oxidized dithiolene is not planar, thus the S-C-C-S dihedral angle of 18° requires the ~30° Θ angle to maximize the S p π orbital overlap (Figure 4B, insert MO contour, and Figure S3). There is also a weak M4s-S3p σ donor bonding interaction that minimizes at the D2d limit which has the same trend as the metal backbonding (Table S4).
On the other hand, the CuII(Me2Dt0)2 complex has a d9 configuration and thus has both a Jahn-Teller stabilization energy and exhibits an increase in covalent M-S bonding toward D2h that also contributes to its geometry. In Figure 4A, the Cu d character is plotted on the right axis. There is little change in Cu d% when Θ decreases from 90° to 75°, therefore an extrapolation along this region of the total stabilization energy (dashed line) gives an estimate of the Jahn-Teller electronic-nuclear coupling force enabling an evaluation of its energy contribution at the D2h limit. This Jahn-Teller stabilization energy is ~17 kcal/mol. Thus, the remaining ~5kcal/mol energy derives from the covalency increase of ~8% in going to the square planar limit.
The Jahn-Teller distortion force derives from the electron-nuclear vibrational coupling. To understand the origin of this strong Jahn-Teller effect in the CuII(Me2Dt0)2 complex, a vibrational perturbation in the non-symmetric ε (in Td symmetry) mode around the D2d geometry was tested using the CuI(Me2Dt0)2 model as there is no spin polarization in d10. At the D2d limit, the HOMO orbitals are doubly degenerate (Figure 6, left). Distortion along the ε vibrational mode by 15° toward the D2h structure results in an energy splitting of these two orbitals by ~0.4 eV. One orbital is stabilized by this non-symmetric distortion having less M-S σ* and the other is destabilized having a larger M-S σ* interaction (Figure 6, right). Because of the strong σ* nature of the M-S bond for the oxidized dithiolenes, the Jahn-Teller distortion force dominates the stabilization energy.
Figure 6.

A) Jahn-Teller distortion near the D2d limit estimated using the CuI(Me2Dt0)2 model. B) Scheme showing that the non-symmetric ε distortion decreases the σ* interaction thus stabilizing the d orbital on the left, and increases the σ* interaction thus destabilizing the orbital on the right.
5. Conclusion
S K-edge XAS combined with DFT calculations have been used to quantify the bonding in ZnII(Me2Dt0)2, CuI(Me2Dt0)2, CuII(Me2Dt0)2 complexes. This has determined the structural distortions of the CuI(Me2Dt0)2 and CuII(Me2Dt0)2 complexes relative to the ZnII(Me2Dt0)2 complex, which is D2d due to ligand-ligand repulsion. For the CuI(Me2Dt0)2, because of its lower Zeff relative to the ZnII complex, a limited amount of backbonding is present that distorts the geometry by ~15° away from D2d (Figure 5, Θ from 90° to 75°).For the CuII(Me2Dt0)2 square planar (D2h) complex, there is additional σ donation that contributes to this structure. However, the Jahn-Teller distortion energy is dominant and derives from the change in sigma antibonding (σ*) interactions with the oxidized dithiolene ligands in the non-symmetric ε vibrational mode (in the Td limit).
Supplementary Material
Highlights:
X-ray absorption spectroscopy (XAS) study of transition metal dithiolene compounds
Sulfur K-edge XAS and DFT are applied to oxidized dithiolene compounds for the first time
Electronic structures determine the corresponding geometric structures
Backbonding in the CuI complexes
The Jahn-Teller effect and change in covalent bonding in the CuII complexes
Acknowledgement
This work was supported by NIH grants (DK031450, E.I.S.; GM061555, P.B.) Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894, K.O. Hodgson). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Reference
- 1.Holm RH; Solomon EI Chem. Rev 2004, 104, 347–348. [DOI] [PubMed] [Google Scholar]
- 2.Holm RH; Kennepohl P; Solomon EI Chem. Rev 1996, 96, 2239–2314. [DOI] [PubMed] [Google Scholar]
- 3.Holm RH; Solomon EI Chem. Rev 2014, 114, 3367–3368. [DOI] [PubMed] [Google Scholar]
- 4.Holm RH Chem. Rev 1987, 87, 1401–1449. [Google Scholar]
- 5.Donahue JP; Goldsmith CR; Nadiminti U; Holm RH J. Am. Chem. Soc 1998, 120, 12869. [Google Scholar]
- 6.Enemark JH; Cooney JJA; Wang J-J; Holm RH Chem. Rev 2004, 104, 1175–1200. [DOI] [PubMed] [Google Scholar]
- 7.Harlan EW; Berg JM; Holm RH J. Am. Chem. Soc 1986, 108, 6992–7000. [DOI] [PubMed] [Google Scholar]
- 8.Lee SC; Holm RH Inorg. Chim. Acta 2008, 361, 1166–1176. [Google Scholar]
- 9.Lim BS; Willer MW; Miao M; Holm RH J. Am. Chem. Soc 2001, 123, 8343–8349. [DOI] [PubMed] [Google Scholar]
- 10.McNaughton RL; Lim BS; Knottenbel SZ; Holm RH; Kirk ML J. Am. Chem. Soc 2008, 130, 4628–4636. [DOI] [PubMed] [Google Scholar]
- 11.Schultz BE; Gheller SF; Muetterties MC; Scott MJ; Holm RH J. Am. Chem. Soc 1993, 115, 2714–2722. [Google Scholar]
- 12.Tucci GC; Donahue JP; Holm RH J. Am. Chem. Soc 1998, 37, 1602–1608. [Google Scholar]
- 13.Venkateswara Rao P; Holm RH Chem. Rev 2004, 104, 527–560. [DOI] [PubMed] [Google Scholar]
- 14.Holm RH; Lo W Chem. Rev 2016, 116, 13685–13713. [DOI] [PubMed] [Google Scholar]
- 15.Musgrave KB; Donahue JP; Lorber C; Holm RH; Hedman B; Hodgson KO J. Am. Chem. Soc 1999, 121, 10297–10307. [Google Scholar]
- 16.Tenderholt AL; Hodgson KO; Hedman B; Holm RH; Solomon EI Inorg. Chem 2012, 51, 3436–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tenderholt AL; Szilagyi RK; Holm RH; Hodgson KO; Hedman B; Solomon EI J. Inorg. Biochem 2007, 101, 1594–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tenderholt AL; Szilagyi RK; Holm RH; Hodgson KO; Hedman B; Solomon EI Inorg. Chem 2008, 47, 6382–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tenderholt AL; Wang J-J; Szilagyi RK; Holm RH; Hodgson KO; Hedman B; Solomon EI J. Am. Chem. Soc 2010, 132, 8359–8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ha Y; Tenderholt AL; Holm RH; Hedman B; Hodgson KO; Solomon EI J. Am. Chem. Soc 2014, 136, 9094–9105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dey A; Glaser T; Moura JJG; Holm RH; Hedman B; Hodgson KO; Solomon EI J. Am. Chem. Soc 2004, 126, 16868–16878. [DOI] [PubMed] [Google Scholar]
- 22.Holm RH; Solomon EI; Majumdar A; Tenderholt A Coord. Chem. Rev 2011, 255, 993–1015. [Google Scholar]
- 23.Sarangi R; George SD; Rudd DJ; Szilagyi RK; Ribas X; Rovira C; Almeida M; Hodgson KO; Hedman B; Solomon EI J. Am. Chem. Soc 2007, 129, 2316–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hille R; Hall J; Basu P Chem. Rev 2014, 114, 3963–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothery RA; Stein B; Solomonson M; Kirk ML; Weiner JH Proc. Natl. Acad. Sci 2012, 109, 14773–14778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basu P; Colston KJ; Mogesa B Coord. Chem. Rev 2020, 409, 213211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nemykin VN; Olsen JG; Perera E; Basu P Inorg. Chem 2006, 45, 3557–3568. [DOI] [PubMed] [Google Scholar]
- 28.Colston KJ; Dille SA; Mogesa B; Astashkin AV; Brant JA; Zeller M; Basu P Eur. J. Inorg. Chem 2019, 2019, 4939–4948. [Google Scholar]
- 29.Ratvasky SC; Mogesa B; van Stipdonk MJ; Basu P Polyhedron 2016, 114, 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dille SA; Colston KJ; Mogesa B; Cassell J; Perera E; Zeller M; Basu P Eur. J. Inorg. Chem 2021, 2021, 914–922. [Google Scholar]
- 31.Yang J; Mogesa B; Basu P; Kirk ML Inorg. Chem 2016, 55, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Solomon EI; Hedman B; Hodgson KO; Dey A; Szilagyi RK Coord. Chem. Rev 2005, 249, 97–129. [Google Scholar]
- 33.Ha Y; Arnold AR; Nuñez NN; Bartels PL; Zhou A; David SS; Barton JK; Hedman B; Hodgson KO; Solomon EI J. Am. Chem. Soc 2017, 139, 11434–11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ha Y; Hu H; Higgins K; Maroney M; Hedman B; Hodgson K; Solomon E Biochem. 2019, 58, 3585–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McQuilken AC; Ha Y; Sutherlin KD; Siegler MA; Hodgson KO; Hedman B; Solomon EI; Jameson GNL; Goldberg DP J. Am. Chem. Soc 2013, 135, 14024–14027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.George GN EXAFSPAK, Stanford Synchrotron Radiaton Laboratory: Menlo Park, CA, 1990. [Google Scholar]
- 37.Tenderholt AL; Hedman B; Hodgson KO In PySpline: A Modern, Cross-Platform Program for the Processing of Raw Averaged XAS Edge and EXAFS Data, AIP conference Proceedings, 2006; pp 105–107. [Google Scholar]
- 38.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ, Gaussian 09, Revision B.01. Wallingford CT, 2009. [Google Scholar]
- 39.Tenderholt AL Stanford University: Stanford, CA, 2007.
- 40.Sun N; Dey A; Xiao Z; Wedd AG; Hodgson KO; Hedman B; Solomon EI J. Am. Chem. Soc 2010, 132, 12639–12647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gorelsky SI; Basumallick L; Vura-Weis J; Sarangi R; Hodgson KO; Hedman B; Fujisawa K; Solomon EI Inorg. Chem 2005, 44, 4947–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.