

Abstract
The immunosuppressive agents sirolimus and everolimus are sensitive CYP3A4 substrates with narrow therapeutic index. Ritonavir is a strong CYP3A inhibitor. A phase 1 study was conducted to evaluate the pharmacokinetics, safety, and tolerability of the co‐administration of sirolimus or everolimus with the ritonavir‐containing 3D regimen of the direct‐acting antiviral agents ombitasvir, ritonavir‐boosted paritaprevir, and dasabuvir in healthy subjects. This study had two independent arms, each with a two‐period, single‐sequence, crossover study design. A single dose of sirolimus 2 mg (N = 12) or everolimus 0.75 mg (N = 12) was administered in Period 1. In Period 2, multiple doses of the 3D regimen (ombitasvir/paritaprevir/ritonavir 25/150/100 mg once daily and dasabuvir 250 mg twice daily) were administered for 34 or 28 days, with a single dose of sirolimus 0.5 mg or everolimus 0.75 mg co‐administered on Day 15. Following co‐administration with the 3D regimen, the sirolimus dose‐normalized maximum observed blood concentration (Cmax) and area under the blood concentration–time curve from time zero to infinity (AUCinf) increased to 6.4‐fold and 38‐fold, respectively. Following co‐administration with the 3D regimen, the everolimus Cmax and AUCinf increased to 4.7‐fold and 27‐fold, respectively. Sirolimus and everolimus half‐lives increased from 96 to 249 h, and 42 to 118 h, respectively. There were no major safety or tolerability issues in this study. The ritonavir‐containing 3D regimen resulted in a significant increase in sirolimus or everolimus exposure, consistent with the known strong inhibitory effect of ritonavir on CYP3A requiring dose and/or frequency modification when co‐administered with each other.
Keywords: CYP3A inhibitor, dasabuvir, everolimus, hepatitis C, ombitasvir, paritaprevir, ritonavir, sirolimus
When co‐administered with the ritonavir‐containing 3D regimen, sirolimus and everolimus exposures (AUC) increased to 38‐fold and 27‐fold, respectively. The study results can be used to guide dose recommendation for sirolimus or everolimus when co‐administered with ritonavir‐containing regimens.
1. INTRODUCTION
Mammalian target of rapamycin (mTOR) inhibitors, such as sirolimus and everolimus, provide an alternative to calcineurin inhibitor (CNI) immunosuppressants which are associated with significant side effects, including nephrotoxicity and neurotoxicity. 1 , 2 Sirolimus is indicated for the prophylaxis of organ rejection in kidney transplant patients aged ≥13 years. Everolimus is indicated for the prophylaxis of organ rejection in adult kidney and liver transplant patients.
Sirolimus and everolimus are substrates of both CYP3A4, the primary enzyme responsible for the metabolism of these agents, and P glycoprotein (P‐gp), a transmembrane transporter. 3 , 4 Both sirolimus and everolimus are narrow therapeutic index drugs and their blood concentrations need to be monitored and maintained within a narrow target concentration range to maintain therapeutic effect and minimize toxicity in post‐transplant patients. Strong CYP3A/P‐gp inhibitors or inducers are expected to significantly increase or decrease the exposures of sirolimus and everolimus. Ritonavir is a strong CYP3A inhibitor that is administered as a pharmacokinetic enhancer in combination with several protease inhibitors of human immunodeficiency virus (HIV, e.g., atazanavir, fosamprenavir, lopinavir), hepatitis C virus (HCV, e.g., paritaprevir), or severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2, e.g., nirmatrelvir [PF‐07321332]). 5 , 6 Therefore, it is important to understand the magnitude of effects of a strong CYP3A inhibitor such as ritonavir on the pharmacokinetics of these mTOR inhibitors.
This report includes the results of the effect of the ritonavir‐containing 3D regimen of the direct‐acting antiviral agents ombitasvir, ritonavir‐boosted paritaprevir, and dasabuvir on the pharmacokinetics of sirolimus or everolimus in healthy adult subjects. The study was conducted as part of clinical development for the treatment of HCV; however, the results can be used to guide dose recommendations for sirolimus or everolimus when co‐administered with other ritonavir‐containing regimens.
2. METHODS
This was a Phase 1, single‐center, multiple‐dose, open‐label study with two independent arms in which subjects were enrolled equally (Figure 1). It was designed to evaluate the safety, tolerability, and pharmacokinetics of the co‐administration of sirolimus or everolimus with the ritonavir‐containing 3D regimen of ombitasvir/paritaprevir/ritonavir plus dasabuvir. The study was conducted at the AbbVie Clinical Pharmacology Research Unit (Grayslake, IL, USA) in accordance with Good Clinical Practice guidelines and ethical principles that have their origin in the Declaration of Helsinki. The study protocol was approved by an independent institutional review board and written informed consent was obtained from each subject before any study‐related procedures were performed.
FIGURE 1.
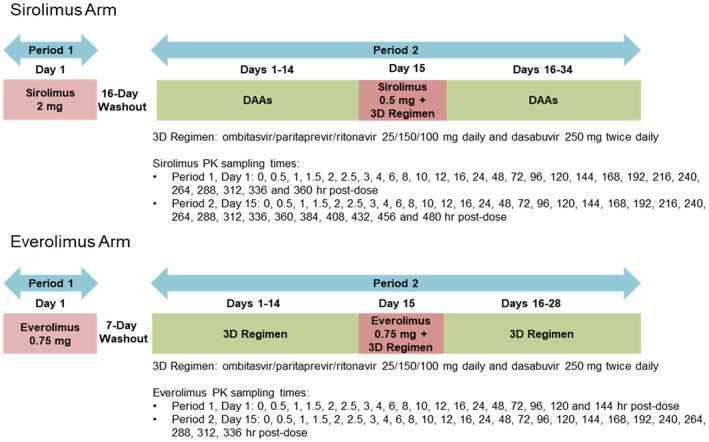
Study design.
Refer to Supplemental Methods for details on subjects, study design, sample collection and bioanalytical methods, pharmacokinetic evaluations, safety and tolerability, and statistical analysis.
2.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, 7 and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 8
3. RESULTS
3.1. Subjects and baseline demographics
A total of 24 subjects were enrolled into the study with 12 subjects in each Arm. Data from all subjects were included in the safety assessment. Data from 23 subjects who had pharmacokinetic data from both Day 1 in Period 1 and Day 15 in Period 2 were included in the statistical analyses of the pharmacokinetic parameters. In the sirolimus Arm, one subject received only a single dose of sirolimus and was discontinued from the study due to an adverse event of abdominal wall abscess on Period 1 Day 16; this subject was excluded from the summary statistics and statistical analyses of the pharmacokinetic parameters.
The demographics for the subjects in this study are presented in Table S1. The mean age of the subjects was approximately 39 years in Arm 1 and 37 years in Arm 2, and the mean BMI was approximately 26 kg/m2 for both study arms. Twenty‐one of the 24 subjects were male. Fifteen subjects were white, and nine subjects were black.
3.2. Effect of the ritonavir‐containing 3D regimen on sirolimus pharmacokinetics
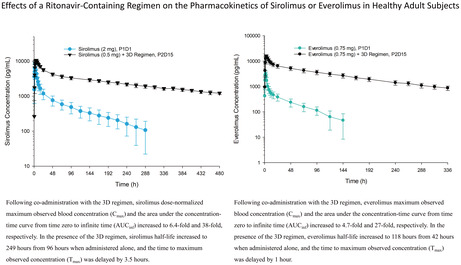
The mean ± standard deviation (SD) blood concentration‐time profiles for sirolimus administered alone (Period 1 Day 1) or with the 3D regimen (Period 2 Day 15) are presented in Figure 2. Sirolimus pharmacokinetic parameters are summarized in Table 1. Following co‐administration with the 3D regimen, sirolimus dose‐normalized maximum observed blood concentration (Cmax) and the area under the concentration‐time curve from time zero to infinite time (AUCinf) increased to 6.4‐fold and 38‐fold, respectively. In the presence of the 3D regimen, sirolimus half‐life increased to 249 h from 96 h when administered alone, and the time to maximum observed concentration (Tmax) was delayed by 3.5 h.
FIGURE 2.
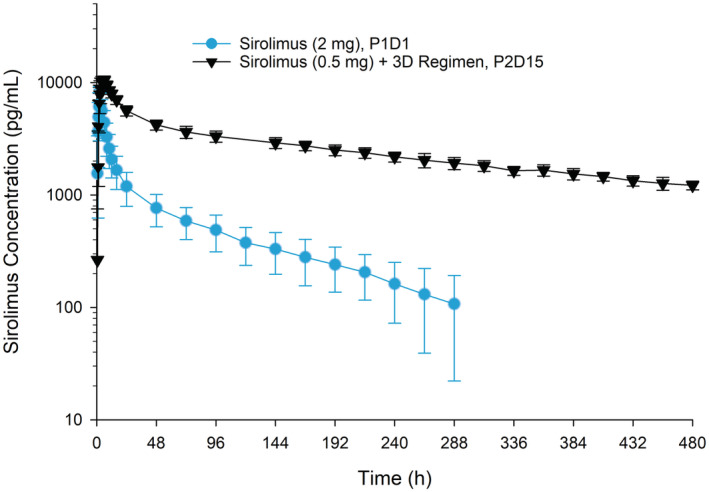
Mean ± SD sirolimus single‐dose blood concentration‐time profiles with or without co‐administration of the ritonavir‐containing 3D regimen. On Period 1 Day 1, a single dose of sirolimus 2 mg was administered. In Period 2, multiple doses of the 3D regimen (ombitasvir/paritaprevir/ritonavir 25/150/100 mg QD plus dasabuvir 250 mg BID) were administered on Day 1 through Day 34, with a single dose of sirolimus 0.5 mg co‐administered on Day 15. P1D1, Period 1 Day 1; P2D15, Period 2 Day 15; SD, standard deviation.
TABLE 1.
Sirolimus pharmacokinetic parameters
| Parameter | Sirolimus 2 mg Period 1 Day 1 (N = 11) | Sirolimus 0.5 mg + 3D regimen Period 2 Day 15 (N = 11) | |
|---|---|---|---|
| Geometric mean (Geometric %CV) | Geometric mean (Geometric %CV) | Geometric mean ratio (90% CI) | |
| Cmax/Dose (ng/ml/mg) | 3.33 (33) | 21.3 (7) | 6.40 (5.34–7.68) c |
| AUCinf/Dose (ng•h/ml/mg) | 89.2 (33) | 3390 (8) | 38.0 (31.5–45.8) c |
| C24/Dose (ng/ml/mg) | 0.57 (29) | 11.2 (11) | 19.5 (16.7–22.9) c |
| Cmax (ng/ml) | 6.66 (33) | 10.7 (7) | – |
| AUCinf (ng•h/ml) | 178 (33) | 1700 (8) | – |
| C24 (ng/m) | 1.14 (29) | 5.59 (11) | – |
| Tmax (h) a | 2.5 (1.5–4.0) | 6.0 (3.0–6.0) | – |
| t 1/2 (h) b | 96.3 (15) | 249 (23) | – |
Note: 3D regimen: ombitasvir/paritaprevir/ritonavir 25/150/100 mg QD plus dasabuvir 250 mg BID.
Abbreviations: AUCinf, area under the blood concentration‐time curve from time zero to infinite time; C24, blood concentration at 24 h after dose; CI, confidence interval; Cmax, maximum observed blood concentration; CV, coefficient of variation; Tmax, time to maximum observed blood concentration; t 1/2, terminal phase elimination half‐life.
Median (range).
Harmonic mean (pseudo‐standard deviation).
p < .0001 based on the repeated measures analysis on the natural logarithms of PK parameters.
3.3. Effect of the ritonavir‐containing 3D regimen on everolimus pharmacokinetics
The mean ± SD blood concentration‐time profiles for everolimus administered alone (Period 1 Day 1) or with the 3D regimen (Period 2 Day 15) are presented in Figure 3. Everolimus pharmacokinetic parameters are summarized in Table 2. Following co‐administration with the 3D regimen, everolimus Cmax, and AUCinf increased to 4.7‐fold and 27‐fold, respectively. In the presence of the 3D regimen, everolimus half‐life increased to 118 h from 42 h when administered alone, and Tmax was delayed by 1 h.
FIGURE 3.
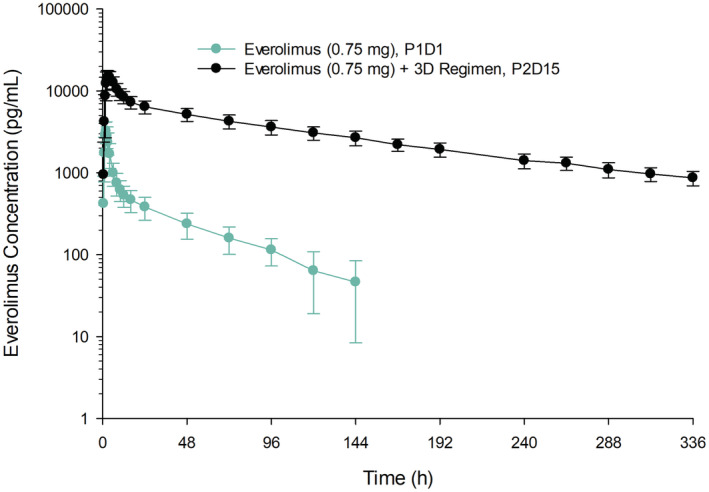
Mean ± SD everolimus single‐dose blood concentration‐time profiles with or without co‐administration of the ritonavir‐containing 3D regimen.On Period 1 Day 1, a single dose of everolimus 0.75 mg was administered. In Period 2, multiple doses of the 3D regimen (ombitasvir/paritaprevir/ritonavir 25/150/100 mg QD plus dasabuvir 250 mg BID) were administered on Day 1 through Day 28, with a single dose of everolimus 0.75 mg co‐administered on Day 15. P1D1, Period 1 Day 1; P2D15, Period 2 Day 15; SD, standard deviation.
TABLE 2.
Everolimus pharmacokinetic parameters
| Parameter | Everolimus 0.75 mg Period 1 Day 1 (N = 12) | Everolimus 0.75 mg +3D regimen Period 2 Day 15 (N = 12) | |
|---|---|---|---|
| Geometric mean (Geometric %CV) | Geometric mean (Geometric %CV) | Geometric mean ratio (90% CI) | |
| Cmax (ng/ml) | 3.33 (31) | 15.8 (19) | 4.74 (4.29–5.25) c |
| AUCinf (ng•h/ml) | 41.9 (31) | 1130 (19) | 27.1 (24.5–30.1) c |
| C12 (ng/ml) | 0.513 (31) | 8.26 (18) | 16.1 (14.5–17.9) c |
| Tmax (h) a | 2.0 (1.5–2.5) | 3.0 (2.0–4.0) | – |
| t 1/2 (h) b | 42.4 (7) | 118 (18) | – |
Note: 3D regimen: ombitasvir/paritaprevir/ritonavir 25/150/100 mg QD plus dasabuvir 250 mg BID.
Abbreviations: AUCinf, area under the blood concentration‐time curve from time zero to infinite time; C12, blood concentration 12 h after dose; CI, confidence interval; Cmax, maximum observed blood concentration; CV, coefficient of variation; Tmax, time to maximum observed blood concentration; t 1/2, terminal phase elimination half‐life.
Median (range).
Harmonic mean (pseudo‐standard deviation).
p < .0001 based on the repeated measures analysis on the natural logarithms of PK parameters.
3.4. Safety and tolerability
All study drugs administered in both study arms were well tolerated by the healthy subjects in the study. Adverse events were infrequent and mild in severity. One subject discontinued from the study prematurely due to an abdominal wall abscess at the site of a spider bite, which was moderate in severity and was considered unrelated to the study drug regimen. No clinically significant vital signs or laboratory measurements were observed during the study. No deaths were reported during the study.
4. DISCUSSION
For a given sensitive CYP3A substrate, all strong CYP3A inhibitors result in at least a five‐fold higher AUC of the substrate; however, the magnitude of the increase can vary. For example, co‐administration of midazolam with a strong CYP3A inhibitor could result in an increase in midazolam AUC ranging from 5.3‐fold for boceprevir 9 to 26.9‐fold for tipranavir/ritonavir. 10 For a sensitive CYP3A substrate such as sirolimus or everolimus that has a narrow therapeutic index, it is important to know the potential magnitude or range of effect of strong CYP3A inhibitors, particularly because the prescribing information for sirolimus and everolimus contain limited data on DDI studies with strong CYP3A inhibitors. 3 , 4 Even though in vivo DDI data are available with several moderate CYP3A inhibitors (e.g., cyclosporine, erythromycin, and verapamil), for strong CYP3A inhibitors, prior to the conduct of the current study, the prescribing information for sirolimus or everolimus only had DDI data with ketoconazole. 3 , 4 To better inform the safe and efficacious use of sensitive CYP3A substrates like sirolimus and everolimus that have a narrow therapeutic index and improve the ability to make appropriate dose and/or frequency modifications, this study evaluated the effect of a regimen that contains ritonavir, another strong CYP3A inhibitor that is used as a pharmacokinetic enhancer for several protease inhibitors of HIV (e.g., atazanavir, fosamprenavir, lopinavir), HCV (e.g., paritaprevir), or SARS‐CoV‐2 (e.g., nirmatrelvir [PF‐07321332]). 5 , 6
Ketoconazole (200 mg BID at steady state) co‐administered with 2 mg single‐dose sirolimus increased Cmax and AUC of sirolimus by 4.3‐fold and 10.9‐fold, respectively. 3 Similarly, ketoconazole (200 mg BID at steady state) co‐administered with 2 mg single‐dose everolimus increased Cmax and AUC of everolimus by 3.9‐fold and 15‐fold, respectively. 4 , 11 Given these significant increases in sirolimus and everolimus exposures in the presence of ketoconazole, 3 , 4 , 11 substantial increases in exposures of these substrates are also expected when co‐administered with another strong CYP3A inhibitor such as ritonavir or a ritonavir‐containing regimen. The single doses of sirolimus (0.5 and 2 mg) selected for this study were considerably lower than the maximum allowed dose of 40 mg/day. 3 The low dose of 0.5 mg sirolimus in Period 2 allowed sufficient exposure coverage for the expected marked increase in sirolimus exposures when administered with ritonavir. For everolimus, the dose of 0.75 mg BID of everolimus is the lowest recommended starting dose in transplant patients. 4 The doses of sirolimus and everolimus chosen for this study had accounted for the expected similar increases in their exposures from co‐administration with ritonavir as with ketoconazole to maximize the translatability of this study.
The present study evaluated the effect of the ritonavir‐containing 3D regimen of ombitasvir, ritonavir‐boosted paritaprevir, and dasabuvir on the pharmacokinetics of sirolimus or everolimus by comparing the exposure of sirolimus or everolimus when co‐administered with the 3D regimen on Day 15 in Period 2 to sole administration on Day 1 in Period 1. The dose of the 3D regimen (ombitasvir/paritaprevir/ritonavir 25/150/100 mg QD and dasabuvir 250 mg BID) used in this study was the approved dose for the treatment of HCV infection. 12 , 13 The 3D regimen was administered for additional days after the co‐administration of sirolimus (Days 16 to 34 in Period 2) or everolimus (Days 16 to 28 in Period 2) to maintain inhibition of CYP3A‐mediated metabolism and to allow adequate characterization of the terminal elimination phase for determination of the half‐life of sirolimus or everolimus in the presence of the continued CYP3A inhibition. A washout of at least 16 days for sirolimus and 7 days for everolimus between dosing periods was sufficient to ensure no drug carryover since the half‐lives of sirolimus and everolimus are approximately 62 h and 30 h, respectively. 3 , 4
The results of the present study showed that multiple doses of the 3D regimen resulted in substantial increases in the exposure of single‐dose sirolimus and everolimus. Sirolimus dose‐normalized Cmax, t 1/2, and dose‐normalized AUCinf increased to 6.4‐fold, 2.6‐fold, and 38‐fold, respectively. Everolimus Cmax, t 1/2, and AUCinf increased to 4.7‐fold, 2.8‐fold, and 27‐fold, respectively. It is worth‐noting that the increases in AUCinf across individual subjects ranged from 19‐ to 61‐fold for sirolimus, and from 20‐ to 36‐fold for everolimus.
Among the components of the 3D regimen, in vitro data suggest that ritonavir, paritaprevir, and dasabuvir are potential inhibitors of P‐gp. 14 However, a dedicated DDI study showed that the 3D regimen had no clinically meaningful effect on the pharmacokinetics of digoxin, a sensitive P‐gp substrate, resulting in ≤16% increase in the Cmax and AUC of digoxin, with essentially no change (ratio of 0.98) in the fraction of unchanged digoxin excreted in the urine. 15 Therefore, the observed effects of the 3D regimen on the pharmacokinetics of sirolimus and everolimus in the present study are attributable to the CYP3A inhibitory effect of ritonavir, with P‐gp inhibition unlikely playing any meaningful role.
Both sirolimus and everolimus are sensitive CYP3A substrates with a narrow therapeutic index that require therapeutic drug monitoring to maintain drug levels within the target range. For safe and efficacious use of these sensitive CYP3A substrates with a narrow therapeutic index, it is important to understand the magnitude of increases in their exposures when given concomitantly with a CYP3A inhibitor. For sirolimus and everolimus, the 38‐fold and 27‐fold increases in their respective AUC from co‐administration with the ritonavir‐containing 3D regimen differed greatly from the 10.9‐fold and 15‐fold increases that were observed with ketoconazole. This is similar to other observations, such as the co‐administration of midazolam with either boceprevir 9 and tipranavir/ritonavir, 10 which also resulted in a wide range of exposures when a sensitive CYP3A substrate is administered in the presence of different strong CYP3A inhibitors. Taken together, these findings underscore the need for more complete DDI information, ideally from clinical studies, to better inform appropriate dose and/or frequency modifications of sensitive CYP3A substrates especially those with narrow therapeutic index. If available dose strengths do not allow for an appropriate dose adjustment, co‐administration should be avoided. 13
In conclusion, the significant increase in sirolimus or everolimus exposures by the ritonavir‐containing 3D regimen is consistent with the known strong inhibitory effect of ritonavir on CYP3A. The magnitude of increase in exposure and half‐life indicates that dose and dosing frequency adjustments will be needed when co‐administered with ritonavir‐containing regimens.
AUTHOR CONTRIBUTIONS
All authors contributed to the study design, study conduct, data analysis/interpretation, writing the original draft, and reviewing/editing subsequent drafts. All authors read and approved the final manuscript.
FUNDING INFORMATION
This study was sponsored by AbbVie.
DISCLOSURE
AbbVie contributed to the study design, research, and interpretation of data, and the writing, reviewing, and approving of the publication. All authors are current or former employees of AbbVie and may hold AbbVie stock or stock options.
ETHICS APPROVAL STATEMENT
The study was conducted in the United States in accordance with Good Clinical Practice guidelines and ethical principles that have their origin in the Declaration of Helsinki.
PATIENT CONSENT STATEMENT
The study protocol was approved by an independent institutional review board and written informed consent was obtained from each subject before any study‐related procedures were performed.
Supporting information
Methods S1.
Table S1.
ACKNOWLEDGMENTS
This study was funded by AbbVie. The authors thank Therese Stickler, a freelance medical writer under contract with AbbVie, for medical writing support of the initial drafting and AbbVie employee, Wesley Wayman, PhD, for copyediting support. The authors also thank AbbVie employees Pamela Watson and Natalie Hycner for assistance with creating the figures and revising the draft.
Zha J, Jiang Q, Yao BB, Cohen DE, Carter DC, Menon RM. Effects of a ritonavir‐containing regimen on the pharmacokinetics of sirolimus or everolimus in healthy adult subjects. Pharmacol Res Perspect. 2022;10:e01024. doi: 10.1002/prp2.1024
DATA AVAILABILITY STATEMENT
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial‐level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html
REFERENCES
- 1. European Association for the Study of the liver . Recommendations on treatment of hepatitis C 2015. http://www.easl.eu/research/our‐contributions/clinical‐practice‐guidelines/detail/recommendations‐on‐treatment‐of‐hepatitis‐c‐2015/report/1. [DOI] [PubMed]
- 2. Wiesner RH, Fung JJ. Present state of immunosuppressive therapy in liver transplant recipients. Liv Transplant. 2011;17(Suppl 3):S1‐S9. [DOI] [PubMed] [Google Scholar]
- 3. Rapamune (sirolimus) [US package insert] . Pfizer; August, 2019. [Google Scholar]
- 4. Zortress (Everolimus) [US Package Insert] . Novartis; August, 2018. [Google Scholar]
- 5. Owen DR, Allerton CMN, Anderson AS, et al. An Oral SARS‐CoV‐2 M(pro) inhibitor clinical candidate for the treatment of COVID‐19. Science. 2021;374:eabl4784. [DOI] [PubMed] [Google Scholar]
- 6. Fact sheet for healthcare providers: emergency use authorization for paxlovid (nirmatrelvir tablets; ritonavir tablets). , Pfizer Inc., April 2022. https://www.fda.gov/media/155050/download (Accessed on June 5, 2022). [Google Scholar]
- 7. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2017;46(D1):D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alexander SPH, Kelly E, Mathie A, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Introduction and other protein targets. Br J Pharmacol. 2019;176(Suppl 1):S1‐s20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kiser JJ, Burton JR, Anderson PL, Everson GT. Review and management of drug interactions with boceprevir and telaprevir. Hepatology. 2012;55(5):1620‐1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dumond JB, Vourvahis M, Rezk NL, et al. A phenotype‐genotype approach to predicting CYP450 and P‐glycoprotein drug interactions with the mixed inhibitor/inducer tipranavir/ritonavir. Clin Pharmacol Ther. 2010;87(6):735‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kovarik JM, Beyer D, Bizot MN, Jiang Q, Shenouda M, Schmouder RL. Blood concentrations of everolimus are markedly increased by ketoconazole. J Clin Pharmacol. 2005;45(5):514‐518. [DOI] [PubMed] [Google Scholar]
- 12. Viekira Pak (ombitasvir, paritaprevir, and ritonavir tablets; dasabuvir tablets) [US package insert] . AbbVie Inc., December 2019. [Google Scholar]
- 13. Viekirax (Ombitasvir/Paritaprevir/Ritonavir Tablets) [Summary of Product Characteristics] . AbbVie Deutschland GmbH & Co. KG, March 2021. https://www.ema.europa.eu/en/documents/product‐information/viekirax‐epar‐product‐information_en.pdf. [Google Scholar]
- 14. Shebley M, Liu J, Kavetskaia O, et al. Mechanisms and predictions of drug‐drug interactions of the hepatitis C virus three direct‐acting antiviral regimen: paritaprevir/ritonavir, ombitasvir, and dasabuvir. Drug Metab Dispos. 2017;45(7):755‐764. [DOI] [PubMed] [Google Scholar]
- 15. Menon RM, Badri PS, Wang T, et al. Drug‐drug interaction profile of the all‐oral anti‐hepatitis C virus regimen of paritaprevir/ritonavir, ombitasvir, and dasabuvir. J Hepatol. 2015;63(1):20‐29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods S1.
Table S1.
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial‐level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html