

Introduction:
In sickle cell disease (SCD), an abnormal beta-globin gene with a missense mutation leads to hemoglobin polymerization and sickling within the red blood cell (RBC)1, associated with extensive morbidity and early mortality2–4. With the advent of newborn screening, penicillin prophylaxis, and pneumococcal vaccination, the mortality rate in children has decreased. However, the median survival for adults in SCD has not improved4, 5. Risk factors for early mortality in adults include elevated tricuspid regurgitant jet velocity (TRV), renal failure, and lung impairment6, 7. Current SCD-directed drug therapies include hydroxyurea (HU), l-glutamine, voxelotor, and crizanlizumab. However, none of these pharmaceutical agents offer a cure8.
Successful hematopoietic cell transplant (HCT) interrupts the progression of SCD-related complications, changes the patient’s genotype from SCD to donor HbAA or HbAS, establishes donor-derived erythropoiesis, and can stabilize or restore organ function9, 10. Hence, HCT is an important curative option for individuals with SCD. SCD HCT can be allogeneic or autologous. In allogeneic HCT, the source of donor hematopoietic cells is from a human leukocyte antigen- (HLA) matched sibling donor (MSD), HLA-haploidentical, umbilical cord blood (UCB), or HLA-matched unrelated donor (MUD).
There is a range of conditioning regimen intensity, such as myeloablative, reduced-intensity conditioning (RIC), and non-myeloablative (NMA) regimens. Myeloablative conditioning includes high-dose chemotherapy, which is generally limited to children or adults without significant organ damage. Infertility is a major concern after myeloablative conditioning. RIC uses lower doses of chemotherapy which may preserve gonadal function. However, myeloablative or RIC can lead to graft-versus-host disease (GVHD)11. Non-myeloablative HCT employs intensive immunosuppression to decrease the risk of graft rejection and GVHD while preserving fertility. While the risk of GVHD may be lower, the graft rejection rate can be higher12 after NMA conditioning.
Patients who undergo gene-modified autologous HCT receive their own hematopoietic stem and progenitor cells (HSPCs). These genetically engineered autologous cells eliminate the need for immunosuppression and, more importantly, the risks of GVHD and immune-mediated graft rejection13, 14. Nevertheless, the need for myeloablative chemotherapy currently limits this treatment to those with suitable organ function.
Four curative options are used most commonly in individuals with SCD: MSD HCT, HLA-haploidentical HCT, and autologous gene therapy and gene editing. Here we review the risks and benefits of these four curative options and summarize progress that has been made in the past five years.
HLA MSD HCT
Pediatric
Indications for MSD HCT in children with SCD have traditionally included stroke, elevated transcranial doppler (>200 m/s), or recurrent VOC despite HU15, 16. In 1996, a pediatric multi-center myeloablative HLA MSD HCT trial for SCD showed overall survival (OS) and EFS of 91% and 73%, respectively at, four-years of follow-up10. Bernaudin et al. in 2007 reported 93.1% OS and 86.1% EFS with at least two years of follow-up. Twenty percent of the patients had grade II or higher acute GVHD (aGVHD), and 12.6% had chronic GVHD (cGVHD). The authors noted that adding anti-thymocyte globulin (ATG) to busulfan and cyclophosphamide decreased the graft rejection rate from 22.6% to 2.9%17.
More recently, a compilation of single-center patient series including more than 800 patients (age range 1.7–20.3 years) reported an OS of 95% and EFS of 92% for pediatric myeloablative MSD HCT16. Still, 7–40 % developed aGVHD, and 5–20% developed cGVHD. Registry data gathered by the Center for International Blood and Marrow Transplant Research showed the risk for aGVHD and cGVHD was higher with increasing age; a similar trend was seen with age and EFS, OS, graft failure, and death18. Further, decreased EFS and higher mortality, aGVHD, and chronic GVHD were shown in patients ≥13 years with RIC compared to NMA regimens19. There was no significant difference in outcome after myeloablative and NMA conditioning. Brazauskas et al. stratified SCD HCT patients according to good, intermediate, and high-risk groups based on their EFS. Patients <12 years old with MSD were in the good-risk group, followed by patients <12 with unrelated donors, then patients >13 years with MSD, and lastly, patients >13 years with unrelated donors20. The 3-year EFS was >90% in the good-risk group compared with ~60% in the high-risk group, which included patients ≤12 years who received mismatched donor HCT. Graft failure was significantly higher in the high-risk group (21–38%), compared with 6% graft failure in the good-risk group.
At least 12 MSD HCT studies in the last five years show improved EFS and OS in the pediatric population (Table 1). These studies include patients with an age range of 0.3–28.9 years. The conditioning regimens vary among studies, and the median follow-up was 41 months across all the studies. OS and EFS range from 71.4–100% and 67–100%. The overall incidence of aGVHD ranged from 0–57%. While the study by Ngwube et al. showed 57% cGVHD, all but one patient had mild/limited cGVHD18. Although, the above historical studies have similar outcomes as the newer studies listed in Table 1, more recent studies report improved EFS and OS as noted in the three large registry studies (with EFS ranging from 90.7% to 93.9% and OS ranging from 95% to 97%)18, 19, 43.
Table 1A.
Summary of recent publications on HLA matched sibling donor hematopoietic cell transplant for sickle cell disease (2017–2022)
| Reference | Size | Median Age (range) years | Graft Source | Conditioning Regimen | Median follow up, months (range) | GVHD | Graft Failure | EFS | OS |
|---|---|---|---|---|---|---|---|---|---|
| * Gluckman et al 2017 | 846 | 8.3 (0.3–16.0) | BM/PBS C/UCB | varied | 56.4 (0.3–324.6) | NA acute, 13.3% chronic | NA | 93% | 95% |
| Marzollo et al 2017 | 7 | 6.5(4–16.3) | BM | Tre/Thio/Flu/ATG | 28.2(9.6–78) | 0% acute, 0% chronic | 0% | 100% | 100% |
| Garcia et al., 2017 | 11 | 7 (2–13) | BM | BU/Alem/CY | 37.2 (12–68.4) | 54% acute, 0% chronic | 0% | 82% | 91% |
| Park et al 2018 | 7 | 12.5 (6.9–16.9) | PBSC | Bu/Cy/ATG | 34 (22–47) | 57% acute, 47.6% chronic | 0% | NA | 71.4% |
| Alonso et al., 2019 | 22 | 8.6(2.09–15) | BM | Bu/Cy | 36 (NR) | 31.8% acute, 13.6% chronic | 0% | 79% | 85% |
| Guilcher et al 2019 | 16 | 12 (3 – 18) | PBSC | Alem/TBI 300cGY | 19.5 (15.2–27.8) | 0% acute, 0% chronic | 12.5% | 100% | 100% |
| * Eapen et al 2019 | 558 | 11 (6–17) | BM/PBS C/UCB | varied | 36 (18–60) | 6.2% acute, 18.1% chronic | 6% | 90.7% | 96.2% |
| * Bernaudin et al 2020 | 234 | 8.4 (2.2–28.9) | BM/PBS C/UCB | Bu/Cy/ATG | 94.8(1.2–331.2) | 20% acute, 10% chronic | 0.90% | 93.9% | 97% |
| Ngwube et al 2020 | 14 | 13 (7–21) | BM | Alem/Flu/Mel ± Thio | 19.2 (12–65.9) | 28.6% acute, 57% chronic | 0.07% | 92.9% | 100% |
| Sahdev et al 2020 | 9 | 10 (5–19) | BM | Late Alem//Flu/Mel | 108 (102–120) | 43% acute, 25% chronic |
33% | 67% | 100% |
| Sahdev et al 2020 | 16 | 6.5 (4–17) | BM | Early Alem/Flu/Mel | 54 (21.6–102) | 11% acute, 0% chronic |
0% | 94% | 94% |
| Oostenbrink et. al 2021 | 12 | 9.7 (1.3–18.0) | BM | Flu/Thio/Treo/ATG | 30 (0.4–36) | NA acute, 16.7% chronic | 13% | 79% | 96% |
| John et. al 2021 | 38 | 8.6 (1.7–19.9). | BM | Bu/Cy/Alem | 57.6 (2.4–204) | 7.8% acute, 15.8% chronic | 0% | 94.7% | 94.7% |
| Carabant et al 2021 | 45 | 9.13 (2.01–19.08) | BM | Bu/Cy or Treo/Flu/Thio | 41 (10–246) | 6.8% acute, 5.4% chronic |
4% | 89.4% | 92.1% |
| Kogel et.al 2021 | 17 | 10.71 (1–20) | BM | Bu/CyorFlu/Thio/Treo, or Flu/Thio/Mel | 90.25 (18.75–208) | NA acute, 0% chronic | 1% | 94% | 100% |
Registry studies, data include cases reported in the individual series.
Abbreviations: BM: bone marrow, PBSC: peripheral blood stem cell, UCB: umbilical cord blood, Treo, treosulfan, Thio thiotepa, Flu: fludarabine, ATG: anti-thymocyte globulin, BU: busulfan, Alem: alemtuzumab, Cy: cyclophosphamide, TBI: total body irradiation, cGY: centigray, Mel: melphalan, NR: not reported, NA: not available
Adult
Early clinical trials of MSD HCT in adults involved myeloablative conditioning. The first attempt to cure adults with MSD HCT involved myeloablative conditioning in two patients who were 40 and 56 years of age21. After prompt donor engraftment, they died of GVHD. Myeloablative chemotherapy also excludes HCT in SCD adults with impaired renal function, decreased pulmonary function, cirrhosis, pulmonary hypertension, and poor Karnofsky Performance Status (KPS <70). Therefore, the National Institutes of Health (NIH), focused on NMA MSD HCT in lieu of myeloablative chemotherapy in adults with irreversible end-organ damage such as stroke, elevated TRV (≥ 2.5 m/s), pulmonary hypertension, chronic renal insufficiency, sickle hepatopathy, and reversible complications not ameliorated by HU. The approach utilized a minimal toxicity regimen of alemtuzumab, 300 cGy total body irradiation (TBI), and sirolimus to elicit an immunoablative effect in lieu of myeloablation to mitigate risks of GVHD and graft failure and to lower the risk of transplant-related morbidity and mortality. The first report in 2009 showed an OS of 100% and an EFS of 90%22 with 30 months follow up. Notably, no one developed GVHD. An expanded update with this regimen included three sites with 122 patients. With a median follow-up of 4 years, the OS was 93%, and EFS was 85%, with no grade III to IV aGVHD and no cGVHD8. Therefore, HCT of adults with SCD is feasible and practical with this regimen.
In the past five years, results from 4 studies of MSD HCT in adults (up to age 65 years) have been reported. The overall median follow-up was 40.2 months (Table 1). One hundred fifty-nine patients received varying conditioning regimens. OS ranged from 93–100%, with 87–100% EFS. The incidence of aGVHD and cGVHD was 2–18% and 0–29%, respectively. Therefore, NMA HLA-matched sibling HCT can be performed safely in adults with comorbidities23. Unfortunately, the constraint of lacking a MSD and an adverse risk profile in older adults have limited access to this curative option in most patients. This underscores the need for new studies to expand donor availability and improve patient outcomes.
Haploidentical HCT
Pediatric
Most patients will have an HLA-haploidentical donor24. In 2013, the first pediatric HLA- haploidentical HCT study for SCD was reported25. Eight patients < 14 years of age received myeloablative conditioning, and all patients had a history of stroke. At seven years of follow-up, OS was 75%, and EFS was 38%. Four patients experienced aGVHD, and 3 had cGVHD25.
In the last five years, approximately 100 pediatric patients have received HLA-haploidentical HCT with an overall median follow-up of 21.5 months (Table 2). The conditioning regimens varied across these patient series; most included ATG, fludarabine, and thiotepa and 4 utilized post-transplant cyclophosphamide (PT-Cy) to abrogate host-versus-graft and graft-versus-host reactions. Overall survival and EFS have improved to 84–100%. However, the incidence of aGVHD after haploidentical HCT is about twice the rate of aGVHD observed after MSD HCT in children (Table 2). The cumulative incidence of haploidentical HCT cGVHD is similar to MSD HCT in the pediatric population. Although the study by Pawlowska et al.26 had a high cGVHD rate at 75%, this series included 3 patients who had mild, limited skin GVHD that responded to therapy. The total sample size of the haploidentical HCT retrospective cohort is smaller than the MSD HCT experience. Hence, there is a need for additional studies to assess outcomes after HLA-haploidentical HCT in children. There is an ongoing multicenter clinical trial conducted through the Blood and Marrow Transplant Clinical Trials Network that is evaluating a reduced intensity regimen with PT-Cy in children with severe SCD.
Table 2A.
Summary of recent publications on Haploidentical hematopoietic cell transplant for sickle cell disease (2017–2022)
| Reference | Size | Median Age years (range) | Graft Source | Conditioning Regimen | Median follow-up, months (range) | GVHD | Graft Failure | EFS | OS |
|---|---|---|---|---|---|---|---|---|---|
| Gilman et al. 2017 | 8 | 16 (8–23) | ex vivo CD34+ cell selected CD3+ depleted PBSC | ATG/Flu/Thio/Mel | 21.5 (9.9–60) | 25% acute, 13% chronic | 13% | 100% | 87% |
| Frangoul et al. 2018 | 4 | 14(12–23) | BM/PBSC | ATG/Thio/Flu Cy/TBI 200 cGy/PT-Cy | 16 (13–34) | 100% acute, 0% chronic | 0% | 100% | 100% |
| Pawlowska et al 2018 | 4 | 19 (13–23) | BM/PBSC | ATG/Flu/Bu/PT-Cy | 9 (5–11) | 25% acute, 75% chronic | 0% | 100% | 100% |
| * Eapen et al 2019 | 137 | 19 (13–27) | BM/PBSC | varied | 25 (12–48) | 8.7% acute, 16% chronic | 26% | 67.2% | 90.5% |
| Cairo et al. 2020 | 19 | 13(3–20) | PBSC | ATG/Flu/Cy/Thio/Bu/TBI 500 cGy | 46.3 (1.9–73.3) | 5% acute, 5% chronic | 0% | 84% | 84% |
| Foell al 2020 | 25 | 13(3–31) | CD3+/CD19+ or TCR αβ/CD19+ depleted PBSC | ATG/Flu/Thio/Treo | 20.0 (NR) | 28% acute, 16% chronic | 8% | 100% | 88% |
| *Gluckman et al 2020 | 71 | 9.3 (2–43) | BM/PBSC | varied | 38 (2–154) | 23% acute, 23% chronic | 16% | 62% | 88% |
| Jaiswal et al 2020 | 5 | 8 (3–19) | PBSC | ATG/Flu/Treo/Mel/PT-Cy & Aba | 28 (13–54) | 0% acute, 0% chronic | 0% | 100% | 100% |
| Kharya et. al 2021 | 25 | 7 (1–27) | PBSC | (Flu/Cy/Dex)/ATG/Flu/Cy/Thio/Bu/TBI 200 cGy//PT-Cy, | 15.9 (6.5–26.3) | 20% acute, 12% chronic | 0% | 88% | 88% |
Registry studies, data include cases reported in the individual series.
Abbreviations: BM: bone marrow, PBSC: peripheral blood stem cell, Treo, treosulfan, Thio thiotepa, Flu: fludarabine, ATG: anti-thymocyte globulin, Bu: busulfan, Alem: alemtuzumab, Cy: cyclophosphamide, TBI: total body irradiation, cGY: centigray, Mel: melphalan, PT-Cy: post-transplant cyclophosphamide Aba: abatacept, Dex: dexamethasone NR: not reported
Adult
The Hopkins group reported the first haploidentical study for adults with SCD in 201212. Indications for HCT included stroke, ACS requiring exchange transfusion or hospitalization, recurrent VOC, stage I or II sickle lung disease, red cell alloimmunization, history of invasive pneumococcal disease, or transfusion-dependence. Fourteen patients, including 2 patients less than 18 years, received conditioning with pre-HCT cyclophosphamide, fludarabine, ATG, TBI 200cGy, and PT-Cy. Importantly, all patients survived, and none developed acute or chronic GVHD 2 years post-HCT. However, 6 patients (43%) experienced acute graft rejection, and a seventh patient with low donor chimerism levels had persistent transfusion-dependent anemia after HCT12.
In parallel, the NIH group enrolled SCD patients with severe comorbidities, including end-stage renal disease requiring dialysis, pulmonary hypertension, heart failure, and cirrhosis23. The NMA conditioning regimen utilized in the MSD HCT setting, based on murine data, was modified to accommodate these patients with severe disease27. The study included three cohorts with increasing doses of PT-Cy. Twenty-three patients who were 20 to 56 years of age were enrolled. Twenty-one had SCD, and two had transfusion-dependent beta-thalassemia. None of the patients died before 100 days post-HCT, and OS was 86%23. The addition of PT-Cy improved the engraftment rate from 33% with no PT-Cy to 83% with 100mg/kg PT-Cy. The EFS increased from 0% after no PT-Cy to 50% with 100mg/kg PT-Cy. While none of the patients experienced moderate to severe GVHD, the graft rejection rate was unacceptably high28.
A compilation of recent studies of HLA- haploidentical HCT in adults with severe SCD over the past five years showed OS at 87–100% and EFS of 50–93%, with an overall median follow-up of 20.6 months (Table 2). The study with the lowest OS and EFS was the initial study referenced above in 2017. While the aGVHD rate ranged from 8.3–33%, most aGVHD was grade II. The cGVHD rate spanned 8.3–16%. An extended period of follow-up will be necessary to assess the efficacy of HLA- haploidentical HCT in adults, but this curative option has the potential to expand the donor pool for SCD patients who might benefit from HCT.
Future of Allogeneic HCT in SCD
Table 3 shows active and planned clinical trials of MSD HCT and haploidentical HCT for SCD. The MSD trials in children focus on decreasing toxicity by administering less intensive conditioning, a strategy that will be monitored carefully for the incidence of graft failure. In adults, efforts aim to improve long-term EFS by reducing GVHD through minimal toxicity regimens (low-dose total body irradiation and alemtuzumab). Three pediatric HLA-haploidentical HCT trials focus on T cell depletion and reduced-intensity conditioning.
Table 3A.
Summary of clinical trials of recruiting and not yet recruiting studies for HLA Haploidentical and Matched Sibling Donor HCT
| Recruiting | ||||||
|---|---|---|---|---|---|---|
| Identifier/Location | Study Title | Age Range (years) | Participant Number | Phase | Study Goal | |
| HLA Matched Related D | ||||||
| Pediatrics | NCT03214354/Calgary | A Phase II Pilot Study of Nonmyeloablative Conditioning Hematopoietic Stem Cell Transplantation in Children With Sickle Cell Disease Wh+C7:H14o Have a Matched Related Major ABO-Incompatible Donor (Sickle-AID) | 1 Year to 19 Years | 12 | 2 | Safety and efficacy of NMA conditioning for matched related major ABO-incompatible donor. |
| NCT04018937/Emory | Early HLA Matched Sibling Hematopoietic Stem Cell Transplantation for Children With Sickle Cell Disease: A Sickle Transplant Advocacy and Research Alliance (STAR) Trial | 2 Years to 13 Years | 58 | 2 | Safety and efficacy of fludarabine, alemtuzumab and melphalan (FAM) conditioning in pediatrics. | |
| NCT03587272/DC | Minimizing Toxicity in HLA-Identical Related Donor Transplantation for Children With Sickle Cell Disease | 2 Years to 25 Years | 30 | 2 | Decrease toxicity while achieving a high cure rate for children using alemtuzumab, low dose total-body irradiation, and sirolimus | |
| NCT01499888/Chicago | Ph I/II Study of Allogeneic SCT for Clinically Aggressive SCD | 16 Years to 60 Years | 15 | 1 / 2 | Use of immune-suppressive agents and low-dose TBI in patients not candidates for or experienced complications from hydroxyurea therapy. | |
| NCT02105766/NIH | Nonmyeloablative Peripheral Blood Mobilized Hematopoietic Precursor Cell Transplantation for Sickle Cell Disease and Beta-Thalassemia in Individuals With Higher Risk of Transplant Failure | 4 Years and older | 162 | 2 | To see if low dose radiation (300 cGy), oral cyclophosphamide, pentostatin, and sirolimus help to better accept donor stem cells. | |
| Children and Adults | NCT03421756/Pittsburgh | A Pilot Study Evaluating the Efficacy of Non-Myeloablative Matched Related Donor Peripheral Blood Stem Cell Transplant in Patients With Severe Sickle Cell Disease | 18 Years and older | 12 | 1 | MRD allogeneic SCT in adults with severe sickle cell disease using alemtuzumab. |
| NCT00061568/NIH | Nonmyeloablative Allogeneic Peripheral Blood Mobilized Hematopoietic Precursor Cell Transplantation For Severe Congenital Anemias Including Sickle Cell Disease (SCD) and Beta-Thalassemia | 2 Years and older | 150 | 1/2. | Low dose radiation, alemtuzumab and sirolimus as a strategy to provide clinical remission with a lower risk of GVHD development | |
| NCT05249452/Amsterdam | Adding Azathioprine/Hydroxyurea Preconditioning to Alemtuzumab/TBI to Reduce Risk of Graft Failure in Matched Sibling Donor Allogeneic HSCT in Adult Sickle Cell Patients | 16 Years to 60 Years | 20 | N/A | Investigate whether 3-month long preconditioning with azathioprine and alemtuzumab/TBI improves disease-free survival and donor chimerism | |
| Identifier | Study Title | Age range (yr) | Participant Number | Phase | Study Goal | |
| HLA Haploidentical | ||||||
| Pediatric | NCT04362293/St. Jude | Reduced Intensity Related Donor Peripheral Blood Derived Hematopoietic Progenitor Cell Transplantation for Patients With Severe SCD | 2 Years to 25 Years | 40 | 2 | RIC of alemtuzumab, thiotepa and low dose total body irradiation and pre-conditioning of hydroxyurea and azathioprine to reduce the risk of graft rejection. |
| NCT04207320/Chicago | Hematopoietic Stem Cell Transplantation for Patients With Severe Sickle Cell Disease Using Myeloablative Conditioning and αβ+ T-cell Depleted Hematopoietic Stem Cells From Partially Matched Familial Donors | 2 Years to 25 Years | 38 | N/A | Safety of haploidentical HSCT using αβ+ T-cell depletion for children and adolescents with severe SCD. | |
| NCT03367546/Minnesota | Haploidentical Donor T-cell Replete Allogeneic Hematopoietic Cell Transplant Following Reducing Intensity Conditioning for Patients With Selected High Risk Non-Malignant Disease | up to 25 Years | 20 | 2 | Use of T-cell replete RIC Haplo HCT for individuals who lack a suitable HLA-matched sibling donor. | |
| NCT03279094/City of Hope | A Pilot Study of Pre-transplant Immunosuppressive Therapy for Haploidentical Transplants in Patients With SCD | 1 Year to 30 Years | 18 | 1 | Haploidentical donors and non-toxic, myeloablative regimen, with the goal of achieving consistent donor chimerism. | |
| NCT03240731/Paris | BMT HLA Haploidentical After RIC and Prevention of GVHD Based on Post-transplant Cyclophosphamide Administration in Patients With Severe SCD | 13 Years to 40 Years | 16 | 2 | RIC haploidentical marrow transplants and prevention of GVHD based on cyclophosphamide administration post-transplantation. | |
| NCT03263559/Wisconsin | Haploidentical BMT in Sickle Cell Patients (BMTCTN1507) | 5 Years to 45 Years | 80 | 2 | Use of pre-conditioning hydroxyurea with Thymoglobulin/Cyclophosp hamide/Fludarabine/Thiotep a and post-transplant high-dose cyclophosphamide. | |
| NCT02757885/Emory | Transplantation Using Reduced Intensity Approach for Patients With SCD From Mismatched Family Donors of Bone Marrow (TRANSFORM) | 15 Years to 40 Years | 15 | 2 | Haploidentical SCT with hydroxyurea, fludarabine, thiotepa, anti-thymocyte globulin, radiation and cyclophosphamide. | |
| NCT02065596/Cleveland | Hematopoietic Stem Cell Transplant for Sickle Cell Disease | 18 Years to 65 Years | 25 | 1/2. | Use of fludarabine to reduce graft rejection. | |
| NCT03077542/NIH | Nonmyeloablative Haploidentical Peripheral Blood Mobilized Hematopoietic Precursor Cell Transplantation for Sickle Cell Disease | 2 Years and older | 93 | 1/2. | Decrease risk of GVHD with low radiation dose, Alemtuzumab, Sirolimus, Cyclophosphamide, and pentostatin. | |
Similarly, trials of HLA-haploidentical HCT in adults are focused on decreasing toxicity and reducing GVHD by applying pre-transplant immunosuppression in lieu of myeloablation, and ex vivo depletion of T cells with CD34+ selection29. Another study compared RIC MSD HCT with standard non-curative disease-modifying therapies. While still in the early phase of clinical development, an antibody-based myeloablative study selectively targeting hematopoietic stem cells using CD117 antibody-drug-conjugates is being pursued and is particularly applicable in non-malignant disorders such as SCD30. If successful, non-chemotherapy conditioning could expand the number of patients eligible for allogeneic HCT and autologous gene-based therapy for SCD.
Unrelated donors (URD) also contribute to expanding the donor pool, where SCD patients have a 57% chance of finding minimally HLA mismatched donors in registries (defined as HLA-matched at 7 of 8 or 8 of 8 HLA alleles)31. However, the risk of aGVHD, cGVHD, and transplant-related mortality in the first URD SCT trial with a 2-year OS of 79% dampened enthusiasm for this particular reduced-intensity regimen32. Efforts to reduce GVHD risks have focused on PT-Cy33 and the use of abatacept. Inhibition of T-cell co-stimulation with abatacept was studied in 14 patients with a median follow-up of 1.6 years34. Seven of the 14 patients had a URD. Abatacept supplemented tacrolimus and methotrexate for GVHD prophylaxis. The median age at HCT was 13 years (range, 7–21 years), and the 2-year probability of OS and EFS was 100% and 92.9%, respectively. At day +100, the incidence of grade II to IV and grades III to IV aGVHD was 28.6% and 7%, respectively. The one-year incidence of cGVHD was 57%, which was mild/limited in all but one patient. Another study reported both OS and EFS were 100% at two years post MSD and MUD HCT51. The median age at HCT was 11 (3–21) years; one patient developed grade III-IV aGVHD and cGVHD in 5 patients. Altogether, these studies are promising for a role in decreasing GVHD after MUD HCT for SCD.
Autologous Gene Addition and Editing Therapies for SCD
Like HLA-haploidentical HCT, autologous gene therapy also expands the donor pool since patients serve as their own donors. However, much less is known about the long-term toxicities and durability of gene therapy compared with allogeneic HCT for SCD. Replacement gene therapy and gene editing rely upon the mobilization and collection of CD34+ HSPCs, isolation of the CD34+ cells followed by random genomic insertion of a gene addition lentiviral vector or modification of a targeted genomic sequence by gene editing reagents introduced by electroporation. The most common strategy utilizes the expression of an anti-sickling globin that generates fetal hemoglobin-like properties. A second strategy targets inactivation of an intronic erythroid-specific enhancer in BCL11a to induce high-level fetal globin expression due to BCL11a repression. BCL11a is a major fetal hemoglobin (HbF) repressor at birth (reviewed in14). Published reports suggest that both strategies have successfully reduced episodes of severe vaso-occlusive pain and markers of hemolysis in SCD35–37. In these clinical trials, the typical toxicity profile mimics that predicted by the administration of myeloablative busulfan that precedes the infusion of the genome-modified HSPCs. There have been no reports of failure of the modified cells to engraft. Bone marrow harvesting has been replaced by using single-agent plerixafor-mobilized HSPCs, which has improved the therapeutic effect of the drug product13, 38. Use of granulocyte-colony stimulating factor (G-CSF), although standard in other settings, is contraindicated in SCD outside of allogeneic HCT due to the risk of triggering VOC, multi-organ failure, and death39.
The gene therapy results in SCD, using the BB305 lentiviral vector, were reported recently37. Thirty-five patients with severe SCD received autologous CD34+ HSPCs after transduction with a lentiviral vector that expresses the anti-sickling hemoglobin, HbAT87Q. Post-infusion levels of the anti-sickling hemoglobin reached a plateau by 1–3 months post-infusion and were stable through 3 years of median follow-up. These levels generated a median total hemoglobin value that increased from 8.5 gm/dL at baseline to 11.0 gm/dL or more at six months, and HbAT87Q contributed to 40% or more of the total hemoglobin. Of interest, the HbAT87Q had a nearly pan-cellular RBC distribution. There was a mean of 85±8% of RBC with at least some HbAT87Q at 24 months balanced by roughly 15% of RBC that contained only HbS, the latter of which would be at risk for sickling. In the HbAT87Q-containing RBC population, the median HbAT87Q ranged from 11.7 to 22.7 pg per RBC, approximating the range of 13 to 18 pg per RBC of HbA present in the sickle trait RBC. This anti-sickling hemoglobin distribution would predict a clinical phenotype of sickle cell trait, which was supported by the clinical outcomes. Markers of hemolysis such as serum bilirubin, haptoglobin, lactate dehydrogenase (LDH), and reticulocyte count all approached normal levels or were improved compared with baseline, post-infusion. In patients who had at least six months of follow-up post-infusion, there was a cessation of severe vaso-occlusive painful events, defined as a hospitalization or administration of intravenous analgesics in an ambulatory setting. There was a median of 3.5 events per year (range, 2.0 to 13.5) 2 years before enrollment that declined to a median of 0 per year (range, 0 to 5.9) (Figure 1). Notably, there were no strokes reported in the follow-up period, including patients at risk of stroke. These preliminary results showed near correction of the SCD phenotype after a one-time infusion of the lentiviral transduced autologous HSPCs.
FIGURE 1:
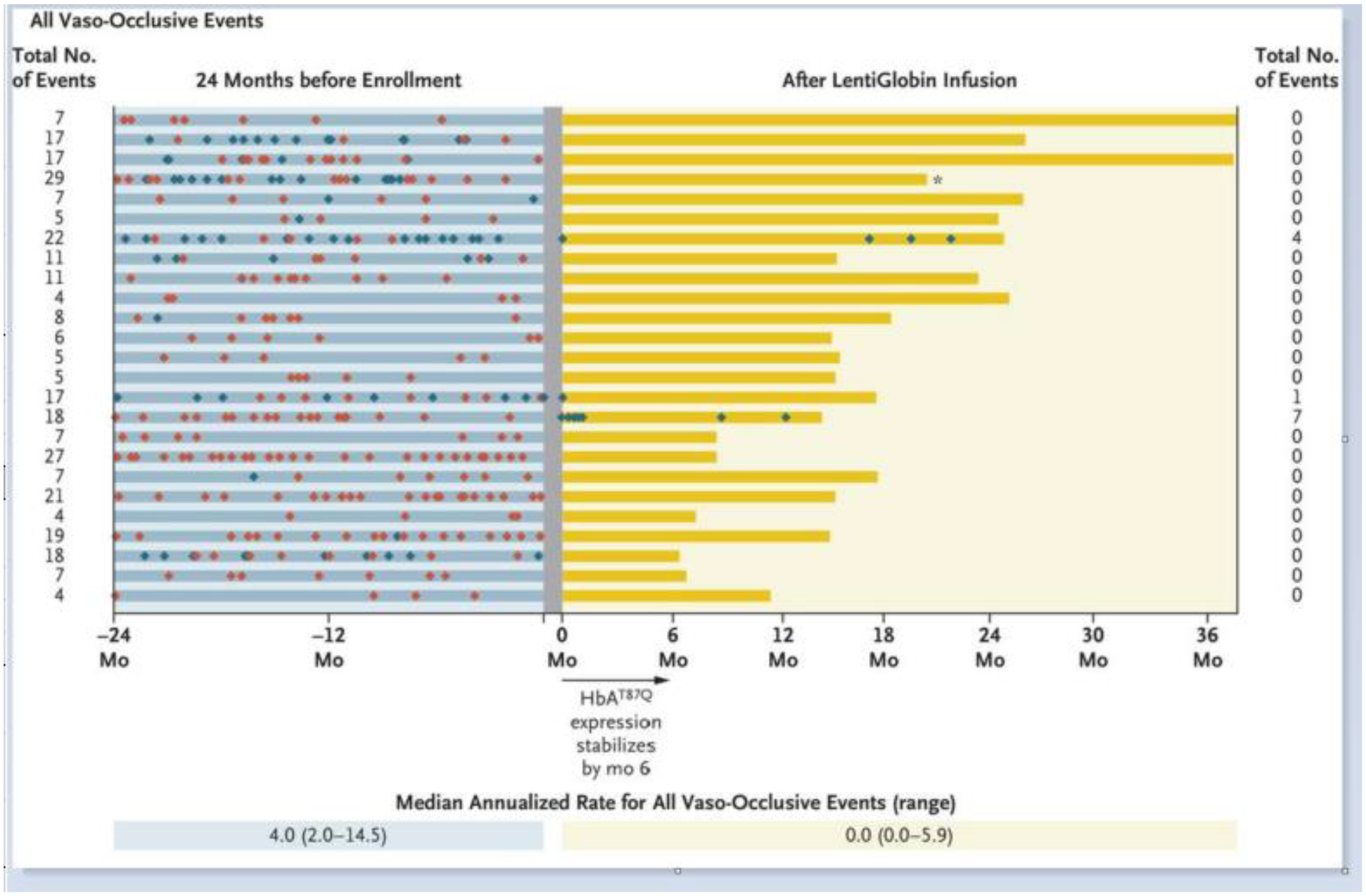
Changes in the Rate of Vaso-Occlusive Events before and after LentiGlobin Infusion. All vaso-occlusive events after the LentiGlobin infusion are presented. The gray shade indicates the period before infusion when transfusions were administered. Red and blue diamonds represent severe and non-severe vaso-occlusive pain events, respectively. From Kanter J, Walters MC, Krishnamurti L, et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N Engl J Med. 2022;386(7):617–628.
Parallel studies in gene therapy/gene editing aimed at inducing HbF have shown a similar toxicity profile after myeloablative busulfan and infusion of the modified HSPCs. One approach for HbF induction utilizes a lentiviral vector to express an inhibitory sequence embedded in a short hairpin microRNA (shmiR) that targets BCL11a mRNA for post-transcriptional silencing35. Because expression of the shmiR is under the control of erythroid-specific regulatory elements, BCL11a inhibition only occurs in the erythroid lineage. Six patients treated with the vector have had high-level, sustained HbF expression post-infusion. The serum hemoglobin increased from 9.3 to 11.4 gm/dL in the five patients not treated by RBC transfusions with a HbF percentage of 30.4% (range, 21.6 to 40.0). Markers of hemolysis also declined from baseline but were not normalized. None of the 6 patients had a severe vaso-occlusive episode, ACS, or stroke post-infusion.
A second approach for inducing HbF utilizes engineered CRISPR gene editing reagents to inactivate the same intronic erythroid-specific enhancer in BCL11a36. In a case report, a patient with SCD had robust HbF induction and stable expression in the peripheral blood, eliminating vaso-occlusive events and the need for RBC transfusions. Together, both strategies for HbF induction by BCL11a inactivation in erythroid cells appear feasible and effective.
Finally, another approach using CRISPR technology to correct the sickle mutation directly by homology-directed repair has generated compelling pre-clinical evidence of safety and efficacy. This approach will also be investigated in early phase clinical trials40, 41.
Risk of Acute Myelogenous Leukemia after Curative Therapy
Myeloid malignancies post NMA allogeneic HCT for SCD were reported in 3 of 76 patients at the NIH42. These arose in the setting of graft failure and, in two out of the three adult patients, pathogenic TP53 mutations detected at baseline prior to HCT increased in size over time until the development of overt malignancy. Pediatric patients have thus far fared better with little to no reports of hematologic malignancies. For example, in a study of 234 patients, with a median age 8.4 years and a median follow-up of about eight years after myeloablative MSD HCT, including 44% with mixed chimerism, no one developed a myeloid malignancy43.
Enthusiasm for autologous gene therapy has been tempered by the observation of myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) in 2 of 47 participants with SCD treated in the lentiviral gene therapy trial with Lentiglobin®44, 45. In both cases, the source of CD34+ cells was bone marrow. In subsequent patients, plerixafor-mobilized peripheral blood has replaced marrow as the source of CD34+ cells, due in part to increased CD34+ cell yield. Both patients developed monosomy 7 and acquired driver mutations in RUNX1 and PTPN11, and an additional KRAS mutation in one case, 3 – 5 years post-infusion. Both patients died of AML. The driver mutations were not detected in the pretreatment samples. In the first case, no vector was seen in the leukemic blast population; thus residual host cells exposed to the alkylating agent busulfan might have conferred treatment-related AML. In the second case, however, vector was present in leukemic blasts with integration near the VAMP4 gene, which is not associated with acute leukemia. The vector did not increase gene expression of VAMP4, and gene expression across the 10-megabase region flanking VAMP4 was not altered by vector insertion. It was determined that vector insertion was not the cause of AML45, but shown that the leukemic clone was in the transfected pool and thus not exposed to high dose busulfan.
The possibility of an association of AML with SCD has been suggested by retrospective registry data that show an increased risk of AML in SCD, although AML is still rare in this population46, 47. There is speculation that this association might be promoted by the inflammatory milieu and oxidative stress of hematopoiesis in the marrow niche that is characteristic of SCD. The selection of clonal hematopoiesis with driver mutations that exert a proliferative advantage in this milieu might also emerge after competitive repopulation of gene-modified HSCs following myeloablation and autologous HCT48. The basis for these observations is under investigation. It will be imperative to better inform patients about the long-term risk of clonal hematopoiesis and AML after gene therapy and, more broadly, after any curative therapy42. There are efforts to incorporate genomic screening to assess risk before gene therapy.
Conclusions
Several new curative therapies have arisen for SCD beyond MSD HCT, including haploidentical HCT, gene therapy, and gene editing. However, compelling comparative information is not yet available for individuals lacking a MSD that would facilitate selecting one curative therapy over another. The success rate of HCT in the pediatric population with HLA-matched donors is promising18, 23, 33, 49, 50. However, GVHD continues to be an obstacle. Continued efforts will be needed to decrease the graft failure rate after MSD HCT for adults and to decrease graft rejection and GVHD in adults and children undergoing HLA-haploidentical HCT. While autologous gene therapy and gene editing avoid immune complications such as GVHD and graft rejection, they rely upon myeloablative chemotherapy, which excludes most older adults who have begun to experience cumulative organ toxicity from SCD from this potentially curative option. Further, enthusiasm for gene therapy and certain allogeneic approaches are tempered by the risk of treatment-related myeloid malignancies. Screening tools might be developed to predict myeloid malignancy development in the future, but screening criteria do not currently exist. There is also considerable uncertainty about how to accomplish access to curative therapies outside of resource-rich countries. Thus, additional research must be conducted, and health policies must be enacted to ensure patient safety and equitable access to these novel curative therapies. Lastly, curative therapy studies are greatly limited by lack of long-term efficacy and safety data. These data will be critical to developing a personalized approach to curative therapy for individuals with SCD.
Table 1B.
Summary of recent publications on HLA matched sibling donor hematopoietic cell transplant for sickle cell disease (2017–2022)
| Reference | Size | Median Age years (range) | Graft Source | Conditioning Regimen | Median Follow up, months (range) | GVHD | Graft Failure | EFS | OS |
|---|---|---|---|---|---|---|---|---|---|
| * Gluckman et al 2017 | 154 | 19.3 (16.0–54.4) | BM/PBSC | varied | 48.0 (2.18–305.9) | NA acute, 19.6% chronic | NA | 81% | 81% |
| Ozdogu et al 2018 | 20 | 33.4 (20–45) | PBSC | Flu/Bu/ATG/TBI 200 cGy/PT-Cy | 13.8 (0.3–50) | 5% acute, 0% chronic | 0% | 100% | 100% |
| Krishnamurti et al 2019 | 17 | (17–36) | PBSC | Flu/Bu/ATG | 32.4 (26.4–60) | 18% acute, 29% chronic | 0% | 94% | 94% |
| Alzahrani et al 2021 | 122 | (10–65) | PBSC | TBI 300 cGy, Alem | (7.2–180) | 2% acute, 0% chronic | 13% | 87% | 93% |
Registry studies, data includes cases reported in the individual series.
Abbreviations: BM: bone marrow, PBSC: peripheral blood stem cell, Flu: fludarabine, ATG: anti-thymocyte globulin, Bu: busulfan, Alem: alemtuzumab, PT-Cy: post-transplant cyclophosphamide, TBI: total body irradiation, cGY: centigray, NA: not available
Table 2B.
Summary of recent publications on haploidentical allogeneic cell transplant for sickle cell disease (2017–2022)
| Adults | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Reference | Size | Median Age (range) yr | Graft Source | Conditioning Regimen | Median follow up, months (range) | GVHD | GRAFT Failure | EFS | OS |
| Fitzhugh et al 2017 * | 12 | 36 (20–56) | PBSC | Alem/TBI 400 cGy PT-Cy | 38.0 (8.1–73.91) | 8.3% acute, 8.3% chronic |
17% | 50% | 92% |
| Saraf et al. 2018 | 8 | 28 (20–38) | PBSC | ATG/Flu/Cy/300 cGy TBI/PT-Cy | 16.8 (12 – 30.6) | 28% acute, 16% chronic |
13% | 75% | 87% |
| Bolanos-Meade et al 2019 | 12 | 26 (6–31) | BM | ATG/Flu/Cy/TBI 400 cGy PT-Cy | 23.2 (11.7–42.5) | 25% acute, 8% chronic |
8% | 83% | 100% |
| de la Fuente et al 2019 | 15 | 20 (7–40) | BM | ATG/Flu/Cy/ATG/T hio/TBI 200 cGy/PT-Cy | 18.0 (5.1–43.6) | 33% acute, 7% chronic |
7% | 93% | 100% |
Results of the third cohort are presented.
Abbreviations: BM: bone marrow, PBSC: peripheral blood stem cell, Thio thiotepa, Flu: fludarabine, ATG: antithymocyte globulin, Alem: alemtuzumab, TBI: total body irradiation, cGY: centigray, Cy: cyclophosphamide, PT-Cy: post-transplant cyclophosphamide
Table 3B.
Summary of clinical trials of recruiting and not yet recruiting studies for HLA Haploidentical and Matched Related Donor HCT
| Identifier | Study Title | Age range (yr) | Participant Number | Phase | Study Goal | |
|---|---|---|---|---|---|---|
| HLA Haploidentical | ||||||
| Children and Adults | NCT03653338/Pittsburgh | T-Cell Depleted, Alternative Donor Transplant in Pediatric and Adult Patients With Severe Sickle Cell Disease (SCD) and Other Transfusion-Dependent Anemias | 5 Years to 40 Years | 5 | 1/2. | CD3/CD19 depletion of mismatched donor grafts in the setting of reduced intensity, immune-ablative conditioning. |
| NCT03121001/Chicago | A Phase II Study of HLA-Haploidentical Stem Cell Transplantation to Treat Clinically Aggressive Sickle Cell Disease | 16 Years to 60 Years | 50 | 2 | HLA-haploidentical HSCT protocol using immunosuppressive agents and low-dose TBI for conditioning and post-transplant cyclophosphamide. | |
| NCT03249831/City of Hope | Pilot Study to Evaluate the Safety and Feasibility of Induction of Mixed Chimerism in Sickle Cell Disease Patients With COH-MC-17: a Non-Myeloablative, Conditioning Regimen and CD4+ T-cell-depleted Haploidentical Hematopoietic Transplant | 18 Years to 45 Years | 6 | 1 | Safety and feasibility of COH-MC-17 (cyclophosphamide, pentostatin and rabbit-anti-thymocyte globulin (ATG)) followed by a CD4+ T-cell-depleted Haplo HCT to induce mixed chimerism. | |
| NCT02675959/NY | Safety and Efficacy of Prophylactic Defibrotide in Children, Adolescents, and Young Adults With Sickle Cell Disease or Beta Thalassemia Following MAC and Haploidentical Stem Cell Transplantation Utili zing CD34 Enrichment and T-Cell (CD3) Addback | 6 Months to 34 Years | 40 | 2 | Follow-up trial to NYMC 526 (NCT01461837) to assess Defibrotide prophylaxis for haploidentical allogeneic SCT with CD34 enrichment and T-cell addback. | |
| NCT04201210/Germany | A Phase II Stratified Trial to Assess Haploidentical T-depleted Stem Cell Transplantation in Patients With Sickle Cell Disease With no Available Sibling Donor | 1 Year to 35 Years | 212 | 2 | Can an α/ß depleted T-Haplo-HCT improve disease-free survival, adverse events and safety which is equivalent to a MSD HCT. | |
| NCT04776850/Texas | Pre-Transplant Immunosuppression and Related Haploidentical Hematopoietic Cell Transplanta tion for Patients With Severe Hemoglobinopathies | 2 Years to 30 Years | 40 | 1 | Pre-transplant immunosuppressi on (PTIS) to lower the risk of GVHD. | |
Table 3C.
Summary of clinical trials of recruiting and not yet recruiting studies for HLA Haploidentical and Matched Sibling Donor HCT
| Not yet recruiting | ||||||
|---|---|---|---|---|---|---|
| Identifier | Study Title | Age range (yr) | Participant number | Phase | Study Goal | |
| NCT04046705/Paris | A Prospective Multicenter Trial Comparing Allogeneic Matched Related HSCT After a RIC Regimen, With Standard of Care in Adolescents and Adults With Severe SCD | 15 Years to 45 Years | 78 | 3 | Compare NMA MSD HCT versus no HSCT (for patients lacking MSD). | |
| NCT04099966/NY | Allo SCT for Malignant and Non-malignant Hematologic Diseases Utilizing Alpha/Beta T Cell and CD19+ B Cell Depletion | up to 30 Years | 20 | 2 | Utilizing α/β CD3+/CD19+ cell depletion to decrease the incidence of GVHD. | |
Key Points.
Review the efficacy of curative therapy for SCD
Study the toxicity associated with curative therapy for SCD
Evaluate the two most utilized allogeneic HCT options, MSD and haploidentical, and gene therapy and gene editing for SCD
Highlight progress made in curative therapy studies over the last 5 years for SCD
Synopsis.
Curative therapies for sickle cell disease (SCD) now encompass human leukocyte antigen (HLA)-haploidentical hematopoietic cell transplant (HCT) and gene therapy, which have the potential to expand access. However, comparative trial data that might facilitate selecting one curative therapy over another are unavailable. While HLA-matched sibling donor (MSD) HCT in children is successful in 90 – 95% of patients, it is rarely available. New strategies to decrease graft rejection and graft-versus-host disease (GVHD) risks are needed to expand HLA-haploidentical HCT. Autologous gene therapy with myeloablation also has limitations, but eliminates problems of GVHD and graft rejection. Herein, we review recent studies on curative therapies in the past 5 years.
Acknowledgements:
This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute (NHLBI) and the Cooperative Study of Late Effects for SCD Curative Therapies (COALESCE, 1U01HL156620-01, NHLBI)), National Institutes of Health.
Glossary
- SCD
sickle cell disease
- RBC
red blood cell
- MSD
matched sibling donor
- HCT
hematopoietic cell transplant
- HLA
human leukocyte antigen
- GVHD
graft-versus-host disease
- aGVHD
acute graft-versus-host disease
- cGVHD
chronic graft-versus-host disease
- RBC
red blood cell
- TRV
tricuspid regurgitant jet velocity
- HU
hydroxyurea
- URD
unrelated donors
- HSPCs
hematopoietic stem and progenitor cells
- AML
acute myeloid leukemia
- VOC
vaso-occlusive crises
- PTIS
pre-transplant immunosuppression
- PT-Cy
post-transplant cyclophosphamide
- OS
overall survival
- EFS
event-free survival
- ATG
anti-thymocyte globulin
- RIC
reduced intensity conditioning
- KPS
Karnofsky Performance Status
- NIH
National Institutes of Health
- NMA
non- myeloablative
- HbF
fetal hemoglobin
- MUD
matched unrelated donor
- UCB
umbilical cord blood
- TBI
total body irradiation
- GCSF
granulocyte-colony stimulating factor
- LDH
lactate dehydrogenase
- shmiR
short hairpin microRNA
- MDS
myelodysplastic syndrome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement- MCW has consulting agreements with All Cells, Inc, BioChip Labs, Inc, Ensoma, Inc and Vertex Pharmaceuticals.
References:
- 1.Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. Jul 15 2017;390(10091):311–323. doi: 10.1016/S0140-6736(17)30193-9 [DOI] [PubMed] [Google Scholar]
- 2.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. Jun 9 1994;330(23):1639–44. doi: 10.1056/NEJM199406093302303 [DOI] [PubMed] [Google Scholar]
- 3.Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. Apr 2010;38(4 Suppl):S512–21. doi: 10.1016/j.amepre.2009.12.022 [DOI] [PubMed] [Google Scholar]
- 4.Lanzkron S, Carroll CP, Haywood C Jr. Mortality rates and age at death from sickle cell disease: U.S., 1979–2005. Public Health Rep. Mar–Apr 2013;128(2):110–6. doi: 10.1177/003335491312800206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeBaun MR, Ghafuri DL, Rodeghier M, et al. Decreased median survival of adults with sickle cell disease after adjusting for left truncation bias: a pooled analysis. Blood. Feb 7 2019;133(6):615–617. doi: 10.1182/blood-2018-10-880575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maitra P, Caughey M, Robinson L, et al. Risk factors for mortality in adult patients with sickle cell disease: a meta-analysis of studies in North America and Europe. Haematologica. Apr 2017;102(4):626–636. doi: 10.3324/haematol.2016.153791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaturvedi S, Ghafuri DL, Jordan N, Kassim A, Rodeghier M, DeBaun MR. Clustering of end-organ disease and earlier mortality in adults with sickle cell disease: A retrospective-prospective cohort study. Am J Hematol. Sep 2018;93(9):1153–1160. doi: 10.1002/ajh.25202 [DOI] [PubMed] [Google Scholar]
- 8.Alzahrani M, Damlaj M, Jeffries N, et al. Non-myeloablative human leukocyte antigen-matched related donor transplantation in sickle cell disease: outcomes from three independent centres. Br J Haematol. Feb 2021;192(4):761–768. doi: 10.1111/bjh.17311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sachdev V, Hsieh M, Jeffries N, et al. Reversal of a rheologic cardiomyopathy following hematopoietic stem cell transplantation for sickle cell disease. Blood Adv. Oct 8 2019;3(19):2816–2824. doi: 10.1182/bloodadvances.2019000387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walters MC, Patience M, Leisenring W, et al. Bone marrow transplantation for sickle cell disease. N Engl J Med. Aug 8 1996;335(6):369–76. doi: 10.1056/NEJM199608083350601 [DOI] [PubMed] [Google Scholar]
- 11.Nakasone H, Fukuda T, Kanda J, et al. Impact of conditioning intensity and TBI on acute GVHD after hematopoietic cell transplantation. Bone Marrow Transplant. Apr 2015;50(4):559–65. doi: 10.1038/bmt.2014.293 [DOI] [PubMed] [Google Scholar]
- 12.Bolanos-Meade J, Fuchs EJ, Luznik L, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. Nov 22 2012;120(22):4285–91. doi: 10.1182/blood-2012-07-438408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esrick EB, Manis JP, Daley H, et al. Successful hematopoietic stem cell mobilization and apheresis collection using plerixafor alone in sickle cell patients. Blood Adv. Oct 9 2018;2(19):2505–2512. doi: 10.1182/bloodadvances.2018016725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orkin SH, Bauer DE. Emerging Genetic Therapy for Sickle Cell Disease. Annual review of medicine. Jan 27 2019;70:257–271. doi: 10.1146/annurev-med-041817-125507 [DOI] [PubMed] [Google Scholar]
- 15.Ozdogu H, Boga C. Hematopoietic Stem Cell Transplantation in Adult Sickle Cell Disease: Problems and Solutions. Turk J Haematol. Sep 2015;32(3):195–205. doi: 10.4274/tjh.2014.0311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walters MC, De Castro LM, Sullivan KM, et al. Indications and Results of HLA-Identical Sibling Hematopoietic Cell Transplantation for Sickle Cell Disease. Biol Blood Marrow Transplant. Feb 2016;22(2):207–211. doi: 10.1016/j.bbmt.2015.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernaudin F, Socie G, Kuentz M, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. Oct 1 2007;110(7):2749–56. doi: 10.1182/blood-2007-03-079665 [DOI] [PubMed] [Google Scholar]
- 18.Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. Mar 16 2017;129(11):1548–1556. doi: 10.1182/blood-2016-10-745711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eapen M, Brazauskas R, Walters MC, et al. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: a retrospective multicentre, cohort study. Lancet Haematol. Nov 2019;6(11):e585–e596. doi: 10.1016/S2352-3026(19)30154-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brazauskas R, Scigliuolo GM, Wang HL, et al. Risk score to predict event-free survival after hematopoietic cell transplant for sickle cell disease. Blood. Jul 30 2020;136(5):623–626. doi: 10.1182/blood.2020005687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Besien K, Bartholomew A, Stock W, et al. Fludarabine-based conditioning for allogeneic transplantation in adults with sickle cell disease. Bone Marrow Transplant. Aug 2000;26(4):445–9. doi: 10.1038/sj.bmt.1702518 [DOI] [PubMed] [Google Scholar]
- 22.Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med. Dec 10 2009;361(24):2309–17. doi: 10.1056/NEJMoa0904971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA. Jul 2 2014;312(1):48–56. doi: 10.1001/jama.2014.7192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de la Fuente J, Dhedin N, Koyama T, et al. Haploidentical Bone Marrow Transplantation with Post-Transplantation Cyclophosphamide Plus Thiotepa Improves Donor Engraftment in Patients with Sickle Cell Anemia: Results of an International Learning Collaborative. Biol Blood Marrow Transplant. Jun 2019;25(6):1197–1209. doi: 10.1016/j.bbmt.2018.11.027 [DOI] [PubMed] [Google Scholar]
- 25.Dallas MH, Triplett B, Shook DR, et al. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. May 2013;19(5):820–30. doi: 10.1016/j.bbmt.2013.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pawlowska AB, Cheng JC, Karras NA, et al. HLA Haploidentical Stem Cell Transplant with Pretransplant Immunosuppression for Patients with Sickle Cell Disease. Biol Blood Marrow Transplant. Jan 2018;24(1):185–189. doi: 10.1016/j.bbmt.2017.08.039 [DOI] [PubMed] [Google Scholar]
- 27.Fitzhugh CD, Weitzel RP, Hsieh MM, et al. Sirolimus and post transplant Cy synergistically maintain mixed chimerism in a mismatched murine model. Bone Marrow Transplant. Oct 2013;48(10):1335–41. doi: 10.1038/bmt.2013.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fitzhugh CD, Hsieh MM, Taylor T, et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical PBSCT. Blood Adv. Apr 25 2017;1(11):652–661. doi: 10.1182/bloodadvances.2016002972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilman AL, Eckrich MJ, Epstein S, et al. Alternative donor hematopoietic stem cell transplantation for sickle cell disease. Blood Adv. Jul 11 2017;1(16):1215–1223. doi: 10.1182/bloodadvances.2017005462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czechowicz A, Palchaudhuri R, Scheck A, et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat Commun. Feb 6 2019;10(1):617. doi: 10.1038/s41467-018-08201-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gragert L, Eapen M, Williams E, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N Engl J Med. Jul 24 2014;371(4):339–48. doi: 10.1056/NEJMsa1311707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shenoy S, Eapen M, Panepinto JA, et al. A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood. Nov 24 2016;128(21):2561–2567. doi: 10.1182/blood-2016-05-715870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bolanos-Meade J, Cooke KR, Gamper CJ, et al. Effect of increased dose of total body irradiation on graft failure associated with HLA-haploidentical transplantation in patients with severe haemoglobinopathies: a prospective clinical trial. Lancet Haematol. Apr 2019;6(4):e183–e193. doi: 10.1016/S2352-3026(19)30031-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ngwube A, Shah N, Godder K, Jacobsohn D, Hulbert ML, Shenoy S. Abatacept is effective as GVHD prophylaxis in unrelated donor stem cell transplantation for children with severe sickle cell disease. Blood Adv. Aug 25 2020;4(16):3894–3899. doi: 10.1182/bloodadvances.2020002236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Esrick EB, Lehmann LE, Biffi A, et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N Engl J Med. Jan 21 2021;384(3):205–215. doi: 10.1056/NEJMoa2029392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. N Engl J Med. Jan 21 2021;384(3):252–260. doi: 10.1056/NEJMoa2031054 [DOI] [PubMed] [Google Scholar]
- 37.Kanter J, Walters MC, Krishnamurti L, et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N Engl J Med. Feb 17 2022;386(7):617–628. doi: 10.1056/NEJMoa2117175 [DOI] [PubMed] [Google Scholar]
- 38.Tisdale JF, Pierciey FJ Jr., Bonner M, et al. Safety and feasibility of hematopoietic progenitor stem cell collection by mobilization with plerixafor followed by apheresis vs bone marrow harvest in patients with sickle cell disease in the multi-center HGB-206 trial. Am J Hematol. Sep 2020;95(9):E239–E242. doi: 10.1002/ajh.25867 [DOI] [PubMed] [Google Scholar]
- 39.Salinas Cisneros G, Thein SL. Research in Sickle Cell Disease: From Bedside to Bench to Bedside. Hemasphere. Jun 2021;5(6):e584. doi: 10.1097/HS9.0000000000000584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeWitt MA, Magis W, Bray NL, et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science translational medicine. Oct 12 2016;8(360):360ra134. doi: 10.1126/scitranslmed.aaf9336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lattanzi A, Camarena J, Lahiri P, et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Science translational medicine. Jun 16 2021;13(598)doi: 10.1126/scitranslmed.abf2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghannam JY, Xu X, Maric I, et al. Baseline TP53 mutations in adults with SCD developing myeloid malignancy following hematopoietic cell transplantation. Blood. Apr 2 2020;135(14):1185–1188. doi: 10.1182/blood.2019004001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bernaudin F, Dalle JH, Bories D, et al. Long-term event-free survival, chimerism and fertility outcomes in 234 patients with sickle-cell anemia younger than 30 years after myeloablative conditioning and matched-sibling transplantation in France. Haematologica. Jan 2020;105(1):91–101. doi: 10.3324/haematol.2018.213207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goyal S, Tisdale J, Schmidt M, et al. Acute Myeloid Leukemia Case after Gene Therapy for Sickle Cell Disease. N Engl J Med. Jan 13 2022;386(2):138–147. doi: 10.1056/NEJMoa2109167 [DOI] [PubMed] [Google Scholar]
- 45.Hsieh MM, Bonner M, Pierciey FJ, et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. May 12 2020;4(9):2058–2063. doi: 10.1182/bloodadvances.2019001330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brunson A, Keegan THM, Bang H, Mahajan A, Paulukonis S, Wun T. Increased risk of leukemia among sickle cell disease patients in California. Blood. Sep 28 2017;130(13):1597–1599. doi: 10.1182/blood-2017-05-783233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seminog OO, Ogunlaja OI, Yeates D, Goldacre MJ. Risk of individual malignant neoplasms in patients with sickle cell disease: English national record linkage study. J R Soc Med. Aug 2016;109(8):303–9. doi: 10.1177/0141076816651037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones RJ, DeBaun MR. Leukemia after gene therapy for sickle cell disease: insertional mutagenesis, busulfan, both, or neither. Blood. Sep 16 2021;138(11):942–947. doi: 10.1182/blood.2021011488 [DOI] [PubMed] [Google Scholar]
- 49.Salinas Cisneros G, Thein SL. Recent Advances in the Treatment of Sickle Cell Disease. Front Physiol. 2020;11:435. doi: 10.3389/fphys.2020.00435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fitzhugh CD, Abraham AA, Tisdale JF, Hsieh MM. Hematopoietic stem cell transplantation for patients with sickle cell disease: progress and future directions. Hematol Oncol Clin North Am. Dec 2014;28(6):1171–85. doi: 10.1016/j.hoc.2014.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chaudhury S, Laskowski J, Rangarajan HG, et al. Abatacept for GVHD prophylaxis after hematopoietic stem cell transplantation (HCT) for pediatric sickle cell disease (SCD): a sickle transplant alliance for research (STAR) trial. Biol Blood Marrow Transplant. 2018;24(3):S91. [Google Scholar]