

Abstract
Approximately one-third of estrogen receptor (ER) positive breast tumors fail to respond to or become resistant to hormonal therapy. Although the mechanisms responsible for hormone resistance are not completely understood, resistance is associated with alterations in ERα; overexpression of proteins that interact with the receptor; and hormone independent activation of the receptor by growth factor signal transduction pathways. Our previous studies show that in estrogen dependent breast cancer cells, activation of the epidermal growth factor signaling pathway increases intracellular calcium which binds to and activates ERα through sites in the ligand binding domain of the receptor and that treatment with extracellular calcium increases the concentration of intracellular calcium which activates ERα and induces hormone independent cell growth. The present study asked whether overexpression of calcium channels contributes to the hormone independent and resistant phenotype of breast cancer cells and whether clinically used calcium channel blockers reverse hormone independence and resistance. The results show that hormone independent and resistant cells overexpress calcium channels, have high concentrations of intracellular calcium, overexpress estrogen responsive genes, and, as expected, grow in the absence of estradiol and that treatment with calcium channel blockers decreased the concentration of intracellular calcium, the expression of estrogen responsive genes, and cell growth. More importantly, in hormone resistant cells, treatment that combined a calcium channel blocker with an antiestrogen reversed resistance to the antiestrogen.
Keywords: breast cancer, hormone resistance, calcium, calcium channels
Introduction
The estrogen receptor (ER) plays a central role in the treatment of breast cancer. At the time of diagnosis, approximately 70% of breast tumors are estrogen receptor positive but one-third of ER positive tumors fail to respond to or become resistant to hormonal therapy (reviewed in (1). Although the mechanisms responsible for hormone resistance are not completely understood, resistance is associated with loss, mutation, or posttranslational modification of ERα; overexpression of proteins that interact with the receptor; hormone independent activation of ERα by growth factor signal transduction pathways as a result of overexpression of the epidermal growth factor (EGF) receptor, HER2, and insulin-like growth factor-1 receptor; mutation of PI3K; activation of AKT; loss of PTEN; DNA damage repair defects; and altered expression of mitotic protein kinases (2–6). The mechanisms by which growth factor signaling pathways activate ERα are also not completely understood but are thought to be due to the phosphorylation of the N-terminal transcription activation function-1 (AF-1) domain. However, phosphorylation of the AF-1 domain does not account for the conformational changes necessary for the activation of the transcription activation function-2 (AF-2) domain in the C-terminal ligand binding domain (LBD) suggesting that additional intracellular events are required for the activation of ERα. Calcium is a second messenger of signal transduction pathways. Our previous studies show that calcium mediates the cross talk between the EGF signaling pathway and the LBD of ERα (7). In contrast to estradiol that binds in the ligand binding pocket of the LBD, calcium binds and activates the receptor through sites on the solvent accessible surface of the LBD (7). The calcium interaction sites are located on helices that are repositioned when the receptor is activated suggesting that the interaction of calcium with these sites induces a conformational change in the LBD similar to the conformational change induced by estradiol. In addition to EGF, the study shows that treatment with extracellular calcium increases the concentration of intracellular calcium which in turn, activates ERα and induces cell growth of estrogen dependent breast cancer cells in the absence of estradiol (7). The ability of extracellular calcium to increase the concentration of intracellular calcium and of calcium to activate ERα suggests a role for calcium and calcium channels in hormone independent and resistant breast cancer.
In addition to utilizing calcium as a second messenger, the mammary gland is a unique organ that transports and concentrates calcium for milk production (8–10). The movement of calcium across mammary epithelial cells is thought to involve two pathways. Calcium is initially taken up from the plasma by calcium channels in the basolateral membrane and then transported through the Golgi and released from secretory vesicles at the apical membrane or transported through the cytoplasm by calcium binding proteins and secreted into the lumen by plasma membrane calcium-ATPases. Channels in the basolateral membrane that are responsible for the influx of calcium are not well characterized but may include the calcium channels CACNA1-A, -B, -C, -G, -H, and -S which are activated by membrane potential; the transient receptor potential (TRP) channels TRPC-1, -4, -5, and 6, TRPV-2, -5, and -6, and TRPM-7 and -8 which respond to heat, cold, stretch, capsacin, vanillin, and noxious chemicals; and the Orai channels which are store activated (8–10). There is some evidence linking these channels with breast cancer (reviewed in(11).
The ability of calcium to activate ERα and increase cell growth in hormone dependent breast cancer cells in the absence of estradiol (7) and the association between expression of calcium channels and breast cancer suggest that calcium channels are potential therapeutic targets for treatment of hormone independent and resistant disease. Calcium channel blockers are drugs that are used to treat hypertension, control heart rate, prevent cerebral vasospasm, and reduce angina. This study asked whether overexpression of calcium channels contributes to the hormone independent and resistant phenotype in breast cancer cells and because calcium channel blockers do not have major side effects, the study also asked whether clinically used calcium channel blockers reverse hormone independence and resistance. The results show that hormone independent and resistant breast cancer cells overexpress calcium channels, have high concentrations of intracellular calcium, and overexpress estrogen responsive genes. In hormone independent cells, treatment with calcium channel blockers decreased the concentration of intracellular calcium and mimicked the effects of an antiestrogen on the expression of estrogen responsive genes and cell growth. In hormone resistant cells, treatment with calcium channel blockers decreased the concentration of intracellular calcium, the expression of estrogen responsive genes, and cell growth and importantly, treatment that combined a calcium channel blocker with an antiestrogen reversed resistance to the antiestrogen. Taken together, the results suggest a potential role for calcium channel blockers in the treatment of hormone independent and resistant breast cancer.
Materials and Methods
Tissue culture
MCF-7 (RRID:CVCL_0031) and MCF7/LCC9 (RRID:CVCL_DP52; LCC9) cells were obtained from the Tissue Culture and Biobanking Shared Resource at Georgetown University. MCF7-2A (RRID:CVCL_4Y53) cells were obtained from Dr. V. Craig Jordan’s Laboratory. All cell lines were authenticated using short tandem repeat profiling within the last three years. All experiments were performed with mycoplasma-free cells. Cells were grown and maintained at 37°C under humidified 5% CO2 conditions. MCF-7 cells were maintained in Improved Minimal Essential Media (IMEM; Crystalgen, Commack, NY) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich). For experiments with MCF-7 cells, the media was changed to phenol red free IMEM containing 5% charcoal stripped calf serum (CCS; Valley Biomedical, Winchester, VA) for 48 hours prior to treatment. MCF7-2A and LCC9 cells were grown and maintained in phenol red free IMEM containing 5% charcoal stripped fetal bovine serum (CFBS; Valley Biomedical, Winchester, VA). ICI-182,780 (catalog number 1047) and 4-hydroxytamoxifen (catalog number 3412) were purchased from Tocris. Methoxyverapamil chloride (catalog number M5644), mibefradil (catalog number M54411), and estradiol (catalog number E8875) were purchased from Sigma-Aldrich.
Calcium measurements
MCF7 cells were plated in IMEM containing phenol red and supplemented with 5% FBS. When 70% confluent, the media was changed to phenol red free IMEM supplemented with 5% CCS for 48 hours. MCF7-2A and LCC9 cells were plated in phenol red free IMEM supplemented with 5% CCS. When 70–80% confluent, the media was changed and the cells were incubated for 72 hours with calcium channel blockers. Cells were treated for 72 hours with methoxyverapamil (75μM) and/or mibefradil (5μM) in the absence and presence of 17β-estradiol (1nM), 4-hydroxy tamoxifen (1μM) or ICI-182,780 (100nM). Following treatment, the cells were trypsinized, washed twice with HBSS and incubated at 37°C for 30 minutes with 5μM calcium dye Fluo-4-AM (in HBSS). Following incubation, the cells (0.5 × 106 or 1 × 106) were washed twice with HBSS, resuspended in 2ml HBSS, and incubated at room temperature for 20 minutes to allow for de-esterification of the dye. The emission at 516nm was determined using an excitation wavelength of 494nm (QuantaMaster 8000) and intracellular calcium concentration was calculated using the equation: [Cain 2+] = Kd × ([F − Fmin])/ ([Fmax −F]) where Kd is the affinity of the dye for calcium, F is the initial fluorescence measured, Fmin is the fluorescence measured in absence of calcium following treatment with 10mM EGTA, and Fmax is the fluorescence measured when calcium is saturated with the dye using 1% Triton-X.
Quantitative real-time reverse transcription-PCR
As described above, MCF7 cells were plated in IMEM containing phenol red and supplemented with 5% FBS. When 70% confluent, the media was changed to phenol red free IMEM supplemented with 5% CCS for 48 hours. MCF7-2A and LCC9 cells were plated in phenol red free IMEM supplemented with 5% CCS. Cells were treated with methoxyverapamil (75μM) and/or mibefradil (5μM) in the absence and presence of 17β-estradiol (1nM), 4-hydroxy tamoxifen (1μM) or ICI-182,780 (100nM). RNA was isolated using the Trizol method and residual DNA was degraded with DNaseI (Invitrogen). For the reverse transcriptase reaction, each 50μL reaction contained 10μL of M-MLV RT 5x buffer (Promega), 11μL 25 mM MgCl2, 10μL deoxynucleotide triphosphates (Applied Biosystems), 2.5μL random hexamers, 1μL RNase inhibitor, 1.25μL MuLV reverse transcriptase, and 1μg RNA. The mixture was incubated in a thermal cycler for 10 minutes at 25°C, 30 minutes at 48°C, and 5 minutes at 95°C. For the real-time PCR reaction, each 10μL reaction contained 5.0μL Universal Master Mix, 0.5μL of 20x Assay on Demand (Applied Biosystems), and 4.5μL cDNA. Samples were run on the 7900HT (Fast Real-Time PCR System (Applied Biosystems) and the data were analyzed by the 2−ΔΔCt method using the SDS 2.3 software (Applied Biosystems).
Cell growth assays
For growth assays, MCF7-2A and LCC9 cells were grown and seeded at a density of 20,000 and 50,000 cells/well, respectively, in 12 well plates containing 1ml of phenol red free IMEM supplemented with 5% CFBS. After 24 hours, the cells were treated in triplicate and allowed to grow for 4 days. Cells were rinsed with PBS, trypsinized, and counted with a Coulter counter. MCF7 cells were seeded at a density of 20,000 cells/well in 96 wells containing 0.2 ml of phenol red free IMEM with 5% CCS. After 48 hours, the cells were treated and allowed to grow for 4 days. Cells were rinsed with PBS and stained with 0.5% w/v crystal violet (25% methanol: 75% water; Sigma-Aldrich) for 10 minutes with continuous shaking at room temperature. The cells were washed 3 times with deionized water and allowed to dry overnight. Sodium citrate buffer (0.1M in 50% ethanol: 50% water; Sigma-Aldrich) was added and the plates were shaken for 5 minutes at room temperature. The absorbance at 560 nm was read on a plate reader.
Statistical analysis
All statistical analyses were performed in Prism 6.0 (GraphPad, San Diego, CA). Data are presented as the mean ± standard error of the mean (SEM). Statistical differences were evaluated by ANOVA followed by Fishers LSD test using PRISM. Statistical significance was defined as a P value of ≤ 0.05. *p≤0.05; **p≤0.01; ***p≤0.001; ****p≤0.0001.
Results
Expression of calcium channels and concentration of intracellular calcium in hormone independent and resistant breast cancer cells
To address whether dysregulation of calcium homeostasis and consequently activation of ERα by the metal contribute to hormone independence and resistance, the expression of calcium channels and the concentration of intracellular calcium were measured in hormone independent and resistant variants of the breast cancer cells, MCF-7. MCF-7 is a hormone dependent cell line that requires estradiol for growth (12). MFC7-2A cells are a hormone independent variant of MCF-7 cells that were selected for growth in the absence of estradiol (13). MFC7/LCC9 cells are a hormone resistant variant of MCF-7 cells. LCC9 cells were selected for growth in the absence of estradiol and subsequently selected for growth in the presence of the pure antiestrogen fulvestrant (ICI-182,780; ICI) (14), also referred to as a selective estrogen receptor degrader (SERD). In addition to resistance to ICI-182,780, LCC9 cells are also resistant to the antiestrogen 4-hydroxy tamoxifen (Tam) known for its selective estrogen receptor modulator (SERM) activity (14). To ask whether acquisition of hormone independence and resistance was due to perturbation in calcium homeostasis, the concentration of intracellular calcium was measured in the MCF-7 variants (Table 1). In the parental MCF-7 cells, the intracellular concentration of calcium was approximately 188.92nM consistent with our previous report (7). In contrast to MCF-7 cells, the hormone independent and resistant variants had an approximately 1.8- to 2.2-fold higher concentration of intracellular calcium. In MCF7-2A and LCC9 cells, the concentration of calcium was approximately 378.65nM and 338.24nM, respectively, similar to the binding affinity of calcium for ERα (Kd = ~500nM +/− 160nM) (7). To identify the calcium channels that contribute to the elevated concentrations of intracellular calcium, the expression of L- and T-type calcium channels as well as other calcium channels that are associated with breast cancer was measured (Table 1). Compared to MCF-7 cells, MCF7-2A cells overexpressed the L-type calcium channel 1C (CANA1C; ~ 6.9-fold) and the T-type calcium channel 1G (CCANA1G; ~ 2.1-fold). The LCC9 cells overexpressed the T-type calcium channel 1G (~ 3.4-fold), the transient receptor potential calcium channels TRPM8 (~2.5-fold) and TRPV6 (~2.3-fold), and the purinergic receptor P2RX6 (~3-fold). The association between the acquisition of hormone independence, overexpression of calcium channels, and elevation of intracellular calcium to concentrations similar to the binding affinity of calcium for ERα suggests that the overexpression of calcium channels and high concentrations of intracellular calcium contribute to the hormone independent and resistant phenotype of breast cancer cells.
Table 1.
Intracellular calcium and calcium channels in hormone independent and resistant breast cancer cells.
| [Ca]i, nM (+/−SEM) | CACNA1C | CACNA1D | CACNA1G | CACNA1H | TRPC6 | TRPM7 | TRPM8 | TRPV6 | P2RX6 | |
|---|---|---|---|---|---|---|---|---|---|---|
| MCF-7 | 188.92 +/−21.5 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| MCF7-2A | 378.65 +/−27.7 | 6.98 | 1.67 | 2.19 | 1.51 | 0.47 | 1.19 | 1.08 | 0.37 | 1.35 |
| MCF7/LCC9 | 338.24 +/−47.2 | 0.71 | 1.56 | 3.39 | 0.58 | 0.32 | 0.46 | 2.51 | 2.34 | 3.01 |
MCF7-2A and MCF7/LCC9 cells were grown in phenol red free IMEM supplemented with 5% CFBS. MCF-7 cells were grown in phenol red containing IMEM supplemented with 10% fetal bovine serum; prior to measurements, the media of MCF-7 cells was changed to phenol red free IMEM containing 5% CCS for 48 hours. To quantify the concentration of intracellular calcium, the cells were trypsinized and incubated for 20 minutes with 5 uM Fluo-4-AM. The emission at 516 nm was determined using an excitation wavelength of 494 nm and the concentration of intracellular calcium was calculated using the equation: [Cain 2+] = Kd × ([F − Fmin])/ ([Fmax −F]) where Kd is the affinity of the dye for calcium. Data are the mean ± SEM (n ≥ 3). To determine the expression of calcium channels, the amount of CACNA1-C, -D, -G, - H; TRPC6; TRPM-7, -8; TRPV6; and P2RX6 mRNA was measured using a quantitative real time-PCR assay and normalized to the amount of ribosomal large protein P0 (RPLP0) mRNA using the 2−ΔΔCt method. Data are presented as fold change compared to MCF-7 cells, (mean ± SEM; n ≥ 3).
Effects of calcium channel blockers in the hormone independent breast cancer cells MCF7-2A
To determine whether calcium channel blockers reverse the hormone independent phenotype, MCF7-2A cells were treated with methoxyverapamil and mibefradil and the effects on the concentration of intracellular calcium, gene expression, and hormone independent growth were measured. Methoxyverapamil and mibefradil are clinically used calcium channel blockers that block both L- and T-type calcium channels. Methoxyverapamil is a nonspecific calcium channel blocker that has greater specificity for L-type channels (Ki/IC50 for L-type channels approximately 10–16μM; Ki/IC50 for T-type channels approximately 100μM) (15,16) and mibefradil has greater specificity for T-type channels (Ki/IC50 for T-type channels approximately 1μM; Ki/IC50 for L-type channels approximately 10–50μM) (16,17). To ask whether the channel blockers decrease the concentration of intracellular calcium, MCF-7 and MCF7-2A cells were treated for 72 hours with methoxyverapamil (75μM) and/or mibefradil (5μM) in the absence and presence of 17β-estradiol (1nM), 4-hydroxy tamoxifen (1μM) or ICI-182,780 (100nM) and the concentration of intracellular calcium was measured (Figure 1A & C). Treatment of MCF7-2A cells with mibefradil or methoxyverapamil plus mibefradil reduced the concentration of intracellular calcium from approximately 378.65nM to 241.39nM (p<0.05) and 266.93nM (p<0.01), respectively, but treatment with methoxyverapamil had no significant effect on the concentration of calcium (313.9nM). Treatment of MCF7-2A cells with tamoxifen or ICI-182,780 also reduced the concentration of intracellular calcium to approximately 249.0nM (p<0.05) and 214.57nM (p<0.01), respectively, and treatments that combined ICI-182,780 with methoxyverapamil, mibefradil or methoxyverapamil plus mibefradil reduced the concentrations of calcium to approximately 182.5nM (p<0.001), 190.7nM (p<0.001), and 224.1nM (p<0.01), respectively. Treatment that combined tamoxifen with mibefradil or methoxyverapamil plus mibefradil reduced the concentration of calcium to 194.5nM (p<0.001) and 195.9nM (p<0.001), respectively, however, treatment that combined tamoxifen with methoxyverapamil had no significant effect on the concentration of intracellular calcium (410.6nM). In contrast to MCF7-2A cells where treatment with methoxyverapamil plus mibefradil significantly reduced the concentration of intracellular calcium, treatment of MCF-7 cells with both channel blockers had no significant effect on the concentration of intracellular calcium (188.92 vs 190.17nM; Figure 1C).
Figure 1.
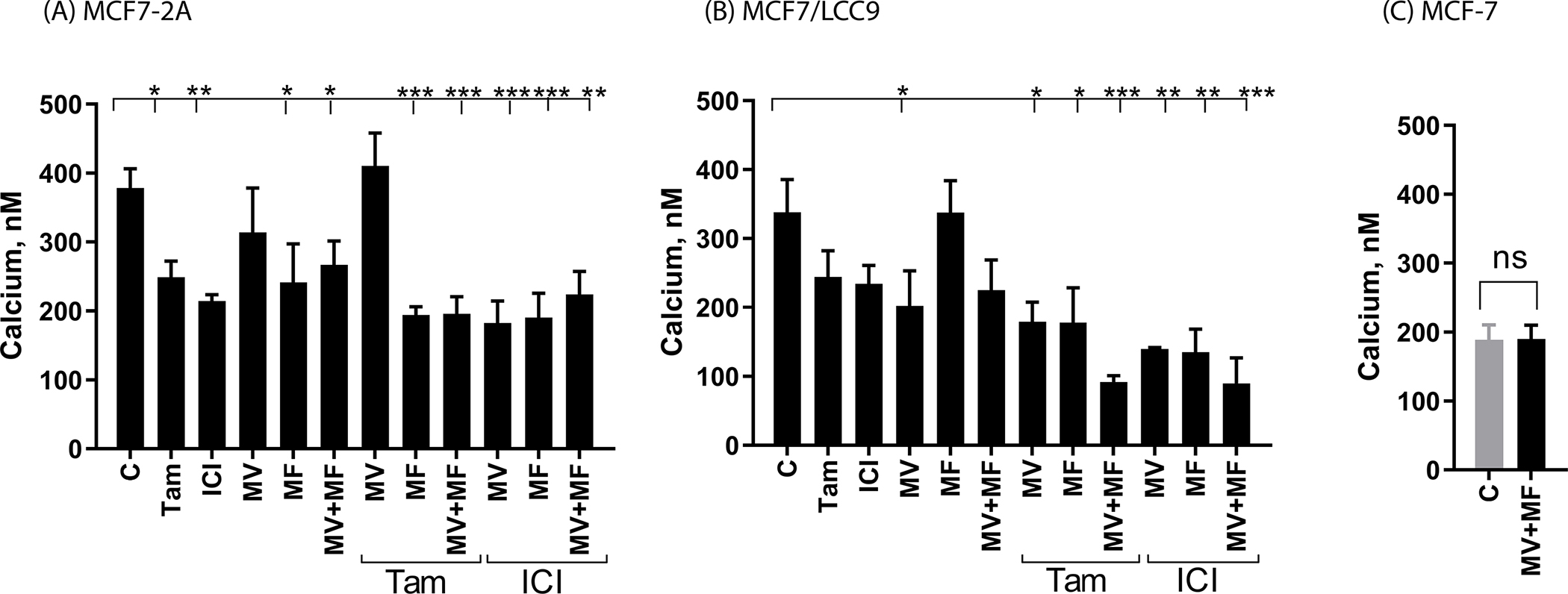
Effects of calcium channel blockers on the concentration of intracellular calcium
MCF7-2A and MCF7/LCC9 cells were grown in phenol red free IMEM supplemented with 5% CFBS. MCF-7 cells were grown in phenol red containing IMEM supplemented with 10% fetal bovine serum; prior to treatment, the media of MCF-7 cells was changed to phenol red free IMEM containing 5% CCS for 48 hours. Cells were treated for 72 hours with methoxyverapamil (MV; 75 μM) and/or mibefradil (MF; 5 μM) in the absence and presence of 17β-estradiol (E2; 1 nM), 4-hydroxy tamoxifen (TAM; 1 μM), or ICI-182,780 (ICI; 100 nM). To quantify the concentration of intracellular calcium, the cells were trypsinized and incubated for 20 minutes with 5 uM Fluo-4-AM. The emission at 516 nm was determined using an excitation wavelength of 494 nm and the concentration of intracellular calcium was calculated using the equation: [Cain 2+] = Kd × ([F − Fmin])/ ([Fmax −F]) where Kd is the affinity of the dye for calcium. Data are the mean ± SEM (n ≥ 3). *, p≤0.05; **, p≤0.01; ***, p≤0.001; ****, p≤0.0001. panel A, MCF7-2A cells; panel B, MCF7/LCC9 cells; panel C, MCF-7 cells.
Compared to MCF-7 cells, MCF7-2A cells also overexpress the estrogen responsive genes progesterone receptor (PgR; ~11-fold; data not shown) and trefoil factor 1 (pS2; ~5-fold; data not shown). To determine whether elevated concentrations of intracellular calcium contribute to hormone independent activation of ERα and expression of ERα regulated genes, MCF7-2A cells were treated with methoxyverapamil (75μM) and/or mibefradil (5μM) in the absence and presence of 17β-estradiol (1nM), 4-hydroxy tamoxifen (1μM) or ICI-182,780 (100nM) and the expression of progesterone receptor (Figure 2A) and pS2 (Figure 3A) was measured at 48 and 72 hours. Treatment with estradiol increased the expression of progesterone receptor and pS2 by approximately 2- to 3-fold (p<0.00001) and treatment with ICI-182,780 decreased expression by approximately 80% to 90%. Treatment with tamoxifen had no effect on the expression of pS2 and increased the expression of progesterone receptor by 2-fold (p<0.05) consistent with its SERM like activity. Treatment with either methoxyverapamil or mibefradil had no effect on the expression of progesterone receptor or pS2 but consistent with the overexpression of both the L- and T-type channels, treatment with methoxyverapamil plus mibefradil decreased expression by approximately 50% and 60%, respectively. Treatment that combined the channel blockers with tamoxifen or ICI-182,780 did not inhibit the estrogen like activity of tamoxifen or further enhance the antiestrogen like activity of ICI-182,780. To ask whether calcium channel blockers interfere with the function of ERα, the cells were treated with estradiol and methoxyverapamil, mibefradil, or methoxyverapamil plus mibefradil. Treatment with the channel blockers did not inhibit estradiol induction of progesterone receptor or pS2 suggesting that the calcium channel blockers do not interfere with the function of ERα. As a control, MCF-7 cells were treated with methoxyverapamil (75μM) and/or mibefradil (5μM) in the absence and presence of 17β-estradiol (1nM), 4-hydroxy tamoxifen (1μM) or ICI-182,780 (100nM) in hormone free media. The expression of progesterone receptor (Figure 2C) and pS2 (Figure 3C) was measured at 48 and 72 hours. In the absence of estradiol, MCF-7 cells have undetectable expression of progesterone receptor and low expression of pS2. Treatment with methoxyverapamil and/or mibefradil, 4-hydroxytamoxifen, or ICI-182-780 had no effect on the expression of progesterone receptor and pS2 but treatment with estradiol increased expression by 22- to 27-fold and 5-fold, respectively. Treatment with the calcium channel blockers did not interfere with the ability of estradiol to increase gene expression. The ability of methoxyverapamil plus mibefradil to decrease the concentration of intracellular calcium and the expression of progesterone receptor and pS2 in MCF7-2A cells but not interfere with the function of ERα suggests that blocking the influx of calcium reduces the concentration of intracellular calcium and reverses the activation of the receptor.
Figure 2.
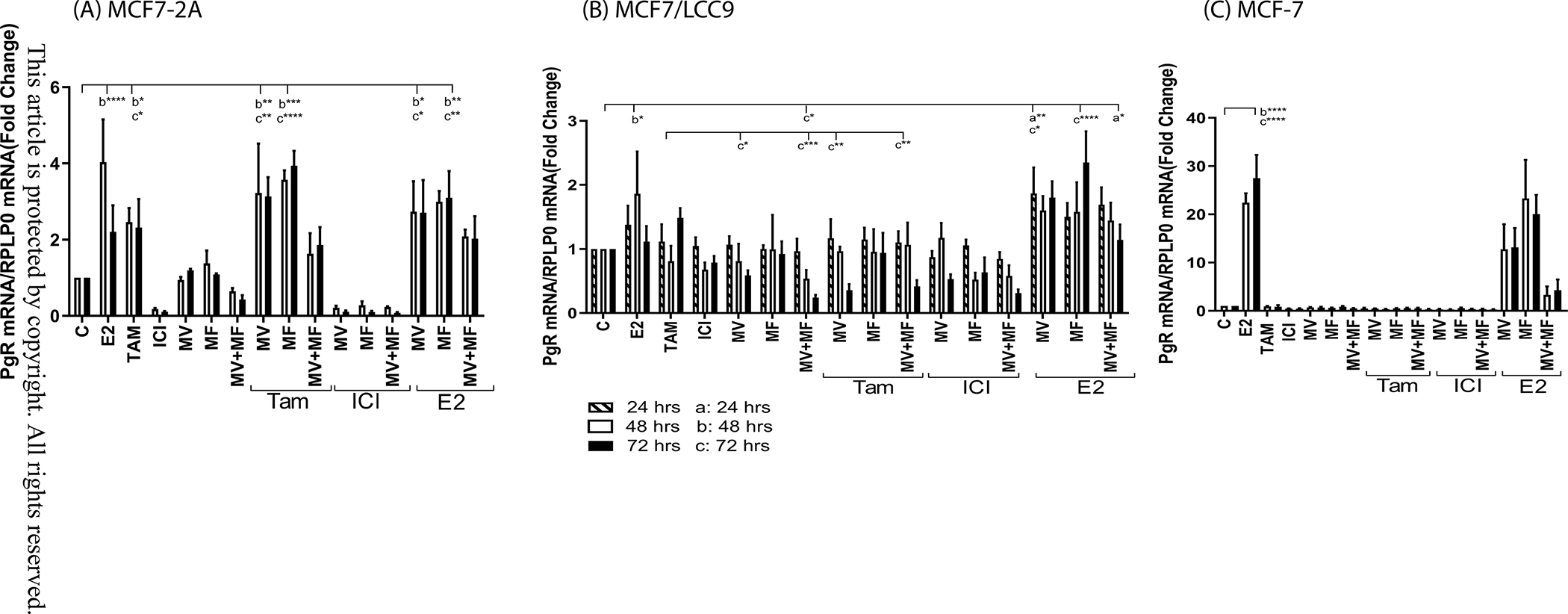
Effects of calcium channel blockers on the expression of progesterone receptor
MCF7-2A and MCF7/LCC9 cells were grown in phenol red free IMEM supplemented with 5% CFBS. MCF-7 cells were grown in phenol red containing IMEM supplemented with 10% fetal bovine serum; prior to treatment, the media of MCF-7 cells was changed to phenol red free IMEM containing 5% CCS for 48 hours. The cells were treated for 24, 48 or 72 hours with methoxyverapamil (MV; 75 μM) and/or mibefradil (MF; 5 μM) in the absence and presence of 17β-estradiol (E2; 1 nM), 4-hydroxy tamoxifen (TAM; 1 μM), or ICI-182,780 (ICI; 100 nM). RNA was isolated. The amount of PgR mRNA was measured using a quantitative real time-PCR assay and normalized to the expression of ribosomal large protein P0 (RPLP0) mRNA using the 2−ΔΔCt method. Data are presented as fold change compared to control. (mean ± SEM; n ≥ 3; *, p≤0.05; **, p≤0.01; ***, p≤0.001; ****, p≤0.0001; a, 24 hours; b, 48 hours; c, 72 hours). panel A, MCF7-2A cells; panel B, MCF7/LCC9 cells; panel C, MCF-7 cells.
Figure 3.
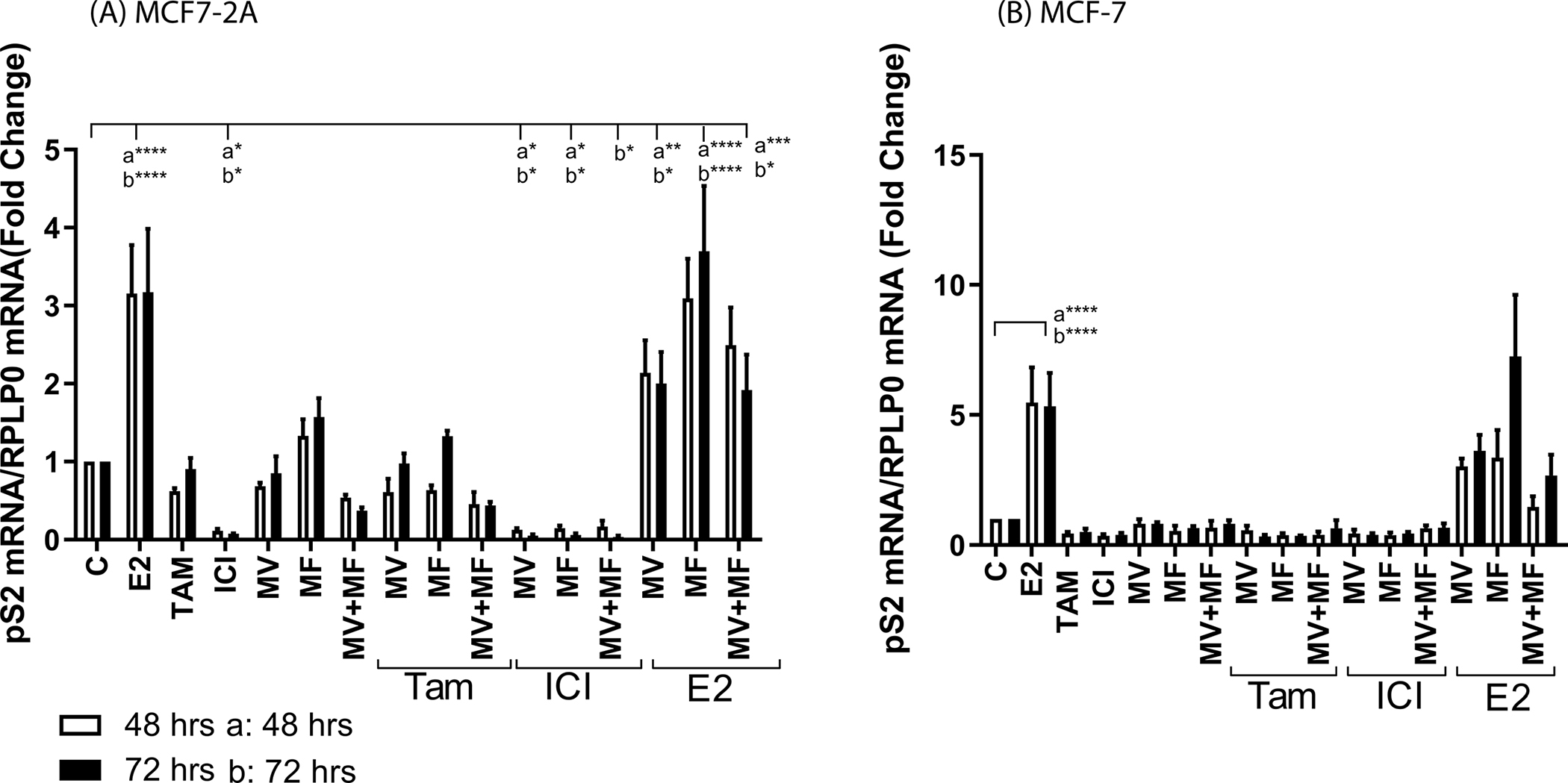
Effects of calcium channel blockers on the expression of pS2
MCF7-2A cells were grown in phenol red free IMEM supplemented with 5% CFBS. MCF-7 cells were grown in phenol red containing IMEM supplemented with 10% fetal bovine serum; prior to treatment, the media of MCF-7 cells was changed to phenol red free IMEM containing 5% CCS for 48 hours. The cells were treated for 48 or 72 hours with methoxyverapamil (MV; 75 μM) and/or mibefradil (MF; 5 μM) in the absence and presence of 17β-estradiol (E2; 1 nM), 4-hydroxy tamoxifen (TAM; 1 μM), or ICI-182,780 (ICI; 100 nM). RNA was isolated. The amount of pS2 mRNA was measured using a quantitative real time-PCR assay and normalized to the expression of ribosomal large protein P0 (RPLP0) mRNA using the 2−ΔΔCt method. Data are presented as fold change compared to control. (mean ± SEM; n ≥ 3; a, 48 hours; b, 72 hours; *, p≤0.05; **, p≤0.01; ***, p≤0.001; ****, p≤0.0001). panel A, MCF7-2A cells; panel B, MCF-7 cells.
To ask whether the calcium channel blockers reverse hormone independent growth, MCF7-2A cells were treated with methoxyverapamil (75μM) and/or mibefradil (5μM) in the absence and presence of 17β-estradiol (1nM), 4-hydroxy tamoxifen (1μM) or ICI-182,780 (100nM) and growth was measured on day 4 (Figure 4A). As expected, MCF7-2A cells were hormone independent; there was an approximately 10- and 9.5-fold increase in the number of cells in the absence or presence of estradiol, respectively. Treatment with tamoxifen or ICI-182,780 reduced cell growth by approximately 50% (p<0.001) and 80% (p<0.0001), respectively. Treatment with methoxyverapamil or mibefradil reduced growth by approximately 40% (p<0.05) and 50% (p<0.001), respectively, and treatment that combined methoxyverapamil and mibefradil reduced growth by approximately 80% (p<0.0001), consistent with the overexpression of both L- and T-type channels. Compared to treatment with tamoxifen, methoxyverapamil, or mibefradil, treatment that combined tamoxifen with methoxyverapamil, mibefradil, or methoxyverapamil plus mibefradil further reduced growth by approximately 70% (p<0.001), 80% (p<0.001), and 90% (p<0.0001), respectively. However, treatment that combined ICI-182,780 with methoxyverapamil, mibefradil, or methoxyverapamil plus mibefradil did not further reduce cell growth (approximately 80%, 90%, and 90% reduction, respectively) compared to treatment with ICI-182,780. The MCF-7 cells were also treated with methoxyverapamil (75μM) and/or mibefradil (5μM) in the absence and presence of 17β-estradiol (1nM), 4-hydroxy tamoxifen (1μM) or ICI-182,780 (100nM) and growth was measured on day 4 (Figure 4B). Treatment with methoxyverapamil and/or mibefradil, 4-hydroxytamoxifen, or ICI-182-780 had no effect on cell growth but treatment with estradiol increased growth by approximately 2.6-fold. Treatment with the calcium channel blockers did not interfere with the ability of estradiol to increase growth. Taken together, the overexpression of calcium channels and the ability of calcium channel blockers to decrease the concentration of intracellular calcium, block the expression of estrogen responsive genes, and inhibit hormone independent growth in the MCF7-2A cells suggest that high concentrations of intracellular calcium contribute to the hormone independent phenotype due, in part, to the ability of calcium to activate ΕRα.
Figure 4.
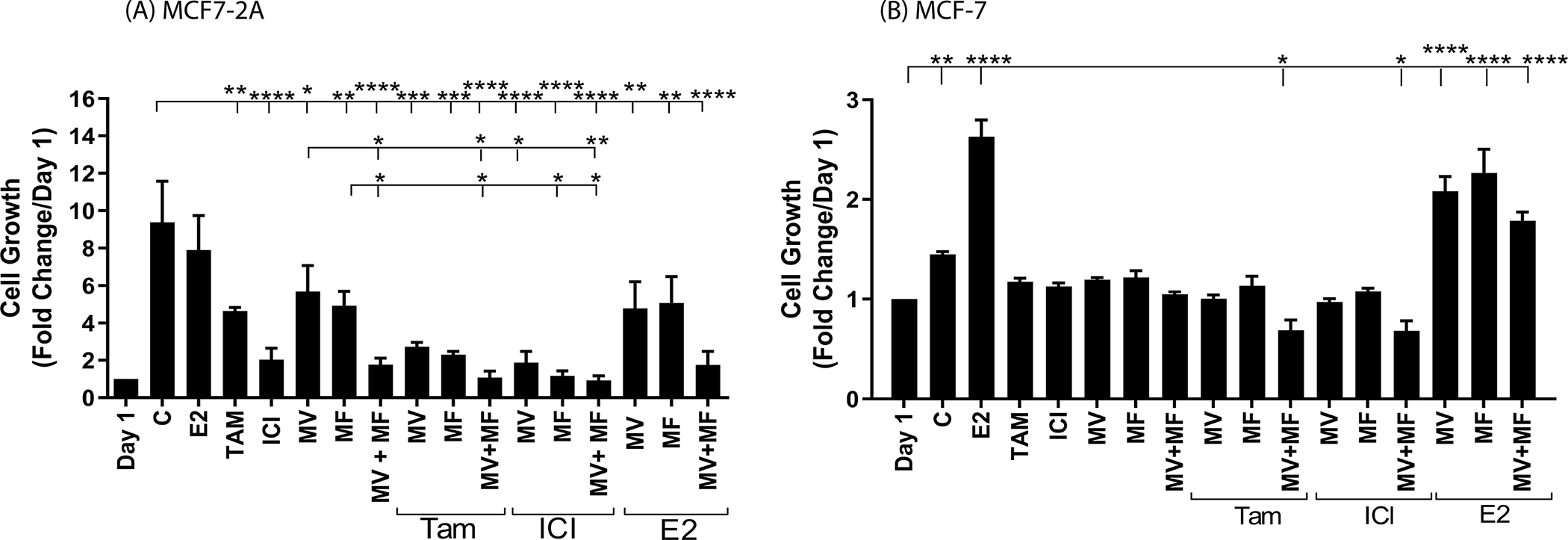
Effects of calcium channel blockers on the growth of MCF7-2A and MCF-7 cells
MCF7-2A cells were grown in phenol red free IMEM supplemented with 5% CFBS. MCF-7 cells were grown in phenol red containing IMEM supplemented with 10% FBS; the media of MCF-7 cells was changed to phenol red free IMEM containing 5% CCS for 48 hours prior to treatment. The cells were not treated (control; C) or treated with methoxyverapamil (MV; 75 μM) and/or mibefradil (MF; 5 μM) in the absence and presence of 17β-estradiol (E2; 1 nM), 4-hydroxy tamoxifen (TAM; 1 μM), or ICI-182,780 (ICI; 100 nM) for four days beginning on day 1. The number of cells was determined using a coulter counter. Data are presented as fold change compared to day 1. (mean ± SEM, n ≥ 3; *, p≤0.05; **, p≤0.01; ***, p≤0.001; ****, p≤0.0001). panel (A), MCF7-2A; panel B, MCF-7 cells.
Effects of calcium channel blockers in the hormone resistant breast cancer cells MCF7/LCC9.
To ask whether intracellular calcium also contributes to the hormone resistant phenotype, LCC9 cells were also treated with calcium channel blockers and the effects on the concentration of intracellular calcium, gene expression, and cell growth were measured. To ask whether the calcium channel blockers decrease the concentration of intracellular calcium, LCC9 cells were treated for 72 hours with methoxyverapamil (75μM) or mibefradil (5μM) in the absence and presence of 17β-estradiol (1nM), 4-hydroxy tamoxifen (1μM) or ICI-182,780 (100nM) and the concentration of intracellular calcium was measured (Figure 1B). Treatment with methoxyverapamil or methoxyverapamil plus mibefradil reduced the concentration of intracellular calcium from 338.24nM to 202.22 (p<0.05) and 225.26nM, respectively, but treatment with mibefradil had no effect on the concentration of calcium (337.6nM). Treatment with tamoxifen or ICI-182,780 reduced the concentration to 244.58 and 234.20nM, respectively. Treatment that combined ICI-182,780 with methoxyverapamil, mibefradil, or methoxyverapamil plus mibefradil reduced the concentration of calcium to approximately 179.30nM (p<0.01), 178.1nM (p<0.01), and 91.7nM (p<0.001), respectively, and treatment that combined tamoxifen with methoxyverapamil, mibefradil, or methoxyverapamil plus mibefradil reduced the concentration of calcium to approximately 139.7nM (p<0.05), 135.2nM (p<0.05), and 89.95nM (p<0.001), respectively.
Compared to MCF-7 cells, LCC-9 cells overexpress progesterone receptor (~2.7-fold; data not shown) but do not overexpress pS2 (data not shown). To ask whether elevated concentrations of intracellular calcium activate ERα in the hormone resistant cells, the LCC-9 cells were treated for 24, 48, or 72 hours with methoxyverapamil (75μM) and/or mibefradil (5μM). Cells were also treated with the calcium channel blockers in the absence and presence of 17β-estradiol (1nM), 4-hydroxy tamoxifen (1μM) or ICI-182,780 (100nM) (Figure 2B). Treatment with estradiol increased the expression of progesterone receptor by approximately 1.8-fold at 48 hours but not at 24 and 72 hours while treatment with tamoxifen, ICI-182,780, methoxyverapamil, or mibefradil had no significant effect on the expression of progesterone receptor. However, treatment with both channel blockers decreased expression of progesterone receptor by approximately 46% and 76% (p<0.05) at 48 and 72 hours, respectively. Although treatment with an antiestrogen or a calcium channel blocker had no significant effect on the expression of progesterone receptor, treatment that combined tamoxifen and methoxyverapamil decreased expression by approximately 64% (p<0.05) and treatment that combined ICI-182,780 with mibefradil decreased expression by approximately 37% to 48% suggesting that the calcium channel blocker sensitized the resistant cells to the antiestrogen. The effects of channel blockers were reversed by estradiol again demonstrating that the channel blockers do not interfere with the function of ERα.
To ask whether elevated concentrations of intracellular calcium also contribute to hormone resistant growth, LCC9 cells were treated for four days with methoxyverapamil (10μM to 100μM) or mibefradil (1μM to 10μM) in the absence and presence of 17β-estradiol (1nM), 4-hydroxy tamoxifen (1μM) or ICI-182,780 (100nM) and growth was measured (Figure 5). As expected, LCC9 cells are hormone independent and resistant and grow in the absence and presence of estradiol, tamoxifen, or ICI-182,780 (Figure 5B). In the absence of estradiol, there was an approximately 5-fold increase in cell growth and in the presence of estradiol, tamoxifen, or ICI-182,780, there was a similar increase in growth (~6.6-, 3.8-, and 3.6-fold increase, respectively). Treatment with 75μM, 90μM and 100μM of methoxyverapamil reduced cell growth by approximately 40% (p<0.05), 46% (p<0.001) and 65% (p<0.001), respectively, while treatment with 3μM, 5μM, 7.5μM, and 10μM of mibefradil reduced cell growth by approximately 35% (p<0.05), 56% (p<0.01), 80% (p<0.0001), and 94% (p<0.0001), respectively (Figure 5A). To ask whether calcium channel blockers reverse hormone resistant growth, the cells were treated with tamoxifen or ICI-182,780 and concentrations of methoxyverapamil (75μM) or mibefradil (5μM) that reduce cell growth by approximately 40%. Treatment with mibefradil decreased growth by approximately 43% (p<0.01) and treatment that combined mibefradil with tamoxifen decreased growth by approximately 50% (p<00.001) while treatment that combined mibefradil with ICI-182,780 decreased growth by approximately 60% (p<0.001). Treatment with methoxyverapamil also decreased growth by approximately 45% (p<0.01) and treatment that combined methoxyverapamil with tamoxifen or ICI-182,780 decreased growth by approximately 65% (p<0.001) and 70% (p<0.001), respectively (Figure 5B), further suggesting the calcium channel blocker sensitized the resistant cells to the antiestrogens. The association between hormone resistance, overexpression of calcium channels, and increased concentrations of intracellular calcium and the ability of calcium channel blockers to decrease the concentration of intracellular calcium, expression of an estrogen responsive gene, and growth suggests that calcium contributes to hormone resistance due, in part, to activation of ERα. The combined effects of calcium channel blockers and antiestrogens further suggests that calcium channel blockers reverse hormone resistance.
Figure 5.
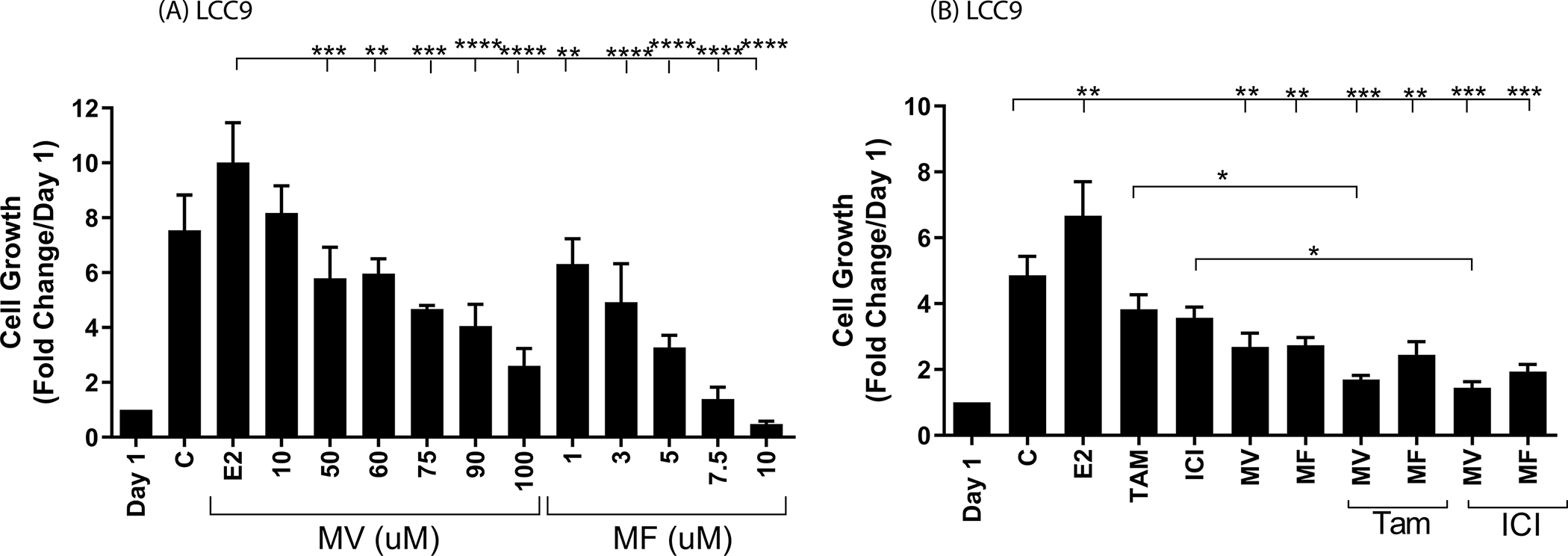
Effects of calcium channel blockers on the growth of MCF7/LCC9 cells
MCF7/LCC9 cells were grown in phenol red free IMEM supplemented with 5% CFBS. The cells were not treated (control; C) or treated with methoxyverapamil (MV; 10–100 μM) or mibefradil (MF; 1–10 μM) (panel A) or with methoxyverapamil (MV; 75 μM) and/or mibefradil (MF; 5 μM) in the absence and presence of 17β-estradiol (E2; 1 nM), 4-hydroxy tamoxifen (TAM; 1 μM), or ICI-182,780 (ICI; 100 nM) (panel B) for four days beginning on day 1. The number of cells was determined and the data are presented as fold change compared to day 1. (mean ± SEM, n ≥ 3; *, p≤0.05; **, p≤0.01; ***, p≤0.001; ****, p≤0.0001)
Discussion
The mechanisms responsible for resistance are not completely understood. Our previous study showed that in the hormone dependent MCF-7 cells, calcium activates ERα (7). The results of this study show that hormone independent and resistant cells overexpress calcium channels; have elevated concentrations of intracellular calcium; overexpress estrogen responsive genes; and as expected, grow in the absence of estradiol and that treatment with calcium channel blockers decreases the concentration of intracellular calcium, expression of estrogen responsive genes, and cell growth in hormone independent cells and treatment combining a calcium channel blocker with an antiestrogen reverses resistance in hormone resistant cells suggesting that hormone independence and resistance may be due, in part, to the overexpression of calcium channels, increase in intracellular calcium, and the subsequent activation of ERα and that treatment with calcium channel blockers may reverse hormone independent and resistant breast cancer.
Estrogen receptor-α is a ligand inducible transcription factor that belongs to the superfamily of nuclear receptors (18). It is divided into regions A through F (19). The A/B region contains the AF-1 domain that plays a role in activation of the receptor; region C is the DNA binding domain that contains two zinc fingers; region D is the region between the DNA binding domain and the ligand binding domain; and region E is the ligand binding domain that contains the ligand binding pocket, a dimerization domain, and the AF-2 domain that is responsible for ligand dependent activation of the receptor. Similar to other nuclear receptors (20–24), the LBD of ERα contains 11 α-helices (H1, H3-H12) folded into a three layered antiparallel α-helical sandwich. The central core of the LBD contains the ligand binding pocket that is formed by helices H5/6, H9, and H10 sandwiched between two layers of helices composed of H1-4, H7, H8, and H11 and flanked by H12. Based upon the crystal structure of the apo- and holo-RXR-α (20,25), it has been proposed that upon binding of estradiol in the pocket, helix H11 is repositioned and forms a continuous bent helix with helix H10. Together with helices H8 and H9, helix H10/H11forms the dimerization domain. In addition to formation of the dimerization domain, the repositioning of helix H11 results in the repositioning of helix H12 over the binding pocket which entraps the hormone. Together with helices H3, H4, and H5, helix H12 forms the AF-2 domain, the binding site for coactivators. In addition to inducing major structural changes, estradiol interacts with hydrophobic amino acids on helices H3, H5, H7, and H11 and in the turn between the β-sheets (21,23,24) resulting in one to two additional turns at the ends of helices H3, H8, H11 and H12 (23). Calcium is a ligand of ERα that also binds to the LBD and activates the receptor. In contrast to estradiol that binds inside the pocket, calcium binds to four sites on the aqueous surface of the LBD (7). One site is located at the interface of helices H10 and H11; the second site is located at the C-terminal end of helix H11 and in loop 11–12; the third site is located at the N-terminal end of helix H12; and the fourth site is located at the bend between helices H4 and H5. In proteins, metals such as calcium have several functions including the formation and stabilization of protein structure. Although the mechanism by which calcium activates ERα in the absence of estradiol is not completely understood, the identification of calcium binding sites at the ends of helices H10, H11, and H12 suggests that, similar to estradiol, one of the functions of calcium is to alter and/or stabilize the ends of the α-helical structures of the LBD (26) thereby promoting a conformational change that results in the formation of the dimerization and AF-2 domains. More specifically, the interaction of calcium at the interface of helices H10 and H11 would promote the formation of the dimerization domain while the interaction of calcium at the C-terminal end of helix H11 and in loop 11–12 and at the N-terminal end of helix H12 would stabilize additional turns in helices H11 and H12 that would shorten and/or induce a conformational change in loop 11–12 resulting in the repositioning of helix H12 over the binding pocket and the formation of the AF-2 domain. Additionally, the interaction of calcium at the interface of helices H4 and H5 would induce a kink in the helix formed by helices H4/H5 to facilitate the binding of coactivators (23). The mechanism by which calcium contributes to hormone resistance is also not understood. The ability of estradiol to activate ERα by binding in the ligand binding pocket of the LBD and calcium to activate the receptor by binding to sites on the outer surface of the LBD suggests that tamoxifen and ICI-182,780 are competitive inhibitors of estradiol but noncompetitive inhibitors of calcium. Hormone resistance may be due, in part, to the interaction of calcium with sites on the aqueous surface of the LBD that repositions helix H12 over the binding pocket and prevents the antiestrogen from entering the pocket. Consistent with this model, treatment of the hormone resistant MCF7/LCC9 cells with an antiestrogen did not decrease expression of the estrogen responsive gene or cell growth; treatment with a calcium channel blocker decreased gene expression and growth; and treatment that combined a calcium channel blocker with an antiestrogen decreased gene expression and growth more than treatment with just the calcium channel blocker. Although the proposed model of hormone resistance suggests that the combined effects of a calcium channel blocker and antiestrogen are due to the ability of calcium to activate ERα, it is also possible that the calcium channel blockers act independently of ERα, e.g., elevated concentrations of intracellular calcium are necessary for cell cycle progression through late G1 and mitosis. This proposed model of hormone resistance remains to be tested.
Calcium may also contribute to the SERM like activity of tamoxifen. Selective estrogen receptor modulators are estrogen receptor ligands that exhibit antiestrogen like activity in some tissues but estrogen like activity in other tissues. Tamoxifen is a SERM that exhibits antiestrogen like activity in breast tissue but estrogen like activity in bone, uterus, and some breast tumors. The estrogen like activity of tamoxifen has been attributed to differences in tissue specific regulation of target genes, the relative expression of coactivators and corepressors, and tamoxifen induced structural changes in ERα (27,28). In addition to its interaction with ERα, tamoxifen increases the concentration of intracellular calcium by increasing calcium release from the endoplasmic reticulum and influx from the extracellular medium (29–32). The ability of tamoxifen to increase intracellular calcium may contribute to its SERM like activity and provide an explanation for its estrogen like activity in the MCF7-2A cells.
Calcium homeostasis is tightly regulated by influx through voltage operated calcium channels, ligand activated channels, and store activated channels; release and uptake by intracellular organelles; and efflux through pumps and exchangers. There is increasing evidence from population studies linking breast cancer with the loss of calcium homeostasis (reviewed in (11)). In a meta-analysis of microarray datasets of breast tumors, overexpression of the L-type calcium channels C and D, the T-type channels G and I, and the N-type channel B is associated with invasive breast cancer (33). In addition to overexpression of membrane potential operated channels, overexpression of ligand activated channels is associated with the disease. Overexpression of TRPM7 and TRPV6 is associated with poorly differentiated and aggressive tumors, respectively, (34–36) and overexpression of the calcium sensing receptor is associated with metastatic disease. In addition to calcium channels, overexpression of the ryanodine receptor is associated with tumor grade (37). There is also evidence that women with hypercalcemia, hypercalcemia associated with hyperparathyroidism, or idiopathic hypercalciurea have a higher risk of developing breast cancer and women with breast cancer associated hypercalcemia have a poor prognosis and metastatic disease (38–41).
Although the causal link between dysregulation of calcium homeostasis and hormone independent and resistant breast cancer remains to be established, the results of the present study show that hormone independent and resistant breast cancer cells overexpress calcium channels, have high concentrations of intracellular calcium, and overexpress estrogen responsive genes. In hormone independent cells, treatment with calcium channel blockers decreased the concentration of intracellular calcium, expression of estrogen responsive genes, and cell growth. In hormone resistant cells, treatment with calcium channel blockers also decreased the concentration of intracellular calcium, expression of estrogen responsive genes, and growth and treatment that combined a calcium channel blocker with an antiestrogen reversed resistance to the antiestrogen. Taken together, the results suggest that calcium channels may be potential targets for the treatment of hormone independent and resistant breast cancer.
Novelty and Impact:
The results of this study suggest a potential role for calcium channel blockers in the treatment of hormone independent and resistant breast cancer.
One-third of oestrogen receptor-positive breast tumours fail to respond to or become resistant to hormonal therapy, due to mechanisms that remain to be fully understood. Here, hormone-independent and resistant breast cancer cells were found to overexpress calcium channels. The cancer cells’ high concentration of intracellular calcium, overexpression of estrogen-responsive genes, and growth in the absence of estradiol were decreased by calcium channel blockers. In hormone-resistant cells, treatment that combined a calcium channel blocker with an antiestrogen reversed resistance. The results suggest a potential role for calcium channel blockers in the treatment of hormone-independent and resistant breast cancer.
Acknowledgements
We thank Dr. G. Yan for helpful discussions.
Funding
This work was supported, in part, by grants from the National Institutes of Health (T32-CA009686 to K.C. & J.B.P., P30-CA51008, and U01-ES026132 to M.B.M. & B.R. H.); Department of Defense (BC122291 to M.B.M. & B.R.H.); Howard Hughes Medical Institute Undergraduate Program (G.N, W.R, W.Y.); Gewirz Foundation (to M.B.M.); King AbdulAziz University, Saudi Arabia (to Z.S.) This research was supported by the Tissue Culture and Biobanking Shared Resource of the Georgetown Lombardi Comprehensive Cancer Center (P30-CA051008).
Abbreviations:
- AF-1
activation function-1
- AF-2
activation function-2
- CCS
charcoal stripped calf serum
- CFBS
charcoal stripped fetal bovine serum
- E2
estradiol
- EGFR
epidermal growth factor receptor
- ERα
estrogen receptor-alpha
- HBSS
Hank’s buffered saline solution
- HER2
human epidermal growth factor receptor 2
- ICI
ICI-182,780
- IMEM
improved minimum essential medium
- LBD
ligand binding domain
- p
probability
- PI3K
phosphoinositide 3-kinase
- PgR
progesterone receptor
- pS2
trefoil factor 1
- PTEN
phosphatase and tensin homolog
- SEM
standard error of the mean
- SERD
selective estrogen receptor degrader
- SERM
selective estrogen receptor modulator
- Tam
4-hydroxytamoxifen
- TRP
transient receptor potential channels
Footnotes
Conflict of interest
The authors do not report any conflicts of interest.
This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1002/ijc.33745
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9(9):631–643. [DOI] [PubMed] [Google Scholar]
- 2.Ignar-Trowbridge DM, Nelson KG, Bidwell MC, Curtis SW, Washburn TF, McLachlan JA, Korach KS. Coupling of dual signaling pathways: epidermal growth factor action involves the estrogen receptor. Proc Natl Acad Sci U S A. 1992;89(10):4658–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270(5241):1491–1494. [DOI] [PubMed] [Google Scholar]
- 4.Power RF, Mani SK, Codina J, Conneely OM, O’Malley BW. Dopaminergic and ligand-independent activation of steroid hormone receptors. Science. 1991;254(5038):1636–1639. [DOI] [PubMed] [Google Scholar]
- 5.Joukov V, De Nicolo A. Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci Signal. 2018;11(543). [DOI] [PubMed] [Google Scholar]
- 6.Lei JT, Anurag M, Haricharan S, Gou X, Ellis MJ. Endocrine therapy resistance: new insights. Breast. 2019;48 Suppl 1:S26–S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Divekar SD, Storchan GB, Sperle K, Veselik DJ, Johnson E, Dakshanamurthy S, Lajiminmuhip YN, Nakles RE, Huang L, Martin MB. The role of calcium in the activation of estrogen receptor-alpha. Cancer Res. 2011;71(5):1658–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mihai R, Stevens J, McKinney C, Ibrahim NB. Expression of the calcium receptor in human breast cancer--a potential new marker predicting the risk of bone metastases. Eur J Surg Oncol. 2006;32(5):511–515. [DOI] [PubMed] [Google Scholar]
- 9.Shennan DB. Calcium transport by mammary secretory cells: mechanisms underlying transepithelial movement. Cell Mol Biol Lett. 2008;13(4):514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.VanHouten JN, Wysolmerski JJ. Transcellular calcium transport in mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2007;12(4):223–235. [DOI] [PubMed] [Google Scholar]
- 11.Lee WJ, Monteith GR, Roberts-Thomson SJ. Calcium transport and signaling in the mammary gland: targets for breast cancer. Biochim Biophys Acta. 2006;1765(2):235–255. [DOI] [PubMed] [Google Scholar]
- 12.Soule HD, Vazguez J, Long A, Albert S, Brennan M. A human cell line from a pleural effusion derived from a breast carcinoma. J Natl Cancer Inst. 1973;51(5):1409–1416. [DOI] [PubMed] [Google Scholar]
- 13.Pink JJ, Jiang SY, Fritsch M, Jordan VC. An estrogen-independent MCF-7 breast cancer cell line which contains a novel 80-kilodalton estrogen receptor-related protein. Cancer Res. 1995;55(12):2583–2590. [PubMed] [Google Scholar]
- 14.Brunner N, Boysen B, Jirus S, Skaar TC, Holst-Hansen C, Lippman J, Frandsen T, Spang-Thomsen M, Fuqua SA, Clarke R. MCF7/LCC9: an antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross-resistance to the nonsteroidal antiestrogen tamoxifen. Cancer Res. 1997;57(16):3486–3493. [PubMed] [Google Scholar]
- 15.Bergson P, Lipkind G, Lee SP, Duban ME, Hanck DA. Verapamil block of T-type calcium channels. Mol Pharmacol. 2011;79(3):411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams ME, Washburn MS, Hans M, Urrutia A, Brust PF, Prodanovich P, Harpold MM, Stauderman KA. Structure and functional characterization of a novel human low-voltage activated calcium channel. J Neurochem. 1999;72(2):791–799. [DOI] [PubMed] [Google Scholar]
- 17.Arnoult C, Villaz M, Florman HM. Pharmacological properties of the T-type Ca2+ current of mouse spermatogenic cells. Mol Pharmacol. 1998;53(6):1104–1111. [PubMed] [Google Scholar]
- 18.Yamamoto KR. Steroid receptor regulated transcription of specific genes and gene networks. Annu Rev Genet. 1985;19:209–252. [DOI] [PubMed] [Google Scholar]
- 19.Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. 1987;51(6):941–951. [DOI] [PubMed] [Google Scholar]
- 20.Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature. 1995;375(6530):377–382. [DOI] [PubMed] [Google Scholar]
- 21.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389(6652):753–758. [DOI] [PubMed] [Google Scholar]
- 22.Renaud JP, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Crystal structure of the RAR-gamma ligand-binding domain bound to all-trans retinoic acid. Nature. 1995;378(6558):681–689. [DOI] [PubMed] [Google Scholar]
- 23.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95(7):927–937. [DOI] [PubMed] [Google Scholar]
- 24.Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc Natl Acad Sci U S A. 1998;95(11):5998–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Egea PF, Mitschler A, Rochel N, Ruff M, Chambon P, Moras D. Crystal structure of the human RXRalpha ligand-binding domain bound to its natural ligand: 9-cis retinoic acid. EMBO J. 2000;19(11):2592–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ordonez E, Thiyagarajan S, Cook JD, Stemmler TL, Gil JA, Mateos LM, Rosen BP. Evolution of metal(loid) binding sites in transcriptional regulators. J Biol Chem. 2008;283(37):25706–25714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dutertre M, Smith CL. Molecular mechanisms of selective estrogen receptor modulator (SERM) action. J Pharmacol Exp Ther. 2000;295(2):431–437. [PubMed] [Google Scholar]
- 28.Szostakowska M, Trebinska-Stryjewska A, Grzybowska EA, Fabisiewicz A. Resistance to endocrine therapy in breast cancer: molecular mechanisms and future goals. Breast Cancer Res Treat. 2019;173(3):489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bollig A, Xu L, Thakur A, Wu J, Kuo TH, Liao JD. Regulation of intracellular calcium release and PP1alpha in a mechanism for 4-hydroxytamoxifen-induced cytotoxicity. Mol Cell Biochem. 2007;305(1–2):45–54. [DOI] [PubMed] [Google Scholar]
- 30.Chang HT, Huang JK, Wang JL, Cheng JS, Lee KC, Lo YK, Liu CP, Chou KJ, Chen WC, Su W, Law YP, Jan CR. Tamoxifen-induced increases in cytoplasmic free Ca2+ levels in human breast cancer cells. Breast Cancer Res Treat. 2002;71(2):125–131. [DOI] [PubMed] [Google Scholar]
- 31.Huang CC, Cheng HH, Lin KL, Cheng JS, Tsai JY, Liao WC, Fang YC, Jan CR. Tamoxifen-induced [Ca2+]i rise and apoptosis in corneal epithelial cells. Toxicology. 2009;255(1–2):58–64. [DOI] [PubMed] [Google Scholar]
- 32.Zhang W, Couldwell WT, Song H, Takano T, Lin JH, Nedergaard M. Tamoxifen-induced enhancement of calcium signaling in glioma and MCF-7 breast cancer cells. Cancer Res. 2000;60(19):5395–5400. [PubMed] [Google Scholar]
- 33.Wang CY, Lai MD, Phan NN, Sun Z, Lin YC. Meta-Analysis of Public Microarray Datasets Reveals Voltage-Gated Calcium Gene Signatures in Clinical Cancer Patients. PLoS One. 2015;10(7):e0125766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolanz KA, Hediger MA, Landowski CP. The role of TRPV6 in breast carcinogenesis. Mol Cancer Ther. 2008;7(2):271–279. [DOI] [PubMed] [Google Scholar]
- 35.Guilbert A, Gautier M, Dhennin-Duthille I, Haren N, Sevestre H, Ouadid-Ahidouch H. Evidence that TRPM7 is required for breast cancer cell proliferation. Am J Physiol Cell Physiol. 2009;297(3):C493–502. [DOI] [PubMed] [Google Scholar]
- 36.Ouadid-Ahidouch H, Dhennin-Duthille I, Gautier M, Sevestre H, Ahidouch A. [TRP calcium channel and breast cancer: expression, role and correlation with clinical parameters]. Bull Cancer. 2012;99(6):655–664. [DOI] [PubMed] [Google Scholar]
- 37.Abdul M, Ramlal S, Hoosein N. Ryanodine receptor expression correlates with tumor grade in breast cancer. Pathol Oncol Res. 2008;14(2):157–160. [DOI] [PubMed] [Google Scholar]
- 38.Michels KB, Xue F, Brandt L, Ekbom A. Hyperparathyroidism and subsequent incidence of breast cancer. Int J Cancer. 2004;110(3):449–451. [DOI] [PubMed] [Google Scholar]
- 39.Nilsson IL, Zedenius J, Yin L, Ekbom A. The association between primary hyperparathyroidism and malignancy: nationwide cohort analysis on cancer incidence after parathyroidectomy. Endocr Relat Cancer. 2007;14(1):135–140. [DOI] [PubMed] [Google Scholar]
- 40.Pal SK, Blazer K, Weitzel J, Somlo G. An association between invasive breast cancer and familial idiopathic hyperparathyroidism: a case series and review of the literature. Breast Cancer Res Treat. 2009;115(1):1–5. [DOI] [PubMed] [Google Scholar]
- 41.Palmer M, Adami HO, Krusemo UB, Ljunghall S. Increased risk of malignant diseases after surgery for primary hyperparathyroidism. A nationwide cohort study. Am J Epidemiol. 1988;127(5):1031–1040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.