

Abstract
To identify potent antiviral compounds, we introduced a high-throughput screen platform that can rapidly classify hit compounds according to their target. In our platform, we performed a compound screen using a lentivirus-based pseudovirus presenting a spike protein of coronavirus, and we evaluated the hit compounds using an amplified luminescence proximity homogeneous assay (alpha) test with purified host receptor protein and the receptor binding domain of the viral spike. With our screen platform, we were able to identify both spike-specific compounds (class I) and broad-spectrum antiviral compounds (class II). Among the hit compounds, thiosemicarbazide was identified to be selective to the interaction between the viral spike and its host cell receptor, and we further optimized the binding potency of thiosemicarbazide through modification of the pyridine group. Among the class II compounds, we found raloxifene and amiodarone to be highly potent against human coronaviruses including Middle East respiratory syndrome coronavirus (MERS-CoV), severe acute respiratory syndrome coronavirus (SARS-CoV), and SARS-CoV-2. In particular, using analogs of the benzothiophene moiety, which is also present in raloxifene, we have identified benzothiophene as a novel structural scaffold for broad-spectrum antivirals. This work highlights the strong utility of our screen platform using a pseudovirus assay and an alpha test for rapid identification of potential antiviral compounds and their mechanism of action, which can lead to the accelerated development of therapeutics against newly emerging viral infections.
Graphical abstract
1. Introduction
Human coronaviruses (HCoV) have infected humans and caused diseases that resulted in endemic and recent pandemic crises (Geller et al., 2012; Paules et al., 2020; Song et al., 2019). Since HCoV 229E and OC43 were first reported in the 1960s, for decades they were considered to be the major human-infecting coronaviruses (Cui et al., 2019). However, the emergence of Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012 (Ksiazek et al., 2003; Zaki et al., 2012), which followed the identification of severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002 (Guan et al., 2003; Rota et al., 2003), proved that these pathogenic viruses frequently cross the species border and may pose a significant risk to public health. Recently, COVID-19-causing SARS-CoV-2 (Andersen et al., 2020; Lam et al., 2020; Shang et al., 2020) resulted in over four-million infection cases within six months, and the WHO declared a pandemic (Hu et al., 2021; Huang et al., 2020; Li et al., 2020). Only six months after this declaration, the virus had caused more than 32 million infection cases and posed an enormous threat to public health, putting most nations on hold (Eurosurveillance Editorial, 2020). Despite the substantial effort placed into the development of vaccines (Krammer, 2020; Tregoning et al., 2021), many nations are still putting effort into implementing social distancing or containment policies to prevent further spread of the viral infection due to the continuous appearance of mutant strains. In addition, because there is a possibility of the appearance of infectious mutants of other coronavirus species, there is a pressing and urgent need to develop broad-spectrum antiviral drugs. Today, a few drugs have been licensed for the treatment of COVID-19, but they all display limited efficacy. Therefore, additional antiviral therapeutics applicable to the clinic need to be developed.
The entry of coronaviruses into the host cell involves a series of steps, and as an enveloped virus, fusion of the coronavirus membrane with the cellular membrane initiates the early process of entry. This can occur either at the plasma membrane or at a distinct endosomal compartment, which is partially governed by pH-dependent viral fusion machineries (Hartenian et al., 2020). Coronaviruses, including SARS-CoV-2 and MERS-CoV, first attach to the heparan sulfate proteoglycans of the host cell (Hartenian et al., 2020). This initial adherence leads to a bridge interaction with the fusion receptors on the host cell. Coronaviruses employ various cellular fusion receptors: SARS-CoV and SARS-CoV-2 are known to bind to angiotensin-converting enzyme 2 (ACE2) (Li et al., 2003, 2005), while MERS-CoV was reported to utilize dipeptidyl peptidase 4 (DPP4) (Raj et al., 2013, 2014). The recognition of host receptors then initiates internalization of the virus via various routes (Fuentes-Prior, 2021; Shang et al., 2020), but the most common is the induction of fusion with cellular membrane followed by clathrin-dependent endocytosis (Burkard et al., 2014; Wang et al., 2008; Zhou et al., 2016). The internalization process and fusion factors have been extensively studied, and therefore the early stage of viral infection has been implicated as one of the most favorable drug targets.
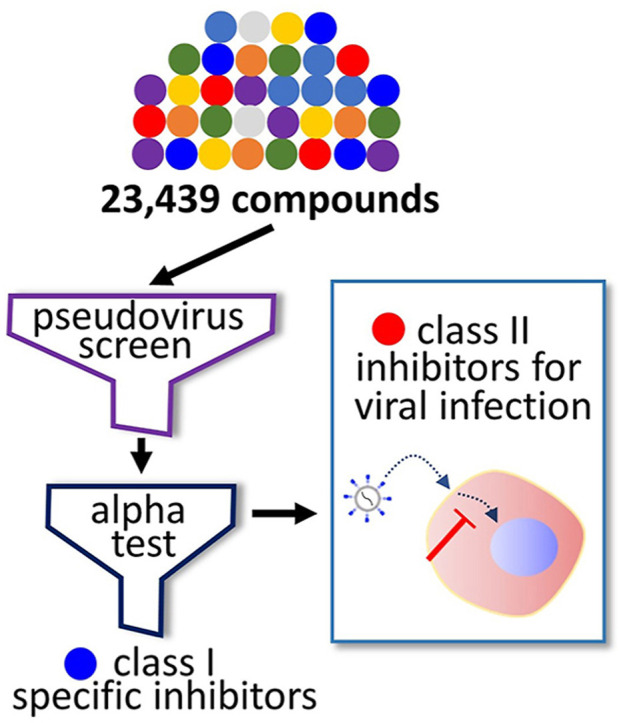
In our effort to identify potent inhibitors of coronavirus infection, we have employed a pseudovirus bearing the full-length spike of MERS-CoV for the compound screen of over 23,000 small molecules. The resulting 1,200 positive hits were further tested by the amplified luminescence proximity homogeneous assay (alpha) that tested the compound efficacy to prevent the spike-DPP4 interaction. The combination of the two assays allowed rapid classification of the hits into two classes: (1) compounds that prevent binding of the spike to its receptor and (2) compounds that inhibit viral infection independent of spike-receptor interaction. In addition, using our cell-based infection assays, we show that thiosemicarbazides are selectively potent against MERS-CoV infection, while several known drugs including amiodarone and raloxifene have broad-spectrum potency against other coronaviruses including SARS-CoV-2. Importantly, the potency of raloxifene against SARS-CoV-2 was validated by an in vivo hamster infection model. In this study, we demonstrate that the pseudovirus-based screen and alpha test combined with other in vitro and in vivo assays are useful for the rapid identification of efficacy measures and mechanism of action of compounds.
2. Materials and methods
2.1. Cell line and virus maintenance
Lenti-X™ 293T cells (Clontech, Mountain View, CA) were cultivated in Dulbecco's Modified Eagle's Medium (DMEM; Sigma-Aldrich, St. Louis, MO) supplemented with 4.5 g/L glucose, 4 mM L-glutamine, 3.7 g/L NaHCO3, 10% fetal bovine serum (FBS; Gibco, Billings, MT), 1 mM sodium pyruvate (Sigma-Aldrich), and 1% penicillin/streptomycin (Sigma-Aldrich). Huh-7 cells (KCLB, Seoul, South Korea) were cultured in Roswell Park Memorial Institute (RPMI-1640; Gibco) medium supplemented with 10% FBS and 1% penicillin/streptomycin. Vero cells (ATCC, Manassas, VA) were cultured in Opti-PRO™ SFM (Gibco) supplemented with 4 mM L-glutamine and 1× Antibiotic-Antimycotic (Gibco). Vero E6 cells were cultured in DMEM (Corning Incorporated, Corning, NY) containing 5% FBS. MRC-5 cells were cultured in Eagle's Minimum Essential Medium (EMEM, Gibco) supplemented with 10% FBS and 1% penicillin/streptomycin. All mammalian cells were maintained at 37°C in a humidified 5% CO2 incubator unless otherwise noted. Serum-free media (SFM)-adapted Spodoptera frugiperda 9 (Sf9) cells (Thermo Fisher Scientific, Waltham, MA) were cultured in Sf-900™ III SFM medium at 27°C with continuous agitation. Patient-derived MERS-CoV (passage 4, MERS-CoV/KOR/KNIH/002_05_2015; GenBank: KT029139.1) was obtained from the Korea Centers for Disease Control and Prevention. SARS-CoV (HKU39849) was obtained from the University of Hong Kong-Pasteur Research Pole. Human coronavirus 229E (HCoV-229E) and human coronavirus OC43 (HCoV-OC43) were obtained from ATCC.
2.2. Chemical compounds and antibodies
The chemical library used for high-throughput screen consists of 978 drugs from the FDA-approved drug library (Selleckchem, Houston, TX) and 1280 compounds from the Library of Pharmacologically Active Compounds (LOPAC®1280; Sigma-Aldrich). All stock compounds were at 10 mM in DMSO. Amiodarone, raloxifene, and chloroquine were purchased from Sigma-Aldrich. The analogs were synthesized as described in the Supplementary Information. The spike antibody was purchased from Sino Biological (R723; Beijing, China). Analogs of compound 2 were purchased as described in Fig. S13.
2.3. Pseudovirus preparation
The pseudovirus was produced as previously described (Zhao et al., 2013). A spike expression vector (pCMV3-Flag-CD5-Spike) was constructed by inserting the CD5 signal sequence upstream of the spike coding sequence of the pCMV3-Flag-Spike (betacoronavirus 2c EMC/2012) vector (VG40069-CF; Sino Biological), after which 4.5 × 106 Lenti-X™ 293T cells were co-transfected with 10 μg of a luciferase expression vector (pLenti-CMV-puro-luc; Addgene, Water Town, MA) (Campeau et al., 2009), 10 μg of a packaging vector (pCMVΔ8.2-dvpr; Addgene) (Stewart et al., 2003), and 600 ng of pCMV3-Flag-CD5-Spike in a 100 cm2 cell culture dish using the calcium phosphate transfection method (Invitrogen, Waltham, MA). After 48-h incubation, the virus-containing supernatant was harvested and concentrated using the Retro-X™ concentrator (Clontech) followed by titration using the Lenti-X™ p24 Rapid Titer kit (Clontech). As a positive control, a pseudovirus of vesicular stomatitis virus (VSV) was generated using pCMV-VSV-G (Sino Biological) as an envelope vector; and as a negative control, a pseudovirus of negative control vector (NCV) was generated using pCMV3FLAG-NCV (Sino Biological) as an envelope vector.
2.4. Pseudovirus infection assay
Huh-7 cells were plated in white 384-well plates at a density of 4 × 104 cells/well. After overnight incubation, compounds at a final concentration of 10 μM and pseudovirus at 5 × 103 IFU were added in sequence. Following a 72-h incubation, 20 μl of Bright-Glo® reagent (Promega, Madison, WI) was dispensed into each well, and the relative luminescence unit (RLU) was measured using an EnVision® multilabel plate reader (PerkinElmer, Waltham, MA). The inhibition of pseudovirus-derived luminescence was presented as % inhibition, which was calculated using the following equation: ([RLUpc – RLUsample]/[RLUpc – RLUnc) × 100], where pc and nc respectively indicate pseudovirus-infected positive control and mock-infected negative control. The cytotoxicity of compounds used in the screen was assessed using the CellTiter-Glo® reagent (Promega). Cytotoxicity was also presented in the manner described above, with pc meaning DMSO treatment and nc meaning media without cells. Activity of the hit compounds were examined by dose-response curve (DRC) analysis. For the high-throughput screening and DRC analyses, we fixed the viral titer and cell number to 5 IFU and 4 × 103 Huh-7 cells, respectively, in a 384-well plate, which resulted in a robust Z’ score of ∼0.6. From the DRC analyses, 50% effective concentrations (EC50) and 50% cytotoxic concentrations (CC50) were calculated using the non-linear regression formula of GraphPad Prism 6 software (GraphPad Software, La Jolla, CA).
2.5. Purification of hDPP4 and spike RBD proteins
For the expression of MERS-CoV spike RBD, we constructed a pFastbac-HBM-RBD(367–606) vector by cloning an RBD gene (residues 367–606) amplified from the pCMV3-Flag-Spike vector using the Bac-to-Bac® HBM TOPO® cloning kit (Invitrogen). To introduce an N-terminal GP67 signal sequence in place of HBM and to insert C-terminal Strep·tag®II, we constructed a pFastbac-GP67-RBD(367–606)-Strep·tag®II vector by cloning a codon-optimized 906 bp gene encoding GP67-RBD(367–606)-Strep·tag®II (GenBank: AKN11075.1) into the pFastbac-HBM-RBD(367–606) vector using BamHI/XhoI restriction sites. For the expression of hDPP4 with a C-terminal hexa-histidine tag, we constructed a plasmid, pFastbac-HBM-hDPP4(39–766), by cloning codon-optimized hDPP4 (residues 39–766) using the Bac-to-Bac® HBM TOPO® cloning kit (Invitrogen). The pFastbac-GP67-RBD(367–606)-Strep·tag®II vector and the pFastbac-HBM-hDPP4(39–766) were each transformed into MAX Efficiency™ DH10Bac™ competent cells (Invitrogen), at which point the recombinant bacmid DNAs were extracted and transfected into Sf9 cells using Cellfectin™ reagent (Invitrogen). After incubation for 7 days, P1 viral stocks were harvested, reinoculated, and further amplified. The resulting P2 viral stocks were titrated with the plaque assay, and a high titer (0.5–1 × 109 pfu) was inoculated in 1 L of Sf9 cells. Supernatant containing the target protein was harvested 3 days after inoculation. The supernatant containing RBD was equilibrated by the addition of 100 mL of 10× buffer W (1 M Tris-Cl, pH 8.0, 1.5 M NaCl, 10 mM EDTA), and endogenous biotin was blocked by incubation with 2.4 mL of BioLock Biotin blocking solution (IBA Lifesciences, Göttingen, Germany). Debris was filtered out using a Nalgene™ Rapid-Flow™ filter (Thermo Scientific). Strep·tag®II (STII)-tagged RBD was purified using Strep-Tactin® Sepharose beads (IBA Lifesciences) according to the manufacturer's instructions. The supernatant containing hDPP4 was filtered with the same filter mentioned earlier, and it was then consecutively purified with affinity chromatography using Ni sepharose® 6 Fast Flow beads (GE Healthcare, Chicago, IL) and ion exchange chromatography using SP sepharose® Fast Flow beads (GE Healthcare). Each protein eluate was concentrated using Vivaspin centrifugal concentrators (Satorious, Göttingen, Germany) and buffer-exchanged in a dialysis buffer (PBS, pH 7.4, 10% glycerol) for 12 h at 4°C.
2.6. Alpha test
In the alpha test, a photosensitizer in the “Donor” bead converts ambient oxygen to a more excited singlet state. The singlet state oxygen molecules diffuse across to react with a thioxene derivative in an "Acceptor" bead, generating chemiluminescence at 370 nm, which further activates the fluorophores to emit light at 520–620 nm. Before screening, we excluded false positives by separately testing the compounds for their ability to quench singlet oxygen or emission light using the AlphaScreen® TrueHits kit and optimized the concentrations of proteins and beads. The optimized assay was performed in assay buffer containing 1× PBS, pH 7.4, 0.01% Tween 20, and 0.1% BSA in a white 384-well OptiPlate® (PerkinElmer) at 25°C. For the reaction, compounds (20 μM) were first incubated with recombinant hDPP4 protein (25 nM) and then with RBD protein (100 nM) for 15 min each, after which 5 μg/mL of each of Strep-Tactin® AlphaLISA® Acceptor bead (PerkinElmer) and Nickel Chelate Alpha Donor bead (PerkinElmer) were added. The Alpha signals were measured using an EnVision® multilabel plate reader (PerkinElmer) after 1-h incubation in the dark. The activity of the hit compounds was determined by the DRC experiments, and the 50% inhibitory concentration (IC50) values were calculated using the GraphPad Prism 6 software (GraphPad Software).
2.7. Pull-down assay of hDPP4 and spike RBD
For the pull-down assay, purified hDPP4 and RBD proteins were incubated at 4°C for 1 h, followed by incubation with Ni Sepharose 6 Fast Flow beads for 2 h. After being washed three times with the assay buffer containing 1× PBS, pH 7.4, 0.01% Tween 20, and 0.1% BSA, the assay was performed in duplicate, out of which one reaction was analyzed by SDS-PAGE and Coomassie Blue staining and the other by immunoblot using the Strep-Tactin®-HRP conjugated antibody (IBA Lifesciences).
2.8. ELISA
An hDPP4 protein (5 μg/mL) was immobilized on a Ni-coated 96-well plate (Thermo Scientific). After 1-h incubation and washing, serially diluted compounds and RBD were added to the plate. Following another 1-h incubation and washing, Strep-Tactin®-HRP conjugate (IBA Lifesciences) was added, and the reaction proceeded for an hour. After being washed three times, 50 μl of 1-Step™ ultra TMB-ELISA (Thermo Scientific) was added, and the chromogenic reaction continued until the appropriate color was developed. Upon termination of reaction by addition of sulfuric acid, absorbance was measured at 450 mM using a FlexStation® 3 multi-mode microplate reader (Molecular devices, San Jose, CA). The IC50 values of the test compounds were calculated using the GraphPad Prism 6 software (GraphPad Software).
2.9. In silico docking
To understand the inhibition mechanism of compound 1, docking analysis of hit compounds was performed using Schrodinger's Maestro 11.6 software. First, structures of the hits were sketched using a 2D sketch module, and a maximum of 32 tautomers that retained specified chiralities and possible ionization states at pH 7.0 were generated for each structure. Then, energy-minimization was conducted under the OPLS4e force field to obtain a stable conformation. Prior to docking, the X-ray co-crystal structure of MERS-CoV RBD in complex with DPP4 (PDB: 4L72) and the apo-structure of MERS-CoV RBD (PDB: 4L3N) were retrieved from the Protein Data Bank (www.rcsb.org). To optimize the structures of the proteins, the assignment of bond orders, removal of original hydrogens, generation of het states, and removal of duplicated side chains and water molecules were performed sequentially. A brief minimization was conducted in the OPLS3e force field using the default option. Several docking sites of protein were defined using a grid generation module by picking key residues and adjacent residues in the hDPP4 binding region of MERS-CoV RBD. To perform extra/precision Glide docking, XP docking was selected, and at least 10 poses per compounds were generated. The binding affinity of each of the docking poses of the ligand was calculated using the Glide docking score function. The most plausible binding mode of compound 1 was selected based on 3D docking poses, glide docking score, and visual inspection of the interactions between the ligand and key residues.
2.10. Electrostatic complementarity analysis
Electrostatic complementarity analysis was conducted using Cresset's Flare 3.0.0 software (Cheeseright et al., 2006). The best docking poses of compound 1 and 1h bound to the target protein (MERS-CoV spike RBD) were exported as SDF files and reloaded into the Flare program. The electrostatic complementary surface and score of each ligand were calculated in terms of the XED molecular mechanics force field using a default setting.
2.11. Cellular cholesterol measurement
Total cholesterol levels were measured using the Cholesterol Ester-Glo™ assay (Promega) according to the manufacturer's instructions. Briefly, A549 cells were seeded in white bottom 96-well plates at a density of 1 × 104 cells and incubated overnight. The cells were then treated with 10 μM raloxifene and incubated for 12 h, after which, a reagent containing cholesterol dehydrogenase, which luminesces under the presence of free cholesterol, is added. The luminescence intensity was measured using the Synergy HTX multi-mode plate reader (BioTek, Winooski, VT).
2.12. qRT-PCR of genes involved in the cholesterol pathway
A549 cells were seeded in 6-well plates at a density of 1 × 106 cells/well and incubated overnight. The cells were then treated with 10 μM raloxifene and incubated for 12 h, after which RNA was isolated using Ambion TRIzol reagent (Thermo Scientific) according to the manufacturer's instructions. RNA (5 μg) was reversely transcribed into cDNA using RNA to cDNA ecoDry Premix (Takara, Kusatsu, Japan), which was further used for real-time polymerase chain reaction using SYBR green 2× Master Mix (Elpis, Daejeon, South Korea). The primers used for the analysis are as follows:
| Target | Sequence | Source |
|---|---|---|
| MSR1 | F: TGCACAAGGCAGCTCACTTTGG | Yang et al. (2020) |
| R: GTGCAAGTGACTCCAGCATCTTC | ||
| FASN | F: GTTCACGGACATGGAGCAC | Che et al. (2020) |
| R: GTGGCTCTTGATGATCAGGTC | ||
| Sqle | F: GTTCGCCCTCTTCTCGGATATT | Che et al. (2020) |
| R: GGTTCCTTTTCTGCGCCTCCT | ||
| CH25H | F: GCTGGCAACGCAGTATATGA | Che et al. (2020) |
| R: ACGGAAAGCCAGATGTTGAC | ||
| Actin | F: TCATGAAGTGTGACGTGGACATC | Che et al. (2020) |
| R: CAGGAGGAGCAATGATCTTGATCT |
2.13. Image-based immunohistochemistry infection assay
Vero cells were seeded at 1.2 × 104 cells per well in black 384-well μClear plates (Greiner bio-one, Kremsmünster, Austria) 24 h prior to experimentation. Test compounds were added to each well at the desired concentrations prior to virus infection. The DMSO concentration was kept at 0.5% or below. For infection with MERS-CoV at an MOI of 0.0625, plates were transferred into the BSL-3 containment laboratory. Infection was arrested at 24 h post-infection by adding 4% PFA followed by immunofluorescence staining. MERS-CoV infection was detected using a rabbit anti-MERS-CoV spike antibody (Sino Biological), and cell viability was evaluated with Hoechst 33342 stain (Jeon et al., 2020). The same procedures were performed for SARS-CoV. Data were acquired by taking images at 20× magnification using an Operetta high-content imaging system (PerkinElmer), and the images were analyzed by an in-house developed software, Image Mining 3.0 (IM 3.0) plug-in. To validate the assay, DRC experiments with two compounds with known antiviral activities against MERS-CoV—chloroquine diphosphate and lopinavir—were performed (de Wilde et al., 2014). Test compounds were evaluated by duplicate, 10-point DRC at a concentration range of 0.0977-50 μM. Percent inhibition (PI) was normalized as follows: PI = [1-(INtest – μINmock)/(μINvehicle – μINmock)] × 100, where INtest is percent infection of test compound, μINmock is average of mock, and μINvehicle is average of infection control. Percent viability (PV) was calculated as follows: PV = (CNtest/μCNmock)] × 100%, where CNtest and μCNmock are the cell numbers in the treatment groups and average cell number in the mock group, respectively. After normalization, the EC50 and CC50 were calculated with the nonlinear regression using GraphPad Prism software. Selective index (SI) was calculated by dividing CC50 with EC50 (Ko et al., 2021).
2.14. Time-of-addition assay
Vero cells seeded in a 96-well microplate (2 × 104 cells/well) were incubated for 24 h prior to compound treatment and virus inoculation. Compounds were treated at 1 h intervals from −1 h pre-infection to 6 h post-infection. At the 0 h time point, cells were inoculated with the virus at an MOI of 5 and incubated at 4°C for 1 h to allow the virus to bind to the cell surface for synchronization. Subsequently, the cells were washed to remove unbound viruses and the temperature was shifted to 37°C to promote viral uptake into the cells. The cells were cultured in a drug-containing medium until the end of the experiment and were fixed at 7 h post-infection and analyzed by immunofluorescence assay as previously described (Daelemans et al., 2011; Jeong et al., 2022; Shin et al., 2022). The images were acquired and analyzed as mentioned in the Image-based immunohistochemistry infection assay section.
2.15. qRT-PCR of viral titer
For analysis of the viral titer of MERS-CoV, Vero cells were seeded at 3 × 105 cells per well in 24-well plates 24 h prior to experimentation. The test compounds were added at the final concentrations of 1, 3, 10, and 30 μM to each well before MERS-CoV infection; the DMSO concentration was kept at 0.5% or lower. The plates were moved to the BSL-3 containment laboratory prior to inoculation with MERS-CoV at an MOI of 0.0625. After 24 h, the cells were washed with PBS and lysed in RLT buffer of the RNeasy® mini Kit (QIAGEN, Valencia, CA) supplemented with 1% β-mercaptoethanol (Sigma-Aldrich). The cell lysates were moved to the BSL-2 containment laboratory, where total RNA was isolated using the kit and eluted in 50 μl of DEPC-treated water. MERS-CoV viral RNA was quantified through multiplex real-time reverse-transcription polymerase chain reaction (RT-PCR) using the TOPscript™ One-step RT-PCR DryMIX kit (Enzynomics, Daejeon, South Korea). The primers used were as follows:
| Target | Sequence | Source |
|---|---|---|
| upE of MERS-CoV | F: GCAACGCGCGATTCAGTT | Corman et al. (2012) |
| R: GCCTCTACACGGGACCCATA | ||
| upE probe | 6-carboxyfluorescein [FAM]-CTCTTCACATAATCGCCCCGAGCTCG-6-carboxy-N,N,N,N'-tetramethylrhodamine [TAMRA] | Corman et al. (2012) |
| GAPDH | F: GAAGGTGAAGGTCGGAGTCAAC | Corman et al. (2012) |
| R: CAGAGTTAAAAGCAGCCCTGGT | ||
| GAPDH probe | 6-carboxy-4′,5′-dichloro-2′,7′-dimethoxyfluorescein [JOE]-TTTGGTCGTATTGGGCGCT-6-TAMRA | Corman et al. (2012) |
For analysis of the viral titer of HCoV-229E and HCoV-OC43, total RNA was isolated from virus- or mock-infected MRC-5 cells using the TRIZol reagent (Invitrogen) at the indicated time points. cDNA synthesis and RT-PCR were conducted using the 2× One Step RT-PCR MasterMix with SYBR Green (MGmed, Seoul, South Korea) according to the manufacturer's instructions. The sequences of target genes and primers used are as follows:
| Target | Sequence | Source |
|---|---|---|
| HCoV-229E | F: CGCAAGAATTCAGAACCAGAG | Niu et al. (2016) |
| R: GGCAGTCAGGTTCTTCAACAA | ||
| HCoV-OC43 | F: ACTCAAATGAATTTGAAATATGC | Niu et al. (2016) |
| R: TCACACTTAGGATAATCCCA | ||
| GAPDH | F: GTCGGAGTCAACGGATT | Niu et al. (2016) |
| R: AAGCTTCCCGTTCTCAG |
2.16. Cytopathic effect (CPE) assay
MRC-5 cells were seeded at 2 × 103 cells per well in a white opaque 384-well plate. After 24 h of incubation, serially diluted compounds were treated 2 h before HCoV-229E infection at an MOI of 5. Following an additional 48 h of incubation, cell viability was measured 20 min after adding CellTiter-Glo reagent containing 1% TX-100 using an EnVision® multilabel plate reader. EC50 was then calculated with the non-linear regression formula of GraphPad Prism 6 software.
2.17. SARS-CoV-2 infection assay
Vero cells were seeded at 2 × 103 cells per well in a white opaque 384-well plate. After 24 h of incubation, the cells were treated with serially diluted compounds 2 h before SARS-CoV-2 infection at an MOI of 0.0125. Following 48 h incubation, cell viability was measured by adding the CellTiter-Glo reagent containing 1% TX-100 and by measuring the luminescence with a plate reader after 20 min. EC50 was calculated using the non-linear regression formula of GraphPad Prism 6 software.
2.18. Hamster experiment
2.18.1. Biosafety and ethics
All experimental procedures were performed in the BSL-3 facility of the Korea Zoonosis Research Institute at Jeonbuk National University. All work using live SARS-CoV-2 was approved by the Institutional Biosafety Committee of Jeonbuk National University. All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee of Jeonbuk National University (JBNU-2020-63) and conducted in accordance with the institutional guidelines.
2.18.2. Production of virus
SARS-CoV-2 virus (NCCP 43326) was provided by the Korea Center for Disease Control and Prevention (Cheongju, South Korea); it was propagated in African green monkey kidney epithelial (Vero E6) cells.
2.18.3. Animals
Male Syrian hamsters (Mesocricetus auratus) aged 13–14 weeks old were purchased from SLC Inc. (Shizuoka, Japan). Hamsters were anaesthetized with isoflurane and intranasally inoculated with 1 × 105.5 TCID50 of SARS-CoV-2 in 50 μl DMEM. Two experimental groups of hamsters (n = 12/group) inoculated with virus were treated orally with either 12 mg/kg (low dose) or 36 mg/kg (high dose) of raloxifene, respectively. Drug treatment was initiated 4 h after infection, and the drug was administered once daily for 5 days. Hamsters inoculated with virus (untreated) and PBS were respectively used as the positive and negative control (n = 6/group). The hamsters were monitored daily for body weight, mortality, and clinical signs. At two days post infection, animals (n = 6 per experimental group and n = 3 for the control group) were euthanized for necropsies, and tissue samples were collected for further analysis. Tissue samples were placed into tissue homogenizing CK14 tubes (Precellys, Bertin Technologies, Montigny-le-Bretonneux, France) prefilled with ceramic beads and DMEM, then homogenized using a Bead blaster 24 (Benchmark Scientific, Sayreville, NJ). Nasal turbinate and right lung lobes were used to determine the viral titer (TCID50) and viral RNA load (qRT-PCR). The other lung lobes were fixed in 10% neutral buffered formalin (NBF) for histopathological examination (Klopfleisch, 2013).
2.18.4. Viral titration and measurement of viral RNA load
Vero E6 cells were plated the day before infection into 96-well plates at 2 × 104 cells/well. After centrifugation of the homogenized tissue samples, the supernatants were inoculated into Vero E6 cells at 10-fold serial dilutions. The cells were monitored for 3 days for recording of CPE, and the TCID50 was calculated using the Spearman & Kärber algorithm. Viral RNA was extracted from the supernatants of homogenized tissue using the QIAamp viral RNA Mini Kit (Qiagen) according to the manufacturer's protocol. Quantitative real-time PCR (qRT-PCR) was performed on a LightCycler96 platform (Roche, Basel, Swiss) with commercial one-step real-time PCR kits for SARS-CoV-2 (Allplex, 2019-nCoV Assay kit, Seegene, Seoul, South Korea). The thermal profile consisted of 1 cycle for 20 min at 50°C, 1 cycle for 15 min at 95°C, and 45 cycles of 15 s at 94°C and 30 s at 58°C. The results were analyzed using 2019-nCoV viewer from Seegene Inc. according to the manufacturer's instructions.
2.19. Statistical analysis
Statistical analyses between multiple experimental groups were conducted by one-way ANOVA followed by Dunnett's multiple comparison tests. The results were expressed in the form of mean ± standard error of mean (SEM). Differences between groups were considered significant for values of p < 0.05. Statistical analyses were conducted using Prism 6 software (GraphPad Software).
3. Results
3.1. Inhibitors of MERS-CoV infection were identified through a phenotypic high-throughput screen
We sought to establish a cell-based phenotypic high-throughput screen (HTS) platform to identify compounds that may inhibit the viral infection using MERS-CoV as a model coronavirus. We generated a chimeric pseudovirus presenting the full-length spike protein of the MERS-CoV as previously demonstrated (Zhao et al., 2013). Then, in a 384-well plate format, Huh-7 cells were infected with the pseudovirus, and the infected cells constitutively expressed luciferase over a two-day period (Fig. 1a and S1a). To validate the pseudovirus infection, we preincubated the Huh-7 cells with the heptad repeat 2 peptide (HR2P), which is a known inhibitor of MERS-CoV infection (Bosch et al., 2004; Liu et al., 2009), and we found that the luciferase signal from the infected Huh-7 cells was reduced in a dose-dependent manner (Fig. S1b). This indicates that the luminescence signal from the infected cells can serve as a marker of pseudovirus infection.
Fig. 1.

Combination of a pseudovirus assay and protein-based assays enabled identification of novel inhibitors targeting the early stage of coronavirus infection. a) Schematic representation of a compound screen using a pseudovirus presenting the spike of MERS-CoV. To generate the pseudovirus, a lentivirus backbone, luciferase gene, and the spike were transfected onto 293T cells. In the compound screen, Huh-7 cells were treated with compounds and infected with the pseudovirus, after which the accumulated luciferase signal was measured. b) Schematic representation of the alpha test. In the alpha test, the receptor binding domain (RBD) of the spike of MERS-CoV was immobilized on the donor bead while its cellular receptor, hDPP4 was attached to an acceptor bead. c) Summary of classification of hits from the pseudovirus screen and subsequent alpha test: ∼16% entry-specific (spike-specific) compounds among 1126 hits. d) Structure of three identified hits: thiosemicarbazide, amiodarone, and raloxifene.
Using our pseudovirus-based screen platform, we tested 23,439 small molecules from either the FDA-approved compound library or pharmacologically active compound library at a single concentration of 10 μM. Hit compounds were identified by their ability to reduce the luciferase signal (Fig. S1c). In parallel, we performed a cytotoxicity screen in the absence of the pseudovirus at a final compound concentration of 20 μM, which allowed us to exclude comparably cytotoxic compounds (>20% cell death) (Fig. S1d). From the screen, we identified 1,126 hits that displayed low cytotoxicity and high inhibition in the range of 70–100% compared to the control (Fig. 1c, S1c, and S1d). The average of the calculated Z’ factors for 80 screening plates was ∼0.62, indicating that the screen was robust (Fig. S1e). Since our pseudovirus is incapable of replicating and generating newly made viral progenies inside the infected cells, we reasoned that the hits identified through the pseudovirus screen would be those capable of preventing the early stages rather than the later stages of viral infection. Importantly, this allows us to narrow down the potential antiviral targets of hit compounds and thus aid in the rapid identification of their molecular mechanism of action.
3.2. Hits were screened for their ability to inhibit hDPP4 and spike interaction by alpha
We sought to further confirm whether our hit compounds inhibit interaction between hDDP4 and the spike of MERS-CoV. To this end, we set up an in vitro protein-based alpha assay (Schorpp et al., 2014; Ullman et al., 1994), as described in Fig. 1 b. Specifically, due to the complexity and insolubility of the full-length spike protein, we used the 28-kDa RBD of the S1 of the MERS-CoV spike (Park et al., 2019; Xia et al., 2014) to attach to phthalocyanine, a singlet oxygen donor, and coupled the hDPP4 to a thioxene derivative as a singlet oxygen acceptor. Since the alpha test allowed us to directly monitor the interaction between the RBD and hDPP4, we reasoned that this alpha test could classify our primary hit compounds into a class of compounds specifically targeting the spike-receptor interaction (class I) and a class of compounds that potentially achieve their antiviral activity through inhibition independent of the binding between the spike and its receptor (class II).
Because we used the RBD of the S1 protein, and not full-length S1, we first determined whether the purified RBD interacts with the purified hDPP4 in our assay conditions (Li, 2016). As shown in Fig. S2a, we were able to capture the hDPP4 protein using the purified RBD protein in our pull-down assay, suggesting that the purified RBD protein retains the ability to interact with hDPP4 in the given conditions. We were able to further optimize the alpha condition by testing combinations of various concentrations of hDPP4 and RBD (Fig. S2b), and when tested with an antibody (R723) of the viral spike, we found that the alpha signal was reduced in a dose-dependent manner (Fig. S2c), which validates our alpha conditions. Subsequently, we examined 1,126 primary hits obtained from the pseudovirus screen and found 188 out of 1,126 compounds were validated for antagonistic activity specifically on the interaction between the RBD and hDPP4, in the range of >40% inhibition relative to the control (Fig. 1c). We note that this group (class I) of compounds displays significance as inhibitors of spike-receptor interaction, and our results demonstrate that the combination of the pseudovirus screen and the in vitro alpha test can be used to rapidly identify the mechanisms of action of inhibitors by specifying their targets.
3.3. Thiosemicarbazide (class I) inhibits MERS-CoV infection by antagonizing the binding of the spike to the hDPP4
Among the 188 class I inhibitors, a thiosemicarbazide compound showed outstanding potency, so we further tested its analogs (Fig. 1, Fig. 2 a). As shown in Fig. 2b, compound J1 showed potency in both the pseudovirus assay (EC50, 1.1 μM) and alpha test (IC50, 6.28 μM). To conduct structure activity relationship (SAR) studies, we synthesized a total of 25 thiosemicarbazide analogs by modifying the compound J1 (Fig. 2a). The results of the SAR studies revealed that the 2-pyridine group on the left portion of the molecule is more crucial for activity than the linker region or phenyl group on the right side. We found that the efficacy of thiosemicarbazide was abolished or reduced when we replaced the 2-pyridine moiety with other groups, including 6-fluoro and 6-hydroxy pyridine (1b-3 and 1b-1). However, when we replaced the p-chlororphenyl on the right side with p-trifluoromethyphenyl (1g-8), 6-methoxypyridine (1j), or 4-methylcyclohexyl (1g-5) groups, their potency in the alpha test was highly improved (1g-8, IC50 = 2.92 μM; 1j, IC50 = 0.85 μM; 1g-5, IC50 = 0.85 μM) (Fig. 2b). As expected, when we modified the thiosemicarbazide backbone, potency was completely abolished (1d and 1e). The SAR analysis suggests that the pyridine moiety is a potential pharmacophore through which thiosemicarbazide analogs may bind to the target. We also found that several modifications of the pyridine group such as in 4-methylcyclohexyl (1g-5) resulted in a significantly increased cytotoxicity (Fig. 2b). Therefore, we excluded the toxic analogs including 1g-5 (CC50, 18.81 μM), and further examined the selected compounds using ELISA designed to capture RBD with immobilized hDPP4 (Fig. 2c). We found that 1 and 1g-8 were validated while 1j showed a lower inhibitory effect (Fig. 2d–f). Since we used the RBD, which is a part of S1 of MERS-CoV, in the alpha test, we retested 1g-8 and 1j with a pseudovirus expressing the full-length MERS-CoV spike to exclude the possibility that the inhibition by thiosemicarbazide analogs is due to promiscuous binding, which may not exist for the full-length spike. The modified thiosemicarbazides strongly inhibited the pseudovirus infection with EC50 values of 0.91 μM and 0.19 μM for 1g-8 and 1j, respectively (Fig. 2b). This data revalidates our alpha conditions and clearly shows that the tested thiosemicarbazide analogs inhibit the interaction between spike and its receptor hDPP4.
Fig. 2.

Thiosemicarbazide (class I) shows target specificity, and its analogs show regions for their antiviral potency. a) Structure of newly-synthesized thiosemicarbazide derivatives. The modified moieties compared to compound 1 or 1f are highlighted in green (2-pyridine), purple (thiosemicarbazide), orange (pyridine), and blue (p-trifuloromethylphenyl). b) Potency (IC50 or EC50 (μM)), cytotoxicity (CC50 (μM)), and selective index (SI = CC50/EC50 (MERS-CoV infection assay)) of thiosemicarbazide derivatives as evaluated in alpha test, pseudovirus assay, and immunofluorescence-based MERS-CoV infection assay. CC50 values that are >50 μM were considered as 50 μM when calculating SI. n.d; not determined. c) Schematic presentation of the ELISA using hDPP4 and the RBD of the S1 of MERS-CoV spike. d-f) Three compounds, 1 (d), 1g-8 (e), and 1j (f) tested for their inhibiting activity on the binding between hDDP4 and RBD. Each data point represents the mean of triplicate assays with ±SEM, and their IC50 values were calculated through curve fitting analysis using Prism 6.0.
Next, we wanted to further test the identified compounds using MERS-CoV. To obtain quantitative values of antiviral activity, we introduced an image-based MERS-CoV infection assay (Ko et al., 2021) in which Vero cells were treated with thiosemicarbazide compounds for 2 h, followed by MERS-CoV infection at an MOI of 0.0625, after which the viral infection was measured using a fluorescence signal from a immunohistochemical reaction using an anti-spike antibody (Fig. 3 a). As shown in Fig. S3, the immunofluorescence signal is quenched in a concentration-dependent manner in the presence of known antiviral drugs, chloroquine and lopinavir (de Wilde et al., 2014). Our compounds were also found to be highly potent against MERS-CoV infection, and 1j showed a particularly outstanding EC50 value of 0.73 μM (Fig. 2b). To further analyze the antiviral activity of 1g-8 and 1j in the host cell, we quantified expression of the E protein gene (upE) of MERS-CoV using quantitative real-time PCR upon treatment with the compounds (Fig. S4). As we expected, the treatment of compounds resulted in a dramatic decrease in the upE gene transcript levels compared to that of the untreated cells (Fig. 3b), suggesting that the compounds inhibited the entry of MERS-CoV, thereby preventing viral proliferation in the host cell.
Fig. 3.

Thiosemicarbazide inhibits MERS-CoV infection by blocking the interaction between RBD and hDPP4. a) Schematic presentation of immunofluorescence-based MERS-CoV infection assay. b) Antiviral potency of compound 1 and 1g-8 against MERS-CoV infection. The cells were treated with individual compounds at the time of MERS-CoV infection at an MOI of 0.0625. The cells were further incubated for 24 h followed by immunofluorescence imaging. MERS-CoV infection was determined using a rabbit anti-MERS-CoV spike antibody (green), and cell viability was measured with Hoechst 33342 (red). Concentrations of 3.125 μM and 50 μM were respectively selected as the low and high doses for each compound. Chloroquine (CHQ) at a concentration of 10 μM was included as a control. c-left) Docking models for compounds 1 and 1j bound to the MERS-CoV receptor binding domain (RBD) (PDB: 4L3N). The molecular surface of MERS-CoV RBD is presented in gray while compound 1 is marked in magenta. c-right) Superimposition of the RBD docked with 1 (the model in left) on the X-ray structure of RBD complexed with hDPP4 (PDB: 4L72). The gray molecular surface is MERS-CoV RBD (PDB id: 4L3N, 4L72) and the green ribbon is hDPP4. d-e) Comparison of docked configurations of 1 (d) and 1j (e) to the binding site in the RBD. Key interactions between ligand and RBD are presented by dashed lines: orange is a hydrogen bond, cyan is a pi-pi interaction, and green is a pi-cation interaction. Ligands are colored by atom types, with carbon as magenta in 1, cyan in 1j, and silver in RBD; nitrogen is blue; oxygen is red; and fluorine is green.
3.4. Docking analysis of thiosemicarbazides reveals mechanism of action of inhibition
To better understand the mechanism of action, molecular docking of the selected analogs, 1g-8 and 1j, along with 1 was performed on the apo-RBD (PDB: 4L3N) (Chen et al., 2013). As shown in Fig. 3c, compound 1 fits well into a pocket of the concave surface of the RBD which was determined as an RBD–hDPP4 binding interface by X-ray crystallography (PDB: 4L72) (Wang et al., 2013). The docking complex of compound 1:RBD was superimposed onto the co-crystal structure of hDPP4:RBD, and interestingly the ligand binding site matched the region of hDPP4 interaction with RBD (Thr288, Ala291, Leu94, and Ile295) (Fig. 3c). This suggests that compound 1 occupies the binding region of hDPP4, interfering with tight protein-protein interaction between RBD and hDPP4, and that its activity could be modulated by varying the substituent on the thioamide NH while keeping the picolinyl hydrazide moiety intact. The para-chloro phenyl group attached to the thioamide NH of 1 interacts with Trp553 by forming a pi-pi contact and synergistic Cl-aromatic pi interaction. Moreover, the picolinyl hydrazide moiety is involved in pi-cation interaction with Lys502 as well as multiple hydrogen bonds with Glu513 and Arg542 (Fig. 3d), which are known to contact both Ala291 and Gln344 of hDPP4. By contrast, the corresponding 2-methoxy pyridine of 1j does not fit into this pocket containing Trp553 due to steric hindrance, as this molecule instead binds in the flipped orientation by forming a pi-cation interaction between 2-methoxy pyridine and Lys502 (Fig. 3e). The binding configurations of 1g-8 are nearly identical to those of compound 1, supporting similar potency between compound 1 and 1g-8 (Fig. 2b). Furthermore, to compare the binding affinity of these compounds, we calculated the docking scores for the top 10 docked poses of each compound using Glide docking outputs (Fig. S5). The median of the docking scores of compounds 1, 1g-8, and 1j were −1.61, −1.12, and −0.42 kcal/mol, respectively, which were consistent with the order of biological activity (1 > 1g-8 » 1j). We also calculated the electrostatic complementarity (EC) scores of the compound-RBD complexes, and compound 1 (EC score = 0.184) showed a more electrostatically favorable interaction with RBD than 1j (EC score = 0.073). Noticeably, the methoxy substituent on the pyridine ring of 1j provides an electrostatic clash, supporting that 1j is less suitable for binding to the RBD (Fig. S6). Based on the binding configurations of the compounds, we concluded that compound 1 has a mechanism of inhibiting RBD binding to hDPP4, and in this process, the picolinyl hydrazide moiety of 1 is crucial for the potency of the thiosemicarbazide compounds.
3.5. Amiodarone (class II) has broad-spectrum antiviral activity against coronaviruses
Among 938 class II compounds, amiodarone (Fig. 1d), which is one of the most commonly used antiarrhythmic drugs (Andreasen et al., 1981; Chatelain and Laruel, 1985), was found to have outstanding antiviral activity in the pseudovirus screen but marginal or no activity in the alpha test (Fig. S7a). We have also tested the potency of amiodarone using the MERS-CoV infection assay (Fig. 4a and S7b) and viral RNA quantification (Fig. 4 b). As mentioned earlier, the results from pseudovirus screen and alpha test indicate that amiodarone may not target the direct binding of the spike to its receptor protein but instead target the infection stage post-binding. As a proof-of-concept, we performed a time-of-addition experiment (Daelemans et al., 2011; Jeong et al., 2022; Shin et al., 2022). As shown in Fig. 4c, when we treated the Huh-7 cells with amiodarone at different time points, amiodarone strongly inhibited the viral infection when added at an early time point (1 h before or at the time of infection), while it showed a marginal antiviral effect when added at later stages of infection (1–6 h post-infection). Such stage-selective activity was also observed upon treatment with chloroquine, an antiviral drug that achieves its activity via increasing lysosomal pH and subsequently prohibiting the release of infected virus from the lysosome, suggesting the possibility of amiodarone acting with a similar mechanism. Lopinavir, which is known to act on the viral protease involved in viral proliferation post-exit from the endosomes, showed efficacy when added at a relatively later stage of infection compared to chloroquine and amiodarone, again suggesting that amiodarone may act early in viral infection. Amiodarone also showed efficacy against other human coronaviruses; an alphacoronavirus HCoV-229E and a betacoronavirus HCoV-OC43 (Fig. 4d and S7c). In total, amiodarone shows potential as a broad-spectrum treatment option for coronaviruses by targeting the early stage of viral infection possibly affecting the endosome (Fig. 4e). This is consistent with previous results that amiodarone, as one of the functional inhibitors of acid sphingomyelinase (FIASMA), has demonstrated antiviral efficacy (Miller et al., 2012; Naser et al., 2020) (Fig. 4e).
Fig. 4.

Amiodarone (class II) inhibits the early stage of coronavirus infection. a) Antiviral activity of amiodarone demonstrated by an immunofluorescence-based MERS-CoV infection assay. In total, ten different concentrations (1–50 μM) of amiodarone were examined, and immunofluorescence images obtained from low (3.12 μM) and high (50 μM) compound concentrations were selected. Green signals represent cells infected with MERS-CoV and red signals indicate cell survival. b) Viral mRNA from cells infected with MERS-CoV quantified by qRT-PCR. Each data point represents the mean ± SEM of triplicate assays. c) Time-of-addition experiment performed by addition of 10 μM amiodarone at different time points during MERS-CoV infection. Drugs were added at 1 h before infection (−1), at the time of infection (0), or at various hours post-infection (+1 ∼ +6), and the extent of inhibition of the MERS-CoV infection was quantified by the immunofluorescence signals. Chloroquine and lopinavir were included at a concentration of 10 μM for comparison. Each data point represents the mean ± SEM of triplicate assays. d) Antiviral activity of amiodarone examined against the HCoV-229E and HCoV-OC43 viruses. Viruses were infected into Vero cells in the presence of amiodarone, and after 24 h of infection, viral mRNA was quantified by qRT-PCR. Each data point represents the mean ± SEM of triplicate assays. e) Graphical presentation of potential mechanisms of the antiviral activity of amiodarone. Amiodarone, as a weak base, could lead to an alkalization of the lysosome and therefore act as a potential inhibitor of lysosomal acid sphingomyelinase. ASM, acid-sphingomyelinase; AM, acid-sphingomyelin.
3.6. Raloxifene and benzothiophene derivatives (class II) inhibit MERS-CoV infection
Another compound that belongs to class II was raloxifene. Raloxifene is a selective estrogen receptor modulator (SERM) that was approved by the FDA in 1997 for the treatment and prevention of postmenopausal osteoporosis and cancer (Hernandez et al., 2003; Lewis and Jordan, 2005). In the pseudovirus assay, raloxifene showed strong potency with an EC50 of 0.2 μM with no significant cytotoxic effects, and was found to be potent against MERS-CoV infection confirmed by the infection assay (Fig. 5 a and b) and viral RNA quantification (Fig. 5c). Raloxifene was also found to be effective against HCoV-229E and HCoV-OC43, indicating its potential as a broad-spectrum coronavirus treatment option (Fig. 5d and S8). These results are in line with those of others published while this work was under preparation (Allegretti et al., 2022). In addition, similar to amiodarone, raloxifene appeared to be more potent when added at the earlier stages while it showed only marginal efficacy when treated at later stages of infection (Fig. 5e). Previous studies have suggested that raloxifene affects host cell lipid metabolism; and as previously reported cholesterol plays a major role in the fluidity of the plasma membrane (Allegretti et al., 2022; Chang et al., 2022; Hong et al., 2021). In addition, recent studies show that antiviral activity can be achieved by the conversion of cholesterol to 25-hyroxycholestrol by CH25H (Schoggins and Randall, 2013; York et al., 2015; Zu et al., 2020). Thus, we reasoned that raloxifene may have broad-spectrum antiviral activity by modulating the host cell membrane through altering cholesterol metabolism. To test this idea, we treated A549 cells with 10 μM raloxifene and found a marginal elevation of total cellular cholesterol species (Fig. 5f). Among the genes in the cholesterol pathway, we found that the SQLE coding squalene epoxidase that converts squalene to 2,3-epoxysqualene was significantly enhanced in its expression upon raloxifene treatment (Fig. 5g). These results indicate that alteration of cholesterol metabolism upon raloxifene treatment may be the mechanism of its antiviral activity.
Fig. 5.

Raloxifene inhibits the early stage of coronavirus infection. a) Antiviral activity of raloxifene demonstrated by an immunofluorescence-based MERS-CoV infection assay as previously noted in Fig. 4a. Green signals represent cells infected with MERS-CoV and red signals represent cell survival. b) In total, ten different concentrations of raloxifene were tested in the immunofluorescence assay, and EC50 and cytotoxicity were calculated through curve fitting analysis using Prism 6. c) Viral mRNA quantified by qRT-PCR from the Vero cells infected with MERS-CoV. d) Potency of raloxifene against HCoV-229E or HCoV-OC43 measured by qRT-PCR as detailed in Fig. 4d. e) Time-of-addition experiment using 10 μM of raloxifene. Experiments were performed as described in Fig. 4c. f) Cellular cholesterol levels in A549 cells treated with 10 μM raloxifene for 12 h. g) Expression levels of selected genes involved in cholesterol metabolism quantified by qRT-PCR after 12 h of treatment with 10 μM raloxifene. CH25H; cholesterol 25-Hydroxylase, FASN; fatty acid synthase, MSR1; macrophage scavenger receptor 1, SQLE; squalene epoxidase. Each data point represents the mean ± SEM of triplicate assays.
Since raloxifene contains a benzothiophene analogue, and since the benzothiophene moiety could be essential for the potency of raloxifene, we decided to further examine other hit compounds with benzothiophene. Importantly, we found that compound 2, like raloxifene, only had marginal potency in the alpha test but a strong efficacy in the MERS-CoV assay. To further characterize 2, we performed an SAR study with a panel of benzothiophene derivatives (Fig. 6 a). As shown in Fig. 6b, our SAR studies revealed that the phenyl moiety was an essential region for improving the potency of benzothiophene. When we replaced the phenyl group with tetrahydronaphthalene or benzyl group, their efficacy in the MERS-CoV assay was not detectable, although it remained active in the pseudovirus assay, which suggests that the phenyl group is essential for potency against MERS-CoV replication in the host cells. These results led us to explore derivatives including a modification on the phenyl group, and we found that derivatives with modifications of para-halogen residues (I, F, Br) showed improved potency in both the MERS-CoV infection and pseudovirus assays. In particular, 2a-4 with p-bromophenyl group showed an improved efficacy against MERS-CoV infection with an EC50 value of 0.95 μM. Interestingly, when we modified the benzothiophene scaffold, the potency of derivative 2b was comparable to that of 2a-1, while the substitution of the p-bromophenyl group of 2a-4 with an ethoxy group completely abolished its potency (Fig. 6c–e). This result suggests that the phenyl group of benzothiophene is crucial for its potency.
Fig. 6.

Benzothiophene analogs (class II) show regions crucial for antiviral potency. a) Structure activity relationship (SAR) analysis performed using newly synthesized benzothiophne derivatives. The modified moieties of the phenyl group of compound 2 are highlighted in green or orange color, and the thiophene moiety is highlighted in purple. b) Potency (EC50 (μM)) of tested benzothiophene derivatives evaluated by MERS-CoV pseudovirus assay and immunofluorescence-based MERS-CoV infection assay, cytotoxicity (CC50 (μM)), and selective index (SI = CC50/EC50 (MERS-CoV infection assay)). CC50 values that are >50 μM were considered as 50 μM when calculating SI. n.d; not determined. c) Antiviral activity of 2a-1 and 2a-4 examined by immunofluorescence-based MERS-CoV infection assay as noted in Fig. 4a. Green signals present the cells infected with MERS-CoV and red signals present all cells. d-e) Antiviral potency of 2a-1 (d) and 2a-4 (e) against HCoV-229E or HCoV-OC43 measured by qRT-PCR as noted in Fig. 4d. Each data point represents the mean of triplicate assays with ±SEM.
3.7. Class II compounds show strong potency against SARS-CoV and SARS-CoV-2
Next, we sought to test whether the class II compounds have antiviral activity against SARS-CoV or SARS-CoV-2. We utilized an image-based SARS-CoV infection assay to test the efficacy of class II compounds against SARS-CoV (Jeon et al., 2020). We first treated the Vero cells with compounds for 2 h and infected the cells with SARS-CoV at an MOI of 0.0625, after which the extent of viral infection was measured by immunofluorescence analysis (Fig. S9). As we expected for class II compounds, raloxifene, amiodarone, and compounds 2a-1 and 2a-4 showed strong potency against SARS-CoV, indicating its potential utility as a broad-spectrum antiviral agent (Fig. 7a and S10). Class II compounds also strongly inhibited SARS-CoV-2 infection as assessed by viral RNA (Fig. 7 b). Among the compounds, amiodarone showed an outstanding EC50 value of 3.1 μM, which is better than the EC50 of remdesivir (7.2 μM) (Fig. S11), a reference antiviral agent for SARS-CoV-2 infection (Eastman et al., 2020). We also found that raloxifene showed strong potency against SARS-CoV-2 infection in a cell-based model, as demonstrated in Fig. 7c. To further assess the potency of raloxifene against SARS-CoV-2, we performed an in vivo SARS-CoV-2 infection experiment using a hamster model (Fig. 7d). We used a sublethal dose of SARS-CoV-2 and measured the mRNA of the virus after a two-day infection period in the presence of 12 or 36 mg/kg raloxifene. As shown in Fig. 7e-7g, we found that raloxifene inhibited the replication of SARS-CoV-2 in both lung and nasal turbinate at the higher dose, which was consistent with other previously published results (Allegretti et al., 2022). During the infection, we also monitored the activity and the weight of the infected animals: no significant changes were observed in the weight (Fig. 7h) and in the activity level or survival rate (Fig. S12). Our in vivo results with the hamster model validated the potency of raloxifene, and combined with the potency of benzothiophene analogs, show potential for further development of the identified chemical classes as effective broad-spectrum antiviral drugs.
Fig. 7.

Compounds inhibiting the early stage of coronavirus infection show potency against both SARS-CoV and SARS-CoV-2 infection. a) Antiviral activity of the selected broad-spectrum compounds (amiodarone, raloxifene, 2a-1, and 2a-4) demonstrated by immunofluorescence-based SARS-CoV infection assay. Among the ten different concentrations (1–50 μM) of each compound examined, images at low (3.12 μM) and high (50 μM) concentrations were selected. Green signals represent cells infected with SARS-CoV and red signals represent cell survival. b-c) Potency of the selected drugs against SARS-CoV2 infection demonstrated with a cell-based infection model. Cells were treated with 10 μM of amiodarone, 2a-1, and 2a-4 (b), raloxifene (c) at the time of infection with SARS-CoV-2, and viral mRNA was quantified by qRT-PCR after 24 h of infection. For comparison, 10 μM of remdesivir was included. d-g) In vivo potency of raloxifene against SARS-CoV-2 demonstrated using a hamster infection model. d) Schematic presentation of SARS-CoV2 infection and sampling. SARS-CoV-2 was infected through nasal inoculation and animals were treated with raloxifene during a two-day infection period at 12 mg/kg or 36 mg/kg doses. On day 2, viral titer in the hamsters was measured by qRT-PCR. e-f) Viral titers of SARS-CoV-2 in lungs quantified on day 2 by qRT-PCR (e) or TCID50 (f). g) Viral titer of nasal turbinate quantified on day 2 by TCID50. h) Body weight of the hamsters was measured daily for the 2-day infection period and they remained normal. Each data point represents the mean ± SEM of triplicate assays.
4. Discussion
In this study, we have used a cell-based high-throughput screen platform to screen more than 20,000 compounds from the FDA-approved compound and pharmacologically active compound libraries for their potency to inhibit infection by the pseudovirus presenting full-length spike of the MERS-CoV. In combination with the alpha test exploiting the RBD of MERS-CoV spike attached to the oxygen donor bead and receptor hDPP4 attached to the oxygen acceptor bead, the identified hits were classified into a group of compounds capable of inhibiting spike-receptor binding (class I), and that of compounds presenting antiviral effects by mechanism other than the binding inhibition (class II). Among class I, thiosemicarbazide, especially its pyridine moiety, was identified as a potent compound that showed efficacy in both our pseudovirus screen and alpha test, for which the mechanism of action was further confirmed by an ELISA test and the cell-based MERS-CoV infection assay.
Among class II compounds, amiodarone, a most used antiarrhythmic drug, showed potential as an effective broad-spectrum antiviral against various coronaviruses possibly by targeting the endosomal survival of the virus. As noted, amiodarone, as one of the FIASMA, has been demonstrated to have inhibitory activity against acid sphingomyelinase in the lysosome, thereby leading to a change in cellular lipid metabolism, which results in the modification of the curvature of the cellular membrane crucial for membrane fusion during viral infection (Kornhuber et al., 2010; Miller et al., 2012; Naser et al., 2020; Schloer et al., 2020).
Another potent class II compound was raloxifene, an FDA-approved selective estrogen receptor modulator. The antiviral efficacy of raloxifene has been also reported by another group while this work was under preparation (Allegretti et al., 2022). Other reports show that the mechanism of antiviral effect of raloxifene is through change in the content of the cytoplasmic membrane or lipid raft, and a recent study shows that the antiviral activity can be achieved by the conversion of cholesterol to 25-hyroxycholestrol by CH25H (Schoggins and Randall, 2013; York et al., 2015; Zu et al., 2020). Although our results do not show alteration in the CH25H gene, we demonstrate change in SQLE also involved in cholesterol metabolism which confirms the role of modulation of cholesterol metabolism in antiviral efficacy of raloxifene. Also, raloxifene and analogs of benzothiophene, a moiety found in raloxifene, showed efficacy against SARS-CoV and SARS-CoV-2 infection. Especially, treatment with the dose of 36 mg/kg of raloxifene showed potency against SARS-CoV-2 in a hamster model. Such in vivo efficacy of raloxifene was also reported by others in a previously published work (Allegretti et al., 2022).
Prophylactic vaccines against SARS-CoV-2 infection are not yet widely available worldwide, thus leading to a concern for the unvaccinated population. Coronaviruses can also easily develop genetic mutations necessary to avoid the current vaccines. Antiviral therapeutics can provide a safer and effective way to cure infections caused by both current and future evolving variants. Currently, several neutralizing antibodies and target-specific drugs against SARS-CoV-2, including ritonavir-boosted nirmatrelvir and molnupiravir, have been introduced in the clinical setting, but clinical failure caused by a rebound problem after treatment with those drugs is becoming increasingly common. Our approach shows that drug repositioning and a pseudovirus assay combined with an alpha test can be used to rapidly identify active compounds that can effectively prevent viral infection. Importantly, through our screening platform, we were able to not only identify several compounds as potential broad-spectrum inhibitors against the coronaviruses but also rapidly specify the potential antiviral mechanisms of action. Especially, our results show that amiodarone, raloxifene, and benzothiophene drugs have the potential to be further developed into broad-spectrum antiviral therapeutic options against coronavirus infection.
Funding source
This work was supported by a grant from an R&D support program of the Gyeonggi provincial government to KJ; grants from Korea Disease Control and Prevention Agency (KCDC 2021-ER1602-00) to WL; grants from National Research Foundation (NRF) of Korea (NRF-2019R1F1A1060071, NRF-2020R1A6A1A03042854) and an Institute for Information & Communication Technology Planning & Evaluation (IITP) grant funded by the Korean government (MSIT) (No. 2020-0-01343) to TN; NRF grants funded by the Korean government (MSIT) (NRF-2017M3A9G6068245 and NRF-2022M3A9J1081343) to SK; NRF grants funded by the Korean government (MSIT) (NRF-2014M3A9D5A01075128, 2020M3A9I2109027, 2021M3H9A1030260) to JKS; and a grant funded by Korea Disease Control and Prevention Agency (KCDC-2020-NI-039-00) to JL.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors thank all members of the Wonsik Lee lab and Bio-center of Gyeonggido Business and Science Accelerator for scientific and experimental discussion.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.antiviral.2022.105473.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- Allegretti M., Cesta M.C., Zippoli M., Beccari A., Talarico C., Mantelli F., Bucci E.M., Scorzolini L., Nicastri E. Repurposing the estrogen receptor modulator raloxifene to treat SARS-CoV-2 infection. Cell Death Differ. 2022;29:156–166. doi: 10.1038/s41418-021-00844-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020;26:450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen F., Agerbaek H., Bjerregaard P., Gotzsche H. Pharmacokinetics of amiodarone after intravenous and oral administration. Eur. J. Clin. Pharmacol. 1981;19:293–299. doi: 10.1007/BF00562807. [DOI] [PubMed] [Google Scholar]
- Bosch B.J., Martina B.E., Van Der Zee R., Lepault J., Haijema B.J., Versluis C., Heck A.J., De Groot R., Osterhaus A.D., Rottier P.J. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard C., Verheije M.H., Wicht O., van Kasteren S.I., van Kuppeveld F.J., Haagmans B.L., Pelkmans L., Rottier P.J., Bosch B.J., de Haan C.A. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E., Ruhl V.E., Rodier F., Smith C.L., Rahmberg B.L., Fuss J.O., Campisi J., Yaswen P., Cooper P.K., Kaufman P.D. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One. 2009;4 doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Kim J., Lee W. Raloxifene prevents intracellular invasion of pathogenic bacteria through modulation of cell metabolic pathways. J. Antimicrob. Chemother. 2022;77:1617–1624. doi: 10.1093/jac/dkac069. [DOI] [PubMed] [Google Scholar]
- Chatelain P., Laruel R. Amiodarone partitioning with phospholipid bilayers and erythrocyte membranes. J. Pharmacol. Sci. 1985;74:783–784. doi: 10.1002/jps.2600740720. [DOI] [PubMed] [Google Scholar]
- Che L., Chi W., Qiao Y., Zhang J., Song X., Liu Y., Li L., Jia J., Pilo M.G., Wang J., Cigliano A., Ma Z., Kuang W., Tang Z., Zhang Z., Shui G., Ribback S., Dombrowski F., Evert M., Pascale R.M., Cossu C., Pes G.M., Osborne T.F., Calvisi D.F., Chen X., Chen L. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut. 2020;69:177–186. doi: 10.1136/gutjnl-2018-317581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseright T., Mackey M., Rose S., Vinter A. Molecular field extrema as descriptors of biological activity: definition and validation. J. Chem. Inf. Model. 2006;46:665–676. doi: 10.1021/ci050357s. [DOI] [PubMed] [Google Scholar]
- Chen Y., Rajashankar K.R., Yang Y., Agnihothram S.S., Liu C., Lin Y.L., Baric R.S., Li F. Crystal structure of the receptor-binding domain from newly emerged Middle East respiratory syndrome coronavirus. J. Virol. 2013;87:10777–10783. doi: 10.1128/JVI.01756-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman V.M., Eckerle I., Bleicker T., Zaki A., Landt O., Eschbach-Bludau M., van Boheemen S., Gopal R., Ballhause M., Bestebroer T.M., Muth D., Muller M.A., Drexler J.F., Zambon M., Osterhaus A.D., Fouchier R.M., Drosten C. Detection of a novel human coronavirus by real-time reverse-transcription polymerase chain reaction. Euro Surveill. 2012;17 doi: 10.2807/ese.17.39.20285-en. [DOI] [PubMed] [Google Scholar]
- Cui J., Li F., Shi Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daelemans D., Pauwels R., De Clercq E., Pannecouque C. A time-of-drug addition approach to target identification of antiviral compounds. Nat. Protoc. 2011;6:925–933. doi: 10.1038/nprot.2011.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilde A.H., Jochmans D., Posthuma C.C., Zevenhoven-Dobbe J.C., van Nieuwkoop S., Bestebroer T.M., van den Hoogen B.G., Neyts J., Snijder E.J. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob. Agents Chemother. 2014;58:4875–4884. doi: 10.1128/AAC.03011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman R.T., Roth J.S., Brimacombe K.R., Simeonov A., Shen M., Patnaik S., Hall M.D. Remdesivir: a review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent. Sci. 2020;6:672–683. doi: 10.1021/acscentsci.0c00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eurosurveillance Editorial T. Note from the editors: world Health Organization declares novel coronavirus (2019-nCoV) sixth public health emergency of international concern. Euro Surveill. 2020;25 doi: 10.2807/1560-7917.ES.2020.25.5.200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Prior P. Priming of SARS-CoV-2 S protein by several membrane-bound serine proteinases could explain enhanced viral infectivity and systemic COVID-19 infection. J. Biol. Chem. 2021;296 doi: 10.1074/jbc.REV120.015980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller C., Varbanov M., Duval R.E. Human coronaviruses: insights into environmental resistance and its influence on the development of new antiseptic strategies. Viruses. 2012;4:3044–3068. doi: 10.3390/v4113044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P.H., Zhang L.J., Guan Y.J., Butt K.M., Wong K.L., Chan K.W., Lim W., Shortridge K.F., Yuen K.Y., Peiris J.S., Poon L.L. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- Hartenian E., Nandakumar D., Lari A., Ly M., Tucker J.M., Glaunsinger B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020;295:12910–12934. doi: 10.1074/jbc.REV120.013930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez E., Valera R., Alonzo E., Bajares-Lilue M., Carlini R., Capriles F., Martinis R., Bellorin-Font E., Weisinger J.R. Effects of raloxifene on bone metabolism and serum lipids in postmenopausal women on chronic hemodialysis. Kidney Int. 2003;63:2269–2274. doi: 10.1046/j.1523-1755.2003.00005.x. [DOI] [PubMed] [Google Scholar]
- Hong S., Chang J., Jeong K., Lee W. Raloxifene as a treatment option for viral infections. J. Microbiol. 2021;59:124–131. doi: 10.1007/s12275-021-0617-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B., Guo H., Zhou P., Shi Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S., Ko M., Lee J., Choi I., Byun S.Y., Park S., Shum D., Kim S. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 2020;64 doi: 10.1128/AAC.00819-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong K., Kim J., Chang J., Hong S., Kim I., Oh S., Jeon S., Lee J.C., Park H.J., Kim S., Lee W. Chemical screen uncovers novel structural classes of inhibitors of the papain-like protease of coronaviruses. iScience. 2022;25 doi: 10.1016/j.isci.2022.105254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfleisch R. Multiparametric and semiquantitative scoring systems for the evaluation of mouse model histopathology--a systematic review. BMC Vet. Res. 2013;9:123. doi: 10.1186/1746-6148-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M., Chang S.Y., Byun S.Y., Ianevski A., Choi I., Pham Hung d'Alexandry d'Orengiani A.L., Ravlo E., Wang W., Bjoras M., Kainov D.E., Shum D., Min J.Y., Windisch M.P. 2021. Screening of FDA-Approved Drugs Using a MERS-CoV Clinical Isolate from South Korea Identifies Potential Therapeutic Options for COVID-19. Viruses 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhuber J., Tripal P., Reichel M., Muhle C., Rhein C., Muehlbacher M., Groemer T.W., Gulbins E. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cell. Physiol. Biochem. 2010;26:9–20. doi: 10.1159/000315101. [DOI] [PubMed] [Google Scholar]
- Krammer F. SARS-CoV-2 vaccines in development. Nature. 2020;586:516–527. doi: 10.1038/s41586-020-2798-3. [DOI] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J., Group S.W. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Lam T.T., Jia N., Zhang Y.W., Shum M.H., Jiang J.F., Zhu H.C., Tong Y.G., Shi Y.X., Ni X.B., Liao Y.S., Li W.J., Jiang B.G., Wei W., Yuan T.T., Zheng K., Cui X.M., Li J., Pei G.Q., Qiang X., Cheung W.Y., Li L.F., Sun F.F., Qin S., Huang J.C., Leung G.M., Holmes E.C., Hu Y.L., Guan Y., Cao W.C. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. 2020;583:282–285. doi: 10.1038/s41586-020-2169-0. [DOI] [PubMed] [Google Scholar]
- Lewis J.S., Jordan V.C. Selective estrogen receptor modulators (SERMs): mechanisms of anticarcinogenesis and drug resistance. Mutat. Res. 2005;591:247–263. doi: 10.1016/j.mrfmmm.2005.02.028. [DOI] [PubMed] [Google Scholar]
- Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev. Virol. 2016;3:237–261. doi: 10.1146/annurev-virology-110615-042301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- Li Q., Guan X., Wu P., Wang X., Zhou L., Tong Y., Ren R., Leung K.S.M., Lau E.H.Y., Wong J.Y., Xing X., Xiang N., Wu Y., Li C., Chen Q., Li D., Liu T., Zhao J., Liu M., Tu W., Chen C., Jin L., Yang R., Wang Q., Zhou S., Wang R., Liu H., Luo Y., Liu Y., Shao G., Li H., Tao Z., Yang Y., Deng Z., Liu B., Ma Z., Zhang Y., Shi G., Lam T.T.Y., Wu J.T., Gao G.F., Cowling B.J., Yang B., Leung G.M., Feng Z. Early transmission dynamics in wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 2020;382:1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu I.J., Kao C.L., Hsieh S.C., Wey M.T., Kan L.S., Wang W.K. Identification of a minimal peptide derived from heptad repeat (HR) 2 of spike protein of SARS-CoV and combination of HR1-derived peptides as fusion inhibitors. Antivir. Res. 2009;81:82–87. doi: 10.1016/j.antiviral.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M.E., Adhikary S., Kolokoltsov A.A., Davey R.A. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J. Virol. 2012;86:7473–7483. doi: 10.1128/JVI.00136-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naser E., Kadow S., Schumacher F., Mohamed Z.H., Kappe C., Hessler G., Pollmeier B., Kleuser B., Arenz C., Becker K.A., Gulbins E., Carpinteiro A. Characterization of the small molecule ARC39, a direct and specific inhibitor of acid sphingomyelinase in vitro. J. Lipid Res. 2020;61:896–910. doi: 10.1194/jlr.RA120000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu P., Shen J., Zhu N., Lu R., Tan W. Two-tube multiplex real-time reverse transcription PCR to detect six human coronaviruses. Virol. Sin. 2016;31:85–88. doi: 10.1007/s12250-015-3653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y.J., Walls A.C., Wang Z., Sauer M.M., Li W., Tortorici M.A., Bosch B.J., DiMaio F., Veesler D. Structures of MERS-CoV spike glycoprotein in complex with sialoside attachment receptors. Nat. Struct. Mol. Biol. 2019;26:1151–1157. doi: 10.1038/s41594-019-0334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paules C.I., Marston H.D., Fauci A.S. Coronavirus infections-more than just the common cold. JAMA. 2020;323:707–708. doi: 10.1001/jama.2020.0757. [DOI] [PubMed] [Google Scholar]
- Raj V.S., Mou H., Smits S.L., Dekkers D.H., Muller M.A., Dijkman R., Muth D., Demmers J.A., Zaki A., Fouchier R.A., Thiel V., Drosten C., Rottier P.J., Osterhaus A.D., Bosch B.J., Haagmans B.L. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495:251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj V.S., Smits S.L., Provacia L.B., van den Brand J.M., Wiersma L., Ouwendijk W.J., Bestebroer T.M., Spronken M.I., van Amerongen G., Rottier P.J., Fouchier R.A., Bosch B.J., Osterhaus A.D., Haagmans B.L. Adenosine deaminase acts as a natural antagonist for dipeptidyl peptidase 4-mediated entry of the Middle East respiratory syndrome coronavirus. J. Virol. 2014;88:1834–1838. doi: 10.1128/JVI.02935-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Schloer S., Brunotte L., Goretzko J., Mecate-Zambrano A., Korthals N., Gerke V., Ludwig S., Rescher U. Targeting the endolysosomal host-SARS-CoV-2 interface by clinically licensed functional inhibitors of acid sphingomyelinase (FIASMA) including the antidepressant fluoxetine. Emerg. Microb. Infect. 2020;9:2245–2255. doi: 10.1080/22221751.2020.1829082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W., Randall G. Lipids in innate antiviral defense. Cell Host Microbe. 2013;14:379–385. doi: 10.1016/j.chom.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorpp K., Rothenaigner I., Salmina E., Reinshagen J., Low T., Brenke J.K., Gopalakrishnan J., Tetko I.V., Gul S., Hadian K. Identification of small-molecule frequent hitters from AlphaScreen high-throughput screens. J. Biomol. Screen. 2014;19:715–726. doi: 10.1177/1087057113516861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2020;117:11727–11734. doi: 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y.H., Jeong K., Lee J., Lee H.J., Yim J., Kim J., Kim S., Park S.B. Inhibition of ACE2-spike interaction by an ACE2 binder suppresses SARS-CoV-2 entry. Angew Chem. Int. Ed. Engl. 2022;61 doi: 10.1002/anie.202115695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z., Xu Y., Bao L., Zhang L., Yu P., Qu Y., Zhu H., Zhao W., Han Y., Qin C. 2019. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart S.A., Dykxhoorn D.M., Palliser D., Mizuno H., Yu E.Y., An D.S., Sabatini D.M., Chen I.S., Hahn W.C., Sharp P.A., Weinberg R.A., Novina C.D. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tregoning J.S., Flight K.E., Higham S.L., Wang Z., Pierce B.F. Progress of the COVID-19 vaccine effort: viruses, vaccines and variants versus efficacy, effectiveness and escape. Nat. Rev. Immunol. 2021;21:626–636. doi: 10.1038/s41577-021-00592-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman E.F., Kirakossian H., Singh S., Wu Z.P., Irvin B.R., Pease J.S., Switchenko A.C., Irvine J.D., Dafforn A., Skold C.N., et al. Luminescent oxygen channeling immunoassay: measurement of particle binding kinetics by chemiluminescence. Proc. Natl. Acad. Sci. U. S. A. 1994;91:5426–5430. doi: 10.1073/pnas.91.12.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Yang P., Liu K., Guo F., Zhang Y., Zhang G., Jiang C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008;18:290–301. doi: 10.1038/cr.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N., Shi X., Jiang L., Zhang S., Wang D., Tong P., Guo D., Fu L., Cui Y., Liu X., Arledge K.C., Chen Y.H., Zhang L., Wang X. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013;23:986–993. doi: 10.1038/cr.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S., Liu Q., Wang Q., Sun Z., Su S., Du L., Ying T., Lu L., Jiang S. Middle East respiratory syndrome coronavirus (MERS-CoV) entry inhibitors targeting spike protein. Virus Res. 2014;194:200–210. doi: 10.1016/j.virusres.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Geng T., Yang G., Ma J., Wang L., Ketkar H., Yang D., Lin T., Hwang J., Zhu S., Wang Y., Dai J., You F., Cheng G., Vella A.T., Flavell R.A., Fikrig E., Wang P. Macrophage scavenger receptor 1 controls Chikungunya virus infection through autophagy in mice. Commun. Biol. 2020;3:556. doi: 10.1038/s42003-020-01285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York A.G., Williams K.J., Argus J.P., Zhou Q.D., Brar G., Vergnes L., Gray E.E., Zhen A., Wu N.C., Yamada D.H., Cunningham C.R., Tarling E.J., Wilks M.Q., Casero D., Gray D.H., Yu A.K., Wang E.S., Brooks D.G., Sun R., Kitchen S.G., Wu T.T., Reue K., Stetson D.B., Bensinger S.J. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell. 2015;163:1716–1729. doi: 10.1016/j.cell.2015.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zhao G., Du L., Ma C., Li Y., Li L., Poon V.K., Wang L., Yu F., Zheng B.J., Jiang S., Zhou Y. A safe and convenient pseudovirus-based inhibition assay to detect neutralizing antibodies and screen for viral entry inhibitors against the novel human coronavirus MERS-CoV. Virol. J. 2013;10:266. doi: 10.1186/1743-422X-10-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N., Pan T., Zhang J., Li Q., Zhang X., Bai C., Huang F., Peng T., Zhang J., Liu C., Tao L., Zhang H. Glycopeptide antibiotics potently inhibit cathepsin L in the late endosome/lysosome and block the entry of ebola virus, Middle East respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV) J. Biol. Chem. 2016;291:9218–9232. doi: 10.1074/jbc.M116.716100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu S., Deng Y.Q., Zhou C., Li J., Li L., Chen Q., Li X.F., Zhao H., Gold S., He J., Li X., Zhang C., Yang H., Cheng G., Qin C.F. 25-Hydroxycholesterol is a potent SARS-CoV-2 inhibitor. Cell Res. 2020;30:1043–1045. doi: 10.1038/s41422-020-00398-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.