

Abstract
The success of Chimeric Antigen Receptor (CAR) T cell therapy against hematological cancers has convincingly demonstrated the potential of using genetically engineered cells as therapeutic agents. While much progress has been achieved in cell therapy, more beneficial capabilities have yet to be fully explored. One of the unique advantages afforded by cell therapies is the possibility to implement genetic control circuits, which enables diverse signal sensing and logical processing for optimal response in the complex tumor microenvironment. In this perspective, we will first outline design considerations for cell therapy control circuits that address clinical demands. We will compare and contrast key design features in some of the latest control circuits development and conclude by discussing potential future directions.
Highlight and eTOC Blurb for Lee et al CELL SYSTEMS review
Lee et al. describe some of the latest progress in genetic circuit design for immune cell therapy, focusing on two classes of circuits; cell-autonomous and exogenous control.
Graphical Abstract
Introduction
Cells are sophisticated information processing systems that can sense diverse environment signals, perform complex computations, and produce a wide array of outputs such as gene expression, signaling molecule secretion, morphological changes, and cell growth.1 Furthermore, a number of cell types have evolved specialized capabilities to survive in different environments and perform various tasks. These features establish cells as excellent candidates for smart therapeutics with enhanced safety and efficacy. Indeed, several cell types have been evaluated for the development of cell therapies, including bacteria and stem cell therapies. In particular, one of the most important classes of cells for therapeutics development is the human immune cells.2 For instance, T cells genetically engineered with a chimeric antigen receptor (CAR) have demonstrated potent anti-cancer cytotoxicity in the clinics, leading to five FDA-approved therapies for B cell malignancies.3–7
While promising, many challenges need to be addressed before we can realize the full potential of immune cell therapies. One of the most pressing concerns for cellular immunotherapy is toxicity caused by the overactivation and off-tumor targeting of the engineered immune cells. Moreover, the heterogeneity and constant evolution of many diseases demand a dynamic intervention rather than a static, one-time treatment.8,9 Furthermore, the efficacy of the immune cell therapies needs further improvement, as exemplified by the challenges CAR T cells face against solid tumors. Advanced therapeutic cell designs with enhanced precision and control are necessary to address these issues. Most importantly, the challenges in safety and efficacy need to be solved simultaneously to create effective treatments.
Cell-autonomous vs exogenous control: design considerations
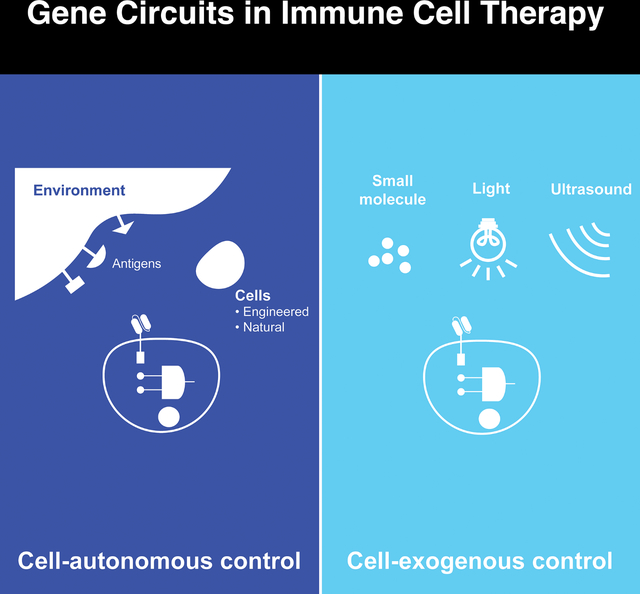
Unlike most therapeutic modalities, cell therapies can be equipped with sophisticated gene circuits to improve their targeting specificity, safety, and efficacy. While there are many different types of gene circuits, they can be broadly classified into two classes; cell-autonomous and exogenous control (Figure 1). Cell-autonomous control gene circuits rely on signals from within the engineered immune cells or the native environment. In contrast, exogenous control gene circuits rely on signals from external reagents such as small molecules, lights, or ultrasound. These circuits are not mutually exclusive and can be deployed together.
Figure 1. Comparison between cell-autonomous and exogenous control.
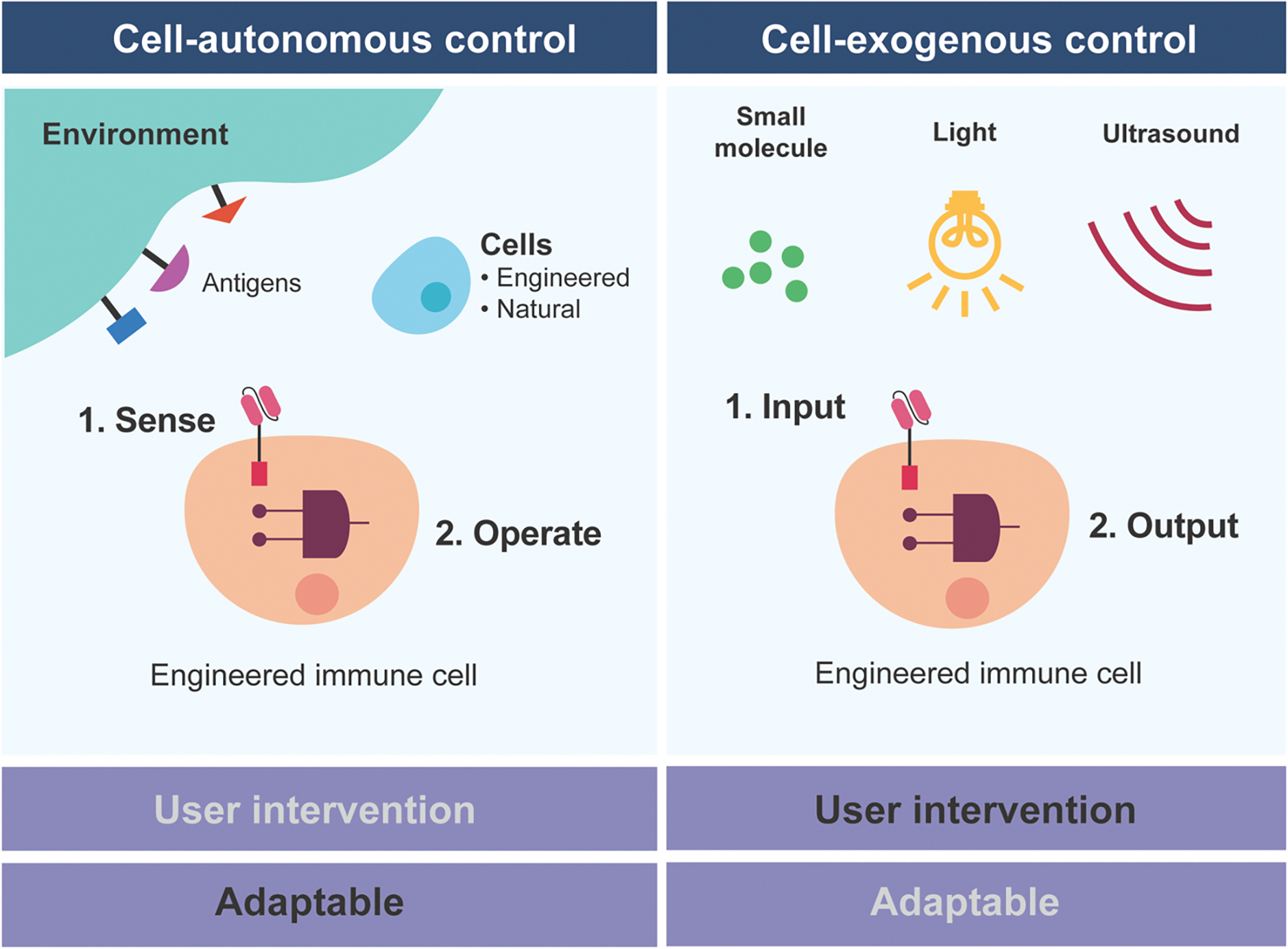
Autonomous control of engineered immune cells can sense and respond to the environment or internal signals, whereas exogenous control enables user intervention of the engineered cells through various types of inputs.
When deciding to employ gene circuits to improve immune cell therapies, it is important to consider the relative strengths and weaknesses of each class of gene circuits. Cell-autonomous circuits are attractive because they can operate without user intervention. This feature may be necessary because some features are not amenable for manual control, such as precisely locating a tumor based on a combination of molecular markers. However, as we have witnessed from autonomous vehicle development, a completely self-operating system may present challenges that require monitoring and adaptation. In clinical settings, unpredictability is far from acceptable. As such, the ability to apply exogenous control to engineer cell therapies will be highly desired.
One of the key considerations for exogenous control circuits is the choice of the input control. The input could be delivered systemically, such as a small molecule, or applied in a highly localized manner, such as light or ultrasound. Small molecules are easy to administer but may have toxicity or poor pharmacokinetic properties. In contrast, light and ultrasound provide non-invasive and precise spatiotemporal control. Still, continuous delivery of light and ultrasound to the patient, which could be required to ensure sustained immune cell function, may not be practical.
A previous review by Lim and June from 2017 has highlighted the pioneering works in this space.10 Here, we would like to bring forth some of the latest developments in genetic circuits for immune cell therapy. We will emphasize discussing the pros and cons of these gene circuits. Finally, we will provide an outlook on how gene circuits can lead to the next generation of smart cell therapies.
Cell-autonomous circuit for therapeutic immune cells
Cell-autonomous gene circuits can sense and respond to input signals within the patient. There are several types of input signals that gene circuits have been designed to sense; the combination of antigens from target and healthy cells, intracellular cell states, and tumor microenvironment. These circuits provide logic and feedback control for more precise temporal and contextual responses of the engineered immune cells.
Receptor logic circuits
Combinatorial antigen recognition is the most logical approach to improve the tumor targeting and reduce the potential toxicity of cancer cell therapies, since often no single antigen exists to uniquely classify cancer cells. Among various receptor logic circuits applied to immune cells,11–17 we will highlight three of the most advanced logic circuits, SUPRA CARs, SynNotch, and Co-LOCKR, that have been applied to perform up to 3-Input AND, NOT and OR logic.18–22
1. CAR circuits
A traditional CAR is comprised of an antigen-binding domain fused to key intracellular signaling domains from the T cell receptor (TCR) (e.g., CD3ζ or CD3ε) and costimulatory receptors (e.g., CD28 or 4–1BB). Signaling from TCR and costimulatory receptor domains are needed for full T cell response. Similarly, for inhibitory CAR (iCAR), intracellular signaling domains from inhibitory receptors have been employed to inhibit the signal from the traditional activating CAR (aCAR).17,23–27
The core design principle of a multi-input CAR logic circuit is to create separate CARs to perform the functions of TCR, costimulatory, and inhibitory receptors separately with different antigen targets (Figure 2A). In essence, a unique CAR is created for each signaling pathway. The signal integration will occur intracellularly through the endogenous signaling network. While conceptually simple, the challenge in implementing the CAR logic circuit is to ensure the signaling strength from each receptor is properly calibrated. For instance, if the aCAR signaling is too strong, the iCAR may not be able to inhibit the signal.
Figure 2. Schematics of chimeric antigen receptors and receptor logic circuits.
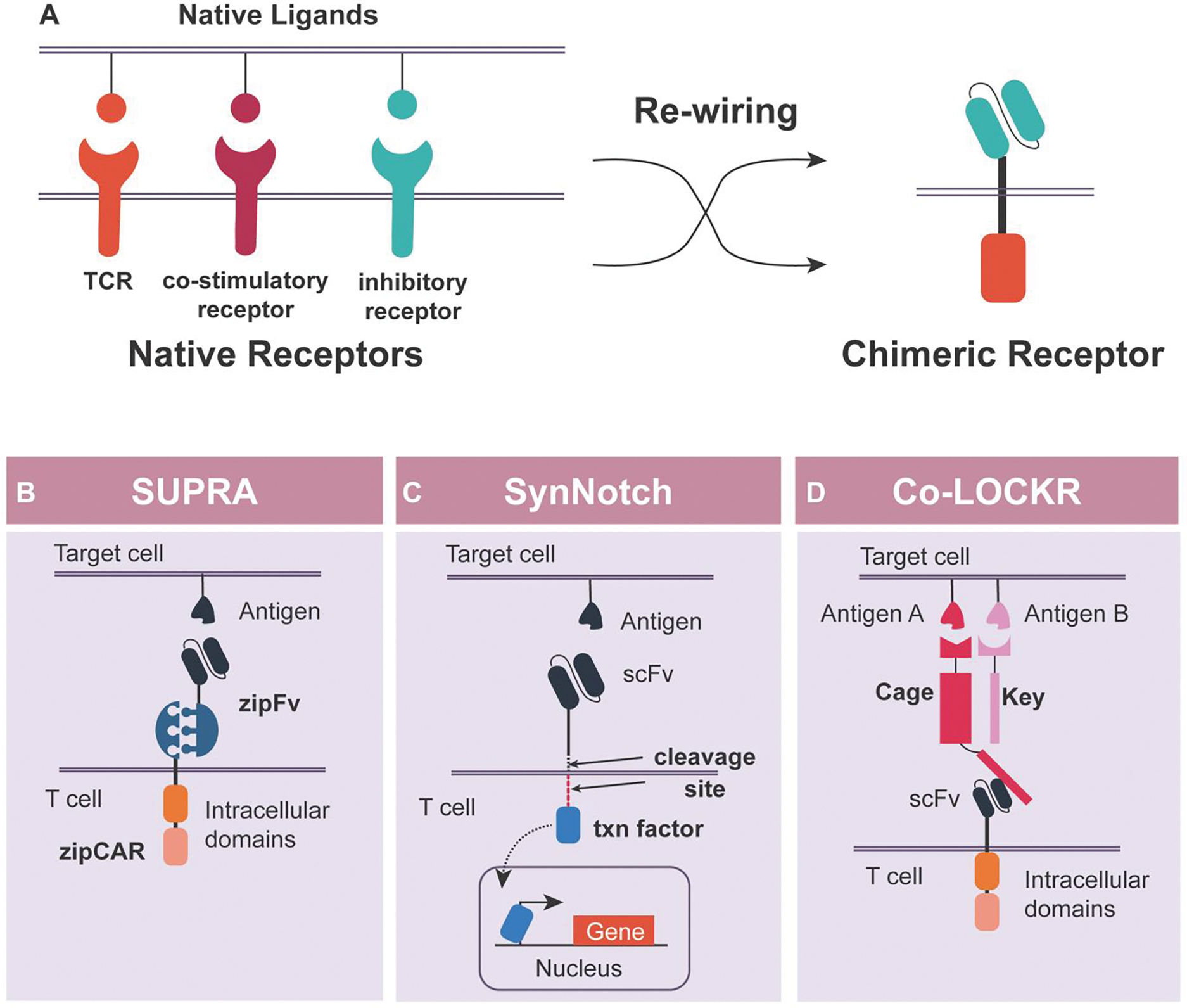
A. Chimeric antigen receptors are derived from native receptors, exchanging their intracellular domains with extracellular domains to rewire their targeting specificity. B. SUPRA CAR consists of zipFv and zipCAR. Swapping the zipFv allows targeting various antigens by the same zipCAR. C. SynNotch receptor can induce gene expression in response to the desired antigen. Once the antigen is bound to the scFv domain, membrane-bound transcription factor (txn factor) will be released to induce the gene expression. D. Co-LOCKR system consists of a CAR and two adaptor proteins; Cage and Key. Only when Cage and Key are bound on the same target cell, the Cage domain can be exposed to activate the CAR.
One of the most direct ways to modulate the CAR signaling strength is to control the number of receptors present on the cell surface. A split universal CAR configuration is the most convenient approach to modulate the number of functional receptors on the cell. A split CAR design is composed of a universal receptor and an adaptor protein that binds both the universal receptor and the target cell. By varying the concentration of the adaptor protein, one can modulate the number of functional CARs and therefore the strength of the signaling.
Many split CAR designs have emerged within the past few years.28–30 However, the most versatile system is the Split, Universal, Programmable CAR (SUPRA CAR) system, which offers the most diverse, orthogonal set of leucine-zipper universal CAR receptors (zipCAR), and leucine-zipper ‘adaptor’ domains that bridge the zipCAR receptors to a variety of antigens specified by an scFv domain (zipFv) (Figure 2B). The SUPRA CAR system showed tunable CAR activation with zipFv titration and antigen-specific activation. Taking advantage of leucine-zipper pairing orthogonality, a variety of logic operations (OR, AND, NOT) with zipFvs of varying affinity against multiple antigens was demonstrated in vitro and in vivo.22
While SUPRA CAR has high modularity, it is a more complicated therapy that consists of protein and cell therapy. Since the adaptor molecule is a protein, zipFv may have less permeability into the desired tissue, a shorter half-life, and potential unknown immune responses. The appropriate indication of the SUPRA CAR will likely be contexts dependent.
2. SynNotch
The synthetic Notch (synNotch) receptor developed by the Lim group represents a distinctive approach to achieve logic in CAR T cells (Morsut et al., 2016; Roybal et al., 2016b). A synNotch receptor is composed of an extracellular antigen-binding domain, followed by a proteolytic transmembrane core from the Notch receptor, and a programmable transcription factor against the target gene promoter. Ligand binding to the SynNotch receptor leads to the cleavage of the core Notch domain and release of the transcription factor and transcription activation (Figure 2C). The synNotch receptor is a programmable surface ligand inducible gene expression system. The Lim lab has previously employed synNotch to reprogram immune cells and design complex tissue patterns.31 Recently, a collection of modular and humanized proteolytic-based receptors similar to the synNotch has been developed by Roybal and colleagues.20 Using mainly human components will minimize immunogenicity and facilitate their clinical translation.
The synNotch-based logic circuit employs an “IF-THEN” logic for which the activation of the synNotch leads to the expression of a CAR or an apoptotic gene to achieve AND or NOT logic respectively.32,33 The synNotch and the CAR can each target different antigens, leading to multi-input logic circuits. The AND logic performance resulting from the synNotch-based circuit seems to enable improved specificity, even against glioblastoma, a solid tumor that is infamous for the high antigen heterogeneity.34
However, a synNotch-based circuit does not require the antigens to be present on the same cell. Once the CAR is expressed, the antigen for the synNotch is not necessary. Therefore, if the off-target healthy cells expressing the antigen for the CAR are near the intended tumor cells, they could also be eradicated.35
3. Co-LOCKR
The colocalization-dependent protein switches (Co-LOCKR) CAR system is also based on a split CAR design. However, the Co-LOCKR system only uses one receptor (Figure 2D). The logic operation is achieved through a set of computationally designed adaptor proteins that can interact with each other and modulate how the adaptor proteins bind to the CAR in the presence of target antigens. The core of the Co-LOCKR system is the “cage” and “key” proteins, each with an antigen-binding domain. The cage protein also contains a peptide that can bind and activate the CAR T cells. The peptide domain of the cage, however, is sequestered by a latch domain. When the key protein binds to the cage protein, it causes a conformational change and exposes the peptide for binding, which allows for activation of the CAR. The cage and key proteins are designed to not interact in solution. Instead, the equilibrium favors cage-key complex formation once they are colocalized to the cell surface by antigen-binding domains. Co-LOCKR switches have been utilized in CAR designs to target up to three different antigens on cancer cells. This split CAR system can also function with AND, OR, and even advanced logic such as A AND B NOT C.21 The Co-LOCKR design does not require the balancing of intracellular signaling domains, but rather requires the presence of the “key” protein to open up the “cage”. However, the employment of decoy “key” protein to generate NOT-logic has a limitation in that the logic will be dependent on the decoy protein abundance.
Cell state-based control
Cell surface antigens are not the only signals capable of redirecting immune cell cytotoxicity. The tumor microenvironment is often immunosuppressive, as it is concentrated with immune inhibitory factors and metabolites to limit cytotoxic immune function.36,37 Immune cells can be engineered to detect some features of the tumor microenvironment, and produce factors to augment antitumor activity, representing a powerful strategy to overcome the tumor microenvironment. Although the interventions based on cell states may enhance the specificity of the treatments, there is a possibility that they could also lead to weaker activity. As the tumor shrinks, the representative cell-states may also be diminished, thus limiting the therapy’s potency or specificity. Therefore, balancing activity and specificity would be crucial for cell-state-based control designs.
1. Oxygen-based CAR control
A hallmark of solid tumors is hypoxia (low oxygen tension), often localized due to irregular vasculature and dense cell mass.38 Therefore, hypoxia can serve as an input signal to further increase tumor targeting specificity for CAR T cell therapy. One strategy to achieve hypoxia-inducible CAR structure is to fuse an oxygen-dependent degradation domain (ODD) to a CAR, rendering the stability of the CAR dependent on hypoxia39 (Figure 3A). This ODD-fused CAR demonstrated hypoxia-induced cancer cell killing in vitro, but substantial basal killing under normal oxygen levels was also observed. An alternative approach that builds upon the ODD-fused CAR concept uses a synthetic hypoxia-inducible promoter to control the ODD-CAR transcription, thus providing two levels of control in CAR activity. The HypoxiCAR T cell40 can infiltrate tumors, leading to partial tumor clearance without cytokine release syndrome (CRS), a known problem associated with some CARs such as anti-Her2.41 Further characterization of the HypoxiCAR performance under hypoxic conditions in normal cells for a prolonged period is needed to establish the safety control of this hypoxia regulatable CAR T cell therapy.
Figure 3. System designs for cell state-based control.
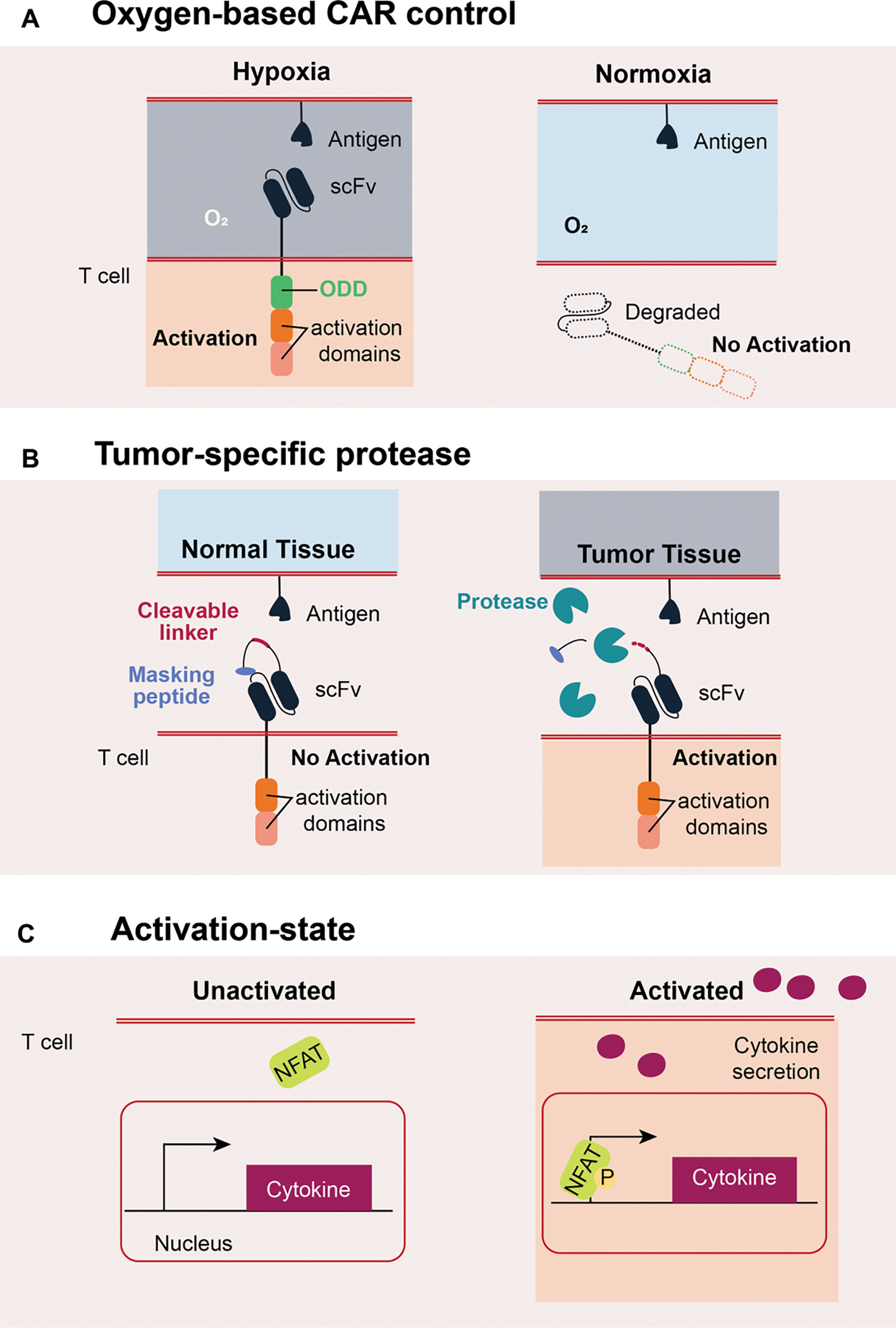
A. CAR with an oxygen-dependent degradation domain (ODD), for which its stability is dependent on hypoxia. The ODD will be degraded under normal oxygen concentration, resulting in the degradation of the CAR. B. A CAR design that can be activated only under the presence of tumor-specific protease. The scFv is masked by a cleavable linker and a masking peptide, and the protease can cleave off the linker to expose the scFv so that the CAR can be activated against the target antigen. C. Activation status of the immune cell can be utilized to control further cytokine generation. Once the T cell is activated, the NFAT can be phosphorylated and translocated into the nuclease to induce the target cytokine transcription.
2. Tumor-specific protease
Tumors often secrete proteases to promote invasion and facilitate various stages of tumor development. As such, tumor-specific proteases can be a marker for cancer diagnostics and therapeutics development. Recently, Han et al. 42 developed a masked anti-EGFR CAR T by adding a masking peptide with a proteolytic site before the scFv domain (Figure 3B). The masking peptide blocks the antigen-binding site by default, thus preventing CAR activation. However, in the presence of a tumor-specific protease, the masking peptide is cleaved, thus exposing the scFv and allowing antigen binding and activation of the CAR T cells. The masked CAR T cells had reduced activity in the absence of proteases despite surrounding target antigens in vitro. Masked CAR T cells demonstrated activity similar to unmasked CAR T cells in a subcutaneous human lung cancer xenograft model, indicating the cleavage of the masking peptide. An analysis of the off-tumor activity of the masked CAR T in relevant animal studies will further support the safety in the clinic.
3. Activation cell state
Immune modulatory factors, such as cytokines, are essential in maintaining immune homeostasis and combating tumors and infection. As such, the application of cytokines like IL-2 and IL-12 as anti-cancer therapies are under investigation. Furthermore, cytokine administration has been explored as a combination therapy to enhance CAR functionality.43–45 However, systemic cytokine administration can cause serious side effects.46–51 Therefore, it would be desirable for the CAR T cells to produce the cytokines only in the tumor microenvironment to minimize systemic toxicity. One approach to ensure localized cytokine production is to make it conditional on CAR activation. The Nuclear Factor of Activated T-cells (NFAT)/IL-2 composite promoter, which has long been used as a reporter of T cell activation,52–54 was employed to control cytokine production in CAR T cells (Figure 3C). IL-12, IL-18, and IL-21 have been explored so far,55–59 establishing the CAR T cells as a cytokine factory.
Exogenous gene control circuits for therapeutic immune cells
One of the most important goals of exogenous gene control circuits is to enhance the safety of the engineered immune cells by limiting T cell activity in the event of adverse side effects or to improve tumor-targeting specificity. Therefore, the pharmacokinetics and safety profile of the inducer are two of the essential parameters in designing the inducible system. From a clinical perspective, implementing a safe, clinically approved inducer has extensive benefits, facilitating the introduction of novel CARs with enhanced safety profiles in the market. In addition to safety, an added benefit of using an inducible switch is increased durability. Weber et al. showed that transiently stopping tonic receptor signaling through a drug-gated CAR can rescue T cells from exhaustion, thus improving their in vivo persistence and ultimately antitumor activity.60
Currently, there are three classes of exogenous gene control circuits, categorized by the type of inducers; small molecules, light, and ultrasound. When implemented in immune cells, each system acts as an ON or OFF switch, with the exogenous inducers modulating this change in response. Most of these systems, with the exception of kill switch or recombinase-based systems, do not have memory. As such, the inducer needs to be present continuously to maintain the ON or OFF state. Therefore, the toxicity and delivery method of the inducer is important. Furthermore, the decision to create an ON or OFF switch is dictated by the property of the components used in the system. Whether an ON or OFF switch is more desirable clinically, however, remains unresolved. We posit that an ON switch, which requires constant induction, is best suited when the output that it controls may become toxic at a high level (e.g., a pleiotropic cytokine or an overactive CAR), thus requiring fine-tuning and careful regulation. In contrast, an OFF switch, which stays ON without any inducer, is best used with an output that is relatively safe (e.g., a well-behaved CAR) and only needs to be turned OFF in case of severe side effects. However, when the output is no longer needed, the ON switch has the advantage that it can be shut off simply by withdrawing the inducer.
1. Small molecules
The simplest way to generate drug-gated CAR T cells is to control the activity of the CAR directly. Common mechanisms used to achieve drug-gated control rely on inducible assembly or stabilization of the receptor. The assembly mechanism typically involves splitting the CAR into antigen recognition and signaling domains. A small molecule is used to either assist (ON switch) or disrupt (OFF switch) the assembly of the components.61–64 The stabilization mechanism involves fusing a small molecule controllable degradation domain (degron) to the CAR. These degrons can unfold or cleave the CAR, and the binding of the inducer can stabilize the degron or inhibit the proteolysis (ON switch; Figure 4A). Some degrons will recruit endogenous proteolysis machinery in the presence of the small molecule inducer (OFF switch; Figure 4B). Recently, inducible CAR systems based on Non-structure 3 (NS3) protease from the hepatitis C virus (HCV) have been developed.61,64,65 The advantage of the NS3 system is that it can be regulated by clinically approved protease inhibitors, which have a favorable safety profile. Usually, a given inducible system can only leverage either the assembly or the stabilization mechanism. However, some systems, such as the Versatile Protease Regulatable CAR (VIPER CAR) or the lenalidomide system,61,62,64 can leverage both mechanisms to create ON and OFF switches using the same inducer. Furthermore, Li et al have shown that the NS3-based system can be combined with other CAR designs (e.g., SUPRA or the lenalidomide system) to create multiplexed control circuits that could improve the safety and specificity of the CAR T cell therapy.
Figure 4. Exogeneous cell control with ON- and OFF- switches.
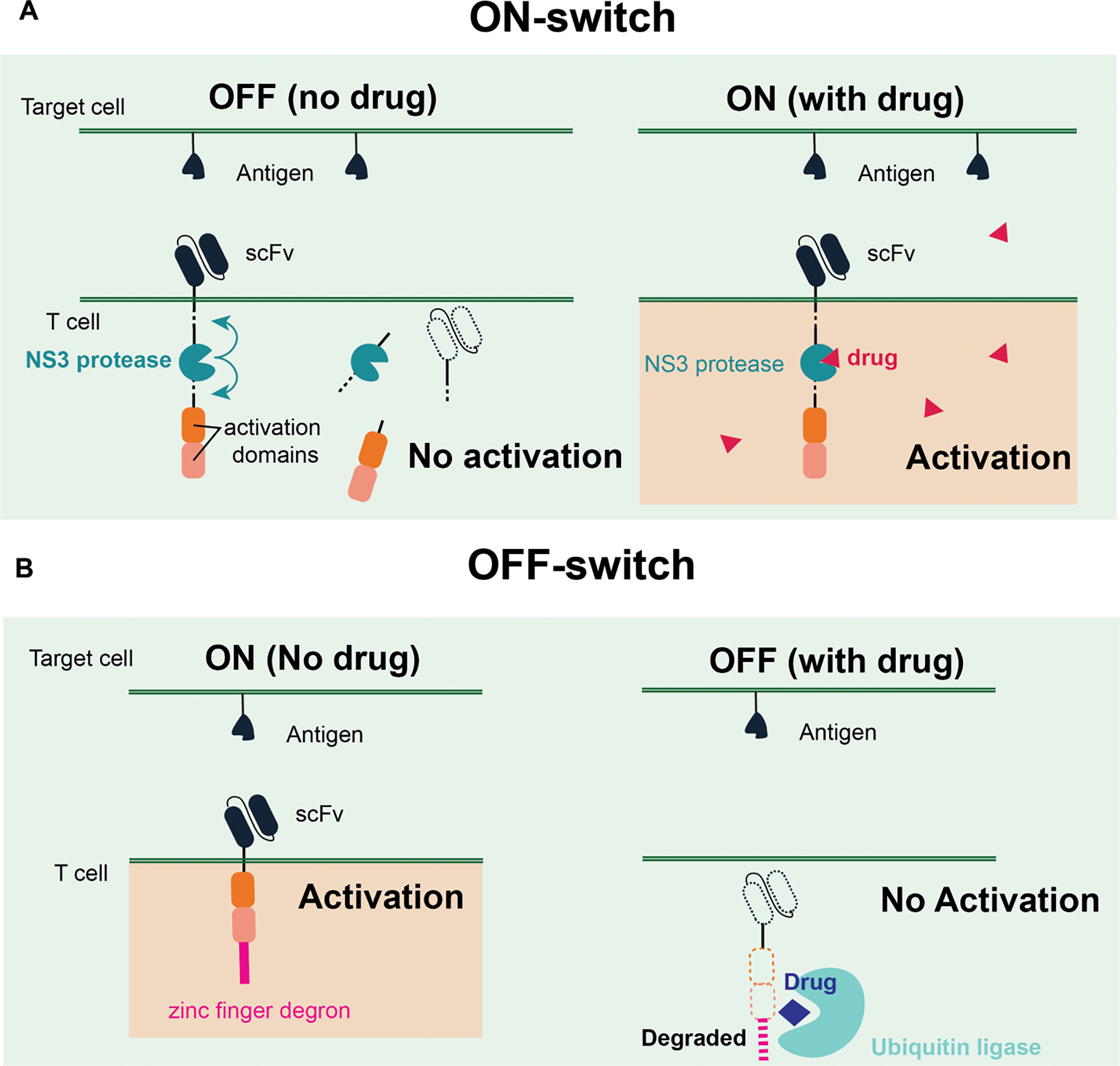
A. Representative ON-switch incorporating an NS3 protease to control the CAR activity. CAR will be stabilized only under the presence of a drug that can inhibit the protease activity to induce the T cell activation. B. Representative OFF-switch using the zinc finger degron motif and a synthetic ubiquitin ligase. A drug that can induce the dimerization between the degron and the ligase will degrade the CAR, so the T cells cannot be activated under the presence of the drug.
An alternative and more flexible approach to creating regulatable immune cell therapies is to deploy drugs for tuning CAR or therapeutic gene expression. The most prominent of such systems is the Tet-ON transcription system.66–68 In this system, CAR is transcribed only in the presence of doxycycline, though some leaky expression has been observed. Moreover, high levels of TetR proteins can be toxic due to off-target binding in the genome.69–71 Programmable synthetic transcription factors, such as those based on zinc-finger or CRISPR, could provide a safer option to mitigate off-target effects. In particular, the synthetic Zinc Finger Transcription Regulators (synZiFTR) has been specifically designed to be orthogonal to the human genome. Multiple inducible synZiFTR systems have been developed using clinically approved drugs as the inducer, leading to the first dual inducible gene expression control system in human primary T cells to regulate CAR and cytokine expression.65 In addition to clinically approved drugs, natural product, such as resveratrol found in red wine, grapes, and berries, has also been used to repress or induce CAR expression, demonstrating its applicability in primary T cells both in vitro and in vivo with high dynamic range.72
Inducible gene switches with memory features will allow for long-term changes in gene expression with transient drug exposure. This feature will minimize the need to continuously administer the drug inducer, which can be beneficial when persistent drug administration is impossible or may result in some toxicity. Using a recombinase-based gene circuit with the FlpO-ERT2 fusion protein, drug-inducible CAR expression with memory was developed to induce CAR expression.73 Depending on the initial design of the target gene, the circuit can be used to either turn ON or OFF CAR expression.
2. Light
Light-inducible dimerization domains have been utilized to make photoactivable CARs in immune cells (Figure 5A).74 Localized CAR expression system in T cells through a blue light-inducible system was demonstrated previously.75,76 A similar optogenetic approach was used to induce cytokine expression in T cells for eliminating cancer cells.77 Using a non-invasive light-inducible system, precise spatiotemporal control is possible with minimal side effects, which is difficult to achieve with a small molecule-inducible gene expression system. However, blue light has minimal tissue penetration depth (less than 1 μm), thus limiting its clinical applications. To address this limitation, a nanoplate technology has been developed that can upconvert near-infrared light (NIR), a more transmittable light in tissue, into blue light. By injecting the nanoplate with blue light-inducible CAR T cells into tumor-bearing mice, reversible and real-time control of the CAR activation was achieved to mitigate the potential cytokine storm.78
Figure 5. Exogenous cell control with light and ultrasound inputs.
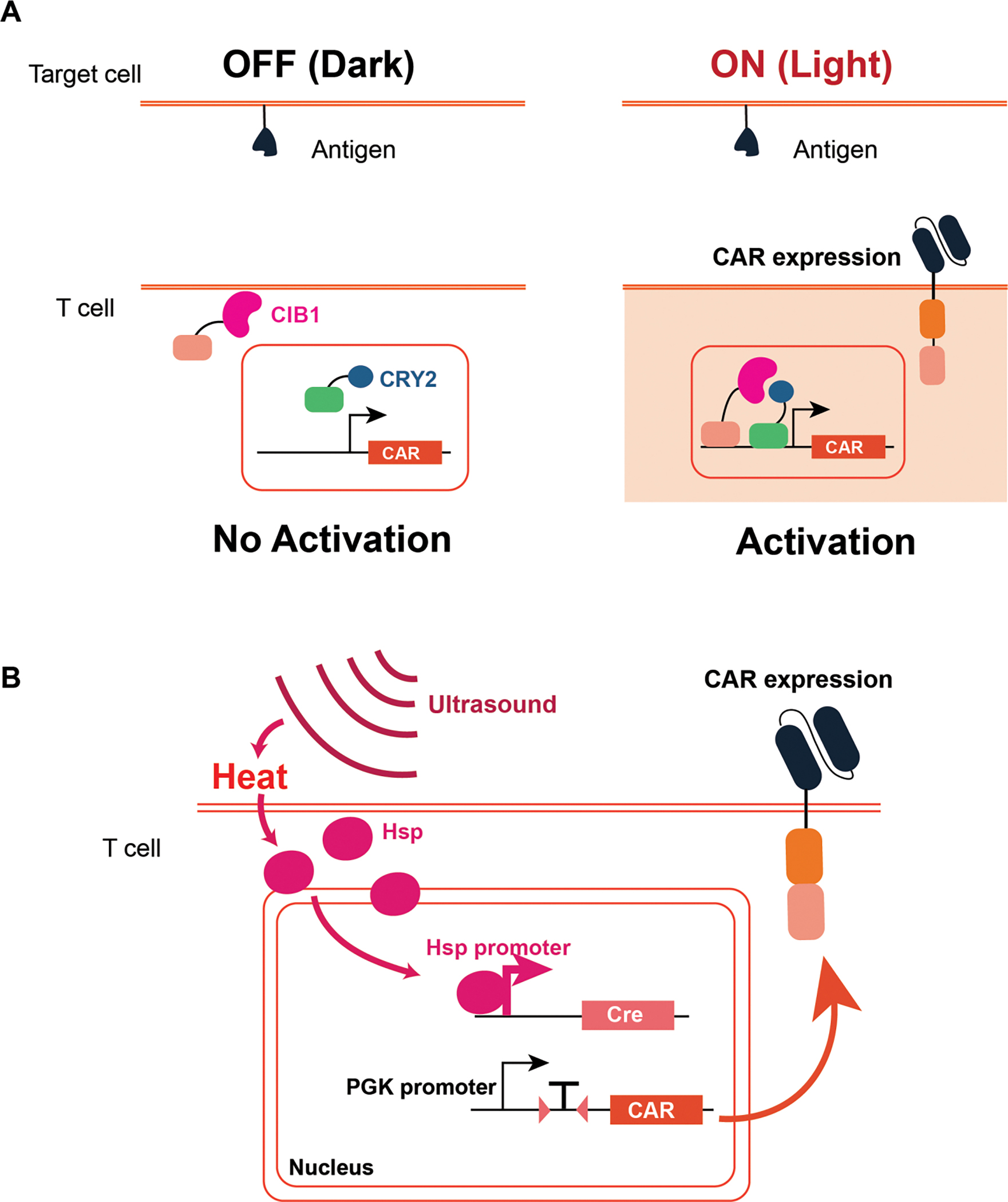
A. Photoactivable CAR activation system is designed with two light-inducible dimerization fused to a transcription factor. Upon light induction, the CAR will be expressed and activate the T cells. B. Ultrasound-inducible CAR system utilizes the heat-shock protein (Hsp) mediated gene expression. Hsp translocation will induce Cre expression, and the Cre will mediate the CAR expression on the T cell.
3. Ultrasound
Given the challenge of using light as the inducer, ultrasound presents an attractive alternative as a physical inducer thanks to its safety and greater penetration depth. Pan et al. utilized a mechanically sensitive Piezo1 calcium channel that can be activated by ultrasound.79 The exposure to ultrasound creates microbubbles, which activate the Piezo1 channel enabling calcium intake into the cell. The influx of calcium activates calcineurin, which leads to downstream dephosphorylation of an NFAT transcription factor. The NFAT responsive promoter was used to induce CAR transcription after ultrasound exposure. However, the requirement for microbubbles hinders their application in vivo. To circumvent this challenge, the same group of researchers developed a heat-induced CAR that responds to ultrasound.80 Focused ultrasound waves increase local temperature, and heat-shock-protein promoter encoding Cre recombinase can initiate and maintain the CAR expression (Figure 5B).
Miller et al. also implemented a heat-responsive element to control T cell activity.81 Instead of directly inducing CAR activity, they used a plasmonic gold nanorod to convert NIR into heat. This system demonstrated successful trafficking of T cells to the antigen-expressing tumor when T cells are expressing CAR. Moreover, they used a plasmonic gold nanorod to convert NIR into heat. They demonstrated that the generated heat can induce both IL-15 superagonist expression to enhance CAR activity in vivo, and bispecific T cell engager (BiTE) expression to mitigate tumor outgrowth due to the antigen escape.
Discussion
One of the most intriguing features of using cells as therapies is the ability of cells to sense the environment and perform many tasks. As such, developing a strategy for engineering multiple features and functions into cell therapies while also addressing the concerns of safety, specificity and efficacy would be highly desirable. As highlighted above, many different powerful systems have been developed to approach these challenges. We introduced two arms of immune cell switches: cell-autonomous and cell-exogenous control. The two arms are not mutually exclusive and they can be used cooperatively. Our ability to rewire receptor machinery and design more ways of controlling the engineered immune cells will further enhance CAR T cell therapies as a whole.
While many of the ideas discussed here are still in their early stage, some of them are closer to the clinics than others. Currently, logic CARs seem to have the most momentum. For instance, 2-input OR gate CARs have already been evaluated in the clinics and shown promising results.82 Several companies are also actively pursuing NIMPLY (A AND NOT B) gate CARs for various cancers.27,83 Many CAR T cell therapies have been designed to also produce factors, such as checkpoint inhibitors,84 immunomodulatory factors,43,85 or prodrug modifying enzymes,86 to augment the anti-tumor activity. Such designs, while necessary, also heighten the risk of severe adverse side effects. Therefore, regulatable control of CAR activity and transgene expression will be needed to balance activity and safety. Some of the drug-inducible CARs and gene switches described here, especially those that use clinically approved drugs, can provide the safety control needed and therefore are likely to move into clinics in the near future.
A major challenge that hinders the field from realizing its full potential is the ability to perform large-scale genetic engineering on human immune cells, especially cells derived from primary sources. Even with the advancement of pioneering genome editing technologies with CRISPR systems,87 the delivery and integration of large DNA payloads represent a major bottleneck that is not readily solvable. An approach to circumvent this complication is to engineer a consortium of immune cells that separately carry the cell-autonomous and cell-exogenous systems, akin to our immune system. These smaller genetic programs could potentially be delivered to T cells in situ,88 thus bypassing the need for the complicated ex vivo manufacturing process and lowering the cost of the therapy. We envision an ideal scenario where complex genetic circuits with enhanced specificity, efficacy, and safety features are delivered in situ into multiple immune cell types, upgrading the patients’ immune systems to combat and protect against a myriad of diseases.
Acknowledgment
We thank Joshua McGee, Arun Nambiar, and Zhuoying Huang for suggestions on the manuscript. WWW acknowledges funding from Senti Biosciences, the NSF Expedition in Computing (1522074), NSF Career (162457), NSF BBSRC (1614642), NIH (R01GM129011, R01EB029483, R01EB031904, U01CA265713, R01DK132576), and the Allen Distinguished Investigator Award, a Paul G. Allen Frontiers Group advised grant of the Paul G. Allen Family Foundation. ASK acknowledges funding from Senti Biosciences, the NSF Expedition in Computing (1522074), the NIH (R01EB029483), NIH Director’s New Innovator Award (1DP2AI131083), DARPA Young Faculty Award (D16AP00142), and DoD Vannevar Bush Faculty Fellowship (N00014-20-1-2825). Competing Interests: WWW is a co-founder and shareholder of Senti Biosciences. ASK is a shareholder of Senti Bioscience and Chroma Medicine. SL is a current employee of Sanofi.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lim WA (2010). Designing customized cell signalling circuits. Nat Rev Mol Cell Biol 11, 393–403. 10.1038/nrm2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey SR, and Maus MV (2019). Gene editing for immune cell therapies. Nat Biotechnol 37, 1425–1434. 10.1038/s41587-019-0137-8. [DOI] [PubMed] [Google Scholar]
- 3.FDA, U.S. (2021). BREYANZI (lisocabtagene maraleucel).
- 4.FDA, U.S. (2021). ABECMA (idecabtagene vicleucel).
- 5.FDA, U.S. (2017). KYMRIAH (tisagenlecleucel).
- 6.FDA, U.S. (2020). TECARTUS (brexucabtagene autoleucel).
- 7.FDA, U.S. (2017). YESCARTA (axicabtagene ciloleucel).
- 8.Marusyk A, Almendro V, and Polyak K (2012). Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer 12, 323–334. 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 9.Meacham CE, and Morrison SJ (2013). Tumour heterogeneity and cancer cell plasticity. Nature 501, 328–337. 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim WA, and June CH (2017). The Principles of Engineering Immune Cells to Treat Cancer. Cell 168, 724–740. 10.1016/j.cell.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, Klichinsky M, Aikawa V, Nazimuddin F, Kozlowski M, et al. (2016). Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest 126, 3814–3826. 10.1172/JCI87366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, Corder A, Schonfeld K, Koch J, Dotti G, et al. (2013). TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids 2, e105. 10.1038/mtna.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, Wakefield A, Fousek K, Bielamowicz K, Chow KK, et al. (2016). Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest 126, 3036–3052. 10.1172/JCI83416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zah E, Lin MY, Silva-Benedict A, Jensen MC, and Chen YY (2016). T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol Res 4, 498–508. 10.1158/2326-6066.CIR-15-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, and Powell DJ Jr. (2013). Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res 1, 43–53. 10.1158/2326-6066.CIR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kloss CC, Condomines M, Cartellieri M, Bachmann M, and Sadelain M (2013). Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 31, 71–75. 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fedorov VD, Themeli M, and Sadelain M (2013). PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med 5, 215ra172. 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, and Lim WA (2016). Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164, 770–779. 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho JH, Collins JJ, and Wong WW (2018). Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 173, 1426–1438 e1411. 10.1016/j.cell.2018.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu I, Liu R, Garcia JM, Hyrenius-Wittsten A, Piraner DI, Alavi J, Israni DV, Liu B, Khalil AS, and Roybal KT (2022). Modular design of synthetic receptors for programmed gene regulation in cell therapies. Cell 185, 1431–1443 e1416. 10.1016/j.cell.2022.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lajoie MJ, Boyken SE, Salter AI, Bruffey J, Rajan A, Langan RA, Olshefsky A, Muhunthan V, Bick MJ, Gewe M, et al. (2020). Designed protein logic to target cells with precise combinations of surface antigens. Science 369, 1637–1643. 10.1126/science.aba6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho JH, Okuma A, Sofjan K, Lee S, Collins JJ, and Wong WW (2021). Engineering advanced logic and distributed computing in human CAR immune cells. Nat Commun 12, 792. 10.1038/s41467-021-21078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards RM, Zhao F, Freitas KA, Parker KR, Xu P, Fan A, Sotillo E, Daugaard M, Oo HZ, Liu J, et al. (2021). NOT-Gated CD93 CAR T Cells Effectively Target AML with Minimized Endothelial Cross-Reactivity. Blood Cancer Discov 2, 648–665. 10.1158/2643-3230.BCD-20-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tao L, Farooq MA, Gao Y, Zhang L, Niu C, Ajmal I, Zhou Y, He C, Zhao G, Yao J, et al. (2020). CD19-CAR-T Cells Bearing a KIR/PD-1-Based Inhibitory CAR Eradicate CD19(+)HLA-C1(−) Malignant B Cells While Sparing CD19(+)HLA-C1(+) Healthy B Cells. Cancers (Basel) 12. 10.3390/cancers12092612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamburger AE, DiAndreth B, Cui J, Daris ME, Munguia ML, Deshmukh K, Mock JY, Asuelime GE, Lim ED, Kreke MR, et al. (2020). Engineered T cells directed at tumors with defined allelic loss. Mol Immunol 128, 298–310. 10.1016/j.molimm.2020.09.012. [DOI] [PubMed] [Google Scholar]
- 26.Hwang MS, Mog BJ, Douglass J, Pearlman AH, Hsiue EH, Paul S, DiNapoli SR, Konig MF, Pardoll DM, Gabelli SB, et al. (2021). Targeting loss of heterozygosity for cancer-specific immunotherapy. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2022410118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandberg ML, Wang X, Martin AD, Nampe DP, Gabrelow GB, Li CZ, McElvain ME, Lee WH, Shafaattalab S, Martire S, et al. (2022). A carcinoembryonic antigen-specific cell therapy selectively targets tumor cells with HLA loss of heterozygosity in vitro and in vivo. Sci Transl Med 14, eabm0306. 10.1126/scitranslmed.abm0306. [DOI] [PubMed] [Google Scholar]
- 28.Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, and Powell DJ Jr. (2012). A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res 72, 1844–1852. 10.1158/0008-5472.CAN-11-3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lohmueller JJ, Ham JD, Kvorjak M, and Finn OJ (2017). mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology 7, e1368604. 10.1080/2162402X.2017.1368604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB, et al. (2016). Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci U S A 113, E450–458. 10.1073/pnas.1524193113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toda S, Blauch LR, Tang SKY, Morsut L, and Lim WA (2018). Programming self-organizing multicellular structures with synthetic cell-cell signaling. Science 361, 156–162. 10.1126/science.aat0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams JZ, Allen GM, Shah D, Sterin IS, Kim KH, Garcia VP, Shavey GE, Yu W, Puig-Saus C, Tsoi J, et al. (2020). Precise T cell recognition programs designed by transcriptionally linking multiple receptors. Science 370, 1099–1104. 10.1126/science.abc6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, Walker WJ, McNally KA, and Lim WA (2016). Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 167, 419–432 e416. 10.1016/j.cell.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, Downey KM, Yu W, Carrera DA, Celli A, et al. (2021). SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med 13. 10.1126/scitranslmed.abe7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, Smythe KS, Dudakov JA, Pierce RH, Rader C, and Riddell SR (2019). Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 35, 489–503 e488. 10.1016/j.ccell.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang T, Huang X, Zhang G, Hong Z, Bai X, and Liang T (2021). Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct Target Ther 6, 72. 10.1038/s41392-020-00449-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zou W (2005). Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 5, 263–274. 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 38.Chang WH, and Lai AG (2020). The hypoxic tumour microenvironment: A safe haven for immunosuppressive cells and a therapeutic barrier to overcome. Cancer Lett 487, 34–44. 10.1016/j.canlet.2020.05.011. [DOI] [PubMed] [Google Scholar]
- 39.Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, Duchateau P, and Poirot L (2017). An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep 7, 39833. 10.1038/srep39833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kosti P, Opzoomer JW, Larios-Martinez KI, Henley-Smith R, Scudamore CL, Okesola M, Taher MYM, Davies DM, Muliaditan T, Larcombe-Young D, et al. (2021). Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep Med 2, 100227. 10.1016/j.xcrm.2021.100227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, and Rosenberg SA (2010). Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 18, 843–851. 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han X, Bryson PD, Zhao Y, Cinay GE, Li S, Guo Y, Siriwon N, and Wang P (2017). Masked Chimeric Antigen Receptor for Tumor-Specific Activation. Mol Ther 25, 274–284. 10.1016/j.ymthe.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bell M, and Gottschalk S (2021). Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front Immunol 12, 684642. 10.3389/fimmu.2021.684642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, and Dotti G (2010). Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 24, 1160–1170. 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, Di S, Shi B, Zhang H, Wang Y, Wu X, Luo H, Wang H, Li Z, and Jiang H (2019). Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3-Targeted Chimeric Antigen Receptor-Engineered T Cells in Hepatocellular Carcinoma. J Immunol 203, 198–207. 10.4049/jimmunol.1800033. [DOI] [PubMed] [Google Scholar]
- 46.Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Perez-Gracia JL, Rodriguez-Ruiz ME, Ponz-Sarvise M, Castanon E, and Melero I (2019). Cytokines in clinical cancer immunotherapy. Br J Cancer 120, 6–15. 10.1038/s41416-018-0328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahmadzadeh M, and Rosenberg SA (2006). IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood 107, 2409–2414. 10.1182/blood-2005-06-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Refaeli Y, Van Parijs L, London CA, Tschopp J, and Abbas AK (1998). Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity 8, 615–623. 10.1016/s1074-7613(00)80566-x. [DOI] [PubMed] [Google Scholar]
- 49.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, and Restifo NP (2005). Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest 115, 1616–1626. 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krenciute G, Prinzing BL, Yi Z, Wu MF, Liu H, Dotti G, Balyasnikova IV, and Gottschalk S (2017). Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Ralpha2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol Res 5, 571–581. 10.1158/2326-6066.CIR-16-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang ZZ, Grote DM, Ziesmer SC, Niki T, Hirashima M, Novak AJ, Witzig TE, and Ansell SM (2012). IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J Clin Invest 122, 1271–1282. 10.1172/JCI59806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shapiro VS, Mollenauer MN, and Weiss A (1998). Nuclear factor of activated T cells and AP-1 are insufficient for IL-2 promoter activation: requirement for CD28 up-regulation of RE/AP. J Immunol 161, 6455–6458. [PubMed] [Google Scholar]
- 53.Jain J, Loh C, and Rao A (1995). Transcriptional regulation of the IL-2 gene. Curr Opin Immunol 7, 333–342. 10.1016/0952-7915(95)80107-3. [DOI] [PubMed] [Google Scholar]
- 54.Riegel JS, Corthesy B, Flanagan WM, and Crabtree GR (1992). Regulation of the interleukin-2 gene. Chem Immunol 51, 266–298. [PubMed] [Google Scholar]
- 55.Koneru M, Purdon TJ, Spriggs D, Koneru S, and Brentjens RJ (2015). IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 4, e994446. 10.4161/2162402X.2014.994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chmielewski M, and Abken H (2017). CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep 21, 3205–3219. 10.1016/j.celrep.2017.11.063. [DOI] [PubMed] [Google Scholar]
- 57.Zimmermann K, Kuehle J, Dragon AC, Galla M, Kloth C, Rudek LS, Sandalcioglu IE, Neyazi B, Moritz T, Meyer J, et al. (2020). Design and Characterization of an “All-in-One” Lentiviral Vector System Combining Constitutive Anti-GD2 CAR Expression and Inducible Cytokines. Cancers (Basel) 12. 10.3390/cancers12020375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo T, Ma D, and Lu TK (2022). Sense-and-Respond Payload Delivery Using a Novel Antigen-Inducible Promoter Improves Suboptimal CAR-T Activation. ACS Synth Biol 11, 1440–1453. 10.1021/acssynbio.1c00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stach M, Ptackova P, Mucha M, Musil J, Klener P, and Otahal P (2020). Inducible secretion of IL-21 augments anti-tumor activity of piggyBac-manufactured chimeric antigen receptor T cells. Cytotherapy 22, 744–754. 10.1016/j.jcyt.2020.08.005. [DOI] [PubMed] [Google Scholar]
- 60.Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, Good Z, Belk JA, Daniel B, Klysz D, et al. (2021). Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 372. 10.1126/science.aba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li HS, Wong NM, Tague E, Ngo JT, Khalil AS, and Wong WW (2022). High-performance multiplex drug-gated CAR circuits. Cancer Cell. 10.1016/j.ccell.2022.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jan M, Scarfo I, Larson RC, Walker A, Schmidts A, Guirguis AA, Gasser JA, Slabicki M, Bouffard AA, Castano AP, et al. (2021). Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci Transl Med 13. 10.1126/scitranslmed.abb6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu CY, Roybal KT, Puchner EM, Onuffer J, and Lim WA (2015). Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 350, aab4077. 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Labanieh L, Majzner RG, Klysz D, Sotillo E, Fisher CJ, Vilches-Moure JG, Pacheco KZB, Malipatlolla M, Xu P, Hui JH, et al. (2022). Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell 185, 1745–1763 e1722. 10.1016/j.cell.2022.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Israni DV, Li H, Gagnon KA, Sander JD, Roybal KT, Joung K, Wong WW, and Khalil AS (2021). Clinically-driven design of synthetic gene regulatory programs in human cells. BioRxiv. [Google Scholar]
- 66.Drent E, Poels R, Mulders MJ, van de Donk N, Themeli M, Lokhorst HM, and Mutis T (2018). Feasibility of controlling CD38-CAR T cell activity with a Tet-on inducible CAR design. PLoS One 13, e0197349. 10.1371/journal.pone.0197349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gu X, He D, Li C, Wang H, and Yang G (2018). Development of Inducible CD19-CAR T Cells with a Tet-On System for Controlled Activity and Enhanced Clinical Safety. Int J Mol Sci 19. 10.3390/ijms19113455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sakemura R, Terakura S, Watanabe K, Julamanee J, Takagi E, Miyao K, Koyama D, Goto T, Hanajiri R, Nishida T, et al. (2016). A Tet-On Inducible System for Controlling CD19-Chimeric Antigen Receptor Expression upon Drug Administration. Cancer Immunol Res 4, 658–668. 10.1158/2326-6066.CIR-16-0043. [DOI] [PubMed] [Google Scholar]
- 69.Kramer JM, and Staveley BE (2003). GAL4 causes developmental defects and apoptosis when expressed in the developing eye of Drosophila melanogaster. Genet Mol Res 2, 43–47. [PubMed] [Google Scholar]
- 70.Pfeiffer BD, Ngo TT, Hibbard KL, Murphy C, Jenett A, Truman JW, and Rubin GM (2010). Refinement of tools for targeted gene expression in Drosophila. Genetics 186, 735–755. 10.1534/genetics.110.119917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rezaval C, Werbajh S, and Ceriani MF (2007). Neuronal death in Drosophila triggered by GAL4 accumulation. Eur J Neurosci 25, 683–694. 10.1111/j.1460-9568.2007.05317.x. [DOI] [PubMed] [Google Scholar]
- 72.Yang L, Yin J, Wu J, Qiao L, Zhao EM, Cai F, and Ye H (2021). Engineering genetic devices for in vivo control of therapeutic T cell activity triggered by the dietary molecule resveratrol. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2106612118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chakravarti D, Caraballo LD, Weinberg BH, and Wong WW (2019). Inducible Gene Switches with Memory in Human T Cells for Cellular Immunotherapy. ACS Synth Biol 8, 1744–1754. 10.1021/acssynbio.8b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tan P, He L, Han G, and Zhou Y (2017). Optogenetic Immunomodulation: Shedding Light on Antitumor Immunity. Trends Biotechnol 35, 215–226. 10.1016/j.tibtech.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Allen ME, Zhou W, Thangaraj J, Kyriakakis P, Wu Y, Huang Z, Ho P, Pan Y, Limsakul P, Xu X, and Wang Y (2019). An AND-Gated Drug and Photoactivatable Cre-loxP System for Spatiotemporal Control in Cell-Based Therapeutics. ACS Synth Biol 8, 2359–2371. 10.1021/acssynbio.9b00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang Z, Wu Y, Allen ME, Pan Y, Kyriakakis P, Lu S, Chang YJ, Wang X, Chien S, and Wang Y (2020). Engineering light-controllable CAR T cells for cancer immunotherapy. Sci Adv 6, eaay9209. 10.1126/sciadv.aay9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao B, Wang Y, Tan X, Zheng X, Wang F, Ke K, Zhang C, Liao N, Dang Y, Shi Y, et al. (2019). An Optogenetic Controllable T Cell System for Hepatocellular Carcinoma Immunotherapy. Theranostics 9, 1837–1850. 10.7150/thno.27051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nguyen NT, Huang K, Zeng H, Jing J, Wang R, Fang S, Chen J, Liu X, Huang Z, You MJ, et al. (2021). Nano-optogenetic engineering of CAR T cells for precision immunotherapy with enhanced safety. Nat Nanotechnol 16, 1424–1434. 10.1038/s41565-021-00982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pan Y, Yoon S, Sun J, Huang Z, Lee C, Allen M, Wu Y, Chang YJ, Sadelain M, Shung KK, et al. (2018). Mechanogenetics for the remote and noninvasive control of cancer immunotherapy. Proc Natl Acad Sci U S A 115, 992–997. 10.1073/pnas.1714900115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu Y, Liu Y, Huang Z, Wang X, Jin Z, Li J, Limsakul P, Zhu L, Allen M, Pan Y, et al. (2021). Control of the activity of CAR-T cells within tumours via focused ultrasound. Nat Biomed Eng 5, 1336–1347. 10.1038/s41551-021-00779-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miller IC, Zamat A, Sun LK, Phuengkham H, Harris AM, Gamboa L, Yang J, Murad JP, Priceman SJ, and Kwong GA (2021). Enhanced intratumoural activity of CAR T cells engineered to produce immunomodulators under photothermal control. Nat Biomed Eng 5, 1348–1359. 10.1038/s41551-021-00781-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Spiegel JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH, Frank MJ, Shiraz P, Sahaf B, Craig J, et al. (2021). CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med 27, 1419–1431. 10.1038/s41591-021-01436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garrison BSD, Han; Yucel Gozde; Frankel Nicholas W.; Guzman-Ayala Marcela; Gordley Russell; Hung Michelle; Lee Derrick; Gainer Marcus; Loving Kathryn; Chien Jenny; Pan Tiffany; Gorman Wesley; Leemans Nelia; Lam Alice; Wood Travis; Wong Wilson; Lee Philip; Lu Tim; Lee Gary (2021). FLT3 OR CD33 NOT EMCN Logic Gated CAR-NK Cell Therapy (SENTI-202) for Precise Targeting of AML. Blood. 10.1182/blood-2021-154201. [DOI] [Google Scholar]
- 84.Zhao Y, Dong Y, Yang S, Tu Y, Wang C, Li J, Yuan Y, and Lian Z (2022). Bioorthogonal Equipping CAR-T Cells with Hyaluronidase and Checkpoint Blocking Antibody for Enhanced Solid Tumor Immunotherapy. ACS Cent Sci 8, 603–614. 10.1021/acscentsci.2c00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li AW, and Lim WA (2020). Engineering cytokines and cytokine circuits. Science 370, 1034–1035. 10.1126/science.abb5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gardner TJ, Lee JP, Bourne CM, Wijewarnasuriya D, Kinarivala N, Kurtz KG, Corless BC, Dacek MM, Chang AY, Mo G, et al. (2022). Engineering CAR-T cells to activate small-molecule drugs in situ. Nat Chem Biol 18, 216–225. 10.1038/s41589-021-00932-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim S, Hupperetz C, Lim S, and Kim CH (2021). Genome editing of immune cells using CRISPR/Cas9. BMB Rep 54, 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rurik JG, Tombacz I, Yadegari A, Mendez Fernandez PO, Shewale SV, Li L, Kimura T, Soliman OY, Papp TE, Tam YK, et al. (2022). CAR T cells produced in vivo to treat cardiac injury. Science 375, 91–96. 10.1126/science.abm0594. [DOI] [PMC free article] [PubMed] [Google Scholar]