

Abstract
Despite an estimated heritability of ~50%, genome-wide association studies of opioid use disorder (OUD) have revealed few genome-wide significant loci. We conducted a cross-ancestry meta-analysis of OUD in the Million Veteran Program (N = 425,944). In addition to known exonic variants in OPRM1 and FURIN, we identified intronic variants in RABEPK, FBXW4, NCAM1 and KCNN1. A meta-analysis including other datasets identified a locus in TSNARE1. In total, we identified 14 loci for OUD, 12 of which are novel. Significant genetic correlations were identified for 127 traits, including psychiatric disorders and other substance use-related traits. The only significantly enriched cell-type group was CNS, with gene expression enrichment in brain regions previously associated with substance use disorders. These findings increase our understanding of the biological basis of OUD and provide further evidence that it is a brain disease, which may help to reduce stigma and inform efforts to address the opioid epidemic.
OUD is a problematic pattern of opioid use that leads to substantial impairment or distress1. In the United States, a tenfold increase in opioid analgesic prescriptions between 1990 and 2010 contributed to an epidemic of opioid misuse, abuse and overdose deaths2–4. By 2019, 3.7% of US adults reported past-year opioid misuse and 0.6% met criteria for an OUD5. Overdose deaths, which continue to increase annually, have reached crisis proportions6, reflecting the limitations of available preventive and treatment efforts.
Genetic studies can help inform our understanding of the biology underlying OUD. However, although the estimated heritability (h2) of OUD based on twin and family studies is ~50%7, few genetic associations have been identified. Genome-wide association studies (GWAS) of OUD, opioid dependence (OD) or related phenotypes have yielded inconsistent results, likely due to the limited sample size of the discovery datasets and different case and control definitions8–11.
The use of data from electronic health records (EHRs) linked to biobanks has provided increasingly large GWAS samples. An EHR-based study of 1,039 OUD cases identified two genome-wide significant (GWS) loci, with a single-nucleotide polymorphism (SNP)-based heritability of 6.0%12. A meta-analysis of African Americans (AAs; 5,212 OUD cases) and European Americans (EAs; 10,544 OUD cases) based largely on data from the Million Veteran Program (MVP), identified a single GWS SNP, rs1799971 in OPRM1, in EAs only, with SNP-based heritability of 11.3%13. In this study, there were no GWS findings in AAs or in a cross-ancestry meta-analysis. A study that combined data from multiple cohorts (20,858 OUD cases), including an earlier release of MVP data, identified two GWS loci—a variant within OPRM1 in a cross-ancestry analysis, and an additional variant in FURIN in a European-ancestry (EUR) meta-analysis14.
Two recent GWAS have increased sample sizes for genetic discovery by examining opioid-related phenotypes other than OUD. A GWAS of prescription opioid misuse in a EUR sample from 23andMe (27,805 cases) identified two novel GWS loci15. A meta-analysis of EUR individuals including 23,367 cases ascertained using either Diagnostic and Statistical Manual of Mental Disorders (DSM) diagnoses or frequency of opioid use16 identified GWS SNPs in OPRM1 and, in gene-based analyses, PPP6C and FURIN.
These studies also identified significant genetic correlations (rgs) with traits well known to co-occur with OUD, suggesting widespread pleiotropy. The strongest positive rgs were with substance-related traits12,13,16 and psychiatric disorders13,16. Negative rgs were seen for educational attainment13,16. and subjective wellbeing16. Causal effects on OUD for some of these traits were identified via Mendelian randomization (MR) analysis13. Positive causal effects on OUD were found for regular tobacco smoking, major depressive disorder (MDD) and neuroticism. A negative causal effect on OUD was seen for educational attainment. It was not possible to examine the causal effect of OUD on these traits due to the limited number of GWS variants.
The different phenotypes used in these studies reflect the difficulty of ascertaining a large, multi-ancestry, well-characterized sample for use in GWAS of opioid-related phenotypes. EHR-based traits generally use International Classification of Disease (ICD) diagnostic codes for phenotyping OUD (for example, see refs. 12,13). Cohorts recruited from some nonclinical biobanks rely on single-item, self-report questionnaires (for example, see ref. 15) or have combined multiple case and control definitions derived as latent variables in genomic structural equation modeling (SEM)16. A key consideration in selecting OUD cases, particularly given the high prevalence of opioid use in the United States, is the stringency of the definition. More stringent case definitions increase confidence in the specificity of the diagnosis and, by reducing heterogeneity, may increase statistical power. However, they also reduce the sample size and have the potential to reduce generalizability by not capturing a disorder’s full range of presentations (for example, by misclassifying cases as subthreshold).
Here, we conducted a cross-ancestry meta-analysis of OUD that included AA, EA and Hispanic American (HA) participants recruited from the MVP that maximized OUD cases by using a less stringent definition (requiring the presence of a single OUD diagnostic code) and compared them to opioid-exposed controls (Ncases = 31,473 and Ncontrols = 394,471). In supplementary analyses, we compared our results to those using a stringent OUD phenotype in MVP (Ncases = 23,459 and Ncontrols = 394,471), and performed a meta-analysis that combined data from the MVP, Yale-Penn (data not shown), the Partners HealthCare Biobank12 and the Psychiatric Genomics Consortium (PGC11; Ncases = 37,761 and Ncontrols = 409,760).
Results
Sample description.
Our MVP sample comprised 425,944 individuals (AA, 88,498; EA, 302,585; HA, 34,861), of whom 90.6% were male (Supplementary Table 1). The less stringent OUD definition yielded 28.8–38.9% more cases across the ancestral groups (AA, 8,968; EA, 19,978; HA, 2,527) than the stringent definition (AA, 6,457; EA, 15,040; HA, 1,962). In total, 2,525 (8%) of the less-stringent cases and 1,926 (8%) of the stringent cases had no opioid prescription fills. Among the individuals with a single OUD code (N = 8,014), 599 (7%) had no opioid prescription fills. Of the remaining individuals with an opioid prescription, less-stringent cases had 77.2 (s.d. = 96.9) opioid prescription fills, stringent cases had 76.5 (s.d. = 97.6) fills, and controls had 25.0 (s.d. = 48.3) fills. Thus, most individuals with an OUD diagnosis had documented prescriptions for opioids. Further, individuals with a single diagnosis code for OUD (that is, less stringent) had a similar number of opioid fills as those with the stringent diagnosis. Finally, the documented exposure to prescription opioids was similar for OUD cases defined using the less stringent diagnosis and those defined using the stringent diagnosis.
Identification of novel loci associated with opioid use disorder.
The cross-ancestry meta-analysis of the less stringent OUD diagnosis within the MVP sample yielded 12 GWS variants, 10 of which were independent after conditioning on the lead variant within each locus (Fig. 1 and Supplementary Table 3). The protein-coding genes nearest these variants are CDKAL1, BTNL2 and OPRM1 (all on chromosome 6), RABEPK (chromosome 9), FBXW4 (chromosome 10; a second locus on chromosome 10 had no protein-coding gene within 500 kb), NCAM1 (chromosome 11), FURIN (chromosome 15), KCNN1 (chromosome 19) and RNF114 (chromosome 20). The most robust signal was in OPRM1 (lead SNP rs1799971, P = 6.78 × 10−10), which replicates the main finding of the previous MVP OUD GWAS13. The variant in FURIN is supported by previous findings in variant-level14 and gene-based16 analyses. In addition, there were 3 ancestry-specific loci (Supplementary Table 4): 1 each in AAs (NNT, chromosome 5), EAs (CDH8, chromosome 16) and HAs (MRS2, chromosome 8).
Fig. 1 |. Manhattan and quantile–quantile plot for cross-ancestry meta-analysis of opioid use disorder (Ncases = 31,480 and Ncontrols = 394,484).
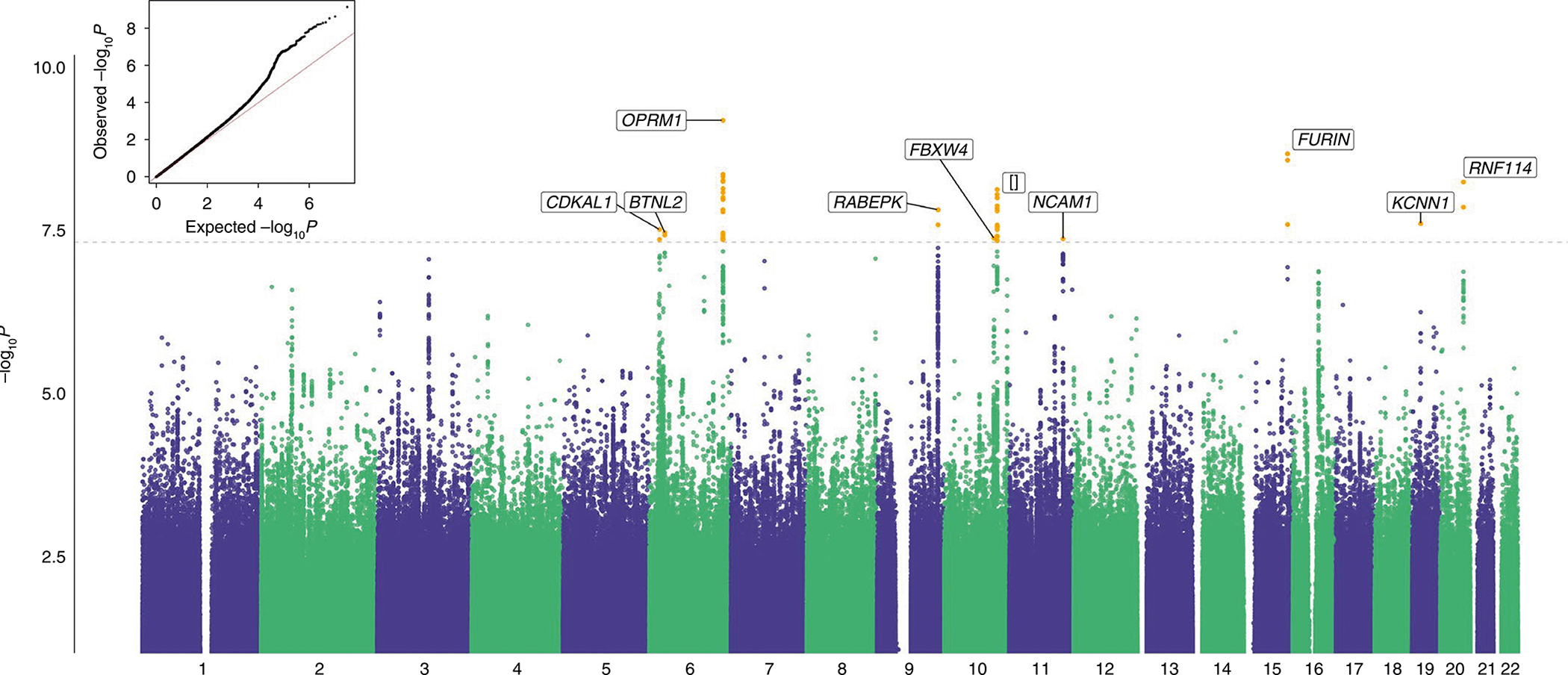
Meta-analyses with effective sample size weighting were performed in METAL. The nearest protein-coding gene (<1 Mb) in each locus is labeled. [] represents an intergenic locus. The dashed line indicates GWS after correction for multiple testing (P < 5 × 10−8).
Replication of loci associated with opioid use disorder.
The cross-ancestry meta-analysis of the stringent OUD diagnosis in the MVP sample also identified the variants in OPRM1 and FURIN and one additional locus (TSNARE1, chromosome 8; Supplementary Table 5). The cross-ancestry meta-analysis of all datasets (MVP, Partners HealthCare Biobank, PGC and Yale-Penn 3 (YP3)) identified no additional loci (Supplementary Table 8). GWS loci from all analyses are presented in Supplementary Tables 3–8. Based on all analyses, we identified a total of 14 GWS loci, 12 of which were novel (Table 1).
Table 1 |.
Summary of the 14 genome-wide significant loci identified in GWAS analyses of opioid use disorder
| Chr | Position (GRCh37/hg19) of lead SNPs | Nearest genes | GWAS analysis |
|---|---|---|---|
| 5 | 43846681 | NNT | AA (less stringent) |
| 6 | 21362610, 21478361 | CDKAL1, SOX4 | Cross-ancestry (less stringent), HA (less stringent), cross-ancestry (meta) |
| 6 | 32383573 | BTNL2 | Cross-ancestry (less stringent) |
| 6 | 154360797, 154380719, 154382139, 154393680, 154396472 | OPRM1 | Cross-ancestry (less stringent), EA (less stringent), cross-ancestry (stringent), EA (stringent), cross-ancestry (meta), EA (meta) |
| 6 | 24394925 | MRS2 | HA (less stringent) |
| 8 | 143312933, 143316970 | TSNARE1 | Cross-ancestry (stringent), EA (stringent), EA (meta) |
| 9 | 127873473, 127959540, 127980426 | SCAI, RABEPK | Cross-ancestry (less stringent), cross-ancestry (meta), EA (meta) |
| 10 | 103414885 | FBXW4 | Cross-ancestry (less stringent), EA (less stringent), EA (meta) |
| 10 | 110504365 | [] | Cross-ancestry (less stringent) |
| 11 | 112869404 | NCAM1 | Cross-ancestry (less stringent) |
| 15 | 91410009, 91406146, 91426560 | FURIN | Cross-ancestry (less stringent), EA (less stringent), cross-ancestry (stringent), EA (stringent), cross-ancestry (meta), EA (meta) |
| 16 | 61631362 | CDH8 | EA (less stringent), EA (stringent) |
| 19 | 18093588 | KCNN1 | Cross-ancestry (less stringent), AA (less stringent), cross-ancestry (meta), AA (meta) |
| 20 | 48540277, 48583726 | RNF114 | Cross-ancestry (less stringent), cross-ancestry (meta) |
Chr, chromosome number.
Single-nucleotide polymorphism heritability and genetic correlations across GWAS datasets.
In MVP, similar estimates of SNP heritability (h2SNP ± s.e.) were obtained for the less stringent phenotype in AAs (0.11 ± 0.03) and EAs (0.12 ± 0.01). Estimates of h2SNP for the stringent OUD phenotype were slightly higher (AA, 0.20 ± 0.05; EA, 0.15 ± 0.01), and estimates were slightly lower for the ancestry-specific meta-analyses across datasets (AA, 0.08 ± 0.03; EA, 0.11 ± 0.01). Variation in these estimates appears to be driven by changes in effective sample size, as estimates using the actual sample size show little variation (Supplementary Table 9). Using a two-sample t-test, we found no significant difference in h2snp across the ancestral groups (MVP less stringent phenotype, P = 0.69; MVP stringent phenotype, P = 0.35; and ancestry-specific meta-analysis, P = 0.40).
The cross-ancestry rg between MVP AA and EA populations was 0.43 (s.e. = 0.21, P = 6.83 × 10−3) for the less stringent diagnosis, and 0.48 (s.e. = 0.23, P = 2.58 × 10−2) for the stringent diagnosis. The within-ancestry rg (± s.e.) between datasets was high, ranging from 0.66 (± 0.3) between the less stringent OUD MVP and Partners HealthCare Biobank datasets in EAs, to 1.2 (± 0.2) between the less stringent OUD MVP and the previous OUD MVP GWAS13 in EAs (which used the same diagnosis definition as the present stringent analysis) (Supplementary Table 10). Because the h2SNP of the PGC and Yale-Penn datasets was low, we did not calculate rgs between MVP and either of these datasets. A sign test showed that the majority of SNPs with P < 1 × 10−5 (N SNPs, AFR = 400 and EUR = 954) had the same direction of effect in both MVP and other datasets, with the exception of MVP AA and PGC AFR (AFR, MVP-PGC 21.7%, P = 2.2 × 10−16; MVP-YP3 60.1%, P = 3.1 × 10−3; EUR, MVP-PGC 61.1%, P = 1.07 × 10−9; MVP-YP3 65.1%, P = 2.2 × 10−16; MVP-Partners 74.5%, P = 2.2 × 10−16).
Considering the similarity in h2SNP between the different OUD GWAS and the greater number of loci captured by the less stringent diagnosis in MVP, all downstream analyses were based on the GWAS for the less stringent OUD case definition in EAs within the MVP sample.
Partitioning heritability enrichment.
We performed partitioning heritability enrichment analyses using linkage disequilibrium score regression (LDSC)17 and examined heritability enrichment for gene expression using Genotype–Tissue Expression (GTEx) data18. In the baseline model, genomic evolutionary rate profiling19 functional annotation was significantly enriched (P = 6.7 × 10−4), suggesting that SNPs included in the analyses are under stronger negative selection (Supplementary Table 11). The only significantly enriched cell-type group was CNS (P = 3.34 × 10−3; Fig. 2a and Supplementary Table 12). We observed significant enrichment for OUD in brain tissues only, including the anterior cingulate cortex (P = 4.71 × 10−6), limbic system (P = 3.25 × 10−5), prefrontal cortex (P = 5.73 × 10−5), cerebral cortex (P = 9.81 × 10−5), cortex (P = 1.11 × 10−4), hypothalamus (P = 1.23 × 10−4), amygdala (P = 1.41 × 10−4) and hippocampus (P = 2.04 × 10−4; Fig. 2b and Supplementary Table 13). There were no significant enrichments for epigenetic annotations after correction for multiple testing (Supplementary Table 14).
Fig. 2 |. Enrichment of opioid use disorder in the brain.
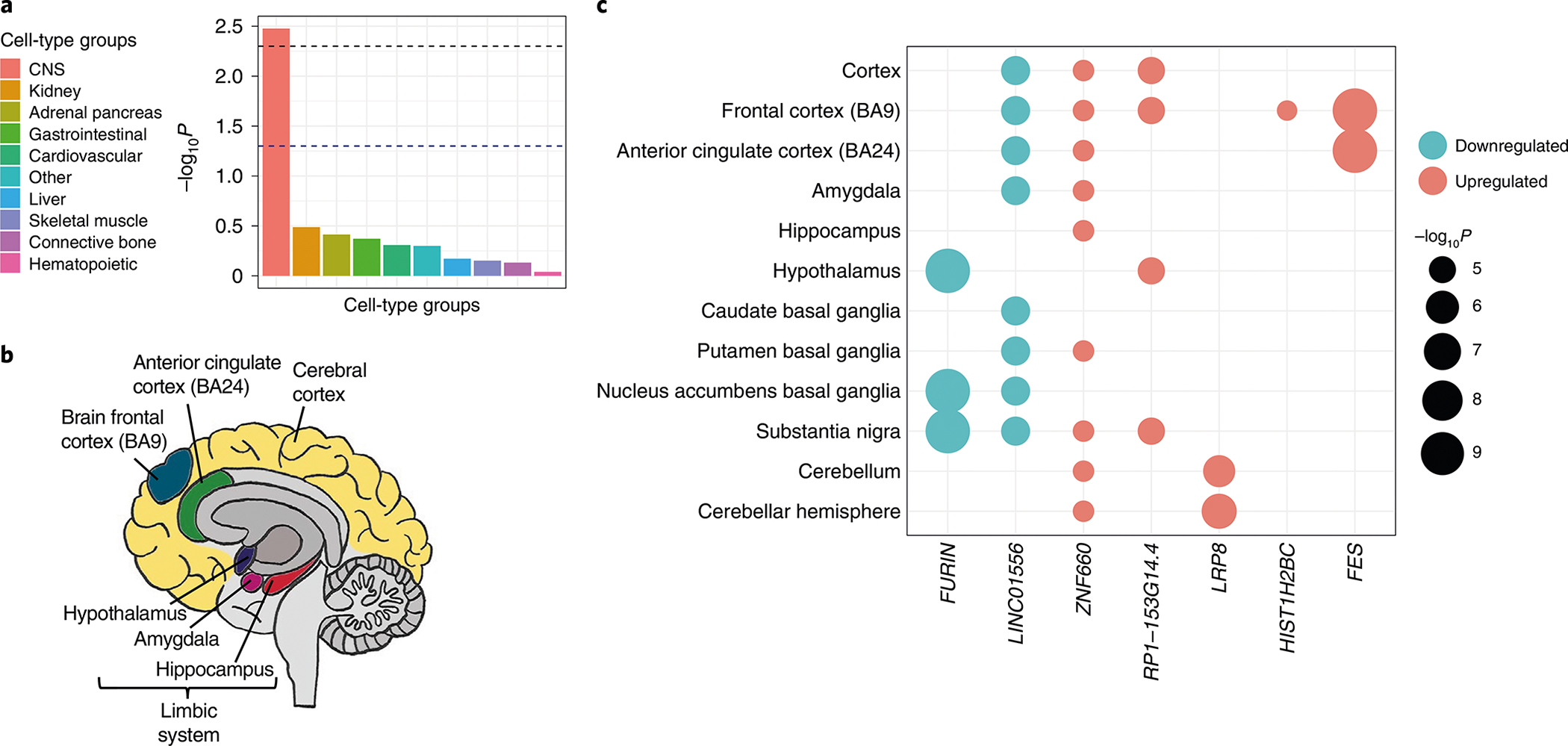
a, Partitioning heritability enrichment analyses using LDSC show enrichment for OUD in the CNS. The dashed black lines indicate Bonferroni-corrected significance (P < 0.005). b, Heritability enrichment analyses for gene expression using GTEx data show enrichment for OUD in brain regions previously associated with addiction. c, Predicted gene expression using S-PrediXcan identified genes with differential expression in brain regions. Color of circle indicates downregulation (blue) or upregulation (red). Size of circle indicates −log10P. Bonferroni correction was applied within each tissue conditioned on the number of genes tested.
Transcriptome-wide analysis.
We used S-PrediXcan20 to predict the effect of genetic variation on gene expression. Significant within-tissue gene expression regulation was identified for 43 tissues, including brain, adipose, gastrointestinal, thyroid and liver (Supplementary Fig. 2 and Supplementary Tables 15 and 16). Significant associations with expression in brain tissues were detected for FURIN, FES, LRP8, LINC01556, ZNF660 and RP1-153G14.4 (Fig. 2c). Some of these genes (FURIN, LINC01556, ZNF660 and RP1-153G14.4) were also expressed in non-brain tissues, such as adipose, gastrointestinal and thyroid (Supplementary Fig. 2), suggesting that OUD-related genetic variation may exert significant transcriptomic changes in the periphery as well as the CNS.
Considering the sharing of expression quantitative trait loci (eQTLs) across multiple tissues, we tested the joint effects of variation in gene expression across tissues using S-MultiXcan21. Significant transcriptomic effects for OUD were detected in eight genes, five of which overlapped with genes detected by S-PrediXcan (FURIN, FES, RP1-153G14.4, LRP8 and RABEPK) and three of which were novel (ZNF391, ZKSCAN4 and MAGOH) (Supplementary Table 17). We also observed that the lead SNP in RABEPK was in high linkage disequilibrium (LD) with PPP6C variants (r2 > 0.8) that are significantly associated with gene expression and chromatin interaction, especially in the prefrontal cortex (Supplementary Fig. 3).
Using summary-based MR (SMR) and Brain-eMeta data22, we found that FURIN (beta = −0.13) and PPP6C (beta = 0.09) passed the SMR (false discovery rate (FDR) q < 0.05) and HEIDI (HEIDI P > 0.05) causality tests, consistent with the genes being associated with OUD via their regulation of brain mRNA expression levels (Supplementary Table 18). We also found that OPRM1 expression was causal for OUD when the variant rs3778151 was used as an instrument (beta = −0.21, FDR q = 0.03, HEIDI P = 0.06). In the cerebellum (which has high levels of OPRM1 expression in GTEx), expression was causal for OUD using either variant as an instrument (FDR q < 0.05). However, the most significant variant (rs1799971) failed the heterogeneity test (HEIDI P = 1.75 × 10−4). This suggests that the effect of rs1799971 is functional rather than mediated by gene expression, consistent with it being a nonsynonymous substitution. This contrasts with rs3778151, which appears to exert its causal effect via gene expression (HEIDI P = 0.07).
Gene-set, functional enrichment and drug repurposing analyses.
MAGMA gene-based analyses identified one GWS gene in AAs (CHRM2, P = 9.52 × 10−7) and three GWS genes in EAs (OPRM1, P = 2.17 × 10−7; FTO, P = 9.52 × 10−7; DRD2, P = 1.67 × 10−6; Supplementary Fig. 4), but none in HAs. GCTA-fastBAT gene-based analyses identified two GWS genes in EAs (OPRM1, P = 3.14 × 10−8; BTRC, P = 3.21 × 10−7), but none in AAs or HAs. Following Bonferroni correction, no biological processes or pathways were significantly enriched, although nominal associations in EAs highlighted pathways of potential relevance, including ‘dopamine receptors’ (P = 1.87 × 10−5) and ‘regulation of adenylate cyclase activating G-protein-coupled receptor signaling pathway’ (P = 4.39 × 10−5; Supplementary Table 19).
Genes identified in the variant-level, gene-based or transcriptome (brain region) analyses (N = 24) are summarized in Supplementary Table 20. Examination of these genes for drug–gene interactions via the Drug Gene Interaction database identified 761 interactions between 8 genes (CHRM2, DRD2, FES, FURIN, KCNN1, NCAM1, OPRM1 and PRL) and 340 unique medications (Supplementary Table 21 and Supplementary Fig. 5). OPRM1 had 193 interactions, mainly with classes of analgesics, anesthetics and drugs for constipation. DRD2 had 376 interactions, most of which were with psycholeptics.
Genetic correlations.
We estimated pairwise rgs with OUD for 40 published phenotypes using LDSC23. OUD showed significant rgs with 21 traits. As expected, the strongest positive correlations were with substance use traits (for example, problematic alcohol use: rg = 0.70, cannabis use disorder: rg = 0.65, ever smoked regularly: rg = 0.44) and psychiatric disorders (for example, bipolar disorder: rg = 0.32, MDD: rg = 0.29). The strongest negative correlation (rg = −0.27) was with educational attainment (Fig. 3a and Supplementary Table 22). We also assessed rgs of OUD with 1,270 complex traits from the UK Biobank using the Complex-Trait Genetics Virtual Lab (CTG-VL)24. After multiple-testing correction (P = 3.94 × 10−5), OUD was significantly associated with 106 traits (Supplementary Fig. 6 and Supplementary Table 23). These included positive correlations with substance use-related traits (for example, current smoking: rg = 0.44; ever addicted to any substance or behavior: rg = 0.67), psychiatric traits (for example, anxiety treatment: rg = 0.41, self-reported depression: rg = 0.35) and pain-related traits (for example, low back pain: rg = 0.44, multisite chronic pain: rg = 0.26), and negative correlations with having secondary education qualifications (rg = −0.34) and the presence of social support (rg = −0.36). Thus, overall, we found that increased risk of OUD is genetically correlated with increased liability for use of substances, psychiatric disorders and experiencing pain, and lower likelihood of educational attainment and social support.
Fig. 3 |. Phenotypic spectrum associated with opioid use disorder.
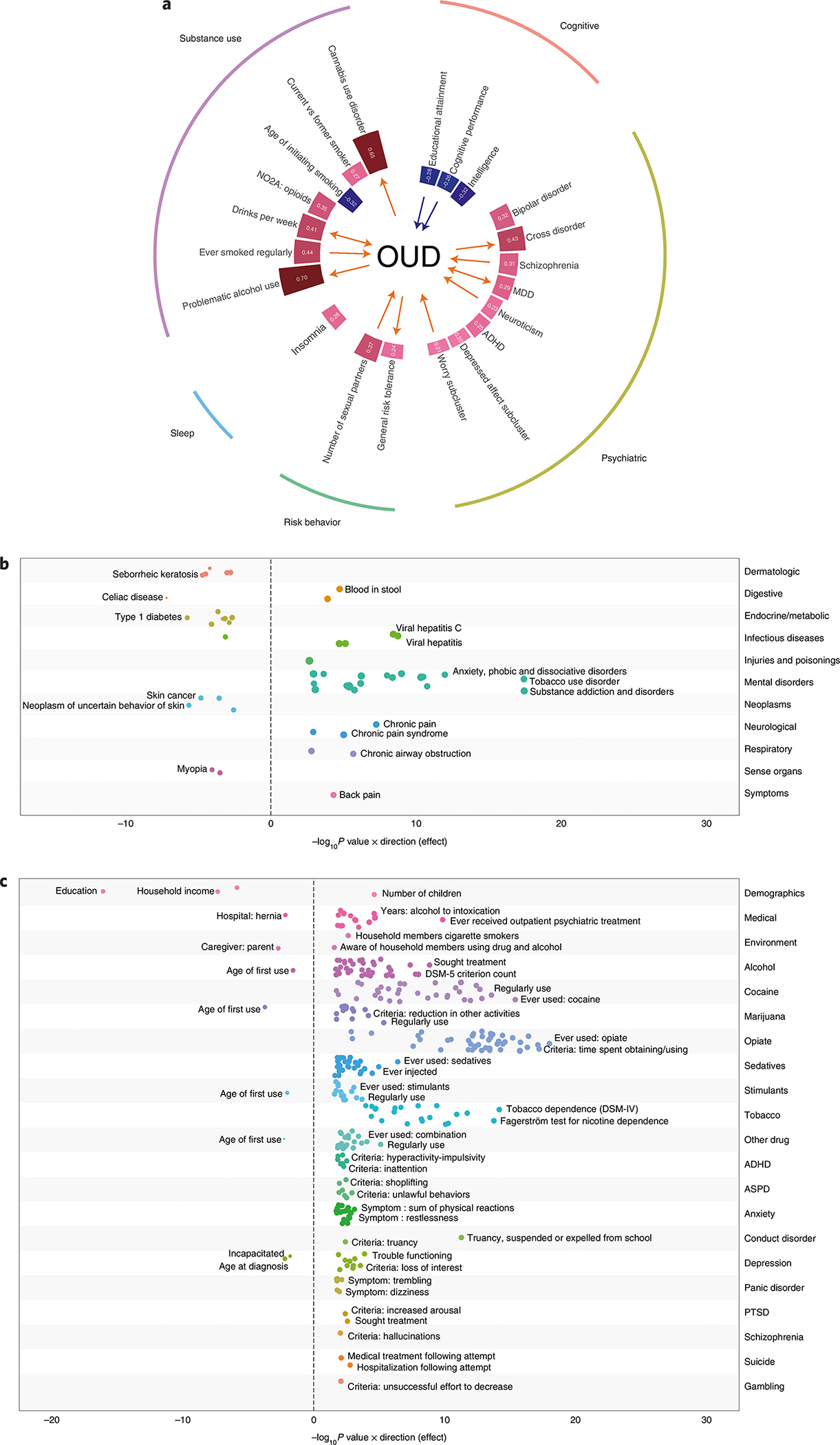
a, Genetic correlation analyses showed multiple traits significantly genetically correlated with OUD following Bonferroni correction (P < 1.25 × 10−3; red bars indicate positively correlated traits, while blue bars indicate negatively correlated traits). MR analyses identify causal associations between OUD and other traits (red arrows denote a positive causal association, blue arrows denote a negative causal association). b,c, PheWAS results in BioVU (b) and Yale-Penn (c) datasets. All phenotypes significant at an FDR of q < 0.05 are plotted. In b, all phenotypes that passed Bonferroni correction (P < 3.7 × 10−5) are labeled. For readability, in c, only the top three traits within each group that passed Bonferroni correction (P < 7.9 × 10−5) are labeled. Circle size denotes effect size. ASPD, antisocial personality disorder; PTSD, post-traumatic stress disorder.
Mendelian randomization.
Using MR, we tested for bidirectional causal effects between OUD and the 21 traits identified as significantly genetically correlated with OUD (Fig. 3a and Supplementary Fig. 7). There was a causal effect of OUD on 6 traits: problematic alcohol use, drinks per week, cannabis use disorder, general risk tolerance, MDD and cross disorder. Among the 21 traits, 9 had a causal effect on OUD, of which 2 showed a negative causal effect on OUD (cognitive performance and educational level) and 7 showed a positive causal effect on OUD (in descending order of magnitude: drinks per week, worry subcluster, neuroticism, number of sexual partners, MDD, cigarettes per day and schizophrenia).
Polygenic risk scores and phenome-wide association studies.
Polygenic risk scores (PRSs) were calculated in two independent datasets to identify phenotypic associations of genetic liability for OUD. In the Yale-Penn sample, PRSs were calculated for 4,918 AA individuals and 5,692 EA individuals. No significant associations were identified for AAs (Supplementary Fig. 8 and Supplementary Table 24). In EAs, phenome-wide association studies (PheWAS) identified 43 phenotypes in the opiate domain and 78 phenotypes in other phenotypic domains that were significantly associated with OUD PRSs (Fig. 3c and Supplementary Table 25). The most significantly associated phenotypes were ‘ever used opioids’ and ‘time spent obtaining/using opioids’. In Vanderbilt University Medical Center’s (VUMC) Biobank (BioVU), PRSs were calculated for 12,384 AAs and 66,903 EAs. No significant associations were found for OUD PRS in AAs (Supplementary Fig. 9 and Supplementary Table 26). In EAs, the OUD PRS was associated with 27 phenotypes, including ‘substance addiction and disorders’ and ‘mood disorders’ (Fig. 3b and Supplementary Table 27).
Genomic structural equation modeling.
We conducted genomic SEM to evaluate how OUD relates to the 3 other substance use traits and the 7 psychiatric disorders identified as the most significantly associated with OUD in genetic correlation analyses. Exploratory factor analysis involving all 11 traits supported a four-factor model with cumulative variance of 0.639. We retained paths with a loading factor ≥ 0.2 and conducted confirmatory factor analysis. In this analysis, the four-factor model fit the data well (comparative fit index of 0.948, Akaike information criterion of 340.840, χ2 = 276.840, d.f. = 34, standard root mean root square error = 0.073). The 4 substance use traits all loaded on factor 1, with a major contribution from OUD (0.84 ± 0.05) and problematic alcohol use (0.91 ± 0.3), and lower contributions from cannabis use disorder (0.58 ± 0.06) and ever smoked regularly (0.40 ± 0.03). Cannabis use disorder (0.37 ± 0.06) and ever smoked regularly (0.42 ± 0.03), together with other psychiatric disorders, also loaded on factor 3. Major psychiatric disorders, including bipolar disorder (0.86 ± 0.04), schizophrenia (0.76 ± 0.03) and MDD (0.43 ± 0.03) loaded on factor 2. Tourette’s syndrome (TS; 0.33 ± 0.07) and obsessive-compulsive disorder (OCD; 1.03 ± 0.21) loaded on factor 4 (Fig. 4 and Supplementary Table 28).
Fig. 4 |. Genomic SEM analysis of ouD with other substance use traits and psychiatric disorders.
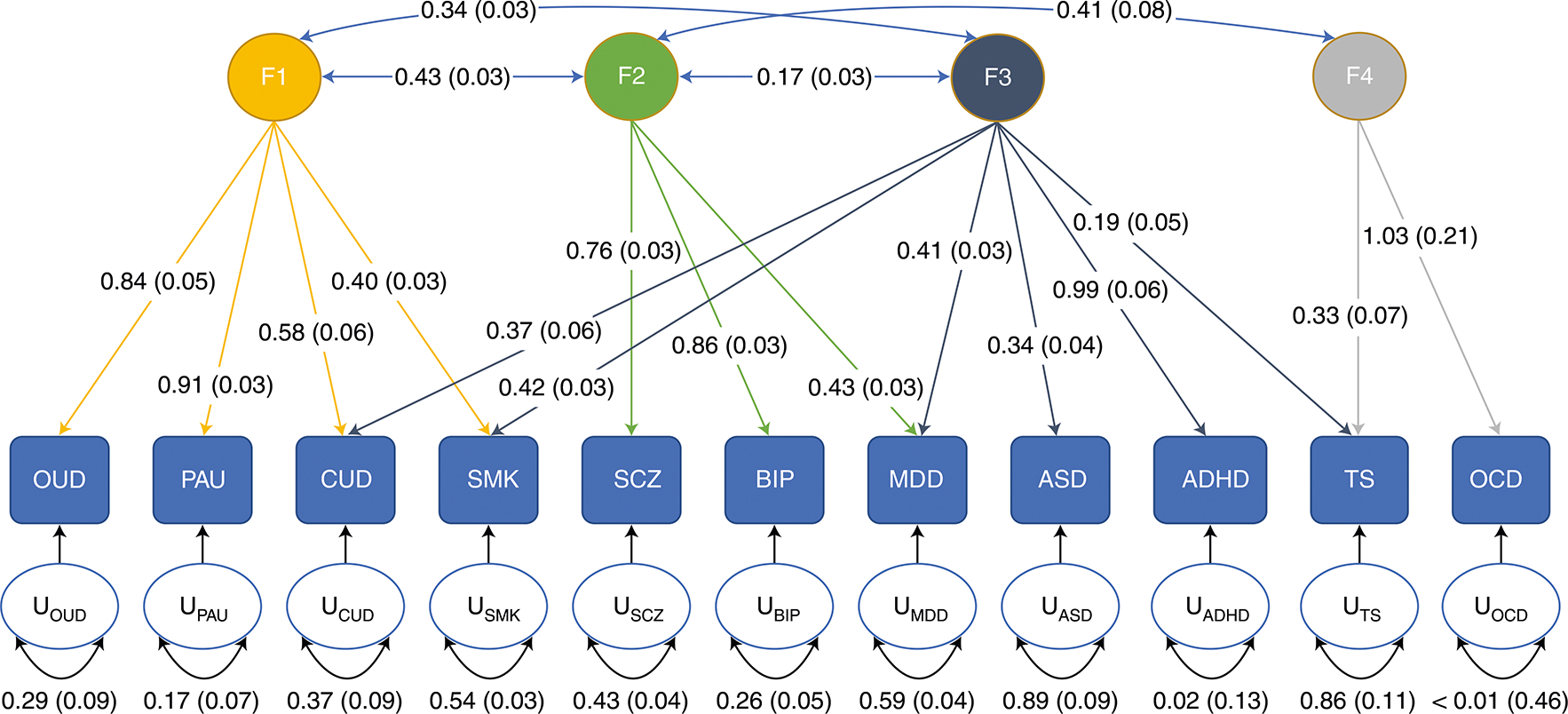
Analysis of OUD with PAU, problematic alcohol use; CUD, cannabis use disorder; SMK, ever smoked regularly; SCZ, schizophrenia; BIP, bipolar disorder; MDD, major depressive disorder; ASD, autism spectrum disorder; ADHD, attention deficit hyperactivity disorder; TS, Tourette’s syndrome; and OCD, obsessive-compulsive disorder identified four factors. Factor loadings for each trait are depicted by arrows between the trait and the factor. Correlation between factors is indicated by arrows between the factors. Residual variance for each trait is indicated by the U-circles. Standard errors are depicted in parentheses.
Discussion
This study, the largest single-sample GWAS of OUD to date, identified 14 loci associated with the disorder, 12 of which are novel findings. Three of these loci were significant in ancestry-specific analyses only, demonstrating that inclusion of diverse ancestral samples in genetic studies of OUD permits the identification of novel genetic variants. Post-GWAS analyses in EAs revealed enrichment for OUD in the CNS, particularly the brain, and an extensive phenotypic spectrum associated with genetic liability for OUD.
Because the effect sizes of common variants contributing to highly polygenic phenotypes such as OUD are small, large sample sizes are required to identify GWS loci. The largest OUD GWAS before the current study greatly increased the effective sample size (Neffective = 88,115) by conducting meta-analyses of the results of studies that used a range of case and control definitions16. Here, we performed GWAS using the stringent definition of OUD used by Zhou et al.13 (Neffective = 88,569) and a less stringent definition requiring the presence of only one ICD Ninth Revision (ICD-9) or Tenth Revision (ICD-10) diagnostic code for opioid abuse or dependence (Neffective = 116,590). Although the less stringent definition lowers the specificity of the case phenotyping (that is, individuals are more likely to be mislabeled as having OUD), it increases the number of cases by more than 8,000, reveals eight more GWS variants than the stringent definition, and as denoted by the high genetic correlation between the two definitions, has a similar polygenic architecture. These results support previous conclusions that the potential variability introduced by broadening phenotypic definitions in genetic studies of OUD is outweighed by substantial increases in sample size16. In contrast, our meta-analysis of the MVP data with other datasets reduced the number of GWS loci identified, potentially because the smaller additional datasets increased the variability in the effect size of variants.
The most significant locus, OPRM1, encodes the mu-opioid receptor, which binds morphine and other opioids and has been the focus of many functional and candidate gene studies of opioid-related phenotypes25–27. In a previous GWAS comprised principally of participants from MVP, OUD was significantly associated only with OPRM1 in EAs13, with the lead SNP being the nonsynonymous, exon 1 variant rs1799971 (A118G). In neither that study, nor the present study, was the SNP associated with OUD in AAs, presumably because the minor (G) allele frequency in this population group is considerably lower than in EAs28. Even so, it is difficult to explain why meta-analysis with AAs does not increase the statistical strength of the association of OUD with this variant if it is truly the lead functional variant, even if based on introgressed EA alleles alone.
We identified a second peak in OPRM1, with the lead SNP rs3778151, a variant in intron 1 that is in high LD with rs9478500 (r2 = 0.56–0.90)29, the variant associated with opioid addiction in a recent meta-analysis16. A previous study in EA alcohol-dependent or drug-dependent cases and controls also identified two independent LD blocks in OPRM1 (ref. 30). Our SMR analyses suggest a plausible role for both variants in OUD: a functional effect for the nonsynonymous substitution rs1799971 and an effect on gene expression by rs3778151.
Our cross-ancestry analysis identified a SNP in FURIN, with transcriptome-wide analyses showing significant downregulation of gene expression in brain-related tissues. These findings support the reported associations of OUD with FURIN both in gene-based analyses14,16 and in a variant-level meta-analysis14. Although FURIN encodes a protease that cleaves some endogenous opioids31, the enzyme has not previously been linked to the effects of exogenous opioids or mu-opioid receptor signaling. Given these findings, further research on the mechanism underlying the gene’s effects on risk of OUD is warranted.
Our analysis also identified 12 novel GWS loci. Two of these, in RABEPK and NCAM1, were GWS in a multi-trait analysis using GWAS of OUD with cannabis use disorder and alcohol use disorder14. Here, we show associations directly with OUD. RABEPK is adjacent to PPP6C, a gene previously implicated in a gene-level analysis of OUD16 that has also been linked to reward-related phenotypes like obesity and smoking32,33. Our analysis shows that the lead SNP in RABEPK is tagging PPP6C variants that affect gene expression and chromatin interaction. Furthermore, SMR analyses show that expression changes in PPP6C are causal for OUD. These lines of evidence suggest that PPP6C is likely the causal gene in this locus. NCAM1 and KCNN1 have been implicated in the neuropharmacology of opioid-related phenotypes. The mouse homolog of KCNN1 is differentially expressed in the nucleus accumbens following chronic morphine exposure34, and downregulated in the rodent prelimbic cortex after exposure to cues associated with morphine withdrawal35. NCAM1 also appears to be involved in the response to morphine exposure. Tolerance in rodents due to repeated morphine injection can be prevented by treatment with an antisense oligodeoxynucleotide that targets Ncam1 (ref. 36). NCAM1 variants have also been significantly associated with other substance use traits33,37–39.
Several other loci contain GWS hits for other traits, suggesting widespread pleiotropy of loci associated with OUD. CDKAL1 and BTNL2 have been associated with metabolic traits such as type 2 diabetes, body mass index40 and obesity-related phenotypes32. FBXW4 and CDH8 have previous associations with cognitive traits such as educational attainment and mathematical ability41, and TSNARE1 has a previous association with schizophrenia42.
Partitioning heritability enrichment analyses showed that CNS cells were the only significantly enriched group. We found significant enrichment for OUD in brain tissues only, including regions previously associated with the underlying neurobiology of the disorder43. These findings underscore the neural basis of OUD and reinforce the conceptualization of substance use disorders, which are often chronic and relapsing, as brain diseases. This notion was novel when proposed nearly 25 years ago44 and although today it is a view widely held by neuroscientists and clinicians, it is not universally understood by politicians or the general public. Improving our understanding of the biological basis of OUD could promote a science-based response to the opioid epidemic.
Consistent with previous findings, OUD showed strong genetic correlations with multiple substance use disorders, psychiatric disorders, cognitive traits and risk behaviors13,16,45. MR analyses demonstrated causal effects of OUD on problematic alcohol use and cannabis use disorder, and a bidirectional causal effect with drinks per week. These findings have both theoretical and clinical implications for the ‘gateway hypothesis’ of addiction liability, which posits that substance use starts with a legal substance and progresses on to the use of hard drugs, such as opioids. A more compelling explanation for the high rate of comorbidity of OUD with other substance-related traits is common genetic liability or pleiotropic effects46, which is supported here by the robust genetic correlations between OUD and other substance-related traits, the causal effects of OUD on other substance use, and the latent addiction factor identified through genomic SEM.
Genetic liability for psychiatric traits, including neuroticism and schizophrenia, was also causally associated with OUD, with a bidirectional causal effect of MDD on OUD. Our findings, along with those of others13, suggest that OUD has a common biological pathway with schizophrenia and MDD. Despite the significant genetic correlations and causal associations between OUD and psychiatric disorders, genomic SEM indicated a common genetic factor representing broad genetic liability for substance use disorders that is distinct from those underlying the psychiatric disorders. The factor structure among psychiatric disorders seen here is consistent with previous findings47 and shows that cannabis use disorder and smoking, unlike OUD, load onto both the substance use disorder factor and the factor underlying MDD, attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder (ASD) and TS.
PheWAS of the genetic liability for OUD in the Yale-Penn sample, which was ascertained for substance use disorders, reproduced the broad association with other substance use. In a clinical dataset using EHR data, the genetic liability for OUD was associated with multiple traits in every phenotypic domain tested, demonstrating the widespread effects of OUD liability on bodily systems. Some of the associations may be due to phenotypic correlation. For instance, associations were found with viral hepatitis and human immunodeficiency virus, potential proxies of injection drug use, and with chronic pain and back pain, potential proxies for the use of analgesic medications. Negative associations with obesity, type 1 diabetes and skin cancer could reflect underreporting or underdiagnosis in individuals with OUD, or they could reflect true biological relationships. The lack of genetic correlation between obesity and OUD (Supplementary Table 22) argues against a biological relationship between the two.
Limitations to the present study should be noted. Although it includes AA, EA and HA individuals, participants of European ancestry make up more than 60% of the total sample, which in large part drove the results of the cross-ancestry analysis. This disparity in sample size is also reflected in analyses of the individual ancestral groups, in which the smaller AA and HA groups provided less statistical power and yielded fewer significant loci. The lower power of the AA GWAS is also reflected in the lack of associations in PRS analysis in AAs. Future GWAS of OUD should focus on expanding sample sizes for populations of non-European ancestry to capture loci that are relevant to specific population groups. The sample for this study is >90% male, reflecting the sex distribution of veterans in the United States. Risk variants relevant only to women may thus have been overlooked due to the lower statistical power. Because the MVP dataset lacks information on the initiation of opioid use among individuals diagnosed with OUD, we could not differentiate participants who developed the disorder only after being prescribed opioid analgesics from those whose first opioid use involved recreational use of analgesics or heroin. Differences in the initiation of opioid use could reflect different genetic risk factors contributing to nonoverlapping intermediate phenotypes (for example, pain threshold/susceptibility in analgesic use versus risk taking in recreational use). Finally, a study of the validity of incident OUD diagnoses in the US Department of Veterans Affairs (VA) EHR data showed that 26% of diagnoses were erroneous, attributable to administrative errors (77%) or clinical ones (23%)48. Such false positive errors, however, are likely to bias the findings to the null, rather than contribute to false positive findings.
In summary, we have identified 14 genetic loci associated with OUD, the majority of which are novel. Many of the loci contain genes with previous associations with substance use or psychiatric disorders, suggesting widespread pleiotropy. The use of a less stringent definition of OUD allowed 25% more OUD cases than a stringent definition in the MVP sample. Downstream analyses validate this approach by demonstrating plausible enrichment of OUD in brain regions, genetic correlations with other substance use disorders and psychiatric disorders, and association between OUD PRSs and OD in an independent sample. Our findings provide insight into the biological underpinnings of OUD, which could inform preventive, diagnostic and therapeutic efforts and thereby help to address the opioid epidemic.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41593-022-01160-z.
Methods
Overview of analyses.
We conducted an ancestry-specific GWAS using a less stringent OUD case definition in AAs, EAs and HAs from the MVP, followed by a cross-ancestry meta-analysis. Cases had received at least one lifetime ICD-9 or ICD-10 diagnosis of OUD and control participants were exposed to opioids. Further details on phenotyping are described below.
In a supplementary analysis, we performed within-ancestry meta-analyses for AAs and EAs from the MVP, Yale-Penn (data not shown), the Partners HealthCare Biobank12 and the PGC11, followed by a cross-ancestry meta-analysis that included all samples. In a second supplementary analysis, we repeated the GWAS in MVP with the more stringent case definition used in the previous MVP OUD GWAS13. Most subsequent downstream analyses are based on the GWAS for the less stringent OUD case definition in EAs within the MVP sample, with the exception being the use of the stringent definition in the same population group to estimate its heritability and to calculate genetic correlations between the less-stringent and stringent traits. An overview of the analyses is provided in Supplementary Fig. 1.
Million Veteran Program cohort.
As of September 2021, the MVP49 had recruited approximately 850,000 veterans at 63 VA medical centers nationwide. All participants provided written informed consent and a blood sample for DNA extraction and genotyping and gave permission to access their EHRs for research purposes. The MVP was approved by the Central Veterans Affairs Institutional Review Board (IRB) and all site-specific IRBs. All relevant ethical regulations for work with human participants were followed in the conduct of the study.
Phenotypes.
OUD diagnostic codes based on ICD-9/10 were obtained from the VA EHRs. The main GWAS used a less stringent definition of OUD (N = 31,473), which required the presence of one inpatient or outpatient ICD-9/10 diagnostic code for OUD (304.0, 304.7, 305.5, F11.1, F11.2) in the EHR. The stringent definition (N = 23,459), used in the supplementary GWAS, required at least one inpatient or two outpatient ICD-9/10 OUD diagnostic codes in the EHR. Controls (N = 394,471) for all GWAS were defined as individuals with at least one outpatient opioid analgesic prescription fill (that is, exposed to opioids, but excluding prescriptions for OUD treatment (for example, buprenorphine or methadone)) and no ICD-9/10 diagnosis code for OUD documented in the EHR. Demographics are presented in Supplementary Table 1.
Genotyping and imputation.
The genotyping of samples in the MVP is ongoing and, in this analysis, we used release 4 imputed data. MVP samples were genotyped with a custom Affymetrix Axiom Biobank Array. Quality control of genotype data and subsequent imputation were performed by the MVP Genomics Working Group. Duplicate samples were removed, as were those with a sex mismatch, seven or more relatives in MVP (kinship > 0.08), excessive heterozygosity or a genotype call rate < 98.5%. Variants were removed if they were monomorphic, had a missing call rate < 0.8 or had a Hardy–Weinberg equilibrium of P < 1 × 10−6 both in the entire sample using a principal-component analysis (PCA)-adjusted method and within one of the three major ancestral groups (AA, EA and HA). Genotypes were phased with SHAPEIT4 (v.4.1.3)50 and imputed using Minimac4 software51, with biallelic SNPs imputed using the African Genome Resources reference panel by the Sanger Institute (which includes all samples from 1000 Genomes Project phase 3, version 5 reference panel52, together with 1,500 unrelated pan-African samples), and non-biallelic SNPs and indels imputed in a secondary imputation using the 1000 Genomes Project phase 3, version 5 reference panel52. Indels and complex variants from the second imputation were merged into the African Genome Resources imputation.
We removed one individual from each pair of related individuals at random (kinship > 0.08, N = 31,010). Genetic ancestry was unified with self-identified race/ethnicity using the HARE (Harmonizing Genetic Ancestry and Self-Identified Race/Ethnicity) method53. Quality control of imputed variants was performed within each ancestral group. Genetic variants were excluded based on minor allele frequency (MAF, AA < 0.005; EA < 0.001; HA < 0.01), genotype call rate < 0.95, and Hardy–Weinberg equilibrium P < 1 × 10−6 or a population-specific imputation quality (INFO) score < 0.7. Genome-wide association analyses were performed using PLINK (v.2.0)54 and a logistic regression model. Covariates included sex, age at enrollment and the first ten genetic principal components (PCs) within each ancestry.
Datasets for meta-analysis.
Supplementary Table 2 summarizes the datasets used for meta-analysis. Summary statistics for GWAS of OUD were obtained from two previously published datasets: (1) Partners HealthCare Biobank, which used the same less stringent case definition and opioid-exposed controls in individuals of European ancestry12, and (2) PGC, which used a DSM-IV OD diagnosis and opioid-exposed controls in individuals of African and European ancestry11. We also included the YP3 unpublished dataset (Yale-Penn 1 and 2 were included in PGC analyses). In YP3, we conducted a GWAS using cases with a DSM-IV OD diagnosis and opioid-exposed controls. For AAs, there were 168 cases and 153 controls; for EAs, there were 578 cases and 219 controls. We used GEMMA to conduct an association analysis to account for relatedness between the individuals. Sex, age at enrollment and the first ten PCs were included as covariates. We used a sign test to compare the direction of effects for SNPs in MVP and the other three datasets. SNPs with P < 1 × 10−5 from MVP AFR results (400 SNPs) or from MVP EUR results (954 SNPs) were evaluated for the direction of their signs in PGC, Partners and YP3 results. We used a binomial test to evaluate the null hypothesis that 50% of SNPs have the same effect direction in independent datasets.
Meta-analysis and independent variants.
Meta-analyses were conducted using a sample size-weighted method in METAL55 due to substantial differences in sample sizes. To compensate for the imbalance in the ratio of cases to controls, effective sample sizes were calculated using the formula: 4/(1/n_case + 1/n_control). Effective sample sizes were used in all meta-analyses and all downstream analyses. Meta-analyses were conducted across the following datasets: (1) cross-ancestry (AA, EA and HA) meta-analysis within MVP, comprising 31,473 less-stringent OUD cases and 394,471 controls and 23,459 stringent cases and 394,471 controls; (2) within-ancestry meta-analysis across datasets (AA: MVP (8,968 less-stringent cases and 79,530 controls), PGC (1,297 less-stringent cases and 1,291 controls), YP3 (168 less-stringent cases and 153 controls); EA: MVP (19,978 less-stringent cases and 282,607 controls), Partners HealthCare Biobank (Partners: 1,038 less-stringent cases and 10,744 controls), PGC (3,272 less-stringent cases and 2,876 controls), YP3 (578 less-stringent cases and 219 controls)); (3) cross-ancestry meta-analysis across all datasets (AA (MVP, PGC, YP3; 10,433 less-stringent cases and 80,974 controls); EA (MVP, Partners HealthCare Biobank, PGC, YP3; 24,866 less-stringent cases and 296,446 controls); HA (MVP; 2,527 less-stringent cases and 32,334 controls)).
To identify independent variants, we performed LD clumping within each ancestry using a range of 3,000 kb, r2 > 0.1, and the matched 1000 Genomes52 reference panel. Following clumping, variants that were located <1 Mb apart were merged into a single locus. For loci that contained multiple variants, we conducted conditional analyses using COJO in GCTA56. Within each locus, we conditioned on the most significant variant. Upon conditioning, variants within the locus that remained significant (P < 5 × 10−8) were considered independent.
Single-nucleotide polymorphism-based heritability analyses and partitioning heritability enrichment.
We used LDSC23 to estimate OUD (less-stringent and stringent case definitions) SNP-based heritability (h2SNP) in AAs and EAs for common SNPs mapped to HapMap3 (ref. 57). To ensure matching of population LD structure, we used pre-computed LD scores based on the African and European 1000 Genomes Project phase 3 (ref. 52). SNPs in the major histocompatibility complex region were excluded. Because of the high degree of genetic admixture in HAs and the smaller size of the sample, we did not estimate h2SNP in that population group.
We used LDSC to partition h2SNP in the OUD EA dataset and examined the enrichment of the partitioned h2SNP based on different functional genomic annotation models17,58. In the baseline model, we examined 75 overlapping functional annotations comprising genomic, epigenomic and regulatory features (see ref. 17 for details). Next, we analyzed ten overlapping cell-type groups derived from 220 cell-type-specific annotations in four histone marks: methylated histone H3 Lys4 (H3K4me1), trimethylated histone H3 Lys4 (H3K4me3), acetylated histone H3 Lys4 (H3K4ac) and H3K27ac (see ref. 17 for details). Finally, enriched cell-type categories were analyzed based on annotations obtained from H3K4me1-imputed, gapped peak data generated by the Roadmap Epigenomics Mapping Consortium59 (see ref. 58. for details). For each h2SNP partitioning model, multi-allelic and major histocompatibility complex region variants were excluded, and Bonferroni correction was applied to identify significant enrichment.
Gene-based, functional enrichment and pathway analyses.
We performed gene-based association testing for OUD in FUMA (v1.3.6a)60, with MAGMA (v1.08)61, which uses multiple regression models to detect multiple marker effects that account for SNP P values and LD between markers, using the matched-ancestry 1000 Genomes Project phase 3 panel52 as LD reference. We used 18,707 protein-coding genes, with P < 2.67 × 10−6 (0.05/18,707) considered GWS. We also conducted a separate gene-based analysis with GCTA-fastBAT, which included 26,292 genes62. We tested the genetic architecture of selected lead SNPs by integrating our GWAS results with brain-related GTEx v7 and chromatin interaction data in FUMA.
To identify gene sets enriched for OUD, we used MAGMA61 to curate gene sets, Gene Ontology terms (obtained from MsigDB c2) and GWAS-catalog enrichment, correcting for gene size, variant density and LD within and between genes. We also used MAGMA to test the association between gene-set properties and tissue-specific gene expression profiles using GTEx (v.7) data from 53 tissues (Bonferroni-corrected P-value threshold of 9.43 × 10−4).
Transcriptome-wide association analyses.
We performed transcriptome-wide association analyses using the MetaXcan framework20 and the GTEx release v.8 eQTL MASHR-M models63. Forty-nine tissues from GTEx v.8 were analyzed comprising 12,951 samples. First, GWAS summary statistics were harmonized for the EA population based on the human genome assembly GRCh38 (hg38) and linked to the 1000 Genomes reference panel using GWAS tools, as previously described20. A transcriptome-wide association analysis of 49 tissues was run using S-PrediXcan20. A Bonferroni correction for statistical significance was applied within each tissue conditioned on the number of genes tested (Supplementary Table 15).
Because eQTLs were correlated across tissues, we integrated gene expression signals for 49 tissue panels using S-MultiXcan21 and tested 10,552 genes in total. Resulting P values were Bonferroni corrected to identify significant gene associations (P-value threshold = 4.74 × 10−6).
To examine whether the effects of GWS variants associated with OUD are mediated by changes in gene expression patterns, we performed SMR analyses64 using brain cis-eQTL summary data (Brain-eMeta22) obtained from a meta-analysis of ten brain regions in GTEx (v6)65, and dorsolateral prefrontal cortex in CMC66 and ROSMAP67. We also conducted SMR analyses for individual brain tissues generated from GTEx (v8)63. We considered causal genes to be those with a P value below an FDR threshold of 5% and no evidence of pleiotropy (HEIDI P > 0.05).
Drug interactions.
To identify drugs that could potentially be repurposed to treat OUD, we examined genes identified in the variant-level or gene-based analyses using the Drug Gene Interaction Database68 (https://www.dgidb.org/). Medications were categorized using the Anatomical Therapeutic Chemical classification system, retrieved from the Kyoto Encyclopedia of Genes and Genomics (https://www.genome.jp/kegg/drug/).
Genetic correlation.
We used LDSC23 to calculate the rg between (a) OUD or OD datasets used for meta-analysis (AA (MVP, PGC, YP3); EA (MVP, Partners HealthCare Biobank, PGC, YP3)) and (b) OUD (MVP EA) and 40 other published psychiatric, substance use, cognitive and anthropometric traits selected based on a priori hypotheses (see Supplementary Table 20 for a full list), using pre-computed LD scores for HapMap3 (ref. 57) SNPs based on the matched-ancestry 1000 Genomes Project phase 3 reference panel52. To explore additional traits in a hypothesis-free manner, we also estimated the rg between OUD and 1,270 traits (comprising published and unpublished traits from the UK Biobank using the CTG-VL (https://genoma.io/). CTG-VL integrates publicly available GWAS summary statistics and utilizes the LDSC framework to calculate rg between complex traits and diseases of interest24. A Bonferroni correction was applied within each LDSC and CTG-VL analysis, and traits with a corrected P < 0.05 were regarded as significantly correlated.
We also estimated the trans-ancestry rgs for OUD in the MVP between the AA and EA populations using the Popcorn package, a computationally efficient method that uses summary-level data from GWAS while accounting for LD69. We used African and European 1000 Genomes Project phase 3 (ref. 52) data as the LD references.
Mendelian randomization.
We performed MR analysis using the MendelianRandomization package in R. Causal relationships between OUD and other traits were tested bidirectionally using three methods: weighted median, inverse-variance weighted and MR-Egger. We tested for pleiotropy using the MR-Egger intercept test. Instrumental variants were those associated with the exposure at P < 1 × 10−5. When the instrumental variants were not present in the outcome data, we identified the best-proxy variant (LD > 0.8). Variants with MAF < 0.01 or with no proxy with LD > 0.8 within 200 kb were removed. Each trait included more than 20 instrumental variables, which provides a robust estimate of causal effects. We considered causal effects as those for which at least two MR tests were significant after Bonferroni correction and that showed no evidence of violation of the horizontal pleiotropy test (MR-Egger intercept P > 0.05).
Polygenic risk scores and phenome-wide association studies.
We calculated PRSs for OUD in two independent datasets (Yale-Penn and BioVU) using PRS-continuous shrinkage (PRS-CS)70, followed by PheWAS. In each dataset, OUD summary statistics from the matched ancestry were used to calculate PRSs. Details for the analysis in each dataset are below.
Yale-Penn.
We removed SNPs with an INFO score < 0.7, a MAF < 0.01, a genotype call rate < 0.95 or an allele frequency difference between genotyping batches > 0.4, which left a total of 8,811,422 SNPs. We removed one individual from each pair of related individuals with pi-hat > 0.25. To estimate genetic ancestry, we calculated PCs on common SNPs between Yale-Penn and the 1000 Genomes Project phase 3 (ref. 52) using the --pca flag in PLINK (v.1.9)54. Participants were assigned to an ancestry based on the distance of 10 PCs from the 1000 Genomes reference populations. The resulting dataset included 4,918 AAs and 5,692 EAs. We excluded binary phenotypes with fewer than either 100 cases or 100 controls, and continuous phenotypes with fewer than 100 individuals. We conducted PheWAS by fitting logistic regression models for binary traits and linear regression models for continuous traits. We used sex, age at enrollment and the top 10 genetic PCs as covariates. We applied a Bonferroni correction to control for multiple comparisons.
BioVU.
We used de-identified clinical data from BioVU. Details on the quality control process have been described elsewhere71. The genotyping information that we used was from the Illumina MEGAEX array. Genotypes were filtered for SNP and individual call rates, sex discrepancies and excessive heterozygosity using PLINK (v1.9)54. Imputation of the autosomes was conducted using the Michigan Imputation Server51 based on the Haplotype Reference Consortium reference panel. PCA using FlashPCA2 combined with CEU, YRI and CHB reference sets from 1000 Genomes Project phase 3 (ref. 52) was conducted to determine participants of African and European ancestry. The sample was then filtered for cryptic relatedness by removing one individual of each pair for which pi-hat > 0.2. This yielded samples from 12,384 individuals of African ancestry and 66,903 individuals of European ancestry for analysis. We conducted PheWAS by fitting a logistic regression for each of the 1,335 disease phenotypes available in BioVU to estimate the odds of a diagnosis of that phenotype given the OUD PRS. Each disease phenotype (commonly referred to as ‘phecode’; https://phewascatalog.org/phecodes/, Phecode Map 1.2) was classified using ICD-9/10 diagnostic codes to establish ‘case’ status. For an individual to be considered a case, they were required to have two ICD codes for the index phenotype, and each phenotype needed at least 100 cases to be included in the analysis. The covariates included in the analyses were sex, median age of the longitudinal EHR measurements and the top 10 genetic PCs. The project was approved by the VUMC IRB (nos. 160302, 172020 and 190418).
Genomic structural equation modeling.
To establish whether there is a shared genetic structure between OUD, other substance use disorders and psychiatric disorders, we performed genomic SEM72 for OUD, three other substance use traits (problematic alcohol use73, cannabis use disorder74 and ever smoked regularly33), and seven psychiatric disorders (schizophrenia75, bipolar disorder76, MDD77, ASD78, ADHD79, TS80 and OCD81). We calculated a genetic covariance matrix using multivariable LDSC and the 1000 Genomes Project phase 3 European samples52 as a reference. An exploratory factor analysis was conducted using the genetic covariance matrix and a four-latent-factor structure with varimax rotation. We used the determined structure containing paths with a loading factor > 0.2 to perform a confirmatory factor analysis implemented in the GenomicsSEM package in R. To prevent negative residual variance after estimation, we restricted the residual variance of OCD and ADHD to greater than 0.
Supplementary Material
Acknowledgements
This work was supported by Merit Review Awards from the US Department of Veterans Affairs Biomedical Laboratory Research and Development Service (no. I01 BX003341 (to A.C.J. and H.R.K.)) and Clinical Science Research and Development Service (no. I01 CX001734 (to K.M.K.)); the VISN 4 Mental Illness Research, Education and Clinical Center (to H.R.K.); NIAAA grant K01 AA028292 (to R.L.K.); and NIDA grant DA046345 (to H.R.K.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The views expressed in this article are those of the authors and do not necessarily represent the position or policy of the Department of Veterans Affairs or the US Government.
Footnotes
Competing interests
H.R.K. is a member of advisory boards for Dicerna Pharmaceuticals, Sophrosyne Pharmaceuticals and Enthion Pharmaceuticals; a consultant to Sobrera Pharmaceuticals; the recipient of research funding and medication supplies for an investigator-initiated study from Alkermes, and a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which was supported in the last three years by Alkermes, Dicerna, Ethypharm, Lundbeck, Mitsubishi and Otsuka. J.G. and H.R.K. are holders of US patent no. 10,900,082 titled: ‘Genotype-guided dosing of opioid agonists,’ issued 26 January 2021. The other authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41593-022-01160-z.
Peer review information Nature Neuroscience thanks Ditte Demontis and Andrew McIntosh for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
Reporting summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
Imputation was performed using Minimac3 (https://genome.sph.umich.edu/wiki/Minimac3). GWAS was performed using PLINK2 (https://www.cog-genomics.org/plink2). Meta-analyses were performed using METAL (https://genome.sph.umich.edu/wiki/METAL_Documentation). GCTA (https://cnsgenomics.com/software/gcta/#Overview) was used for identification of independent loci (GCTA-COJO) and gene-based analysis (GCTA-fastBAT). FUMA (https://fuma.ctglab.nl/) was used for gene association, functional enrichment and gene-set enrichment analyses. Transcriptomic analyses were performed using S-PrediXcan and S-MultiXcan (https://github.com/hakyimlab/MetaXcan). LDSC (https://github.com/bulik/ldsc) was used for heritability estimation, genetic correlation analysis (also using the CTG-VL; https://genoma.io) and heritability enrichment analyses. Trans-ancestry genetic correlation was estimated using Popcorn (https://github.com/brielin/Popcorn). PRS analyses were performed using PRS-CS (https://github.com/getian107/PRScs). PheWAS analyses were run using the PheWAS R package (https://github.com/PheWAS/PheWAS). The MendelianRandomization R package (https://cran.r-project.org/web/packages/MendelianRandomization/index.html) was used for MR analyses. Genomic SEM was conducted using the GenomicsSEM R package (https://github.com/GenomicSEM/GenomicSEM).
Data availability
The full summary-level association data from the meta-analysis are available through dbGaP at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001672.
References
- 1.American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders (American Psychiatric Association, 2013); 10.1176/appi.books.9780890425596 [DOI] [Google Scholar]
- 2.Strang J et al. Opioid use disorder. Nat. Rev. Dis. Primers 6, 3 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Vowles KE et al. Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis. Pain 156, 569–576 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention Vital signs: overdoses of prescription opioid pain relievers and other drugs among women–United States, 1999–2010. MMWR Morb. Mortal. Wkly Rep. 62, 537–542 (2013). [PMC free article] [PubMed] [Google Scholar]
- 5.Key Substance Use and Mental Health Indicators in the United States: Results from the 2019 National Survey on Drug Use and Health (Substance Abuse and Mental Health Services Administration, 2020); https://www.samhsa.gov/data/sites/default/files/reports/rpt29393/2019NSDUHFFRPDFWHTML/2019NSDUHFFR090120.htm [Google Scholar]
- 6.Wilson N Drug and opioid-involved overdose deaths—United States, 2017–2018. MMWR Morb. Mortal. Wkly Rep. 69, 290–297 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kendler KS, Jacobson KC, Prescott CA & Neale MC Specificity of genetic and environmental risk factors for use and abuse/dependence of cannabis, cocaine, hallucinogens, sedatives, stimulants, and opiates in male twins. Am. J. Psychiatry 160, 687–695 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Gelernter J et al. Genome-wide association study of opioid dependence: multiple associations mapped to calcium and potassium pathways. Biol. Psychiatry 76, 66–74 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng Z et al. Genome-wide association study identifies a regulatory variant of rgma associated with opioid dependence in European Americans. Biol. Psychiatry 84, 762–770 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson EC et al. Evidence of CNIH3 involvement in opioid dependence. Mol. Psychiatry 21, 608–614 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polimanti R et al. Leveraging genome-wide data to investigate differences between opioid use vs. opioid dependence in 41,176 individuals from the Psychiatric Genomics Consortium. Mol. Psychiatry 25, 1673–1687 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song W et al. Genome-wide association analysis of opioid use disorder: a novel approach using clinical data. Drug Alcohol Depend. 217, 108276 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou H et al. Association of OPRM1 functional coding variant with opioid use disorder: a genome-wide association study. JAMA Psychiatry 77, 1072–1080 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deak JD et al. Genome-wide association study and multi-trait analysis of opioid use disorder identifies novel associations in 639,709 individuals of European and African ancestry. Mol. Psychiatry 10.1038/s41380-022-01709-1 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez-Roige S et al. Genome-wide association study of problematic opioid prescription use in 132,113 23andMe research participants of European ancestry. Mol. Psychiatry 10.1038/s41380-021-01335-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaddis N et al. Multi-trait genome-wide association study of opioid addiction: OPRM1 and beyond. Preprint at medRxiv 10.1101/2021.09.13.21263503v1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davydov EV et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6, e1001025 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barbeira AN et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbeira AN et al. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 15, e1007889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi T et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat. Commun. 9, 2282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bulik-Sullivan BK et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuellar-Partida G et al. Complex-Trait Genetics Virtual Lab: a community-driven web platform for post-GWAS analyses. Preprint at bioRxiv 10.1101/518027 (2019). [DOI] [Google Scholar]
- 25.Crist RC & Berrettini WH Pharmacogenetics of OPRM1. Pharmacol. Biochem. Behav. 123, 25–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crist RC, Reiner BC & Berrettini WH A review of opioid addiction genetics. Curr. Opin. Psychol. 27, 31–35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moningka H, Lichenstein S & Yip SW Current understanding of the neurobiology of opioid use disorder: an overview. Curr. Behav. Neurosci. Rep. 6, 1–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gelernter J, Kranzler H & Cubells J Genetics of two mu opioid receptor gene (OPRM1) exon I polymorphisms: population studies, and allele frequencies in alcohol- and drug-dependent subjects. Mol. Psychiatry 4, 476–483 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Howe KL et al. Ensembl 2021. Nucleic Acids Res. 49, D884–D891 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang H et al. Association between two μ-opioid receptor gene (OPRM1) haplotype blocks and drug or alcohol dependence. Hum. Mol. Genet. 15, 807–819 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breslin MB et al. Differential processing of proenkephalin by prohormone convertases 1(3) and 2 and furin. J. Biol. Chem. 268, 27084–27093 (1993). [PubMed] [Google Scholar]
- 32.Christakoudi S, Evangelou E, Riboli E & Tsilidis KK GWAS of allometric body-shape indices in UK Biobank identifies loci suggesting associations with morphogenesis, organogenesis, adrenal cell renewal and cancer. Sci. Rep. 11, 10688 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu M et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 51, 237–244 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grice DE et al. Transcriptional profiling of C57 and DBA strains of mice in the absence and presence of morphine. BMC Genomics 8, 76 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu L et al. Activity in projection neurons from prelimbic cortex to the PVT is necessary for retrieval of morphine withdrawal memory. Cell Rep. 35, 108958 (2021). [DOI] [PubMed] [Google Scholar]
- 36.Fujita-Hamabe W, Nakamoto K & Tokuyama S Involvement of NCAM and FGF receptor signaling in the development of analgesic tolerance to morphine. Eur. J. Pharmacol. 672, 77–82 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Yang B-Z et al. Association of haplotypic variants in DRD2, ANKK1, TTC12 and NCAM1 to alcohol dependence in independent case control and family samples. Hum. Mol. Genet. 16, 2844–2853 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Yang B-Z et al. Haplotypic variants in DRD2, ANKK1, TTC12 and NCAM1 are associated with comorbid alcohol and drug dependence. Alcohol. Clin. Exp. Res. 32, 2117–2127 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasman JA et al. GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia. Nat. Neurosci. 21, 1161–1170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vujkovic M et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 52, 680–691 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JJ et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 50, 1112–1121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amare AT et al. Bivariate genome-wide association analyses of the broad depression phenotype combined with major depressive disorder, bipolar disorder or schizophrenia reveal eight novel genetic loci for depression. Mol. Psychiatry 25, 1420–1429 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koob GF Neurobiology of opioid addiction: opponent process, hyperkatifeia and negative reinforcement. Biol. Psychiatry 87, 44–53 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Leshner AI Addiction is a brain disease, and it matters. Science 278, 45–47 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Karlsson Linnér R et al. Multivariate analysis of 1.5 million people identifies genetic associations with traits related to self-regulation and addiction. Nat. Neurosci. 24, 1367–1376 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vanyukov MM et al. Common liability to addiction and ‘gateway hypothesis’: Theoretical, empirical and evolutionary perspective. Drug Alcohol Depend. 123, S3–S17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic relationships, novel loci and pleiotropic mechanisms across eight psychiatric disorders. Cell 179, 1469–1482 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Howell BA et al. Validity of incident opioid use disorder (OUD) diagnoses in administrative data: a chart verification study. J. Gen. Intern. Med. 36, 1264–1270 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 49.Gaziano JM et al. Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 70, 214–223 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Delaneau O, Zagury J-F, Robinson MR, Marchini JL & Dermitzakis ET Accurate, scalable and integrative haplotype estimation. Nat. Commun. 10, 5436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Das S et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Auton A et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fang H et al. Harmonizing genetic ancestry and self-identified race/ethnicity in genome-wide association studies. Am. J. Hum. Genet. 105, 763–772 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J, Lee SH, Goddard ME & Visscher PM GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Altshuler DM et al. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Finucane HK et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bernstein BE et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 28, 1045–1048 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bakshi A et al. Fast set-based association analysis using summary data from GWAS identifies novel gene loci for human complex traits. Sci. Rep. 6, 32894 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barbeira AN et al. Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 22, 49 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu Z et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Aguet F et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fromer M et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 19, 1442–1453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ng B et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat. Neurosci. 20, 1418–1426 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Freshour SL et al. Integration of the Drug–Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 49, D1144–D1151 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brown BC, Asian Genetic Epidemiology Network Type 2 Diabetes Consortium, Ye CJ, Price AL & Zaitlen N Transethnic genetic-correlation estimates from summary statistics. Am. J. Hum. Genet. 99, 76–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ge T, Chen C-Y, Ni Y, Feng Y-CA & Smoller JW Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dennis JK et al. Clinical laboratory test-wide association scan of polygenic scores identifies biomarkers of complex disease. Genome Med. 13, 6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grotzinger AD et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat. Hum. Behav. 3, 513–525 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou H et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat. Neurosci. 23, 809–818 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson EC et al. A large-scale genome-wide association study meta-analysis of cannabis use disorder. Lancet Psychiatry 7, 1032–1045 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mullins N et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet. 53, 817–829 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Howard DM et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grove J et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Demontis D et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 51, 63–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu D et al. Interrogating the genetic determinants of Tourette’s syndrome and other tic disorders through genome-wide association studies. Am. J. Psychiatry 176, 217–227 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.International Obsessive Compulsive Disorder Foundation Genetics Collaborative (IOCDF-GC) and OCD Collaborative Genetics Association Studies (OCGAS). Revealing the complex genetic architecture of obsessive-compulsive disorder using meta-analysis. Mol. Psychiatry 23, 1181–1188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The full summary-level association data from the meta-analysis are available through dbGaP at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001672.