

Abstract
Investigation of the marine sponge Agelas dispar MeOH fractions using feature-based molecular networking, dereplication, and isolation led to the discovery of new bromopyrrole-derived metabolites. An in-house library of bromopyrrole alkaloids previously isolated from A. dispar and Dictyonella sp. was utilized, along with the investigation of an MS/MS fragmentation of these compounds. Our strategy led to the isolation and identification of the disparamides A−C (1–3), with a novel carbon skeleton. Additionally, new dispyrins B–F (4–8) and nagelamides H2 and H3 (9 and 10) and known nagelamide H (11), citrinamine B (12), ageliferin (13), bromoageliferin (14), and dibromoageliferin (15) were also isolated and identified by analysis of spectroscopic data. Analysis of MS/MS fragmentation data and molecular networking analysis indicated the presence of hymenidin (16), oroidin (17), dispacamide (18), monobromodispacamide (19), keramadine (20), longamide B (21), methyl ester of longamide B (22), hanishin (23), methyl ester of 3-debromolongamide B (24), and 3-debromohanishin (25). Antibacterial activity of ageliferin (13), bromoageliferin (14), and dibromoageliferin (15) was evaluated against susceptible and multi-drug-resistant ESKAPE pathogenic bacteria Klabsiella pneumoniae, Escherichia coli, Staphylococcus aureus, Pseudomonas aeruginosa, Acinetobacter baumannii, and Enterococcus faecalis. Dibromoageliferin (15) displayed the most potent antimicrobial activity against all tested susceptible and MDR strains. Compounds 13–15 presented no significant hemolytic activity up to 100 μM.
Graphical Abstract
The isolation and identification of minor secondary metabolites from biological sources is now becoming routine, since the isolation of major metabolites frequently leads to known compounds.1 Also, the isolation of minor metabolites is required to investigate the structure of compounds that present significant biological activities2 or structural novelty.3,4 Recent examples of chemically unique and/or biologically active natural products isolated in sub-milligram scale include the guanidine-bearing polyacetylenes mollenynes from the marine sponge Spirastrella mollis,5 borolithochromes, pink-colored boron-containing pigments discovered from specimens of the Jurassic putative red alga Solenopora jurassica older than 150 million years,6 and the new aureolic acid derivative metathramycin, discovered by heterologous expression from an environmental metagenome sample.7 Not only do structural analyses of sub-milligram samples constitute a challenge, the isolation of pure samples in minute amounts requires considerable care in the manipulation of samples.3,8
We are interested in revisiting the chemistry of previously investigated biological species using contemporary approaches for dereplication and analysis of prefractionated extracts. The aim is the detection of minor compounds that may constitute either novel bioactive chemical scaffolds or putative bio-synthetic intermediates that may clarify the biosynthesis of metabolites. The last approach has been intensively explored as a strategy toward the understanding of the biosynthesis of tetrodotoxin.9–13 A recent example from our group resulted in the discovery of minor amino-acid-bearing tambjamines that may represent the putative precursors of bis-pyrrole alkaloids isolated from bryozoans, ascidians, and nudibranchs.14
Bromopyrrole alkaloids isolated from marine sponges constitute one of the first groups of natural products discovered from these very primitive animals.15 More than 200 bromopyrrole alkaloids have been so far isolated from marine sponges.15 Even though such compounds have been extensively investigated, new members continue to be isolated, presenting unprecedented structures and potent biological activities. Such is the case of new bromopyrrole derivatives recently discovered from the sponges Agelas oroides,16 Agelas spp.,17 A. nemoechinata,18,19 A. kosrae,20 and Dictyonella sp.21
Dereplication is considered essential for the discovery of new natural products.22 Several chemoinformatics tools and strategies have been utilized toward this aim.23,24 Molecular networking (MN) is a currently well-consolidated chemoinformatic tool that organizes MS/MS data sets and automates database searches for metabolite identification in complex mixtures.25 Feature-based molecular networking (FBMN) is a tool that allies data preprocessing tools for LC-MS2 and MN analysis on GNPS.26 This tool has been recently utilized to guide the isolation of new stylissamide L,27 caffeic acids esters,28 and peptides.29
A recent collection of an A. dispar sample prompted us to develop a method for the investigation of minor bromopyrrole alkaloids, as this species has been extensively investigated.30–33 We herein report a dereplication method developed for the analysis of bromopyrrole alkaloids, based on the reliable detection of bromine isotopes by FBMN, using both in-house and in silico CFM-ID libraries, described in detail in the Supporting Information. Molecular networking analysis allowed the detection of clusters with known and unknown compounds 1–25 (Supporting Information, Figures S2–S7). Novel disparamides A–C (1–3) and new dispyrins B–F (4–8) and nagelamides H2 and H3 (9 and 10) were subsequently isolated and identified by analysis of spectroscopic data. Known nagelamide H (11), citrinamine B (12), ageliferin (13), bromoageliferin (14), dibromoageliferin (15), hymenidin (16), oroidin (17), dispacamide (18), monobromodispacamide (19), keramadine (20), longamide B (21), methyl ester of longamide B (22), hanishin (23), methyl ester of 3-debromolongamide B (24), and 3-debromohanishin (25) were also annotated. The fragmentation study of hymenidin (16), 4-debromooroidin (26), 4-debromougibohlin (27), 5-debromougibohlin (28), and monobromoisophakellin (29) is also reported (the structures of 26–29 are included in the Supporting Information). Cytotoxic activities of the alkaloids 1, 2, 4–6, and 9–15 were evaluated on an ovarian cancer cell line (OVCAR3). Ageliferin (13), bromoageliferin (14), and dibromoageliferin (15) were tested against a panel of ESKAPE Gram-positive and -negative susceptible and multi-drug-resistant bacteria and for potential hemolytic activity in mice erythrocytes.
RESULTS AND DISCUSSION
A sample of A. dispar was collected from Fernando de Noronha Island, Northeast Brazil. The EtOH/MeOH extract of the sponge was defatted with hexane, and the polar fraction was partitioned between H2O and EtOAc. The EtOAc fraction was subjected to a separation by C8 reversed-phase column chromatography, generating five fractions from 100% H2O (M4A1) to 100% MeOH (M4A5). HPLC-UV-MS analysis of these fractions indicated brominated compounds in fractions M4A2, M4A3, and M4A4. Each of these fractions was further separated by chromatography on Sephadex LH-20, generating 63 fractions. Separation by C8 RP column chromatography and by size exclusion chromatography provided a wide chemical space distributed in different fractions, which improved the quality of the untargeted metabolic profile observed by UPLC-QToF-MS/MS analysis.26 FBMN generated in the GNPS platform provided 432 nodes interconnected by 737 edges, organized into 39 clusters (Figure S1).
HRMS/MS analysis of bromopyrrole alkaloids previously isolated and identified from Dictyonella sp.,21 namely, hymenidin (16), 4-debromooroidin (26), 4-debromougibohlin (27), 5-debromougibohlin (28), and monobromoisophakellin (29), constituted the starting point of our strategy, because compounds 16 and 26–29 are isobaric. HRMS/MS acquisition was optimized using 4-debromooroidin (26) (see the Supporting Information for analysis details and structures of compounds).
Linear isomers 16 and 26 presented different MS/MS fragmentation patterns from the cyclic isomers 27–29 (Supporting Information). Fragmentations of 16 and 26 are characterized by the cleavage of the amide bond, fragment ions m/z 171 (B) and m/z 139 (C), and subsequent decompositions to form the fragment ions D–G. The fragmentation patterns of 27–29 were similar to other guanidine alkaloids,23,34–36 with typical loss of CH2N2, forming fragment ions m/z 250 (L), m/z 189 (S), and m/z 188 (T). For compounds 27–29, the base skeleton is maintained in the fragments, with the exception of the loss of CO for fragment ions m/z 144 (Q) and m/z 143 (R), generating fragments M–T (Supporting Information, pp S8–S21). Some fragment ions are proposed as radicals, the existence of which are justified by the presence of bromine in the structures of 16 and 26–29 and in agreement with previous MS analyses.36–38 Analysis of the fragmentation patterns of 16 and 26–29 was important for the dereplication process and enabled the construction of an inhouse library that supported the annotation confidence of related compounds within GNPS and in silico libraries.
The GNPS library search annotated hymenidin (16, m/z error = 9 ppm), oroidin (17, m/z error = 4 ppm), dihydrohymenialdisine (m/z error = 0 ppm), and hymenamide B (m/z error = 5 ppm) (see the Supporting Information, pp S21 and S22, for details of the network construction). Identification of 16 was unambiguously confirmed by an inhouse MS/MS library search (Figure S18). A similar fragmentation pattern was observed for oroidin (17) (Figures S19–S21).
Integration of an in silico database (ISDB) and MS/MS molecular networking has been reported as an efficient dereplication strategy.39–41 Similarly, network annotation propagation (NAP) combines the MetFrag in silico fragmentation prediction of MS/MS spectra with a network topological consensus and reranking the in silico annotations.42–44 Polybrominated compounds are not trivially dereplicated, because the precursor ion usually does not correspond to the monoisotopic mass of the compounds due to the inherent isotopic composition of bromine, making it difficult to use dereplication tools such as SIRIUS.45
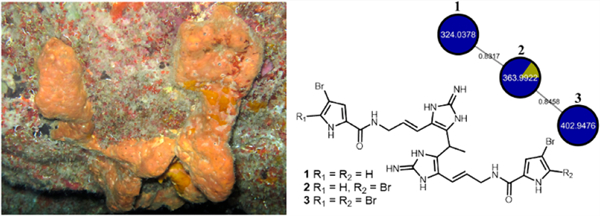
Our in silico library was constructed using a CFM-ID platform, with taxonomic information and manual annotation (see Supporting Information for analysis details) and allowed us to annotate dispacamide (18),33 monobromodispacamide (19),33 keramadine (20),32 and longamide B (21)32 (Figures S22–S36). Manual annotation propagation of 21 allowed the putative identification of unreported compounds from A. dispar, including the methyl ester of longamide B (22),46 hanishin (23),47 the methyl ester of 3-debromolongamide (24), and 3-debromohanishin (25) (Figures S37–S52). This is the first report of natural 24 and 25. The dereplication procedure used to annotate bromopyrrole alkaloids in the FBMN allowed us to select clusters of undescribed compounds in A. dispar fractions. Novel disparamides A–C (1–3) displayed FBMN nodes at m/z 324.0378, m/z 363.9922, and m/z 402.9476, respectively (Figure S53), but MS1 data showed that these m/z values were detected for the [M + 2H]+2 ions. Alkaloids 1–3 have been subsequently isolated for unambiguous structure assignment.
Disparamide A (1) presented a 1:2:1 [M + H]+ cluster at m/z 645.0662, m/z 647.0651, and m/z 649.0635 by HRESI-QToF-MS, indicating that 1 contained two bromine atoms, corresponding to the molecular formula C24H27Br2N10O2. Dereplication using the Dictionary of Natural Products48 showed no results for this molecular weight. The 1H NMR spectrum of 1 showed a singlet at δH 11.79 (NH-1 and –1′), a triplet at δH 8.37 (NH-7 and –7′), three singlets at δH 7.20 (NH-12 and –12′), δH 7.03 (NH-14 and –14′), and δH 7.11 (NH-16 and –16′), two double doublets of hydrogens attached to an aromatic moiety at δH 6.98 (H-2 and –2′) and δH 6.85 (H-4 and –4′), signals for olefinic hydrogens at δH 6.03 (H-9 and –9′) and δH 6.33 (H-10 and –10′), one CH methine signal at δH 4.47 (H-17), one methylene signal at δH 3.93 (H-8 and –8′), and one methyl signal at δH 1.49 (H-18). The integration of the proton signals and the molecular formula suggested that the compound had an internal symmetry plane. The 13C NMR spectrum revealed 12 signals, one amide carbonyl signal at δC 159.7 (C-6 and –6′), one signal for a guanidine carbon embedded in a 2-aminoimidazole group at δC 147.7 (C-13 and 13′), eight aromatic or double-bond carbon signals at δC 127.5–95.2, one methine signal at δC 25.1 (C-17), and one methyl signal at δC 18.7 (C-18) (Table 1). Analysis of 1H NMR and 13C NMR data indicated chemical shifts typically observed for 3-bromopyrrole carbonyl moieties.49 The COSY spectrum disclosed the connectivity from NH-7 to H-10 and from NH-7′ to H-10′. The configuration of the double bond between C-9 and C-10/C-9′ and C-10′ was defined as E considering the coupling constant observed for the olefinic hydrogens (15.8 Hz). HMBC correlations from H-9 to C-11, from H-9′ to C-11′, as well as from H-10 to C-15 and from H-10′ to C-15′ established the connections between the double bonds and the 2-aminoimidazole moieties. Key correlations observed in the HMBC spectrum from H-17 and H-18 to C-15/C-15′ revealed an ethylidene group connection between the two hymenidin (16) substructures (Figure 1). HRMS/MS analysis of 1 is in agreement with this structure, indicating brominated fragments 1A–1C, as well as 2-aminoimidazole dimer fragments 1D–1H (Figures S67 and S68 and Table S3). The structure of disparamide A (1) defines a new carbon skeleton for bromopyrrole alkaloids.
Table 1.
NMR Data for Disparamides A (1) and B (2) in DMSO-d6
| 1 |
2 |
|||
|---|---|---|---|---|
| Position | δC,a type | δH (J in Hz)b | δC,a type | δH (J in Hz)b |
| NH-1 | 11.79, s | 11.79, s | ||
| NH-1′ | 11.79, s | 12.59, s | ||
| 2 | 121.6, CH | 6.98, dd (1.6, 2.9) | 121.5, CH | 6.98, dd (1.6, 2.9) |
| 2′ | 121.6, CH | 6.98, dd (1.6, 2.9) | 104.9, C | |
| 3 | 95.2, C | 95.1, C | ||
| 3′ | 95.2, | C 98.1, C | ||
| 4 | 111.8, CH | 6.85, dd (1.6, 2.6) | 111.7, CH | 6.85, dd (1.6, 2.6) |
| 4′ | 111.8, CH | 6.85, dd (1.6, 2.6) | 112.9, CH | 6.93, d (2.5) |
| 5 | 126.8, C | 126.8, C | ||
| 5′ | 126.8, C | 128.1, C | ||
| 6 | 159.7, C | 159.6, C | ||
| 6′ | 159.7, C | 158.2, C | ||
| NH-7 | 8.37, t (5.7) | 8.37, t (5.7) | ||
| NH-7′ | 8.37, t (5.7) | 8.40, t (5.8) | ||
| 8 and 8′ | 40.3, CH2 | 3.93, dd (5.7, 6.0) | 40.6, CH2 | 3.94, dd (5.3, 5.7) |
| 9 and 9′ | 127.5, CH | 6.03, dt (6.0, 15.8) | 127.4, CH | 6.03, m |
| 10 | 115.7, CH | 6.33, d (15.8) | 115.8, CH | 6.33, dd (5.7, 15.9) |
| 10 | 115.7, CH | 6.33, d (15.8) | 115.7, CH | 6.33, dd (5.7, 15.9) |
| 11 and 11′ | 120.2, C | 120.2, C | ||
| NH-12 and NH-12′ | 7.20,c s | 7.21,c s | ||
| 13 and 13′ | 147.7, C | 147.8, C | ||
| NH-14 and NH-14′ | 7.03,c s | 7.04,c s | ||
| 15 and 15′ | 123.9, C | 123.9, C | ||
| NH-16 and NH-16′ | 7.11,c s | 7.13,c s | ||
| 17 | 25.1, CH | 4.47, q (7.2) | 25.0, CH | 4.47, q (7.2) |
| 18 | 18.7, CH3 | 1.49, d (7.2) | 18.7, CH3 | 1.48, d (7.2) |
150 MHz.
600 MHz.
Signals for NH-12, NH-14, NH-16, NH-12′, NH-14′, and NH-16′ may be interchangeable.
Figure 1.

HMBC and COSY correlations for disparamides A and B (1 and 2).
Disparamide B (2) displayed a 1:3:3:1 [M + H]+ cluster at m/z 722.9767, m/z 724.9753, m/z 726.9734, and m/z 728.9716 by HRESI-QToF-MS, indicating three bromine atoms and the molecular formula C24H26Br3N10O2. 1H NMR and 13C NMR spectra of 2 were very similar to NMR data of 1. Differences in its 13C NMR spectrum included signals at δC 98.1 (C-3′), δC 104.9 (C-2′), and δC 112.9 (C-4′), while the 1H NMR spectrum showed a doublet at δH 6.93 (H-4′), indicating one 2,3-dibromopyrrole carbonyl and one 3-bromopyrrole moiety. COSY and HMBC data showed the same pattern observed for 1. Therefore, the structure of 2 was assigned as a combination of one hymenidin (16) and one oroidin (17) substructure connected by an ethylidene group. This hypothesis was confirmed by analysis of HRMS/MS fragmentation data recorded for 2 (Figures S68 and S81, Table S4). Although disparamide B (2) presents a stereogenic center at C-17, its small value of specific rotation [α]D –0.89 (c 0.18, MeOH) and a flat ECD spectrum (Figure S165) indicate that either 2 was isolated in too small a quantity to obtain reliable data to assign its absolute configuration or it has been isolated as a racemic mixture.
Disparamide C (3) displayed a 1:4:6:4:1 [M + H]+ cluster at m/z 800.8874, m/z 802.8864, m/z 804.8847, m/z 806.8830, and m/z 808.8806 by HRESI-QToF-MS, indicating four bromine atoms and the molecular formula C24H25Br4N10O2. While it was not possible to isolate compound 3 for its complete characterization due to the small amount available, the structure of 3 could be proposed based on the manual annotation propagation in the disparamide cluster (Figure S53) as well as by analyzing the compound 3 HRMS/MS fragmentation pattern (Figures S68 and S84, Table S5). Disparamides A–C (1–3) are the first dimers of hymenidin/oroidin with both subunits connected through C-15 and C15′.
Dispyrin B (4) displayed a 1:3:3:1 [M + H]+ cluster at m/z 549.9340, m/z 551.9324, m/z 553.9310, and m/z 555.9318 in its HRESI-QToF-MS analysis, indicating three bromine atoms, allowing the assignment of the molecular formula C18H22Br3N3O2 . Inspection of 1H and 13C NMR data of 4 suggested a close relationship with dispyrin.30 NMR data showed the presence of a 3-bromopyrrole carbonyl moiety.49 Analysis of 1H and 13C NMR data indicated a 1,2,3,5-tetrasubstituted benzene ring, with chemical shifts at δC 139.6 (C-10), δC 133.2 (C-11 and C-15), δC 117.3 (C-12 and C-14), and δC 150.4 (C-13). HMBC data indicated that carbons at δC 117.3 (C-12 and C14) showed only a correlation with δH 7.53 (H-11 and H-15), while the 1H signal at δH 7.53 showed a correlation with the methylene at δC 33.8 (C-9). These correlations positioned the two bromine atoms at C-12 and C-14 of the benzene ring. HMBC correlations from H-17 to C-13 and COSY correlations from H-17 to H-22 allowed the unambiguous identification of dispyrin B (4) as a new dispyrin (Figure 2). Analysis of HRMS/MS fragmentation data of 4 (Figures S98 and S99, Table S6) is in agreement with the proposed structure, which corresponds to 14-bromodispyrin, and we named it as dispyrin B.
Figure 2.

HMBC and COSY correlations for dispyrins B−D (4−6).
Dispyrin C (5) displayed a 1:2:1 [M + H]+ cluster at m/z 597.9208, m/z 599.9189, and m/z 601.9170 by HRESI-QToF-MS, with two bromine atoms corresponding to the molecular formula C18H22Br2IN3O2. Analysis of the 13C NMR data of 5 suggested a difference in the benzene ring when compared to 4. Analysis of HSQC and HMBC spectra indicated the substitution by one bromine (δC 115.8, C-12) and one iodine (δC 93.2, C-14) on the benzene ring, a structure proposal that was confirmed by analysis of HRMS/MS fragmentation data (Figures S99 and S112, Table S7). Compound 5 is 14-iododispyrin, and we named it as dispyrin C.
Dispyrin D (6) displayed a 1:1 [M + H]+ at m/z 645.9067 and m/z 647.9048 by HRESI-QToF-MS, including one bromine atom and corresponding to the molecular formula C18H22BrI2N3O2. The 13C NMR data of 6 indicated a similarly 1,2,3,5-tetrasubstituted ring as 4, differing only by the presence of two iodine atoms instead of the two bromines. The structure proposal of 6 was completely confirmed by analysis of HRMS/MS fragmentation data (Figures S99 and S125, Table S8) as being 12-debromo-12,14-diiododispyrin, named as dispyrin D.
The structures of dispyrins E (7) and F (8) were proposed by manual annotation propagation comparing the HRMS/MS fragmentation patterns of 7 and 8 with those of 4–6, as 4–8 are correlated with each other in the same cluster (Figure S85). Dispyrin E (7) displayed a 1:4:6:4:1 [M + H]+ cluster at m/z 627.8464, m/z 629.8443, m/z 631.8421, m/z 633.8402, and m/z 635.8385 by HRESI-QToF-MS, with four bromine atoms and the molecular formula C18H21Br4N3O2. HRMS/MS data of 7 indicated two bromine atoms in the benzene ring (fragment 7A) and a 2,3-bromopyrrole carbonyl moiety (fragment 7D, Figures S99 and S128, Table S9). Dispyrin F (8) displayed a 1:3:3:1 [M + H]+ cluster at m/z 675.8316, m/z 677.8296, m/z 679.8279, and m/z 681.8259 by HRESI-QToF-MS, corresponding to the molecular formula C18H21Br3IN3O2 (Δ 2.07 ppm). Analysis of HRMS/MS fragmentation data of 8 indicated one bromine and iodine substituents at the benzene ring (fragment 8A) and a 2,3-bromopyrrole carbonyl moiety (fragment 8D, Figures S99 and S131, Table S10). Compound 7 is 2,14-dibromodispyrin, named as dispyrin E, while alkaloid 8 is 2-bromo-14-iododispyrin, named as dispyrin F.
Nagelamide H (11)49,50 and citrinamine B (12)50 were isolated and identified by comparing MS and NMR data (Figures S133–S141) as well as by analysis of HRMS/MS fragmentation data (Figures S137, S138, and S142, Tables S11 and S12). Nagelamide H2 (9) was isolated in a 1:2 mixture with nagelamide H3 (10), which displayed a [M + H]+ cluster at m/z 817.9448, m/z 819.9431, m/z 821.9412, and m/z 823.9390 by HRESI-QToF-MS, consistent with the molecular formula C24H26Br3N11O5S. The 1:2 mixture of 9/10 was optically active, with [α]D +61.66 (c 0.12, MeOH). Analysis of NMR data of 9 and 10 indicated high similarity to 11 and 12. In the case of 9, 1H and HMBC data showed couplings between δH 7.04/δC 121.9 (CH-2′) and δH 6.88/δC 112.3 (CH-4′), along with couplings between δH 7.01 (H-4) and δC 105.3 (C-2), indicating a 2,3-bromopyrrole carbonyl moiety in the upper monomer (NH-1–C-6) and a 3-bromopyrrole carbonyl moiety in the lower monomer (NH-1′–C-6′) for 9. As for alkaloid 10, it was possible to observe 1H and HMBC couplings between δH 6.99/δC 121.8 (CH-2) and δH 6.92/δC 112.0 (CH-4), in addition to couplings between δH 6.97 (H-4′) and δC 105.3 (C-2′), indicating a 3-bromopyrrole carbonyl moiety in the upper monomer (NH-1–C-6) and a 2,3-bromopyrrole carbonyl moiety in the lower monomer (NH-1′–C-6′). Analysis of HRMS/MS fragmentation data for the mixture of 9 and 10 was fully consistent with the proposed structures (Figures S138 and S151, Tables S13 and S14).
Ageliferin (13),51 bromoageliferin (14),51 and dibromoageliferin (15)51 were also isolated and identified by comparison of HRMS and NMR data (Figures S152–S160) and analysis of HRMS/MS fragmentation data (Figures S161–S164, Tables S15–S17).
The cytotoxicities of alkaloids 1, 2, 4–6, and 9–15 were screened in an ovarian cancer cell line, OVCAR3, but all were inactive. Alkaloids 13–15 have been previously tested against lung carcinoma (A549), colon carcinoma (HT29), and breast (MDA-MB-231) cancer cell lines, but no cytotoxic activity was observed below 10 μg/mL.52 The same alkaloids were not cytotoxic against monkey kidney cells at 200 μg/disk.53 Ageliferin (13) displayed no cytotoxic activity against the lymphoma cell line (L5178Y); only at 50 μM was 13 able to inhibit 50% of the cellular viability.54
Multi-drug-resistant bacteria are now considered as a world health threat by the World Health Organization (WHO). WHO listed ESKAPE pathogens in a list of 12 bacteria against which new antibiotics are urgently needed. The term “ESKAPE” encompasses six of these pathogens with increasing multidrug resistance: Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp. These pathogens are responsible for most nosocomial infections and can “escape” the action of available antibiotics.55
In the present investigation, ageliferin (13), bromoageliferin (14), and dibromoageliferin (15) were tested against a panel of susceptible and multi-drug-resistant Gram-positive and Gram-negative bacteria, E. coli, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and E. faecalis. Results are summarized in Table 4. Dibromoageliferin (15) showed the most promising activity against all tested Gram-positive and -negative strains, being active against susceptible and resistant strains of K. pneumoniae (carbapenems-, aminoglycosides-, and fluoroquinolones-resistant) and vancomycin-resistant E. faecalis. Ageliferin A (13) displayed antibiotic activity against susceptible and resistant E. coli and against antibiotic-susceptible P. aeruginosa. Ageliferin B (14) was effective against susceptible and resistant strains of E. coli, with MIC values 12.5 and 25 μM, respectively. It showed weak activity against carbapenem-resistant A. baumannii with an MIC value of 100 μM and against susceptible and carbapenem-resistant P. aeruginosa, with MIC values of 12.5 and 50 μM, respectively. Ageliferin B (14) also showed activity against both susceptible and oxacillin-resistant S. aureus, with an MIC value of 12.5 μM. Several bromopyrrole alkaloids display antibacterial activity.53,56–59 Synthesis of bromopyrrole alkaloid analogues has been explored toward the development of new antibiotics as well.60 Moreover, several syntheses have been described for ageliferins,61 alkaloids that can be, therefore, considered as potential lead scaffolds for the development of new antibiotics.
Table 4.
Antibacterial Activities of Ageliferin (13), Bromogeliferin (14), and Dibromoageliferin (15) against Multi-Drug-Resistant Gram-Positive and Gram-Negative Bacteria
| strain (Gram +/−) | strain | resistance marker | 13a | 14a | 15a |
|---|---|---|---|---|---|
| E. coli (−) | ATCC 25922 | susceptible | 100 | 12.5 | 12.5 |
| ATCC 35218 | ESBL-producing | 100 | 25 | 12.5 | |
| K. pneumoniae (−) | ATCC 700603 | susceptible | >100 | >100 | 12.5 |
| id-146/19 | carbapenemresistant | >100 | >100 | 25 | |
| A. baumannii (−) | ATCC 19606 | susceptible | >100 | >100 | 100 |
| id-261/16 | carbapenemresistant | >100 | 100 | 100 | |
| P. aeruginosa (−) | ATCC 27853 | susceptible | 50 | 12.5 | 25 |
| S-6065/06 | carbapenemresistant | >100 | 50 | 25 | |
| E. faecalis (+) | ATCC 29212 | susceptible | >100 | >100 | 6.2 |
| ATCC 51299 | vancomycin resistant | >100 | >100 | 6.2 | |
| S. aureus (+) | ATCC 25923 | susceptible | >100 | 12.5 | 6.2 |
| ATCC 33591 | oxacillinresisant | >100 | 12.5 | 6.2 |
Concentration in μM. ATCC, American Type Culture Collection; ESBL, β-lactamase broad-spectrum.
Additionally, drug-induced hemolysis is a frequent complication associated with chemotherapy treatment, inducing toxicity due to the interaction of drugs with the erythrocyte membrane.62 Considering the antibiotic activities of ageliferin A (13), ageliferin B (14), and dibromoageliferin (15), we analyzed the mammalian toxicity of these compounds on mice erythrocytes. No significant damage could be observed after 2 h of incubation to the highest tested concentration of 100 μM, resulting in marginal hemolysis promoted by 13–15 of 2.3% (±0.2), 1.5% (±0.6), and 0.7% (±0.08), respectively, when compared to untreated cells (Figure S166).
Previous investigations on the chemistry of A. dispar led to the isolation of dispyrin and dibromoagelaspongin methyl ether,28 longamide B, and clathramides C and D,31 as well as of dispacamide and monobromodiscapamide.32,33 Our approach, which relied on feature-based molecular networking, enabled us to unveil the metabolome of a sample of A. dispar, comprised of alkaloids 1–25. Disparamides A–C (1–3) represent new carbon skeletons, and dispyrins B–F (4–8) and nagelamides H2 and H3 (9 and 10) are new bromopyrrole derivatives. Iodinated dispyrins C (5), D (6), and F (8) belong to the rather small group of iodine-bearing natural products,63 of which the large majority are marine-derived.64 Only agelanesin B and agelanesin D65 are iodinated bromopyrrole alkaloids related to dispyrin.30 The discovery of such minor metabolites could be directed by an efficient dereplication procedure. While the small amounts of disparamides A–C hampered a more comprehensive assessment of the biological activities of these alkaloids, total syntheses have been developed for the related “dimeric oroidin” alkaloids nagelamides.61d A total synthesis of dispyrin has also been reported, as this compound displays activities as an antagonist of α1D and α2A adrenergic receptors and H2 and H3 histamine receptors.66 It is clear to us that reinvestigation of marine invertebrates using contemporary spectrometry-based metabolomics and dereplication analysis should lead to the isolation of an array of novel and new metabolites, which may contribute to the discovery of potentially bioactive natural products aiming to meet society’s needs for new pharmaceuticals.
In conclusion, the application of FBMN proved useful to visualize LC-MS2 data of a fractionated extract from A. dispar. MS/MS fragmentation investigation of bromopyrrole alkaloids 16 and 26–29 was seminal to distinguish linear from cyclic skeletons. Construction of in-house and in silico libraries, together with the GNPS library, improved the dereplication process. Using this strategy, we have been able to annotate compounds 18–25 and focus on the isolation of previously undescribed bromopyrrole alkaloids disparamides A (1) and B (2), dispyrins B–D (4–6), and nagelamides H2 (9) and H3 (10). FBMN and MS/MS fragmentation pattern analysis led us also to propose the putative structures of disparamide C (3) and dispyrins E and F (7 and 8). Our results demonstrate the power of chemoinformatic tools which, together with the unambiguous identification of new alkaloids, constitute a reliable strategy to unveil minor metabolites which can be of chemical and biological interest for further investigations.
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotations were recorded on a Polartronic H Schmidt+Haensch polarimeter. Ultraviolet spectra were obtained on a UV-3600 Shimadzu UV spectrophotometer. The samples were diluted in MeOH to a concentration of 0.01 mg mL−1. Circular dichroism spectra were measured on a JASCO J-810 spectropolarimeter using quartz cells (1, 2, or 5 mm path length) at 23 °C. Infrared (IR) spectra were obtained using the Shimadzu model IRAffinity on a silica plate. NMR spectra were obtained at 25 °C. NMR spectra were recorded on a Bruker AVANCE III spectrometer (9.4 T) operating at either 600 MHz (1H) or 150 MHz (13C). The 1H chemical shifts were referenced to the residual DMSO-d6 (δ 2.49), whereas 13C chemical shifts were referenced to the DMSO-d6 solvent peak (δ 39.5). HPLC-PDA-MS analysis was carried out on a Waters chromatography system consisting of a Waters 2695 Alliance control system coupled to a Waters 2696 UV−visible spectrophotometric detector with a photodiode array detector, connected sequentially to a Waters Micromass ZQ 2000 mass spectrometry detector operated using an Empower platform. Analyses were performed using a Waters XTerra RP18 column (250.0 × 4.6 mm, 5.0 μm) and eluting with H2O + 0.1% formic acid, MeOH + 0.1% formic acid, and MeCN + 0.1% formic acid using a gradient from 90:5:5 to 0:50:50 of H2O/MeOH/MeCN over 22 min, maintaining in 0:50:50 H2O/MeOH/MeCN for 8 min, from 0:50:50 to 90:5:5 in 1 min, and maintaining at 90:5:5 for 9 min, using a flow rate of 1.0 mL min−1. The PDA detector scanned between λ 200 and 800 nm. The mass spectrometer detector was optimized using the following conditions: capillary voltage 3 kV; temperature of the source 100 °C; desolvation temperature 350 °C; ESI mode, acquisition range m/z 250–2000; gas flow without cone 50 L h−1; and desolvation gas flow 350 L h−1. Samples were diluted in MeOH at a concentration of 2 mg mL−1. HRMS and HRMS/MS analyses of pure compounds were performed using a UPLC H-class liquid chromatograph coupled to a Waters Xevo G2-XS QToF mass spectrometer. A BEH C18 column (dimensions: 2.1 × 100 mm, 1.7 μm; Waters) and a mobile phase consisting of H2O + 0.1% formic acid and MeCN + 0.1% formic acid were used. The elution gradient used was from 9:1 to 8:2 H2O/MeCN for 4 min, from 2:8 to 9:1 H2O/MeCN for 0.1 min, and maintained in 9:1 H2O/MeCN for 0.9 min, totaling 5.0 min with a flow rate of 0.5 mL min−1. The injection volume used was 0.5 μL. The column was maintained at 40 °C, and the samples were maintained at 15 °C. The positive mode ESI conditions were 1.2 kV capillary voltage, 30 V cone voltage, 100 °C source temperature, 450 °C desolvation temperature, 50 L h−1 cone gas flow, and 750 L h−1 desolvation gas flow. For internal calibration, a solution of leucine enkephalin (Sigma, 200 pg mL−1), infused by the lock-mass probe with a flow rate of 10 μL min−1, was used. MS or MS/MS spectra were acquired using positive, resolution, centroid mode, with an acquisition time of 0 to 5 min, ESI source, mass range of m/z 50−1200 for MS mode and variable to MS/MS mode, scan time of 0.2 s−1, and collision energy of 25 eV for 1−3 and 9−15 and 50 eV for 4−8. Samples of pure compounds were diluted in MeOH at a concentration of 0.01 mg mL−1.
Animal Material.
The sponge Agelas dispar was collected on April 21, 2016, on Ilha do Meio localized at Fernando de Noronha-PE, Brazil (3°49′17.08″ S, 32°23′45.39″ W). A voucher sample has been deposited at Museu Nacional, Universidade Federal do Rio de Janeiro, under registry code MNRJ 20217.
Extraction and Isolation.
The marine sponge A. dispar was collected and immediately stored in 96% EtOH. The biological material (1.5 kg) was subsequently extracted with MeOH. The EtOH and MeOH extracts were pooled and evaporated, generating the extract (M4). Extract M4 was resuspended in MeOH (500 mL) and partitioned with hexane (3 × 500 mL). The MeOH fraction was evaporated, suspended in EtOAc (500 mL), and partitioned against H2O (3 × 500 mL), resulting in 12 g of EtOAc fraction (M4A). Fraction M4A was separated in 12 subfractions of 1 g, all of which were subjected to reversed-phase column chromatography on C8 cartridges (Waters, 10 g), eluted with a gradient of MeOH (with 0.5% TFA) in H2O (with 0.5% TFA). Five fractions of 150 mL were collected: 100% H2O (M4A1, 1.2 g), 75:25 H2O/MeOH (M4A2, 3.2 g), 50:50 H2O/MeOH (M4A3, 3.8 g), 25:75 H2O/MeOH (M4A4, 1.9 g), and 100% MeOH (M4A5, 2.2 g). Analysis by HPLC-PDA-MS indicated bromopyrrole alkaloids in fractions M4A2−M4A4, which were separately subjected to a separation by column chromatography on a Sephadex LH-20 column (dimensions: 5.0 × 185 cm), generating a total of 63 fractions. FBMN analysis and dereplication indicated fractions M4A3G, M4A4J, M4A4K ,and M4A4L of interest.
Fraction M4A4K (164.6 mg) was separated by HPLC using a C18 InertSustain column (10.0 × 250.0, μm, GL Science), H2O + 0.5% TFA, MeCN + 0.5% TFA, and MeOH + 0.5% TFA, in gradient mode, from 50:25:25 to 40:30:30 H2O/MeCN/MeOH in 24 min, from 40:30:30 to 0:50:50 H2O/MeCN/MeOH in 1 min, maintaining in 0:50:50 H2O:MeCN/MeOH for 4 min, and from 0:50:50 to 50:25:25 H2O:MeCN/MeOH in 1 min and maintaining in 50:25:25 H2O:MeCN/MeOH for 10 min, using flow of 4.0 mL min−1 and detection in λ 277 and 300 nm, to give 10 fractions (M4A4KA− M4A4KJ). Fraction M4A4KB (12.8 mg) was further separated by HPLC using an XTerra RP18 column (7.8 × 150.0 mm, 5 μm Waters) and 75:25 H2O (+0.5% TFA)/MeCN (+0.5% TFA) as eluent, a flow rate of 3.0 mL min−1, and detection at λmax 277 and 300 nm to give disparamide A (1, 1.5 mg). Fraction M4A4KD (19.8 mg) was separated by HPLC using an XTerra RP18 column (7.8 × 150.0 mm, 5 μm, Waters), using 72:28 H2O (+0.5% TFA)/MeCN (+0.5% TFA), a flow rate of 3.0 mL min−1, and detection at λmax 277 and 300 nm to give disparamide B (2, 1.8 mg).
Fraction M4A3G (1.2 g) was separated by C18 column chromatography (15 g of stationary phase), with H2O + 0.5% TFA and MeOH + 0.5% TFA, in gradient mode, collecting eight fractions: 90:10 H2O/MeOH (M4A3GEA), 20:80 H2O/MeOH (M4A3GB), 30:70 H2O/MeOH (M4A3GC), 35:65 H2O/MeOH (M4A4GD), 40:60 H2O/MeOH (M4A3GE), 45:55 H2O/MeOH (M4A3GF), 50:50 H2O/MeOH (M4A3GG), and 100:0 H2O/MeOH (M4A3GH). Fractions M4A3GE (106.3 mg) and M4A3GF (121.2 mg) were reunited and separated by HPLC using an XBridge BEH amide (10.0 × 250.0 mm, 5 μm, Waters), H2O + 0.5% TFA and MeCN + 0.5% TFA in gradient mode, maintained in 8:92 H2O/MeCN for 10 min, from 8:92 to 16:84 H2O/MeCN in 10 min, maintained in 16:84 H2O/MeCN for 5 min, from 16:84 to 28:72 H2O/MeCN in 7 min, and from 28:72 to 8:92 H2O/MeCN in 3 min and maintained in 8:92 H2O/MeCN for 10 min, using a flow of 4.0 mL min−1 and detection at λ 277 and 300 nm, to give 13 fractions (M4A3GEA−M4A3GEM). Fraction M4A3GEA (40.1 mg) was further separated by HPLC using an XTerra RP18 column (7.8 × 150.0 mm, 5 μm, Waters) and 73:27 H2O (+ 0.5% TFA)/MeCN (+0.5% TFA) as eluent, with a flow rate of 2.5 mL min−1 and detection at λ 277 and 300 nm, to give dispyrin B (4, 2.5 mg), dispyrin C (5, 1.8 mg), and dispyrin D (6, 1.0 mg).
Fraction M4A4L (150.8 mg) was separated by HPLC using an XBridge BEH amide (10.0 × 250.0 mm, 5 μm, Waters), H2O + 0.5% TFA and MeCN + 0.5% TFA in gradient mode, maintaining in 8:92 H2O:MeCN for 10 min, from 8:92 to 16:84 H2O/MeCN in 10 min, maintained in 16:84 of H2O/MeCN for 5 min, from 16:84 to 35:65 H2O/MeCN in 14 min, and from 35:65 to 8:92 H2O/MeCN in 1 min and maintained in 8:92 H2O/MeCN for 10 min, using a flow rate of 4.0 mL min−1 and detection at λ 277 and 300 nm, to give nine fractions (M4A4L1−M4A4L9). Fraction M4A4L6 (9.6 mg) was further separated by HPLC using an XTerra RP18 column (7.8 × 150.0 mm, 5 μm, Waters), with H2O, MeCN, and MeOH in a linear gradient mode, from 67:17:16 to 56:24:20 of H2O/MeCN/MeOH, a flow rate of 2.5 mL min−1, and detection at λ 260 and 290 nm, to give a 1:2 mixture of nagelamides H2/H3 (9/10, 1.2 mg).
Disparamide A (1): brown, glassy solid; UV (MeOH) λmax (log ε) 273 (4.12) nm; IR (film on silica plate) νmax 3630−2600, 1714, 1429, 1199, 1024, 1002, 921, 835, 800, 721, 599 cm−1; 1H and 13C NMR data, Table 1; HRESI-QToF-MS m/z 645.0662 [M + H]+ (calcd for C24H27Br2N10O2+, m/z 645.0680, Δ −2.79 ppm).
Disparamide B (2): brown, amorphous solid; [α]D −0.89 (c 0.18, MeOH); UV (MeOH) λmax (log ε) 275 (4.11), 269 (4.36) nm; IR (film on silica plate) νmax 3660−2540, 1714, 1633, 1435, 1319, 1199, 1024, 837, 763, 723, 518 cm−1; 1H and 13C NMR data, Table 1; HRESI-QToF-MS m/z 722.9767 [M + H]+ (calcd for C24H26Br3N10O2+, m/z 722.9785, Δ −2.49 ppm).
Dispyrin B (4): brown, amorphous solid; UV (MeOH) λmax (log ε) 269 (4.41) nm; IR (film on silica plate) νmax 3500−2900, 2885, 2713, 2486, 1693, 1631, 1568, 1531, 1384, 1323, 1255, 1172, 1128, 1006, 921, 829, 798, 738, 719, 605, 553, 518 cm−1; 1H and 13C NMR data, Table 2; HRESI-QToF-MS m/z 549.9341 [M + H]+ (calcd for C18H23Br3N3O2+, m/z 549.9335, Δ −0.07 ppm).
Table 2.
NMR Data for Dispyrins B (4), C (5), and D (6) in DMSO-d6
| 4 |
5 |
6 |
||||
|---|---|---|---|---|---|---|
| position | δC,a type | δH (J in Hz)b | δC,a type | δH (J in Hz)b | δC,a type | δH (J in Hz)b |
| NH-1 | 11.79, s | 11.77, s | 11.78, s | |||
| 2 | 121.3, CH | 6.95, dd (1.6, 3.0) | 121.4, CH | 6.95, dd (1.6, 3.0) | 121.4, CH | 6.95, dd (1.6, 2.9) |
| 3 | 95.1, C | 95.1, C | 95.1, C | |||
| 4 | 111.6, CH | 6.80, dd (1.6, 2.6) | 111.6, CH | 6.79, dd (1.6, 2.6) | 111.6, CH | 6.79, dd (1.6, 2.6) |
| 5 | 127.0, C | 127.0, C | 127.0, C | |||
| 6 | 159.8, C | 159.8, C | 159.8, C | |||
| NH-7 | 8.16, t (5.7) | 8.15, t (6.0) | 8.13, t (5.7) | |||
| 8 | 39.6, CH2 | 3.42, td (5.7, 6.8) | 39.8, CH2 | 3.40, td (6.0, 6.4) | 39.7, CH2 | 3.38, m |
| 9 | 33.8, CH2 | 2.77, t (6.8) | 33.6, CH2 | 2.74, t (6.4) | 33.3, CH2 | 2.71, t (6.5) |
| 10 | 139.6, C | 140.2, C | 140.5, C | |||
| 11 | 133.2, CH | 7.53, s | 134.1, CH | 7.53, d (2.0) | 140.0, CH | 7.71, s |
| 12 | 117.3, C | 115.8, C | 91.6, C | |||
| 13 | 150.4, C | 152.9, C | 155.3, C | |||
| 14 | 117.3, C | 93.2, C | 91.6, C | |||
| 15 | 133.2, CH | 7.53, s | 139.2, CH | 7.70, d (2.0) | 140.0, CH | 7.71, s |
| 17 | 70.3, CH2 | 3.97, t (5.8) | 70.1, CH2 | 3.94, t (5.8) | 69.9, CH2 | 3.92, t (5.6) |
| 18 | 24.9, CH2 | 2.15, m | 25.0, CH2 | 2.17, m | 25.1, CH2 | 2.19, m |
| 19 | 54.5, CH2 | 3.33, m | 54.6, CH2 | 3.34, t (8.3) | 54.7, CH2 | 3.36, m |
| NH-20 | 9.91, s | 9.74, s | 9.65, s | |||
| 21 and 22 | 45.5, CH3 | 2.83, s | 42.5, CH3 | 2.83, s | 42.5, CH3 | 2.84, s |
600 MHz.
150 MHz.a
Dispyrin C (5): brown, amorphous solid; UV (MeOH) λmax (log ε) 214 (4.77), 269 (4,36) nm; IR (film on silica plate) νmax 3450−3060, 2935, 2879, 2708, 2385, 2127, 1789, 1710, 1568, 1531, 1469, 1384, 1323, 1249, 1199, 1026, 920, 833, 721, 605, 518 cm−1; 1H and 13C NMR data, Table 2; HRESI-QToF-MS m/z 597.9210 [M + H]+ (calcd for C18H23Br2IN3O2+, m/z 597.9197, Δ 1.05 ppm).
Dispyrin D (6): brown, amorphous solid; UV (MeOH) λmax (log ε) 223 (4.42), 269 (4.13); IR (film on silica plate) νmax 3360−3196, 2935, 2873, 2713, 2522, 2254, 1693, 1568, 1529, 1440, 1384, 1327, 1180, 1128, 1026, 921, 833, 721, 603 cm−1; 1H and 13C NMR data, Table 2; HRESI-QToF-MS m/z 645.9065 [M + H]+ (calcd for C18H23BrI2N3O2+, m/z 645.9058, Δ 0.61 ppm).
Nagelamides H2/H3 (9/10): brown, amorphous solid; [α]D +61.66 (c 0.12, MeOH); UV (MeOH) λmax (log e) 275 (4.54) nm; IR (film on silica plate) νmax 3649, 3309, 3130, 2362, 2339, 2258, 1652, 1560, 1521, 1458, 1436, 1419, 1365, 1319, 1132, 1024, 977, 920, 864, 821, 738, 609, 565 cm−1; 1H and 13C NMR data, Table 3; HRESI-QToF-MS m/z 817.9448 [M + H]+ (calcd for C24H27Br3N11O5S+, m/z 817.9462, Δ −1.71 ppm).
Table 3.
NMR Data for Nagelamides H2 (9) and H3 (10) in DMSO-d6
| 9 |
10 |
|||
|---|---|---|---|---|
| position | δC,a type | δH (J in Hz)b | δC,a type | δH (J in Hz)b |
| NH-1 | 12.68, s | 12.68, s | ||
| NH-1′ | 11.80, s | 11.80, s | ||
| 2 | 105.3, C | 7.04, dd (2,8, 1,3) | 121.8, CH | 6.99, m |
| 2′ | 121.9, CH | 105.3, C | ||
| 3 | 98.3, C | 94.5, C | ||
| 3′ | 95.4 | 98.4, C | ||
| 4 | 113.2, CH | 7.01, d (2,8) | 112.0, CH | 6.92, dd (2,6, 1,9) |
| 4′ | 112.3, CH | 6.89, dd (2,5, 1,3) | 113.4, CH | 6.98, m |
| 5 | 128.1, C | 126.8, C | ||
| 5′ | 126.7, C | 127.9, C | ||
| 6 | 160.1, C | 159.4, C | ||
| 6′ | 158.9, C | 159.9, C | ||
| NH-7 | 8.52, m | 8.46, m | ||
| NH-7′ | 8.52, m | 8.46, m | ||
| 8 | 39.7, CH2 | 3.90, m | 39.7, CH2 | 3.90, m |
| 8′ | 40.4, CH2 | 3.90, m | 40.4, CH2 | 3.90, m |
| 9 | 4.08, m | 4.08, m | ||
| 9′ | 130.9, CH | 5.93, m | 130.9, CH | 5.93, m |
| 10 | 130.6, CH | 6.17, m | 130.6, CH | 6.17, m |
| 10′ | 124.9, CH | 5.93, m | 124.9, CH | 5.93, m |
| 11 | 115.4, CH | 6.17, m | 115.4, CH | 6.17, m |
| 11′ | 68.0, C | 68.0, C | ||
| NH-12 | 124.2, C | 124.2, C | ||
| NH-12′ | ||||
| 13 | ||||
| 13′ | 167.6, C | 167.6, C | ||
| NH-14 | 147.9, C | 147.9, C | ||
| NH-14′ | 10.04,c s | 10.04,c s | ||
| 15 | 177.9, C | 177.9, C | ||
| 15′ | 124.2, C | 124.2, C | ||
| NH-16 | 8.60,c, s | 8.60,c s | ||
| 9.11,c s | ||||
| 9.11,c s | ||||
| 9.11,c s | ||||
| NH-16′ | 7.73,c s | 7.73,c, s | ||
| NH-1″ | 9.86, brs | 9.86, brs | ||
| 2″a | 40.6, CH2 | 3.54, m | 40.6, CH2 | 3.54, m |
| 2″b | 3.67, m | 3.67, m | ||
| 3″a | 48.9, CH2 | 2.78, m | 48.9, CH2 | 2.85, m |
| 3″b | 2.78, m | 2.85, m | ||
600 MHz.
150 MHz.
No HMBC correlation observed. Assignments were based on the literature assignments for 11 and 12.49
LC-HRMS and LC-HRMS/MS Analysis of Agelas dispar Fractions.
The 63 fractions obtained after the fractionation by column chromatography on Sephadex LH-20 were analyzed by UPLC-QToF-MS. Analysis was performed in a Waters UPLC H-class liquid chromatograph coupled to a Waters Xevo G2-XS QToF mass spectrometer. A BEH C18 column (dimensions: 2.1 × 50.0 mm, 1.7 μm; Waters) was maintained at 40 °C, and the mobile phase consisted of H2O + 0.1% formic acid and MeCN + 0.1% formic acid. The elution gradient used was from 9:1 to 8:2 H2O/MeCN in 4 min, from 2:8 to 9:1 H2O/MeCN in 0.1 min, and maintained in 9:1 H2O/MeCN for 0.9 min, totaling 5.0 min, with a flow rate of 0.5 mL min−1. The volume of injection used was 4.0 μL of a 1.0 mg mL−1 solution, maintained at 15 °C. The positive mode ESI conditions were a 1.2 kV capillary voltage, 30 V cone voltage, 100 °C source temperature, 450 °C desolvation temperature, 50 L h−1 cone gas flow, and 750 L h−1 desolvation gas flow. For internal calibration, a solution of leucine enkephalin (Sigma, 200 pg mL−1) was infused by the lock-mass probe with a flow rate of 10 μL min−1 every 20 s with a scan time of 0.1 s and real time mass correction. Data were acquired in data-dependent acquisition (DDA), using positive, centroid, resolution mode, 5 min of acquisition time, m/z range 50−2000, scan time of 0.2 s, MS survey threshold of 50.000 intensity s−1, 3 MS/MS per MS survey, mass exclusion between ±10 ppm for MS/MS in real time for 3 s, and ramp of collision energy for MS/MS 10−30 eV for the lowest mass (m/z 50) to 30−50 eV for the highest mass (m/z 2000) (average: m/z 1025, collision energy: 20−40 eV).
Cytotoxicity Assay.
The cytotoxicity assay using human ovarian cancer cells OVCAR3 has been performed as previously described.67
Antibacterial Assays.
The minimum inhibitory concentrations of ageliferin A (13), ageliferin B (14), and dibromoageliferin (15) were determined using a version of the Clinical Laboratory and Standard Institute (CLSI) broth microdilution method. Pure bacterial suspensions were adjusted to the 0.5 McFarland standard containing approximately (1−2) × 108 CFU·mL−1 and properly diluted. The three compounds were tested in 2-fold dilution (100 to 0.78 μM). The final test bacterial concentration was approximately 5 × 105 CFU/mL (5 × 104 CFU/well). The plate was incubated at 35 °C for 24 h. MIC was considered as the lowest concentration of the compound that completely inhibits the bacteria growth in the microdilution well by the unaided eye.
Hemolytic Activity.
Erythrocytes were collected from BALB/c mice, seeded at a 3% suspension in 96-well U-shape microplate, and incubated with ageliferin A (13), ageliferin B (14), and dibromoageliferin (15) (100 to 6.25 μM) in 1× PBS (Sigma-Aldrich), for 2 h at 24 °C. The hemolytic activity was determined in the cell supernatant by optical density reading at 570 nm (FilterMax F5 multi-mode microplate reader, Molecular Devices). Maximum hemolysis was obtained using ultrapure distilled water, and untreated erythrocytes were used as negative control.68
Supplementary Material
Figure 3.

Key HMBC and COSY correlations for nagelamides H2 (9) and H3 (10).
Chart 1.

ACKNOWLEDGMENTS
Financial support was provided by a FAPESP BIOTA/730 BIOparospecTA grant (2013/50228–8 and 2019/17721–9) to R.G.S.B. and FAPESP (2021/04464–8) to A.G.T. E.H. thanks the following funding agencies for providing financial support: CAPES (23038.001427/2014–15), CNPq (425839/2016–8), FAPERJ (102.292/2013; 200.124/2019; 202.624/2019), and FAPESP (2013/50228–8). V.F.F. thanks CAPES (1681546) and CNPq (141464/2017–8) for Ph.D. scholarships. J.R.G. and E.V.C. thank FAPESP for postdoctoral scholarships (2017/06014–4 and 2020/03637–3, respectively).
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jnatprod.2c00094
The authors declare no competing financial interest.
Contributor Information
Vítor F. Freire, Instituto de Química de São Carlos, Universidade de São Paulo, CEP 13560-970 São Carlos, SP, Brazil.
Juliana R. Gubiani, Instituto de Química de São Carlos, Universidade de São Paulo, CEP 13560-970 São Carlos, SP, Brazil
Tara M. Spencer, Department of Pharmaceutical Sciences, Center for Biomolecular Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States
Eduardo Hajdu, Museu Nacional, Universidade Federal do Rio de Janeiro, CEP 20940-040 Rio de Janeiro, RJ, Brazil.
Antonio G. Ferreira, Departamento de Química, Universidade Federal de São Carlos, CEP 13565-905 São Carlos, SP, Brazil
Dayana A. S. Ferreira, Instituto Adolfo Lutz, Secretaria de Saúde do Estado de São Paulo, CEP 01246-000 Sao Paulo, Brazil
Erica V. de Castro Levatti, Instituto Adolfo Lutz, Secretaria de Saúde do Estado de São Paulo, CEP 01246-000 Sao Paulo, Brazil
Joanna E. Burdette, Department of Pharmaceutical Sciences, Center for Biomolecular Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Carlos Henrique Camargo, Instituto Adolfo Lutz, Secretaria de Saúde do Estado de São Paulo, CEP 01246-000 Sao Paulo, Brazil.
Andre G. Tempone, Instituto Adolfo Lutz, Secretaria de Saúde do Estado de São Paulo, CEP 01246-000 Sao Paulo, Brazil.
Roberto G. S. Berlinck, Instituto de Química de São Carlos, Universidade de São Paulo, CEP 13560-970 São Carlos, SP, Brazil.
REFERENCES
- (1).Berlinck RGS; Monteiro AF; Bertonha AF; Bernardi DI; Gubiani JR; Slivinski J; Michaliski LF; Tonon LAC; Venancio VA; Freire VF Nat. Prod. Rep 2019, 36, 981–1004. [DOI] [PubMed] [Google Scholar]
- (2).Murata M; Oishi T; Yoshida M In Compounds Antifouling, Marine Molecular Biotechnology; Fusetani N; Clare AS, Eds.; Springer: Berlin, Heidelberg, 2006; Vol. 42, pp 203–220. [Google Scholar]
- (3).Molinski TF Nat. Prod. Rep 2010, 27, 321–329. [DOI] [PubMed] [Google Scholar]
- (4).Molinski TF Curr. Opin. Biotechnol 2010, 21, 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wang X; Duggan BM; Molinski TF J. Am. Chem. Soc 2015, 137, 12343–12351. [DOI] [PubMed] [Google Scholar]
- (6).Wolkenstein K; Sun H; Falk H; Griesinger CJ Am. Chem. Soc 2015, 137, 13460–13463. [DOI] [PubMed] [Google Scholar]
- (7).Stevenson LJ; Bracegirdle J; Liu L; Sharrock AV; Ackerley DF; Keyzers RA; Owen JG RSC Chem. Biol 2021, 2, 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Claridge TD W. High-Resolution NMR Techniques in Organic Chemistry; Elsevier: Amsterdam, 2016; pp 89–95. [Google Scholar]
- (9).Kudo Y; Yamashita Y; Mebs D; Cho Y; Konoki K; Yasumoto T; Yotsu-Yamashita M Angew. Chemie - Int. Ed 2014, 53, 14546–14549. [DOI] [PubMed] [Google Scholar]
- (10).Kudo Y; Yasumoto T; Mebs D; Cho Y; Konoki K; Yotsu-Yamashita M Angew. Chemie - Int. Ed 2016, 55, 8728–8731. [DOI] [PubMed] [Google Scholar]
- (11).Kudo Y; Yotsu-Yamashita MJ Nat. Prod 2019, 82, 1656–1663. [DOI] [PubMed] [Google Scholar]
- (12).Ueyama N; Sugimoto K; Kudo Y; Onodera KI; Cho Y; Konoki K; Nishikawa T; Yotsu-Yamashita M Chem. - A Eur. J 2018, 24, 7250–7258. [DOI] [PubMed] [Google Scholar]
- (13).Kudo Y; Hanifin CT; Kotaki Y; Yotsu-Yamashita MJ Nat. Prod 2020, 83, 2706–2717. [DOI] [PubMed] [Google Scholar]
- (14).Takaki M; Freire VF; Nicacio KJ; Bertonha AF; Nagashima N; Sarpong R; Padula V; Ferreira AG; Berlinck RG S. J. Nat. Prod 2021, 84, 790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lindel T Alkaloids: Chemistry and Biology 2017, 77, 177–219. [DOI] [PubMed] [Google Scholar]
- (16).Kovalerchik D; Singh RP; Schlesinger P; Mahajni A; Shefer S; Fridman M; Ilan M; Carmeli SJ Nat. Prod 2020, 83, 374–384. [DOI] [PubMed] [Google Scholar]
- (17).Lee S; Tanaka N; Takahashi S; Tsuji D; Kim SY; Kojoma M; Itoh K; Kobayashi J; Kashiwada Y Mar. Drugs 2020, 18, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Li T; Tang X; Luo X; Wang Q; Liu K; Zhang Y; De Voogd NJ; Yang J; Li P; Li G Org. Lett 2019, 21, 9483–9486. [DOI] [PubMed] [Google Scholar]
- (19).Li T; Li PL; Luo XC; Tang XL; Li GQ Tetrahedron Lett 2019, 60, 1996–1998. [Google Scholar]
- (20).Kwon O-S; Kim D; Kim H; Lee Y-J; Lee H-S; Sim CJ; Oh D-C; Lee SK; Oh K-B; Shin J Mar. Drugs 2018, 16, 10–13.29301345 [Google Scholar]
- (21).De Souza RTMP; Freire VF; Gubiani JR; Ferreira RO; Trivella DBB; Moraes FC; Paradas WC; Salgado LT; Pereira RC; Amado Filho GM; Ferreira AG; Williams DE; Andersen RJ; Molinski TF; Berlinck RG S. J. Nat. Prod 2018, 81, 2296–2300. [DOI] [PubMed] [Google Scholar]
- (22).Hubert J; Nuzillard JM; Renault JH Phytochem. Rev 2017, 16, 55–95. [Google Scholar]
- (23).Kind T; Fiehn O Phytochem. Lett 2017, 21, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Blaženović I; Kind T; Ji J; Fiehn O Metabolites 2018, 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang M; Carver JJ; Phelan VV; Sanchez LM; Garg N; Peng Y; Nguyen DD; Watrous J; Kapono CA; Luzzatto-Knaan T; Porto C; Bouslimani A; Melnik AV; Meehan MJ; Liu WT; Crüsemann M; Boudreau PD; Esquenazi E; Sandoval-Calderón M; Kersten RD; Pace LA; Quinn RA; Duncan KR; Hsu C-C; Floros DJ; Gavilan RG; Kleigrewe K; Northen T; Dutton RJ; Parrot D; Carlson EE; Aigle B; Michelsen CF; Jelsbak L; Sohlenkamp C; Pevzner P; Edlund A; McLean J; Piel J; Murphy BT; Gerwick L; Liaw C-C; Yang Y-L; Humpf H-U; Maansson M; Keyzers RA; Sims AC; Johnson AR; Sidebottom AM; Sedio BE; Klitgaard A; Larson CB; Boya P CA; Torres-Mendoza D; Gonzalez DJ; Silva DB; Marques LM; Demarque DP; Pociute E; O’Neill EC; Briand E; Helfrich EJN; Granatosky EA; Glukhov E; Ryffel F; Houson H; Mohimani H; Kharbush JJ; Zeng Y; Vorholt JA; Kurita KL; Charusanti P; McPhail KL; Nielsen KF; Vuong L; Elfeki M; Traxler MF; Engene N; Koyama N; Vining OB; Baric R; Silva RR; Mascuch SJ; Tomasi S; Jenkins S; Macherla V; Hoffman T; Agarwal V; Williams PG; Dai J; Neupane R; Gurr J; Rodríguez AMC; Lamsa A; Zhang C; Dorrestein K; Duggan BM; Almaliti J; Allard P-M; Phapale P; Nothias L-F; Alexandrov T; Litaudon M; Wolfender J-L; Kyle JE; Metz TO; Peryea T; Nguyen D-T; VanLeer D; Shinn P; Jadhav A; Müller R; Waters KM; Shi W; Liu X; Zhang L; Knight R; Jensen PR; Palsson BØ; Pogliano K; Linington RG; Gutiérrez M; Lopes NP; Gerwick WH; Moore BS; Dorrestein PC; Bandeira N Nat. Biotechnol 2016, 34, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Nothias LF; Petras D; Schmid R; Dührkop K; Rainer J; Sarvepalli A; Protsyuk I; Ernst M; Tsugawa H; Fleischauer M; Aicheler F; Aksenov AA; Alka O; Allard PM; Barsch A; Cachet X; Caraballo-Rodriguez AM; Da Silva RR; Dang T; Garg N; Gauglitz JM; Gurevich A; Isaac G; Jarmusch AK; Kameník Z; Kang KB; Kessler N; Koester I; Korf A; Le Gouellec A; Ludwig M; Martin H C; McCall LI; McSayles J; Meyer SW; Mohimani H; Morsy M; Moyne O; Neumann S; Neuweger H; Nguyen NH; Nothias-Esposito M; Paolini J; Phelan VV; Pluskal T; Quinn RA; Rogers S; Shrestha B; Tripathi A; van der Hooft JJJ; Vargas F; Weldon KC; Witting M; Yang H; Zhang Z; Zubeil F; Kohlbacher O; Böcker S; Alexandrov T; Bandeira N; Wang M; Dorrestein PC Nat. Methods 2020, 17, 905–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Scarpato S; Teta R; della Sala G; Pawlik JR; Costantino V; Mangoni A Mar. Drugs 2020, 18, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Padilla-González GF; Sadgrove NJ; Ccana-Ccapatinta GV; Leuner O; Fernandez-Cusimamani E Metabolites 2020, 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chen S; Huang G; Liao W; Gong S; Xiao J; Bai J; Wendy Hsiao WL; Li N; Wu JL Food Chem 2021, 347, 129008. [DOI] [PubMed] [Google Scholar]
- (30).Piña IC; White KN; Cabrera G; Rivero E; Crews P J. Nat. Prod 2007, 70, 613–617. [DOI] [PubMed] [Google Scholar]
- (31).Cafieri F; Fattorusso E; Taglialatela-Scafati OJ Nat. Prod 1998, 61, 122–125. [DOI] [PubMed] [Google Scholar]
- (32).Cafieri F; Fattorusso E; Mangoni A; Taglialatela-Scafati O Tetrahedron Lett 1996, 37, 3587–3590. [Google Scholar]
- (33).Cafieri F; Fattorusso E; Taglialatela-Scafati OJ Nat. Prod 1998, 61, 1171–1173. [DOI] [PubMed] [Google Scholar]
- (34).Berlinck RG S. Progr. Chem. Org. Nat. Prod 1995, 66, 119–295. [DOI] [PubMed] [Google Scholar]
- (35).Nikolić D; Gödecke T; Chen S-N; White J; Lankin DC; Pauli GF; van Breemen RB Fitoter 2012, 83, 441–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Phytochem Nikolić D.. Lett 2017, 21, 292–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Cai T; Guo Z-Q; Xu X-Y; Wu Z-J. Mass Spectrom. Rev 2018, 37, 202–216. [DOI] [PubMed] [Google Scholar]
- (38).Vessecchi R; Zocolo GJ; Gouvea DR; Hübner F; Cramer B; Marchi MRR; Humpf H-U; Lopes NP Rapid Commun. Mass Spectrom 2011, 25, 2020–2026. [DOI] [PubMed] [Google Scholar]
- (39).Allard PM; Péresse T; Bisson J; Gindro K; Marcourt L; Pham VC; Roussi F; Litaudon M; Wolfender JL Anal. Chem 2016, 88, 3317–3323. [DOI] [PubMed] [Google Scholar]
- (40).Péresse T; Jézéquel G; Allard PM; Pham VC; Huong DTM; Blanchard F; Bignon J; Lévaique H; Wolfender JL; Litaudon M; Roussi FJ Nat. Prod 2017, 80, 2684–2691. [DOI] [PubMed] [Google Scholar]
- (41).Klein-Juńior LC; Cretton S; Allard PM; Genta-Jouve G; Passos CS; Salton J; Bertelli P; Pupier M; Jeannerat D; Heyden Y; Vander; Gasper AL; Wolfender JL; Christen P; Henriques AT J. Nat. Prod 2017, 80, 3032–3037. [DOI] [PubMed] [Google Scholar]
- (42).da Silva RR; Wang M; Nothias LF; van der Hooft JJJ; Caraballo-Rodríguez AM; Fox E; Balunas MJ; Klassen JL; Lopes NP; Dorrestein PC PLoS Comput. Biol 2018, 14, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lee J; da Silva RR; Jang HS; Kim HW; Kwon YS; Kim JH; Yang H Food Chem 2019, 295, 368–376. [DOI] [PubMed] [Google Scholar]
- (44).Kang KB; Park EJ; Da Silva RR; Kim HW; Dorrestein PC; Sung SH J. Nat. Prod 2018, 81, 1819–1828. [DOI] [PubMed] [Google Scholar]
- (45).Dührkop K; Fleischauer M; Ludwig M; Aksenov AA; Melnik AV; Meusel M; Dorrestein PC; Rousu J; Böcker S Nat. Methods 2019, 16, 299–302. [DOI] [PubMed] [Google Scholar]
- (46).Umeyama A; Ito S; Yuasa E; Arihara S; Yamada TJ Nat. Prod 1998, 61, 1433–1434. [DOI] [PubMed] [Google Scholar]
- (47).Mancini I; Guella G; Amade P; Roussakis C; Pietra F Tetrahedron Lett 1997, 38, 6271–6274. [Google Scholar]
- (48).Dictionary of Natural Products. https://dnp.chemnetbase.com/faces/chemical/ChemicalSearch.xhtml.
- (49).Endo T; Tsuda M; Okada T; Mitsuhashi S; Shima H; Kikuchi K; Mikami Y; Fromont J; Kobayashi JJ Nat. Prod 2004, 67, 1262–1267. [DOI] [PubMed] [Google Scholar]
- (50).Cychon C; Lichte E; Köck M Beilstein J. Org. Chem 2015, 11, 2029–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kobayashi J; Tsuda M; Murayama T; Nakamura H; Ohizumi Y; Ishibashi M; Iwamura M; Ohta T; Nozoe S Tetrahedron 1990, 43, 5579–5586. [Google Scholar]
- (52).Regalado EL; Laguna M; Mendiola J; Thomas OP; Nogueiras C Quimica Nova 2011, 34, 289–291. [Google Scholar]
- (53).Keifer PA; Schwartz RE; Koker MES; Hughes RG; Rittschof D; Rinehart KL J. Org. Chem 1991, 56, 2965–2975. [Google Scholar]
- (54).Hamed ANE; Schmitz R; Bergermann A; Totzke F; Kubbutat M; Müller WEG; Youssef DTA; Bishr MM; Kamel MS; Edrada-Ebel R; Wätjen W; Proksch P Zeitschrift fur Naturforschung. Section C: Journal of Biosciences 2018, 73, 199–210. [DOI] [PubMed] [Google Scholar]
- (55).Mulani MS; Kamble EE; Kumkar SN; Tawre MS; Pardesi KR Front Microbiol 2019, 10, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Endo T; Tsuda M; Okada T; Mitsuhashi S; Shima H; Kikuchi K; Mikami Y; Fromont J; Kobayashi J J. Nat. Prod 2004, 67, 1262–1267. [DOI] [PubMed] [Google Scholar]
- (57).Pech-Puch D; Pérez-Povedano M; Martinez-Guitian M; Lasarte-Monterrubio C; Vázquez-Ucha JC; Bou G; Rodríguez J; Beceiro A; Jimenez C Mar. Drugs 2020, 18, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Zhang H; Khalil Z; Conte MM; Plisson F; Capon RJ Tetrahedron Lett 2012, 53, 3784–3787. [Google Scholar]
- (59).Araki A; Kubota T; Aoyama K; Mikami Y; Fromont J; Kobayashi J Org. Lett 2009, 11, 1785–8. [DOI] [PubMed] [Google Scholar]
- (60).Rane RA; Gutte SD; Sahu NU Bioorg. Med. Chem. Lett 2012, 22, 6429–32. [DOI] [PubMed] [Google Scholar]
- (61) (a).Wang X; Wang X; Tan X; Lu J; Cormier KW; Ma Z; Chen CJ Am. Chem. Soc 2012, 134, 18834–18842. Errata: [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang X; Wang X; Tan X; Lu J; Cormier KW; Ma Z; Chen CJ Am. Chem. Soc 2013, 135, 1163–1163. [Google Scholar]; (c) Wang X; Wang X; Tan X; Lu J; Cormier KW; Ma Z; Chen CJ Am. Chem. Soc 2016, 138, 14505–14505. [DOI] [PubMed] [Google Scholar]; (d) Bhandari MR; Herath AK; Rasapalli S; Yousufuddin M; Lovely CJ J. Org. Chem 2020, 85, 12971–12987. [DOI] [PubMed] [Google Scholar]
- (62).Jeswani G; Alexander A; Saraf S; Saraf S; Qureshi AJ Controlled Release 2015, 211, 10–21. [DOI] [PubMed] [Google Scholar]
- (63).Wang L; Zhou X; Fredimoses M; Liao S; Liu Y RSC Adv 2014, 4, 57350. [Google Scholar]
- (64).Woolner VH; Jones CM; Field JJ; Fadzilah NH; Munkacsi AB; Miller JH; Keyzers RA; Northcote PT J. Nat. Prod 2016, 79, 463–469. [DOI] [PubMed] [Google Scholar]
- (65).Hertiani T; Edrada-Ebel R; Ortlepp S; van Soest RWM; de Voogd NJ; Wray V; Hentschel U; Kozytska S; Müller WEG; Proksch P Bioorg. Med. Chem 2010, 18, 1297–1311. [DOI] [PubMed] [Google Scholar]
- (66) (a).Kennedy JP; Brogan JT; Lindsley CW J. Nat. Prod 2008, 71, 1783–1786. [DOI] [PubMed] [Google Scholar]; (b) Yoshida M; Yamaguchi K Chem. Pharm. Bull 2008, 56, 1362–1363. [DOI] [PubMed] [Google Scholar]; (c) Kennedy JP; Conn PJ; Lindsley CW Bioorg. Med. Chem. Lett 2009, 19, 3204–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Gubiani JR; Bernardi DI; De Paula CCP; Seleghim MHR; Ferreira AG; Batista ANL; Batista JM Jr.; Oliveira LFP; Lira SP; Burdette JE; Berlinck RG S. Arch. Pharm 2022, 355, e2100441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Djoumbou-Feunang Y; Pon A; Karu N; Zheng J; Li C; Arndt D; Gautam M; Allen F; Wishart DS Metabolites 2019, 9, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.