

Abstract
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic inflammation in synovial joints and protease-induced cartilage degradation. Current biologic treatments for RA can effectively reduce symptoms, primarily by neutralizing the proinflammatory cytokine TNFα; however, continued, indiscriminate overinhibition of inflammatory factors can significantly weaken the host immune system, leading to opportunistic infections and interrupting treatment. We hypothesize that localizing anti-TNFα therapeutics to denatured collagen (dCol) present at arthritic joints, via conjugation with collagen-hybridizing peptides (CHPs), will reduce off-site antigen binding and maintain local immunosuppression. We isolated the antigen-binding fragment of the clinically approved anti-TNFα therapeutic infliximab (iFab) and prepared iFab-CHP conjugates via lysine-based conjugation with an SMCC linker. After successful conjugation, confirmed by LC-MS, the binding affinity of iFab-CHP was characterized by ELISA-like assays, which showed comparable antigen binding relative to infliximab, comparable dCol binding relative to CHP, and the hybrid ability to bind both dCol and TNFα simultaneously. We further demonstrated localization of Fab-CHP to areas of high dCol in vivo and promising therapeutic efficacy, assessed by histological staining (Safranin-O and H&E), in a pilot mouse study.
Graphical Abstract
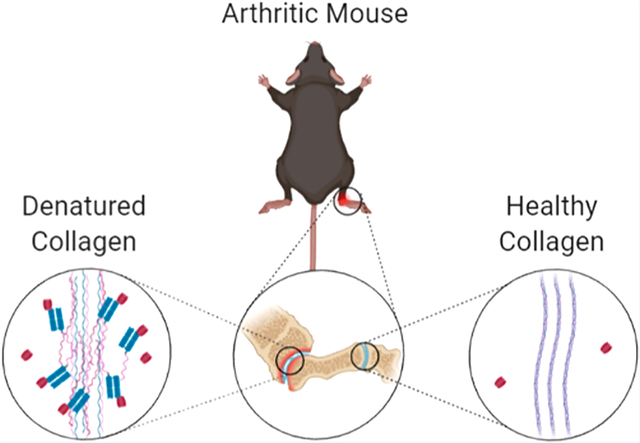
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic, autoimmune joint inflammatory disease that affects approximately 0.3–1.0% of the global population.1,2 Left untreated, RA progressively worsens, causing pain, limited mobility, and irreversible joint damage. As most patients are diagnosed around 60 years of age, sufficient treatment is required to prevent decades of pain and disability.2,3 While the underlying cause of RA is poorly understood, it has been shown to be a combination of genetic and environmental factors.4 Further, disease progression is characterized by chronic inflammation in the synovial joints, cartilage destruction yielding damaged collagen, and premature death.2,4,5 Collagen, when denatured (dCol), unwinds from its native triple-helical structure and can be targeted by collagen-hybridizing peptides (CHPs), providing a potential mechanism for the localization of therapeutic antibodies to arthritic joints.6 Current biologic treatments for RA primarily focus on neutralizing proinflammatory tissue disease mediators such as TNFα and CD80.2 While these therapies are effective in reducing RA symptoms, continued, indiscriminate overinhibition of inflammatory factors can significantly weaken the host immune system, paving the way for opportunistic infections.7–10 Patient dosing can be restricted to mediate negative side effects, leading to incomplete treatment.11 Recent studies have shown that the factors driving inflammation in RA are principally ectopic lymphoid and immune cells at the disease site.12–14 These studies indicate that a localized, anti-inflammatory therapeutic will not only reduce systemic immunosuppression but may also improve the efficacy of RA treatments.
While monoclonal antibody (mAb) therapies have significantly improved the treatment of chronic conditions, they can present with severe on-target toxicity limiting the eligible treatment population and potentially the length of treatment.11,15,16 Common side effects of anti-TNFα therapies, like Remicade (infliximab), include opportunistic infections (commonly tuberculosis), congestive heart failure, and pancytopenia.7–10 Here, we present a therapeutic antibody conjugate that can potentially reduce patient suffering and undesirable on-target effects of current anti-TNFα biologics by limiting cytokine neutralization to primarily disease sites through targeting of denatured collagen with CHPs.
While collagen remodeling occurs during natural tissue development and maintenance, excessive collagen degradation is associated with a variety of diseases and conditions including rheumatoid arthritis.5,17–19 Autoantibodies against citrullinated collagen type II are commonly found in RA patients and may contribute to pathogenesis.20–22 CHP [sequence: (GPO)9; G: glycine, P: proline, O: hydroxyproline] is advantageous relative to other collagen-binding peptides, as it binds specifically with dCol through triple-helical hybridization. The triple helix structure is a defining characteristic of collagen, existing outside of collagen only in the form of small subdomains, facilitating highly specific targeting with CHP.23 The unwound triple helix of dCol allows for strong, exclusive hybridization of monomeric CHPs to the denatured strands to form a new triple-helical structure, which is similar to a short single-strand DNA primer binding to its complementary DNA strand to form a double-helical structure.6 Additionally, CHPs are charge neutral, hydrophilic, and proline-rich, resulting in high serum stability and low nonspecific binding.24,25 Although CHPs are prone to self-trimerization, this is nullified by either heating CHPs to 80 °C to denature them before use or, as used for this study, by attaching a photocleavable nitrobenzyl (NB) protecting group to the glycine at the central GPO repeat to negate folding through steric hindrance.18 The protecting group is removed just prior to use by exposure to UV light (365 nm, approximately 10 mW/cm2). As CHPs are generated through standard solid-phase peptide synthesis (SPPS) methods, they can be easily modified to facilitate various conjugation methods to native proteins.
We hypothesize that conjugating CHP to infliximab will improve localization of TNFα neutralization to diseased joints through targeting of dCol. We conjugated CHP to the Fab region of infliximab (iFab) through a two-step, lysine-based conjugation with the heterobifunctional linker, sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC). While this type of conjugation is common for antibody–drug conjugates, in which potent small molecule drugs are used to potentiate antibody lethality, in our work, we flipped this paradigm by using CHP as the targeting moiety toward the novel target dCol and the antibody as the primary therapeutic agent. Further, we chose to use only the Fab fragment of infliximab to maintain TNFα binding while minimizing unnecessary immune cell activation by eliminating the Fc region, preventing interaction with Fcγ receptors.26 Targeting of dCol in RA is further facilitated by the enhanced permeability of inflamed tissue to macromolecules promoting the exposure of iFab-CHP to disease sites.27–29
RESULTS AND DISCUSSION
Preparation of Fab-CHP Conjugates.
The effect of CHP conjugation on therapeutic efficacy was assessed using the Fab fragment from infliximab (iFab), and the effect of CHP conjugation on biodistribution was assessed using the Fab fragment from bevacizumab (bFab). Neither bFab nor iFab significantly neutralize the murine versions of their respective antigen.30,31 Bevacizumab, while not used for the treatment of RA, was readily available to us and provided foundational insight into the pharmacokinetics of Fab-CHP conjugates. Both iFab-CHP and bFab-CHP constructs were synthesized via a two-step, lysine-based conjugation method (Scheme 1) similar to that used for the production of the clinically approved antibody–drug conjugate Kadcyla.32
Scheme 1.
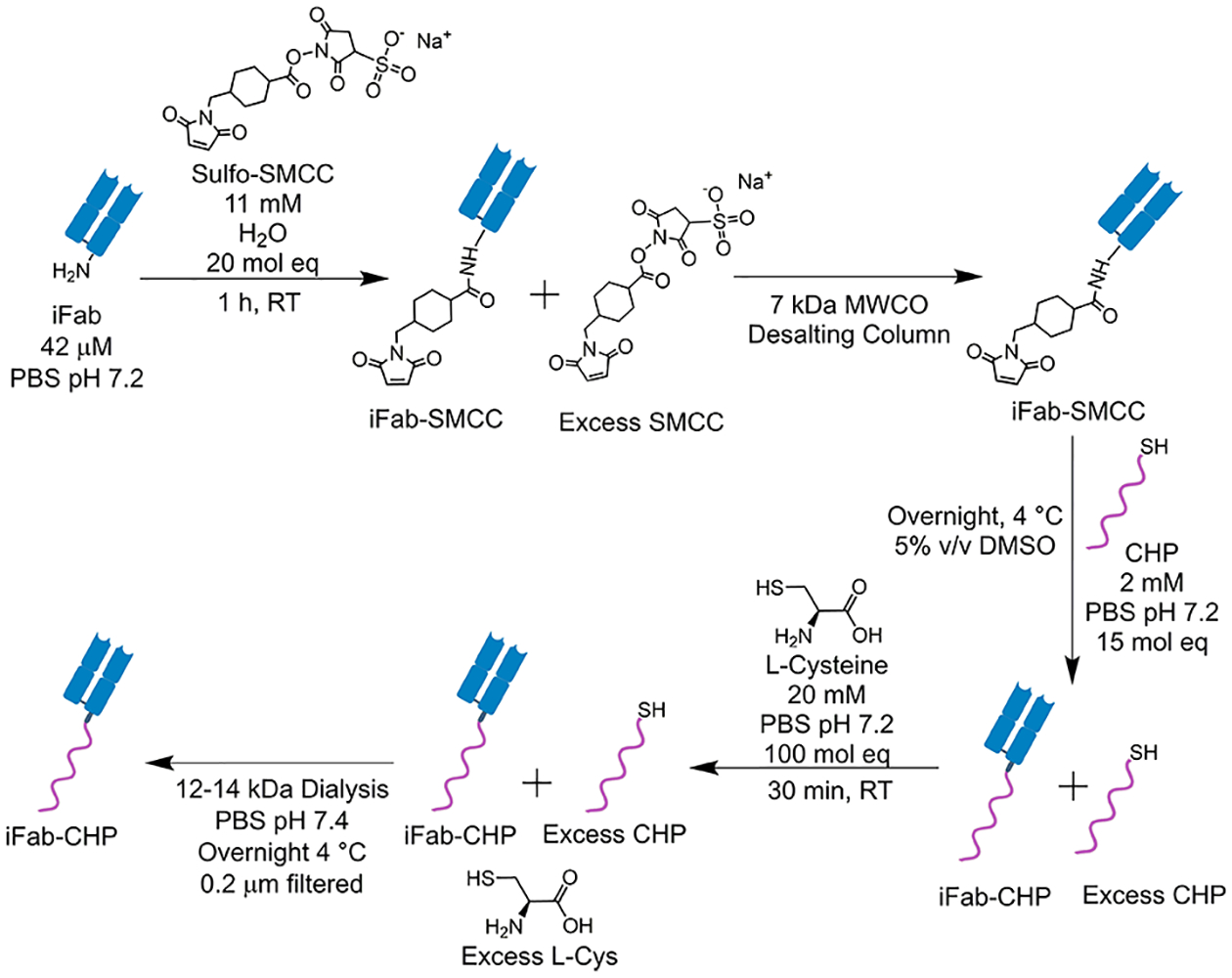
iFab Was Produced by Papain Digestion of the Clinical Product, Remicadea
aSubsequent conjugation of iFab to CHP was achieved by a two-step, lysine-based conjugation via a sulfo-SMCC heterobifunctional linker.
CHPs were synthesized as described previously using standard SPPS techniques.18,25 The full sequence of CHP is Ac-Cys-Ahx-NB(GPO)9, where Ac is an acetyl capped N-terminus, Ahx is aminohexanoic acid, and NB is a photocleavable nitrobenzyl group attached to the glycine on the central GPO repeat to prevent self-trimerization (Figure S1). CHPs were cleaved and purified by HPLC on a C18 column. Purified CHPs were lyophilized and characterized by MALDI to confirm purity. Fluorophore-tagged CHPs [sequence: Ac-Cys-Ahx-K(Fluoro)-Ahx-NB(GPO)9] were synthesized by the same SPPS technique and fluorophore tagged on resin with either 5(6)-carboxyfluorescein (CF) or IR680 (NIRF).
Fabs were isolated from the parent antibody using papain digestion, followed by Protein A affinity chromatography to remove the crystallizable fragment (Fc) and undigested antibody. Purified Fab was then modified with the heterobifunctional linker, sulfo-SMCC, converting native lysine residues to maleimide groups. Chemical conjugation of CHP was then achieved through a thiol-maleimide reaction between the partially conjugated SMCC and N-terminal cysteine of CHP. l-Cysteine was then added to cap unreacted maleimides, followed by 0.2 μm filtration to remove any aggregates and final purification by dialysis into PBS pH 7.4.
While many “next-generation” antibody-conjugates have transitioned to site-specific conjugation methods to improve product homogeneity,33 we chose a lysine-based conjugation method for flexibility and applicability to a wide range of therapeutic antibodies. In particular, numerous antibodies targeting different components of the inflammatory pathway are currently in use, such as infliximab, TNFα; tocilizumab, IL-6 receptor; abatacept, CD80/CD86; rituximab, CD20.2
Infliximab was chosen for this study because of its well-established therapeutic efficacy, and availability of an RA mouse model driven by human TNFα (hTNFα). However, of concern during the development of this platform was whether a Fab-CHP conjugate, when injected intravenously, could potentially have a negative effect by capturing proinflammatory cytokines in circulation and transporting them to the disease tissue, at which point they may be slowly released, creating a proinflammatory depot. Based on the relative binding affinities of infliximab (4.2 pM sTNFα, 468 pM mTNFα)34 and CHP (~0.1–0.9 μM) for their respective targets, we considered this negative outcome unlikely, but by creating a flexible platform, we can easily substitute the therapeutic antibody to target different cytokines if needed.
Characterization of iFab-CHP Conjugates.
Native lysine conjugation produces a heterogeneous product with a distribution in the number of conjugated peptides per Fab, herein referred to as the conjugation ratio (CR). Product heterogeneity is caused both by the differences in the number of SMCCs attached during the original modification and by the subsequent efficiency of the thiol-maleimide reaction during CHP conjugation. Different CR species within a sample can have profoundly different pharmacokinetic characteristics.35 Once the validity of the iFab-CHP platform is confirmed, alternative conjugation strategies can be used to improve product homogeneity however that was not within the scope of this work. Further, the optimal average CR (CRavg) was not investigated, rather the goal of conjugation was to minimize the amount of unconjugated iFab in the final product (percent unmodified).
Purity of the papain-digested infliximab and successful conjugation of iFab-CHP was first confirmed by SDS-PAGE (Figure S2). Papain digestion of infliximab resulted primarily in iFab as indicated by the dark band at ~50 kDa in lane c; however, full IgG infliximab was present to a lesser degree after purification as indicated by the high MW band, ~150 kDa. In lane a, the ~50 kDa band that corresponds to unmodified iFab was less intense than the iFab stock. Further, multiple bands were observed above the iFab band, similar to rungs on a ladder, which indicated successful conjugation that produced multiple CR species.
Intact LC-MS analysis was then used to characterize the specific conjugation profile, including relative amounts of each CR species and CRavg, in greater detail. The simplified LC-MS spectra overlay is shown in Figure 1, with the more detailed, deconvoluted spectra found in Figure S3. The molecular weight of unconjugated iFab was found to be 47 904 Da. Using unconjugated iFab as a baseline, CR of each peak was determined by the known mass of SMCC–CHP (3035 Da). As discussed previously, the two-step synthesis method resulted in partially conjugated iFab-SMCC species.32 These are observed in the LC-MS spectra as secondary peaks with an average MW difference of approximately 219 Da. As CR is a representation of the number of CHPs per Fab, partially conjugated iFab-SMCC species were combined with the corresponding primary peak to calculate CRavg (i.e., iFab-CHP with 2 fully conjugated CHPs and 1 partially conjugated SMCC is treated as CR 2). The CRavg of iFab-CHP was calculated to be 3.4, and no unmodified iFab was detected in the final product. bFab-CHP conjugates were prepared using the same methods.
Figure 1.
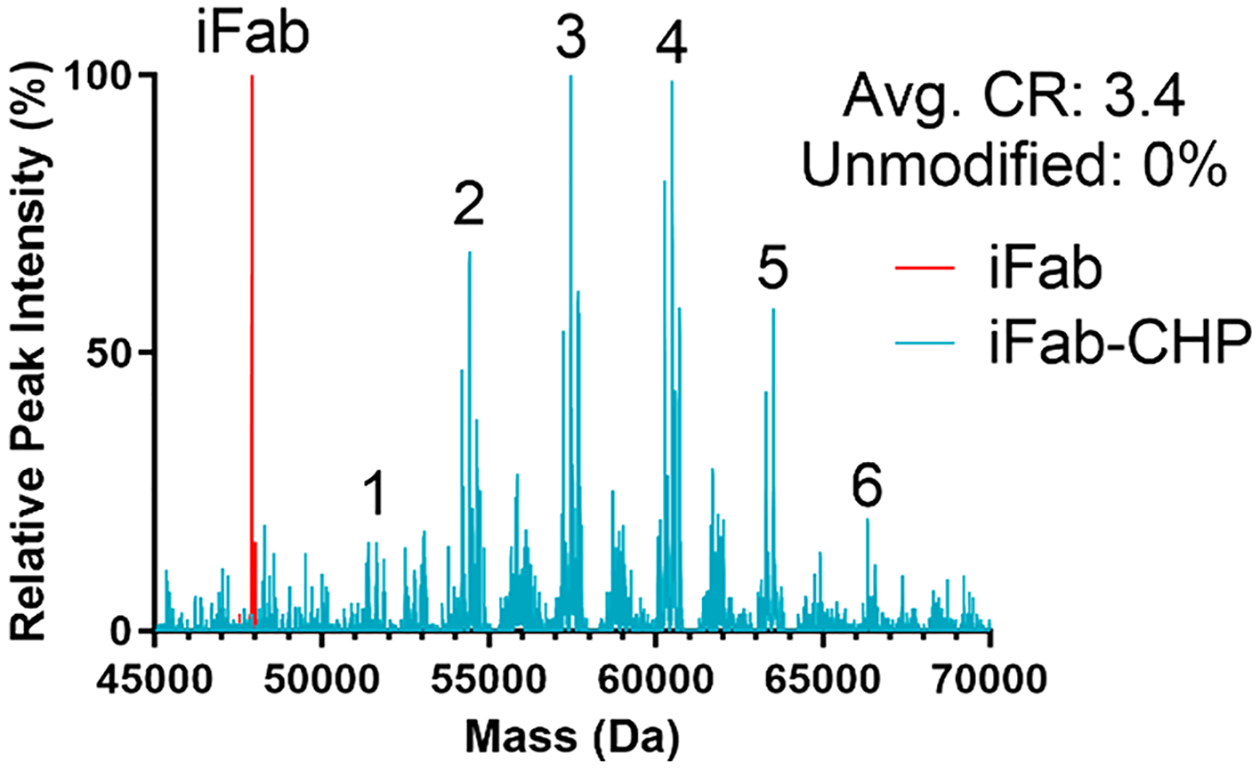
Intact LC-MS analysis of unmodified iFab (red) overlapped with analysis of iFab-CHP (blue) conjugates. Integer values indicate the conjugation ratio (CR) of the species in that peak. No unmodified iFab was present after conjugation.
CHPs are well suited for antibody conjugation because they are hydrophilic and serum stable.24,25 As reported by Buecheler et al.,36 when CR is held equal, more hydrophobic payloads induce a significantly greater decrease in antibody-conjugate stability, with a corresponding increase in the propensity to self-associate and aggregate. Since CHPs are hydrophilic, conjugation to a Fab will not increase the overall hydrophobicity of the Fab-CHP conjugate and should not negatively impact stability or induce aggregation. This claim is supported by previous work that showed the addition of CHP minimized aggregation of β-sheet derived nanofibers.37 While not specifically investigated, no aggregation or other stability issues were observed for Fab-CHP conjugates.
While the serum stability of iFab-CHP was not specifically investigated, the individual components of iFab-CHP have been previously characterized. In vitro studies from Bennink et al.25 characterized the serum stability of monomeric CHP and found that the absence of charged and hydrophobic residues in the core (GPO)9 sequence minimized degradation from common serum proteases. Additionally, N-terminal modifications, like those used in the CHP sequence described herein, improved resistance to proline specific proteases.25 In that study, 96.6% of IR680-Ahx-NB(GPO)9, similar to the CHP used for the biodistribution study described below, remained intact after incubation in dilute mouse serum (25% in PBS) at 37 °C for 24 h.25 An in vitro study of infliximab stability by Perry et al.38 demonstrated no decrease in infliximab concentration after 7 days at room temperature in either human serum or whole blood taken from Crohn’s disease patients. Other studies have shown that infliximab can be degraded by matrix metalloproteinases (MMPs), particularly MMP-3 which is overexpressed in RA patients and thought to play a significant role in cartilage erosion.39–42 Importantly, analysis by Biancheri et al.42 indicated MMP-3 cleavage occurs in the Fc region of infliximab, which may not impact the efficacy of iFab used here. SMCC linkers, while noncleavable, are susceptible to premature payload release from the thiosuccinimide moiety via oxidation and sulfoxide elimination, or retro-Michael reaction and exchange with native thiol containing proteins (i.e., albumin).43–45 Ponte et al.43 studied this phenomena in the context of antibody–drug conjugates and determined that approximately 7.9% of the conjugated payload was released after 7 days at 37 °C in buffered mouse plasma. Release of CHP from the SMCC linker is unlikely to significantly affect the therapeutic efficacy of iFab-CHP over the time course of our study, as further supported by the clinical success of Kadcyla, which also uses an SMCC linker.32 Considering the stability of the individual iFab-CHP components in serum, we do not expect any major instability issues for Fab-CHP conjugates in serum.
Characterization of iFab-CHP Binding.
Following successful conjugation, ELISA-like assays were used to assess the affinity of iFab-CHP for hTNFα and dCol, relative to free iFab, infliximab, caged iFab-CHPNB, and free CHP (Figure 2). To confirm that iFab-CHP maintained binding affinity for hTNFα, hTNFα was immobilized, binders were added, and anti-Human IgG (H+L) antibody–horseradish peroxidase (HRP) conjugates were used for detection (Figure 2A). All three binders (iFab, iFab-CHP, and infliximab) demonstrated significantly greater hTNFα binding relative to the negative control, bovine serum albumin (BSA). No difference was seen between iFab and iFab-CHP, while the greater signal produced by infliximab can be explained as an artifact of the detection antibody. As anti-human IgG (H+L) is a polyclonal detection antibody with multiple binding sites, more sites can be simultaneously occupied on a full IgG (~150 kDa) relative to the much smaller Fab (~50 kDa). When more sites of the 1° antibody are bound, a greater fluorescence signal is produced. In particular, the fluorescence intensity of the infliximab sample was roughly 3× that of iFab or iFab-CHP, corresponding perfectly with their relative size. These results are consistent with our previous work demonstrating that conjugation of an antibody does not significantly affect binding affinity.46
Figure 2.
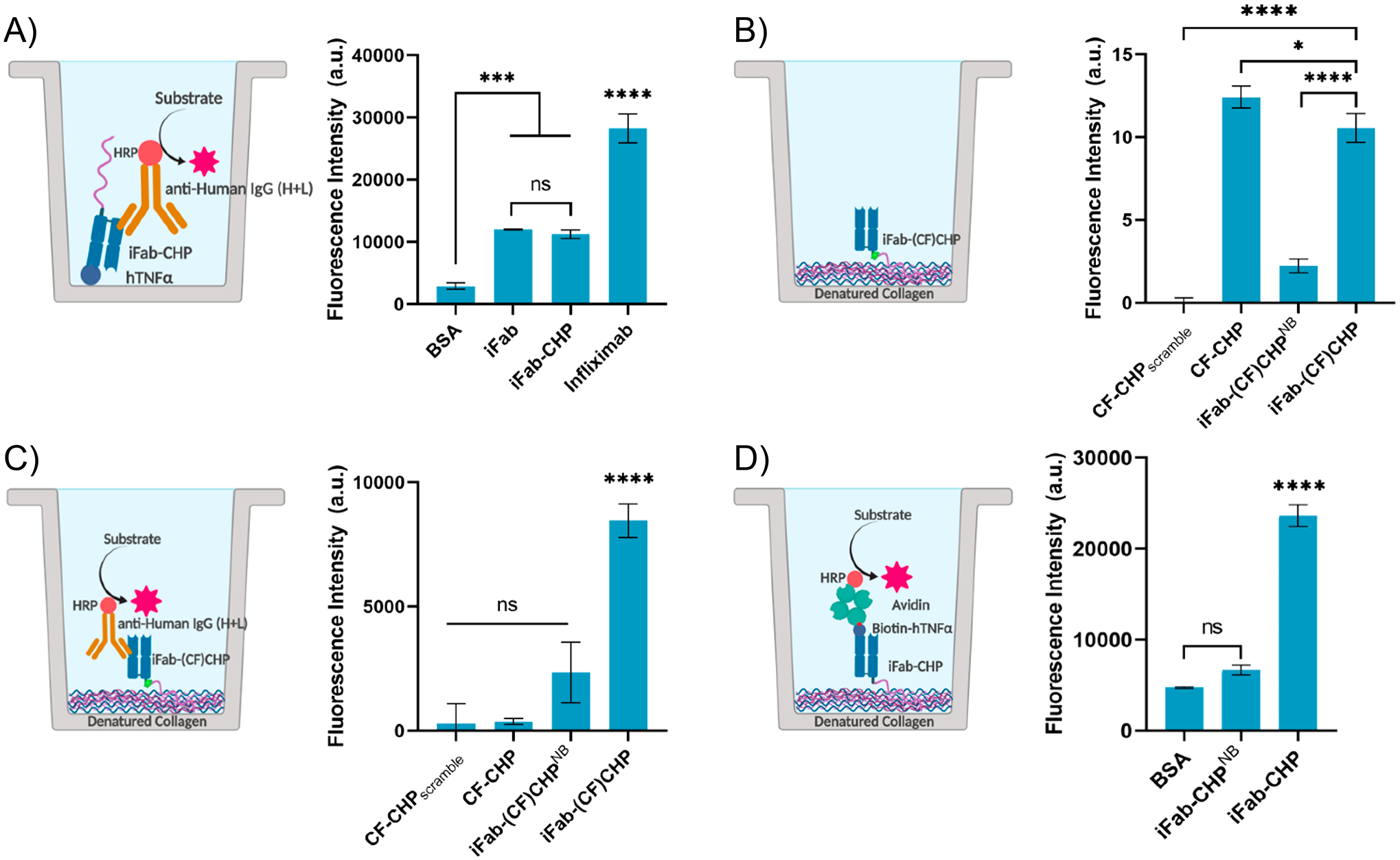
ELISA-like binding assays were used to confirm the ability of iFab-CHP to bind (A) immobilized hTNFα, (B/C) dCol, and (D) dCol and hTNFα simultaneously. (A) No significant difference in hTNFα binding was measured between iFab-CHP and nonconjugated iFab, indicating that CHP conjugation did not affect iFab binding affinity. (B) Direct fluorescence measurements of dCol binding by iFab-(CF)CHP were significantly greater than those of both negative controls (CF-CHPscramble, iFab-(CF)CHPNB); however, a slight decrease was observed relative to free CF-CHP. (C) iFab-(CF)CHP binding to dCol was significantly greater than all negative controls when detected by anti-human IgG (H+L)-HRP. (D) A statistically significant increase in fluorescence intensity was observed for iFab-CHP binding to dCol, followed by the addition and detection of hTNFα, relative to both negative controls, which confirmed simultaneous binding of both targets. Values are shown as a mean ± standard deviation (n = 3); statistical significance is defined as follows: ns = P > 0.05; *P ≤ 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Binding of iFab-(CF)CHP for dCol was then measured by a similar assay with immobilized dCol, relative to CF-tagged free CHP (CF-CHP) and scrambled sequence (CF-CHPscramble), representing the positive and negative controls, respectively. Caged iFab-(CF)CHPNB, in which the NB photocleavable protecting group is not removed, was also used as a negative control for this set of assays. Binders were then detected either by direct fluorescence measurements (Figure 2B) or by anti-human IgG (H+L) antibody-HRP (Figure 2C). A slight decrease in dCol binding was observed for iFab-(CF)CHP relative to CF-CHP when measured by direct fluorescence, but was still significantly greater than either negative control [CF-CHPScramble and iFab-(CF)CHPNB]. As expected, when detected by anti-human IgG (H+L) antibody-HRP, CF-CHP signal was negligible. Nonsignificant differences in fluorescence were measured for CF-CHPScramble, CF-CHP, and iFab-(CF)CHPNB, with a significant increase in signal observed for iFab-(CF)CHP.
For iFab-CHP to produce a therapeutic effect in vivo, it must be able to bind both hTNFα and dCol simultaneously. Simultaneous binding was tested in vitro by an ELISA-like assay in which dCol was immobilized prior to the addition of the binders (negative controls: BSA, iFab-CHPNB). Samples were washed, and biotinylated hTNFα was added. Bound hTNFα was then detected by an avidin–HRP conjugate. Nonsignificant differences in signal were observed for both negative controls with a significant increase in fluorescence signal measured for iFab-CHP relative to both, indicating successful, simultaneous binding of both targets (Figure 2D). Comparable assays (hVEGFA replacing hTNFα) were used to assess the relative binding of bFab-CHP to unmodified bFab (Figure S4). Outcomes were equivalent to those seen for iFab-CHP, indicating that simultaneous binding of two domains, as well as maintained antigen binding affinity, was not exclusive to iFab.
Biodistribution of bFab-CHP.
To explore the ability of Fab-CHP conjugates to localize to areas of dCol in vivo, IR680 (NIRF) tagged bFab-CHP underwent a comparative biodistribution study against caged bFab-(NIRF)CHPNB in nude mice (n = 5) (Figure 3). bFab-(NIRF)CHP conjugates were prepared and characterized using the same methods described above for iFab-CHP. While immunodeficient arthritic mouse models do exist,47 healthy nude mice were chosen for this study to establish baseline biodistribution and for technical reasons, as NIRF measurement is inhibited by fur.48 Although not as elevated as seen in arthritic mice, healthy mouse joints undergo continual tissue remodeling by MMPs, producing dCol.18 As observed in Figure 3A, IV tail vein injection of bFab-(NIRF)CHP led to an initial systemic distribution (t = 0.5 h), followed by prolonged residence in areas of higher dCol (i.e., spine, ribs, joints) relative to the caged control. These results were similar to those seen for unconjugated (NIRF)–CHP and nanofiber conjugated (NIRF)–CHP, indicating that conjugation to a Fab does not significantly alter CHP binding to dCol in vivo.18,37,49 Previous studies of monomeric (NIRF)–CHP in healthy mice showed localization to the spine, hind paws, and tail at 24 and 48 h after injection, similar to the results seen for bFab-(NIRF)CHP at the 24 h time point (Figure 3A).25,49 Previous work investigating prefolded (NIRF)–CHP and caged (NIRF)–CHPNB demonstrated poor skeletal localization, significant signal reduction after 6 h, and negligible signal after 96 h, which correlates with the results observed for caged bFab-(NIRF)CHPNB (Figure 3A.B).37,49
Figure 3.
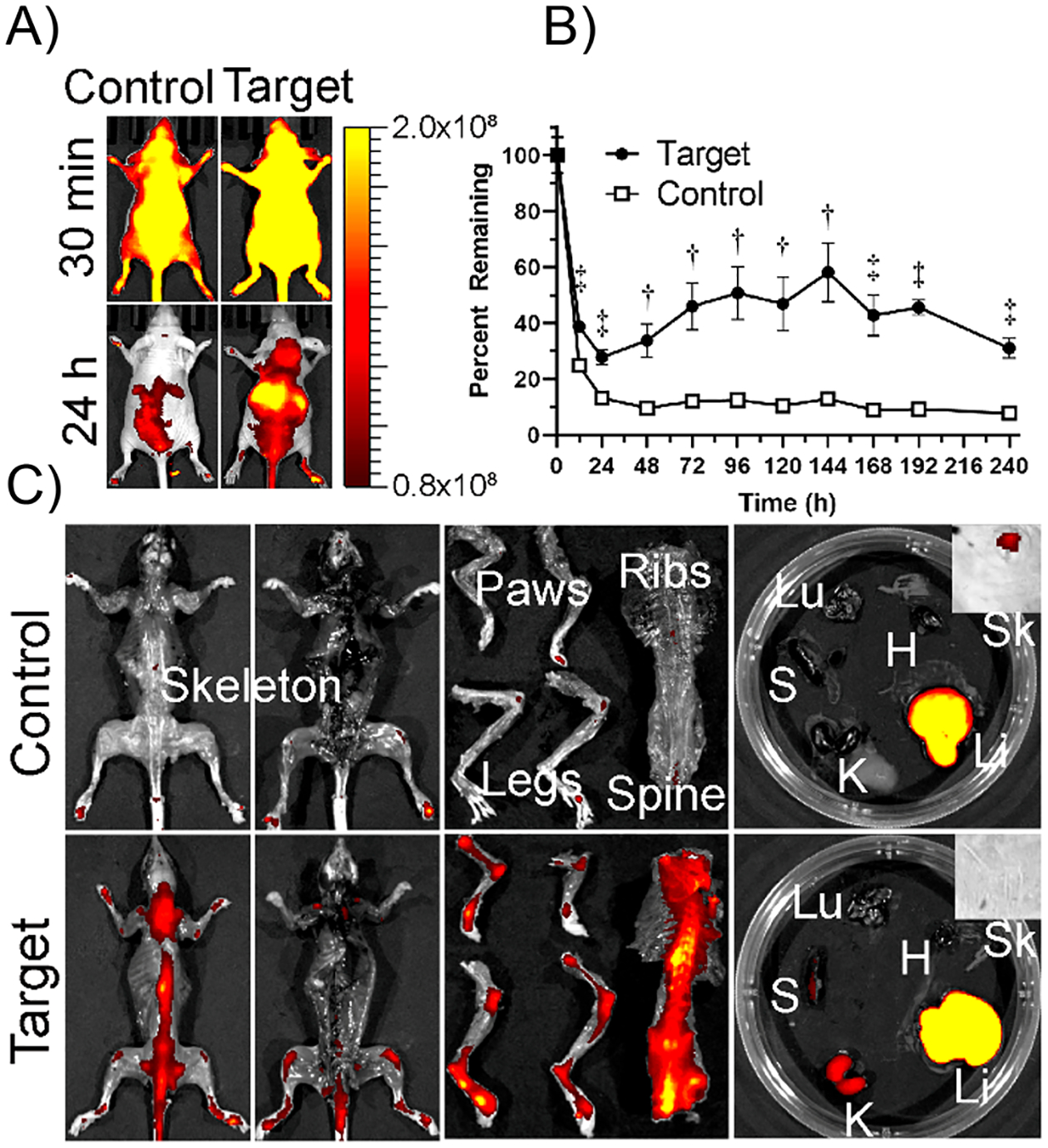
(A) Biodistribution of IR680 labeled bFab-(NIRF)CHP (Target) and caged bFab-(NIRF)CHPNB (Control) IV injected into the tail vein of nude mice (n = 5). (B) Fluorescence intensities were quantified over 10 days by whole-body scan. Data reported as mean ± standard deviation. (C) Mice were sacrificed and NIRF intensity was measured for isolated organs. Both target and control were processed primarily by the liver, while bFab-(NIRF)CHP showed significantly greater skeletal localization. (Li: liver, K: kidney, Sk: skin, H: heart, Lu: lung, S: spleen). Statistical significance is defined as follows: †P < 0.001; ‡P < 0.0001.
Significantly greater levels of targeted bFab-(NIRF)CHP, as measured by overall fluorescence intensity, were present throughout the 10-day study relative to the caged bFab-(NIRF)CHPNB control (Figure 3B). The percent fluorophore remaining was calculated by dividing the average fluorescence intensity at a given time point by the average fluorescence reading at t = 0.5 h. Interestingly, for the untargeted control, the percent remaining rapidly decreased to approximately 13% after 24 h, which was then sustained with little change over the course of the study resulting in 8% remaining at day 10; however, targeted bFab-(NIRF)CHP decreased to roughly 28% remaining after 24 h followed by an increase in fluorescence signal until day 6, and subsequent decrease resulting in 31% remaining after 10 days. We posit that this trend was caused by highly localized bFab-(NIRF)CHP masking the fluorescence signal, thus artificially reducing the apparent percent remaining. As bFab-(NIRF)CHP dissociated from dCol over the course of the study, fluorescence signal was distributed, allowing the sensor to accurately measure the entire fluorescence signal. Regardless, the consistent and statistically significant increase in percent bFab-(NIRF)CHP remaining relative to the caged control at all time points indicated that bFab-(NIRF)CHP binding of dCol partially compensated for the reduction in half-life caused by the elimination of the Fc region.
After 10 days, mice were sacrificed and organs extracted to further assess bFab-(NIRF)CHP localization and clearance pathways (Figure 3C). As expected, bFab-(NIRF)CHP remained bound to areas of high dCol (i.e., spine, ribs, joints), while caged bFab-(NIRF)CHPNB remaining outside of the liver was negligible. This strongly indicates that biodistribution of Fab-CHP in arthritic models will heavily favor the areas of highest dCol. Additionally, we suspect that Fab-CHP retention will be improved with an arthritic model as more dCol is present to bind. This is supported by previous work investigating free (NIRF)-CHP localization in mice suffering from Marfan syndrome relative to healthy mice18 and nonpublished work with (NIRF)-CHP in arthritic and healthy mice (data not shown). Clearance of bFab-CHP appeared to be primarily mediated by the liver and, to a lesser degree, the kidneys, likely after nonspecific protease degradation, which is consistent with the clearance pathways of infliximab.50 Free CHP is primarily eliminated through renal pathways, with an approximate half-life of 2 days in healthy nude mice.25,51 bFab-(NIRF)CHP demonstrated an apparent increase in half-life relative to monomeric CHP, likely as a result of its increased size which reduced renal clearance. This is consistent with previous, unpublished work that demonstrated increased half-life of targeted PEG-CHP conjugates relative to free CHP.51 Each moiety of the Fab-CHP conjugate contributes synergistically to its pharmacokinetic profile, as demonstrated by improved half-life relative to (NIRF)-CHP, and improved half-life and dCol localization relative to bFab-(NIRF)-CHPNB.49
Deora et al.52 demonstrated that antibody binding to mTNFα (referred to as TmTNFα) triggers rapid clathrin-mediated endocytosis and degradation in the lysosome of mTNFα-presenting dendritic cells. Once degraded, peptides from the anti-TNFα IgG were presented on the surface potentially inducing the development of anti-drug antibodies. While not within the scope of this study, it is possible that by binding dCol, iFab-CHP is less susceptible to internalization and degradation than free infliximab, potentially minimizing immunogenicity and improving long-term efficacy. Regardless, this biodistribution study demonstrates improved longevity and localization to sites of dCol than the nontargeted control.
Therapeutic Efficacy of iFab-CHP.
Infliximab does not bind to murine TNFα;31 as such, to test therapeutic efficacy in vivo, a transgenic mouse model driven by overexpression of hTNFα was required. hTNFα expressing transgenic female mice, originally characterized by Hayward et al.,53 were acquired at 8 weeks of age (Taconic Biosciences), acclimated for 1 week, and treated at disease onset (~9 weeks of age), as reported by the supplier. Mice (n = 3) were IV tail vein injected with either a clinically relevant dose of iFab-CHP (85 nmol/kg)54–56 or equal volume of PBS at 9 and 16 weeks of age. Mice were observed 3 times per week to blindly score joint swelling (0–8 per paw, increments of 0.5, increasing severity),57 measure joint width and thickness, as well as monitor weight, lethargy, and signs of distress. No significant toxicity or weight loss was observed (Figure S5).
Real-time assessment of disease progression by both joint scoring and measurements was inconclusive. Based on previous experience with mycoplasma arthritidis mitogen induced arthritis models, we expected to see a much greater degree of joint swelling as the disease progressed.58 However, likely as a combination of both the slower disease progression and the sex of the mice, the joints did not swell significantly, even at severe disease states, as confirmed by histology. At 19 weeks of age, iFab-CHP and PBS treated mice had average summed paw scores of 8 and 8.7, respectively, out of maximum score of 32. As such, it was extremely difficult to monitor slight nuances of disease progression in real-time, and therefore no significant therapeutic effect by either scoring or joint measurements was observed (Figure S6).
All mice were sacrificed at 10 weeks post-initial injection (19 weeks of age), hind paw tissue was collected and prepared for histological staining, with representative histological sections shown in Figure 4. Hematoxylin & eosin (H&E) staining revealed significant synovial hyperplasia with periarticular inflammatory cell infiltration, and articular cartilage degradation in PBS treated mice.59 iFab-CHP treated mice showed a lesser degree of synovial hyperplasia with no inflammatory cell infiltration, and largely intact articular cartilage. Safranin-O (Saf-O), which stains sulfated proteoglycans (red) present in healthy cartilage, was used to more specifically evaluate the integrity of articular cartilage.55,60,61 Saf-O staining revealed cartilage degradation in PBS-treated mice demonstrated by negative staining at the superficial joint interface. iFab-CHP treated mice maintained healthy cartilage with minimal matrix degradation as demonstrated by uniformly positive Saf-O staining at the joint interface.
Figure 4.
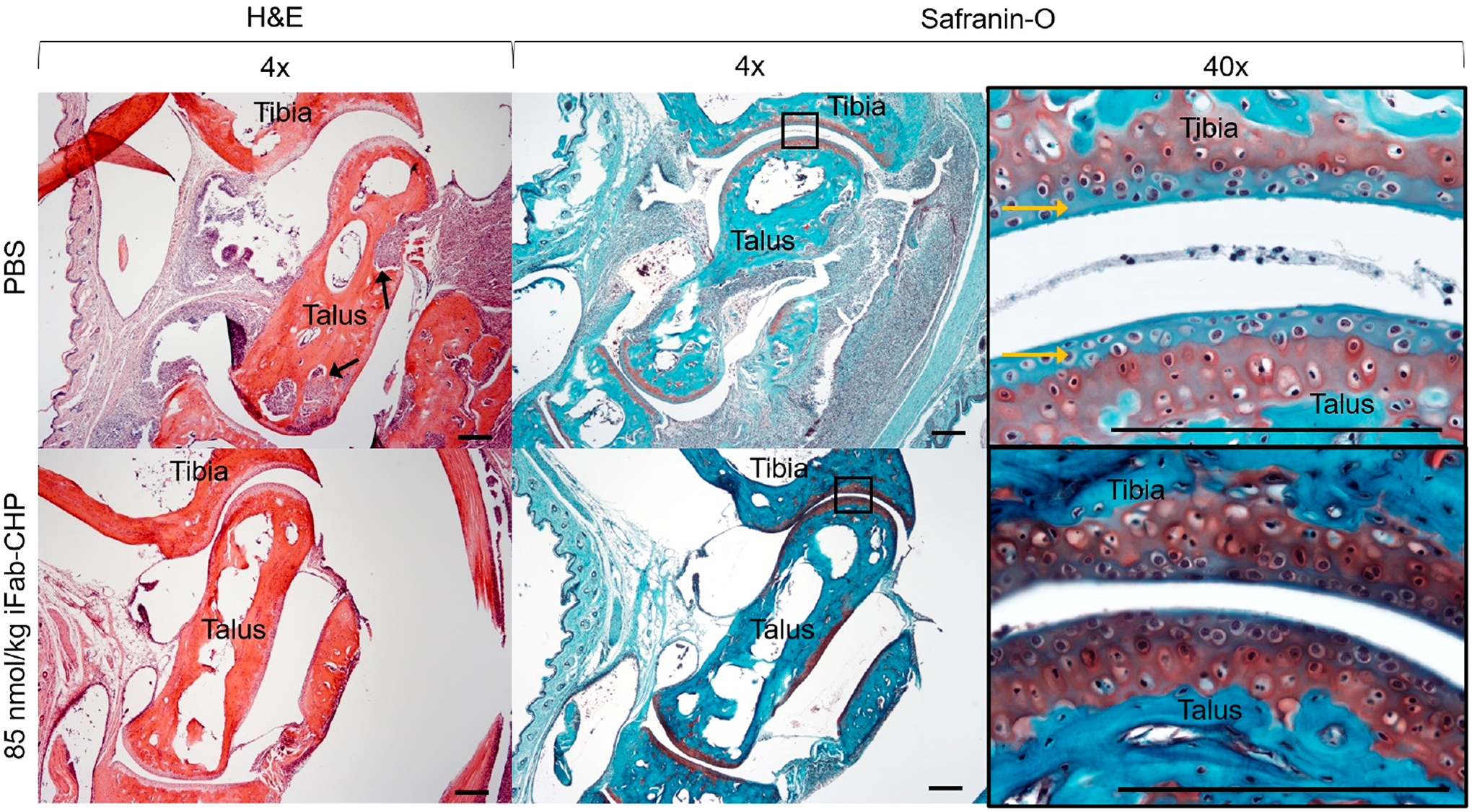
Histological staining of hTNFα transgenic mice tibia-talus joints with either hematoxylin and eosin (H&E) (first column) or Safranin-O (red) with Fast Green (blue) (second and third columns). Mice were injected at disease onset with either 85 nmol/kg iFab-CHP or PBS, with an additional injection 7 weeks post-initial injection, and sacrificed at 10 weeks post-initial injection. H&E staining revealed a greater degree of synovial hyperplasia and inflammatory cell infiltration (black arrows) in PBS treated mice. Additionally, Saf-O/Fast Green staining revealed more intact hyaline articular cartilage (sulfated proteoglycans stained red) in iFab-CHP vs PBS treated mice. Yellow arrows point to negative hyaline cartilage staining. Scale bars = 200 μm.
While traditional joint scoring methods were insufficient to characterize disease progression in this model, terminal histology provided strong evidence that iFab-CHP slowed disease progression and maintained healthy cartilage. Importantly, a negative “proinflammatory depot effect”, as described above, was not observed. While further studies are needed to optimize dosing of iFab-CHP, as well as compare therapeutic efficacy and immunosuppression directly to infliximab, this pilot study was an important verification and validation of the iFab-CHP platform for the treatment of RA.
CONCLUSION
Cytokine specific biologic treatments have revolutionized the treatment of RA over the past 2 decades;2 however, improved treatment localization, augmented through conjugation with CHP, has the potential to reduce on-target toxicity and improve therapeutic outcomes. Here, we have synthesized and characterized iFab-CHP conjugates using simple, native amino acid conjugation methods that can be readily applied to any clinically approved therapeutic antibody. ELISA-like assays validated the binding ability of iFab and CHP moieties to their respective targets both individually and simultaneously in vitro. In vivo experiments showed that Fab-CHP conjugates localized to areas of high dCol in healthy mice, and demonstrated therapeutic efficacy, by slowing disease progression and cartilage degradation, in a transgenic mouse RA model. Future work aims to optimize the iFab-CHP platform, through improved homogeneity and determination of optimal CRavg, and compare therapeutic outcomes to unmodified infliximab. Additionally, while this study focused specifically on RA, the Fab-CHP platform is applicable to many pathological conditions with excessive collagen degradation (e.g., cancer, fibrosis) that are currently treated by therapeutic antibodies.62,63
MATERIALS AND METHODS
CHP Synthesis.
CHP was synthesized as described previously using standard SPPS methods.18,37 All peptides were synthesized on a Focus XC peptide synthesizer (AAPPTec) using TentaGel R Ram resins and standard Fmoc chemistry with HBTU/DIEA activation. Nonfluorescent CHP was prepared with the sequence Ac-C-Ahx-NB(GPO)9. Fluorescent CHP was prepared with the sequence Ac-C-Ahx-K(Fluoro)-Ahx-NB(GPO)9, where Fluoro was either IR680 dye (NIRF) (Li-COR) or 5(6)-carboxyfluorescein (CF) (Sigma-Aldrich). Fluorescent tags were conjugated on resin as described previously.18 Cleaved peptides were purified by RP-HPLC (Agilent) on a C18 column with a 5–50% gradient of Water with 0.1% trifluoroacetic acid (TFA) and acetonitrile. Peptide mass and purity was confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) (UltrafleXtreme, Bruker Daltronics) and HPLC.
Fab-CHP Synthesis.
Infliximab Fab was prepared from infliximab (Janssen Biotech) stock with Pierce Fab Preparation Kit (ThermoFisher Scientific) following the protocol provided with the kit. Following Protein A purification, flowthrough and bound fractions were analyzed with SDS-PAGE with 4–12% Bis-Tris gel. Infliximab Fabs were buffer exchanged into PBS pH 7.2 using 5 kDa MWCO spin columns to a final concentration between 20.9 and 83.5 μM (1–4 mg/mL). Sulfo-SMCC (ThermoFisher Scientific) was prepared at 11 mM in Milli-Q water. 20 mol equiv of Sulfo-SMCC was mixed with iFab for 1 h at room temperature (RT). Excess Sulfo-SMCC was removed from the solution using a 7 kDa MWCO desalting column. Desalted iFab-SMCC was transferred to a new tube to which dimethyl sulfoxide (DMSO) was added such that the final reaction contained 5% DMSO by volume. CHP was prepared at 2 mM in PBS pH 7.2. 15 mol equiv of 2 mM CHP was added to the iFab-SMCC solution while mixing at 4 °C, covered in foil. Reaction proceeded overnight. 100 mol equiv of 20 mM l-cysteine was freshly prepared and added to the reaction and mixed for 30 min at RT to cap unreacted maleimides. Excess l-cysteine and CHP was removed via dialysis with 12–14 kDa MWCO dialysis membrane tubing in PBS (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4) pH 7.4. iFab-CHP was dialyzed in 1 L PBS pH 7.4 with buffer replaced after 2 h, followed by overnight dialysis with 2 L PBS pH 7.4. Bevacizumab Fab-CHP conjugates were prepared using the same method.
LC-MS.
Intact LC-MS analysis of Fab-CHP conjugates was performed as described previously.32,46 Briefly, Fab-CHP samples were buffer exchanged into ammonium acetate buffer (50 mM ammonium acetate, pH 7.0) using a 10 kDa 0.5 mL centrifugal filter. Intact protein LC-MS analysis was performed on an ACQUITY UPLC I-Class with a Xevo G2-S QToF mass spectrometer (Waters Corporation). Fab-CHP samples were further desalted with a MassPREP microdesalting column (Waters Corporation) using a 6 min linear gradient run at flow rate of 0.3 mL/min, 80 °C. The gradient was programmed as follows: 5% B from 0 to 2 min, 5–90% B from 2 to 5 min, then 90–5% B from 5 to 6 min. The mobile phase A was water with 0.1% formic acid and the mobile phase B was acetonitrile with 0.1% formic acid. The mass spectrometer was operated in positive electrospray ionization (ESI) mode. The capillary voltage was 3 kV and the sampling cone voltage was 150 V. The source temperature was 150 °C and the desolvation temperature was 500 °C. The source desolvation gas flow and cone gas flow was 800 L/h and 10 L/h, respectively. The recorded mass spectra were combined and deconvoluted using MassLynx 4.1 (Waters Corporation).
CR for each species peak was identified by dividing the difference between the MW of the peak and the unmodified Fab peak (iFab: 47904 Da) by the expected MW of the CHP-linker species (SMCC–CHP: 3035 Da). MWs of each component were as follows: SMCC, 219 Da; CHP, 2816 Da; CF-CHP, 3416 Da; NIRF-CHP, 3946 Da. Partially conjugated SMCC species were identified by a MW difference of approximately 219 Da. Analysis of MS results assumed equal ionization efficiencies between all CR species of the Fab-CHP conjugates. The relative peak intensity of each CR species was calculated by dividing the peak area of that species (including all CR + partially conjugated linker) by the total peak area of that sample. Average CR was calculated by summation of the product of each CR species multiplied, but it is relative peak intensity within the sample.
hTNFα Binding Assay.
hTNFα binding assays were performed in a 96-well plate. All wash steps consisted of three washes with 1× PBS. All samples were run in triplicate. Wells were coated with 50 μL of 0.2 mg/mL avidin for 2 h at RT, while shaking. Wells were washed and biotin-hTNFα (2 μg/mL, 50 μL) was added for 1 h at RT. Wells were washed, blocked with 5% BSA for 1 h at RT, and then washed again. Binders (BSA, iFab-CHP, iFab, infliximab) were added (5 μM, 50 μL) for 1 h at RT. Wells were washed and samples were detected using a rabbit anti-human IgG (H+L)-HRP conjugate (Bethyl Laboratories, Inc.) (1/2000 dilution, 50 μL) for 1 h at RT. Signal was then generated adding 100 μL QuantaBlu Fluorogenic Peroxidase Substrate working solution (ThermoFisher Scientific) for 5 min prior to addition of 100 μL stop solution. Signal was measured (Ex: 325 nm, Em: 420 nm) on SpectraMax M-2 microplate reader (Molecular Devices).
dCol Binding Assay.
Denatured collagen binding assays were performed as described previously.37 All samples were run in triplicate. Briefly, dCol (Gelatin from Bovine Skin Type B, Sigma-Aldrich) (50 μL, 0.5 mg/mL) was immobilized on a 96-well plate for 2 h at RT. All wash steps consisted of three washes with 1× PBS. Wells were washed, blocked with 5% BSA for 1 h at RT, and washed again. 50 μL of 10 μM binders (CF-CHPscramble, CF-CHP, iFab-(CF)CHPNB, iFab-(CF)CHP) were added to their respective wells and incubated at 37 °C for 2 h. Wells were washed and fluorescence based on CF was measured as described above (Ex: 489 nm, Em: 533 nm). Immobilized iFab was subsequently detected by rabbit anti-human IgG (H+L)-HRP conjugate (1/2000 dilution, 50 μL) for 1 h at RT. Signal was then generated adding 100 μL QuantaBlu Fluorogenic Peroxidase Substrate working solution for 5 min prior to addition of 100 μL stop solution. Signal was measured as described above (Ex: 325 nm, Em: 420 nm).
Simultaneous hTNFα-dCol Binding Assay.
Simultaneous binding assays were performed in triplicate, on a 96-well plate. All wash steps consisted of three washes with 1× PBS. dCol (Gelatin from Bovine Skin Type B, Sigma-Aldrich) (50 μL, 0.5 mg/mL) was immobilized on a 96-well plate for 2 h at RT. Wells were washed, blocked with 5% BSA for 1 h at RT, and washed again. 50 μL of 5 μM binders (BSA, iFab-CHPNB, iFab-CHP) were added to their respective wells and incubated at RT for 2 h. Wells were washed and biotin-hTNFα (R&D Systems) (2 μg/mL, 50 μL) was added to each well for 1 h at RT. Wells were washed, and avidin-HRP (ThermoFisher Scientific) (1/2000 dilution, 50 μL) was added for 1 h at RT. Wells were washed and signal was generated by addition of 100 μL QuantaBlu Fluorogenic Peroxidase Substrate working solution for 20 min prior to addition of 100 μL stop solution. Signal was measured as described above (Ex: 325 nm, Em: 420 nm).
Biodistribution Study.
All animal experiments were performed in accordance with the University of Utah Institutional Animal Care and Use Committee (IACUC). As described previously,37 healthy female Nu/Nu mice (n = 5), age 5–6 weeks, were used for this study. 2 nmol (10 μM, 200 μL) of either bFab-(IR680)CHP or bFab-(IR680)CHPNB was injected into each mouse via IV tail vein injection. Mice were visualized at 0.5, 12, 24, 48, 72, 96, 120, 144, 168, 192, and 240 h postinjection using an IVIS imager (PerkinElmer) with an excitation/emission wavelength 675/720 nm and exposure time of 1 s. Fluorescence measurements were averaged at each time point and divided by the average fluorescence measurement from the 0.5 h time point, to calculate a percent remaining. Mice were sacrificed at 240 h post-injection. Organs and skeletons were collected for further analysis and scanned using the same IVIS imager.
In Vivo Therapeutic Efficacy Study.
hTNFα expressing transgenic mice (tg/wt) were purchased from Taconic [model #: 1006-F, B6.Cg-Tg(TNF)#Xen] and delivered at 8 weeks of age. Mice were allowed to acclimate for 1 week prior to any procedures in accordance with approved IACUC protocols. At 9 weeks of age (disease onset), mice (n = 3) were IV tail vein injected with 85 nmol/kg (5 mg/kg) iFab-CHP or equivalent volume of PBS pH 7.4. Mice were observed 3 times per week to blindly score joint swelling, measure joint width and thickness, as well as monitor weight, lethargy, and signs of distress. Each mouse paw was assessed by a blinded observer and given two scores (paw and digits) between 0 and 4 (0.5 increments, increasing severity) for a maximum score of 32 per mouse. Mouse hind paw width and thickness were measured by calipers (two measurements per hind paw, four measurements per mouse) three times per week. Score and measurements were summed for each mouse and averaged among that treatment group for each week. Mice were given food and water ad libitum. Second injections of equal dose were administered 7 weeks post-initial injection. At 10 weeks post-initial injection, mice were sacrificed and hind paw tissue was collected and fixed overnight at 4 °C in 10% v/v formalin. Samples were then sent to the ARUP Laboratories Histology Core Facility at the Huntsman Cancer Institute for paraffin embedding and sectioning. Due to a miscommunication, tissue samples were not decalcified prior to embedding. To compensate, paraffin blocks were surface decalcified (~1.5 h) prior to sectioning. Sections were cut along the midsagittal plane at 5 μm thickness.
Histology Staining – Hematoxylin & Eosin (H&E).
Tissue sections were deparaffinized and rehydrated by overnight incubation at 65 °C followed by 5 min incubations in xylene (3×), 100% EtOH (2×), 70% EtOH (2×), and running deionized (DI) water. Slides were stained by 5 min incubation in Mayer’s hematoxylin, 5 min in running DI water, and 3 min in eosin. After this step, slides were transferred to DI water and checked under a microscope to assess stain intensity. If overstained, slides were then incubated in 80% EtOH for 5 min. Next, slides were dipped (10–15 s) in 70% EtOH, 80% EtOH, 90% EtOH, 100% EtOH (3×), xylene (3×), and final incubation in xylene for 5 min. Stained tissue sections were then mounted using glass coverslips.
Histology Staining – Safranin-O with Fast Green.
Tissue sections were deparaffinized and rehydrated as described above. Sections were then stained with Weigert’s iron hematoxylin (1:1 dilution) for 4 min, followed by a 10 min incubation in running DI water. Sections were then incubated in 0.05% Fast Green for 5 min, dipped (10–15 s) in 1% acetic acid, and incubated in 0.1% Safranin-O for 13 min. After this step, slides were transferred to DI water and checked under a microscope to assess stain intensity. Slides were trapped by dipping (10–15 s) in 95% EtOH, followed by 30 s incubations in 100% EtOH (4×), and then 2 min in xylene (2×). Slides were mounted as described above.
Data Analysis.
All statistical analysis was performed using GraphPad Prism ver. 8.2.1 (GraphPad Software). Unless otherwise stated, data is presented as a mean ± standard deviation. Analysis of ELISA-like assay data was performed using a one-way ANOVA with Bonferroni post hoc correction to identify statistical significance. In vivo data was analyzed by multiple t tests using the Holm-Sidak method. α = 0.05, ns = P > 0.05; *P ≤ 0.05; **P < 0.01; ***/†P < 0.001; ****/‡P < 0.0001.
Supplementary Material
ACKNOWLEDGMENTS
We thank Connor Meaney and Tim Moore for their expertise and assistance in scoring the mice. We also thank Dr. Eileen Hwang for providing bevacizumab, as well as Drs. Yang Li and Hendra Wahyudi for providing caged CHPs. TOC figure as well as Figures 2, S4, and Scheme 1 were prepared using BioRender.
Funding
Support was provided by the National Institutes of Health R21-EY029430 and by The University of Utah Graduate Research Fellowship.
ABBREVIATIONS
- RA
rheumatoid arthritis
- CHP
collagen hybridizing peptide
- iFab
infliximab Fab fragment
- bFab
bevacizumab Fab fragment
- dCol
denatured collagen
- TNFα
tumor necrosis factor-α
- CR
conjugation ratio
- CRavg
average conjugation ratio
- VEGFA
vascular endothelial growth factor A
- Fc
crystallizable fragment
- SPPS
solid-phase peptide synthesis
- MWCO
molecular weight cutoff
- BSA
bovine serum albumin
- CF
5(6)-carboxyfluorescein
- NIRF
IR680 fluorophore
- sulfo-SMCC
sulfosuccinimidyl 4-(N-maleimidomethyl)-cyclohexane-1-carboxylate
- HRP
horseradish peroxidase
- TFA
trifluoroacetic acid
- MALDI-TOF MS
matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
- RT
room temperature
- DMSO
dimethyl sulfoxide
- mol eq
molar equivalents
- IACUC
Institutional Animal Care and Use Committee
- DI
deionized
- MMP
matrix metalloproteinase
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.0c00324.
Figure S1: Chemical structures of CHP compounds. Figure S2: SDS-PAGE analysis of iFab-CHP conjugation. Figure S3: Intact LC-MS analysis of iFab and iFab-CHP conjugates. Figure S4: ELISA-like assays to assess binding of bFab-CHP conjugates. Figure S5: Mouse weight during therapeutic efficacy study. Figure S6: Real-time monitoring of disease progression by joint scoring and thickness. (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.bioconjchem.0c00324
The authors declare the following competing financial interest(s): S.M.Y. is a cofounder of 3Helix, which commercializes CHP.
Contributor Information
Keith J. Arlotta, Department of Biomedical Engineering, University of Utah, Salt Lake City, Utah 84112, United States
Boi Hoa San, Department of Biomedical Engineering, University of Utah, Salt Lake City, Utah 84112, United States.
Hong-Hua Mu, Department of Internal Medicine, University of Utah, Salt Lake City, Utah 84132, United States.
S. Michael Yu, Department of Biomedical Engineering and Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, Utah 84112, United States.
Shawn C. Owen, Department of Biomedical Engineering and Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, Utah 84112, United States.
REFERENCES
- (1).Chaudhari K, Rizvi S, and Syed BA (2016) Rheumatoid arthritis: current and future trends. Nat. Rev. Drug Discovery 15, 305–306. [DOI] [PubMed] [Google Scholar]
- (2).Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, Kavanaugh A, McInnes IB, Solomon DH, Strand V, and Yamamoto K (2018) Rheumatoid arthritis. Nat. Rev. Dis. Primers 4, 1 DOI: 10.1038/nrdp.2018.1. [DOI] [PubMed] [Google Scholar]
- (3).Rheumatoid Arthritis, Center for Disease Control and Prevention. https://www.cdc.gov/arthritis/basics/rheumatoid-arthritis.html (accessed April 10th, 2020). [Google Scholar]
- (4).Malmstrom V, Catrina AI, and Klareskog L (2017) The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting. Nat. Rev. Immunol 17, 60–75. [DOI] [PubMed] [Google Scholar]
- (5).Keyszer G, Redlich A, Häupl T, Zacher J, Sparmann M, Ungethüm U, Gay S, and Burmester GR (1998) Differential expression of cathepsins B and L compared with matrix metalloproteinases and their respective inhibitors in rheumatoid arthritis and osteoarthritis: A parallel investigation by semiquantitative reverse transcriptase-polymerase chain reaction and immunohistochemistry. Arthritis Rheum 41, 1378–1387. [DOI] [PubMed] [Google Scholar]
- (6).Li Y, and Yu SM (2013) Targeting and mimicking collagens via triple helical peptide assembly. Curr. Opin. Chem. Biol 17, 968–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, and Braun MM (2001) Tuberculosis Associated with Infliximab, a Tumor Necrosis Factor a–Neutralizing Agent. N. Engl. J. Med 345, 1098–1104. [DOI] [PubMed] [Google Scholar]
- (8).Cush JJ (2004) Safety overview of new disease-modifying antirheumatic drugs. Rheum. Dis. Clin. North. Am 30, 237–255. [DOI] [PubMed] [Google Scholar]
- (9).Hochberg MC, Lebwohl MG, Plevy SE, Hobbs KF, and Yocum DE (2005) The benefit/risk profile of TNF-blocking agents: findings of a consensus panel. Semin. Arthritis Rheum 34, 819–836. [DOI] [PubMed] [Google Scholar]
- (10).Lisman KA, Stetson SJ, Koerner MM, Farmer JA, and Torre-Amione G (2002) The Role of Tumor Necrosis Factor Alpha Blockade in the Treatment of Congestive Heart Failure. Congestive Heart Failure 8, 275–279. [DOI] [PubMed] [Google Scholar]
- (11).Hoekstra M, van Ede AE, Haagsma CJ, van de Laar MA, Huizinga TW, Kruijsen MW, and Laan RF (2003) Factors associated with toxicity, final dose, and efficacy of methotrexate in patients with rheumatoid arthritis. Ann. Rheum. Dis 62, 423–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, Donlin LT, Henderson LA, Wei K, Mizoguchi F, Teslovich NC, Weinblatt ME, Massarotti EM, Coblyn JS, Helfgott SM, Lee YC, Todd DJ, Bykerk VP, Goodman SM, Pernis AB, Ivashkiv LB, Karlson EW, Nigrovic PA, Filer A, Buckley CD, Lederer JA, Raychaudhuri S, and Brenner MB (2017) Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Fonseka CY, Rao DA, and Raychaudhuri S (2017) Leveraging blood and tissue CD4+ T cell heterogeneity at the single cell level to identify mechanisms of disease in rheumatoid arthritis. Curr. Opin. Immunol 49, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jones GW, and Jones SA (2016) Ectopic lymphoid follicles: inducible centres for generating antigen-specific immune responses within tissues. Immunology 147, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cohen M, Omair MA, and Keystone EC (2013) Monoclonal Antibodies in Rheumatoid Arthritis. Int. J. Clin. Rheumatol 8, 541–556. [Google Scholar]
- (16).Hansel TT, Kropshofer H, Singer T, Mitchell JA, and George AJ (2010) The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discovery 9, 325–338. [DOI] [PubMed] [Google Scholar]
- (17).Hwang J, Huang Y, Burwell TJ, Peterson NC, Connor J, Weiss SJ, Yu SM, and Li Y (2017) In Situ Imaging of Tissue Remodeling with Collagen Hybridizing Peptides. ACS Nano 11, 9825–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Li Y, Foss CA, Summerfield DD, Doyle JJ, Torok CM, Dietz HC, Pomper MG, and Yu SM (2012) Targeting collagen strands by photo-triggered triple-helix hybridization. Proc. Natl. Acad. Sci. U. S. A 109, 14767–14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wahyudi H, Reynolds AA, Li Y, Owen SC, and Yu SM (2016) Targeting collagen for diagnostic imaging and therapeutic delivery. J. Controlled Release 240, 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Burkhardt H, Sehnert B, Bockermann R, Engstrom A, Kalden JR, and Holmdahl R (2005) Humoral immune response to citrullinated collagen type II determinants in early rheumatoid arthritis. Eur. J. Immunol 35, 1643–1652. [DOI] [PubMed] [Google Scholar]
- (21).Liang B, Ge C, Lonnblom E, Lin X, Feng H, Xiao L, Bai J, Ayoglu B, Nilsson P, Nandakumar KS, Zhao M, and Holmdahl R (2019) The autoantibody response to cyclic citrullinated collagen type II peptides in rheumatoid arthritis. Rheumatology 58, 1623–1633. [DOI] [PubMed] [Google Scholar]
- (22).Yoshida M, Tsuji M, Kurosaka D, Kurosaka D, Yasuda J, Ito Y, Nishizawa T, and Yamada A (2006) Autoimmunity to citrullinated type II collagen in rheumatoid arthritis. Mod. Rheumatol 16, 276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Engel J, and Bächinger HP (2005) Structure, Stability and Folding of the Collagen Triple Helix, in Collagen: Primer in Structure, Processing and Assembly (Brinckmann J, Notbohm H, and Müller PK, Eds.) pp 7–33, Springer Berlin Heidelberg, Berlin, Heidelberg. [Google Scholar]
- (24).Li Y, Ho D, Meng H, Chan TR, An B, Yu H, Brodsky B, Jun AS, and Yu SM (2013) Direct detection of collagenous proteins by fluorescently labeled collagen mimetic peptides. Bioconjugate Chem 24, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bennink LL, Smith DJ, Foss CA, Pomper MG, Li Y, and Yu SM (2017) High Serum Stability of Collagen Hybridizing Peptides and Their Fluorophore Conjugates. Mol. Pharmaceutics 14, 1906–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Arlotta KJ, and Owen SC (2019) Antibody and antibody derivatives as cancer therapeutics. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol 11, e1556. [DOI] [PubMed] [Google Scholar]
- (27).Katsumata K, Ishihara J, Mansurov A, Ishihara A, Raczy MM, Yuba E, and Hubbell JA (2019) Targeting inflammatory sites through collagen affinity enhances the therapeutic efficacy of anti-inflammatory antibodies. Sci. Adv 5, eaay1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Fang J, Nakamura H, and Maeda H (2011) The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Delivery Rev 63, 136–151. [DOI] [PubMed] [Google Scholar]
- (29).Maeda H, Wu J, Sawa T, Matsumura Y, and Hori K (2000) Tumor Vascular Permeability and the EPR Effect in Macromolecular Therapeutics: a Review. J. Controlled Release 65, 271–284. [DOI] [PubMed] [Google Scholar]
- (30).Yu L, Wu X, Cheng Z, Lee CV, LeCouter J, Campa C, Fuh G, Lowman H, and Ferrara N (2008) Interaction between bevacizumab and murine VEGF-A: a reassessment. Invest. Ophthalmol. Visual Sci 49, 522–527. [DOI] [PubMed] [Google Scholar]
- (31).Assas BM, Levison SE, Little M, England H, Battrick L, Bagnall J, McLaughlin JT, Paszek P, Else KJ, and Pennock JL (2017) Anti-inflammatory effects of infliximab in mice are independent of tumour necrosis factor alpha neutralization. Clin. Exp. Immunol 187, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Gandhi AV, Arlotta KJ, Chen HN, Owen SC, and Carpenter JF (2018) Biophysical Properties and Heating-Induced Aggregation of Lysine-Conjugated Antibody-Drug Conjugates. J. Pharm. Sci 107, 1858–1869. [DOI] [PubMed] [Google Scholar]
- (33).Beck A, Goetsch L, Dumontet C, and Corvaia N (2017) Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discovery 16, 315–337. [DOI] [PubMed] [Google Scholar]
- (34).Kaymakcalan Z, Sakorafas P, Bose S, Scesney S, Xiong L, Hanzatian DK, Salfeld J, and Sasso EH (2009) Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin. Immunol 131, 308–316. [DOI] [PubMed] [Google Scholar]
- (35).Sun X, Ponte JF, Yoder NC, Laleau R, Coccia J, Lanieri L, Qiu Q, Wu R, Hong E, Bogalhas M, Wang L, Dong L, Setiady Y, Maloney EK, Ab O, Zhang X, Pinkas J, Keating TA, Chari R, Erickson HK, and Lambert JM (2017) Effects of Drug-Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody-Maytansinoid Conjugates. Bioconjugate Chem 28, 1371–1381. [DOI] [PubMed] [Google Scholar]
- (36).Buecheler JW, Winzer M, Tonillo J, Weber C, and Gieseler H (2018) Impact of Payload Hydrophobicity on the Stability of Antibody–Drug Conjugates. Mol. Pharmaceutics 15, 2656–2664. [DOI] [PubMed] [Google Scholar]
- (37).San BH, Hwang J, Sampath S, Li Y, Bennink LL, and Yu SM (2017) Self-Assembled Water-Soluble Nanofibers Displaying Collagen Hybridizing Peptides. J. Am. Chem. Soc 139, 16640–16649. [DOI] [PubMed] [Google Scholar]
- (38).Perry M, Bewshea C, Brown R, So K, Ahmad T, and McDonald T (2015) Infliximab and adalimumab are stable in whole blood clotted samples for seven days at room temperature. Ann. Clin. Biochem 52, 672–674. [DOI] [PubMed] [Google Scholar]
- (39).Rengel Y, Ospelt C, and Gay S (2007) Proteinases in the joint: clinical relevance of proteinases in joint destruction. Arthritis Res. Ther 9, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Rose BJ, and Kooyman DL (2016) A Tale of Two Joints: The Role of Matrix Metalloproteases in Cartilage Biology. Dis. Markers 2016, 4895050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sun S, Bay-Jensen A-C, Karsdal MA, Siebuhr AS, Zheng Q, Maksymowych WP, Christiansen TG, and Henriksen K (2014) The active form of MMP-3 is a marker of synovial inflammation and cartilage turnover in inflammatory joint diseases. BMC Musculoskeletal Disord 15, 1 DOI: 10.1186/1471-2474-15-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Biancheri P, Brezski RJ, Di Sabatino A, Greenplate AR, Soring KL, Corazza GR, Kok KB, Rovedatti L, Vossenkamper A, Ahmad N, Snoek SA, Vermeire S, Rutgeerts P, Jordan RE, and MacDonald TT (2015) Proteolytic cleavage and loss of function of biologic agents that neutralize tumor necrosis factor in the mucosa of patients with inflammatory bowel disease. Gastroenterology 149, 1564–1574. [DOI] [PubMed] [Google Scholar]
- (43).Ponte JF, Sun X, Yoder NC, Fishkin N, Laleau R, Coccia J, Lanieri L, Bogalhas M, Wang L, Wilhelm S, Widdison W, Pinkas J, Keating TA, Chari R, Erickson HK, and Lambert JM (2016) Understanding How the Stability of the Thiol-Maleimide Linkage Impacts the Pharmacokinetics of Lysine-Linked Antibody-Maytansinoid Conjugates. Bioconjugate Chem 27, 1588–1598. [DOI] [PubMed] [Google Scholar]
- (44).Fishkin N, Maloney EK, Chari RV, and Singh R (2011) A novel pathway for maytansinoid release from thioether linked antibody-drug conjugates (ADCs) under oxidative conditions. Chem. Commun 47, 10752–10754. [DOI] [PubMed] [Google Scholar]
- (45).Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, and Senter PD (2008) Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjugate Chem 19, 759–765. [DOI] [PubMed] [Google Scholar]
- (46).Arlotta KJ, Gandhi AV, Chen H-N, Nervig CS, Carpenter JF, and Owen SC (2018) In-Depth Comparison of Lysine-Based Antibody-Drug Conjugates Prepared on Solid Support Versus in Solution. Antibodies 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Schinnerling K, Rosas C, Soto L, Thomas R, and Aguillon JC (2019) Humanized Mouse Models of Rheumatoid Arthritis for Studies on Immunopathogenesis and Preclinical Testing of Cell-Based Therapies. Front. Immunol 10, 1 DOI: 10.3389/fimmu.2019.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Tseng J-C, Vasquez KV, and Peterson JD (2015) Optical Imaging on the IVIS Spectrum CT System: General and Technical Considerations for 2D and 3D Imaging. Perkin Elmer Technical Notes [Google Scholar]
- (49).Li Y, Foss CA, Pomper MG, and Yu SM (2014) Imaging Denatured Collagen Strands In vivo and Ex vivo via Photo-triggered Hybridization of Caged Collagen Mimetic Peptides. J. Visualized Exp, e51052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Klotz U, Teml A, and Schwab M (2007) Clinical Pharmacokinetics and Use of Infliximab. Clin. Pharmacokinet 46, 645–660. [DOI] [PubMed] [Google Scholar]
- (51).Bennink LL (May 2019) Department of Biomedical Engineering, The University of Utah, Salt Lake City, UT. [Google Scholar]
- (52).Deora A, Hegde S, Lee J, Choi CH, Chang Q, Lee C, Eaton L, Tang H, Wang D, Lee D, Michalak M, Tomlinson M, Tao Q, Gaur N, Harvey B, McLoughlin S, Labkovsky B, and Ghayur T (2017) Transmembrane TNF-dependent uptake of anti-TNF antibodies. mAbs 9, 680–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hayward MD, Jones BK, Saparov A, Hain HS, Trillat AC, Bunzel MM, Corona A, Li-Wang B, Strenkowski B, Giordano C, Shen H, Arcamone E, Weidlick J, Vilensky M, Tugusheva M, Felkner RH, Campbell W, Rao Y, Grass DS, and Buiakova O (2007) An extensive phenotypic characterization of the hTNFalpha transgenic mice. BMC Physiol 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).de Vries HS, van Oijen MG, Driessen RJ, de Jong EM, Creemers MC, Kievit W, and de Jong DJ (2011) Appropriate infliximab infusion dosage and monitoring: results of a panel meeting of rheumatologists, dermatologists and gastroenterologists. Br. J. Clin. Pharmacol 71, 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Semerano L, Biton J, Delavallee L, Duvallet E, Assier E, Bessis N, Bernier E, Dhellin O, Grouard-Vogel G, and Boissier MC (2013) Protection from articular damage by passive or active anti-tumour necrosis factor (TNF)-alpha immunotherapy in human TNF-alpha transgenic mice depends on anti-TNF-alpha antibody levels. Clin. Exp. Immunol 172, 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Shealy D, Cai A, Staquet K, Baker A, Lacy ER, Johns L, Vafa O, Gunn G, Tam S, Sague S, Wang D, Brigham-Burke M, Dalmonte P, Emmell E, Pikounis B, Bugelski PJ, Zhou HZ, Scallon B, and Giles-Komar J (2010) Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor alpha. mAbs 2, 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Brand DD, Latham KA, and Rosloniec EF (2007) Collagen-induced arthritis. Nat. Protoc 2, 1269–1275. [DOI] [PubMed] [Google Scholar]
- (58).Cole BC, Mu H-H, and Sawitzke AD (2000) The mycoplasma superantigen MAM: Role in arthritis and immune-mediated disease. Int. J. Med. Microbiol 290, 489–490. [DOI] [PubMed] [Google Scholar]
- (59).Caplazi P, and Diehl L (2015) Histopathology in Mouse Models of Rheumatoid Arthritis, in Molecular Histopathology and Tissue Biomarkers in Drug and Diagnostic Development (Potts SJ, Eberhard DA, and Wharton JKA, Eds.) pp 65–78, Springer New York, New York, NY. [Google Scholar]
- (60).Bolon B, Stolina M, King C, Middleton S, Gasser J, Zack D, and Feige U (2011) Rodent preclinical models for developing novel antiarthritic molecules: comparative biology and preferred methods for evaluating efficacy. J. Biomed. Biotechnol 2011, 569068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Malfait AM, Tortorella M, Thompson J, Hills R, Meyer DM, Jaffee BD, Chinn K, Ghoreishi-Haack N, Markosyan S, and Arner EC (2009) Intra-articular injection of tumor necrosis factor-alpha in the rat: an acute and reversible in vivo model of cartilage proteoglycan degradation. Osteoarthritis Cartilage 17, 627–635. [DOI] [PubMed] [Google Scholar]
- (62).Oggionni T, Morbini P, Inghilleri S, Palladini G, Tozzi R, Vitulo P, Fenoglio C, Perlini S, and Pozzi E (2006) Time course of matrix metalloproteases and tissue inhibitors in bleomycin-induced pulmonary fibrosis. Eur. J. Histochem 50, 317–325. [PubMed] [Google Scholar]
- (63).Tlsty TD, and Coussens LM (2006) Tumor stroma and regulation of cancer development. Annu. Rev. Pathol.: Mech. Dis 1, 119–150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.