

SUMMARY
RNA polymerase II (RNAPII) pausing in early elongation is critical for gene regulation. Paused RNAPII can be released into productive elongation by the kinase P-TEFb or targeted for premature termination by the Integrator complex. Integrator comprises endonuclease and phosphatase activities, driving termination by cleavage of nascent RNA and removal of stimulatory phosphorylation. We generated a degron system for rapid Integrator endonuclease (INTS11) depletion to probe the direct consequences of Integrator-mediated RNA cleavage. Degradation of INTS11 elicits nearly universal increases in active early elongation complexes. However, these RNAPII complexes fail to achieve optimal elongation rates and exhibit persistent Integrator phosphatase activity. Thus, only short transcripts are significantly upregulated following INTS11 loss, including transcription factors, signaling regulators, and non-coding RNAs. We propose a uniform molecular function for INTS11 across all RNAPII-transcribed loci, with differential effects on particular genes, pathways, or RNA biotypes reflective of transcript lengths rather than specificity of Integrator activity.
Graphical Abstract

eTOC Blurb:
Stein et al. show that Integrator broadly balances levels of RNAPII pause release with premature termination. Acute depletion of Integrator endonuclease allows for increased RNAPII release into coding and non-coding RNAs. However, RNAPII elongation is inefficient, and only short RNAs are upregulated, including several transcription factors that initiate signaling pathways.
INTRODUCTION
Precise regulation of gene expression is critical for organismal development and cellular adaption to the environment. Transcription regulation in metazoans centers on the pausing of RNAPII during early elongation (Core and Adelman, 2019), which provides a checkpoint for controlling gene activity. Promoter-proximally paused RNAPII, bound by SPT5 and the NELF complex (Vos et al., 2018), remains associated with the DNA template and nascent RNA while awaiting release into productive elongation (Henriques et al., 2013). Pause release involves the kinase P-TEFb, which phosphorylates RNAPII and SPT5 to dissociate NELF and promote efficient transcription elongation (Core and Adelman, 2019). Alternatively, promoter paused RNAPII can be terminated by the Integrator complex, resulting in the removal of RNAPII from the DNA and degradation of the nascent RNA (Beckedorff et al., 2020; Elrod et al., 2019; Tatomer et al., 2019).
Integrator is a metazoan-specific complex with subunits arranged in discrete modules. The core of Integrator associates with RNAPII, SPT5, and NELF in the paused elongation complex, positioning the Integrator endonuclease near the RNA exit channel to cleave nascent RNA (Fianu et al., 2021). INTS11 harbors the active endonuclease and resides in a subcomplex with INTS4 and INTS9 (Albrecht et al., 2018; Pfleiderer and Galej, 2021; Wu et al., 2017). The Integrator phosphatase module includes INTS6 and Protein Phosphatase 2A (PP2A) A and C subunits, which associate with the core of Integrator through a conserved motif within INTS8 (Huang et al., 2020; Zheng et al., 2020). Integrator-bound PP2A removes stimulatory phosphorylation from the RPB1 C-terminal domain (CTD) and SPT5 to slow RNAPII elongation and prevent the transition towards full elongation competence. Thus, Integrator facilitates promoter-proximal termination using both endonuclease and phosphatase activities, raising parallels with the cleavage and polyadenylation (CPA) machinery that functions at mRNA 3’ ends (Cortazar et al., 2019; Huang et al., 2020; Kamieniarz-Gdula and Proudfoot, 2019; Parua et al., 2018; Vervoort et al., 2021; Zheng et al., 2020).
Integrator was first described for its role in 3’ end formation at U-rich snRNAs (Baillat et al., 2005), but recent results imply a broader role. Integrator has been shown to act at sets of messenger RNAs (mRNA) (Beckedorff et al., 2020; Dasilva et al., 2021; Elrod et al., 2019; Gardini et al., 2014; Huang et al., 2020; Skaar et al., 2015; Stadelmayer et al., 2014; Tatomer et al., 2019; Yue et al., 2017) and long non-coding RNAs (lncRNA) (Barra et al., 2020; Nojima et al., 2018a), as well as upstream antisense RNAs (uaRNAs, or PROMPTs), which are short transcripts that originate near active mRNA promoters and are rapidly targeted for degradation by the RNA exosome (Liu et al., 2022; Lykke-Andersen et al., 2020; Preker et al., 2008; Seila et al., 2008). Similarly, Integrator has been implicated in the biogenesis of enhancer RNAs (eRNAs) (Elrod et al., 2019; Lai et al., 2015). The expanded repertoire of Integrator targets raises questions about the scope and specificity of the Integrator endonuclease and how Integrator localization and cleavage might be controlled. Further, Integrator has been suggested to function differently at mRNA genes than at short, unstable uaRNAs or eRNAs (Lykke-Andersen et al., 2020) though mechanistic explanations for such differences are unknown.
The consequences of Integrator-mediated termination at mRNAs also remains unclear. We and others have suggested that Integrator binds paused RNAPII and drives termination to prevent polymerase release into gene bodies, thereby attenuating gene activity (Elrod et al., 2019; Fianu et al., 2021; Huang et al., 2020; Lykke-Andersen et al., 2020). Contrasting models assert that Integrator recognizes RNAPII that becomes arrested or ‘stuck’ in early elongation. In this model, Integrator removes non-productive RNAPII obstructing the DNA template, stimulating gene activity (Beckedorff et al., 2020; Gardini et al., 2014; Yue et al., 2017). Questions thus remain concerning whether Integrator predominantly represses gene expression, stimulates it, or plays a locus-specific role (Kirstein et al., 2021). Interestingly, regardless of the species used or approach for Integrator depletion, genes within growth and stress-responsive pathways have emerged as recurrent targets of Integrator activity (Beckedorff et al., 2020; Dasilva et al., 2021; Elrod et al., 2019; Gardini et al., 2014; Huang et al., 2020; Skaar et al., 2015; Stadelmayer et al., 2014; Tatomer et al., 2019; Yue et al., 2017). Consistent with the importance of these pathways in cellular homeostasis and disease, Integrator subunits have been implicated in cancer and a number of developmental disorders (Rienzo and Casamassimi, 2016), highlighting the importance of understanding Integrator function.
Studies of Integrator activity to date have relied on long-term (48–96 h) RNAi-based perturbations, such that some observed changes in gene expression might reflect indirect or compensatory effects (Jaeger and Winter, 2021). Thus, to dissect Integrator’s direct targets and effects, we developed a system to deplete INTS11 rapidly. We fuse a HaloTag to endogenous INTS11 in mouse embryonic stem cells (mESCs), enabling the use of HaloPROTAC3, a Proteolysis Targeting Chimera (PROTAC) that recruits the E3 ubiquitin ligase VHL to HaloTagged proteins for their rapid degradation (Buckley et al., 2015; Caine et al., 2020). Using this system, we demonstrate the role of Integrator as a universal terminator of promoter-proximal transcription that exerts the same attenuating activity at all RNAPII-transcribed loci regardless of biotype. Further, our results highlight the modular nature of the Integrator complex and show that endonuclease and phosphatase activities are separable, distinct functional units.
RESULTS
Establishment of INTS11 degron line
To interrogate the direct effects of INTS11 loss, we generated and validated a rapid INTS11 depletion system in mESCs. A HaloTag was integrated into the N terminus of the endogenous INTS11 gene using CRISPR-mediated genome editing (Figure 1A). PCR of genomic DNA confirmed homozygous insertion of the HaloTag (Figure S1A), and western blotting showed expression of INTS11Halo at levels comparable to untagged INTS11 (Figure 1B). Tagging of INTS11 did not affect cell viability or the ability of INTS11Halo mESCs cells to differentiate to epiblast-like cells (Figure S1B). Furthermore, labeling INTS11Halo with a fluorophore confirmed that INTS11Halo is localized primarily in the nucleus (Figure S1B) as anticipated, and we observed good agreement between ChIP-seq localization of endogenous INTS11 and INTS11Halo (Pearson’s R2=0.95). Homozygous tagging of INTS11 therefore does not detectably affect cell health or protein localization.
Figure 1. Generation and validation of a rapid INTS11 depletion system in mouse embryonic stem cells.

(A) Schematic depicting insertion of a HaloTag at the N terminus of endogenous INTS11 (top) and the PROTAC system (bottom).
(B) Western blots of INTS11 (67.8 kDa) and HaloTagged-INTS11 (104.3 kDa). Histone H3 is a loading control.
(C) Western blots of Integrator subunits from lysates of INTS11Halo cells treated with 500 nM PROTAC for the indicated time. Histone H3 is a loading control.
(D) RT-qPCR of total RNA isolated from cells treated as indicated for 4 h (n=3 per condition). Primer pairs amplify regions downstream of the snRNA TES. Bar graphs depict averages and standard deviations. P values from paired, two-sided t test. DMSO was set to 1.
(E) Metagene analysis of TT-seq coverage around active snRNA genes (N=37). Samples were treated for 4 h (n=2 per condition). Data outside gene bodies are shown as average reads per gene in 50 nt bins; bins within gene bodies are scaled to gene length, with 10 bins/gene.
(F) Boxplots depict TT-seq read density in the indicated regions for snRNAs shown in E. Boxes show 25th–75th percentiles and whiskers depict 1.5 times the interquartile range. P values from Wilcoxon matched-pairs signed rank test. Gene Body = TSS to TES; Readthrough = TES to +1kb downstream.
We next characterized the kinetics of INTS11 loss following treatment of INTS11Halo cells with a Halo-targeted PROTAC. We observe substantial depletion of INTS11Halo protein within 4 h of PROTAC treatment (Figure 1C); thus, this timepoint is used in further experiments unless otherwise noted. Due to the modular architecture of the Integrator complex, we wondered if other subunits are degraded concomitantly with INTS11. INTS4, which interacts with INTS11 in the endonuclease module, is depleted with similar kinetics upon PROTAC treatment. However, subunits in the Integrator core (e.g., INTS1) or other modules (e.g., INTS10, INTS3) show no notable change in protein levels during PROTAC treatment (Figure 1C and S1C) (Albrecht et al., 2018; Pfleiderer and Galej, 2021; Sabath et al., 2020). Thus, our experimental system acutely depletes the Integrator endonuclease module but not the remaining components of the Integrator complex.
To initially assess the effects of INTS11 loss, we evaluated snRNA 3’ end processing. We employed a reporter plasmid encoding GFP downstream of the U7 snRNA. In this system, loss of Integrator causes readthrough of RNAPII into GFP (Ezzeddine et al., 2011; Wu et al., 2017). PROTAC treatment of INTS11Halo cells increased GFP RNA and protein levels (Figures S1D–F). Furthermore, RT-qPCR reveals significant readthrough at endogenous snRNAs in INTS11Halo cells but not in parental cells upon PROTAC treatment (Figure 1D). We conclude that PROTAC treatment causes rapid and efficient loss of INTS11 activity.
To more broadly probe the consequences of INTS11 loss on snRNA transcription, we performed Transient Transcriptome Sequencing (TT-seq), a measure of recently synthesized RNA (Schwalb et al., 2016). INTS11Halo cells isolated after 4 h of DMSO or PROTAC treatment were spiked with 4sU-labeled Drosophila cells to enable absolute normalization. Analysis of TT-seq read coverage at active snRNA genes shows the anticipated increase in readthrough signal beyond the snRNA 3’ end after PROTAC treatment (Figure 1E, 1F). Intriguingly, the TT-seq signal is also significantly increased within snRNA gene bodies in PROTAC-treated INTS11Halo cells (Figure 1F), suggesting that loss of INTS11 broadly increases the transcription of snRNAs.
INTS11 globally attenuates promoter-proximal transcription
To analyze the consequences of acute INTS11 loss on nascent RNA production, we monitored actively engaged RNAPII at single-nucleotide resolution using Precision Run-on Sequencing (PRO-seq) (Kwak et al., 2013). INTS11Halo cells treated with DMSO or PROTAC for 4 h were spiked with Drosophila cells before library preparation to allow absolute normalization (Reimer et al., 2021). Metagene profiles of PRO-seq reads from the transcription start site (TSS) to the transcript end site (TES, where RNA cleavage occurs) of all active non-snRNA genes revealed a dramatic increase in promoter-proximal RNAPII levels upon PROTAC treatment (Figure 2A). To probe the breadth of this effect, we calculated the fold change in PRO-seq reads upon INTS11 degradation and generated heatmaps of the difference in signal (Figure 2B). These analyses indicate that INTS11 loss causes a global increase in RNAPII near active, annotated TSSs (Figures 2B and S2A). The changes observed within gene bodies are more variable: the most highly upregulated genes (Figure 2B, top of heatmap) exhibit increased PRO-seq reads across the entire gene body, while genes with more modest changes have increased RNAPII levels primarily at the 5’ end (Figure 2B).
Figure 2. Acute loss of INTS11 increases nascent RNA production near TSSs.

(A) Metagene analysis of PRO-seq signal for active annotated genes over 400 nt in length (N=16,036, does not include snRNAs) after 4 h of indicated treatment. Data outside gene bodies are shown as average reads per gene in 50 nt bins; bins within gene bodies are scaled to gene length, with 90 bins/gene.
(B) Heatmap representation of difference in PRO-seq signal after PROTAC treatment (Δ = PROTAC - DMSO) for genes shown in A. Genes are ranked by the fold change in PRO-seq signal from TSS (arrow) to TES (red octagon). Signal across gene bodies is shown in 90 bins, while 1 kb upstream and downstream regions are shown in 200 nt bins.
(C) Metagene analysis of INTS11 ChIP-seq signal for active annotated genes. Data outside gene bodies are shown as average reads per gene in 50 bp bins; bins within gene bodies are scaled to gene length, with 100 bins/gene.
(D) Readthrough Index (RI) was calculated from TT-seq data as indicated. Heatmaps depict normalized TT-seq signals in DMSO and PROTAC treated cells (left and middle) or relative difference (right) in 100 nt bins for mRNA genes with no significant differences in TT-seq signal in the 2kb window upstream of the TES (N=7,560). Genes with increased RI in INTS11-depleted cells are at the top.
Acute loss of INTS11 does not impact mRNA 3’ end formation
Studies using RNAi-mediated INTS11 depletion have proposed that Integrator works in conjunction with the CPA machinery at specific mRNAs to prevent transcription readthrough (Dasilva et al., 2021; Nojima et al., 2018b; Rosa-Mercado et al., 2021). However, the above data show no global difference between control and INTS11-degraded cells in PRO-seq signal near the TES or downstream (Figures 2A, 2B, and S2A). In agreement with this, ChIP-seq localization of INTS11 confirms a substantial peak of INTS11 occupancy near promoters, with much lower levels in gene bodies or 3’ ends (Figure 2C). Notably, long-term depletion of INTS11 can reduce the expression of CPSF73, the endonuclease subunit in the canonical CPA machinery (Dasilva et al., 2021; Davidson et al., 2020), suggesting the caveat of indirect effects. To rule out this possibility in our system, we confirmed using western blotting that no changes to CPSF73 levels are observed following 4 h of INTS11 depletion (Figure S2B), allowing us to focus on direct targets of the Integrator complex.
We calculated Readthrough Index (RI) for mRNA genes as a ratio of TT-seq reads downstream of the TES to reads upstream (Figure 2D). To avoid changes in gene body RNA synthesis from biasing our analysis, we focused on mRNAs with no significant changes in TT-seq signal 2 kb upstream of the TES (Figure 2D, STAR methods). Comparing RI in control and INTS11-degraded cells revealed very few genes with increased readthrough upon INTS11 loss (<3%), with similarly few genes showing decreased readthrough (<1%, Figure 2D, S2C). These findings are consistent with recent work indicating that similarly small proportions of genes displayed altered readthrough upon Integrator depletion (Lykke-Andersen et al., 2020). Furthermore, the modest changes we observed did not persist more than 2 kb downstream of TESs (Figure 2D, S2C), contrasting with observations following acute depletion of CPSF73, where readthrough continues for >10 kb (Eaton et al., 2020). Of note, even in the subset of genes where INTS11 loss affects TT-seq signal upstream of the TES (e.g., at upregulated genes), the signal downstream of the TES is the same in DMSO and PROTAC conditions (Figure S2D). We conclude that rapid degradation of INTS11 does not cause substantial defects in canonical RNA cleavage or termination near mRNA 3’ ends. Taken together, our assays of nascent transcription following rapid INTS11 degradation indicate that Integrator affects promoter-proximal polymerase at nearly all RNAPII-transcribed loci but has minimal effects around TESs.
Universal increase in early elongation complexes upon INTS11 degradation
As noted above, it remains unclear whether Integrator drives termination of actively engaged but paused RNAPII to attenuate gene transcription, or removes non-productive, arrested RNAPII to enable new initiation and successful transcription elongation (Beckedorff et al., 2020; Dasilva et al., 2021; Elrod et al., 2019; Gardini et al., 2014; Huang et al., 2020; Lykke-Andersen et al., 2020; Skaar et al., 2015; Stadelmayer et al., 2014; Tatomer et al., 2019; Yue et al., 2017). Importantly, these models make opposing predictions concerning the levels of actively engaged RNAPII capable of running-on in the PRO-seq assay following INTS11 loss. If Integrator-mediated termination removes active transcription complexes, then INTS11 depletion should increase PRO-seq signal near mRNA promoters. If, by contrast, INTS11 removes inactive RNAPII to make way for active polymerase, then INTS11 depletion should decrease PRO-seq signal near mRNA promoters and across gene bodies.
To address these questions, we first investigated the activity of INTS11 at protein-coding genes. Metagene plots of PRO-seq signal around mRNA promoters revealed a significant increase in actively engaged RNAPII upon PROTAC treatment (Figure 3A). Heatmaps of the difference in signal at all active mRNAs (Figure 3B) demonstrated that PRO-seq read densities were broadly and significantly increased upon PROTAC treatment in both promoter regions (TSS to +150 nt downstream) and early gene bodies (+250 to +2250 nt downstream) (Figure 3C). The promoter-proximal increase in signal upon INTS11 degradation occurs at 94% of genes, strongly supporting models wherein termination of RNAPII by Integrator reduces actively engaged RNAPII available to enter gene bodies. Notably, the increase in early elongation complexes and transcribing RNAPII upon PROTAC treatment persists for the first several kb of genes, but control and INTS11-degraded PRO-seq signals converge ~5 kb downstream of the TSS (Figure 3A). Heatmaps rank-ordered by gene length indicate that this pattern is consistent across protein-coding genes regardless of length (Figure 3B).
Figure 3. INTS11 loss increases promoter-proximal transcription at all RNAPII-transcribed loci.

(A) Metagene analysis of PRO-seq signal at active mRNA genes (N=13,057) upon 4 h of indicated treatment. Data are shown as average reads in 25 nt bins.
(B) Heatmap representation of difference in PRO-seq signal (Δ=PROTAC - DMSO) for mRNA genes (N=13,057). TSS is indicated by arrow. Data are shown in 100 nt bins.
(C) Boxplots depict PRO-seq read density in the indicated gene region for mRNAs (N=13,057). P values from Wilcoxon matched-pairs signed rank test.
(D-F) Same as A-C, but for 1,855 active, annotated lncRNA genes.
(G-I) Same as A-C, but for 8,284 uaRNA loci.
(J-L) Same as A-C, but for 9,571 intergenic sites of unannotated transcription identified as putative enhancers.
We next determined how INTS11 loss affected engaged RNAPII at promoters of active, annotated lncRNAs. Metagene profiles of PRO-seq signal show a substantial increase in RNAPII near lncRNA promoters upon INTS11 degradation, with elevated levels of RNAPII entering these genes (Figure 3D). Heatmaps reflecting the difference in PRO-seq signal upon PROTAC treatment reveal a widespread increase in RNAPII elongating into lncRNA genes (Figure 3E). The breadth of this effect is confirmed by quantitative analysis of PRO-seq reads in the promoter and early gene body windows (Figure 3F). Overall, no significant differences were observed in the effect of INTS11 loss on lncRNAs as compared to mRNAs.
As noted above, Integrator has been reported to act on short, unstable transcripts including upstream antisense RNAs (uaRNAs) and enhancer RNAs (eRNAs) (Lai et al., 2015; Lykke-Andersen et al., 2020). To extend our analyses to these unannotated RNA species, we identified TSSs for uaRNAs using our PRO-seq data (see STAR Methods, Figure S3A) and generated similar metagene plots, heatmaps, and quantitation of PRO-seq signal at these loci. These analyses demonstrated that RNAPII transcribing uaRNAs (Figure 3G–I) was affected by INTS11 loss in a manner that is indistinguishable from that seen at mRNAs. We next identified putative enhancers using PRO-seq data and the dREG algorithm (Danko et al., 2015) and found these sites to be enriched in features of active regulatory regions (Figure S3B). These sites, too, were broadly upregulated by INTS11 degradation (Figure 3J–L). Although these unannotated RNA loci do not have defined gene bodies, we nonetheless observe a significant and widespread increase of RNAPII within the downstream region that is comparable to that observed at annotated RNA species (Figures 3I, 3L).
These results strongly argue that INTS11 functions similarly at all RNAPII-transcribed RNA biotypes, serving as a promoter-proximal termination complex that restricts the level of paused RNAPII that escapes into the transcript body. We observed no differences in the behavior of RNAPII following acute depletion of INTS11 between protein-coding vs. non-coding loci or between loci that produce stable vs. unstable RNAs. The consistent observation at all loci investigated is that INTS11 degradation allows more RNAPII to actively elongate downstream.
RNAPII released into elongation upon INTS11 degradation fails to achieve full productivity
We next probed the fate of RNAPII that enters gene bodies in the absence of INTS11-mediated termination, focusing on mRNAs. We stratified genes >10 kb in length into four intervals and calculated the fold change in PRO-seq signal upon PROTAC treatment in each window (Figure 4A). As anticipated, the most significant increase in PRO-seq occurs near the promoter, and each subsequent gene region shows a diminished effect, with the window representing the most TES-proximal region exhibiting no substantive change in PRO-seq signal. These results suggest that RNAPII released into gene bodies upon depletion of INTS11 is competent to transcribe a short distance but fails to productively elongate across longer intervals. Inspection of both PRO-seq and TT-seq data at individual genes supports this model, with increased signal extending across the short gene Gstt2 (Figure 4B, top). In contrast, the longer gene Lrriq3 shows an elevated signal only within the 5’ region (Figure 4B, bottom).
Figure 4. Loss of INTS11 stimulates transcription of short RNAs.
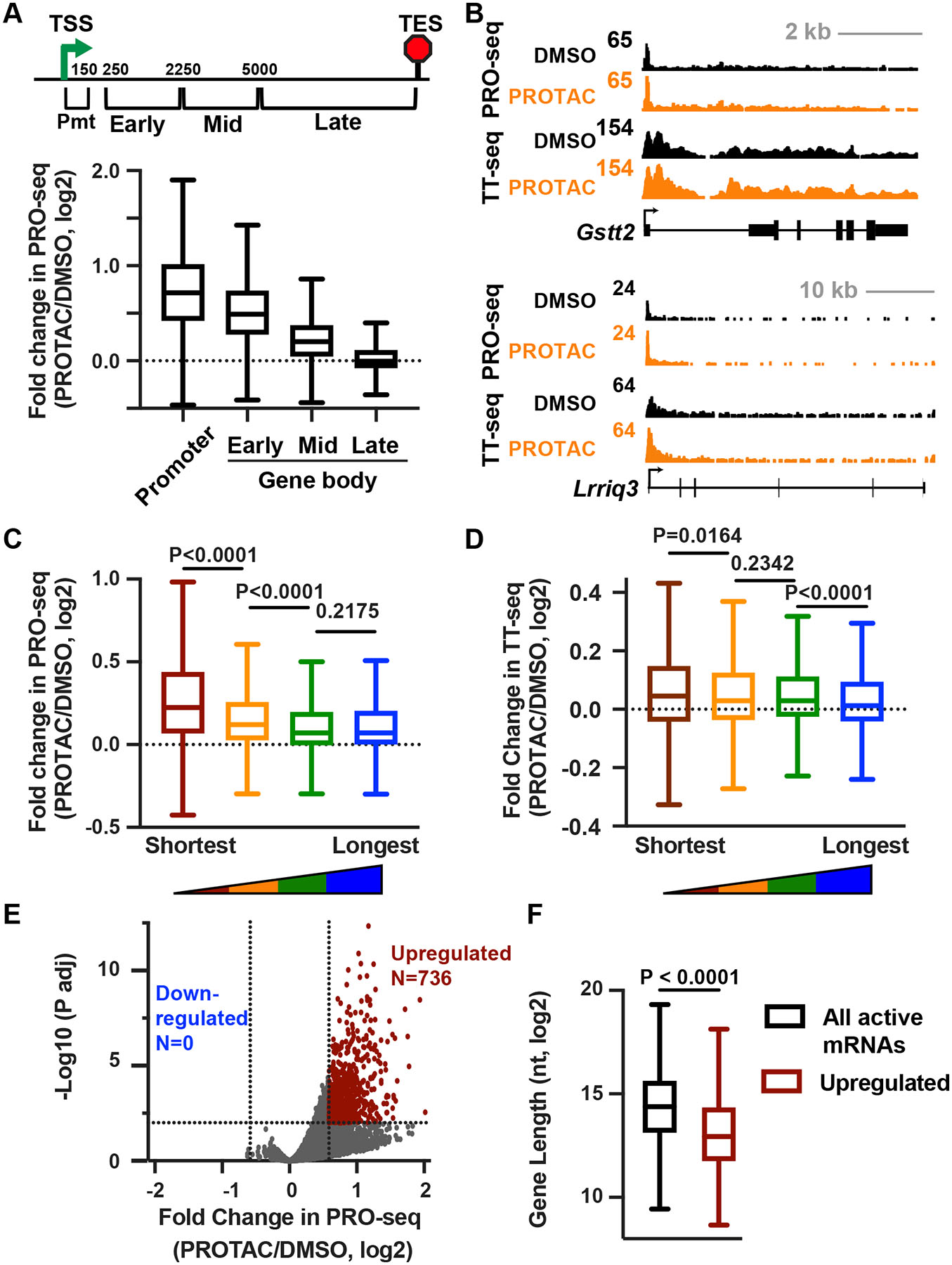
(A) Schematic at top depicts locations of windows across mRNA genes >10 kb (N=9,468) with respect to the TSS. Boxplots show the fold change in PRO-seq reads in each window.
(B) Browser shots of example genes that are short (top) or long (bottom).
(C) Shown is the change in gene body (+250 nt to TES) PRO-seq signal after PROTAC treatment for mRNAs (N=13,057) divided into length quartiles. P values from Mann-Whitney test.
(D) The change in gene body TT-seq signal (exonic reads from +250 nt to TES) after PROTAC treatment for mRNAs divided into length quartiles as in C. P values from Mann-Whitney test.
(E) Volcano plot shows fold changes and adjusted P values for active mRNA genes (N=13,057), counting PRO-seq reads from TSS to TES. Affected genes are those with fold change > 1.5 and P adj < 0.01.
(F) Boxplots depict the gene lengths of all (N=13,057) or significantly upregulated (N=736) mRNAs. P value from Mann-Whitney test.
In A, C, and D, boxes show 25th–75th percentiles and whiskers depict 1.5 times the interquartile range.
The above data predict that acute INTS11 loss would specifically upregulate short genes but that longer transcripts would display diminished effects. To test this idea, we separated active mRNA genes into quartiles by length and graphed the fold change in gene body PRO-seq signal for each quartile (Figure 4C, from TSS+250 to TES; see Figure S4A for TSS to TES). As anticipated, we find that short genes are the most highly upregulated by INTS11 loss, with longer length quartiles showing reduced effects. To confirm this result, we evaluated TT-seq signal upon INTS11 loss across the gene length quartiles (Figure 4D). These data reveal a pattern similar to PRO-seq.
To stringently define a set of transcripts significantly affected in PROTAC-treated cells, we used PRO-seq data to define differentially expressed genes. We identified 736 strongly upregulated genes and zero downregulated genes upon INTS11 degradation (Figure 4E), underscoring that Integrator serves primarily to repress gene transcription. Importantly, significantly upregulated genes are much shorter than typical mRNA genes (Figure 4F). We find that Gene Ontology (GO) terms enriched in upregulated genes are often comprised of particularly short genes compared to all active mRNAs (Figure S4C). Moreover, within these GO categories, genes upregulated upon INTS11 degradation are significantly shorter than the median of the category. Taken together, our data suggest that the genes rapidly and directly affected by INTS11 loss might be impacted because of their short gene length, rather than sequence or epigenetic features.
Enriched GO terms among genes upregulated following acute degradation of INTS11 include pathways widely accepted to be regulated by Integrator based on long-term depletion studies (Kirstein et al., 2021) (Figure S4B). These results suggest that signaling molecules that contribute to the activation of these pathways (e.g., transcription factors, kinases) might be encoded by short transcripts. To test this idea, we performed GO analysis on genes in the shortest length quartile. This analysis reveals a striking enrichment in factors that bind DNA and RNA (Figure S4D). Accordingly, we found 48 transcription factors (TFs) among the short transcripts upregulated by INTS11 loss, including master regulators of several distinct pathways (Table S1). Upregulated TFs include multiple targets of the MAPK pathway, such as FOS, JUN, and MYC (Wagner and Nebreda, 2009; Whitmarsh and Davis, 1996). These data imply that TFs and regulators encoded by short genes activated upon rapid INTS11 degradation could initiate signaling cascades that are perpetuated across longer experimental time scales.
Effects on gene activity of short-term vs. long-term INTS11 depletion are different
To probe the connection between the short transcripts upregulated upon rapid loss of INTS11 with the gene expression changes observed following long-term depletion of INTS11, we knocked down INTS11 in mESCs using siRNA. INTS11 was effectively depleted by 48 h of siRNA treatment (Figure 5A) compared to a non-targeting control siRNA (siNT). The loss of INTS11 results in lower levels of INTS4, which is also in the endonuclease module, but not core Integrator subunits or members of other modules (Figure 5A). Cells harvested at this time point were spiked to allow absolute normalization, and total RNA-seq performed. Differential expression analysis of mRNA genes identified 1,694 genes upregulated and 1,681 genes downregulated in cells treated with INTS11 siRNA compared to siNT (Figure 5B). The equivalent number of upregulated and downregulated genes observed following siINTS11 is consistent with previous mammalian cell culture studies using long-term depletion strategies (Beckedorff et al., 2020; Huang et al., 2020), but differs markedly from our results using acute INTS11 degradation (Figure 4E). Examination of individual loci indicates that many genes affected during long-term depletion of INTS11 are unaffected in shorter-term degradation experiments (e.g., Lif, Figure 5C, top), suggesting the potential for indirect effects in our siRNA experiments. In agreement with this, gene expression changes following short-term and long-term loss of INTS11 show little overall correlation (Pearson’s r2=0.03), with no meaningful overlap of genes downregulated following INTS11 siRNA with genes affected by INTS11 degradation (Figure 5D, P=0.9). However, there was a modest overlap between genes upregulated following short-term vs. long-term loss of INTS11 (Figure 5D, P=1.74 × 10−05; e.g., Id2, Figure 5C, bottom), suggesting a connection between the rapid gene activation observed in our degron system and the longer-term effects of INTS11 loss.
Figure 5. Similar pathways affected by acute INTS11 loss and long-term INTS11 depletion.
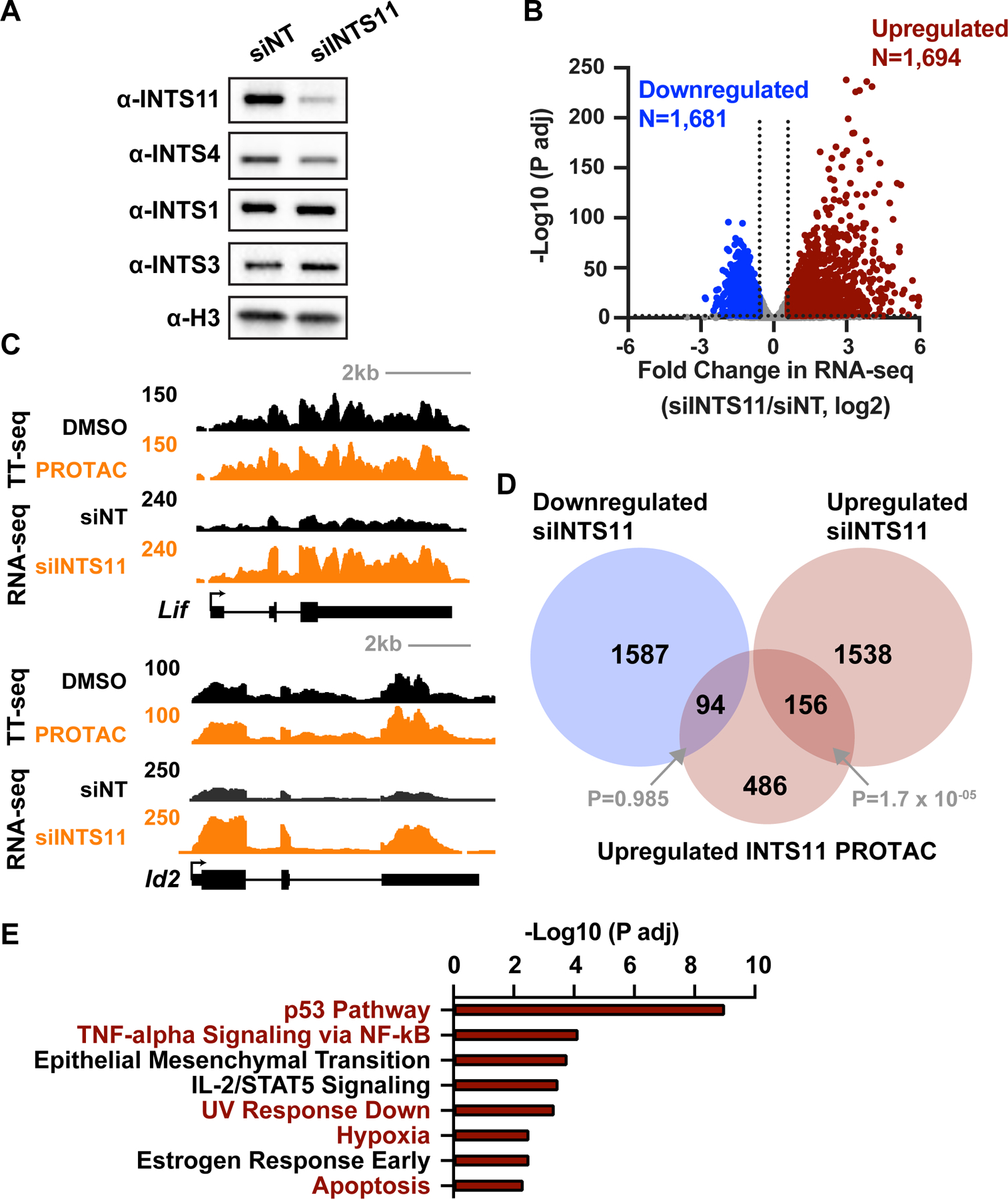
(A) mESCs were treated with non-targeting (siNT) or INTS11-targeting (siINTS11) siRNA for 48 h and harvested for western blots. Histone H3 is a loading control.
(B) Gene expression levels in cells depleted of INTS11 with siRNA as compared to siNT using total RNA-seq (n=3 per condition). Volcano plot shows fold changes and adjusted P values for active mRNA genes that did not display changes in RNA splicing (N=10,871). Affected genes are those with a fold change > 1.5 and P adj < 0.01.
(C) Example browser shots of (top) Lif, which is upregulated in RNA-seq from cells treated with INTS11 siRNA (48 h), but not TT-seq following INTS11 PROTAC treatment (4 h), and (bottom) Id2, which is upregulated under both conditions.
(D) Venn diagram depicting the overlap of genes affected by acute (4 h) INTS11 degradation vs. longer term INTS11 loss (48 h siRNA). P values for overlap were determined by hypergeometric test.
(E) Gene Ontology (GO) analysis of 1,694 mRNA genes upregulated upon siINTS11, as defined in B. MSigDB terms with P adj < 0.05 are shown. Terms in red were also found in GO analysis of upregulated genes from PRO-seq, as in Figure S4B.
To understand how increased transcription of a set of short mRNAs might lead to the entire repertoire of gene expression changes observed using RNAi strategies, we considered that the TFs, RNA binding proteins, and kinases upregulated rapidly after INTS11 protein loss might initiate cellular responses that lead to enduring pathway activation. In this manner, the GO categories associated with genes upregulated within 4 h of INTS11 depletion might remain enriched among genes with elevated expression in siINTS11-treated cells. To evaluate this possibility, we determined GO term enrichment of the 1,694 genes upregulated in cells following siINTS11 (Figure 5E), using the same parameters for analysis of PRO-seq data from INTS11Halo cells. Of the eight GO categories with significant enrichment among genes upregulated following siINTS11, five are also enriched upon acute INTS11 degradation (Figure 5E, compared to S4B), including the p53, TNF-α/NFκb, and hypoxia pathways, apoptosis, and UV response. Additionally, five GO terms with significant enrichment among genes downregulated upon siINTS11 were enriched after acute INTS11 loss (Figure S5A). Notably, many of these pathways show little basal activity in wildtype ESCs, suggesting that loss of INTS11 causes aberrant activation of signaling networks. For example, we find evidence of estrogen signaling in ESCs treated with siINTS11, in agreement with an elevated synthesis of FOS, JUN, HES1, MYC, and ARID5A upon rapid loss of INTS11 (Figure S5B, S5C, S5D).
Signaling pathways directly activated upon rapid INTS11 degradation instigate changes in gene expression observed during longer-term INTS11 depletion
We propose that the constellation of genes affected by long-term depletion of INTS11 includes both direct targets of Integrator and indirect effects arising from aberrant activation of signaling pathways. To test this model further, we performed total RNA-seq on INTS11Halo cells subjected to PROTAC treatment for an extended time (16 h). This time frame allows the persistent effects of INTS11 PROTAC to accumulate and functionally affect steady-state RNA levels. Differential expression analysis demonstrates that hundreds of genes are upregulated and downregulated under these conditions (Figure 6A). Importantly, the genes encoding TFs FOS and JUN remain upregulated (Figure 6B), indicating that INTS11 loss causes prolonged activation of these TFs. Accordingly, western blots for FOS and JUN proteins in cells depleted of INTS11 by extended PROTAC treatment (24 h, Figure 6C) showed marked increases in expression.
Figure 6. Sustained upregulation of FOS and JUN transcription factors activates AP-1 transcriptional program in INTS11 depleted cells.
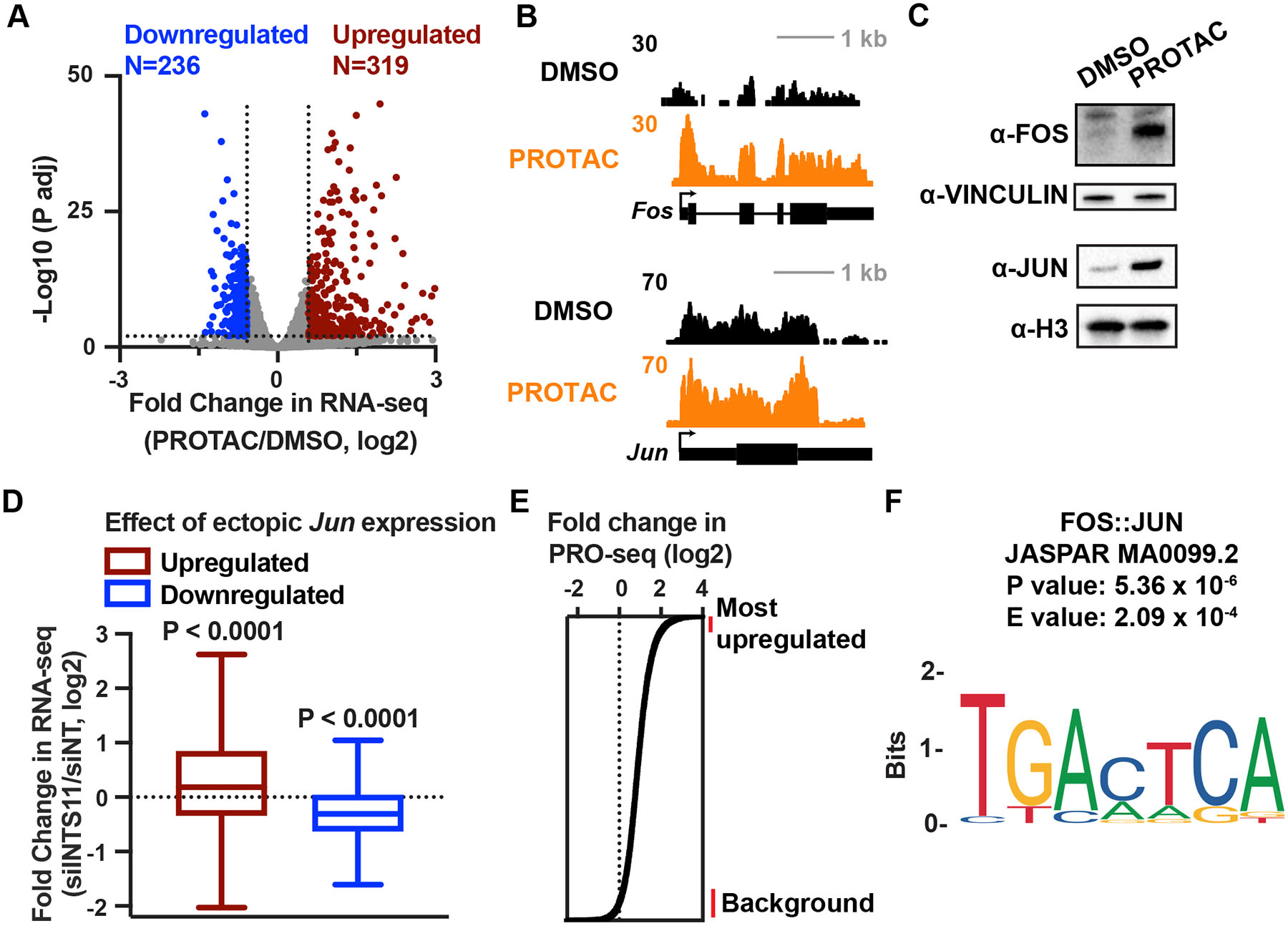
(A) Gene expression levels in cells depleted of INTS11 with PROTAC for 16 h as compared to DMSO, using total RNA-seq (n=3 per condition). Volcano plot shows fold changes and adjusted P values for active mRNA genes shown in Figure 5B (N=10,871). Affected genes are those with a fold change > 1.5 and P adj < 0.01.
(B) RNA-seq browser shots of Fos and Jun from cells treated with DMSO or PROTAC for 16 h.
(C) Western blot from lysates of INTS11Halo cells treated with PROTAC or DMSO for 24 h. Vinculin and Histone H3 are loading controls.
(D) Boxplots depict fold change in RNA-seq after 48 h of siINTS11 for genes upregulated (N=1,000) or downregulated (N=903) by ectopic Jun expression in mESCs (Liu et al. 2015). Boxes show 25th–75th percentiles and whiskers depict 1.5 times the interquartile range. P value from Wilcoxon signed-rank test using a theoretical median of 0.
(E) Enhancers are rank ordered based on fold change in PRO-seq in the eTSS to +150 nt window. Lines at right indicate groups of enhancers used for motif search, comparing the 500 most upregulated enhancers to 1,000 unaffected enhancers.
(F) The motif most significantly enriched at highly upregulated enhancers after INTS11 depletion is for AP-1. Sequences 200 bp upstream of each enhancer TSS were used.
To address whether gene expression changes in INTS11-depleted cells might reflect the increased activity of FOS and/or JUN proteins we evaluated RNA-seq data from mESCs after the ectopic overexpression of Jun (Liu et al., 2015). Greater than half of genes upregulated by ectopic Jun expression are also upregulated in INTS11 depleted cells. Moreover, more than 75% of genes downregulated by ectopic Jun activity are downregulated in INTS11 depleted cells (Figure 6D). These findings imply that a substantial portion of gene expression changes following prolonged INTS11 loss, especially gene repression, is a consequence of JUN activation.
Since FOS and JUN often affect gene activity by binding at distal enhancers, we asked whether changes in enhancer transcription in PRO-seq (after 4 h of PROTAC treatment) might reflect increased FOS/JUN binding. We searched for enriched TF motifs at the most upregulated enhancers, as determined by PRO-seq signal for eRNA synthesis (Figure 6E). Notably, at the highly activated enhancers following INTS11 PROTAC, the most enriched motif was for a FOS/JUN heterodimer (Figure 6F). Thus, our data support a prominent role for these TFs in shaping the transcriptional landscape in cells depleted of INTS11.
Loss of INTS11 causes reduced RNAPII elongation rate
A key feature of the transcriptional response to rapid INTS11 degradation is an increase of active RNAPII in the promoter-proximal region and the release of RNAPII into gene bodies (Figure 7A). However, increased levels of RNAPII entering genes in the absence of INTS11 are not sustained to the end of long genes, suggesting defects in processive elongation. First, we evaluated if Integrator facilitates RNAPII passage through the first (+1) nucleosome (Beckedorff et al., 2020). We identified the position of nucleosomes using MNase-seq data, yielding high confidence localization of the +1 nucleosome at 11,214 active mRNA genes (Figure 7B). We plotted the PRO-seq signal from DMSO- and PROTAC-treated INTS11Halo cells aligned around these TSSs (Figure 7A) or the +1 nucleosome dyad (Figure 7B). Despite substantially more early elongation complexes in PROTAC-treated cells than in control cells, the peak of paused RNAPII is at the same position with respect to the TSS (Figure 7A) and differs only by 1 nt with respect to the +1 dyad (Figure 7B). Thus, acute depletion of INTS11 does not alter the predominant position of RNAPII pausing or the profile of transcription into the first nucleosome. To measure whether INTS11 affects the ability of RNAPII to traverse the +1 nucleosome, we calculated a Passage Index as the ratio of PRO-seq signal downstream of the +1 dyad to signal upstream (Figure S7A). As suggested qualitatively by the metagene plots (Figure 7B), this analysis reveals no quantitative difference in Passage Index between control and INTS11-degraded conditions (Figure S7A), indicating that Integrator does not play a central role in enabling passage of RNAPII through the first nucleosome.
Figure 7. INTS11 depletion reduces elongation rate.
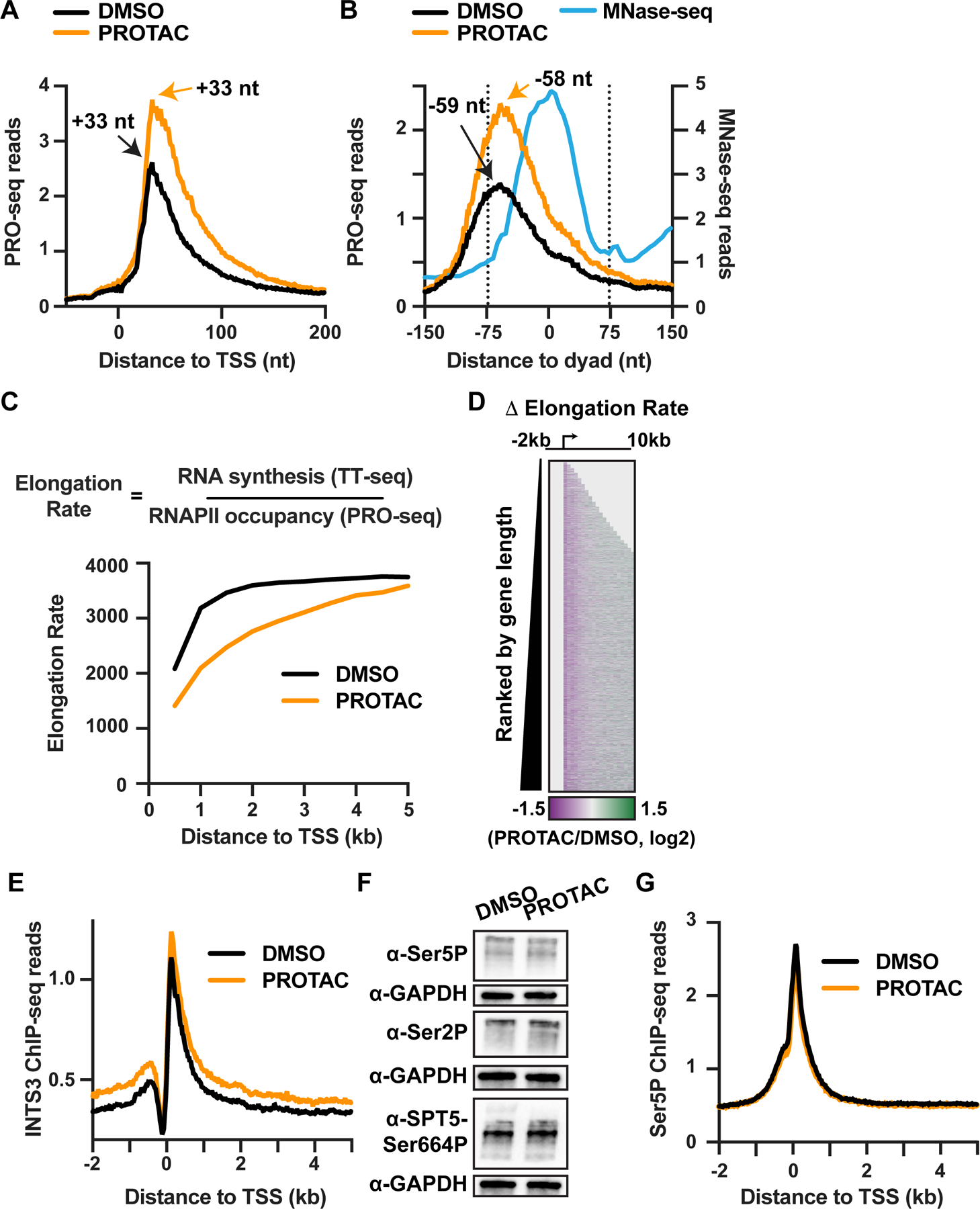
(A and B) Metagene analyses of PRO-seq signal from INTS11Halo cells treated with DMSO or PROTAC for 4 h at mRNA genes with well-defined +1 nucleosomes (N=11,214). Data are shown as average reads per gene aligned around (A) TSSs or (B) the +1 nucleosome dyad, at single nucleotide resolution for PRO-seq and in 5 nt bins for MNase-seq (with reads depicted at read center). The peak location of PRO-seq reads, corresponding to (A) the position of paused RNAPII or (B) the initial position of stalling in the +1 nucleosome, are shown.
(C) Analysis of elongation rate from INTS11Halo cells treated with DMSO or PROTAC for 4 h. Data are shown as average elongation rate in 500 nt bins.
(D) Heatmap representation of fold change in elongation rate after PROTAC treatment for active mRNA genes (N=13,055). TSS is indicated by arrow. Data are shown in 500 nt bins. Bins upstream of the TSS and downstream of the TES are shaded in light gray.
(E) Metagene analysis of average INTS3 ChIP-seq signal around mRNA TSSs (N=13,055), from INTS11Halo cells treated with DMSO or PROTAC for 4 h. Data are shown in 25 nt bins.
(F) Western blots for phosphorylated forms of the RNAPII CTD (Ser5 or Ser2) and SPT5 after 4 h DMSO or PROTAC treatment of INTS11Halo cells. GAPDH is a loading control.
(G) Metagene analysis of Ser5P ChIP-seq signal around mRNA TSSs (N=13,055), from INTS11Halo cells treated as in (F). Data are shown in 25 nt bins.
To probe other possibilities for the defect in elongation observed in INTS11-depleted cells, we investigated the nature of the decline in PRO-seq reads observed across early gene bodies (Figure 3A). The reduction in PRO-seq signal within this region, seen in both DMSO- and PROTAC-treated cells (Figure S7B, bottom), is often attributed to premature termination (Kamieniarz-Gdula and Proudfoot, 2019). However, a lower PRO-seq signal at each position could also reflect faster elongation rates achieved as RNAPII transitions to productive elongation (Vos et al., 2018). We used our TT-seq data to measure RNA output in control and INTS11-degraded cells to discriminate between these possibilities, focusing on genes >5 kb in length. Interestingly, we found that TT-seq read coverage was comparable between control and INTS11-depleted cells across the first 5kb of average genes (Figures S7B and S7C, top), suggesting that similar amounts of RNA were synthesized across this window. This result is not consistent with high termination levels in either PROTAC or DMSO conditions and instead suggests that RNAPII elongation rate increases across the first several kb of the gene body in both conditions.
To derive a proxy for RNAPII elongation rate, we calculated the ratio of TT-seq signal (RNA synthesis) to PRO-seq signal (RNAPII occupancy) as described previously (Caizzi et al., 2021; Žumer et al., 2021). We find that the RNAPII elongation rate increases substantially over the first 2 kb in control cells, reaching a plateau after this position (Figure 7C). In INTS11-degraded cells, however, the elongation rate starts lower and increases more slowly, failing to reach a plateau within 5 kb (Figure 7C). Indeed, the overall RNAPII elongation rate in INTS11-degraded cells is significantly lower than in control cells (Figure S7D). To confirm the breadth of this effect, we generated a heatmap of the fold change in elongation rate upon INTS11 loss for all active mRNAs. We find that the RNAPII elongation rate is broadly lower in PROTAC-treated cells (Figure 7D) in the promoter-proximal region and early gene body, independent of gene length. These data imply that the increase in RNAPII elongation rate that generally accompanies the transition from pausing to productive elongation fails to occur in cells lacking INTS11, rendering RNAPII less efficient at RNA synthesis.
Rapid degradation of INTS11 does not dissociate other Integrator subunits from RNAPII
To probe why RNAPII would fail to achieve productive RNA synthesis upon loss of INTS11, we considered that Integrator contains at least two catalytic activities to reduce RNAPII processivity and elongation: the endonuclease and the phosphatase. We wondered whether these Integrator modules might be functionally independent. In support of this idea, recent studies demonstrated that depletion of different Integrator subunits yields distinct transcriptional outcomes (Pan et al., 2022; Replogle et al., 2022). Indeed, while degradation of INTS11 reduces levels of INTS4 within the endonuclease module (Figure 1C), several other INT proteins are not sensitive to INTS11 loss (e.g., INTS3, INTS1, Figure S1C). Thus, to gain insight into the localization and action of remaining Integrator subunits following INTS11 depletion, we performed spike normalized ChIP-seq for INTS3, a subunit that associates with INTS6 in the phosphatase module (with which it also participates in the SOSS1 complex, Fianu et al., 2021; Jia et al., 2021; Li et al., 2021). INTS3 ChIP-seq signal around mRNA TSSs shows a peak in occupancy near the TSSs of mRNA genes and the nearby upstream antisense RNA (Figure 7E), as observed for INTS11 (Figure 2C). Interestingly, we observed an increase, rather than a decrease, in the INTS3 ChIP signal in PROTAC-treated cells (Figure 7E), suggesting that INTS3 remains associated with RNAPII. To account for the increase in RNAPII levels observed in INTS11 PROTAC cells, we calculated the ratio of INTS3 and INTS11 ChIP-seq signals near promoters to the PRO-seq levels in this region for DMSO and PROTAC conditions. This confirms a large reduction of INTS11 signal upon PROTAC treatment with a significantly smaller effect on INTS3 (Figure S7E). Thus, we conclude that INTS3, and by extension, other subunits of the Integrator complex, can continue to associate with RNAPII even after INTS11 is lost.
Since INTS3 localizes to promoters in INTS11-degraded cells, we wondered if the Integrator phosphatase module might also remain associated and retain its activity on polymerases that would otherwise be targeted for Integrator-mediated termination. Previous reports showed that Integrator-associated PP2A dephosphorylates the elongation factor SPT5 and RNAPII CTD (Huang et al., 2020; Vervoort et al., 2021; Zheng et al., 2020), removing stimulatory phosphate groups added by transcriptional kinases. We could not obtain antibodies against mouse INTS6 or INTS8 that worked reliably. Therefore, as a proxy for the activity of PP2A on early elongation complexes, we tested whether INTS11 degradation alters the phosphorylation status of SPT5 and the RNAPII CTD using western blots. Strikingly, despite increased levels of paused RNAPII released into genes upon PROTAC treatment, we observed no concomitant increases in phosphorylated SPT5 or RNAPII CTD (Figure 7F). In agreement with this, ChIP-seq revealed no increase in the levels of Ser5P RNAPII around mRNA TSSs after INTS11 loss (Figures 7G and S7F). These findings imply that the Integrator phosphatase module interacts with RNAPII and is functional even in the absence of the endonuclease. Consequently, without appropriate phosphorylation on SPT5 and the CTD, the RNAPII complexes released into gene bodies by loss of INTS11 would fail to associate with factors such as SPT6 (Chun et al., 2019), preventing fully productive elongation (Narain et al., 2021; Žumer et al., 2021).
DISCUSSION
Using acute protein degradation, we demonstrate that INTS11 attenuates promoter-proximal transcription at nearly all RNAPII-transcribed loci, driving premature transcription termination to suppress release of paused RNAPII into productive RNA synthesis. While our data are consistent with aspects of previous work using long-term depletion or perturbation strategies, our use of a rapid depletion system has yielded new insights, as detailed below. First, long-term loss of Integrator subunits leads to both up- and downregulation of mRNA genes (Figure 5B), suggesting both inhibitory and activating functions (Beckedorff et al., 2020; Gardini et al., 2014; Huang et al., 2020; Lykke-Andersen et al., 2020; Stadelmayer et al., 2014; Vervoort et al., 2021). In contrast, our rapid depletion of INTS11 exclusively causes upregulation of transcription (Figure 4E), indicating that the direct function of INTS11 in this system is to repress, rather than facilitate, transcription.
Second, it has been suggested that specific mRNA genes, such as stress and signal-responsive genes, are selective Integrator targets, despite a lack of evidence for gene-specific Integrator recruitment (Gardini et al., 2014; Huang et al., 2020; Kirstein et al., 2021). Our results confirm that rapid degradation of INTS11 upregulates many TFs, kinases, and regulators of signal responses. However, rather than this upregulation reflecting gene-specific activities of Integrator, increased transcription levels are a general feature of short genes. Thus, we posit that the apparent selectivity of Integrator for particular signaling pathways is primarily due to gene length. In support of this model, we find that there is a genomic bias for rapidly inducible and stress-responsive factors to be encoded by short transcripts with short introns (Lopes et al., 2021, Figure S4D). Notably, we demonstrate that much of the downregulation of transcription observed upon long-term INTS11 depletion could be an indirect consequence of activation of JUN.
Third, current models suggest that Integrator activity is guided by interactions with sequence-specific TFs and motifs in nascent RNA. These models stem from early work on Integrator at snRNAs, where it was proposed that the complex is recruited by the TF SNAPc and INTS11 cleavage activated by the 3’ box motif in RNA (Baillat and Wagner, 2015; Baillat et al., 2005). However, sequences resembling the 3’ box could not be detected near most Integrator target genes (Lykke-Andersen et al., 2020) and recent structural studies found no evidence of this specificity (Fianu et al., 2021; Sabath et al., 2020). Furthermore, Integrator does not form contacts with DNA in the structures, and modelling of RNA does not provide evidence for an RNA sequence-specific recognition element near the RNAPII exit channel (Fianu et al., 2021). Integrator forms stable complexes in vitro with purified paused RNAPII including SPT5 and NELF, and additional interactions with TFs would be sterically challenging to accommodate. Together with our findings that Integrator acts globally at RNAPII loci, these data imply that no specific association with TFs, DNA, or RNA elements is required for Integrator recruitment. Instead, we suggest that Integrator is broadly recruited to paused RNAPII as a mechanism to prevent pause release and limit the transition to productive elongation. Importantly, this suggests that gene activation could involve mechanisms to prevent Integrator activity and promote pause release. In agreement with this model, recent work reveals that proteins that bind m6A-modified RNA can prohibit Integrator-mediated termination (Xu et al., 2022).
Fourth, Integrator was suggested to function differently at distinct RNA biotypes although the mechanisms underlying such differences were elusive (Gardini et al., 2014; Lai et al., 2015; Lykke-Andersen et al., 2020). However, our data support a unified Integrator function across all RNA species transcribed by RNAPII (Figure 3). In this regard, we note that even snRNA genes show a similar transcriptional response to INTS11 degradation as other genes, with an increase in transcription near promoters that continues across gene bodies (Figure 1E, 1F). Furthermore, TT-seq from INTS11-degraded cells shows evidence of accurate RNA cleavage at the snRNA TES, consistent with recent work demonstrating that snRNA 3’ end formation is not as reliant on Integrator as previously assumed (Davidson et al., 2020). Further work is warranted to clarify the role of INTS11 in snRNA biogenesis.
Finally, our data support the emerging concept that Integrator displays functional and physical modularity (Pan et al., 2022; Replogle et al., 2022). We find that RNAPII released into elongation in the absence of INTS11 activity fails to accumulate phosphorylation on RNAPII and SPT5 or transition to productive elongation. This is in stark contrast to the depletion of the Integrator phosphatase module, which leads to hyperphosphorylation of RNAPII and SPT5 and facilitated elongation (Hu et al., 2021; Huang et al., 2020; Vervoort et al., 2021; Zheng et al., 2020). More work will be required to understand the assembly of different Integrator modules and how their activities are coordinated. We envision that in the absence of stimulatory phosphate groups, elongation factors like SPT6 or PAF1 are unable to associate with RNAPII, reducing RNAPII elongation potential. Indeed, recent reports suggest a physical and functional connection between Integrator and the PAF complex, though the exact nature of such interactions remains unclear (Fianu et al., 2021; Liu et al., 2022; Wang et al., 2022). We note that the kinetic competition between elongation and termination could render the slowly elongating, hypo-phosphorylated RNAPII susceptible to termination by other factors, for example the CPA machinery (Davidson et al., 2020).
In summary, our work reveals Integrator as a central regulator of promoter-proximal transcription, governing the balance between pause release and premature termination to tune gene activity. Its role is consistent across RNAPII-transcribed loci, although the outcome of this activity on each RNA species may differ. For example, INTS11 activity at mRNAs and lncRNAs attenuates the synthesis of these long transcripts, allowing Integrator to repress expression of these genes. In contrast, inherently short transcripts such uaRNAs and eRNAs may be impacted differently by promoter-proximal termination, with Integrator potentially stimulating transcription re-initiation at these loci. As clearer roles for such non-coding RNA species emerge, a better understanding the role of INTS11 in the biogenesis of these RNAs will be an important area of study.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Karen Adelman (Karen_Adelman@hms.harvard.edu).
Materials Availability
Unique and stable reagents generated in this study are available upon request.
Data and Code Availability
All genomic datasets generated in this manuscript have been deposited to GEO under accession GSE200702.
Original scripts described have been deposited to Zenodo and DOIs are provided in the Key Resources Table.
Further information required for reanalysis of data reported here will be available upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| INTS1 | Bethyl Labs | RRID: AB 2127258; Cat # A300–361A | |
| INTS3 | Bethyl Labs | RRID: AB 2127274; Cat # A302–051A | |
| INTS4 | Bethyl Labs | RRID: AB 937909; Cat # A301–296A | |
| INTS10 | Proteintech | RRID: AB 2127260; Cat # 15271–1-AP | |
| INTS11 | Sigma | RRID: AB 10600425; Cat # HPA029025 | |
| HaloTag | Promega | RRID: AB 2688011; Cat # G9211 | |
| CPSF73 | Bethyl Labs | RRID: AB 2084528; Cat # A301–091A | |
| c-Jun (60A8) | Cell Signaling Technology | RRID: AB 2130165; Cat # 9165 | |
| c-Fos (9F6) | Cell Signaling Technology | RRID: AB 2247211; Cat # 2250 | |
| RNA Pol II CTD Ser2-p (3E10) | Millipore | RRID: AB 10627998; Cat # 04–1571 | |
| RNA Pol II CTD Ser5P (3E8) (for Western) | Active Motif | RRID: AB 2687451; Cat # 61085 | |
| RNA Pol II CTD Ser5P (for ChIP-seq) | Abcam | RRID: AB 449369; Cat # AB5131 | |
| Spt5-Ser664P | (Huang et al., 2020) | N/A | |
| Histone H3 | Abcam | RRID: AB 302613; Cat # AB1791 | |
| GAPDH | Proteintech | RRID: AB 2263076; Cat # 10494–1-AP | |
| Vinculin (EPR8158) | Abcam | RRID: AB 11144129; Cat # AB129002 | |
| Chemicals, Peptides, and Recombinant Proteins | |||
| LIF | Cell Guidance Systems | Cat # GFM200 | |
| PD0325901 | Reprocell | Cat # 04–0006 | |
| CHIR99021 | Reprocell | Cat # 04–0004 | |
| Fibronectin | Millipore | Cat # FC010 | |
| bFGF | PeproTech | Cat # 100–18B | |
| Biotin-11-NTPs | Perkin Elmer | Cat # NEL54(2/3/4/5)001 | |
| 4-thiouridine | Tocris | Cat # 37005 | |
| HaloPROTAC3 | Promega | Cat # GA3110 | |
| Janelia Fluor 646 | Gift from Talley Lambert (HMS) | N/A | |
| Janelia Fluor X549 | Gift from Stephen Blacklow (HMS) | N/A | |
| Phosphatase Inhibitor Cocktail 2 | Sigma | Cat # P5726 | |
| Phosphatase Inhibitor Cocktail 3 | Sigma | Cat # P0044 | |
| Critical Commercial Assays | |||
| Illumina TruSeq Stranded Total RNA Library Prep Gold | Illumina | Cat # 20020598 | |
| NEB Next Ultra II DNA library kit | NEB | Cat # E7103S | |
| Deposited Data | |||
| Raw and analyzed data | This paper | GEO: GSE200702 | |
| MNase-seq | (Henriques et al., 2018) | GEO: GSE85191 | |
| H3K27Ac ChIP-seq | (Vlaming et al., 2022) | GEO: GSE178230 | |
| H3K4me3 ChIP-seq | (Vlaming et al., 2022) | GEO: GSE178230 | |
| H3K4me1 ChIP-seq | (Buecker et al., 2014) | GEO: GSE56098 | |
| Experimental Models: Cell Lines | |||
| F121–9 | Jaenisch/Gribnau labs | 4DNSRMG5APUM | |
| INTS11Halo | This paper | N/A | |
| Experimental Models: Organisms/Strains | |||
| Oligonucleotides | |||
| Table S2 | This paper | N/A | |
| siGENOME Non-Targeting siRNA #2 | Dharmacon | D-001210-02-05 | |
| siGENOME Mouse INTS11 | Dharmacon | MQ-062233-01-0002 | |
| Recombinant DNA | |||
| pGEM-3Z N-terminal HaloTag | Gift from Danette Daniels and Elizabeth Caine (Promega) | N/A | |
| pGEM-3Z-mINTS11-CAST | This paper | N/A | |
| pGEM-3Z-mINTS11–129 | This paper | N/A | |
| pUC-U7-GFP | (Albrecht and Wagner, 2012) | N/A | |
| pCAG-eCas9-GFP-U6-gRNA | Addgene | 79145 | |
| pCAG-eCas9-GFP-U6-mINTS11-gRNA | This paper | N/A | |
| Software and Algorithms | |||
| bowtie 1.2.2 | (Langmead et al., 2009) | N/A | |
| STAR 2.7.3a | (Dobin et al., 2013) | N/A | |
| R | www.r-project.org | N/A | |
| Rstudio | www.rstudio.com | N/A | |
| featureCounts | (Liao et al., 2014) | N/A | |
| DESeq2 | (Love et al., 2014) | N/A | |
| rMATS | (Shen et al., 2014) | N/A | |
| Prism | GraphPad | N/A | |
| Partek Genomics Suite | www.partek.com | N/A | |
| get_gene_annotations.sh | DOI 10.5281/zenodo.5519927 | N/A | |
| make_heatmap | DOI 10.5281/zenodo.5519914 | N/A | |
| trim_and_filter_PE.pl | DOI 10.5281/zenodo.5519914 | N/A | |
| bowtie2stdBedGraph.pl | DOI 10.5281/zenodo.5519914 | N/A | |
| bedgraphs2stdBedGraph | DOI 10.5281/zenodo.5519914 | N/A | |
| cutadapt | DOI:10.14806/ej.17.1.200 | N/A | |
| samtools | (Li et al., 2009) | N/A | |
| bedtools | (Quinlan and Hall, 2010) | N/A | |
| UCSCtools | (Kent et al., 2010) | N/A | |
| Enrichr | (Kuleshov et al., 2016) | N/A | |
| ImageJ | (Schneider et al., 2012) | N/A | |
| dREG | (Danko et al., 2015) | N/A | |
| Enhancer annotation pipeline: dRIP-Filter, CoGENT, TSScentR | DOI 10.5281/zenodo.6654472 | N/A | |
| DANPOS | (Chen et al., 2013) | N/A | |
| MEME Suite | (Bailey et al., 2015) | N/A | |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
For routine maintenance, F121–9 mouse embryonic stem cells (mESCs) (CASTx129 hybrid, female) were cultured at 37°C with 5% CO2 on gelatinized plates in serum free ES media (SFES) + 2i+LIF media (Neurobasal and DMEM F/12 supplemented with 0.5x N2, 0.5x B27, 2mM L-Glutamine, 0.05% BSA, 0.15 mM 1-thioglycerol, 1000 U/mL LIF, 1 μM PD0325901, and 3 μM CHIR99021).
Prior to genome editing, cells were grown in KO-DMEM containing 15% Knock-Out Serum Replacement (KOSR), 1 mM Sodium Pyruvate, 1x GlutaMAX, 1x Penicillin-Streptomycin, 1x non-essential amino acids, 1% 2-mercaptoethanol, 1000 U/mL LIF, 1 μM PD0325901, and 3μM CHIR99021. Single cell sorting was performed into 96-well plates coated with 5 μg/mL fibronectin in PBS. INTS11Halo cells were then adapted to and maintained in SFES + 2i+LIF media.
Cultures were routinely checked for mycoplasma contamination.
HaloPROTAC3 (referred to as PROTAC throughout the manuscript) was obtained from Promega (GA3110) and used at a final concentration of 500 nM in all experiments. Unless otherwise noted, treatments lasted four hours.
METHOD DETAILS
Generation of INTS11Halo mESC cells
To generate repair templates for CRISPR, homology arms corresponding to genomic regions surrounding the INTS11 start codon were cloned into pGEM-3Z (a gift of Danette Daniels and Elizabeth Caine, Promega) using Gibson assembly. To reflect the hybrid genome of F121–9, two repair templates were generated, one for each allele. These were assembled into pGEM-3Z to make pGEM-3Z-mINTS11-CAST and pGEM-3Z-mINS11–129. A synonymous mutation at the PAM site was introduced to avoid re-cleavage by Cas9. Oligos containing an sgRNA sequence cleaving 10 bp downstream of the INTS11 start codon were annealed and phosphorylated, then ligated into BbsI-digested pCAG-eCas9-GFP-U6-gRNA (Addgene #79145) to generate pCAG-eCas9-GFP-U6-mINTS11-gRNA. See Table S2 for oligonucleotide sequences used.
Using Lipofectamine 2000 (ThermoFisher #11668019), 2 × 105 F121–9 cells were cotransfected with 875 ng of each repair template and 750 ng of the eCas9-GFP/sgRNA-containing vector. Two days later, GFP-positive were isolated using fluorescence-activated cell sorting (FACS). To enrich for cells that integrated the repair template in least one allele, the following week, cells were labeled with Janelia Fluor 646 HaloTag ligand (a gift of Talley Lambert, Nikon Imaging Core at HMS) by adding it to cells at a final concentration of 200 nM for 20 minutes. Excess ligand was removed by washing cells once with PBS. FACS was then used to collect cells that were positive for the ligand. After expanding single-cell clones, homozygous integration was confirmed using PCR of genomic DNA (Table S2) and Western blotting. Multiple clones were obtained, and experiments presented in this work focused on one. Cells were passaged in SFES for at least two passages before using for other experiments.
RNA interference
A pool of four INTS11-targeting siRNAs were diluted to 10 μM each in water; non-targeting siRNA was also diluted to 10 μM in water. 10 μL of 10 μM siRNA was added to 200 μL SFES media. 9 μL RNAiMax (ThermoFisher #13778150) reagent was diluted into 200 μL SFES media and allowed to incubate at room temperature for 2–3 minutes. The siRNA and RNAiMax dilutions were combined and incubated at room temperature for 20 minutes. 4 × 105 mESCs were seeded into gelatin-coated 6-well plates and the INTS11 or nontargeting siRNA mixes were added to the wells and mixed by pipetting. siRNAs were present at a final concentration of 40 nM (10 nM each in the case of siINTS11). At 24 hours post-transfection, cells were rinsed with PBS and fed new media. At 48 hours post-transfection, cells were harvested for protein lysates and RNA preparation.
Western blotting
Protein lysates were prepared by resuspending cell pellets in 1× Laemmli Sample Buffer (Bio-Rad #1610737) with freshly added β-mercaptoethanol (2.5% final concentration). Lysates were boiled for 10 minutes and spun at room temperature at maximum speed for 10 minutes. Samples were resolved on 8–16% (Bio-Rad, #4561103; c-Fos and phosphoepitopes) or 4–20% (Bio-Rad #4561095; all others) gradient gels. Protein was then transferred to a nitrocellulose (Amersham #10600001; all except c-Fos) or PVDF membrane (Bio-Rad 1620177; c-Fos). Membranes were blocked and probed in TBS + 0.1% Tween 20 supplemented with 5% nonfat milk powder (w/v), or in the case of phosphoepitopes, TBS + 0.1% Tween 20 supplemented with 5% BSA (w/v). Signal was detected using SuperSignal West Pico PLUS Chemiluminescent Substrate (ThermoFisher #34577) according to the manufacturer’s instructions and blots were imaged using a BioRad ChemiDoc.
RT-qPCR
Cell pellets were resuspended in Trizol and RNA was isolated according to the manufacturer’s instructions. Genomic DNA was removed with DNase (either RQ1 [Promega #M6101] or Amplification Grade DNase I [ThermoFisher #18068015]) before chloroform extraction. cDNA was synthesized using Superscript IV Reverse Transcriptase (ThermoFisher #18090200) according to the manufacturer’s instructions using random hexamers. RT-qPCR was performed using homemade SYBR Green master mix (1.7% v/v glycerol, 12.76 mM Tris-HCl pH 8, 53.2 mM KCl, 5.32 mM MgCl2, 0.21% v/v Tween 20, 212.7 μg/mL BSA [Life Technologies AM2616), 0.71 X SYBR Green [Sigma T8531]) and run on a Bio-Rad CFX384. RT-qPCR primer sequences are provided in Table S2.
Chromatin Immunoprecipitation (ChIP)
For ChIP of Integrator subunits, INTS11Halo cells were treated with DMSO or 500 nM PROTAC for four hours; F121–9 cells were untreated. Room temperature Accutase was added to plates of cells followed by dilution in PBS. A single-cell suspension was created by pipetting up and down. Formaldehyde solution was added to cells (1% final concentration) and the plate was agitated for 10 minutes at room temperature. Glycine was added (0.125 M final concentration) and the plate was agitated at room temperature for 5 minutes. Cells were moved to a conical tube and the plate was washed with cold PBS. After combining cell mixture and PBS, cells were spun for 5 minutes at 4°C at 300 × g. The supernatant was discarded, and the pellet was resuspended in cold PBS. Cells were counted using an automated cell counter (BioRad TC20), and the mixture was spun again for 5 minutes at 4°C at 300 × g. After a second wash with cold PBS, the pellet was lysed in Sonication Buffer (20 mM Tris-HCl [pH 8], 2 mM EDTA, 0.5 mM EGTA, 0.5% w/v SDS, 0.5 mM PMSF, cOmplete EDTA-free protease inhibitor cocktail [Sigma #11873580001]) at a concentration of 1 × 108 cells/mL on ice for 10 minutes before flash freezing in liquid nitrogen. Chromatin was sheared using the Qsonica Q800R3 system (70% power; 15 s ON/45 s OFF cycles; 10 minutes total; 100 μL chromatin per tube). 20% input material was set aside for each experiment.
Chromatin was pre-cleared in IP buffer (20 mM Tris-HCl [pH 8], 2 mM EDTA, 0.5% v/v Triton X-100, 150 mM NaCl, 10% v/v glycerol) with Protein A agarose beads (Millipore 16–125) before further dilution in IP buffer and overnight incubation with appropriate antibody: 2 independent replicates were carried out with 10 μL (F121–9) or 12.5 μL (INTS11Halo) anti-INTS11 per 2.5 × 106 cells (different volumes due to antibody batch) and 10 μL anti-INTS3 per 2.5 × 106 cells. Antibody-epitope complexes were allowed to bind to Protein A agarose beads for two hours at 4°C. Beads were then washed once with low salt buffer (20 mM Tris-HCl [pH 8], 2 mM EDTA, 1% v/v Triton X-100, 150 mM NaCl), three times with high salt buffer (20 mM Tris-HCl [pH 8], 2 mM EDTA, 1% v/v Triton X-100, 500 mM NaCl), once with LiCl buffer (20 mM Tris-HCl [pH 8], 2 mM EDTA, 1% v/v IGEPAL, 250 mM LiCl), and twice with TE. Beads were then eluted twice with elution buffer (1% w/v SDS, 100 mM NaHCO3) and crosslinks were reversed overnight at 65°C. Immunoprecipitated material was then phenol-chloroform purified.
ChIP of RNA Pol II Ser5P was performed as above with the following modifications: Cells were crosslinked for 5 minutes. After the first PBS wash, phosphatase inhibitors were included in all buffers (Phosphatase Inhibitor Cocktails 2 and 3, both 1:000). Total sonication time was 5 minutes. 10 μL of antibody was used per IP. 0.1% w/v SDS was added to low salt buffer and high salt buffer. 1% w/v sodium deoxycholate was added to LiCl buffer.
Imaging
For imaging of HaloTag-INTS11, Glass bottom culture dishes (MatTek P35G1.514C) were coated with 5 μg/mL fibronectin (Millipore Sigma FC01010MG) in PBS at room temperature. 1 × 105 F121–9 or INTS11Halo cells were added to the plates and allowed to adhere for two hours. 2i+LIF-containing media was aspirated and Differentiation Media (SFES, 12 ng/mL bFGF, 1% v/v KOSR) was added. Media was changed the next day. Prior to imaging, Hoechst 33258 (final concentration 5 μg/mL) and JFX549 (final concentration 100 nM) was added to cells for 15 minutes in fresh Differentiation Media. Excess label was removed by washing four times with Imaging Media (Fluorobrite DMEM, 2 mM L-Glutamine, 0.05% BSA, 0.15 mM 1-thioglycerol, 12 ng/mL bFGF, 1% KOSR, 25 mM HEPES). Images were taken while cells were in Imaging Media on a Nikon Eclipse Ti and processed in ImageJ.
For detection of GFP, images were obtained on a Bio-Rad ZOE Fluorescent Cell Imager and not processed further.
PRO-seq library preparation and analysis
INTS11Halo cells were plated in 15 cm tissue culture dishes one day prior to treatment. The day of treatment, HaloPROTAC3 was added to a final concentration of 500 nM to SFES media; an equal volume of DMSO was added to media for control treatments.
Cells were permeabilized essentially as in (Reimer et al., 2021). Briefly, cells were washed with PBS and detached from the plate with trypsin. Cold DMEM + 10% FBS was used to quench the trypsin and the suspension was pipetted up and down to make single cells. Cells were pelleted at 400 × g for 4 minutes at 4°C and washed by resuspending in cold PBS. After pelleting again, cells were resuspended in 250 μL ice cold Buffer W (10 mM Tris HCl [pH 8], 10 mM KCl, 250 mM sucrose, 5 mM MgCl2 1 mM EGTA, 10% v/v glycerol, 0.5 mM DTT). Permeabilization was performed by slowly adding 10 mL ice cold Buffer P (Buffer W supplemented with 0.05% v/v Tween 20 and 0.1% v/v IGEPAL) to the side of the tube and incubating on ice for 5 minutes. Cells were spun at 400 × g for 4 minutes at 4°C. The supernatant was aspirated and the pellet gently resuspended in 10 mL Buffer W. Cells were again spun at 400 × g for 4 minutes at 4°C and the pellet was resuspended in 500 μL ice cold Buffer F (50 mM Tris HCl [pH 8], 5 mM MgCl2, 1.1 mM EDTA, 40% v/v glycerol, 0.5 mM DTT, SuperaseIN RNase Inhibitor [ThermoFisher AM2694]). Cells were counted and permeabilization efficiency confirmed with trypan blue staining. Permeabilized cells were flash frozen and stored until use.
Aliquots of frozen (−80°C) permeabilized cells were thawed on ice and pipetted gently to fully resuspend. Aliquots were removed and permeabilized cells were counted using a Luna II, Logos Biosystems instrument. For each sample, 1 million permeabilized cells were used for nuclear run-on, with 50,000 permeabilized Drosophila S2 cells added to each sample for normalization. Nuclear run-on assays and library preparation were performed essentially as described in Reimer et al. (Reimer et al., 2021) with modifications noted: 2X nuclear run-on buffer consisted of (10 mM Tris (pH 8), 10 mM MgCl2, 1 mM DTT, 300mM KCl, 40 μM/ea biotin-11-NTPs (Perkin Elmer NEL54(2/3/4/5)001), 0.8U/μL SuperaseIN (ThermoFisher AM2694), 1% sarkosyl). Run-on reactions were performed at 37°C. Adenylated 3’ adapter was prepared using the 5’ DNA adenylation kit (NEB, #E2610) and ligated using T4 RNA ligase 2, truncated KQ (NEB # M0373), per manufacturer’s instructions with 15% PEG-8000 final) and incubated at 16°C overnight. 180 μL of betaine blocking buffer (1.42 g of betaine brought to 10mL with binding buffer supplemented to 0.6 μM blocking oligo (TCCGACGATCCCACGTTCCCGTGG/3InvdT/)) was mixed with ligations and incubated 5 min at 65°C and 2 min on ice prior to addition of streptavidin beads. After T4 polynucleotide kinase (NEB M0201) treatment, beads were washed once each with high salt, low salt, and blocking oligo wash (0.25 X T4 RNA ligase buffer (NEB B0216), 0.3 μM blocking oligo) solutions and resuspended in 5’ adapter mix (10 pmol 5’ adapter, 30 pmol blocking oligo, water). 5’ adapter ligation was per Reimer but with 15% PEG-8000 final. Eluted cDNA was amplified 5-cycles (NEBNext Ultra II Q5 master mix (NEB M0544) with Illumina TruSeq PCR primers RP-1 and RPI-X) following the manufacturer’s suggested cycling protocol for library construction. A portion of preCR was serially diluted and for test amplification to determine optimal amplification of final libraries. Pooled libraries were sequenced using the Illumina NovaSeq platform.
All custom scripts described herein are available on the AdelmanLab Github (https://github.com/AdelmanLab/NIH_scripts). Using a custom script (trim_and_filter_PE.pl), FASTQ read pairs were trimmed to 41bp per mate, and read pairs with a minimum average base quality score of 20 retained. Read pairs were further trimmed using cutadapt 1.14 to remove adapter sequences and low-quality 3’ bases (--match-read-wildcards -m 20 -q 10). R1 reads, corresponding to RNA 3’ ends, were then aligned to the spiked in Drosophila genome index (dm3) using Bowtie 1.2.2 (-v 2 -p 6 --best --un), with those reads not mapping to the spike genome serving as input to the primary genome alignment step (using Bowtie 1.2.2 options -v 2 --best). Reads mapping to the mm10 reference genome were then sorted using samtools 1.3.1 (-n), and subsequently converted to bedGraph format using a custom script (bowtie2stdBedGraph.pl). Because R1 in PRO-seq reveals the position of the RNA 3’ end, the “+” and “-“ strands were swapped to generate bedGraphs representing 3’ end position at single nucleotide resolution.
As described, permeabilized Drosophila S2 cells were added to all samples prior to the run-on reaction based on cell count. We observed consistent differences in Drosophila reads between conditions, with PROTAC-treated samples returning fewer spike reads per mouse genomic read across both replicates. The reproducible decrease in spike return in PROTAC-treated conditions indicates a small global increase in RNA synthesis in these cells. Thus, per-sample spike normalization factors were calculated by dividing reads mapped to dm3 by 1,000,000, as shown below.
Replicate 2 was spiked with a different batch of permeabilized Drosophila cells than Replicate 1, and this second batch yielded a 1.33x higher percentage spike reads on average. To avoid Replicate 2 being underrepresented in analyses combining the two replicates (e.g. bigWigs, metagenes, heatmaps), we divided the Replicate 2 spike factors by 1.33 prior to combining the two replicates. The final number of spike-normalized reads for each sample is displayed below:
| Sample | Total Reads | Reads Mapping to mm10 | Reads Mapping to dm3 | Per-sample Normalization Factor | Normalization factor after accounting for spike batch | Final spike-normalized reads mapping to mm10 |
|---|---|---|---|---|---|---|
| DMSO Replicate 1 | 77,623,395 | 55,775,379 | 3,189,566 | 3.19 | 3.19 | 17,486,824 |
| PROTAC Replicate 1 | 70,023,318 | 52,203,115 | 2,531,649 | 2.53 | 2.53 | 20,620,202 |
| DMSO Replicate 2 | 123,575,803 | 74,259,815 | 5,635,785 | 5.64 | 4.25 | 17,485,191 |
| PROTAC Replicate 2 | 114,975,085 | 84,886,620 | 5,597,308 | 5.60 | 4.22 | 20,124,771 |
BigWigs and bedGraphs used for generating UCSC Genome Browser tracks and metagene analyses represent the 3′ end of each mapped read from combined replicates per condition, normalized as in the table above.
Generation of Transcript Annotations
The Get Gene Annotation pipeline was used to generate high-confidence gene annotations based on PRO-seq and RNA-seq (https://github.com/AdelmanLab/GetGeneAnnotation_GGA; DOI 10.5281/zenodo.5519927). A hybrid Ensembl/RefSeq GTF was used as a basis for gene annotations. Unnormalized reads from all four PRO-seq samples (n=2 DMSO, n=2 PROTAC) were used to refine TSS position for annotated TSSs based on 5’ ends of PRO-seq data, and parental mESC RNA-seq was used to define TESs. A minimum 5’ PRO-seq read count of 8 and a search window of 1kb was required for a gene to be considered active and for re-alignment of the annotated TSS to the position with maximal nascent RNA 5’ end reads. Genes with fewer than 30 PRO-seq reads in the TSS-to-150 window using all four samples combined (PRO-seq 3’ end reads) were removed. To avoid analyzing low-confidence annotations, the following gene biotypes were not considered in downstream analyses: miscRNA, miRNA, ribozyme, scaRNA, TEC. Two genes annotated as lncRNAs (ENSMUSG00000097971 and ENSMUSG00000098178) that were found to overlap an rRNA locus were removed to avoid analyzing RNAPI transcripts. To avoid analyzing potential contamination from highly abundant mature RNA species, nucleotide positions were masked that fell within 4 nt of an annotated miRNA, miscRNA, snRNA, or snoRNA TES and contained greater than 200 reads when summed across all four samples.
Differentially expressed genes in PRO-seq
Read counts were calculated per gene, in a strand-specific manner, based on the annotations described above, using the custom script make_heatmap (available at https://github.com/AdelmanLab/NIH_scripts; DOI 10.5281/zenodo.5519914). This quantification procedure includes signal from the dominant TSS to TES. Differentially expressed genes were identified using DESeq2 (Love et al., 2014). Spike normalization factors were enforced. At an adjusted p value threshold of < 0.01 and fold change > 1.5, 1,320 genes (736 mRNAs) were identified as differentially expressed upon INTS11 depletion in INTS11Halo cells.
Enhancer calling, filtering, and analysis
Unnormalized, single-nucleotide PRO-seq bedGraphs representing 3’ ends from four datasets (n=2 DMSO, n=2 PROTAC) were merged and converted to bigWig files, one representing each strand. These bigWigs were uploaded to the dREG server (Danko et al., 2015) and all resulting files were downloaded.
Refinement of enhancer calls was performed with a custom enhancer annotation pipeline (DOI 10.5281/zenodo.6654472). dRIP-filter was used to select for high-confidence peaks: peaks were filtered for a dREG score of 0.5 and p value of 0.025. To remove dREG peaks with low PRO-seq counts, the only peaks with at least 10 reads on each strand in at least one sample were considered. For this manuscript, only promoter-distal (>1 kb from TSSs), intergenic enhancers were analyzed as determined by the CoGENT annotation script. Assignment of peaks to TSSs was performed using TSScentR based on overlap between dREG peak coordinates with unannotated TSSs from GGA output. As enhancers are typically characterized by the histone modification H3K27Ac, peaks were finally filtered to require greater than 40 H3K27Ac ChIP-seq reads in the eTSS +/−1 kb window. A final list of 9,571 dREG peak TSSs were used for analysis.
TT-seq library preparation and analysis
INTS11Halo cells were plated in 10 cm tissue culture dishes one day prior to treatment. The day of treatment, HaloPROTAC3 was added to a final concentration of 500 nM to SFES media; an equal volume of DMSO was added to media for control treatments. During the final 20 minutes of the four-hour depletion, 4sU was added to each plate to a final concentration of 500 μM. After 20 minutes, cells were rinsed with room-temperature PBS and harvested using trypsin and quenched with cold DMEM + 10% FBS. After an additional wash with PBS, cells were counted then resuspended in 1 mL Trizol.
Prior to addition of chloroform to the lysates, samples were spiked at 5% based on cell counts with 4sU-labeled Drosophila S2 Trizol lysate (cells labeled with 4sU for 2 h and resuspended a concentration of 10 million cells/mL in Trizol). RNA was then isolated per the manufacturer’s protocol. The aqueous phase was precipitated by addition of 2.5 volumes of 100% ethanol, incubation at −20°C for 2h. Pellets were collected by centrifugation at 20,000 × g for 30 min at 4°C and washed twice with 500 μL of 75% ethanol before resuspension in 180 μL of nuclease free water. Aliquots were removed for quantification by spectrophotometry and analysis of RNA integrity by Agilent TapeStation 4200 using RNA high sensitivity tapes. Samples with RIN > 9.0 were used for further processing.
Total RNA from the previous step was treated to remove residual DNA by addition of 20 μL of 10X DNase I buffer and 2 μL of Amplification Grade DNase I (ThermoFisher #18068015), followed by incubation at room temperature for 30min. Reactions were transferred to prepared MaXtract High Density gel tubes (Qiagen #129056), shaken with equal volume of chloroform:isoamyl alcohol and spun at 12,000 × g at 4°C for 5 min to separate phases. The aqueous phase was recovered to fresh tubes, quantified, and analyzed by TapeStation as above. 60 μg of RNA was brought to a volume of 80 μL with nuclease-free water and placed on ice. RNA was then lightly fragmented by addition of 20 μL cold 5X fragmentation solution (375 mM Tris-HCl (pH 8.3), 562.5 mM KCl, 22.5 mM MgCl2) and incubation at 94°C for 2.5 minutes. At the end of the fragmentation time, RNA was placed immediately on ice and 25 μL of cold 250 mM EDTA was added. RNA was precipitated by addition of 1/10 volume of 5 M NaCl, 2.5 volumes of 100% ethanol and incubation at −20°C overnight. RNA was pelleted, washed, quantified, and analyzed again as above.
Fragmented RNA was biotinylated essentially as described in (Duffy et al., 2015) with the following modifications: the biotinylation reaction was performed in a total volume of 200 μL and allowed to incubate for 45 min in the dark. Excess biotin was removed using chloroform:isoamyl alcohol and MaXtract Heavy gel tubes (Qiagen) were used to separate organic and aqueous phases. Biotinylated RNA was resuspended in 100 μL of nuclease-free water and aliquots taken to use as the total RNA input fraction. In parallel, Dynabeads M-280 Streptavidin (ThermoFisher #11205D) were prepared for binding to render them RNase-free: for each sample, 75 μL of beads were used and treated in batch to render them RNase free. The beads were incubated 10min in a solution of 100 mM NaOH, 50 mM NaCl, placed on a magnetic stand, and then washed, resuspending the beads fully for each wash, twice with 500 μL of 100 mM NaCl, twice with 1 X TTseq wash solution (100 mM Tris-HCl (pH 7.5), 10mM EDTA, 1 M NaCl, 0.05% Tween 20 in nuclease free water to which 1 μL SuperaseIN RNase Inhibitor (ThermoFisher AM2694) per 5mL solution is added prior to use), once in 0.3 X TTseq wash solution, and finally resuspended in 52 μL/sample of 0.3 X TTseq wash solution + 1 μL/sample SuperaseIn RNase Inhibitor (ThermoFisher AM2694).
Biotinylated RNA was heated at 65°C for 5min, placed on ice for 2 min, and mixed with 50 μL of prepared beads. Samples were rotated at room temperature in the dark for 30 min. After binding, the tubes were placed on a magnetic rack and the beads were washed 4 times with 500 μL of 1 X TT-seq wash solution to remove unbound RNA, fully resuspending for each wash. The wash solution was removed, and the beads resuspended in 50 μL of 0.1 M DTT (freshly diluted from 1 M stock) and rolled in the dark for 15 min at room temp. The eluted RNA was recovered and the elution step repeated with an additional 50 μL of 0.1 M DTT. The combined eluates were purified using Norgen RNA clean-up and concentration microElute kit (Norgen #61000) following the manufacturer’s instructions for small RNA enriched samples. Final elution was performed in 14 μL of nuclease-free water and the eluate was reapplied to the column for a total of 2 elution steps. A Qubit RNA high sensitivity reagent kit was used to quantify the input RNA and enriched RNA. Yields of 1–2% were typical. 180 ng of enriched RNA was used for library construction with the Illumina TruSeq stranded total RNA kit and Illumina CD index plate. After 5 cycles of PCR, samples were removed from the thermal cycler and a test PCR was performed to determine the optimal number of final cycles. Libraries were pooled and sequenced paired-end 150 on a Novaseq S1 full flowcell.
Sequencing reads were filtered (requiring a mean quality score ≥ 20) and trimmed to 100 nt. Reads were first mapped the dm3 version of the Drosophila genome using STAR 2.7.31. Reads not mapping to spike were then used for alignment to mm10 using parameters --quantMode TranscriptomeSAM GeneCounts --outMultimapperOrder Random --outSAMattrIHstart 0 --outFilterType BySJout --outFilterMismatchNmax 4 --alignSJoverhangMin 8 --outSAMstrandField intronMotif --outFilterIntronMotifs RemoveNoncanonicalUnannotated --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --outFilterScoreMinOverLread 0 --outFilterMatchNminOverLread 0. Duplicates were also removed using STAR. The total number of TT-seq reads aligned in the DMSO- and PROTAC-treated samples is described in the table below.
| Sample | Total Reads | Reads Mapping to mm10 | Normalization Factor |
|---|---|---|---|
| DMSO Replicate 1 | 128,058,250 | 118,852,748 | 1.10 |
| DMSO Replicate 2 | 120,172,903 | 112,316,476 | 1.00 |
| DMSO Replicate 3 | 149,250,802 | 138,820,533 | 1.48 |
| PROTAC Replicate 1 | 115,150,827 | 107,478,313 | 1.00 |
| PROTAC Replicate 2 | 134,295,046 | 124,929,756 | 1.10 |
| PROTAC Replicate 3 | 135,521,330 | 126,130,242 | 1.31 |
Samples displayed highly comparable recovery of spike-in reads. Thus, normalization based on the DESeq2 size factors (see above) was used for each bedGraph. To determine size factors, read counts were calculated per gene, in a strand-specific manner, based on annotations described in the transcript annotations section. This quantification procedure includes signal from the dominant TSS to TES. Combined bedGraphs were generated by summing coverage from all replicates for each condition.
Analysis of readthrough by TT-seq:
For analysis of snRNAs in Figure 1 and S1, gene body TT-seq signal was determined using featureCounts in the dominant TSS-to-TES region and divided by length (in kb) to produce a read density. For readthrough signal, read coverage was determined in the same way using the TES-to-1kb region. To remove genes where the annotated TES used in our analyses was not efficiently or uniquely recognized in control cells, we focused on genes with a Readthrough Index of 0.8 or lower in DMSO conditions. Furthermore, to avoid analysis of genes not well-represented in TT-seq, snRNAs with fewer than 100 reads/kb in control cells were removed. Finally, to avoid counting signal from non-snRNA genes, those that were found to be overlap other active genes by manual inspection were removed. This resulted in a final list of 37 high-confidence snRNAs for analysis. A pseudocount of one read was added to both upstream and downstream windows when calculating Readthrough Index (TES-to-1kb density / TSS-to-TES density) to avoid dividing by zero.
For analysis of mRNAs in Figure 2 and S2, only genes longer than 2kb were considered (N=12,598). Read counts in the upstream and downstream regions were determined using featureCounts. To remove genes not well represented in our TT-seq data, genes with fewer than 20 reads in the 2 kb upstream of the TES in DMSO or PROTAC were removed (leaving N=11,521). To remove genes where the annotated TES used in our analyses was not efficiently or uniquely recognized in control cells, we focused on genes with a Readthrough Index of 0.8 or lower in DMSO conditions (N=9,890). To prevent changes in upstream TT-seq signal from dominating analysis of readthrough, genes with a log2 fold change greater than 0.15 in the 2 kb upstream region upon INTS11 depletion were removed for Figure 2D and S2C. However, the TT-seq signal at these genes is depicted in Figure S2D as reference. The final list of mRNAs used in Figure 2D and S2C consisted of the remaining 7,560 genes. A pseudocount of one read was added to both upstream and downstream windows when calculating Readthrough Index (TES-to-2kb / 2kb-to-TES) to avoid dividing by zero.
Analysis of elongation rate
PRO-seq reads were summed (make_heatmap -v t) in 500 nt bins downstream of annotated mRNA TSSs. TT-seq mean per-nucleotide coverage (make_heatmap -v c) was calculated in the same windows, then multiplied by 500 to convert to mean coverage across the bin. To avoid analysis of low-confidence regions, bins containing 0 reads in any dataset and those that contain the gene TES or are downstream of the TES were converted to values of 0. Elongation rate was then calculated as the ratio of TT-seq to PRO-seq.
For metagene and boxplot analysis of the early gene body, only genes longer than 5 kb were plotted. To avoid analysis of low-confidence regions, only bins that contain at least 10 reads/kb in all datasets were included. The first bin in a gene was excluded (Schwalb et al. 2016).
ChIP-seq library preparation and analysis
ChIP material was prepared as described above, with two biological replicates per condition. To allow absolute quantification of INTS11 and INTS3 in INTS11Halo cells, immunoprecipitated DNA was spiked with a constant amount of MNase-digested Drosophila DNA (0.0208 ng) and ChIP-seq libraries were prepared using the NEBNext Ultra II DNA library kit (NEB) according to the manufacturer’s instructions. Libraries were then pooled and sequenced using a paired-end 80 bp cycle run on the Illumina NextSeq system for F121–9 and 50 bp cycle run on the Illumina NovaSeq system for INTS11Halo samples, all with standard sequencing protocols.
For RNA Pol II Ser5P, a constant amount of Drosophila chromatin was added (100,000 Drosophila cells to 2.5 million mouse cells) prior to pre-clearing with agarose beads. After IP, libraries were then pooled and sequenced using a paired-end 50 bp cycle on the Illumina NovaSeq system with standard sequencing protocols.
For all samples, sequences were quality filtered and trimmed to 50bp.
F121–9 samples were immediately aligned against the mm10 version of the mouse genome using Bowtie version 1.2.2 (Langmead et al., 2009) with a maximum allowed mismatch of 2 (-v2 -k1 - X1000 −best). INTS11Halo samples were first mapped to the dm3 version of the Drosophila genome and reads not mapping to this were subsequently mapped to mm10 using the same parameters. The yield of reads mapping to mm10 is listed below.
Due to the difficulty of Integrator ChIP, high background signal was observed. To allow for the quantitative comparison of signal rather than background in DMSO and PROTAC-treated INTS11Halo samples, the following normalization strategy was used: samples were first depth normalized using the depth normalization factors listed in the table below. Replicates were then merged. Average signal from 15,999 random intergenic regions in 5 bp bins was calculated and considered genomic background; this value (listed below) was subtracted from all 5 bp bins. Finally, due to consistent changes in spike read returns for INTS11Halo-derived samples between PROTAC and DMSO, reads mapping to dm3 were used for normalization. Finally, spike normalization factors (listed below) were determined by choosing the condition with the fewest Drosophila reads for each antibody and dividing each bedGraph by a factor that would cause the samples to have the same number of Drosophila reads.
| Cell line/Condition | Antibody | Total reads | Deduplicated reads mapping to mm10 | Reads mapping to dm3 | Depth norm. factor | Background signal | Spike norm. factor | Agreement between replicates (Pearson’s r) |
|---|---|---|---|---|---|---|---|---|
| F121–9 Replicate 1 | INTS11 | 45,087,205 | 32,978,564 | N/A | N/A | N/A | N/A | >0.99 |
| F121–9 Replicate 2 | INTS11 | 52,171,958 | 38,321,645 | |||||
| INTS11Halo/DMSO Replicate 1 | INTS11 | 131,202,996 | 62,935,010 | 41,926 | 1.16 | 0.176 | 1.00 | >0.99 |
| INTS11Halo/DMSO Replicate 2 | INTS11 | 109,944,866 | 56,793,447 | 38,042 | 1.05 | |||
| INTS11Halo/PROTAC Replicate 1 | INTS11 | 143,158,380 | 69,687,029 | 60,305 | 1.28 | 0.175 | 1.54 | >0.99 |
| INTS11Halo/PROTAC Replicate 2 | INTS11 | 109,126,747 | 54,239,223 | 64,980 | 1.00 | |||
| INTS11Hato/DMSO Replicate 1 | INTS3 | 145,129,425 | 72,736,187 | 39,670 | 1.71 | 0.135 | 1.22 | >0.99 |
| INTS11Halo/DMSO Replicate 2 Replicate 2 | INTS3 | 105,573,272 | 56,348,152 | 29,408 | 1.32 | |||
| INTS11Halo/PROTAC Replicate 1 | INTS3 | 125,956,094 | 63,665,886 | 26,854 | 1.49 | 0.136 | 1.00 | >0.99 |
| INTS11Halo/PROTAC Replicate 2 | INTS3 | 79,847,597 | 42,651,940 | 19,176 | 1.00 |
To calculate the correlation coefficient between endogenous INTS11 and INTS11Halo, reads were summed using TSS+/−1kb region for all active genes, and the spike normalized, background-subtracted DMSO number was compared to INTS11 from F121–9 cells in this same window.
For RNA Pol II Ser5P, samples displayed highly comparable recovery of spike-in reads. Thus, normalization based on sequencing depth was used for each sample:
| Condition | Total reads | Deduplicated reads mapping to mm10 | Reads mapping to dm3 | Depth norm. factor | Agreement between replicates (Pearson’s r) |
|---|---|---|---|---|---|
| DMSO Replicate 1 | 38,703,205 | 21,304,168 | 348,557 | 1.41 | >0.99 |
| DMSO Replicate 2 | 39,459,724 | 21,150,752 | 323,241 | 1.40 | |
| PROTAC Replicate 1 | 27,605,528 | 15,146,174 | 247,757 | 1.00 | >0.99 |
| PROTAC Replicate 2 | 30,006,421 | 16,496,917 | 233,376 | 1.09 |
To calculate the correlation coefficient between replicates, reads were summed using TSS to 150 bp region for all active genes.
H3K4me3, H3K27Ac, and ChIP-seq were downloaded from GEO and mapped to mm10 with Bowtie 1.2.2 (-v2 -X1000 −best -k1 for H3K4me3/me1; -v2 -X1000 −best -m1 for H3K27Ac).
Mapped reads were converted to bedGraphs representing deduplicated fragment midpoints using extract_fragments.pl (DOI 10.5281/zenodo.5519914), which were visually examined using the UCSC genome browser after conversion to bigWig format. BedGraphs were used for all downstream analysis.
RNA-seq library preparation and analysis
For data to be used for GGA annotation, F121–9 cells were harvested in Trizol 48 hours after plating.
For control versus siINTS11 conditions, cells were treated as described in the RNA Interference section above. After 48 hours of depletion, cells were harvested using trypsin, quenched with cold DMEM + 10% FBS, counted, and equal cell numbers were resuspended in Trizol. For DMSO versus PROTAC conditions, cells were treated with media containing DMSO or 500 nM PROTAC the day after plating for 16 hours and harvested as above. To allow for absolute quantification of changes in transcription, 1 μL of a 1:10 dilution of ERCC spike RNAs were added per 1 million cells.
For all samples, RNA was extracted using Direct-zol columns (Zymo Research R2052). Genomic DNA was removed with DNase off-column (RQ1 or Amplification Grade DNase I), and RNA was re-purified with either a second Direct-zol column or the RNeasy Mini clean-up kit (Qiagen #74106). Libraries were prepared using the Illumina TruSeq Stranded Total RNA kit with 500 ng of RNA as input. The manufacturer’s instructions were followed with two exceptions: SuperScript III was used rather than SuperScript II for reverse transcription for RNAi and PROTAC samples, and the number of PCR cycles were empirically determined. GGA annotation libraries were sequenced on the Illumina NextSeq 500; siRNA and PROTAC samples were sequenced on the NovaSeq platform.
Reads were quality filtered (requiring a mean quality score ≥ 20) and trimmed (100 nt for siRNA; 50 nt for PROTAC). Reads were first mapped to ERCC spike sequences using STAR 2.7.3a. Reads not mapping to spike were used for alignment to mm10 using parameters --quantMode TranscriptomeSAM GeneCounts --outMultimapperOrder Random --outSAMattrIHstart 0 --outFilterType BySJout --outFilterMismatchNmax 4 --alignSJoverhangMin 8 --outSAMstrandField intronMotif --outFilterIntronMotifs RemoveNoncanonicalUnannotated --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --outFilterScoreMinOverLread 0 --outFilterMatchNminOverLread 0. Duplicates were also removed using STAR. The total number of RNA-seq reads aligned for siRNA and PROTAC treatments are described in the table below.
Samples displayed highly comparable recovery of spike-in reads between conditions (siNT compared to siINTS11 and DMSO compared to PROTAC). Thus, normalization based on the DESeq2 size factors (see below) were used for each sample. To determine size factors, featureCounts was used to calculate coverage over exons for each gene in a strand-specific manner based on annotations described in the modified transcript annotations section above from get_gene_annotations.sh. Combined bedGraphs were generated by summing coverage from all replicates for each condition.
| Sample | Total Reads | Reads Mapping to mm10 | Normalization Factor |
|---|---|---|---|
| siNT Replicate 1 | 85,082,344 | 72,263,883 | 1.03 |
| siNT Replicate 2 | 78,243,589 | 68,479,194 | 0.98 |
| siNT Replicate 3 | 80,597,983 | 67,888,840 | 0.98 |
| siINTS11 Replicate 1 | 77,847,141 | 65,114,768 | 0.95 |
| siINTS11 Replicate 2 | 82,250,852 | 69,078,801 | 1.05 |
| siINTS11 Replicate 3 | 83,996,398 | 70,556,807 | 1.07 |
| 16 h DMSO Replicate 1 | 102,106,718 | 83,305,007 | 1.39 |
| 16 h DMSO Replicate 2 | 72,949,752 | 58,503,964 | 0.90 |
| 16 h DMSO Replicate 3 | 70,398,856 | 58,278,259 | 0.91 |
| 16 h PROTAC Replicate 1 | 79,019,627 | 63,058,019 | 0.97 |
| 16 h PROTAC Replicate 2 | 76,766,582 | 62,302,374 | 1.03 |
| 16 h PROTAC Replicate 3 | 72,362,829 | 59,376,163 | 0.89 |
UCSC Genome Browser tracks representing read coverage were generated from the combined replicates in each condition after normalizing using the factors above.
Differentially expressed genes in RNA-seq
Differentially expressed genes were identified with DESeq2 (Love et al., 2014) using coverage counts from above. For siNT versus siINTS11 and DMSO versus PROTAC comparisons, an adjusted p-value threshold of 0.01 and fold change > 1.5 was used. Further filtering (see below) was performed before a final differentially expressed gene set was determined.
rMATS analysis
To avoid analyzing genes whose splicing patterns were changed upon long-term INTS11 depletion as a consequences of snRNA misprocessing, we used rMATS (Shen et al., 2014) to quantify splicing events in RNA-seq. SAM files from STAR alignment were converted to BAM files using samtools (v1.9). BAM files for control and siINTS11 were input into rMATS as separate groups. The GTF output from get_gene_annotations.sh was used for annotation. The following parameters were used for running rMATS: -t paired --readLength 100 --libType fr-secondstrand --tstat 5 --novelSS --variable-read-length. Overlapping events were collapsed, retaining those with the most reads per gene (subset_rmats_junctioncountonly.py, available at https://github.com/YeoLab/rbp- maps/blob/master/preprocessing_scripts/subset_rmats_junctioncountonly.py). A threshold of 25 reads, 0.1 change in PSI (corresponding to a 10% change), and FDR of 0.001 was applied. 2,184 genes (of which 2,154 were mRNAs) met these criteria and were thus not included in analysis. This resulted in a final list of 2,339 upregulated genes (1,694 of which were mRNAs) and 1,764 downregulated (1,681 mRNAs) genes after 48 hours of siINTS11 treatment using the thresholds described above. The same list of filtered genes was used for analysis of PROTAC-treated samples.
Gene expression after ectopic Jun expression
Genes differentially expressed after ectopic Jun expression in mESCs were downloaded from (Liu et al., 2015). The list of genes was overlapped with genes meeting the above criteria based on gene name.
MNase-seq data analysis
MNase-seq FASTQ files corresponding to control mESCs were downloaded from GSE85191. The trim_and_filter_PE.pl (DOI 10.5281/zenodo.5519914) custom script was used to trim FASTQ read pairs to 70nt per mate and retain read pairs above a minimum quality threshold of 20. Trimmed and retained read pairs were mapped to mm10 using Bowtie 1.2.2 (-m1 -v2 -X1000 -- best -p 5). To retain reads corresponding to mono-nucleosome size fragments, read pairs spanning <120bp or >180bp were excluded using extract_fragments.pl (DOI 10.5281/zenodo.5519914) (o a -b 1 -min 120 -max 180). Bedgraphs reporting the fragment center were generated using extract_fragments.pl. The bedgraphs2stdBedGraph (DOI 10.5281/zenodo.5519914) custom script was used to merge replicate bedgraphs.
DANPOS3 (https://github.com/sklasfeld/DANPOS3) was used to identify nucleosome positions (start, dyad, and end) found in the control mESC MNase-seq dataset (default parameters). To define the +1 nucleosome, bedtools intersect was used to pair identified nucleosomes by DANPOS with active gene models in control mESCs. For each gene, the start of the DANPOS identified nucleosome was required to be downstream of the dominant TSS. The +1 nucleosome was classified as the first identified nucleosome downstream of the TSS.
Gene ontology analysis
The Enrichr web platform was used to define enriched gene ontology terms (Kuleshov et al., 2016). The ontologies used for each analysis are indicated in the figure legend and/or text. An adjusted P value of 0.05 was used as a threshold for significantly enriched terms.
To define upregulated transcription factor (TF) genes, a list of Ensembl gene IDs representing mouse TFs was downloaded from AnimalTFDB3.0 (Hu et al., 2019), a web server that contains annotations of TF genes from animals. 1,623 unique mouse TF gene annotations were downloaded. TFs active in INTS11Halo cells were determined by overlapping active genes from GGA with the gene IDs from AnimalTF3.0. Of 13,057 active mRNA genes, 1,163 were found to be TFs. This list was then overlapped with genes upregulated by PRO-seq, resulting in 48 upregulated TFs.
Motif analysis
Distal dREG peaks were rank ordered based on fold change in PRO-seq in the eTSS to 150 window after four hours of PROTAC treatment. The 500 most upregulated peaks were used for motif searches. A list of 1,000 peaks representing the 500 least upregulated and 500 least downregulated was used as a background set. Sequences in the eTSS minus 200 nt window were downloaded from the UCSC genome browser in FASTA format and input into the XSTREME module of the MEME Suite (Bailey et al., 2015). AP-1 was returned as the top hit.
To further refine the most significantly enriched AP-1 motif in the set of 500 most upregulated peaks compared to the 1,000 least affected, 39 AP-1 motif logos were downloaded from JASPAR (Castro-Mondragon et al., 2022) and input into AME (Buske et al., 2010). The top motif is displayed in Figure 6F.
Metagene and heatmap analysis
Count matrices were generated based on the assay as described in above sections. Metagene plots were generated by summing reads (PRO-seq), averaging coverage per nucleotide (TT-seq), or summing fragment centers (ChIP-seq) at each indicated position with respect to the indicated loci and dividing by the number of loci within each group. Values were graphed across a range of distances as indicated in figure legends. Heatmaps were generated using Partek Genomics Suite. To avoid interpretation of changes in regions with low signal, bins with 0 reads in either DMSO or PROTAC were assigned a value of 0 in unscaled Δ PRO-seq (Figure 3B, 3E, 3H, 3K), fold change in elongation rate (Figure 7D), and Δ TT-seq heatmaps (Figure 2D).
Boxplot analysis
Boxes show 25th–75th percentiles and whiskers depict 1.5 times the interquartile range. All figures were made in Graphpad Prism.
QUANTIFICATION AND STATISTICAL ANALYSIS
Parametric and nonparametric comparisons were performed in GraphPad Prism using tests described in figure legends. The hypergeometric test was performed in R using the phyper function.
Supplementary Material
Highlights:
Rapid degradation of INTS11 globally increases early elongation complexes
In the absence of INTS11, RNAPII released into genes is deficient in elongation
Loss of INTS11 endonuclease does not disrupt Integrator-PP2A phosphatase function
Predominantly short genes and non-coding RNAs are upregulated upon INTS11 degradation
ACKNOWLEDGMENTS
We thank members of the Adelman and Wagner labs and Marie Bao for critical feedback on the manuscript. Lena Tveriakhina and Julia Rogers helped with microscopy, and Susanna Dang for preparing the lysate used in Figure 1C. We thank the HMS Nascent Transcriptomics Core for assistance with PRO-seq and TT-seq, the HMS Immunology Flow Cytometry Core for help with flow cytometry, and the Harvard Bauer Core Facility and HMS Biopolymers Facility for next-generation sequencing. This work was supported by NIH R01GM134539 to K.A. and E.J.W. and NIH F31AI160672 to C.B.S.
DECLARATION OF INTERESTS
K.A. received research funding from Novartis not related to this work, is on the SAB of CAMP4 Therapeutics and is a member of the Advisory Board of Molecular Cell. All other authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albrecht TR, and Wagner EJ (2012). snRNA 3’ End Formation Requires Heterodimeric Association of Integrator Subunits. [DOI] [PMC free article] [PubMed]
- Albrecht TR, Shevtsov SP, Wu Y, Mascibroda LG, Peart NJ, Huang K-L, Sawyer IA, Tong L, Dundr M, and Wagner EJ (2018). Integrator subunit 4 is a “Symplekin-like” scaffold that associates with INTS9/11 to form the Integrator cleavage module. Nucleic Acids Res. 46, 4241–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, and Noble WS (2015). The MEME Suite. Nucleic Acids Res. 43, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat D, and Wagner EJ (2015). Integrator: surprisingly diverse functions in gene expression. Trends Biochem. Sci 40, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat D, Hakimi M-A, Näär AM, Shilatifard A, Cooch N, and Shiekhattar R (2005). Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell 123, 265–276. [DOI] [PubMed] [Google Scholar]
- Barra J, Gaidos GS, Blumenthal E, Beckedorff F, Tayar MM, Kirstein N, Karakac TK, Jensen TH, Impens F, Gevaert K, et al. (2020). Integrator restrains paraspeckles assembly by promoting isoform switching of the lncRNA NEAT1. Sci. Adv 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckedorff F, Blumenthal E, daSilva LF, Aoi Y, Cingaram PR, Yue J, Zhang A, Dokaneheifard S, Valencia MG, Gaidosh G, et al. (2020). The Human Integrator Complex Facilitates Transcriptional Elongation by Endonucleolytic Cleavage of Nascent Transcripts. Cell Rep. 32, 107917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran T, Pahita E, Ghosh S, Lenhard B, and Sarkies P (2021). Integrator is recruited to promoter-proximally paused RNA Pol II to generate Caenorhabditis elegans piRNA precursors. EMBO J. 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Raina K, Darricarrere N, Hines J, Gustafson JL, Smith IE, Miah AH, Harling JD, and Crews CM (2015). HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol 10, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buecker C, Srinivasan R, Wu Z, Calo E, Acampora D, Faial T, Simeone A, Tan M, Swigut T, and Wysocka J (2014). Reorganization of Enhancer Patterns in Transition from Naive to Primed Pluripotency. Cell Stem Cell 14, 838–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buske FA, Bodén M, Bauer DC, and Bailey TL (2010). Assigning roles to DNA regulatory motifs using comparative genomics. Bioinformatics 26, 860–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caine EA, Mahan SD, Johnson RL, Nieman AN, Lam N, Warren CR, Riching KM, Urh M, and Daniels DL (2020). Targeted Protein Degradation Phenotypic Studies Using HaloTag CRISPR/Cas9 Endogenous Tagging Coupled with HaloPROTAC3. Curr. Protoc. Pharmacol 91, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caizzi L, Monteiro-martins S, Chen Y, Lidschreiber M, Cramer P, Lysakovskaia K, Schmitzova J, and Sawicka A (2021). Article Efficient RNA polymerase II pause release requires U2 snRNP function Article Efficient RNA polymerase II pause release requires U2 snRNP function. 1–15. [DOI] [PubMed]
- Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Berhanu Lemma R, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Perez NM, et al. (2022). JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, and Li W (2013). DANPOS: Dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 23, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun Y, Joo YJ, Suh H, Batot G, Hill CP, Formosa T, and Buratowski S (2019). Selective Kinase Inhibition Shows That Bur1 (Cdk9) Phosphorylates the Rpb1 Linker In Vivo. Mol. Cell. Biol 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core L, and Adelman K (2019). Promoter-proximal pausing of RNA polymerase II: a nexus of gene regulation. [DOI] [PMC free article] [PubMed]
- Cortazar MA, Sheridan RM, Erickson B, Fong N, Glover-Cutter K, Brannan K, and Bentley DL (2019). Control of RNA Pol II Speed by PNUTS-PP1 and Spt5 Dephosphorylation Facilitates Termination by a “Sitting Duck Torpedo” Mechanism. Mol. Cell 76, 896–908.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danko CG, Hyland SL, Core LJ, Martins AL, Waters CT, Lee HW, Cheung VG, Kraus WL, Lis JT, and Siepel A (2015). Identification of active transcriptional regulatory elements from GRO-seq data. Nat. Methods 12, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasilva LF, Blumenthal E, Beckedorff F, Cingaram PR, Santos H.G. Dos, Edupuganti RR, Zhang A, Dokaneheifard S, Aoi Y, Yue J, et al. (2021). Integrator enforces the fidelity of transcriptional termination at protein-coding genes. Sci. Adv 7, 3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson L, Francis L, Eaton JD, and West S (2020). Integrator-Dependent and Allosteric/Intrinsic Mechanisms Ensure Efficient Termination of snRNA Transcription. Cell Rep. 33, 108319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy EE, Rutenberg-Schoenberg M, Stark CD, Kitchen RR, Gerstein MB, and Simon MD (2015). Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol. Cell 59, 858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JD, Francis L, Davidson L, and West S (2020). A unified allosteric/torpedo mechanism for transcriptional termination on human protein-coding genes. Genes Dev. 34, 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod ND, Henriques T, Huang KL, Tatomer DC, Wilusz JE, Wagner EJ, and Adelman K (2019). The Integrator Complex Attenuates Promoter-Proximal Transcription at Protein-Coding Genes. Mol. Cell 76, 738–752.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzeddine N, Chen J, Waltenspiel B, Burch B, Albrecht T, Zhuo M, Warren WD, Marzluff WF, and Wagner EJ (2011). A Subset of Drosophila Integrator Proteins Is Essential for Efficient U7 snRNA and Spliceosomal snRNA 3’-End Formation. Mol. Cell. Biol 31, 328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fianu I, Chen Y, Dienemann C, Dybkov O, Linden A, Urlaub H, and Cramer P (2021). Structural basis of Integrator-mediated transcription regulation. Science (80-. ) 374, 883–887. [DOI] [PubMed] [Google Scholar]
- Gardini A, Baillat D, Cesaroni M, Hu D, Marinis JM, Wagner EJ, Lazar MA, Shilatifard A, and Shiekhattar R (2014). Integrator regulates transcriptional initiation and pause release following activation. Mol. Cell 56, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Gilchrist DA, Nechaev S, Bern M, Muse GW, Burkholder A, Fargo DC, and Adelman K (2013). Stable Pausing by RNA Polymerase II Provides an Opportunity to Target and Integrate Regulatory Signals. Mol. Cell 52, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Scruggs BS, Inouye MO, Muse GW, Williams LH, Burkholder AB, Lavender CA, Fargo DC, and Adelman K (2018). Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 32, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Miao YR, Jia LH, Yu QY, Zhang Q, and Guo AY (2019). AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 47, D33–D38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Peng L, Xu C, Wang Z, Song A, and Chen FX (2021). SPT5 stabilizes RNA polymerase II, orchestrates transcription cycles, and maintains the enhancer landscape. Mol. Cell 81, 4425–4439.e6. [DOI] [PubMed] [Google Scholar]
- Huang K-L, Jee D, Stein CB, Elrod ND, Henriques T, Mascibroda LG, Baillat D, Russell WK, Adelman K, and Wagner EJ (2020). Integrator Recruits Protein Phosphatase 2A to Prevent Pause Release and Facilitate Transcription Termination. Mol. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger MG, and Winter GE (2021). Fast-acting chemical tools to delineate causality in transcriptional control. Mol. Cell 81, 1617–1630. [DOI] [PubMed] [Google Scholar]
- Jia Y, Cheng Z, Bharath SR, Sun Q, Su N, Huang J, and Song H (2021). Crystal structure of the INTS3/INTS6 complex reveals the functional importance of INTS3 dimerization in DSB repair. Cell Discov. 2021 71 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamieniarz-Gdula K, and Proudfoot NJ (2019). Transcriptional Control by Premature Termination: A Forgotten Mechanism. Trends Genet. 35, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein N, Gomes Dos Santos H, Blumenthal E, and Shiekhattar R (2021). The Integrator complex at the crossroad of coding and noncoding RNA. Curr. Opin. Cell Biol 70, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujirai T, and Kurumizaka H (2020). Transcription through the nucleosome. Curr. Opin. Struct. Biol 61, 42–49. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, and Lis JT (2013). Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science (80-. ) 339, 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Gardini A, Zhang A, and Shiekhattar R (2015). Integrator mediates the biogenesis of enhancer RNAs. Nature 525, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ma X, Banerjee S, Baruah S, Schnicker NJ, Roh E, Ma W, Liu K, Bode AM, and Dong Z (2021). Structural basis for multifunctional roles of human Ints3 C-terminal domain. J. Biol. Chem 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu J, Han Q, Peng T, Peng M, Wei B, Li D, Wang X, Yu S, Yang J, Cao S, et al. (2015). The oncogene c-Jun impedes somatic cell reprogramming. Nat. Cell Biol 17, 856–867. [DOI] [PubMed] [Google Scholar]
- Liu X, Guo Z, Han J, Hu X, David CJ, Correspondence C, Peng B, Zhang B, Li H, and Chen M (2022). The PAF1 complex promotes 3′ processing of pervasive transcripts. Cell Rep. 38, 110519. [DOI] [PubMed] [Google Scholar]
- Lopes I, Altab G, Raina P, and de Magalhães JP (2021). Gene Size Matters: An Analysis of Gene Length in the Human Genome. Front. Genet 12, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen S, Žumer K, Molska EŠ, Rouvière JO, Wu G, Demel C, Schwalb B, Schmid M, Cramer P, and Jensen TH (2020). Integrator is a genome-wide attenuator of non-productive transcription. Mol. Cell 1–16. [DOI] [PubMed] [Google Scholar]
- Narain A, Bhandare P, Adhikari B, Backes S, Eilers M, Dölken L, Schlosser A, Erhard F, Baluapuri A, and Wolf E (2021). Targeted protein degradation reveals a direct role of SPT6 in RNAPII elongation and termination. Mol. Cell 81, 3110–3127.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T, Tellier M, Foxwell J, Ribeiro de Almeida C, Tan-Wong SM, Dhir S, Dujardin G, Dhir A, Murphy S, and Proudfoot NJ (2018a). Deregulated Expression of Mammalian lncRNA through Loss of SPT6 Induces R-Loop Formation, Replication Stress, and Cellular Senescence. Mol. Cell 72, 970–984.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T, Tellier M, Foxwell J, Ribeiro de Almeida C, Tan-Wong SM, Dhir S, Dujardin G, Dhir A, Murphy S, and Proudfoot NJ (2018b). Deregulated Expression of Mammalian lncRNA through Loss of SPT6 Induces R-Loop Formation, Replication Stress, and Cellular Senescence. Mol. Cell 72, 970–984.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Kwon JJ, Talamas JA, Borah AA, Vazquez F, Boehm JS, Tsherniak A, Zitnik M, McFarland JM, and Hahn WC (2022). Sparse dictionary learning recovers pleiotropy from human cell fitness screens. Cell Syst. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parua PK, Booth GT, Sansó M, Benjamin B, Tanny JC, Lis JT, and Fisher RP (2018). A Cdk9-PP1 switch regulates the elongation-termination transition of RNA polymerase II. Nature 558, 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleiderer MM, and Galej WP (2021). Structure of the catalytic core of the Integrator complex. Mol. Cell 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, and Jensen TH (2008). RNA exosome depletion reveals transcription upstream of active human promoters. Science (80-. ) 322, 1851–1854. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer KA, Mimoso CA, Adelman K, and Neugebauer KM (2021). Co-transcriptional splicing regulates 3′ end cleavage during mammalian erythropoiesis. Mol. Cell 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Replogle JM, Saunders RA, Pogson AN, Hussmann JA, Lenail A, Guna A, Mascibroda L, Wagner EJ, Adelman K, Lithwick-Yanai G, et al. (2022). Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell 185, 2559–2575.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rienzo M, and Casamassimi A (2016). Integrator complex and transcription regulation: Recent findings and pathophysiology. Biochim. Biophys. Acta - Gene Regul. Mech 1859, 1269–1280. [DOI] [PubMed] [Google Scholar]
- Rosa-Mercado NA, Zimmer JT, Apostolidi M, Rinehart J, Simon MD, and Steitz JA (2021). Hyperosmotic stress alters the RNA polymerase II interactome and induces readthrough transcription despite widespread transcriptional repression. Mol. Cell 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsova MP, Vasilkova DP, Moshareva MA, Malyavko AN, Meerson MB, Zatsepin TS, Naraykina YV, Beletsky AV, Ravin NV, and Dontsova OA (2019). Integrator is a key component of human telomerase RNA biogenesis. Sci. Reports 2019 91 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabath K, Stäubli ML, Marti S, Leitner A, Moes M, and Jonas S (2020). INTS10–INTS13–INTS14 form a functional module of Integrator that binds nucleic acids and the cleavage module. Nat. Commun 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012 97 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Hauf KF, Demel C, Tresch A, Gagneur J, and Cramer P (2016). TT-seq maps the human transient transcriptome. Science (80-. ) 352, 1225–1228. [DOI] [PubMed] [Google Scholar]
- Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, and Sharp PA (2008). Divergent transcription from active promoters. Science (80-. ) 322, 1849–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U. S. A 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Ferris AL, Wu X, Saraf A, Khanna KK, Florens L, Washburn MP, Hughes SH, and Pagano M (2015). The Integrator complex controls the termination of transcription at diverse classes of gene targets. Cell Res. 25, 288–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadelmayer B, Micas G, Gamot A, Martin P, Malirat N, Koval S, Raffel R, Sobhian B, Severac D, Rialle S, et al. (2014). Integrator complex regulates NELF-mediated RNA polymerase II pause/release and processivity at coding genes. Nat. Commun 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatomer DC, Elrod ND, Liang D, Xiao M-S, Jiang JZ, Jonathan M, Huang K-L, Wagner EJ, Cherry S, and Wilusz JE (2019). The Integrator complex cleaves nascent mRNAs to attenuate transcription. Genes Dev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoort SJ, Welsh SA, Devlin JR, Barbieri E, Knight DA, Offley S, Bjelosevic S, Costacurta M, Todorovski I, Kearney CJ, et al. (2021). The PP2A-Integrator-CDK9 axis fine-tunes transcription and can be targeted therapeutically in cancer. Cell 184, 3143–3162.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlaming H, Mimoso CA, Field AR, Martin BJE, and Adelman K (2022). Screening thousands of transcribed coding and non-coding regions reveals sequence determinants of RNA polymerase II elongation potential. Nat. Struct. Mol. Biol 29, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SM, Farnung L, Boehning M, Wigge C, Linden A, Urlaub H, and Cramer P (2018). Structure of activated transcription complex Pol II–DSIF–PAF–SPT6. Nature 560, 607–612. [DOI] [PubMed] [Google Scholar]
- Wagner EF, and Nebreda ÁR (2009). Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009 98 9, 537–549. [DOI] [PubMed] [Google Scholar]
- Wang Z, Song A, Xu H, Hu S, Tao B, Peng L, Wang J, Li J, Yu J, Wang L, et al. (2022). Coordinated regulation of RNA polymerase II pausing and elongation progression by PAF1. Sci. Adv 8, 5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ, and Davis RJ (1996). Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med 1996 7410 74, 589–607. [DOI] [PubMed] [Google Scholar]
- Wu Y, Albrecht TR, Baillat D, Wagner EJ, and Tong L (2017). Molecular basis for the interaction between Integrator subunits IntS9 and IntS11 and its functional importance. Proc. Natl. Acad. Sci. U. S. A 114, 4394–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, He C, Kaye EG, Li J, Mu M, Nelson GM, Dong L, Wang J, Wu F, Shi YG, et al. (2022). Dynamic control of chromatin-associated m6A methylation regulates nascent RNA synthesis. Mol. Cell 82, 1156–1168.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J, Lai F, Beckedorff F, Zhang A, Pastori C, and Shiekhattar R (2017). Integrator orchestrates RAS/ERK1/2 signaling transcriptional programs. Genes Dev. 31, 1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Qi Y, Hu S, Cao X, Xu C, Yin Z, Chen X, Li Y, Liu W, Li J, et al. (2020). Identification of Integrator-PP2A complex (INTAC), an RNA polymerase II phosphatase. Science (80-. ) 370. [DOI] [PubMed] [Google Scholar]
- Žumer K, Maier KC, Farnung L, Jaeger MG, Rus P, Winter G, and Cramer P (2021). Two distinct mechanisms of RNA polymerase II elongation stimulation in vivo. Mol. Cell 81, 3096–3109.e8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic datasets generated in this manuscript have been deposited to GEO under accession GSE200702.
Original scripts described have been deposited to Zenodo and DOIs are provided in the Key Resources Table.
Further information required for reanalysis of data reported here will be available upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| INTS1 | Bethyl Labs | RRID: AB 2127258; Cat # A300–361A | |
| INTS3 | Bethyl Labs | RRID: AB 2127274; Cat # A302–051A | |
| INTS4 | Bethyl Labs | RRID: AB 937909; Cat # A301–296A | |
| INTS10 | Proteintech | RRID: AB 2127260; Cat # 15271–1-AP | |
| INTS11 | Sigma | RRID: AB 10600425; Cat # HPA029025 | |
| HaloTag | Promega | RRID: AB 2688011; Cat # G9211 | |
| CPSF73 | Bethyl Labs | RRID: AB 2084528; Cat # A301–091A | |
| c-Jun (60A8) | Cell Signaling Technology | RRID: AB 2130165; Cat # 9165 | |
| c-Fos (9F6) | Cell Signaling Technology | RRID: AB 2247211; Cat # 2250 | |
| RNA Pol II CTD Ser2-p (3E10) | Millipore | RRID: AB 10627998; Cat # 04–1571 | |
| RNA Pol II CTD Ser5P (3E8) (for Western) | Active Motif | RRID: AB 2687451; Cat # 61085 | |
| RNA Pol II CTD Ser5P (for ChIP-seq) | Abcam | RRID: AB 449369; Cat # AB5131 | |
| Spt5-Ser664P | (Huang et al., 2020) | N/A | |
| Histone H3 | Abcam | RRID: AB 302613; Cat # AB1791 | |
| GAPDH | Proteintech | RRID: AB 2263076; Cat # 10494–1-AP | |
| Vinculin (EPR8158) | Abcam | RRID: AB 11144129; Cat # AB129002 | |
| Chemicals, Peptides, and Recombinant Proteins | |||
| LIF | Cell Guidance Systems | Cat # GFM200 | |
| PD0325901 | Reprocell | Cat # 04–0006 | |
| CHIR99021 | Reprocell | Cat # 04–0004 | |
| Fibronectin | Millipore | Cat # FC010 | |
| bFGF | PeproTech | Cat # 100–18B | |
| Biotin-11-NTPs | Perkin Elmer | Cat # NEL54(2/3/4/5)001 | |
| 4-thiouridine | Tocris | Cat # 37005 | |
| HaloPROTAC3 | Promega | Cat # GA3110 | |
| Janelia Fluor 646 | Gift from Talley Lambert (HMS) | N/A | |
| Janelia Fluor X549 | Gift from Stephen Blacklow (HMS) | N/A | |
| Phosphatase Inhibitor Cocktail 2 | Sigma | Cat # P5726 | |
| Phosphatase Inhibitor Cocktail 3 | Sigma | Cat # P0044 | |
| Critical Commercial Assays | |||
| Illumina TruSeq Stranded Total RNA Library Prep Gold | Illumina | Cat # 20020598 | |
| NEB Next Ultra II DNA library kit | NEB | Cat # E7103S | |
| Deposited Data | |||
| Raw and analyzed data | This paper | GEO: GSE200702 | |
| MNase-seq | (Henriques et al., 2018) | GEO: GSE85191 | |
| H3K27Ac ChIP-seq | (Vlaming et al., 2022) | GEO: GSE178230 | |
| H3K4me3 ChIP-seq | (Vlaming et al., 2022) | GEO: GSE178230 | |
| H3K4me1 ChIP-seq | (Buecker et al., 2014) | GEO: GSE56098 | |
| Experimental Models: Cell Lines | |||
| F121–9 | Jaenisch/Gribnau labs | 4DNSRMG5APUM | |
| INTS11Halo | This paper | N/A | |
| Experimental Models: Organisms/Strains | |||
| Oligonucleotides | |||
| Table S2 | This paper | N/A | |
| siGENOME Non-Targeting siRNA #2 | Dharmacon | D-001210-02-05 | |
| siGENOME Mouse INTS11 | Dharmacon | MQ-062233-01-0002 | |
| Recombinant DNA | |||
| pGEM-3Z N-terminal HaloTag | Gift from Danette Daniels and Elizabeth Caine (Promega) | N/A | |
| pGEM-3Z-mINTS11-CAST | This paper | N/A | |
| pGEM-3Z-mINTS11–129 | This paper | N/A | |
| pUC-U7-GFP | (Albrecht and Wagner, 2012) | N/A | |
| pCAG-eCas9-GFP-U6-gRNA | Addgene | 79145 | |
| pCAG-eCas9-GFP-U6-mINTS11-gRNA | This paper | N/A | |
| Software and Algorithms | |||
| bowtie 1.2.2 | (Langmead et al., 2009) | N/A | |
| STAR 2.7.3a | (Dobin et al., 2013) | N/A | |
| R | www.r-project.org | N/A | |
| Rstudio | www.rstudio.com | N/A | |
| featureCounts | (Liao et al., 2014) | N/A | |
| DESeq2 | (Love et al., 2014) | N/A | |
| rMATS | (Shen et al., 2014) | N/A | |
| Prism | GraphPad | N/A | |
| Partek Genomics Suite | www.partek.com | N/A | |
| get_gene_annotations.sh | DOI 10.5281/zenodo.5519927 | N/A | |
| make_heatmap | DOI 10.5281/zenodo.5519914 | N/A | |
| trim_and_filter_PE.pl | DOI 10.5281/zenodo.5519914 | N/A | |
| bowtie2stdBedGraph.pl | DOI 10.5281/zenodo.5519914 | N/A | |
| bedgraphs2stdBedGraph | DOI 10.5281/zenodo.5519914 | N/A | |
| cutadapt | DOI:10.14806/ej.17.1.200 | N/A | |
| samtools | (Li et al., 2009) | N/A | |
| bedtools | (Quinlan and Hall, 2010) | N/A | |
| UCSCtools | (Kent et al., 2010) | N/A | |
| Enrichr | (Kuleshov et al., 2016) | N/A | |
| ImageJ | (Schneider et al., 2012) | N/A | |
| dREG | (Danko et al., 2015) | N/A | |
| Enhancer annotation pipeline: dRIP-Filter, CoGENT, TSScentR | DOI 10.5281/zenodo.6654472 | N/A | |
| DANPOS | (Chen et al., 2013) | N/A | |
| MEME Suite | (Bailey et al., 2015) | N/A | |