

Abstract
The fate of influenza A virus (IAV) infection in the host cell depends on the balance between cellular defence mechanisms and viral evasion strategies. To illuminate the landscape of IAV cellular restriction, we generated and integrated global genetic loss-of-function screens with transcriptomics and proteomics data. Our multi-omics analysis revealed a subset of both IFN-dependent and independent cellular defence mechanisms that inhibit IAV replication. Amongst these, the autophagy regulator TBC1 domain family member 5 (TBC1D5), which binds Rab7 to enable fusion of autophagosomes and lysosomes, was found to control IAV replication in vitro and in vivo and to promote lysosomal targeting of IAV M2 protein. Notably, IAV M2 was observed to abrogate TBC1D5–Rab7 binding through a physical interaction with TBC1D5 via its cytoplasmic tail. Our results provide evidence for the molecular mechanism utilised by IAV M2 protein to escape lysosomal degradation and traffic to the cell membrane, where it supports IAV budding and growth.
Influenza A virus (IAV) depends on the host cell machinery and signalling pathways to complete its replication cycle. From viral entry to budding, multifunctional IAV proteins simultaneously engage host factors that provide essential activities and subvert cellular mechanisms that inhibit viral replication. To dissect the complex interplay between IAV and the host cell, multiple large-scale loss-of-function and proteomics datasets have been generated1–9. Meta-analysis and integration of these screens have identified multiple proviral factors and pathways that are essential for IAV replication and pathogenesis and are relevant targets for host-directed therapies10,11. However, the global landscape of IAV cellular restriction remains less understood1,12.
Macroautophagy (hereafter referred to as autophagy) is among the cellular pathways identified by genetic screening to impact IAV replication11. Autophagy is a catabolic process involved in the maintenance of cellular homoeostasis and also functions as a crucial cellular defence mechanism for clearance of pathogens by lysosomal degradation13. Cellular, bacteria or viral cargo are engulfed into autophagosomes, which are double-membrane vesicles decorated with Atg8/LC3 proteins, where they are degraded on fusion with late endosomes and lysosomes14,15. The small GTPase Rab7 is a major regulator of this pathway, being responsible for the directionality of late endosome and autophagosome trafficking and their fusion with lysosomes16–18. Rab7 activation depends on its nucleotide state (GTP-bound active or GDP-bound inactive) and is regulated by the interaction with GTPase-activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs) that reside in different cellular compartments19. TBC1D5 localizes in autophagosomes and late endosomes, where it functions as a GAP for Rab7 (ref.20). Association of Rab7 with TBC1D5 favours the recruitment of the homotypic fusion and protein-sorting (HOPS) complex and promotes SNARE-dependent fusion of late endosomes and autophagosomes with lysosomes21. Many viruses induce autophagy upon infection22. To antagonize autophagy and escape lysosomal degradation, pathogens have evolved two general strategies: either interference with autophagosomal biogenesis (for instance, herpes simplex virus (HSV-1)) or by blocking the fusion of autophagosomes with lysosomes (for instance, human immunodeficiency virus (HIV-1) and IAV)23,24. However, for many viruses, the molecular strategy that is used to circumvent this crucial host defence mechanism remains to be elucidated.
Here, we report a global analysis of the host restrictome for IAV assembled by integrating loss-of-function screening, transcriptomics and proteomics data. Mechanistically, we show that IAV M2 protein evades lysosomal degradation by abrogating TBC1D5–Rab7 interaction thus enabling trafficking to the cell surface and budding of IAV.
Results
Global landscape of IAV cellular restriction.
The systematic identification of cellular factors that constrain viral replication can provide valuable insights into IAV pathogenesis7. Towards this end, we carried out arrayed genome-wide small interfering RNA (siRNA) screens in human macrophage-like THP-1 cells challenged with the seasonal influenza A/Wyoming/03/03 (Wyom/03, H3N2) virus or the highly pathogenic influenza A/Vietnam/1203/04 HALo (Viet/04, H5N1) virus (Methods; Fig. 1a). These cells were selected because macrophages mount an antiviral response to IAV (ref.25) and THP-1 cells support productive replication of IAV (Extended Data Fig. 1a). Screens were conducted twice each using mock- or IFN-treated cells to explore the contribution of IFN-stimulated genes (ISGs) in limiting IAV replication. The impact of each human gene knockdown on viral replication was determined by high-content imagining of IAV nucleoprotein (NP) and calculated as the percentage of IAV NP+ cells. The average Pearson correlation coefficient between screen replicates was 0.69 (Extended Data Fig. 1b) and the average z prime (z′), a statistical indicator of assay quality26, between scrambled siRNA (negative control) and immune regulator IRF9 siRNA (positive control, IFN-treated screen) or viral-targeting NP siRNA (positive control, mock-treated screen) was 0.51, suggesting favourable screening conditions26. Using a cut-off criteria for infection z-score (≥1.5 for antiviral or ≤−1.5 for proviral) and cell viability (at least 70% of scrambled siRNA), we identified 333 factors that exerted proviral activities for H3N2 and/or H5N1 IAVs and 105 that restricted viral replication (Fig. 1b and Supplementary Table 1). These factors were cross-referenced with hits from previous loss-of-function studies for IAV and found a modest but statistically significant overlap (antiviral factors, P = 0.029) (Extended Data Fig. 1c)1,3–7,11. Antiviral hits identified in the genome-wide siRNA screen were further evaluated for their ability to inhibit IAV replication. Knockdown of 33 cellular factors increased the replication of H3N2 and/or H5N1 IAVs by 1.25-fold or more without altering cell viability and are summarized in Supplementary Table 2.
Fig. 1 |. Multi-omics analysis of IAV cellular restriction.
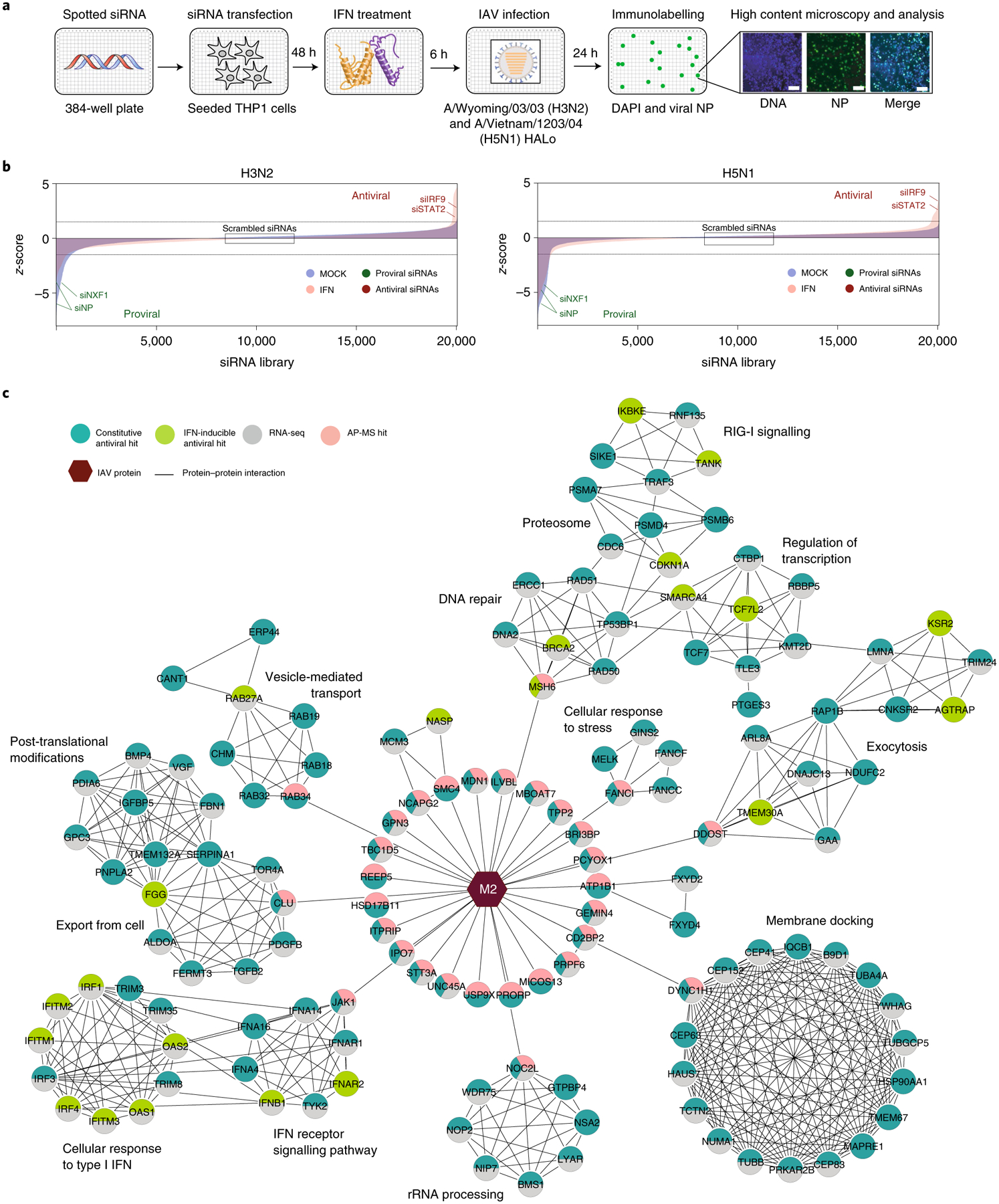
a, Schematic representation of the genome-wide siRNA screen to identify cellular factors affecting IAV replication. b, Ranked z-scores from the genome-wide siRNA screen (blue, mock-treated cells; pink, IFN-treated cells). Dashed lines illustrate z-score cut-off: z-score >1.5 indicates antiviral factors, z-score <–1.5 indicates proviral factors. Controls are shown (scrambled, negative; siIRF9 and siNP, positive), as well as known negative (STAT2) and positive (NXF1) regulators of IAV replication1. c, Integration of antiviral siRNA hits (light green, IFN-inducible; dark green, constitutive), RNA-seq (grey) and IAV M2 protein AP-MS (pink) reveals cellular networks associated with IAV restriction supported by orthogonal measurements. Nodes indicate proteins and edges indicate interactions from STRING. Hexagons represent IAV M2 viral protein. Network visualization generated by Cytoscape v.3.8.0.
Integration of loss-of-function studies with orthogonal datasets can highlight cellular factors and pathways that have multi-omics support and are probably relevant for viral infection. Using network propagation, we integrated the identified antiviral factors with published transcriptomics data of IAV-infected macrophages27. First, we calculated the cut-off values that provide maximum overlap between datasets with an enrichment factor >1 (Methods; siRNA screen cut-off z-score ≥0.5 and RNA-seq cut-off log2FC (where FC is fold change) ≥1.0 or ≤−1.0, Padj < 0.01). Then, the resulting factors were sorted as IFN-inducible or constitutively expressed on the basis of available databases28,29. Next, we applied community detection algorithms to identify densely interconnected clusters of factors that show significant membership to biological pathways and built a hierarchical ontology network that depicts the subset of cellular mechanisms that were found to inhibit IAV replication (Extended Data Fig. 2 and Supplementary Table 3; Methods).
Analysis of the IFN-inducible factors revealed enrichment in immune response pathways, including type I IFN signalling (P = 1.85 × 10−48) and cytokine and chemokine signalling (P = 1.17 × 10−12 and 2.77 × 10−27, respectively) but also coagulation and homeostasis pathways (P = 5.14 × 10−20 and 1.89 × 10−22), important for in vitro activation of TLR3 by double-stranded RNA30 and vesicle transport and exocytosis (P = 2.78 × 10−8 and 7.84 × 10−6), which are known to be required for TLR and MyD88 signalling31 (Extended Data Fig. 2a). Importantly, this analysis revealed ISGs previously associated with IAV restriction, including members of the IFITM or OAS family1, as well as nine ISGs not previously associated with IAV restriction—ARAP2, BHLHE22, BRCA2, FAM189B/COTE1, GNG5, HLA-DOB, KDM6A/UTX, SAMSN1 and TRPS1. The inhibitory effect of all nine ISGs in IAV replication was further validated (Supplementary Table 2) suggesting that these factors warrant further investigation.
Analysis of the constitutively expressed antiviral factors revealed six major networks. These were cell cycle (P = 4.50 × 10−127), G-alpha signalling (P = 9.46 × 10−268), metabolism (P = 1.58 × 10−157), RNA processing (P = 4.50 × 10−227), which included the subnetwork non-sense mediated decay (NMD)32, and protein modifications (P = 4.49 × 10−132), which included the subnetworks ubiquitination and proteasomal degradation, histone modifications, and SMAD proteins phosphorylation, which is known to inhibit Zika and hepatitis C virus (HCV) replication33 (Extended Data Fig. 2b). The last major network was vesicle-mediated transport (P = 7.91 × 10−140), which included the subnetworks endocytosis, macroautophagy and phagocytosis, previously linked to macrophage clearance of invading pathogens in vitro34 (Extended Data Fig. 2b). The complete list of factors included in each pathway and their P values are shown in Supplementary Table 3.
IAV M2 protein has been implicated in the regulation of several pathways represented in this network including vesicle transport, endocytosis and autophagy (Extended Data Fig. 2b). However, the interplay between M2 and these pathways has thus far eluded molecular understanding35. To address this gap, we sought to systematically identify M2–host restriction protein–protein interactions (PPIs) using affinity purification mass spectrometry (AP-MS). These experiments were conducted using IFN-stimulated and unstimulated human lung epithelial cells (A549) to also evaluate interaction of M2 with ISGs. Strep-tagged Viet/04 H5N1 M2 (M2-Strep) or negative controls (Strep-tag only and GFP-Strep), were stably expressed in A549 cells treated or untreated with IFN, and bait proteins were pulled down by Strep-Tactin affinity purification (n = 3 replicates per condition and bait). Samples were then digested by trypsin, proteins were identified by MS and then scored for the probability of being a true interactor using the MiST scoring algorithm36 (Methods). Following the network propagation approach described above, putative M2 interactors with a MiST score ≥0.55 were integrated with the restriction network generated using the siRNA and RNA-seq data to provide insight into the interplay between M2 protein and the host restrictome (Supplementary Table 4). Though this low-stringency MiST threshold included medium confidence interactors, by combining these three datasets we illuminated factors and subnetworks that have multi-omics support and are thus more likely to be relevant negative regulators of IAV infection (Fig. 1c).
The resulting network was also integrated with manually curated bioinformatic resources to create a visual model that displays the identified restriction factors (Fig. 2 and Supplementary Table 5; Methods). As expected, autophagy and vesicle trafficking were amongst the antiviral pathways found to interact with M2, which encompassed restriction factors not previously associated with IAV inhibition including the Golgi-bound member of the secretory pathway Rab34 (ref.37), the inhibitor of SARS-CoV-2 egress Rab27a (ref.38) or the regulator of Rab7-mediated lysosomal degradation TBC1D5 (ref.21) (Figs. 1c and 2). In addition, these analyses revealed interaction of M2 with several pathways not previously associated with M2 or IAV restriction, including Fanconi anaemia (FA) genes (FANCI, FANCC and FANCF), important for mitophagy, viral clearance and reduction of reactive oxygen species (ROS)39, several regulators of cell cycle progression and factors involved in DNA repair including that of mitochondria origin (mtDNA), namely MSH6, RAD50/51 and BRCA2 (Figs. 1c and 2). M2 was also found to interact with several restriction factors involved in lipid metabolism, immune signalling, including JAK1 and ITPRIP, the latter of which triggers IFN-independent antiviral activities through MDA-5 association40, as well as factors exerting anti-apoptotic activities, such as the growth factor PDGF-B (ref.41) (Figs. 1c and 2). In addition, M2 was found to interact with PCYOX1, a factor involved in degradation of prenylated proteins that restrain innate immunity42 and the deubiquitinase protein USP9X, which has been associated with restriction of HSV-1 (ref.43) (Figs. 1c and 2). These factors were prioritized on the basis of membership to pathway, omics support and siRNA validation, and are summarized in Extended Data Fig. 3.
Fig. 2 |. Integrated model of IAV cellular restriction.
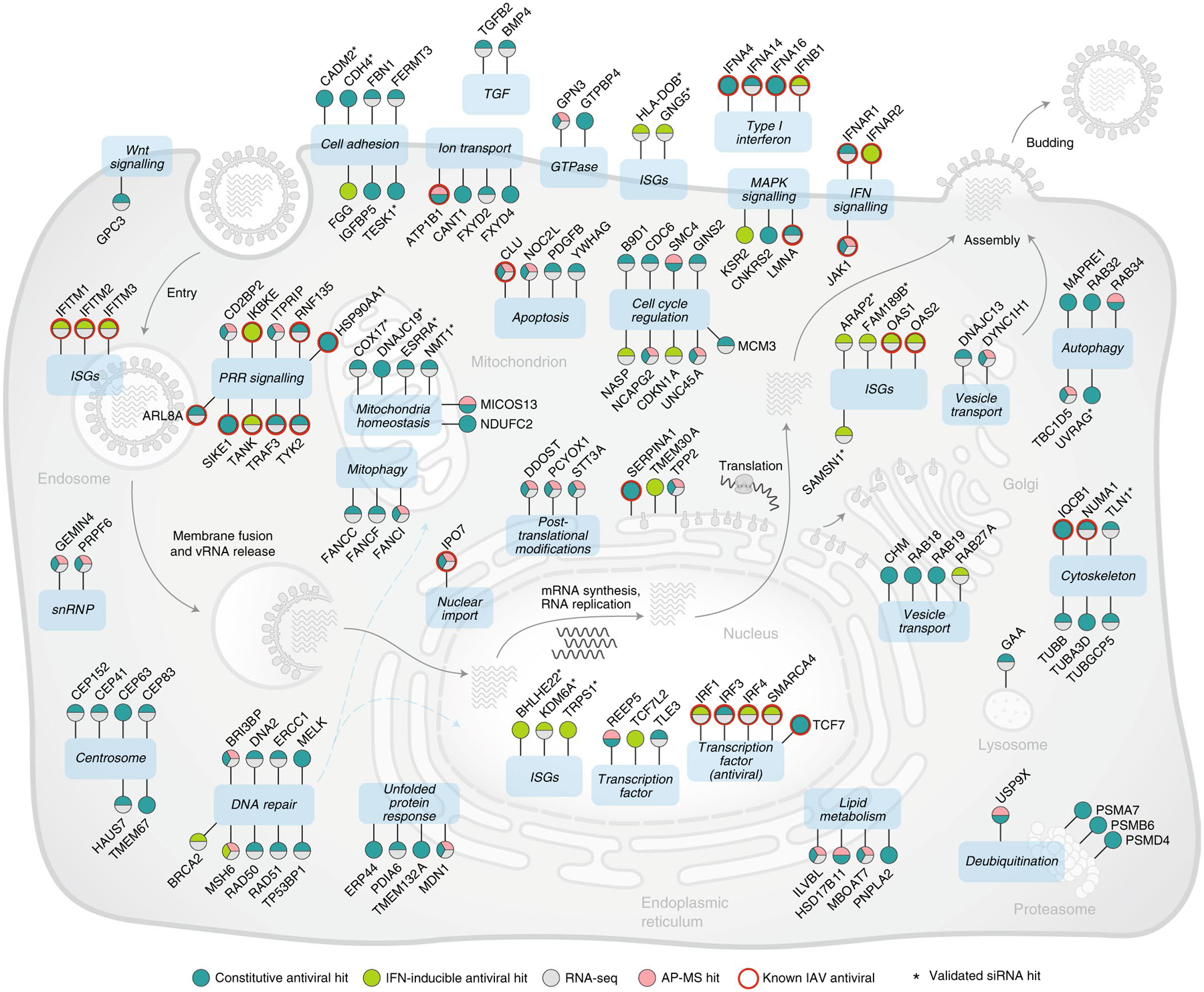
IAV restriction factors identified in this study were placed at specific subcellular localizations on the basis of curated literature research as well as Gene Ontology, KEGG and Reactome databases (Methods). Antiviral siRNA hits are shown in green (light green, IFN-inducible; dark green, constitutive), RNA-seq in grey and IAV M2 protein interactors in pink. Red circles denote antiviral factors previously associated with IAV restriction, and * shows subsequently validated siRNA hits.
TBC1D5 restricts IAV replication in vitro and ex vivo.
The regulator of Rab7-mediated lysosomal degradation TBC1D5 was identified through the integrative analysis as a relevant host-restriction factor supported by all orthogonal datasets, including physical interaction with IAV M2 protein (Fig. 2). Since M2 was previously found to escape lysosomal degradation by a molecular mechanism that remains to be elucidated24,44, we thus pursued the hypothesis that TBC1D5 could be targeted by M2 to evade autophagy-mediated degradation.
First, we validated TBC1D5 effect controlling IAV replication using several viral strains and cell types to exclude strain and/or cell-type specific effects. Of note, M2 sequence is relatively well conserved across different IAV strains (72.8% conservation across Wyom/03 H3N2, Viet/04 H5N1, A/WSN/33 H1N1 and A/PR8/33 H1N1 viruses). Efficient, non-cytotoxic siRNA-mediated knockdown of TBC1D5 (Extended Data Fig. 4a,b) resulted in an increase in the amount of released infectious influenza H1N1 A/WSN/33 (WSN/33) virus by ten-fold in the human lung cell line A549 (Fig. 3a). Conversely, ectopic expression of TBC1D5 significantly reduced WSN/33 virus release by fivefold in these cells (Fig. 3b and Extended Data Fig. 4c). We then investigated the effect of TBC1D5 depletion on IAV replication in human tracheobronchial epithelial (HTBE) cells, a relevant ex vivo cell model for influenza replication, and found a significant increase in WSN/33 IAV replication (Fig. 3c,d and Extended Data Fig. 4d). To further confirm TBC1D5 antiviral activity, we generated A549 CRISPR-Cas9 TBC1D5 knockout (KO) cells (Fig. 3e) and observed that WSN/33 growth was augmented by over tenfold in these cells (Fig. 3f), while cell viability was unaltered (Extended Data Fig. 4e). This effect was not due to off-target effects, as complementation of TBC1D5 KO cells recapitulated IAV infection to levels of parental cells (Fig. 3g). Taken together, these data provide functional evidence that TBC1D5 exerts antiviral activity in multiple cell types.
Fig. 3 |. TBC1D5 restricts IAV replication in vitro, ex vivo and in vivo.
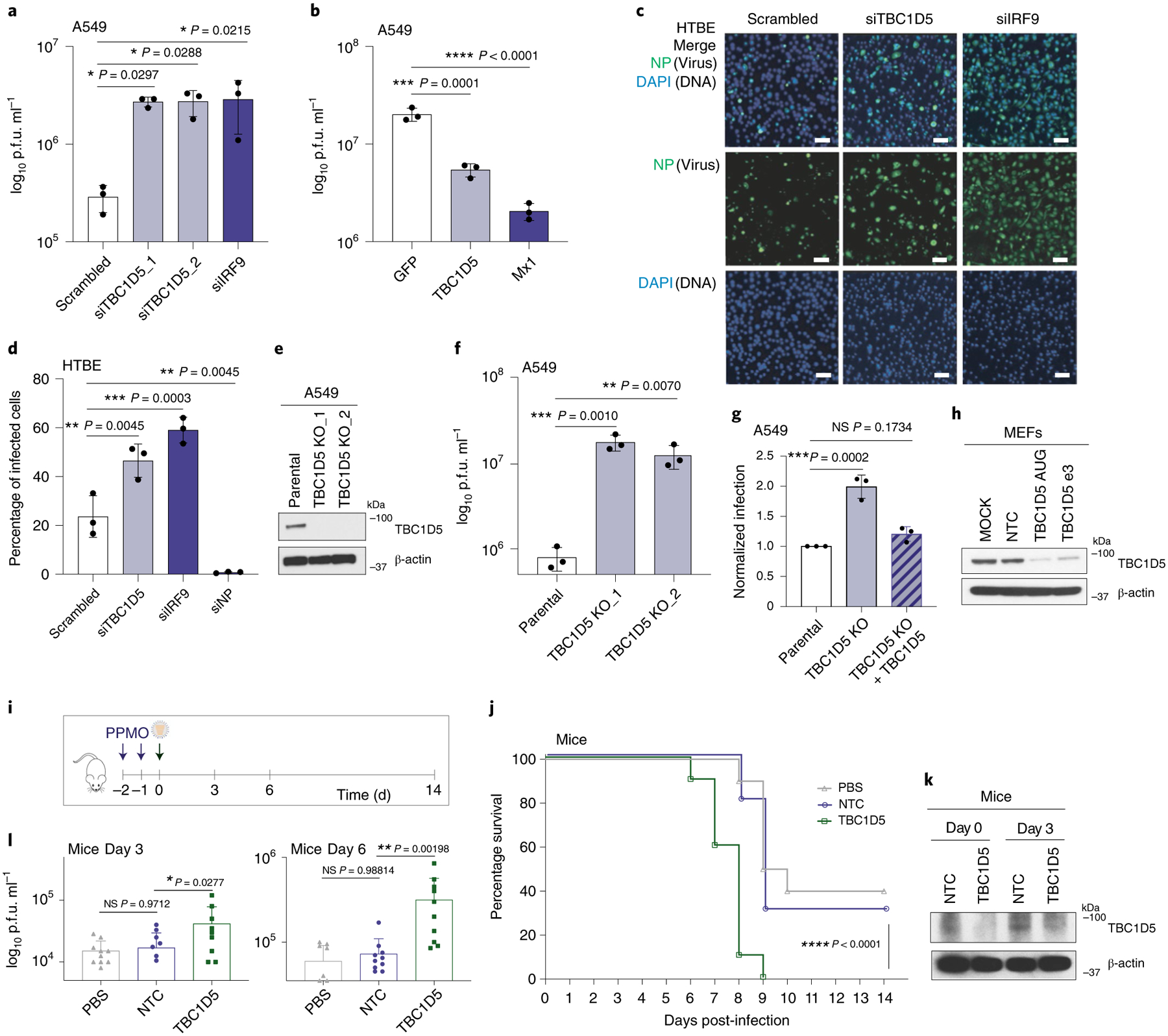
a, A549 cells were transfected with indicated siRNAs for 48 h before infection with A/WSN/33 (MOI 0.01). Supernatants were analysed at 48 h postinfection (h p.i.) by plaque assay. Data represent mean ± s.d. of three independent experiments (n = 3). b, A549 cells were transfected with indicated plasmids (10 ng) for 36 h and infected with A/WSN/33 (MOI 0.01). At 48 h p.i., supernatants were analysed by plaque assay. Data show mean ± s.d. from one representative experiment in triplicate (n = 3). c,d, HTBE cells were transfected for 36 h with indicated siRNAs before A/WSN/33 infection (MOI 1). At 24 h p.i., cells were immunolabelled with NP antibody. Representative images are shown in c. Scale bar, 10 μm. Quantification (d) shows mean ± s.d. of three independent experiments (n = 3). e, Protein analysis of A549 parental and TBC1D5 KO clones. Blot is representative of three independent experiments. f, A549 parental and TBC1D5 KO cells were infected with A/WSN/33 (MOI 0.01) for 48 h and supernatants analysed by plaque assay. Data show mean ± s.d. from one representative experiment in triplicate (n = 3). g, Parental, TBC1D5 KO and TBC1D5 KO + 10 ng of TBC1D5 cells were infected with A/WSN/33 (MOI 0.1) for 24 h before immunolabelling with NP antibody. Data show mean ± s.d. normalized infection from one representative experiment in triplicate (n = 3). h, MEFs were treated with indicated PPMOs or PBS for 72 h and subjected to SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and immunoblotting. Blot is a representative from two independent experiments. i, Schematic representation of mouse experiments. j, Mice survival was monitored for 14 d p.i. Shown is percentage survival ± s.d. from two independent experiments, each with five mice per condition (n = 10). k,l, On days 3 and 6 p.i., mice were euthanized to harvest the lungs and determine TBC1D5 expression (k) and virus titre (l). Blot is a representative from two independent experiments (k). Graph shows mean lung virus titre ± s.d. from two independent experiments each with five mice per condition (n = 10) (l). Statistical significance was calculated using one-way analysis of variance (ANOVA) with Dunnett’s post hoc test (a,b,d,f,g) or log rank Mantel–Cox test (j). l, Two-way ANOVA test and Dunnett post hoc test were conducted by adding experiment batch as a covariate along with PPMO treatment effect. NS, not significant.
TBC1D5 knockdown increases IAV growth and lethality in vivo.
To understand the impact of TBC1D5 depletion on IAV replication in vivo, we used peptide-conjugated phosphorodiamidate morpholino oligomers (PPMOs). PPMOs are sequence-specific antisense agents that can be administered intranasally to elicit a transient reduction in the expression of the targeted gene product in the lungs of treated mice45. Two PPMOs targeting TBC1D5 were designed and synthetized (TBC1D5 AUG and TBC1D5 e3i3) and their efficacy was validated in vitro (Fig. 3h). TBC1D5 PPMO treatment itself was non-toxic as no significant differences in body weight were detected compared to control mice (Extended Data Fig. 4f). Mice were treated with PBS, a non-targeting PPMO control (NTC) or TBC1D5-targeting PPMOs for two consecutive days, then infected with 40 p.f.u. of A/Puerto Rico/8/34 H1N1 (PR8) virus and monitored for 14 d (Fig. 3i). Treatment with TBC1D5-targeting PPMOs resulted in a significant decrease in survival as compared to PBS or NTCs (P < 0.0001) (Fig. 3j), though no differences in body weight were observed (Extended Data Fig. 4g). Lungs of mice treated with PPMOs for TBC1D5 exhibited reduced TBC1D5 expression (Fig. 3k) and significantly heightened viral titres at both day 3 and day 6 post-infection (Fig. 3l). Overall, these data are consistent with our in vitro and ex vivo findings, providing additional in vivo genetic evidence that supports a role for TBC1D5 in restricting IAV replication and growth.
TBC1D5 interacts with the cytoplasmic tail of IAV M2 protein.
To validate the physical interaction between M2 and TBC1D5, co-immunoprecipitations (co-IPs) were performed in 293T cells in the context of GST-M2 pull-down (Fig. 4a). Reciprocal pull-downs of GFP-TBC1D5 in IAV-infected 293T cells confirmed an association of TBC1D5 with M2 during IAV infection (Fig. 4b). To further validate these observations, we performed a proximity ligation assay (PLA). This approach enables in situ detection of endogenous PPIs at a single molecule resolution46. 293T cells were infected with WSN/33 virus or mock-infected and then subjected to PLA staining (Fig. 4c). A significant number of PLA events were measured in the infected but not mock-infected cells, confirming that endogenous TBC1D5 and M2 interact during IAV infection (Fig. 4c,d). M2 protein consists of three domains: an ectodomain (ED), an ion-channel containing transmembrane domain (TM) and a C-terminal cytoplasmic tail (CT)35 (Fig. 4e). To further define the region of M2 that interacts with TBC1D5, we transfected 293T cells with a series of N-terminal GST-tagged M2 deletion constructs that were characterized in a previous publication7, using GST-M2 full length and GST as positive and negative control, respectively. After validating that these constructs are expressed to a similar extent in the lysates of transfected 293T cells (Extended Data Fig. 5a), we conducted PLA staining for TBC1D5 and GST to evaluate the interaction between TBC1D5 and the M2 domains. Quantification of the PLA events across all conditions revealed significant interaction of TBC1D5 with GST-M2 Full, GST-TMCT and GST-CT but not GST-ED or GST-EDTM, suggesting that M2 ED was dispensable for TBC1D5 binding but the M2 CT domain was required for the interaction (Fig. 4f,g). This is consistent with the proposed topology of M2 at the endosome, with its ED facing the endosome lumen and the CT facing the cytosol, as well as TBC1D5 localization anchored onto endosomes35,47. We further investigated the cellular localization of TBC1D5 and M2 in IAV-infected cells and found that M2 colocalizes with endogenous TBC1D5 in the perinuclear region of IAV-infected 293T cells with a Pearson correlation coefficient (PCC) of 0.51 (Fig. 4h). A similar colocalization between TBC1D5 and M2 was observed in A549 cells (PCC = 0.66, representative images not shown). TBC1D5 is recruited to Rab7-positive late endosomes and autophagosomes either by direct interaction with Rab7 or through its two LC3-interacting regions (LIRs)20,48. Since IAV M2 protein has also been found to colocalize with autophagosomal markers during IAV infection24, we proposed that M2 and TBC1D5 may colocalize in vesicles that are positive for Rab7. Immunolabelling of Rab7, TBC1D5 and M2 suggested that these three proteins are in close proximity in IAV-infected cells (Fig. 4i). Taken together, these data confirm TBC1D5 as a physical interactor of IAV M2 protein.
Fig. 4 |. TBC1D5 interacts with the cytoplasmic tail of IAV M2 protein.
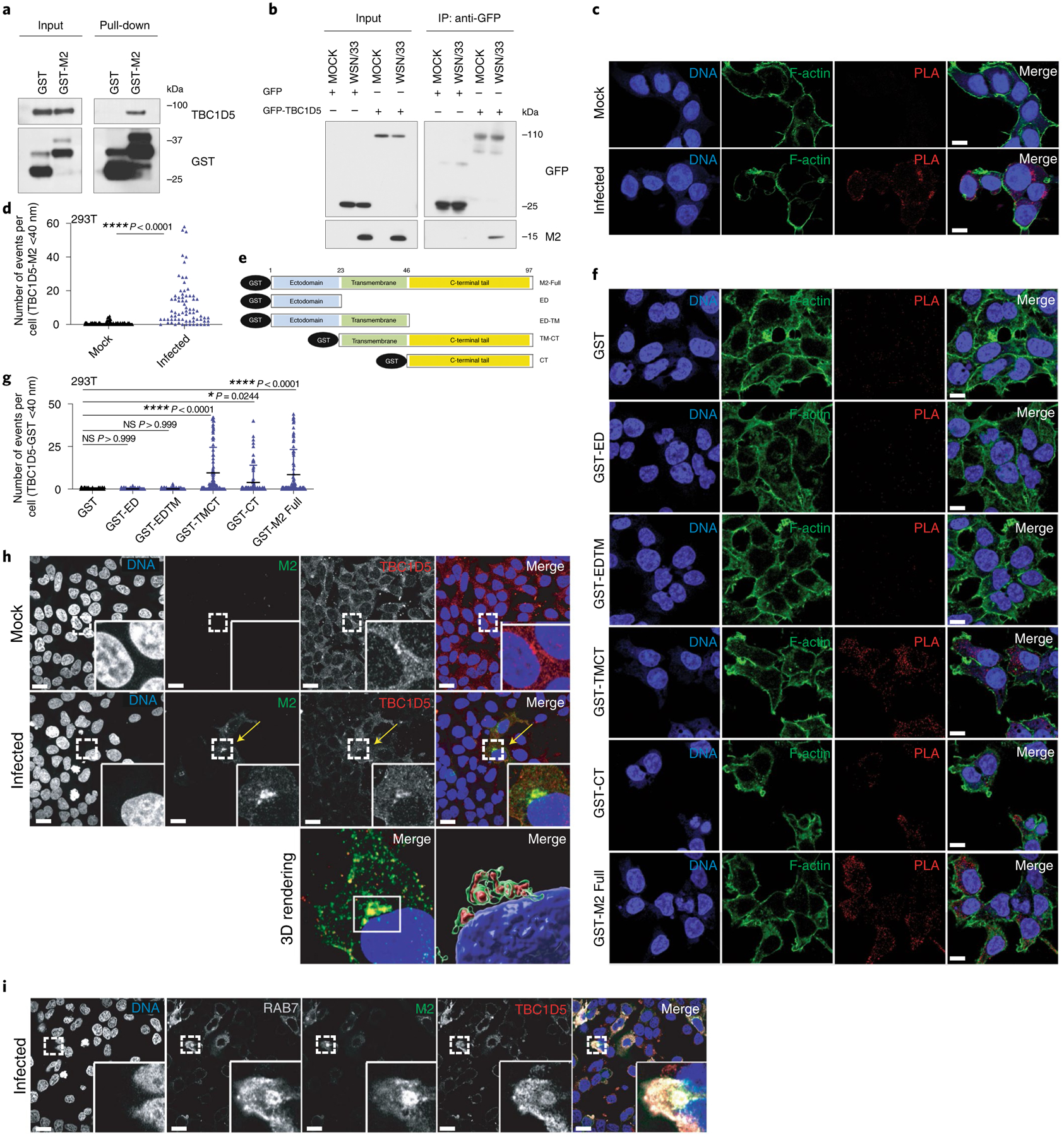
a, 293T cells were transfected with GST or GST-M2 and subjected to GST-affinity pull-down. Input and pull-down samples were analysed by SDS–PAGE using indicated antibodies. Blot is a representative from two independent experiments. b, 293T cells were transfected with indicated plasmids and infected with A/WSN/33 (MOI 3) for 24 h. Immunoprecipitation (IP) was carried out using GFP-trap resin. Inputs and IP samples were analysed by SDS–PAGE using indicated antibodies. Blot is a representative from three independent experiments. 293T cells were mock-treated or infected with A/WSN/33 (MOI 3) for 1 h on ice. At 12 h p.i., cells were subjected to PLA staining. c, Representative images show PLA signal events (red) where TBC1D5 and M2 proteins interact, Phalloidin (F-actin, green) and Hoechst (DNA, blue). Scale bar, 10 μm. d, Quantification of PLA signal events per cell. Data show mean ± s.d. from one representative experiment where at least 80 cells per condition (n = 80) were quantified. Statistical significance was calculated using unpaired two-tailed Student’s t-test. e, GST-tagged M2 constructs. 293T cells were transfected with indicated constructs for 16 h before PLA staining using TBC1D5 and GST primary antibodies. f, Representative images are shown. Scale bar, 10 μm. g, Quantification of PLA signal events per cell. Data show mean ± s.d. from one representative experiment where at least 100 cells per condition (n = 100) were quantified. Statistical significance was calculated using one-way ANOVA with Dunnett’s post hoc test. h, 293T cells were infected with A/WSN/33 (MOI 0.5) for 9 h. Cells were then fixed, stained with DAPI (blue) and immunostained with anti-M2 (green) and anti-TBC1D5 (red). Arrow locates areas of colocalization between TBC1D5 and M2. Scale bar, 10 μm. Images are representative of two independent experiments where at least 100 cells per condition were measured. i, 293T cells were infected with A/WSN/33 (MOI 1) for 9 h. Cells were then stained with DAPI (blue), anti-M2 (green), anti-TBC1D5 (red) and anti-Rab7A (white). Images are representative of two independent experiments. Scale bar, 10 μm.
TBC1D5 promotes lysosomal targeting of IAV M2 protein.
We next examined which step in the IAV replication cycle was negatively impacted by TBC1D5. To assess if TBC1D5 was involved in early stages of IAV replication, we used an IAV minigenome reporter driven by WSN/33 virus infection49. Cells overexpressing TBC1D5, MX1, IFITM3 or the negative control vector GFP, in combination with a viral minigenome firefly (F) luciferase reporter and transfection control renilla (R) luciferase, were infected with WSN/33 virus and the F/R ratio was measured. While the overexpression of known IAV restriction factors involved in entry (IFITM3) and replication (MX1) significantly lowered the amount of F/R relative light units (RLU), TBC1D5 overexpression showed no difference compared to the negative control, suggesting that TBC1D5 does not affect viral entry, replication or transcription (Fig. 5a). TBC1D5 also did not affect translation of early or late IAV proteins, as NP and NS1 levels in cells depleted for TBC1D5 were comparable to those of negative control cells at 8 and 16 h postinfection (Extended Data Fig. 6a). Since our data revealed an interaction between TBC1D5 and M2 (Fig. 4a–g), we next evaluated M2 levels in TBC1D5-depleted cells. While NS1 levels were unchanged, increased M2 protein levels were detected at 16 and 24 h post-infection (Fig. 5b). Conversely, TBC1D5 overexpression significantly reduced both total and membrane-bound M2 (Fig. 5c). These data suggest that TBC1D5 affects M2 protein levels.
Fig. 5 |. TBC1D5 promotes lysosomal targeting of IAV M2 protein.
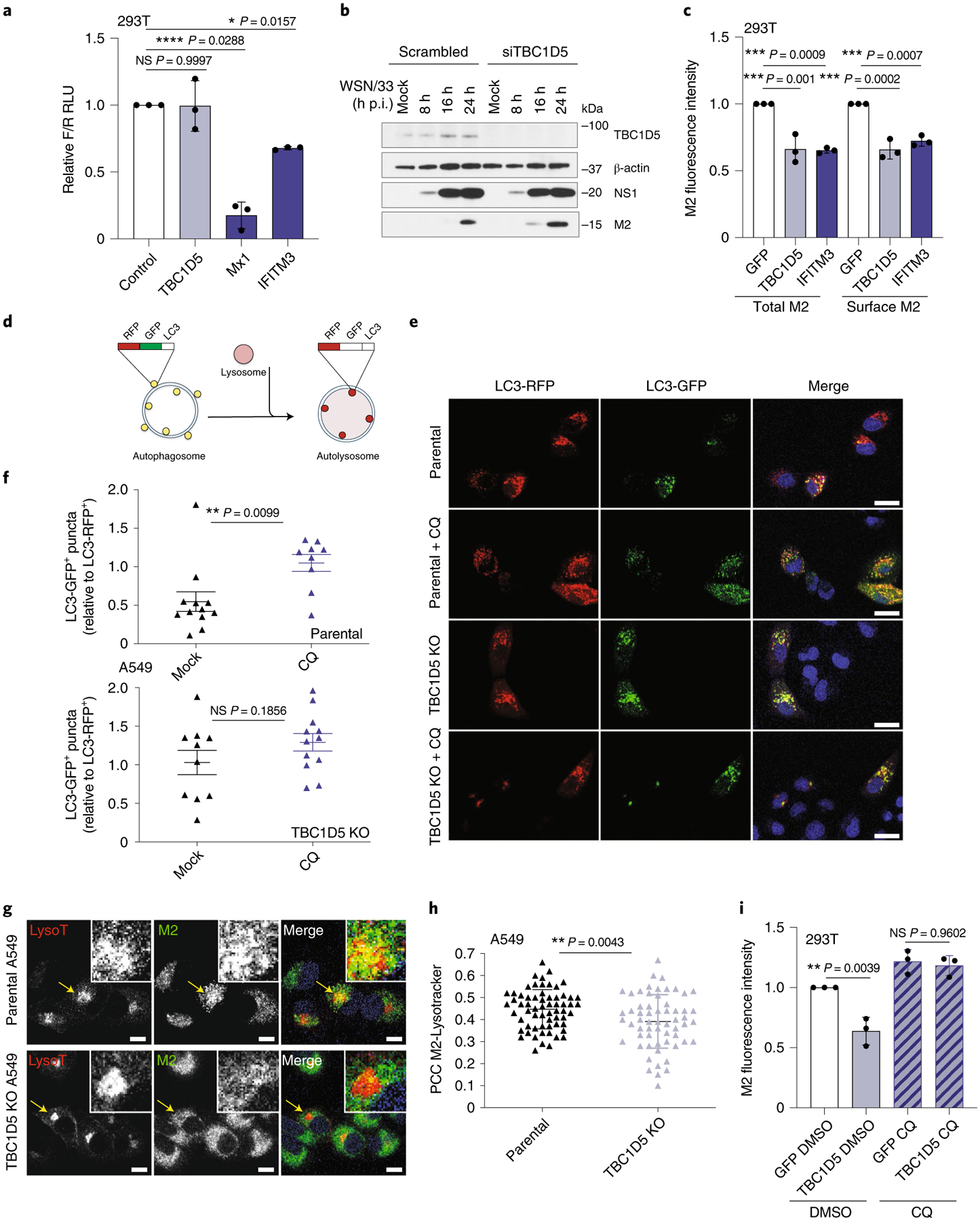
a, 293T cells were transfected with indicated plasmids, a firefly (F) influenza minigenome reporter and transfection control renilla (R). At 24 h post-transfection, cells were infected with A/WSN/33 (MOI 5) for 16 h and the levels of F/R measured. Data represent mean ± s.d. of F/R RLU from three independent experiments (n = 3). b, 293T cells were treated with indicated siRNAs for 48 h before infection with A/WSN/33 (MOI 2). At indicated h p.i., protein levels were analysed using SDS–PAGE. Blot is representative of two independent experiments. c, 293T cells were transfected with indicated expression constructs and infected with A/WSN/33 (MOI 1). At 18 h p.i., cells were immunolabelled with anti-M2 in the presence (total M2) or absence (surface M2) of cell permeabilization agent and M2 fluorescence levels measured by flow cytometry. Data represent mean ± s.d. of three independent experiments (n = 3). d, RFP-GFP-LC3B reporter. e, Parental and TBC1D5 KO cells were transduced with BacMam 2.0 RFP-GFP-LC3B for 24 h, infected with A/WSN/33 (MOI 3) or treated with 100 μM CQ and, 16 h later, the relative number of LC3-GFP+ /LC3-RFP+ puncta per cell were quantified. Images are representative of three independent experiments. Scale bar, 10 μm. f, Data show mean ± s.e.m. LC3-GFP+ /LC3-RFP+ puncta per cell from one representative experiment where at least nine cells per condition were quantified (n = 9). g, A549 parental and TBC1D5 KO cells were infected with A/WSN/33 (MOI 3). At 7 h p.i., cells were treated with 70 nM Lysotracker, incubated for 1 h and then labelled with anti-M2. Images are representative of two independent experiments. Scale bar, 10 μm. h, PCC of M2-Lysotracker colocalization. Data show mean ± s.d. from one representative experiment where at least 50 cells per condition were quantified (n = 50). i, 293T cells were transfected with indicated expression constructs and infected with A/WSN/33 (MOI 1). At 4 h p.i., cells were treated with 100 μM CQ for 12 h p.i. and subjected to M2 immunolabelling in the absence of permeabilization agent. M2 mean fluorescence levels were recorded by flow cytometry. Data represent mean ± s.e.m. of three independent experiments (n = 3). Statistical significance was calculated using one-way ANOVA with Dunnett’s (a,c) or Tukey’s multiple comparisons post hoc (i) or two-tailed unpaired Student’s t-test (f,h).
Both M2 and TBC1D5 can regulate autophagy. IAV M2 protein reduces fusion of autophagosomes with lysosomes and redirects the autophagosomal marker LC3 to the plasma membrane, where M2 drives budding of IAVs24,44. Failure to evade lysosomal degradation reduces M2 levels and results in decreased viral growth50. TBC1D5 regulates Rab7 activation, which in turn controls fusion of late endosomes and autophagosomes with lysosomes51 (Discussion). To explore if TBC1D5 depletion reduces fusion of autophagosomes with lysosomes, we measured autophagic flux using a fluorescent tandem reporter coupled to the autophagosome marker LC3B (RFP-GFP-LC3B)52. Using this system, autophagosomes display both RFP and GFP fluorescence. However, upon fusion with lysosomes, the GFP signal is quenched and only the acid-insensitive RFP signal can be detected (Fig. 5d). First, to evaluate the basal autophagic flux in these cells, parental cells were transduced with the tandem RFP-GFP-LC3B reporter and either mock-treated or treated with chloroquine (CQ), an inhibitor of autophagosomes and lysosomes fusion. Then the ratio of LC3-GFP+/LC3-RFP+ puncta per cell was calculated. The relative number of LC3-GFP+ puncta was significantly reduced in mock-treated compared to CQ-treated cells, suggesting that parental cells support fusion of autophagosomes with lysosomes (Fig. 5e,f, upper panel). In contrast, TBC1D5 KO cells that were mock-treated showed no significant differences in the relative number of LC3-GFP+ puncta compared to those treated with CQ, suggesting that in TBC1D5 KO cells the fusion of autophagosomes with lysosomes is reduced (Fig. 5e,f, bottom panel).
Because TBC1D5-depleted cells displayed reduced fusion of autophagosomes with lysosomes and supported increased M2 protein levels (Fig. 5b,f), we proposed that TBC1D5 could promote lysosomal degradation of M2. To test this, we assessed the colocalization of M2 and lysosomes in parental and TBC1D5 KO cells, using the acidic organelle dye Lysotracker. The colocalization of M2 and Lysotracker was significantly reduced in TBC1D5-depleted cells (P < 0.0043; Fig. 5g,h), suggesting that lysosomal targeting of M2 is reduced in the absence of TBC1D5. Conversely, consistent with previous M2 protein levels measurements (Fig. 5c), cells depleted for TBC1D5 showed a significant enhancement of cell surface-bound M2 over time (Extended Data Fig. 6b). To further confirm that TBC1D5 promotes lysosomal targeting of M2, control cells or cells overexpressing TBC1D5 were treated with DMSO or CQ and the levels of surface M2 were measured. TBC1D5 overexpression showed reduced M2 levels at the plasma membrane in DMSO-treated but not CQ-treated cells (Fig. 5i), further indicating that TBC1D5 promotes lysosomal targeting of M2 protein.
IAV M2 protein abrogates TBC1D5 and Rab7 interaction.
Since TBC1D5 promotes autophagic flux and M2 reduces fusion of autophagosomes with lysosomes24, we hypothesised that M2 antagonizes TBC1D5 activity to evade lysosomal degradation. Binding of TBC1D5 regulates Rab7 activation state, which is essential for vesicle trafficking and fusion of autophagosomes with lysosomes16. Therefore, we investigated Rab7–TBC1D5 physical interaction in the presence and absence of IAV infection. Upon Rab7 immunoprecipitation, we observed reduced Rab7–TBC1D5 association in WSN/33 infected cells as compared to uninfected cells (Fig. 6a). Notably, Rab7–TBC1D5 interaction was reduced in a multiplicity of infection (MOI)-dependent manner (Fig. 6b). Similar results were observed when we immunoprecipitated TBC1D5, as IAV infection resulted in lower Rab7 association (Fig. 6c). To further validate these observations in the context of endogenous TBC1D5 and Rab7, we performed PLA assays. The ability of this system to capture differences in the number of Rab7–TBC1D5 interaction events was validated using cells depleted for TBC1D5 (Extended Data Fig. 7a). 293T cells were then infected with A/Puerto Rico/8/34 H1N1 (PR8) or mock-infected and then subjected to PLA staining to detect if endogenous TBC1D5 and Rab7 interact in the presence of IAV infection. Consistent with the biochemical data, we observed a significant reduction of TBC1D5 and Rab7 interaction in PR8-infected cells as compared to mock-infected cells (P < 0.0001; Fig. 6d,e). This difference was not due to reduced TBC1D5 or Rab7 protein levels in IAV-infected cells, as shown by protein levels analyses carried out in parallel (Extended Data Fig. 7b). We then investigated if M2 is necessary to abrogate this interaction by using a PR8 virus that contains a stop codon on the M segment to prevent M2 expression (PR8 ΔM2)24. While the infectivity of this mutant virus was not affected (Extended Data Fig. 7c), we validated that M2-deficient viruses were unable to block fusion of autophagosomes and lysosomes, as reflected by increased LC3-II accumulation (Extended Data Fig. 7d)24. Critically, the interaction between TBC1D5 and Rab7 was not disrupted in 293T cells infected with the M2-deficient virus, since we observed comparable number of PLA events to those in uninfected conditions (Fig. 6d,e).
Fig. 6 |. M2 protein abrogates TBC1D5 and Rab7 interaction.
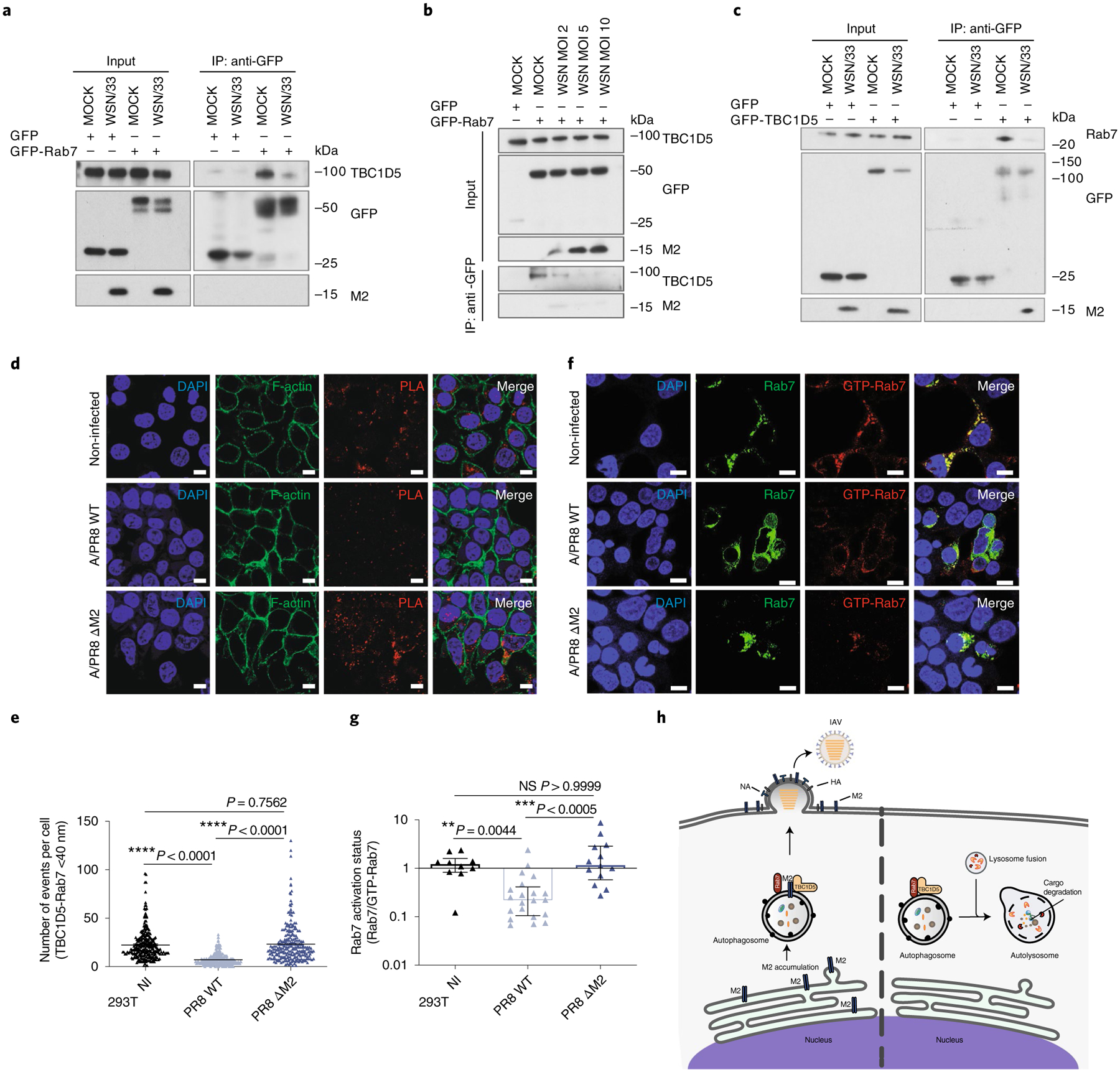
a, 293T cells were transfected with GFP or GFP-Rab7 and infected with A/WSN/33 (MOI 3) for 24 h. IP was carried out using GFP-trap resin. Inputs and IP samples were analysed by SDS–PAGE using indicated antibodies. b, 293T cells were transfected with GFP or GFP-Rab7 and infected with A/WSN/33 (MOI 2, 5 and 10) for 24 h. IP was carried out using GFP-trap resin, and inputs and IP samples were analysed by SDS–PAGE using indicated antibodies. c, 293T cells were transfected with GFP or GFP-TBC1D5 and infected with A/WSN/33 (MOI 3). At 24 h p.i. cell lysates were subjected to IP using GFP-trap resin and inputs and IP samples were analysed by SDS–PAGE using indicated antibodies. In a–c, blots are representatives from at least two independent experiments. In d and e, 293T cells were infected with A/PR8 WT or A/PR8 ΔM2 (MOI 3) and subjected to PLA staining. d, Representative images from three independent experiments show PLA signal events (red) where TBC1D5 and Rab7 proteins interact, Phalloidin (F-actin, green) and Hoechst (DNA, blue). Scale bar, 10 μm. e, Quantification of PLA signal events. Data show mean ± s.d. from one representative experiment of at least three independent experiments where at least 100 cells per condition (n = 100) were quantified. Statistical significance was calculated using one-way ANOVA with Tukey’s multiple comparisons test. f, Representative images from two independent experiments show Rab7 (green) and GTP-Rab7 (red) staining across 293T cells that express GFP-Rab7 WT and are either mock-infected or infected with A/PR8 WT or A/PR8 ΔM2 (MOI 3) for 14 h. Scale bar, 10 μm. g, Quantification of GTP-Rab7/total Rab7 ratio. Data show mean ± s.d. from one representative experiment of at least two independent experiments where at least 100 cells per condition (n = 100) were quantified. Statistical significance was calculated using one-way ANOVA with Kruskal–Wallis multiple comparisons test. h, Proposed model. IAV M2 protein abrogates interaction of TBC1D5 with Rab7, which in turn prevents fusion of autophagosomes with lysosomes. By escaping degradation at the lysosome, M2 can now assist IAV budding at the plasma membrane and support viral growth.
Rab7 activation is essential for vesicle trafficking and fusion of autophagosomes with lysosomes16 and depends on its nucleotide state (GTP-bound active or GDP-bound inactive), which is controlled by association with GAP and GEF regulators. We therefore suggested that IAV M2 could abrogate Rab7–TBC1D5 interaction to reduce Rab7 activation. To test this, we took advantage of an antibody that specifically recognizes active GTP-bound Rab7. We validated the specificity of this antibody by detecting immunofluorescence signal in cells transfected with wild-type (WT) Rab7 or the constitutively active GTP-bound Q67L-Rab7 mutant but not in the T22N-Rab7 mutant, which harbours higher GDP affinity (Extended Data Fig. 7e)53. Using this antibody, we measured Rab7 activation status (GTP-Rab7/total Rab7 ratio) in mock-infected cells or cells infected with WT or the M2-deficient virus. While M2-deficient IAV-infected cells did not alter Rab7 nucleotide-binding state (P > 0.999 compared to non-infected), cells infected with WT-PR8 virus showed a significant reduction in Rab7 activation levels (**P = 0.0044 compared to non-infected and ***P < 0.0005 compared to M2-deficient PR8) (Fig. 6f,g). Taken together, these data support a model wherein in steady-state conditions, TBC1D5 interacts with Rab7, which enables fusion of late endosomes and autophagosomes with lysosomes and thus cargo degradation (Fig. 6h, right panel). In IAV-infected cells, extensive accumulation of M2 at the ER triggers autophagy and results in the incorporation of M2 in autophagosomes (Fig. 6h, left panel). These M2 proteins abrogate TBC1D5–Rab7 interaction, which reduces Rab7 activation and arrests lysosomal fusion, thus enabling M2 to traffic to the plasma membrane to support IAV budding and growth (Fig. 6h, left panel).
Discussion
Successful completion of the IAV replication cycle involves co-option of essential cellular factors and antagonism of antiviral activities. Previous genome-wide loss-of-function screens have provided considerable insight into cellular factors that are essential for IAV replication7,11. However, much less is known about host factors involved in IAV restriction at a global scale, particularly in the context of immune cells that are responsible for mounting immune responses against IAV25. Here, we conducted a genome-wide siRNA screen using human macrophage-like cells and integrated the identified antiviral factors with global transcriptomics of IAV-infected macrophages. These analyses provided insight into the global landscape of IAV cellular restriction, which was clustered into IFN-inducible or constitutive mechanisms. Analysis of the IFN-inducible factors revealed, as expected, enrichment in the type I IFN response, including ISGs with known roles in IAV restriction and nine validated ISGs not previously linked to IAV, including the guanine nucleotide-binding protein GNG5, recently reported interactor of SARS-CoV-2 (refs.54,55) (Extended Data Fig. 3). While the IFN response is generally fast and efficient, providing an important first-line defence against invading pathogens, it can also trigger excessive inflammation and cause immunopathology56. Analysis of the constitutive antiviral factors found in this study show that cells also rely on an extensive set of cell-intrinsic mechanisms to control IAV infection, including metabolic pathways such as glycolysis and fatty acid synthesis, whose intermediate metabolites have been previously linked to viral restriction57,58, NMD, which can promote decay of viral messenger RNAs32, the ubiquitin/proteosome pathway, known to target viral proteins for proteolysis59, vesicle sorting and autophagy (Extended Data Fig. 2b).
Regulation of vesicle sorting and autophagy have been previously associated with IAV M2 protein. However, the molecular regulators of these activities remained to be elucidated. Integration of these antiviral clusters with an IAV M2 PPI network provided potential insight into the interplay of M2 with these antiviral mechanisms. M2 interactors were predominantly enriched in pathways that included vesicle transport, exocytosis and membrane docking, which are consistent with M2 trafficking using the secretory and/or autophagy pathways35 (Fig. 1c). In addition, factors and pathways not previously associated with M2 interaction or IAV antiviral activity were identified, including those involved in mtDNA repair, mitophagy, mitochondrial homoeostasis and apoptosis, suggesting that the mitochondrion is a crucial platform for cell-intrinsic restriction of IAV and that M2 is either the target or the antagonist of these cellular defence mechanisms.
The identification of these pathways, complexes and factors that are supported by orthogonal datasets suggest that they may be more likely to be physiologically relevant antiviral regulators of IAV replication. For instance, this study uncovers the role of the autophagy regulator TBC1D5 as a restriction factor of IAV infection. TBC1D5 was not amongst the most potent hits from the functional genetic screen, as it would be expected for a factor targeted for viral evasion, but it was prioritized on the basis of orthogonal data including the M2 AP-MS. Therefore, multi-omics integration enables the identification of factors that may be missed due to prioritization based only on activity from a single data source but have important roles in host–pathogen interactions. Taken together, we report 111 host-restriction factors not previously reported to impinge on IAV replication (Fig. 2 and Supplementary Table 5) and a list of prioritized factors including those with multiple levels of orthogonal support (Extended Data Fig. 3). Additional studies will be required to illuminate their role in IAV pathogenesis in an ex vivo, in vivo and clinical context.
IAV infection induces both canonical and non-canonical autophagy, which represent critical cellular defence mechanisms for pathogen clearance through fusion of the pathogen-engulfed vesicles with lysosomes60,61. To escape degradation, M2 protein has been shown to reduce fusion of autophagosomes with lysosomes and instead traffic to the plasma membrane where it supports IAV budding24,44. However, the molecular mechanism behind this evasion strategy remained to be elucidated. Data generated in this study indicate that M2 abrogates TBC1D5 and Rab7 interaction to reduce Rab7 activation and thus fusion of autophagosomes with lysosomes16. However, this viral evasion strategy seems to be dependent on the stoichiometry of TBC1D5 and M2 proteins in the cell (Fig. 6b). Increased TBC1D5 expression probably overwhelms the ability of M2 to block TBC1D5 interaction with Rab7, resulting in M2 degradation. Conversely, TBC1D5 depletion probably reduces the need for M2 to inhibit TBC1D5 interaction with Rab7, resulting in enhanced M2 protein levels at the cellular surface.
Rab7 activation is essential for both the recruitment of SNARE-like proteins that fuse the membranes of late endosomes and autophagosomes with lysosomes21 and for dictating the directionality of vesicle trafficking (anterograde to the plasma membrane or retrograde to the perinuclear region where lysosomes reside)17,18,62,63. Therefore, displacement of Rab7–TBC1D5 physical interaction might be a prerequisite for arresting fusion with lysosomes and redirecting the M2-containing vesicles to the plasma membrane, where M2 initiates viral budding and promotes IAV growth. Notably, two bacterial pathogens have been previously shown to target Rab7 activation to promote survival. Legionella pneumophila secretes the effector protein RidL, which prevents recruitment of TBC1D5 to Rab7 (refs.64,65). In addition, Salmonella enterica SopD2 protein blocks Rab7 nucleotide exchange, preventing its association with effectors Rab-interacting lysosomal protein (RILP) and FYVE and coiled-coil (CC) domain-containing protein (FYCO1), which govern endosome, lysosome and autophagosome trafficking16,66. Induction of autophagy is a crucial feature of infection by multiple viral families, including flaviviruses and coronaviruses22. Since the manipulation of Rab7 activation represents a critical node exploited by both bacteria and viruses to escape lysosomal degradation, targeting of this molecular circuit probably represents a convergent evolutionary evasion strategy. Therefore, therapeutic targeting of this mechanism may be an important strategy for the development of broad-spectrum antivirals that act to restore lysosomal degradation of viral cargo.
Methods
Cells and viruses.
A549 (ATCC CCL-185), MDCK (ATCC CCL-34), HEK293T (ATCC CRL-3216) and MEF (ATCC CRL-2991) cells were cultured in DMEM (Fisher Scientific) supplemented with 10% FBS (Gibco), 1 mM sodium pyruvate (Life Technologies), 2 mM l-glutamine (Fisher Scientific), 10 mM HEPES (Fisher Scientific),100 U ml−1 of penicillin and 100 μg ml−1 of streptomycin (Fisher Scientific). THP-1 cells (ATCC TIB-202) were cultured in RPMI-1640 (Fisher Scientific) supplemented as described above. Cells were grown at 37 °C in 5% CO2. HTBE cells (ATCC PCS-300–010) were cultured in commercially available airway epithelial cell basal medium following manufacturer’s protocol (ATCC). HTBE cells were derived from one donor and all tissues used for isolation of these cells were obtained under informed consent and conform to HIPAA standards to protect the privacy of the donors’ personal health information. All cells were tested and were confirmed to be free of mycoplasma contamination. A/Vietnam/1203/04 (H5N1) HALo mutant virus is an attenuated H5N1 influenza A virus generated using wild-type influenza A/Vietnam/1203/04 (H5N1) virus67,68. A/Wyoming/3/03 (H3N2) and A/Vietnam/1203/05-HALo were generated using reverse genetics and propagated in the allantoic cavity of embryonated eggs (Charles River Laboratories). A/WSN/33 (H1N1), A/Puerto Rico/8/34 (H1N1) WT and ΔM2 were propagated in MDCK cells (WSN and PR8 WT) or MDCK-M2 (PR8ΔM2). Titre was determined by plaque assay on MDCK cells using agar overlay medium. A549-doxycycline(dox)-Cas9 cells were generated by transduction of Lenti-dox-Cas9 (Dharmacon) into A549 cells. Cells were then plated for colony formation and screened for Cas9 expression after dox treatment.
Antibodies.
The antibodies used in this study include the following. Immunofluorescence: rabbit polyclonal anti-TBC1D5 (Atlas, catalogue no. HPA035125, 1:200), mouse monoclonal anti-Rab7A antibody (Cell Signaling, catalogue no. 95746, 1:100), goat polyclonal anti-Rab7A (LSBio catalogue no. B13237, 1:200), mouse monoclonal anti-GTP-Rab7 (Neweastbio, catalogue no. 26923, 1:100), mouse monoclonal anti-GST (Cell Signaling, catalogue no. 2624, 1:500), mouse monoclonal anti-M2 E10 (Mount Sinai, in-house antibody, 1:2,000), mouse monoclonal anti-NP HT103 (Mount Sinai, in-house antibody, 1:10,000), Alexa Fluor 488-conjugated anti-mouse secondary antibody (Thermo Fisher Scientific, no. A-11001, 1:1,000) and Alexa Fluor 568-conjugated anti-mouse secondary antibody (Thermo Fisher Scientific, no. A-11004, 1:1,000). Western blotting: mouse monoclonal anti-TBC1D5 (Santa Cruz, catalogue no. sc-376296, 1:1,000), mouse monoclonal anti-Rab7A (Cell Signaling, catalogue no. 95746, 1:1,000), mouse monoclonal anti-M2 (Santa Cruz, catalogue no. sc-32238, 1:1,000), rabbit monoclonal anti-β-actin (Cell Signaling, catalogue no. 4970, 1:10,000), rabbit anti-GFP (Cell Signaling, catalogue no. 2555, 1:10,000), rabbit polyclonal anti-NP (a kind gift of A. Nieto, 1:10,000), rabbit polyclonal anti-LC3A/B (Cell Signaling, catalogue no. 4108, 1:1,000) and rabbit monoclonal anti-Cox IV (Cell Signaling, catalogue no. 4850, 1:10,000). Flow cytometry: mouse monoclonal anti-M2 E10 (Mount Sinai, in-house antibody, 1:2,000).
Genome-wide siRNA screen.
A genome-wide siRNA screen was carried out in human macrophage-like THP-1 cells to identify host cell factors that affect the replication of IAV. The screen was performed using the arrayed genome-wide ON-TARGETplus SMARTpool siRNA library (Dharmacon), where each pool contains four unique siRNAs targeting each human gene. In addition, non-targeting siRNAs (scrambled) were added to each plate as negative controls and siRNAs targeting IRF9 and IAV NP were included as positive controls. The siRNA sequences are listed in Supplementary Table 6. THP-1 cells were differentiated using 10 ng ml−1 phorbol-12-myristate-13-acetate (PMA) for 72 h at 37 °C in 5% CO2. Pooled siRNAs were arrayed in 384-well plates at a concentration of 12.5 nM siRNA per well. To enable the formation of siRNA-transfection reagent complexes, 0.075 μl of Lipofectamine RNA interference MAX (RNAiMAX) transfection reagent diluted in 9.925 μl of Opti-MEM media (both reagents Thermo Fisher Scientific) were added to each well. Following a 20 min incubation period at room temperature, 15,000 differentiated THP-1 cells diluted in 20 μl of RPMI media supplemented with 10% FBS, 1% PenStrep and 1% HEPES were seeded on top of the complexes and incubated for 48 h at 37 °C in 5% CO2. Cells were then mock-treated or treated with 100 IU ml−1 universal interferon beta (IFN, R&D Systems) diluted in 10 μl of serum-free RPMI media. After 6 h of incubation at 37 °C in 5% CO2, media were removed and cells were infected with IAV A/Wyoming/3/2003 H3N2 (MOI 0.50) or A/Vietnam/1203/2004 H5N1 HALo (MOI 0.25) diluted in 20 μl of serum-free RPMI media. After 1 h of incubation at room temperature, the inoculum was removed and replaced with 40 μl of serum-free RPMI media and cells were incubated for 24 h at 37 °C in 5% CO2. Cells were then fixed with 4% PFA (Boston BioProducts) for 30 min at room temperature and washed twice with PBS. Cells were permeabilized with 0.5% Triton X-100 for 20 min, followed by blocking with 3% BSA (Sigma) for 1 h at room temperature. Primary anti-NP mouse monoclonal (HT103, in-house antibody) was added for 2 h at room temperature, followed by three washes with PBS and a 1-h incubation with Alexa Fluor 488-conjugated anti-mouse secondary antibody (Thermo Fisher Scientific, catalogue no. A-11001) diluted in 3% BSA. Following three washes with PBS, cells were stained with DAPI (4,6-diamidine-2-phenylindole, KPL) and plates were sealed and stored at 4 °C until imaging.
High-content imaging and data analysis.
Viral replication was assessed using high-throughput microscopy. The assay plates were imaged using the IC200 imaging system (Vala Sciences) located at the Conrad Prebys Center for Chemical Genomics (CPCCG). The analysis software Columbus v.2.5 (Perkin Elmer) was used to calculate infectivity (number of Alexa 488+ objects per number of DAPI+ objects). Screens were run twice and the infectivity values for each well were normalized to the median of each plate. The hit calling strategy was based on z-score values. The z-score is a statistical value that evaluates the number of standard deviations from the mean of the population and is calculated as the (log2FC siRNA x − mean log2FC of the sample)/ s.d. of the sample. In context of siRNA screening, it ranks the effects of each siRNA across the entire population. Factors with a corresponding z-score <−1.5 were considered host-dependency factors and those with a z-score >1.5 host-restriction factors. Cytotoxicity resulting from siRNA transfection was evaluated by counting the total number of cells per well and normalizing to the value for the negative control siRNA. Targeting siRNAs resulting in cytotoxicity measurements <70% compared to scrambled siRNA were considered cytotoxic and removed from the hit list.
Oseltamivir treatment.
A total of 15,000 PMA-differentiated THP-1 cells (Genome-wide siRNA screen) were seeded overnight. Cells were then treated with 100 IU ml−1 of universal interferon or mock-treated for 6 h and then treated with 1 μM of oseltamivir (Sigma) for 2 h before infection with A/Wyoming/03/03 H3N2 (MOI 0.50). At 24 h postinfection the percentage of infected cells was calculated on the basis of DAPI staining and viral NP immunostaining.
GO overrepresentation analysis.
The genetic screen hits were tested for pathway and process enrichment using the following ontology sources: KEGG Pathway, gene ontology (GO) biological processes, Reactome Gene Sets, Canonical Pathways, CORUM, TRRUST, DisGeNET and PaGenBase, using the whole genome as the enrichment background. Terms with P < 0.01, a minimum count of 3 and a ratio between the observed and the expected by chance counts >1.5, were collected and grouped into clusters on the basis of their membership similarities. Here, P values are calculated on the basis of the accumulative hypergeometric distribution. The most statistically significant term and higher membership within a cluster is chosen to represent the cluster.
AP-MS.
The complete coding sequences for H5N1 A/Vietnam/1203/05-HALo M2 or eGFP (GQ404376.1) were cloned into pLVX-TetOne-Puro (Clontech) with a C-terminal 2×Strep-tag. To generate lentiviruses, these constructs were cotransfected with Gag-Pol packaging construct and VSV-G envelope (pMD2.G, Addgene) into HEK293T cells. A549 cells were transduced with the generated lentiviruses and selected under 1 μg ml−1 of puromycin. The expression of viral proteins or GFP was confirmed by western blot. For immunoprecipitation, expression of M2 or GFP was induced with 1 μg ml−1 of dox for 12 h and cells were then treated with 1,000 IU ml−1 of IFN for an additional 12 h. Cells were then lysed, cleared of cellular debris and bound with Strep-Tactin Sepharose beads (IBA Lifesciences) in IP buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl and 1 mM EDTA). Beads were washed four times (twice with 0.05% NP-40 and twice without) before on-bead protein digest. Strep-Tactin-purified proteins were reduced and alkylated on beads with reduction-alkylation buffer (50 mM Tris-HCl, pH 8.0, 2 M urea, 1 mM dithiothreitol (DTT), 3 mM iodoacetamide). Subsequently, 3 mM DTT was added to quench the reaction and proteins were digested with 0.75 μg of trypsin (Invitrogen). Formic acid (1%) was added to acidify the peptides. Peptides were desalted using Agilent OMIX C18 tips. Digested peptides were subjected to LC-MS/MS analysis using an Easy-nLC 1000 coupled to a dual-pressure linear ion trap (Velos Pro) Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). Peptides were eluted by a gradient of 5–30% acetonitrile in 0.1% formic acid in 110 min delivered at a flow rate of 300 nl min−1. For each cycle, one full MS scan (150–1,500 m/z, resolution of 120,000) in the Orbitrap was followed by 20 data-dependent MS/MS scans fragmented by normalized collision energy (setting of 35%) and acquired in the linear ion trap. Raw MS files were analysed by MaxQuant v.1.3.0.3 and MS/MS spectra searched by the Andromeda search engine against a database containing reviewed SwissProt human and influenza protein sequences (20,226 total). For each bait and control, putative interactors were scored for their probability of being a true interactor using the MiST algorithm, which provides a composite score taking into account the abundance, specificity and reproducibility of each identified protein36. To increase confidence in each interaction, all runs from each bait were analysed together regardless of condition and to better estimate specificity of these interactions for the MiST algorithm, these data were scored relative to other IAV baits pulled down also in A549 cells under these same conditions (access to files including the additional baits available at: https://massive.ucsd.edu/ProteoSAFe/private-dataset.jsp?task=82154043f1ca4b0c9e22c10aa091f476, password: pandemic).
siRNA, RNA-seq and AP-MS data integration and network analysis.
To select the cut-off criteria that will result in the highest number of overlapping factors across the siRNA and RNA-seq datasets, while maintaining a larger overlap than expected by chance (enrichment factor >1), the number of observed overlapping genes between genetic data, RNA-seq and AP-MS were calculated across a range of z-score, log2FC and MiST thresholds. The enrichment factor was calculated as the number of observed divided by the number of expected overlapping genes. Expected values were computed by randomly permuting the genetic data and measuring the overlap on the permuted values. The expected value was the mean value over 100 random permutations. On the basis of these analyses, the following cut-off values were selected which optimized the enrichment factor and number of overlapping genes over the range of thresholds tested: siRNA z-score ≥0.5, RNA-seq log2FC ≥1.0 or ≤−1.0 and P <0.005, MiST ≥0.55. To explore the highest confidence interactions of the input factors list, we selected the STRING - Human Protein Links - High Confidence (score ≥0.7) protein–protein interaction network available on the Network Data Exchange (NDEx). We then identified densely interconnected regions (‘communities’) amongst the input list, using the community detection algorithm HiDeF via the Community Detection Application and Service (CDAPS)69 (app available at http://apps.cytoscape.org/apps/cycommunitydetection). The resulting HiDeF from CDAPS was a ‘hierarchy’ network where each node represented a community of proteins and edges denoted containment of one community (the ‘child’) by another (the ‘parent’). Finally, the hierarchy network was styled, communities were labelled by functional enrichment using gProfiler (via CDAPS) and a layout was applied. The STRING - Human Protein Links - High Confidence (score ≥0.7) network is available in NDEx at http://ndexbio.org/#/network/275bd84e-3d18-11e8-a935-0ac135e8bacf. Molecular complex detection (MCODE) and GO analyses were applied to the resulting overlapping factors to highlight densely connected subnetworks. Nodes with over ten members (IFN-inducible) or over 20 members (constitutive) are shown (Extended Data Fig. 2). To provide more stringency on the network and provide biochemical context to the identified nodes, those factors identified in two or more siRNA screens were integrated with the IAV M2–host AP-MS hits identified using a low stringency MiST score ≥0.55 and the resulting network was subjected to MCODE analyses (Cytoscape) to find highly interconnected regions (Fig. 1c). Network visualization was based on Metascape (http://www.metascape.org) and Cytoscape (http://www.cytoscape.org, v.3.8.0)70,71.
Generation of the integrated model of IAV cellular restriction.
The identified restriction factors were evaluated for pathway enrichment using the following ontology sources: KEGG Pathway, Reactome Gene Sets, GO biological processes, CORUM, Canonical Pathways, TRRUST, DisGeNET and PaGenBase. Resulting data were reviewed and manually curated using published literature. Factors were then placed on the basis of their reported subcellular localization, clustered into functional categories shown in light blue boxes and labelled accordingly.
Virus infection.
Cells were seeded overnight and then infected with indicated IAV strains in 1x phosphate buffered saline (PBS) supplemented with 0.3% BSA, 0.01 mM Ca2+ and 0.02 mM Mg2+. After 1 h of incubation at room temperature, cells were washed and culture medium containing 1 μg ml−1 of TPCK-treated trypsin (Sigma) was added. Cells were then incubated at 37 °C and supernatant samples collected at indicated times. Viral titre in supernatants was determined by plaque assay using MDCK cells.
Generation of CRISPR-Cas9 TBC1D5 KO cells.
To generate CRISPR-Cas9 TBC1D5 KO cells, we targeted the TBC1D5 locus sequence CTTGGCAACAATA AGGCAGA with the commercial gRNA CRISPRevolution srRNA EZ-17406474 (Synthego). The gRNA was transfected into A549-dox-Cas9 cells previously treated with doxycycline (1 μg ml−1; Clontech) for 48 h. At 48 h post-transfection, cells were plated for colony formation. Colonies derived from single cells were screened for TBC1D5 knockout using western blot analysis.
IAV minigenome assays.
Minigenome-based assays were performed as described previously72. Briefly, 293T cells were transfected with an influenza-like minigenome encoding a negative-sense firefly and the internal control renilla (kindly provided by W. Barclay, Imperial College London, UK) using Fugene (Promega) transfection reagent. At 24 h post-transfection, cells were infected with A/WSN/33 (H1N1) at MOI 5 for 16 h. Firefly and renilla RLU were measured using Dual-Glo Luciferase assay (Promega).
Flow cytometry analysis of M2 expression.
293T cells were mock-treated or infected with A/WSN/33 at an MOI of 2 and incubated for indicated times. Cells were trypsinized, washed in 1× PBS (Gibco) with 3% FBS and subjected to permeabilization with 0.1% Tween 20 (total M2). Cells were then blocked in 1× PBS with 1:200 dilution of normal rabbit serum (Abcam, ab7487) for 2 h at room temperature. Membrane-bound (surface) and total M2 were labelled using an anti-IAV M2 (E10, Mount Sinai) overnight at 4 °C in 1× PBS with 3% FBS. Cells were then stained with a goat anti-mouse Alexa Fluor 568 secondary antibody (Thermo Fisher) for 2 h at room temperature, washed and fixed in 4% paraformaldehyde in 1× PBS for 20 min at room temperature. Fluorescence M2 mean intensities were recorded using Attune NxT Flow Cytometer (Thermo Fisher) and analysed using FlowJo software v.10.0 (Tree Star).
GST pull-down assays.
A total of 1 × 107 293T cells were seeded in poly-d-lysine (Corning) 15-cm dishes and incubated overnight. Cells were then transfected with GST or GST-M2 (A/WSN/33) using Fugene (Promega) as transfection reagent. At 24 h post-transfection, cells were washed with ice-cold 1× PBS and lysed using Pierce IP Lysis Buffer (Thermo Fisher) supplemented with protease inhibitor cocktail (Roche). Protein lysates were cleared by centrifugation at 4,000g for 15 min at 4 °C and input samples were collected and stored at −20 °C. The remaining lysates were incubated with Gluthatione Sepharose beads (Thermo Fisher) in a rotator overnight at 4 °C. Beads were washed four times and proteins collected by addition of 2× NuPAGE sample buffer (Thermo Fisher) supplemented with 50 mM DTT and heated for 10 min at 95 °C. Samples were analysed by immunoblotting.
GFP-trap immunoprecipitation assays.
A total of 1 × 107 293T cells were seeded in poly-d-lysine (Corning) 15-cm dishes and incubated overnight. Cells were then transfected with GFP, GFP-TBC1D5 or GFP-Rab7. At 24 h post-transfection, cells were infected with A/WSN/33 (H1N1) at MOI 5 (unless otherwise stated) for 18 h. Cells were then washed with ice-cold 1× PBS and lysed using Pierce IP Lysis Buffer (Thermo Fisher) supplemented with protease inhibitor cocktail (Roche). Protein lysates were cleared by centrifugation at 4,000g for 15 min at 4 °C and an input sample was collected and stored at −20 °C. The remaining lysates were incubated with GFP-trap beads (Chromotek) in a rotator for 2 h at 4 °C. Beads were washed four times and proteins collected by addition of 2× SDS NuPAGE sample buffer (Thermo Fisher) supplemented with 50 mM DTT and heated for 10 min at 95 °C. Samples were analysed by immunoblotting.
RFP-GFP-LC3B tandem reporter studies.
A549 parental and TBC1D5 KO cells were seeded in glass-bottom 24-well plates (Cellvis) and incubated overnight at 37 °C in 5% CO2. Cells were then treated with BacMam 2.0 RFP-GFP-LC3B reagent (Thermo Fisher, P36239) (MOI 50) for 24 h and then infected with A/WSN/33 (MOI 3) or treated with 100 μM chloroquine (CQ). At 16 h p.i., cells were treated with Hoechst 33342 (Thermo Fisher, H3570) for 20 min at room temperature and then imaged using the Nikon A1R HD confocal microscope. The number of LC3-RFP+ and LC3-GFP+ puncta per cell was quantified using Fiji73. Finally, the ratio LC3-GFP+/LC3-RFP+ ratio was calculated.
Confocal imaging and colocalization studies.
A549 or 293T cells were seeded in glass-bottom plates (Cellvis) and incubated overnight at 37 °C in 5% CO2. Cells were washed twice with 1× PBS and infected with A/WSN/33 at indicated MOI for 1 h on ice to synchronize infection. Inoculum was then removed and replaced with fresh serum-free media. At indicated time points, cells were washed twice with 1× PBS and fixed using 4% paraformaldehyde (Boston BioProducts) for 30 min at room temperature. Cells were permeabilized using 0.5% Triton X-100 for 15 min, followed by 1 h of blocking with 3% BSA (Sigma) at room temperature. Cells were stained with DAPI (KPL) and immunolabelled with indicated antibodies. Images were acquired using the Nikon A1R HD confocal microscope and colocalization was assessed using PCC. PCC was calculated using Fiji’s software73.
Proximity ligation assays.
293T cells were seeded in 96-well glass-bottom plates (Cellvis) and incubated overnight at 37 °C. Cells were either transfected with GST constructs (M2 domain mapping interaction studies) or infected with either A/WSN/33 (M2-TBC1D5 interaction studies), A/PR8 WT or A/PR8 ΔM2 (TBC1D5–Rab7 interaction studies) at MOI 3. After 1 h of incubation on ice to enable viral infection synchronization, the inoculum was removed and replaced with fresh serum-free media. At 16 h post-transfection or 12 h postinfection, cells were washed twice with 1× PBS, fixed using 4% paraformaldehyde (Boston BioProducts) and permeabilized with 0.5% Triton X-100 for 15 min at room temperature. Cells were then subjected to Duolink PLA Fluorescence protocol (Sigma). Briefly, cells were blocked with Duolink Blocking solution (Sigma) in a heated humidity chamber for 60 min at 37 °C. Rabbit anti-TBC1D5 (Atlas, HPA035125), mouse monoclonal anti-GST (Cell Signaling, no. 2624), mouse monoclonal anti-M2 E10 (Mount Sinai, in-house antibody) or mouse anti-Rab7 (Cell Signaling, no. 95746) were diluted in Duolink Antibody Diluent (Sigma), applied to the samples and incubated overnight at 4 °C. Cells were washed 3 × 10 min with PLA Wash Buffer A (Sigma) and incubated with Rabbit-PLUS and Mouse-MINUS PLA probes (Sigma) in a preheated humidity chamber for 1 h at 37 °C. Cells were washed 3 × 10 min with PLA Wash Buffer A (Sigma) and incubated with 1× Ligation solution (Sigma) in a preheated humidity chamber for 30 min at 37 °C. After 3 × 10 min with PLA Wash Buffer A (Sigma), cells were incubated with Amplification Solution (Sigma) in a preheated humidity chamber for 1 h at 40 min at 37 °C. Cells were washed 3 × 10 min with PLA Wash Buffer B (Sigma) and stained with Phalloidin-iFluor 488 (Abcam, 176753) and Hoechst 33342 (Thermo Fisher, H3570) for 20 min at room temperature. Images were acquired using the Nikon A1R HD confocal microscope and analysed using Fiji73. Single cells were defined based on Phalloiding staining and the number of dots within each region of interest (ROI) was quantified using Fiji’s analyse particle feature.
Rab7 activation status measurement.
293T cells were seeded in 96-well glass-bottom plates (Cellvis), transfected with GFP-Rab7 WT and incubated overnight at 37 °C. Cells were washed twice with 1× PBS and infected with A/PR8 WT or A/PR8 ΔM2 at MOI 3 or left uninfected. After 1 h of incubation at room temperature to enable virus absorption, the inoculum was removed and replaced with fresh serum-free media. At 16 h p.i., cells were washed twice with 1× PBS, fixed using 4% paraformaldehyde (Boston BioProducts) and permeabilized with 0.5% Triton X-100 for 15 min at room temperature. Cells were then treated with mouse monoclonal anti-Rab7 GTP overnight at 4 °C. Cells were washed for 3 × 10 min with PBS and incubated with anti-mouse Alexa 568 for 1 h at room temperature. Cells were then washed for 3 × 10 min with PBS and stained with Phalloidin-iFluor 488 (Abcam, 176753) and Hoechst 33342 (Thermo Fisher, H3570) for 20 min at room temperature. Images were acquired using the Nikon A1R HD confocal microscope. Images were projected in Z and analysed for vesicle segmentation and quantification with Icy v.2.0.3.0 (http://icy.bioimageanalysis.com) with the procedures published in ref.74. Briefly, individual cell nuclei were identified using the HK-means tool to create nuclear masks, following which, we used the active contours plugin to enlarge the ROIs/ masks around the individual nucleus in the Rab7-GFP channel to enlarge the masks to their intracellular vesicles. This second method dilates the masks only for Rab7-GFP positive cells (Extended Data Fig. 7f). The created masks in both HK-means step and active contours were visually inspected for correct segmentation. The individual cell, individual fluorescence channel quantification of total cells and Rab7-GFP positive cells was exported from the software then plotted as normalized (to minimum, maximum per biological condition) Rab7/GTP-Rab7 ratio.
PPMO synthesis and design.
Peptide-conjugated PPMOs were synthesized in the Moulton Lab at Oregon State University. For each PPMO, the cell-penetrating peptide (RXR)4 (where R is arginine and X is 6-aminohexanoic acid) was covalently conjugated to a morpholino oligomer (Gene Tools) at the 3′ end through a non-cleavable linker, by methods described previously75. Two PPMOs targeting TBC1D5 were generated: (1) TBC1D5 AUG, designed to target the translation start site region of the TBC1D5 mRNA (morpholino sequence: TCAGACACAGACTTATACATTGCAT) and (2) TBC1D5 e3i3, which targets the splice site between exon 3 and intron 3 of the TBC1D5 pre-mRNA (morpholino sequence: CTGCATCTGCACAGAAAACTTACCT). In addition, an NTC PPMO was designed (morpholino sequence: CCTCTTACCTCAGTTACAATTTATA), having little homology to mouse transcripts or influenza viral sequences.
Animal experiments.
Mice used in this study were purchased from the Jackson Laboratories. Mice were 5 weeks old, belonging to the BALB/c strain and all female mice were housed at 21 °C, with humidity of 35% and 12 h dark/ 12 h light cycle. Mice were anaesthetized by intraperitoneal injection of a mixture of ketamine and xylazine (100 μg and 5 μg per gram of body weight), before intranasal administration of either PBS or 100 μg of PPMO combination (50 μg of PPMO TBC1D5 AUG and 50 μg of PPMO TBC1D5 e3i3) in 40 μl of 1× PBS (the equivalent of ~5 mg kg−1) on day 2 and day 1. On day 0, mice were challenged intranasally with 40 p.f.u. A/PR8 IAV (LD50 = 50 p.f.u.) in 40 μl of PBS. Mice were monitored daily for weight loss and clinical signs. Mouse lungs were harvested on day 3 and day 6 p.i. to evaluate viral titres (n = 5). Lung homogenates were prepared using a FastPrep24 system (MP Biomedicals). After addition of 800 μl of PBS containing 0.3% BSA, lungs were subjected to two rounds of mechanical treatment for 10 s each at 6.5 m s−1. Tissue debris was removed by low-speed centrifugation and virus titres in supernatants were determined by plaque assay. A group of mice (n = 5) was monitored for survival at 14 d p.i.
Ethics statement.
All research studies involving the use of animals were reviewed and approved by the Institutional Animal Care and Use Committees of the Icahn School of Medicine at Mount Sinai, New York and were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals.
Statistics.
Data distribution was assumed to be normal but this was not formally tested. Statistical parameters including the exact value of n, dispersion and precision measures (mean ± s.d. or s.e.m.) and statistical significance are reported in the figures and figure legends. Statistical significance between groups was determined using GraphPad Prism v.8.0 (GraphPad) and the test used is indicated in the figure legends.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Global genetic screen to identify host-restriction factors for IAV.
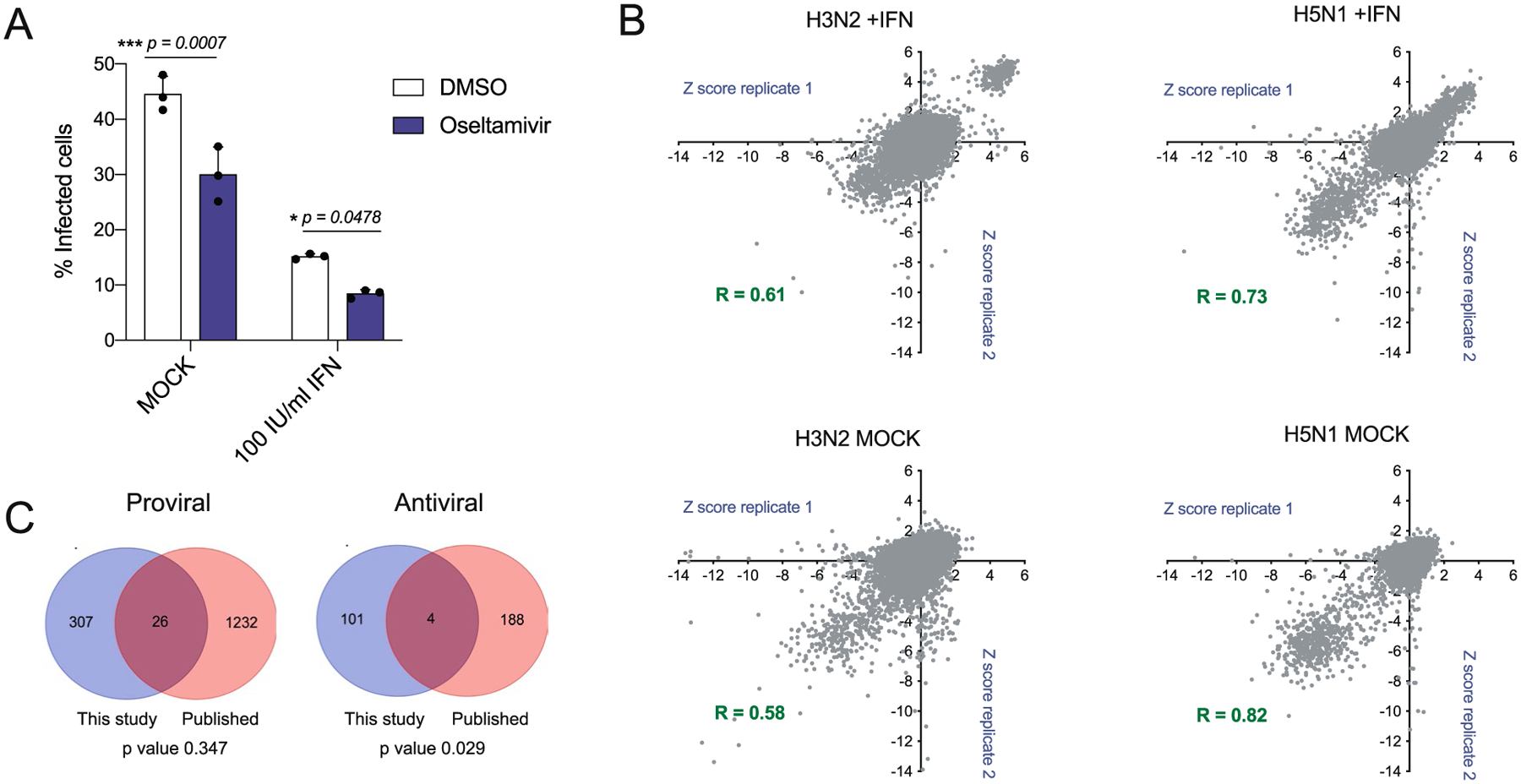
(a) THP-1 cells were seeded overnight and then treated with 100 IU/ml universal interferon or mock-treated for 6 h. Cells were then treated with 1 μM oseltamivir for 2 h before infection with A/Wyoming/03/03 H3N2 (MOI = 0.50). At 24 h post-infection the percentage (%) of infected cells was calculated based on DAPI staining and viral nucleoprotein (NP) immunostaining. Data show mean ± SD from one representative experiment in triplicate (n = 3) of at least two independent experiments. Statistical significance was calculated using two-way ANOVA with Sidak’s multiple comparison test. (b) Correlation plots of z-score values for genome-wide siRNA screens of IFN- or mock-treated THP-1 cells infected with H3N2 or H5N1 IAVs. (c) Venn diagram shows the overlap between the genome-wide siRNA screen conducted in this study and previously published proviral (left) or antiviral (right) cellular factors identified by Tripathi, Pohl et al.7, Karlas et al.,4, Brass et al.,1, Konig et al.5, 2010, Shapira et al.6, 2010, Han et al.2 and Watanabe et al.,8.
Extended Data Fig. 2 |. Global analysis of IAV cellular restriction.
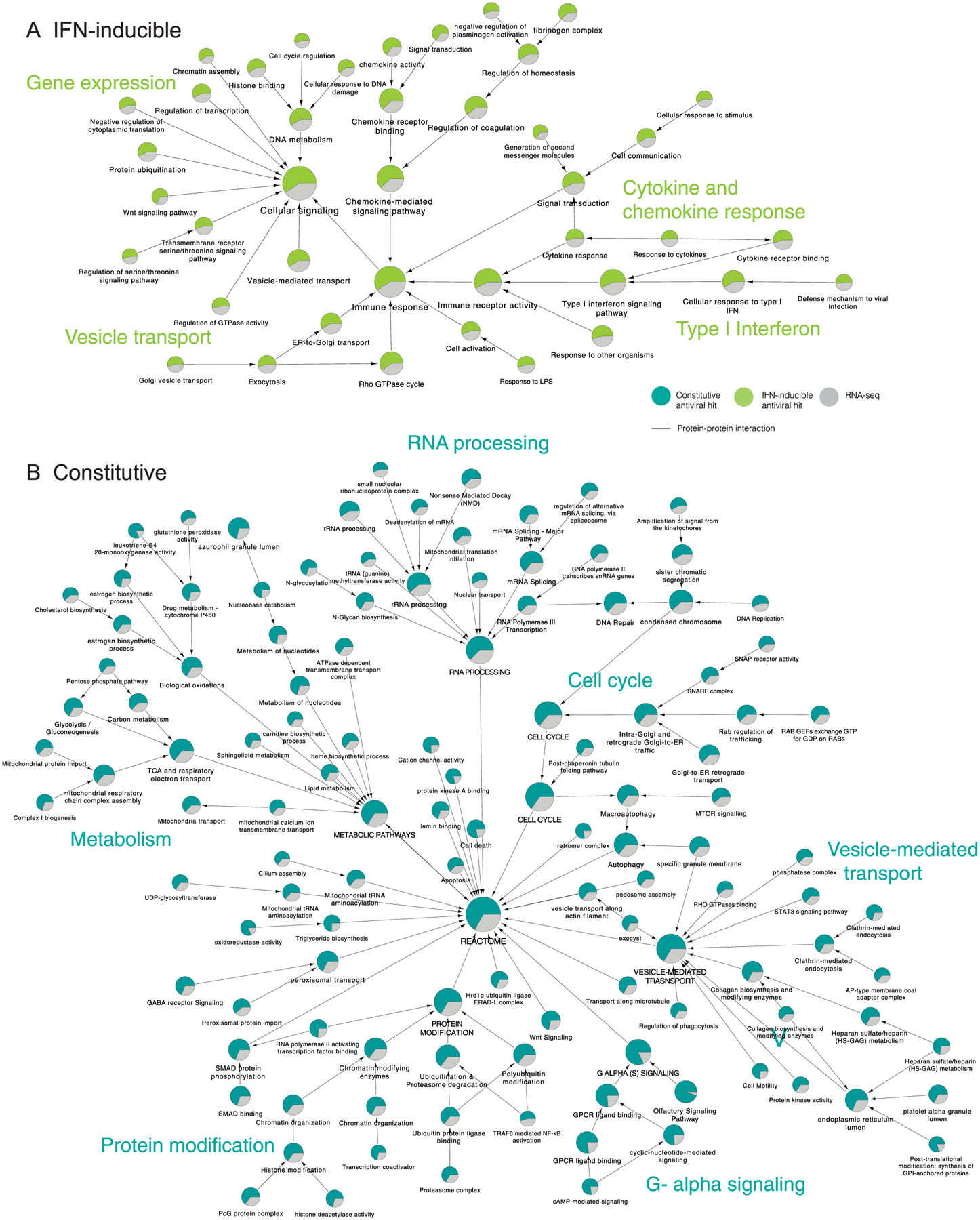
The list of factors identified by siRNA screening (z-score ≥ 0.5), or RNA-seq (log2FC ≥ 1.0 or ≤ −1.0 and P value < 0.005) were subjected to supervised community detection71,76. The resultant hierarchy is shown. Here, each node represents a community of densely interconnected proteins, and each edge (arrow) denotes containment of one community (edge target) by another (edge source). Enriched biological processes are indicated. The percentage of each community that corresponds to siRNA hits is shown in green, and RNA-seq in grey. Nodes indicate proteins, and edges indicate interactions as defined by STRING (High Confidence (Score ≥ 0.7), available at NDEx. (a) Hierarchy of IFN-inducible antiviral factors. (b) Hierarchy of constitutive expressed antiviral factors.
Extended Data Fig. 3 |. Prioritized antiviral factors.
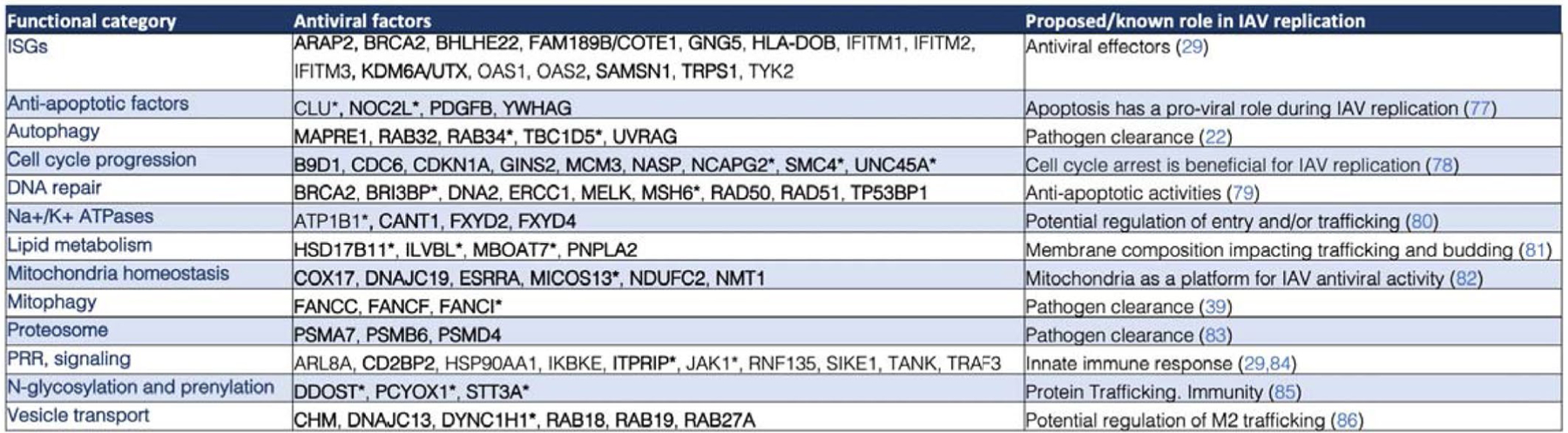
Prioritized antiviral factors clustered by functional category and proposed/known role in IAV replication. Factors with * represent those identified as M2 interactors. Factors in bold represent those not previously associated with IAV restriction24,29,39,42,77–86.
Extended Data Fig. 4 |. TBC1D5 restricts IAV replication and growth in vitro, ex vivo and in vivo.
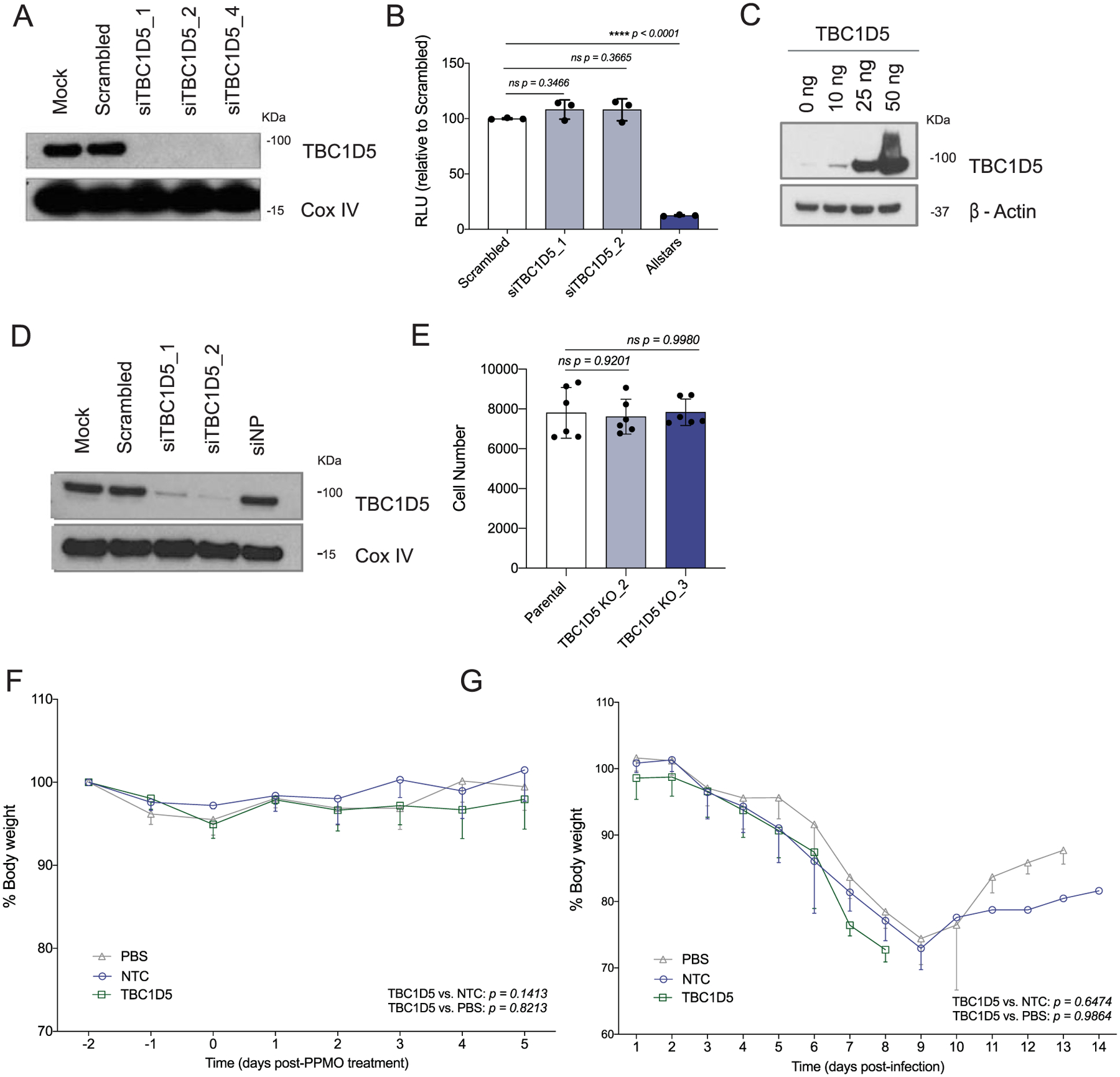
A549 cells were transfected with indicated siRNAs. At 48 h post-transfection, cells were (a) subjected to SDS–PAGE and immunoblotted using antibodies specific for TBC1D5 and β-actin (loading control). Blot is representative of two independent experiments, or (b) Cell viability was assessed using Cell Titer Glo and compared to scrambled (non-targeting, negative control) and Allstars (toxic siRNA, positive control). Data show mean ± SD from one representative experiment in triplicate (n = 3) of at least two independent experiments. (c) A549 cells were transfected with a plasmid encoding TBC1D5 (0–50 ng) for 36 h. Cells were then subjected to SDS–PAGE and immunoblotted using antibodies specific for TBC1D5 and β-actin. Blot is representative of two independent experiments. (d) HTBE cells were transfected for 36 h with indicated siRNAs. At 48 h post-transfection, cell lysates were subjected to SDS–PAGE and immunoblotted using antibodies specific for TBC1D5 and Cox IV (loading control). Blot is representative of two independent experiments. (e) A549 parental and two TBC1D5 KO clones cell numbers were determined 48 h post-seeding using image-based analysis of DAPI (nuclei) staining. Data show mean ± SD from one representative experiment in triplicate (n = 3) of at least two independent experiments. Five-week old female BALB/c mice were administered 100 μg PBS, NTC or TBC1D5 PPMOs (equivalent of 5 mg/kg) intranasally for 2 consecutive days and (f) Mice were monitored over the course of 5 days in the absence of IAV infection to evaluate PPMO-derived cytotoxicity. Data represent percent body weight ± SD from 2 independent experiments each with 3 mice per condition (n = 6). (g) Mice were infected with A/Puerto Rico/8/34 (40 p.f.u.) intranasally and were monitored for body weight over the course of 14 days. Graphs show percent body weight ± SD from 2 independent experiments each with 15 mice per condition (n = 30). (b, e) Statistical significance was calculated using one-way ANOVA with Dunnett’s post hoc test, or Tukey’s multiple comparison post hoc (f, g).
Extended Data Fig. 5 |. SDS–PAGe analysis of GST-tagged M2 constructs.
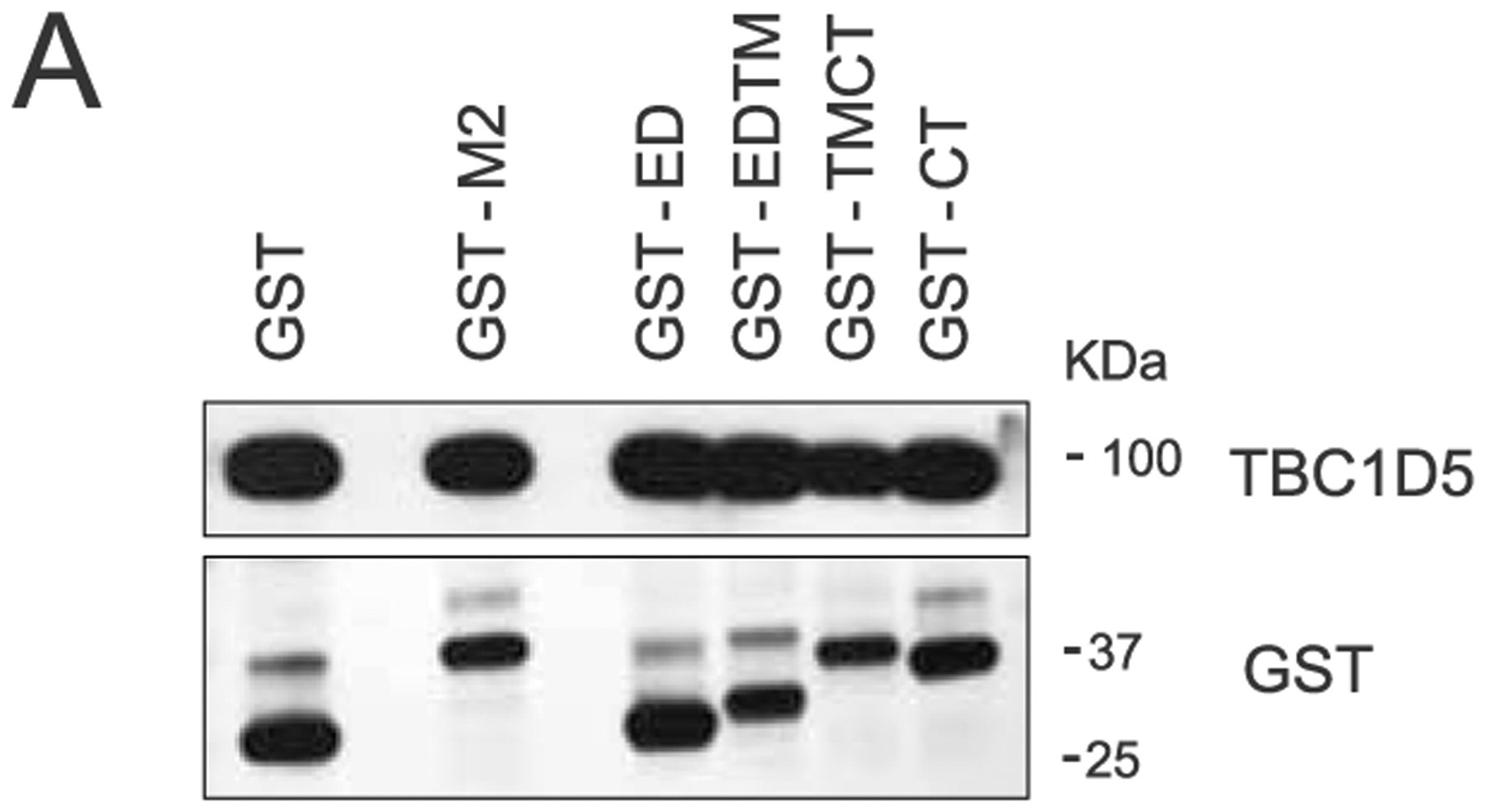
(a) 293 T cells were transfected with a series of N-terminally GST-tagged M2 constructs. At 16 h post-transfection, cells were lysed and levels of TBC1D5, and GST were analysed using SDS–PAGE. Blot is representative of two independent experiments.
Extended Data Fig. 6 |. TBC1D5 promotes lysosomal targeting of M2 protein.
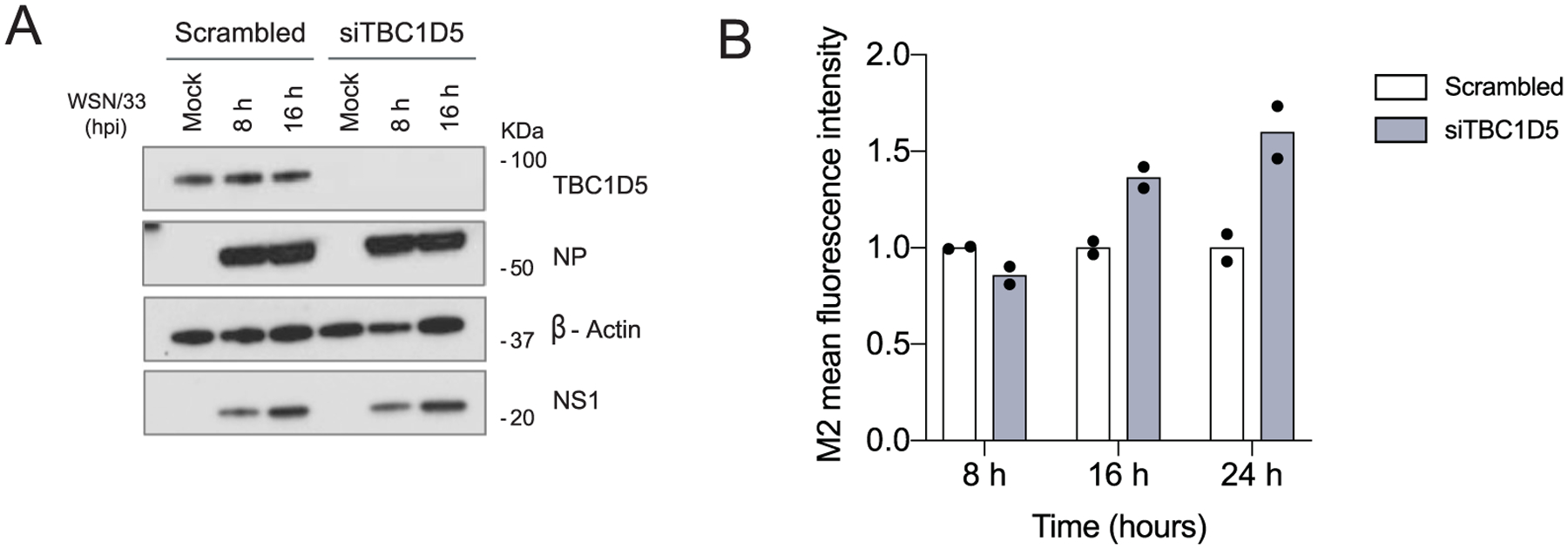
(a) 293 T cells were treated with negative control scrambled siRNA or siTBC1D5 for 48 h. Cells were then infected with A/WSN/33 (MOI 2), and at 8 and 16 h p.i. cells were lysed and levels of TBC1D5, NS1, and β-actin were analysed using SDS–PAGE. Blot is representative of two independent experiments. (b) 293 T cells were transfected with indicated siRNAs followed by infection with A/WSN/33 (MOI 1). At 8, 16 and 24 h p.i., cells were subjected to immunolabeling with anti-M2 in the absence of permeabilization agent (surface M2), and M2 relative fluorescence mean intensity levels were recorded by flow cytometry. Data represent mean ± s.d. of two independent experiments (n = 2). Statistical significance was calculated using two-way ANOVA with Bonferroni’s multiple comparisons post hoc test.
Extended Data Fig. 7 |. IAV M2 protein abrogates TBC1D5 and Rab7 interaction.
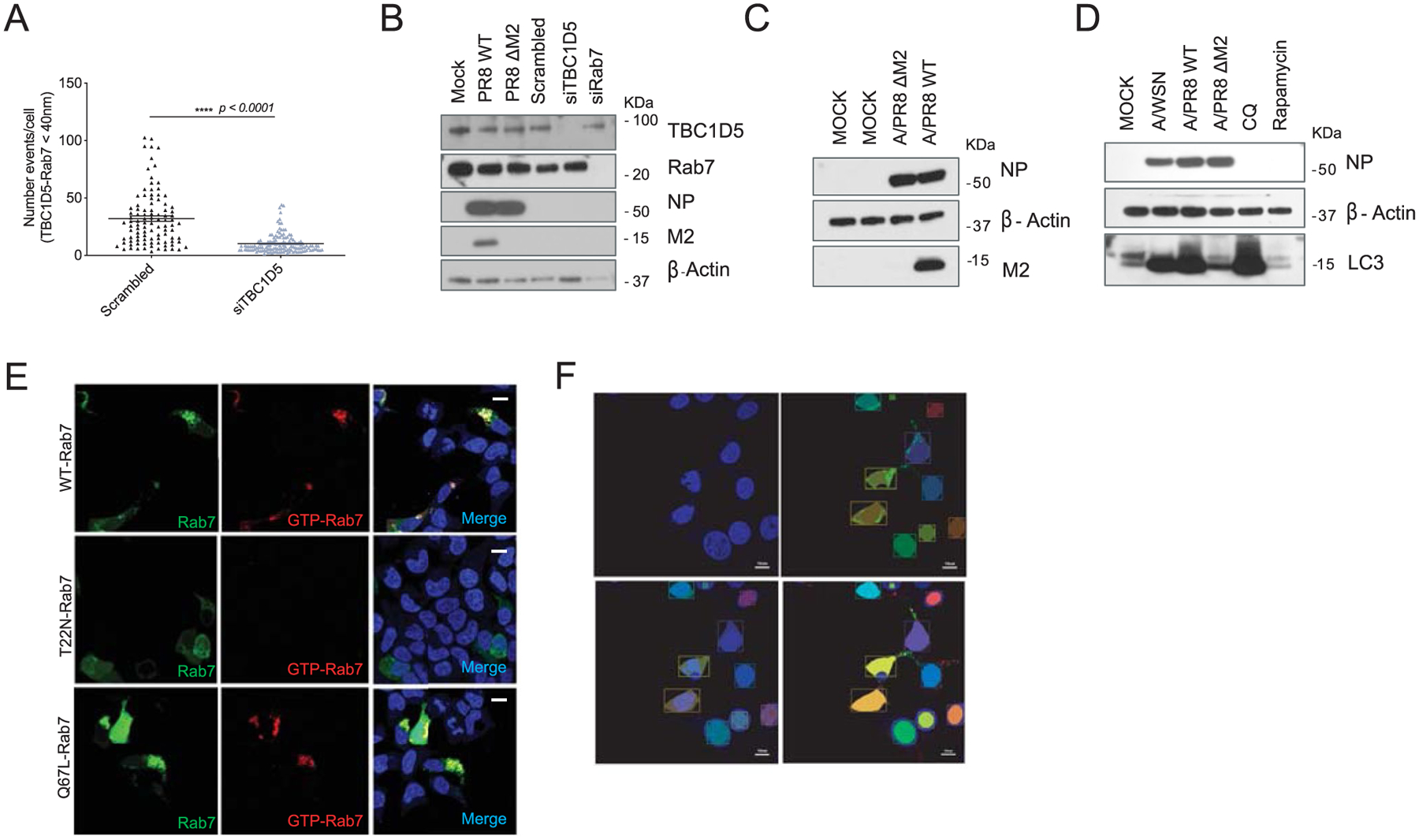
(a) 293 T cells were transfected with scrambled or TBC1D5 siRNAs. At 48 h post-transfection, cells were subjected to proximity ligand assays (PLA) staining. Quantification of number of PLA signal events where TBC1D5 proteins interact with Rab7. Data show mean ± s.d. from one representative experiment of at least two independent experiments where at least 50 cells per condition (n = 50) were quantified. Statistical significance was calculated using unpaired two-tailed Student’s t-test. (b) In parallel to PLA experiments (Fig. 6d,e) 293 T cells were subjected to transfection with indicated siRNAs for 48 h, or infected with A/PR8 WT or A/PR8 ΔM2 (MOI 3) for 18 h. Cells were then lysed and levels of TBC1D5, Rab7, NP, M2 and β-actin analysed using SDS–PAGE. Blot is representative of two independent experiments. (c) 293 T cells were mock treated, infected with A/PR8 WT or A/PR8 ΔM2 (MOI 3). At 18 h p.i. cells were lysed and levels of NP, β-actin and M2 were analysed using SDS–PAGE. Blot is representative of two independent experiments. (d) 293 T cells were mock-treated, infected with A/WSN/33, A/PR8 WT or A/PR8 ΔM2 (MOI 3), or treated with 100 μM chloroquine (CQ) or 1 μM Rapamycin for 18 h. Cells were then lysed and levels of NP, β-actin and LC3 were analysed using SDS–PAGE. Blot is representative of two independent experiments. (e) To test the specificity of the GTP-Rab7 antibody, Total- and GTP-bound Rab7 intensities were simultaneously acquired in cells that are either transfected with eGFP-Rab7 WT, a dominant negative Rab7 mutant with higher GDP affinity (eGFP-Rab7 T22N), or a constitutively active GTP-bound Rab7 mutant (eGFP-Rab7 Q67L) for 24 h. Representative images from two independent experiments show Rab7 (GFP, green) and GTP-Rab7 (red) staining. Scale bar = 10 μm. (f) Representation of generation of mask to detect nuclei and cells positive for eGFP-Rab7 signal (see material and methods). Images are representative of two independent experiments. Scale bar = 10 μm.
Supplementary Material
Acknowledgements
We would like to thank I. Dikic and C. Progida for providing the GFP-TBC1D5 constructs and W. Barclay for providing the influenza-like minigenome reporter. We also thank A. M. O’Neill for his valuable support with the microscopy images. We thank D. Chen and S. Heynen-Genel in the Sanford Burnham Prebys High Content Microscopy Core for their technical assistance with the IC200 imaging system. Thanks to M. Hansen and I. Marazzi for their insightful comments on the manuscript. This work was supported by NIH/NIAID research grant nos. U19AI106754 and U19AI135972 to C.B., R.A.A., N.J.K., A.G.-S. and S.K.C. This work was also partly supported by NIAID grant no. U19AI117873 and by CRIP (Centre for Research on Influenza Pathogenesis), a NIAID-funded Center of Excellence for Influenza Research and Surveillance (CEIRS, contract no. HHSN272201400008C) to A.G.-S. This work was also supported by a generous grant from the James B. Pendleton Charitable Trust. K.M.H. was supported by the National Science Foundation (1650113). Support for J.F.H. was provided by the Gilead Sciences Research Scholars Program in HIV and NIH grant no. K22 AI136691.
Competing interests
The N.J.K. Laboratory has received research support from Vir Biotechnology and F. Hoffmann-La Roche. N.J.K. has consulting agreements with the Icahn School of Medicine at Mount Sinai, New York, Maze Therapeutics and Interline Therapeutics, is a shareholder of Tenaya Therapeutics and has received stocks from Maze Therapeutics and Interline Therapeutics. The remaining authors declare no competing interests.
Footnotes
Extended data is available for this paper at https://doi.org/10.1038/s41564-021-00964-2.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41564-021-00964-2.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
The MDM RNA-seq data used in this study are deposited in the Gene Expression Omnibus (GEO) database repository under the accession number GSE97672. The genome-wide siRNA screen data generated in this study have been deposited to figshare (https://doi.org/10.6084/m9.figshare.13826429). AP-MS data generated in this study have been deposited into the Center for Computational MS (Massive): https://massive.ucsd.edu/ProteoSAFe/private-dataset.jsp?task=82154043f1ca4b0c9e22c10aa091f476 (password, pandemic). Source data are provided with this paper.
References
- 1.Brass AL et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and Dengue virus. Cell 139, 1243–1254 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han J et al. Genome-wide CRISPR/Cas9 screen identifies host factors essential for influenza virus replication. Cell Rep. 23, 596–607 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hao L et al. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454, 890–893 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karlas A et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463, 818–822 (2010). [DOI] [PubMed] [Google Scholar]
- 5.König R et al. Human host factors required for influenza virus replication. Nature 463, 813–817 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shapira SD et al. A physical and regulatory map of host–influenza interactions reveals pathways in H1N1 infection. Cell 139, 1255–1267 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tripathi S et al. Meta- and orthogonal integration of influenza ‘OMICs’ data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe T et al. Influenza virus–host interactome screen as a platform for antiviral drug development. Cell Host Microbe 16, 795–805 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li B et al. Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nat. Commun 11, 164 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lesch M et al. RNAi-based small molecule repositioning reveals clinically approved urea-based kinase inhibitors as broadly active antivirals. PLoS Pathog. 15, e1007601 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watanabe T, Watanabe S & Kawaoka Y Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 7, 427–439 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heaton BE et al. A CRISPR activation screen identifies a pan-avian influenza virus inhibitory host factor. Cell Rep. 20, 1503–1512 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Randow F & Youle RJ Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 15, 403–411 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He C & Klionsky DJ Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet 43, 67–93 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizushima N et al. A protein conjugation system essential for autophagy. Nature 395, 395–398 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Stroupe C This is the end: regulation of Rab7 nucleotide binding in endolysosomal trafficking and autophagy. Front. Cell Dev. Biol 6, 129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerra F & Bucci C Multiple roles of the small GTPase Rab7. Cells 5, 34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutierrez MG, Munafó DB, Berón W & Colombo MI Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci 117, 2687–2697 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Pan X, Eathiraj S, Munson M & Lambright DG TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature 442, 303–306 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Seaman MNJ, Harbour ME, Tattersall D, Read E & Bright N Membrane recruitment of the cargo-selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-GAP TBC1D5. J. Cell Sci 122, 2371–2382 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purushothaman LK, Arlt H, Kuhlee A, Raunser S & Ungermann C Retromer-driven membrane tubulation separates endosomal recycling from Rab7/Ypt7-dependent fusion. Mol. Biol. Cell 28, 783–791 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi Y, Bowman JW & Jung JU Autophagy during viral infection—a double-edged sword. Nat. Rev. Microbiol 16, 341–354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deretic V & Levine B Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5, 527–549 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gannagé M et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6, 367–380 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakabe S et al. Cytokine production by primary human macrophages infected with highly pathogenic H5N1 or pandemic H1N1 2009 influenza viruses. J. Gen. Virol 92, 1428–1434 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J-H, Chung TDY & Oldenburg KR A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen 4, 67–73 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Heinz S et al. Transcription elongation can affect genome 3D structure. Cell 174, 1522–1536 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rusinova I et al. INTERFEROME v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res 41, D1040–D1046 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schoggins JW et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antoniak S The coagulation system in host defense. Res. Pract. Thromb. Haemost 2, 549–557 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhatnagar S, Shinagawa K, Castellino FJ & Schorey JS Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood 110, 3234–3244 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wada M, Lokugamage KG, Nakagawa K, Narayanan K & Makino S Interplay between coronavirus, a cytoplasmic RNA virus, and nonsense-mediated mRNA decay pathway. Proc. Natl Acad. Sci. USA 115, E10157–E10166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eddowes LA et al. Antiviral activity of bone morphogenetic proteins and activins. Nat. Microbiol 4, 339–351 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu M-Y & Lu J-H Autophagy and macrophage functions: inflammatory response and phagocytosis. Cells 9, 70 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manzoor R, Igarashi M & Takada A Influenza A virus M2 protein: roles from ingress to egress. Int. J. Mol. Sci 18, 2649 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jäger S et al. Global landscape of HIV–human protein complexes. Nature 481, 365–370 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldenberg NM, Grinstein S & Silverman M Golgi-bound Rab34 is a novel member of the secretory pathway. Mol. Biol. Cell 18, 4762–4771 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin-Sancho L et al. Functional landscape of SARS-CoV-2 cellular restriction. Mol. Cell 81, 2656–2668 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sumpter R et al. Fanconi anemia proteins function in mitophagy and immunity. Cell 165, 867–881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Onomoto K, Onoguchi K & Yoneyama M Regulation of RIG-I-like receptor-mediated signaling: interaction between host and viral factors. Cell. Mol. Immunol 18, 539–555 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Au PYB et al. The oncogene PDGF-B provides a key switch from cell death to survival induced by TNF. Oncogene 24, 3196–3205 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Akula MK et al. Protein prenylation restrains innate immunity by inhibiting Rac1 effector interactions. Nat. Commun 10, 3975 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sato Y et al. Ubiquitin-specific protease 9X in host cells interacts with herpes simplex virus 1 ICP0. J. Vet. Med. Sci 78, 405–410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beale R et al. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe 15, 239–247 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rajsbaum R Intranasal delivery of peptide-morpholinos to knockdown influenza host factors in mice. Methods Mol. Biol 1565, 191–199 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Zhu Y, Choi SH & Shah K Multifunctional receptor-targeting antibodies for cancer therapy. Lancet Oncol. 16, e543–e554 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Jimenez-Orgaz A et al. Control of RAB7 activity and localization through the retromer–TBC1D5 complex enables RAB7-dependent mitophagy. EMBO J. 37, 235–254 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Popovic D et al. Rab GTPase-activating proteins in autophagy: regulation of endocytic and autophagy pathways by direct binding to human ATG8 modifiers. Mol. Cell Biol 32, 1733–1744 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moncorgé O, Mura M & Barclay WS Evidence for avian and human host cell factors that affect the activity of influenza virus polymerase. J. Virol 84, 9978–9986 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren Y et al. Proton channel activity of influenza A virus matrix protein 2 contributes to autophagy arrest. J. Virol 90, 591–598 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jia D et al. Structural and mechanistic insights into regulation of the retromer coat by TBC1d5. Nat. Commun 7, 13305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimura S, Noda T & Yoshimori T Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Toyofuku T, Morimoto K, Sasawatari S & Kumanogoh A Leucine-rich repeat kinase 1 regulates autophagy through turning on TBC1D2-dependent Rab7 inactivation. Mol. Cell Biol 35, 3044–3058 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Z et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 27, 883–890 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gordon DE et al. A SARS-CoV-2 protein interaction map reveals drug targets for drug repurposing. Nature 583, 459–468 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rouse BT & Sehrawat S Immunity and immunopathology to viruses: what decides the outcome? Nat. Rev. Immunol 10, 514–526 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Isaacs CE & Xu W Theaflavin-3,3′-digallate and lactic acid combinations reduce herpes simplex virus infectivity. Antimicrob. Agents Chemother 57, 3806–3814 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tyssen D et al. Anti-HIV-1 activity of lactic acid in human cervicovaginal fluid. Msphere 10.1128/mSphere.00055-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hauler F, Mallery DL, McEwan WA, Bidgood SR & James LC AAA ATPase p97/VCP is essential for TRIM21-mediated virus neutralization. Proc. Natl Acad. Sci. USA 109, 19733–19738 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fletcher K et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. 37, e97840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tooze SA, Abada A & Elazar Z Endocytosis and autophagy: exploitation or cooperation? Cold Spring Harb. Perspect. Biol 6, a018358 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen K, Healy MD & Collins BM Towards a molecular understanding of endosomal trafficking by retromer and retriever. Traffic 20, 465–478 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Jäger S et al. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci 117, 4837–4848 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Bärlocher K et al. Structural insights into Legionella RidL-Vps29 retromer subunit interaction reveal displacement of the regulator TBC1D5. Nat. Commun 8, 1543 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Romano-Moreno M et al. Molecular mechanism for the subversion of the retromer coat by the Legionella effector RidL. Proc. Natl Acad. Sci. USA 114, E11151–E11160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pankiv S et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end–directed vesicle transport. J. Cell Biol 188, 253–269 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Steel J et al. Live attenuated influenza viruses containing NS1 truncations as vaccine candidates against H5N1 highly pathogenic avian influenza. J. Virol 83, 1742–1753 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rodriguez-Frandsen A et al. Viral determinants in H5N1 influenza A virus enable productive infection of HeLa cells. J. Virol 10.1128/JVI.01410-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singhal A et al. Multiscale community detection in Cytoscape. PLoS Comput. Biol 16, e1008239 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shannon P Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cauldwell AV, Moncorgé O & Barclay WS Unstable polymerase–NP interaction is not responsible for avian influenza polymerase restriction in human cells. J. Virol 10.1128/JVI.02597-12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dufour A, Thibeaux R, Labruyere E, Guillen N & Olivo-Marin J-C 3-D active meshes: fast discrete deformable models for cell tracking in 3-D time-lapse microscopy. IEEE Trans. Image Process 20, 1925–1937 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Abes S et al. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J. Control. Release 116, 304–313 (2006). [DOI] [PubMed] [Google Scholar]
- 76.Carlin DE, Demchak B, Pratt D, Sage E & Ideker T Network propagation in the Cytoscape cyberinfrastructure. PLoS Comput. Biol 13, e1005598 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Atkin-Smith GK, Duan M, Chen W & Poon IKH The induction and consequences of influenza A virus-induced cell death. Cell Death Dis. 9, 1002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.He Y et al. Influenza A virus replication induces cell cycle arrest in G0/G1 phase. J. Virol 84, 12832–12840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Norbury CJ & Zhivotovsky B DNA damage-induced apoptosis. Oncogene 23, 2797–2808 (2004). [DOI] [PubMed] [Google Scholar]
- 80.Clausen MJV & Poulsen H in Metallomics and the Cell (ed. Banci L) 41–67 (Springer, 2013). [Google Scholar]
- 81.Heaton NS & Randall G Multifaceted roles for lipids in viral infection. Trends Microbiol. 19, 368–375 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tiku V, Tan M-W & Dikic I Mitochondrial functions in infection and immunity. Trends Cell Biol. 30, 263–275 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McCarthy MK & Weinberg JB The immunoproteasome and viral infection: a complex regulator of inflammation. Front. Microbiol 10.3389/fmicb.2015.00021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mogensen TH Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev 22, 240–273 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vigerust DJ & Shepherd VL Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 15, 211–218 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Robinson M, Schor S, Barouch-Bentov R & Einav S Viral journeys on the intracellular highways. Cell Mol. Life Sci 75, 3693–3714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MDM RNA-seq data used in this study are deposited in the Gene Expression Omnibus (GEO) database repository under the accession number GSE97672. The genome-wide siRNA screen data generated in this study have been deposited to figshare (https://doi.org/10.6084/m9.figshare.13826429). AP-MS data generated in this study have been deposited into the Center for Computational MS (Massive): https://massive.ucsd.edu/ProteoSAFe/private-dataset.jsp?task=82154043f1ca4b0c9e22c10aa091f476 (password, pandemic). Source data are provided with this paper.