

Keywords: alpha-sheet, amyloid, toxic oligomer
Abstract
Amyloid diseases are linked to protein misfolding whereby the amyloidogenic protein undergoes a conformational change, aggregates and eventually forms amyloid fibrils. While the amyloid fibrils and plaques are hallmarks of these diseases, they typically form late in the disease process and do not correlate with disease. Instead, there is growing evidence that smaller, soluble toxic oligomers form prior and appear to be early triggers of the molecular pathology underlying these diseases. Nearly 20 years ago, we proposed the α-sheet hypothesis after discovering that the early conformational changes observed during atomistic molecular dynamics simulations involve the formation of a non-standard protein structure, α-sheet. Furthermore, we proposed that toxic oligomers contain α-sheet structure and that preferentially targeting this structure could neutralize the toxicity, prevent further aggregation and serve as the basis for early detection of disease. Here, we present the origin of the α-sheet hypothesis and describe α-sheet structure and the corresponding mechanisms of conversion. We discuss experimental studies demonstrating that both mammalian and bacterial amyloid systems form α-sheet oligomers before converting to conventional β-sheet fibrils. Furthermore, we show that the process can be inhibited with de novo designed α-sheet peptides complementary to the structure in the toxic oligomers.
1. Introduction
Amyloidogenic proteins undergo conformational changes from their native structure, misfold and self-aggregate to form amyloid fibrils [1]. These self-aggregating proteins have been identified in both mammalian and bacterial species [1–3]. Mammalian amyloid proteins are associated with over 50 diseases, including Alzheimer's disease (AD), Parkinson's disease (PD) and Type 2 diabetes (T2D) [4,5]. Bacteria can also form amyloid fibrils using programmed machinery and incorporate the fibrils into their extracellular biofilms [2,3]. Biofilms protect the organism from the surrounding environment, including the host immune response and antibiotics [2,3]. Regardless of the function and native structure of an amyloidogenic protein, the corresponding fibrils form cross-β sheet structure [6]. Recent studies show that toxicity associated with amyloid diseases is due to the soluble oligomeric species rather than fibrillar plaques [7–11]. Furthermore, there is increasing evidence that both mammalian and bacterial toxic oligomers are comprised of a non-standard protein secondary structure known as α-sheet [9,12,13]. By targeting the α-sheet structure in the toxic oligomers, it may be possible to combat the pathology associated with human amyloid diseases, as well as infections associated with bacterial amyloid.
2. Amyloid proteins and their aggregation properties
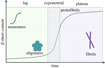
The amyloid aggregation pathway has been studied extensively, primarily in the context of human disease pathology. Aggregation occurs via a nucleation-dependent mechanism with three main phases of aggregation: lag, exponential and plateau (figure 1) [14]. The lag phase begins when a protein or peptide undergoes conformational changes to form an aggregation competent monomer [15]. This aggregation competent species alone has a low propensity for oligomerization, although small aggregates have the capacity to form over time [1,15]. The lag phase ends with the formation of an aggregation nucleus, the structure from which amyloid fibrils assemble [1,16]. The exponential phase is characterized by rapid oligomerization, resulting in the formation of fibrils composed of β-sheet structure [15]. The plateau phase describes the end of the aggregation pathway when fibrils, or plaques, are deposited in surrounding tissues [5].
Figure 1.
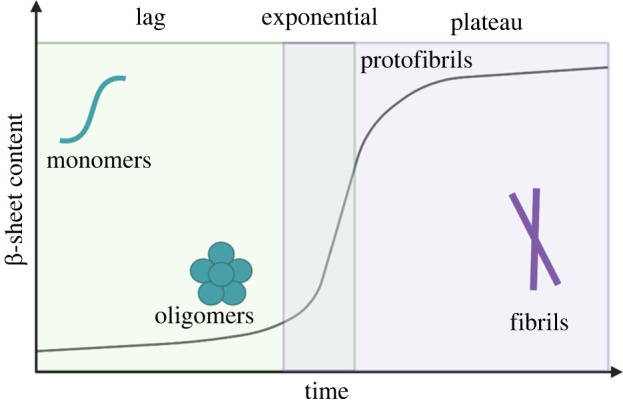
Amyloid aggregation pathway. The first step is known as the lag phase and is characterized by the formation of an amyloid-competent monomer and oligomerization. In the exponential phase, the amyloid protein changes structure and forms β-sheet protofibrils. Finally, the plateau phase describes the stage at which fibrils have rearranged to adopt highly ordered cross-β-sheet structure.
Amyloid fibrils and plaques are the pathological hallmarks of amyloid diseases. As such, amyloid research has historically focused on the study and characterization of the fibrils, rather than the oligomers or the misfolded monomers. One of the most extensively studied amyloid systems is the amyloid-β peptide (Aβ), an intrinsically disordered peptide that is associated with AD [17]. Notably, extensive studies of Aβ and other amyloid fibrils show that the fibrils, or amyloid state, are not responsible for disease-related pathogenicity [8–10,17,18]. Instead, studies of Aβ oligomers indicate that the low molecular weight oligomers (approx. 8–70 kDa) are the toxic species linked to hippocampal long-term potentiation impairment, microglial activation and synaptic dysfunction [8,10,18]. Higher-molecular-weight oligomers, protofibrils and plaques display little to no cytotoxicity in biological assays [9,10]. In fact, plaque formation may serve a protective function by removing toxic oligomers from surrounding tissue [8].
The toxicity of low molecular weight soluble oligomers is not unique to Aβ and AD; studies show that the insoluble fibrils of many amyloid proteins are stable and non-toxic, while oligomers are the primary toxic species [8–10,19–22]. Islet amyloid polypeptide (IAPP), for example, is an intrinsically disordered peptide [23] whose amyloid formation is associated with T2D [24–26]. Electron microscopy experiments in which an aqueous solution of IAPP was applied to cultured islet cells demonstrated small IAPP oligomers disrupting the cell membranes, resulting in apoptosis [27]. This finding suggests that β-cell apoptosis associated with T2D is also likely caused by small, low molecular weight IAPP oligomers, rather than fibrils [27]. Given that oligomers are thought to be the trigger responsible for downstream pathology in amyloid-associated diseases, a deeper understanding of the structural characteristics of these low molecular weight species is necessary.
3. Cross-reactivity of A11 oligomer antibody suggests a conserved oligomeric structure
An attempt to probe the structural properties of Aβ oligomers by Glabe, Kayed and co-workers revealed the existence of a conserved conformation between amyloid oligomers, thereby introducing a novel tool through which to target and isolate this toxic species [19,28,29]. With knowledge of the structure of both Aβ monomers and mature fibrils, they sought to characterize the intermediary species which had previously eluded researchers by raising a polyclonal antibody, now known as A11, against molecular mimics of Aβ soluble oligomers. This was done to produce a molecule that targets the soluble toxic aggregates of Aβ with no specificity toward the monomeric or fibrillar forms [19,28,29]. While they successfully produced an antibody with specificity for Aβ oligomers, they found that A11 also binds to many oligomeric species, regardless of the amyloid protein's native structure or amino acid sequence [19,28,29]. A11 does not recognize the corresponding monomers or amyloid fibrils of these proteins, thereby exhibiting specificity for a particular structure or motif in toxic oligomers [19,28,29].
The cross-reactivity of the A11 antibody suggests that oligomers adopt a conserved structure, regardless of the amyloidogenic protein's native structure or sequence. In addition, A11 inhibits the toxicity of oligomers, including IAPP, Aβ, human insulin, lysozyme and more, when co-incubated and applied to live cells [19,28,29]. This suggests that not only do oligomers share a conserved conformation, they also share a conserved mechanism of toxicity that is inherently related to oligomeric structure [19,28,29]. Therefore, the production of the A11 antibody revealed that targeting oligomeric structure may serve as a strategic method to ameliorate toxicity and potentially combat amyloid-associated diseases.
4. Toxic oligomers are composed of α-sheet structure
Directly observing the conserved toxic oligomeric structure implicated by the cross-reactivity of the A11 antibody in vivo is challenging. This is due to the heterogeneous and interconverting mixture of misfolded monomers and oligomers that is present during the lag phase of aggregation [30]. Furthermore, elucidating the molecular mechanisms associated with amyloid formation is a challenging task because the process can span multiple orders of magnitude of time [31]. Molecular dynamics (MD) simulations serve as an alternative to experimental isolation; they can be used to predict the conformations sampled by a protein or peptide as it transitions from its native structure to an amyloid-competent monomer.
The Daggett lab conducted MD simulations of many structurally unrelated amyloidogenic proteins in order to study the conformational changes that occur over the course of aggregation. Proteins simulated included transthyretin (TTR), which has β-sandwich structure and is implicated in senile systemic amyloidosis and peripheral polyneuropathy [32–36]; the prion protein, a primarily helical protein involved in transmissible spongiform encephalopathies [37–44]; β2-microglobulin, a β-sheet protein implicated in hereditary renal amyloidosis [45]; lysozyme variants, small globular proteins involved in autosomal dominant hereditary amyloidosis [46]; polyglutamine repeats involved in Huntington's disease [47]; and superoxide dismutase-1, associated with amyotrophic lateral sclerosis [48,49]. Simulations were conducted under known amyloidogenic conditions to observe conformational changes that drive aggregation, including low pH and mutations associated with familial forms of the diseases [32–49].
A non-standard secondary structure, now referred to as α-sheet structure, was identified as an unfolding intermediate in simulations of each amyloid protein. We learned later that Linus Pauling and Robert Corey had predicted α-sheet structure in 1951, then referred to as ‘polar pleated sheet’ [50]. However, Pauling and Corey found that the structure was not an energetic minimum in their dihedral potential function, and it was therefore rejected. Until its independent ‘rediscovery’ in the Daggett lab, the structure was dismissed and considered to be a rare and alternative conformation to the energetically favourable β-sheet, which is correct for normal, native proteins. Following its observation in MD simulations conducted by the Daggett lab, α-sheet structure has been observed in a number of other MD simulations of amyloid proteins [51–56].
5. α-Sheet geometry and features of the structure
α-Sheet structure is stabilized by hydrogen bonding between individual α-strands, similar to the formation of standard β-sheet secondary structure [32,33,51,52,57,58]. Amino acids that form the strands are locally helical, occupying the left- (αL) and right-handed (αR) helical regions of Ramachandran space (figure 2a) [32,57–59]. Sequential alternation between αL and αR backbone (Φ, Ψ) dihedral angles results in the formation of an elongated strand (figure 2b) [32,52,57,59]. Sheets are formed with bifurcated hydrogen bonding between individual residues in adjacent strands (figure 2c) [32,57,60]. Bifurcated hydrogen bonding is also seen in α-helices, but not in β-sheet structures, and it can significantly increase structural stability [32,57]. α-Sheet is also unique in that the NH groups are aligned along one side of the sheet, while the carbonyl groups are located along the other [12,32,35,53,57]. The alignment of NH groups on one side of the sheet and CO groups on the opposite forms a strong molecular dipole across the sheet, which may assist in oligomerization through monomer–monomer interactions [32,57,61,62].
Figure 2.
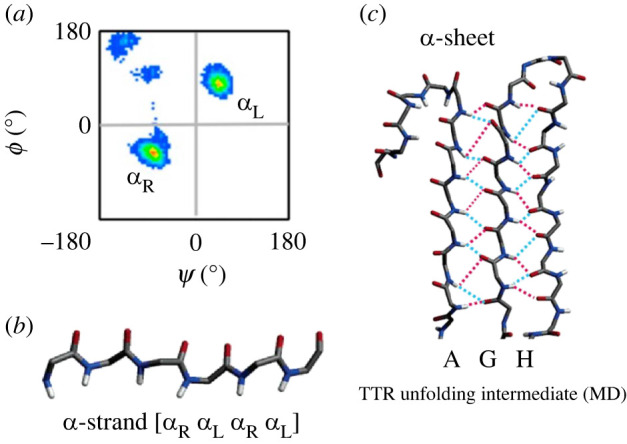
α-sheet structure. (a) Sequential amino acids in the α-strand conformation have backbone Φ and Ψ angles that alternately occupy the αR and αL regions of Ramachandran space. (b) The sequential alternation between αR and αL conformation results in the alignment of carbonyl groups on one side of the peptide backbone, and the alignment of the peptide's amide groups on the other side of the backbone. (c) Bifurcated hydrogen bonding between the A, G and H strands of TTR stabilize the peptide in α-sheet conformation. Reproduced from [12] and [32].
6. Mechanisms driving α-sheet formation
Beyond identifying a conserved oligomeric structure, MD simulations can also be used to determine the mechanism of structure formation in a particular amyloid system. Currently, the most well-studied α-sheet transition is that of TTR.
TTR exists in the body as a homotetramer and forms a β-sandwich structure composed of two β-sheets, referred to as the DAGH and CBEF sheets [33,35,63–67]. Both familial mutations and specific amyloidogenic conditions can lead to the destabilization of the tetramer, causing dissociation into an amyloid-competent monomer; notably, most cases are sporadic (WT) [33,35,68]. To further probe the mechanism of conversion from native β-sheet structure to α-sheet, 21 simulations were conducted on monomeric TTR under amyloidogenic conditions for 0.5 µs each [35]. Because both WT and mutant TTR monomers have been implicated in amyloid diseases, six pathogenic mutants (D18G, A36P, L58H, Y69H, L111M and V122I) in addition to the WT monomer were simulated in triplicate [35].
A variety of analyses were used to determine both the extent and molecular mechanisms of α-sheet formation (figure 3a) [35]. The conversion of the DAGH native β-sheet structure to α-sheet structure was consistent among the seven simulated monomers, while the CBEF sheet largely maintained a stable β-sheet conformation (figure 3b) [35]. It was determined that peptide plane flipping causes α-sheet conversion in the DAGH sheet of both mutant and WT monomeric TTR (figure 3c) [35]. This change in secondary structure is preceded by three main events: distortion of main-chain geometries, referred to as ‘pleated main-chain geometry’, loss of the hydrogen bonding network between the main chain, and a reorientation of solvent-exposed side chain interaction networks (figure 3c) [35]. In addition to these phenomena, MD simulations revealed that hydrogen bonding employed by polar side chains and water molecules actively assists in pulling the peptide's backbone into α-sheet structure (figure 3c). Transitions not mediated by hydrogen bonding were driven by electrostatic repulsion of oxygen molecules present in the carbonyl groups in neighbouring strands [35].
Figure 3.

Conversion from native β-sheet to α-sheet structure in the DAGH sheet of TTR is observed in MD simulations of both wild-type and mutant TTR. (a) Secondary structure of each residue over the course of the simulation shows that residues 1–20 and 100–127 of the DAGH sheet (red) maintain α-sheet structure for approximately 60% of the total simulation time. (b) α-sheet content of the DAGH sheet increases linearly from 0 to 24% over the course of the simulations, while the CBEF sheet demonstrates little propensity toward α-sheet conversion. (c) Peptide plane flipping causes α-sheet conversion in the DAGH sheet of monomeric TTR. Once the peptide groups lose their native structure, water molecules participate in hydrogen bonding with the peptide's main-chain atoms to actively pull the backbone into the α-sheet conformation. Reproduced from [34].
This study outlines the molecular interactions that lead to the destabilization of the native β-sheet structure in TTR and subsequent conversion to α-sheet. The elucidation of these mechanisms further supports the α-sheet hypothesis and provides a blueprint through which to determine the molecular mechanisms involved in the conversion to α-sheet structure. The identification of the α-sheet conformation in TTR suggests that this structure may be responsible for the preferential oligomer binding behaviour exhibited by the A11 antibody. Indeed, synthetic de novo peptides that adopt stable α-sheet structure specifically bind to toxic TTR oligomers and inhibit amyloid formation [12], as discussed below, which supports the hypothesis that A11 recognizes and binds to α-sheet structure.
7. De novo designed synthetic model α-sheet peptides inhibit amyloid aggregation and toxicity
To experimentally probe the structure of soluble oligomers, de novo hairpin peptides were designed and engineered through MD simulations and produced by solid-phase peptide synthesis. These peptides were designed to adopt a conformation complementary to the non-standard α-sheet structure that was observed in MD simulations, and the designed peptides are themselves α-sheet. The designed peptides were hypothesized to selectively bind to the toxic oligomers of amyloid proteins regardless of the protein's native structure to facilitate isolation and characterization of the nonstandard structure [12].
The design process for these small, stable hairpins began with the computational development of a backbone template in an optimized α-sheet structure. The next step was to determine how to design the sequence to ensure alternation of sequential amino acids between αR and αL backbone conformations, which is the hallmark of α-sheet structure. To do this, the Structural Library of Intrinsic Residue Properties associated with the Dynameomics project was used to determine the propensities for various combinations of amino acids to occupy the desired regions of conformational space [69–75]. It was determined that sequential alternation between L- and D-amino acids would produce an extended sheet structure that mimics the α-sheet conformational signature observed in MD simulations of amyloid proteins containing all L-amino acids [12]. A variety of sequences were chosen according to these criteria and were used to engineer a library of de novo peptides predicted to adopt stable α-sheet structure. Finally, MD simulations of the α-sheet hairpins were conducted in conjunction with designed random coil and β-sheet control peptides to determine the stability of the de novo designs. The designs were then ranked, and the highest ranked designs were chemically synthesized and evaluated. The sequences corresponding to some of these designs and the controls can be found in table 1.
Table 1.
The amino acid sequences of the de novo α-sheet peptides (AP). Residues that are denoted by a capital letter reference L-chirality amino acids, and D-chirality amino acid residues are depicted by lowercase letters. The underlined cysteine residues in AP407 refer to disulfide linkage sites. Each peptide has C-terminus amidation (shown as ‘NH2’), and ‘Ac’ refers to acetylation on the N-terminus.
| AP sequences | |
|---|---|
| AP5 | Ac-RGNwNeSkMNEYSGWmLmLtMGR-NH2 |
| AP90 | Ac-RGEmNlSwMNEYSGWtMnLkMGR-NH2 |
| AP193 | Ac-RGEmNyFwMNEYYGWtMnCkMGR-NH2 |
| AP401 | Ac-rGeMnLsWmneysGwTmNlKmGr-NH2 |
| AP407 | Ac-RGEmNlCwMNEYSGWcMnLkMGR-NH2 |
| AP421 | Ac-RGEcNlSwMNEYSGWtMnLkCGR-NH2 |
| P1 | Ac-KLKpLLTSENTL-NH2 |
| P90 | SWTWEpNKWTWK-NH2 |
8. Determination of spectroscopic signatures for α-sheet
The most stable designs as determined through MD were synthesized. As the structure is new and non-standard, it was necessary to determine the spectroscopic signature of α-sheet using the designed model compounds. The results from the model compounds were then used to determine whether amyloid intermediates and soluble oligomers contain α-sheet. Various techniques including circular dichroism (CD) spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, Fourier transform infrared spectroscopy (FTIR) and microfluidic modulation spectroscopy in the infrared region (MMS-IR) were conducted on the α-sheet hairpin peptides in an effort to determine the spectroscopic signature of α-sheet secondary structure.
8.1. Circular dichroism spectroscopy
CD spectroscopy is a technique that uses left- and right-handed circularly polarized light to evaluate the structure of chiral molecules. Due to the chirality of amide bonds, CD can effectively be used to study protein secondary structure. Because L- and D- amino acids absorb oppositely circularly polarized light, we predicted that CD of our de novo α-sheet peptides would produce relatively featureless spectra. As shown in figure 4a, the CD spectra of an α-sheet peptide, AP90, is indeed flat and primarily featureless with some random coil structure due to the influence of the tails and a hairpin turn composed of L-amino acids. In fact, AP401, which has inverse chirality of AP90, shows an inverse spectrum due to the dominance of D-amino acids in the hairpin turn and ends [13]. By contrast, the all L-amino acid version of AP90, P90, displayed a hallmark β-sheet spectrum with a minimum around 218 nm (figure 4a). Also of note is that AP90 and P90 have the same amino acid sequence, but AP90 contains 6 D-amino acids (6 of 23) to produce α-sheet, and P90 is comprised solely of L-amino acids. Despite having the same sequence, the peptides have different structures, solubilities and functions [61]. These results confirm that our de novo peptides adopt a structure that is distinct from random coil, β-sheet, and α-helical conformations.
Figure 4.

Spectroscopic characterization of de novo α-sheet peptides. (a) CD of an α-sheet peptide (AP90) displays relatively flat and featureless spectra due to the alternation of αR and αL residues, leading to cancellation of the CD signal. The dip around 200 nm is from the L-amino acids in the turn and at the ends of the hairpin. The all L-amino acid P90 peptide produced standard β-sheet spectra with a dip around 220 nm. AP90 and P90 have the same sequence, but AP90 contains six D-amino acids to template the α-sheet structure. (b) NMR of AP407 showed that the main-chain NOEs did not correlate to β-sheet or α-helical structure. The NMR ensemble of conformers determined for AP407, dominant conformer shown, confirms the two α-strands connected by a hairpin turn. (c) MMS-IR spectra of AP407, a β-hairpin control (P411) and an α-helix control (PSMα1) show that AP407 adopts a structure that is neither α-helical nor β-sheet. (d) Despite having the same amino acid sequence, AP90 and P90 produce distinct FTIR spectra, with P90 adopting β-sheet structure and AP90 displaying two bands associated with α-sheet, noting that the strength of the signals can change with 1675 cm−1 becoming less dominant in some AP designs [76]. Reproduced from [9] and [61].
8.2. Nuclear magnetic resonance structure of de novo designed α-sheet peptide
Homonuclear NMR spectroscopy was conducted on several of our de novo peptides [9,12]. One design in particular, AP407, was well characterized. The NMR studies of AP407 resulted in 455 nuclear Overhauser effect (NOE) crosspeaks between protons, a considerably large number considering the peptide length of only 23 residues (approx. 20 NOEs per residue). AP407 contains a disulfide bond linking the two α-strands, and this structural constraint likely contributed to the high number of NOEs. We expected to observe sequential dNN NOEs along the backbone of our peptide, as predicted for α-sheet structure [12]. Further, α-sheet should not exhibit long-range dNN or dαN NOEs, which would indicate α-helical or β-sheet structure [12].
As expected, standard main-chain NOE patterns that are typical of α-helix and β-sheet structure were not observed (figure 4b) [9]. Instead, the NMR showed sequential HN-HN NOEs that were predicted for α-sheet structure in addition to crosspeaks across the hairpin [9]. Further, the observed coupling constants suggest β-sheet structure while secondary chemical shifts suggest α-helical structure [9]. The combination of the coupling constant and secondary chemical shift results supports the extended sheet structure in which each residue is locally helical, which is precisely what we had hypothesized and designed into our de novo α-sheet peptides [9]. The NOEs were used to construct an NMR ensemble, with the dominant conformer of this small, dynamic peptide presented in figure 4b. NMR structural data have been presented and discussed in depth elsewhere [9], and they are available from the Biological Magnetic Resonance Data Bank (BMRB Entry 27873).
8.3. Microfluidic modulation spectroscopy
Next, we sought to further validate the presence of α-sheet structure in our de novo peptides through MMS-IR, which reports on amide I band absorption (1714 cm−1 to 1590 cm−1) [52]. Amide I band absorption is correlated with shifts in hydrogen bonding patterns and dipole–dipole interactions, and can therefore aid in determining protein secondary structure [77]. We conducted MMS-IR on a number of our de novo α-sheet peptides, including AP5, AP90, AP407 and AP421, in addition to β-sheet (P411) and α-helical (PSMα1) control peptides [9]. Comparing the MMS-IR spectra of the α-sheet, β-sheet and α-helical peptides confirmed that each of the three classes of peptides has distinct conformations and spectroscopic signatures, supporting and validating our original MD prediction of α-sheet structure (figure 4c).
8.4. Fourier transform infrared spectroscopy
FTIR was also employed on AP90 and its all L-amino acid structural isomer, P90, to help develop a spectroscopic signature for the non-standard α-sheet structure. Although these two peptides are composed of identical amino acid sequences, they produce very different FTIR spectra due to the presence of alternating L- and D- amino acids in AP90 (figure 4d) [12,61,76]. The aligned amide groups present in α-sheet structure (AP90) result in electrostatic interactions that are expected to produce strong FTIR signals, while these electrostatic interactions are not present in the structural isomer, P90 [12]. AP90 shows the strongest absorbance at 1675 and 1640 cm−1 [12,76], but the relative strength of these two bands depends on the sequence of the α-sheet peptides [76]. Conversely, the FTIR spectra of P90 is consistent with β-sheet structure with a strong absorbance at 1620 cm−1 (figure 4d) [61,76]. We can conclude from our FTIR experiments that the alternation of L- and D-chirality amino acids results in a unique and non-standard secondary structure that is spectroscopically distinct from its corresponding structural isomer.
9. α-Sheet in amyloid systems
9.1. Direct observation of α-sheet in amyloid proteins
As previously mentioned, isolation of a single conformer during the amyloid aggregation process is challenging due to the heterogeneous mixture of interconverting monomers, oligomers and protofibrils present prior to the deposition of stable fibrils [11]. However, aggregation conditions can be optimized to facilitate analysis of the various conformations sampled by an amyloidogenic peptide in vitro while maintaining a physiologically relevant environment [9,78,79]. The following studies were conducted using diligently engineered conditions to effectively lengthen the aggregation timeline and promote individual isolation of α-sheet and β-sheet containing species for spectroscopic characterization.
The engineered conditions include the use of more physiologically relevant buffers such as phosphate-buffered saline (PBS), rather than organic solvents, to modulate native aggregation [9,78,79]. Additionally, peptide concentration was carefully optimized for each system to ensure successive transitions between the lag, exponential and plateau phases of aggregation [9,78,79]. Using these engineered conditions, we can isolate essentially ‘pure’, or enriched, conformers for further experimental characterization, as described below.
9.1.1. Circular dichroism spectroscopy
By engineering stable α-sheet peptides and subsequently characterizing the structure through a variety of spectroscopic techniques, we determined the spectroscopic signatures of α-sheet through which to observe and validate the existence of the structure in toxic oligomers in different amyloid systems. This spectroscopic signature has been used to directly probe α-sheet structure in the toxic oligomers of a number of mammalian amyloid proteins including IAPP and the 42-residue fragment of Aβ (Aβ42), in addition to multiple bacterial amyloid proteins, such as PSMα1, the amyloid protein in Staphylococcus aureus (S. aureus) biofilms, and CsgA, the amyloid protein in E. coli biofilms.
CD spectroscopy was conducted on each of the aforementioned amyloid species over the course of aggregation to determine how secondary structure evolves throughout amyloidogenesis (figure 5a–d). At the beginning of incubation (t = 0 h), CD spectra of IAPP, Aβ, and CsgA indicated random coil structure, which is the expected structure for the monomeric species that exist at the beginning of aggregation for these peptides (figure 5a,b,d). By contrast, PSMα1 is natively α-helical, which is reflected in its CD spectrum at t = 0 h (figure 5c). Each of the four peptides have different aggregation kinetics, and therefore they form oligomers and adopt α-sheet structure after varying incubation times. While IAPP, Aβ, PSMα1 and CsgA form toxic oligomers at different times, each of these species produced a flat and relatively featureless CD spectrum at the end of their respective lag phase (figure 5a–d). Based on the spectroscopic signature derived from our de novo α-sheet peptides, we know that a flat, null spectrum is characteristic of α-sheet structure. Finally, during the plateau phase of aggregation, IAPP, Aβ, PSMα1 and CsgA all adopt β-sheet structure as shown by the strong CD signal around 218 nm (figure 5a–d). These CD studies allowed us to track secondary structure during aggregation, as well as observe a conserved secondary structure in four structurally and physiologically unrelated amyloid proteins.
Figure 5.

CD spectra of (a) islet amyloid polypeptide (IAPP), (b) Aβ42, (c) phenol soluble modulin a1 (PSMα1) from S. aureus and (d) CsgA from E. coli over the course of aggregation. IAPP, Aβ, and CsgA begin in a random coil state and PSMα1 is α-helical. Over time, each species produces a relatively flat and featureless CD spectrum, indicative of α-sheet conformation. At the end of aggregation, the amyloid species have β-sheet structure as shown by CD. All spectra are presented as MRE. Reproduced from [9,13,78,79].
9.1.2. Microfluidic modulation spectroscopy
A combination of kinetic assays and size exclusion chromatography (SEC) data was used to inform MMS-IR studies of Aβ42 aggregates that were incubated for various durations. A toxic, late-lag phase Aβ sample (24 h incubation) was analysed by SEC and determined to be a low-molecular-weight oligomeric sample containing primarily hexamer aggregates with some dodecamer (figure 6a) [9]. This species was analysed by MMS-IR, and it was found that this oligomeric Aβ sample closely aligned with the secondary derivative of MMS-IR spectra of our de novo α-sheet peptide, AP407 [9]. Subtraction of the AP407 spectra from the spectra of each of the α-sheet and control peptides, as well as the Aβ samples, confirms that there is little to no variance between the second derivative of MMS-IR spectra of the 24 h Aβ sample and the de novo α-sheet peptides (figure 6b) [9]. By contrast, an Aβ sample that had a longer incubation period (120 h) was closely aligned with our β-sheet control, P411, and the shifted wavelength commonly associated with amyloid fibrils (figure 6b). This 120 h sample was shown by SEC to contain primarily higher molecular weight, β-sheet oligomers (figure 6b) [9]. This further confirms that amyloid peptides and proteins, including Aβ, form low molecular weight oligomers with α-sheet structure before transitioning to higher molecular weight aggregates that adopt β-sheet structure.
Figure 6.

Selective binding of α-sheet peptide to toxic oligomers and spectroscopic identification of α-sheet in Aβ toxic oligomers. Aβ was incubated at 25°C for 24 h to obtain a sample enriched in toxic oligomers [9]. (a) AP407 binds specifically to oligomeric Aβ, as shown by SEC, depicted by the shift to higher molecular weight (lower elution volume) for the hexameric and dodecameric oligomers and the lack of binding to monomeric Aβ. (b) The subtraction of the second derivative of the MMS-IR spectrum of AP407 from each of the other signals, including three other α-sheet designs (shown as a band of three light blue toned lines), illustrates the similarities between the de novo α-sheet peptides. Furthermore, during the lag phase (24 h, in red) Aβ has a similar structure to the AP designs as shown by the second derivative of MMS-IR, confirming the presence of α-sheet conformation in the Aβ oligomers, as also observed by CD (figure 5b). In addition, toxic oligomeric Aβ displays a spectrum distinct from the other secondary structures, including the α-helical and monomeric β-hairpin model compounds as well as the non-toxic 120 h incubated Aβ protofibrils/fibrils with β-structure by CD (figure 5b) from the plateau of the ThT aggregation assay [9]. Reproduced from [9].
9.2. Indirect support of α-sheet in amyloid proteins
9.2.1. Inhibition of amyloid formation by targeting α-sheet oligomers
With the knowledge that binding the A11 antibody inhibits the toxicity of amyloid oligomers by recognizing a conserved conformation, we sought to determine whether our de novo peptides could selectively bind to and inhibit both the aggregation and the toxicity of these species. We tested a number of amyloid species, including both mammalian and bacterial proteins [3,9,12,61,76,78,79]. We found that our de novo α-sheet peptides significantly inhibited both the aggregation and toxicity of the amyloid proteins, while the control peptides had no effect on either the aggregation or the toxicity (figure 7a–e) [3,9,12,62,76,78,79].
Figure 7.

α-Sheet peptides inhibit amyloid formation and oligomeric toxicity. (a) Incubation of Aβ with excess (4 : 1) α-sheet peptide resulted in up to 96% inhibition, while control peptides (P1 and P411) had no effect on aggregation. (b) Incubation of TTR with excess (10 : 1) AP90 and AP5 resulted in 63% and 37% inhibition, respectively. (c) Exposure to oligomeric Aβ resulted in almost 20% loss of viability of SH-SY5Y cells. Incubation with AP5 and AP421 resulted in complete recovery of cell viability. AP5 and AP421 themselves were not toxic to the cells. (d) Incubation of PSMα1 with excess (4 : 1) AP90 resulted in 90% aggregation inhibition. The random coil control had little effect. (e) Incubation with excess CsgA (2 : 1) and AP193 resulted in approximately 60% inhibition, while the random coil control (P1) had no effect on aggregation. While the extent of inhibition differs among the various systems, α-sheet designs inhibit aggregation and toxicity in all systems independent of the sequence of the AP peptide and the sequence of the amyloid peptides/proteins. Statistical significance: *p < 0.05, **p < 0.01, and ***p < 0.001. Reproduced from [9], [13] and [78].
SEC was employed to confirm that the observed inhibition with α-sheet peptides was due to the inhibition of amyloid formation through preferential binding to the oligomeric species with the same conformation, rather than through interactions with the corresponding random coil monomers [9]. Monomeric Aβ samples analysed both in the presence and in the absence of AP407 produced essentially identical SEC data, suggesting that the α-sheet peptide does not bind specifically to monomeric, random coil Aβ (figure 6a) [9]. By contrast, an oligomeric (24 h incubation) Aβ sample with AP407 showed an approximately 0.4 ml peak shift to a higher molecular weight with respect to the same Aβ sample alone (figure 6a) [9]. This indicates that AP407 binds preferentially to oligomeric Aβ, as hypothesized [9].
After confirming that de novo α-sheet peptides bind preferentially to α-sheet containing toxic oligomers, we used a Thioflavin T (ThT) assay to probe whether this binding could lead to inhibition of aggregation [3,9,12,76]. ThT is a fluorescent dye that is often used to track amyloid formation [80]. Because it binds to β-sheet rich structures, it can serve as a proxy for the extent of fibrillization [80]. Low ThT signals are emitted during the lag phase of aggregation because there is little to no β-sheet in solution. The ThT signal exponentially increases following the lag phase as the peptides rapidly oligomerize and form fibrils. Finally, the ThT signal plateaus once highly ordered β-sheet containing fibrils have formed.
The extent of amyloid inhibition by our α-sheet peptides was quantified by measuring the ThT signal both in the presence and in the absence of the α-sheet designs. The incubation of Aβ with excess (4 : 1) α-sheet peptide resulted in up to 96% aggregation inhibition as shown by the reduced ThT signal (figure 7a) [9]. Random coil (P1) and β-sheet (P411) control peptides had no significant effect on aggregation (figure 7a) [9]. Incubation of TTR with excess (10 : 1) AP5 and AP90 resulted in approximately 37% and 65% inhibition, respectively (figure 7b) [76]. As with the A11 antibody, α-sheet peptides also inhibit toxicity of amyloid oligomers [9,12,61,62]. The addition of α-sheet peptides (AP5 and AP421) to toxic Aβ oligomers immediately prior to plating with SH-SY5Y neuroblastoma cells resulted in complete recovery of cellular viability, thereby inhibiting oligomeric toxicity (figure 7c) [9]. The α-sheet peptides themselves were not toxic to the cells (figure 7c) [9].
α-Sheet peptides have also proved effective at inhibiting the aggregation of bacterial amyloid. PSMα1, the amyloid protein present in S. aureus biofilms, incubated with excess (4 : 1) AP90 resulted in 81% amyloid inhibition (figure 7d) [13]. Incubation of AP193 with excess CsgA (1 : 2), the amyloid protein that composes E. coli biofilms, resulted in approximately 50% inhibition, while incubation with equal (1 : 1) random coil control (P1) had no effect on aggregation (figure 7e) [78]. Combined with the SEC results, we conclude that aggregation inhibition is due to the binding of α-sheet peptides to α-sheet containing oligomers, thereby preventing subsequent fibrillization.
Inhibition of bacterial amyloid can also be observed by comparing β-sheet content and cell density of biofilms grown in the presence and in absence of α-sheet peptides. For these studies, bacteria are grown in biofilm-forming conditions and assayed once the biofilm has matured. Planktonic, free-floating cells are removed following biofilm maturation and cell density is estimated through optical density measurements at 600 nm (OD600). The biofilm is then resuspended in ThT and measurements are taken both for ThT signal and OD600. These in vivo studies have been conducted on a number of both gram-positive and gram-negative bacteria, including E. coli, P. aeruginosa, S. aureus and S. mutans [13,78,81]. In each of these studies, incubation with α-sheet peptides resulted in a reduced ThT signal and decreased biofilm OD600 measurements [13,78,81]. For example, incubation of E. coli with 2 pg/colony forming unit (CFU) of AP90 and AP401 resulted in 37% and 65% reduction in biofilm amyloid content, respectively (figure 8a). Biofilm cell density, as measured by OD600, was reduced 15% and 47% when incubated with AP90 and AP401, respectively (figure 8b). Furthermore, OD600 measurements of the planktonic phase increase when grown in the presence of α-sheet peptides, indicating an increase in the number of free, non-biofilm-protected bacteria (figure 8b). We can therefore conclude that not only does exposure to α-sheet peptides inhibit bacterial amyloid formation, it also disrupts the biofilms, thereby causing more cells to remain in the planktonic phase rather being incorporated into the protective extracellular matrix that is the biofilm. It is also important to note that by comparing total OD600 measurements of the biofilm and planktonic phases, we can conclude that α-sheet peptides do not cause cell death, but rather inhibit amyloid aggregation and robust biofilm formation (figure 8b). This is crucial because if α-sheet peptides caused bacterial cell death, it would be possible for mutations to arise that would confer resistance to the peptides, as has occurred with antibiotics.
Figure 8.

α-Sheet peptides inhibit biofilm amyloid content and cell density, leading to an increase in the susceptibility of the bacteria to antibiotics. (a) Incubation with 2 pg CFU−1 AP90 and AP401 resulted in 37% and 65% reduction of amyloid in E. coli biofilms, respectively, as measured by ThT. (b) AP90 and AP401 reduced the cell density of E. coli biofilms by 15% and 47%, respectively, as measured by OD600. Planktonic cell density increased 23% with AP90 and 75% with AP401, while the total cell density remained statistically unchanged. (c) PSMa1 amyloid fibrils are visible as deposits in spaces between cells of S. aureus SH1000 WT biofilms. (d) No extracellular matrices, or fibrils, were visible upon the addition of AP90. (e) Incubation of E. coli with 2 pg CFU−1 AP401 and 300 ug ml−1 gentamicin resulted in a reduction of CFUs by a factor of over 104 with respect to the control conditions. (f) This CFU reduction corresponds to a 13 000× increase in susceptibility to gentamicin, calculated by comparing the ratio of CFUs following antibiotic administration, with and without the addition of AP401. CFU ratios are calculated based on cell density at the time of plating. Statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001 and #p< 0.0001. Reproduced from [13] and [78].
Bacterial biofilm inhibition with α-sheet peptides can also be observed in vivo through various imaging techniques. For example, S. aureus, S. mutans and E. coli form robust biofilms, but when these same bacteria are grown in the presence of α-sheet peptides, the biofilms are visibly less robust and the amyloid content drops dramatically, as illustrated in figures 8c,d for S. aureus [13,78,81]. This is in agreement with ThT and OD600 measurements that show that α-sheet peptides inhibit biofilm formation and force more cells to remain free-floating rather than be incorporated into the biofilm [13,78,81].
9.2.2. Antibiotic susceptibility and α-sheet inhibitors of amyloidogenesis
Biofilms can significantly reduce the efficacy of antibiotics and can confer antibiotic resistance by providing structural support and allowing for communication between individual cells [82,83]. Targeting bacterial biofilms is one strategy through which to increase the susceptibility of bacteria to antibiotics without the added risk of increased resistance due to selective pressure. Having previously shown that incubation with α-sheet peptides significantly decreases biofilm amyloid content, weakens the biofilm, and reduces the number of cells present in and protected by the biofilm, we investigated whether this biofilm inhibition could lead to increased antibiotic susceptibility to resistant strains. Uropathogenic E. coli was used as our model system; we found that cells incorporated into mature E. coli biofilms were 13 000× more susceptible to gentamicin when grown in the presence of AP401 (figure 8e,f) [78]. Therefore, exposing E. coli to α-sheet peptides can render apparent antibiotic-resistant bacterial strains susceptible, and they may also aid in reducing antibiotic resistance, which has major implications on the future of targeting drug-resistant bacteria.
9.2.3. Soluble oligomer binding assay for detection of α-sheet oligomers
The soluble oligomer binding assay (SOBA) is a novel ELISA-like assay that detects α-sheet structure in toxic oligomers and is used to quantify toxic α-sheet oligomers in solution [9,62]. By using a de novo α-sheet peptide in place of an antibody as the capture agent, SOBA selects for α-sheet structure with high specificity [9,62], providing an indirect readout of α-sheet structure. Also, we note that matched cell toxicity assays with SOBA confirm the strong correlation between α-sheet content as measured by SOBA and toxicity, as inferred from cell viability assays (R2 = 0.94, figure 9a) [9].
Figure 9.

Quantification of soluble Aβ oligomers using SOBA. (a) SOBA shows that oligomer content peaks during the late-lag phase of aggregation of Aβ42, and SOBA signal correlates with toxicity, confirming that SOBA has specificity for the toxic, oligomeric form of Aβ. (b) Aβ42 spiked into PBS and evaluated by SOBA. Monomer is at beginning of aggregation reaction; toxic oligomers, incubated 24 h; and protofibrils, incubated 120 h (times correspond to (a)). (c) CD spectra for time points tested on SOBA assay [84]. Figures are taken from [9] and [84].
SOBA can reproducibly differentiate between toxic Aβ oligomers and the monomeric and protofibrillar/fibrillar forms of the peptide (figure 9b) [9,62]. Based on this discrimination between different Aβ conformers (figure 9b,c), we explored the detection of toxic oligomers in biological fluids, including cerebrospinal fluid (CSF) and blood. SOBA was able to distinguish between individuals with mild cognitive impairment and moderate to severe AD from non-cognitively impaired controls in both CSF and plasma [84]. The generality of the approach was tested by modifying SOBA to detect α-sheet containing α-synuclein oligomers in samples from patients with PD with excellent discrimination between control and PD cases [84].
10. Conclusion
Throughout this review, we have outlined the role of the non-standard α-sheet structure in amyloid aggregation and toxicity. De novo α-sheet peptides bind specifically to α-sheet oligomers of a variety of amyloid systems, including both bacterial and mammalian peptides and proteins. This conserved structure reveals a strategy through which to combat amyloid-associated diseases as well as to design diagnostics that can be used to identify a myriad of amyloid diseases in the early stages. This proof of concept has recently been demonstrated for the detection of α-sheet containing oligomers in AD and PD patient samples.
Data accessibility
This article has no additional data.
Authors' contributions
V.D.: conceptualization, funding acquisition, project administration, resources, supervision, writing—original draft and writing—review and editing; T.P.: funding acquisition, visualization, writing—original draft and writing—review and editing.
All authors gave final approval for publication and agreed to be held accountable for the work performed therein.
Conflict of interest declaration
We declare we have no competing interests.
Funding
Research reported in this publication was supported by the National Institutes of Health (NIH) General Medical Sciences (GMS) (grant no. R01 GM958987 to V.D.), NIH National Institute of Aging (grant no. R01 AG067476 to V.D.), NIH GMS (grant no. T32GM008268 to T.P.), as well as the US Army Medical Research Acquisition Activity (USAMRAA), Department of Defense Office of the Congressionally Directed Medical Research Programs (CDMRP), through the Peer Reviewed Medical Research Program (PRMRP) under award No. W81XWH-19-0050 (to V.D.).
References
- 1.Aliyan A, Cook NP, Martí AA. 2019. Interrogating amyloid aggregates using fluorescent probes. Chem. Rev. 119, 11 819-11 856. ( 10.1021/acs.chemrev.9b00404) [DOI] [PubMed] [Google Scholar]
- 2.DePas WH, Chapman MR. 2012. Microbial manipulation of the amyloid fold. Res. Microbiol. 163, 592-606. ( 10.1016/j.resmic.2012.10.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bleem A, Daggett V. 2017. Structural and functional diversity among amyloid proteins: agents of disease, building blocks of biology, and implications for molecular engineering. Biotechnol. Bioeng. 114, 7-20. ( 10.1002/bit.26059) [DOI] [PubMed] [Google Scholar]
- 4.Knowles TP, Vendruscolo M, Dobson CM. 2014. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol Cell Biol. 15, 384-396. ( 10.1038/nrm3810) [DOI] [PubMed] [Google Scholar]
- 5.Chiti F, Dobson CM. 2017. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27-68. ( 10.1146/annurev-biochem-061516-045115) [DOI] [PubMed] [Google Scholar]
- 6.Chuang E, Hori AM, Hesketh CD, Shorter J. 2018. Amyloid assembly and disassembly. J Cell Sci. 131, jcs189928. ( 10.1242/jcs.189928) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahmed M, et al. 2010. Structural conversion of neurotoxic amyloid-β(1–42) oligomers to fibrils. Nat. Struct. Mol. Biol. 17, 561-567. ( 10.1038/nsmb.1799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haass C, Selkoe DJ. 2007. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101-112. ( 10.1038/nrm2101) [DOI] [PubMed] [Google Scholar]
- 9.Shea D, et al. 2019. α-Sheet secondary structure in amyloid β-peptide drives aggregation and toxicity in Alzheimer's disease. Proc. Natl Acad. Sci. USA 116, 8895-8900. ( 10.1073/pnas.1820585116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang T, Li S, Xu H, Walsh DM, Selkoe DJ. 2017. Large soluble oligomers of amyloid β-protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J. Neurosci. 37, 152-163. ( 10.1523/JNEUROSCI.1698-16.2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shea D, Daggett V. 2022. Amyloid-β oligomers: multiple moving targets. Biophysica 2, 91-110. ( 10.3390/biophysica2020010) [DOI] [Google Scholar]
- 12.Hopping G, et al. 2014. Designed α-sheet peptides inhibit amyloid formation by targeting toxic oligomers. Elife 3, e01681. ( 10.7554/eLife.01681) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bleem A, Francisco R, Bryers JD, Daggett V. 2017. Designed α-sheet peptides suppress amyloid formation in Staphylococcus aureus biofilms. NPJ Biofilms Microb. 3, 1-10. ( 10.1038/s41522-017-0025-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen SI, et al. 2013. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl Acad. Sci. USA 110, 9758-9763. ( 10.1073/pnas.1218402110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiti F, Dobson CM. 2006. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333-366. ( 10.1146/annurev.biochem.75.101304.123901) [DOI] [PubMed] [Google Scholar]
- 16.Jarrett JT, Lansbury PT Jr. 1993. Seeding ‘one-dimensional crystallization’ of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73, 1055-1058. ( 10.1016/0092-8674(93)90635-4) [DOI] [PubMed] [Google Scholar]
- 17.Hardy J, Selkoe DJ. 2002. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353-356. ( 10.1126/science.1072994) [DOI] [PubMed] [Google Scholar]
- 18.Tomic JL, Pensalfini A, Head E, Glabe CG. 2009. Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol. Dis. 35, 352-358. ( 10.1016/j.nbd.2009.05.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glabe CG, Kayed R. 2006. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 66(suppl. 1), S74-S78. ( 10.1212/01.wnl.0000192103.24796.42) [DOI] [PubMed] [Google Scholar]
- 20.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. 1999. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860-866. () [DOI] [PubMed] [Google Scholar]
- 21.Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. 1999. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 48, 491-498. ( 10.2337/diabetes.48.3.491) [DOI] [PubMed] [Google Scholar]
- 22.Salahuddin P, Fatima MT, Uversky VN, Khan RH, Islam Z, Furkan M. 2021. The role of amyloids in Alzheimer's and Parkinson's diseases. Int. J. Biol. Macromol. 190, 44-55. ( 10.1016/j.ijbiomac.2021.08.197) [DOI] [PubMed] [Google Scholar]
- 23.Akter R, et al. 2016. Islet amyloid polypeptide: structure, function, and pathophysiology. J. Diabetes Res. 2016. ( 10.1155/2016/2798269) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin CY, Gurlo T, Kayed R, Butler AE, Haataja L, Glabe CG, Butler PC. 2007. Toxic human islet amyloid polypeptide (h-IAPP) oligomers are intracellular, and vaccination to induce anti-toxic oligomer antibodies does not prevent h-IAPP-induced β-cell apoptosis in h-IAPP transgenic mice. Diabetes 56, 1324-1332. ( 10.2337/db06-1579) [DOI] [PubMed] [Google Scholar]
- 25.Gurlo T, et al. 2010. Evidence for proteotoxicity in β cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am. J. Pathol. 176, 861-869. ( 10.2353/ajpath.2010.090532) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El Saghir A, Farrugia G, Vassallo N. 2021. The human islet amyloid polypeptide in protein misfolding disorders: mechanisms of aggregation and interaction with biomembranes. Chem. Phys. Lipids 234, 105010. ( 10.1016/j.chemphyslip.2020.105010) [DOI] [PubMed] [Google Scholar]
- 27.Haataja L, Gurlo T, Huang CJ, Butler PC. 2008. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 29, 303-316. ( 10.1210/er.2007-0037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. 2003. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486-489. ( 10.1126/science.1079469) [DOI] [PubMed] [Google Scholar]
- 29.Kayed R, Glabe CG. 2006. Conformation-dependent anti-amyloid oligomer antibodies. Methods Enzymol. 413, 326-344. ( 10.1016/S0076-6879(06)13017-7) [DOI] [PubMed] [Google Scholar]
- 30.Nag S, et al. 2011. Nature of the amyloid-β monomer and the monomer-oligomer equilibrium. J. Biol. Chem. 286, 13 827-13 833. ( 10.1074/jbc.M110.199885) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dear AJ, et al. 2020. Kinetic diversity of amyloid oligomers. Proc. Natl Acad. Sci. USA 117, 12 087-12 094. ( 10.1073/pnas.1922267117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Armen RS, DeMarco ML, Alonso DO, Daggett V. 2004. Pauling and Corey's α-pleated sheet structure may define the prefibrillar amyloidogenic intermediate in amyloid disease. Proc. Natl Acad. Sci. USA 101, 11 622-11 627. ( 10.1073/pnas.0401781101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armen RS, Alonso DO, Daggett V. 2004. Anatomy of an amyloidogenic intermediate: conversion of β-sheet to α-sheet structure in transthyretin at acidic pH. Structure 12, 1847-1863. ( 10.1016/j.str.2004.08.005) [DOI] [PubMed] [Google Scholar]
- 34.Childers MC, Daggett V. 2020. Edge strand dissociation and conformational changes in transthyretin under amyloidogenic conditions. Biophys. J. 119, 1995-2009. ( 10.1016/j.bpj.2020.08.043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Childers MC, Daggett V. 2019. Drivers of α-sheet formation in transthyretin under amyloidogenic conditions. Biochemistry 58, 4408-4423. ( 10.1021/acs.biochem.9b00769) [DOI] [PubMed] [Google Scholar]
- 36.Steward RE, Armen RS, Daggett V. 2008. Different disease-causing mutations in transthyretin trigger the same conformational conversion. Protein Eng. Design Selection 21, 187-195. ( 10.1093/protein/gzm086) [DOI] [PubMed] [Google Scholar]
- 37.Alonso DO, DeArmond SJ, Cohen FE, Daggett V. 2001. Mapping the early steps in the pH-induced conformational conversion of the prion protein. Proc. Natl Acad. Sci. USA 98, 2985-2989. ( 10.1073/pnas.061555898) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng CJ, Daggett V. 2014. Molecular dynamics simulations capture the misfolding of the bovine prion protein at acidic pH. Biomolecules 4, 181-201. ( 10.3390/biom4010181) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alonso DO, An C, Daggett V. 2002. Simulations of biomolecules: characterization of the early steps in the pH-induced conformational conversion of the hamster, bovine and human forms of the prion protein. Phil. Trans. R. Soc. Lond. A 360, 1165-1178. ( 10.1098/rsta.2002.0986) [DOI] [PubMed] [Google Scholar]
- 40.Van der Kamp MW, Daggett V.. 2010. Pathogenic mutations in the hydrophobic core of the human prion protein can promote structural instability and misfolding. J. Mol. Biol. 404, 732-748. ( 10.1016/j.jmb.2010.09.060) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van der Kamp MW, Daggett V.. 2010. Influence of pH on the human prion protein: insights into the early steps of misfolding. Biophys. J. 99, 2289-2298. ( 10.1016/j.bpj.2010.07.063) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeMarco ML, Daggett V. 2009. Characterization of cell-surface prion protein relative to its recombinant analogue: insights from molecular dynamics simulations of diglycosylated, membrane-bound human prion protein. J. Neurochem. 109, 60-73. ( 10.1111/j.1471-4159.2009.05892.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeMarco ML, Daggett V. 2007. Molecular mechanism for low pH triggered misfolding of the human prion protein. Biochemistry 46, 3045-3054. ( 10.1021/bi0619066) [DOI] [PubMed] [Google Scholar]
- 44.DeMarco ML, Silveira J, Caughey B, Daggett V. 2006. Structural properties of prion protein protofibrils and fibrils: an experimental assessment of atomic models. Biochemistry 45, 15 573-15 582. ( 10.1021/bi0612723) [DOI] [PubMed] [Google Scholar]
- 45.Armen RS, Daggett V. 2005. Characterization of two distinct β2-microglobulin unfolding intermediates that may lead to amyloid fibrils of different morphology. Biochemistry 44, 16 098-16 107. ( 10.1021/bi050731h) [DOI] [PubMed] [Google Scholar]
- 46.Kazmirski SL, Daggett V. 1998. Non-native interactions in protein folding intermediates: molecular dynamics simulations of hen lysozyme. J. Mol. Biol. 284, 793-806. ( 10.1006/jmbi.1998.2192) [DOI] [PubMed] [Google Scholar]
- 47.Armen RS, Bernard BM, Day R, Alonso DO, Daggett V. 2005. Characterization of a possible amyloidogenic precursor in glutamine-repeat neurodegenerative diseases. Proc. Natl Acad. Sci. USA 102, 13 433-13 438. ( 10.1073/pnas.0502068102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidlin T, Ploeger K, Jonsson AL, Daggett V. 2013. Early steps in thermal unfolding of superoxide dismutase 1 are similar to the conformational changes associated with the ALS-associated A4V mutation. Protein Eng. Design Selection 6, 503-513. ( 10.1093/protein/gzt030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidlin T, Kennedy BK, Daggett V. 2009. Structural changes to monomeric CuZn superoxide dismutase caused by the familial amyotrophic lateral sclerosis-associated mutation A4V. Biophys. J. 97, 1709-1718. ( 10.1016/j.bpj.2009.06.043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pauling L, Corey RB. 1951. Configurations of polypeptide chains with favored orientations around single bonds: two new pleated sheets. Proc. Natl Acad. Sci. USA 37, 729. ( 10.1073/pnas.37.11.729) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hayward S, Milner-White EJ. 2008. The geometry of α-sheet: implications for its possible function as amyloid precursor in proteins. Proteins 71, 415-425. ( 10.1002/prot.21717) [DOI] [PubMed] [Google Scholar]
- 52.Balupuri A, Choi KE, Kang NS. 2020. Aggregation mechanism of Alzheimer's amyloid β-peptide mediated by α-strand/α-sheet structure. Int. J. Mol. Sci. 21, 1094. ( 10.3390/ijms21031094) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balupuri A, Choi KE, Kang NS. 2019. Computational insights into the role of α-strand/sheet in aggregation of α-synuclein. Sci. Rep. 9, 1-13. ( 10.1038/s41598-018-37276-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hayward S, James Milner-White E. 2011. Simulation of the β-to α-sheet transition results in a twisted sheet for antiparallel and an α-nanotube for parallel strands: implications for amyloid formation. Proteins 79, 3193-3207. ( 10.1002/prot.23154) [DOI] [PubMed] [Google Scholar]
- 55.Wu H, Canfield A, Adhikari J, Huo S. 2010. Quantum mechanical studies on model α-pleated sheets. J. Comput. Chem. 31, 1216-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Babin V, Roland C, Sagui C. 2011. The α-sheet: a missing-in-action secondary structure? Proteins 79, 937-946. ( 10.1002/prot.22935) [DOI] [PubMed] [Google Scholar]
- 57.Daggett V. 2006. α-Sheet: the toxic conformer in amyloid diseases? Accounts Chem. Res. 39, 594-602. ( 10.1021/ar0500719) [DOI] [PubMed] [Google Scholar]
- 58.Milner-White JE, Watson JD, Qi G, Hayward S. 2006. Amyloid formation may involve α-to β sheet interconversion via peptide plane flipping. Structure 14, 1369-1376. ( 10.1016/j.str.2006.06.016) [DOI] [PubMed] [Google Scholar]
- 59.DeMarco ML, Daggett V. 2004. From conversion to aggregation: protofibril formation of the prion protein. Proc. Natl Acad. Sci. USA 101, 2293-2298. ( 10.1073/pnas.0307178101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Di Blasio B, Saviano M, Fattorusso R, Lombardi A, Pedone C, Valle V, Lorenzi GP.. 1994. A crystal structure with features of an antiparallel α-pleated sheet. Biopolymers 34, 1463-1468. [DOI] [PubMed] [Google Scholar]
- 61.Maris NL, Shea D, Bleem A, Bryers JD, Daggett V. 2018. Chemical and physical variability in structural isomers of an L/D α-sheet peptide designed to inhibit amyloidogenesis. Biochemistry 57, 507-510. ( 10.1021/acs.biochem.7b00345) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bi TM, Daggett V. 2018. Focus: medical technology: the role of α-sheet in amyloid oligomer aggregation and toxicity. Yale J. Biol. Med. 91, 247. [PMC free article] [PubMed] [Google Scholar]
- 63.Schneider F, Hammarström P, Kelly JW. 2001. Transthyretin slowly exchanges subunits under physiological conditions: a convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein Sci. 10, 1606-1613. ( 10.1110/ps.8901) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prapunpoj P, Leelawatwattana L. 2009. Evolutionary changes to transthyretin: structure–function relationships. FEBS J. 276, 5330-5341. ( 10.1111/j.1742-4658.2009.07243.x) [DOI] [PubMed] [Google Scholar]
- 65.Saldaño TE, Zanotti G, Parisi G, Fernandez-Alberti S. 2017. Evaluating the effect of mutations and ligand binding on transthyretin homotetramer dynamics. PLoS ONE 12, e0181019. ( 10.1371/journal.pone.0181019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guo X, et al. 2020. Review on the structures and activities of transthyretin amyloidogenesis inhibitors. Drug Design Dev. Therapy 14, 1057. ( 10.2147/DDDT.S237252) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wieczorek E, et al. 2021. Destabilisation of the structure of transthyretin is driven by Ca2+ Int. J. Biol. Macromol. 166, 409-423. ( 10.1016/j.ijbiomac.2020.10.199) [DOI] [PubMed] [Google Scholar]
- 68.Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. 2012. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J. Mol. Biol. 421, 185-203. ( 10.1016/j.jmb.2011.12.060) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beck DA, Alonso DO, Inoyama D, Daggett V. 2008. The intrinsic conformational propensities of the 20 naturally occurring amino acids and reflection of these propensities in proteins. Proc. Natl Acad. Sci. USA 105, 12259-12264. ( 10.1073/pnas.0706527105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Childers MC, Towse CL, Daggett V. 2016. The effect of chirality and steric hindrance on intrinsic backbone conformational propensities: tools for protein design. Protein Eng. Design Selection 29, 271-280. ( 10.1093/protein/gzw023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Towse CL, Hopping G, Vulovic I, Daggett V. 2014. Nature versus design: the conformational propensities of D-amino acids and the importance of side chain chirality. Protein Eng. Design Selection 27, 447-455. ( 10.1093/protein/gzu037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Towse CL, Rysavy SJ, Vulovic IM, Daggett V. 2016. New dynamic rotamer libraries: data-driven analysis of side-chain conformational propensities. Structure 24, 187-199. ( 10.1016/j.str.2015.10.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Childers MC, Towse CL, Daggett V. 2018. Molecular dynamics-derived rotamer libraries for D-amino acids within homochiral and heterochiral polypeptides. Protein Eng. Design Selection 31, 191-204. ( 10.1093/protein/gzy016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van der Kamp MW, et al. 2010. Dynameomics: a comprehensive database of protein dynamics. Structure 18, 423-435. ( 10.1016/j.str.2010.01.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beck DA, et al. 2008. Dynameomics: mass annotation of protein dynamics and unfolding in water by high-throughput atomistic molecular dynamics simulations. Protein Eng. Design Selection 21, 353-368. ( 10.1093/protein/gzn011) [DOI] [PubMed] [Google Scholar]
- 76.Kellock J, Hopping G, Caughey B, Daggett V. 2016. Peptides composed of alternating L-and D-amino acids inhibit amyloidogenesis in three distinct amyloid systems independent of sequence. J. Mol. Biol. 428, 2317-2328. ( 10.1016/j.jmb.2016.03.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shah K. 2018. Applications of microfluidic modulation spectroscopy for antibody-drug conjugate structural characterization. Am. Pharmaceut. Rev. 9, 120-125. [Google Scholar]
- 78.Bleem A, Prosswimmer T, Chen R, Hady T, Li J, Bryers J, Daggett V. 2022. Designed α-sheet peptides disrupt uropathogenic Escherichia coli biofilms by inhibiting amyloid fibril formation rendering the bacteria more susceptible to antibiotics and immune cells. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hsu CC, Templin AT, Shea D, Prosswimmer T, Li J, Brooks-Worrell B, Kahn SE, Daggett V. Submitted. Human islet amyloid polypeptide-induced b-cell cytotoxicity is linked to the formation of α-sheet structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Biancalana M, Koide S. 2010. Molecular mechanism of thioflavin-T binding to amyloid fibrils. Biochim. et Biophys. Acta (BBA) 1804, 1405-1412. ( 10.1016/j.bbapap.2010.04.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Paranjapye N, Daggett V. 2018. De novo designed α-sheet peptides inhibit functional amyloid formation of Streptococcus mutans biofilms. J. Mol. Biol. 430, 3764-3773. ( 10.1016/j.jmb.2018.07.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Penesyan A, Nagy SS, Kjelleberg S, Gillings MR, Paulsen IT. 2019. Rapid microevolution of biofilm cells in response to antibiotics. NPJ Biofilms Microb. 5, 1-14. ( 10.1038/s41522-019-0108-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Manna S, Ghanty C, Baindara P, Barik TK, Mandal SM. 2020. Electrochemical communication in biofilm of bacterial community. J. Basic Microbiol. 60, 819-827. [DOI] [PubMed] [Google Scholar]
- 84.Shea D, et al. In press. SOBA: development and testing of a soluble oligomer binding assay for detection of amyloidogenic toxic oligomers. Proc. Natl Acad. Sci. USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hayward S, Milner-White EJ. 2021. Determination of amino acids that favour the αL region using Ramachandran propensity plots. Implications for α-sheet as the possible amyloid intermediate. J. Struct. Biol. 213, 107738. ( 10.1016/j.jsb.2021.107738) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.