

Abstract
Chemical probes for epigenetic proteins are essential tools for dissecting the molecular mechanisms for gene regulation and therapeutic development. The bromodomain and extra-terminal (BET) proteins are master transcriptional regulators. Despite promising therapeutic targets, selective small molecule inhibitors for a single bromodomain remain an unmet goal due to their high sequence similarity. Here, we address this challenge via a structure–activity relationship study using 1,4,5-trisubstituted imidazoles against the BRD4 N-terminal bromodomain (D1). Leading compounds 26 and 30 have 15 and 18 nM affinity against BRD4 D1 and over 500-fold selectivity against BRD2 D1 and BRD4 D2 via ITC. Broader BET selectivity was confirmed by fluorescence anisotropy, thermal shift, and CETSA. Despite BRD4 engagement, BRD4 D1 inhibition was unable to reduce c-Myc expression at low concentration in multiple myeloma cells. Conversely, for inflammation, IL-8 and chemokine downregulation were observed. These results provide new design rules for selective inhibitors of an individual BET bromodomain.
Keywords: 7RXR, 4; 7RXS, 20; 7RXT, 22; 7R9C, 32
Graphical Abstract
INTRODUCTION
Bromodomains are conserved, approximately 110 amino acid structural motifs found in many epigenetic regulatory proteins from humans to flies.1 The canonical function of bromodomains is to bind to N-ε-acylated-lysine residues on histones and transcription factors. These often-transient interactions facilitate localization of bromodomain-containing proteins to chromatin and within transcriptional complexes for regulating gene transcription.2,3 Several bromodomain-containing proteins have established roles in aberrant transcription, leading to a wide range of pathologies including cancer, heart disease, and inflammation.2,4–6 Thus, inhibiting the protein–protein interactions between bromodomains and acylated proteins has become a heavily investigated research area to both understand the molecular mechanisms associated with these interactions and to treat disease.
There are 61 human bromodomains classified into eight subgroups based on the structure and sequence similarity.3 The bromodomain and extra-terminal (BET) family proteins, BRD2, 3, 4, and the testis-specific protein BRDT, are some of the most broadly studied bromodomain-containing proteins. BET proteins minimally contain two tandem bromodomains, D1 and D2, and an extra-terminal domain.3 Within the BET family, genetics studies have associated BRD4 with multiple disease states. Examples of BRD4-dependent mechanisms of disease include forming protein–protein interactions with acetylated RelA of NF-κB and recruiting P-TEFb to activate the expression of related inflammatory cytokines and chemokines.7 Additionally, BRD4 can regulate MYC oncogene expression via formation of transcriptional complexes at superenhancer regions.8 However, clinical studies in oncology have revealed pan-BET inhibition leads to dose-limiting toxicity, primarily through gastrointestinal effects and thrombocytopenia,9 the latter of which can be affected by BRD2 and BRD3 which play compensatory roles.10–12 As such, selective BRD4 inhibition over pan-BET inhibition could be a potential therapeutic strategy for inflammatory diseases and cancers with fewer side effects. However, the high structure and sequence similarity across the BET bromodomains has made such inhibitors challenging to develop to test this hypothesis.
To address this challenge, structure-based design approaches have identified several useful design rules. GSK778, a potent pan-D1 inhibitor, was reported by Gilan et al.13 The pyrrolidinyl group interacts with a nonconserved Asp on D1s (His on D2s) via a water-bridged hydrogen bond (Figure 1A,B).13 Similar interactions were found by our recently reported triazole-based inhibitors, including DW34, which exhibit pan-D1 selectivity, with the exception of a high affinity interaction with BRD4 D2 (Figure 1A,C).14 Meanwhile, pan-BD2 inhibitors have been developed, including RVX-208,15 ABBV-744,16 and GSK046,13 and are effective in different disease models. These inhibitors are being pursued in clinical trials with reduced toxic side effects. RVX-208 showed efficacy in decreasing the major adverse cardiovascular event in patients with high-risk type 2 diabetes mellitus in a phase III clinical trial and received Breakthrough Therapy Designation from the U.S. Food and Drug Administration. ABBV-744 (Figure 1A) is a pan-D2 BET inhibitor.16 The 2,6-dimethylphenyl ether motif was installed to target Ile (D1) and Val (D2) differences. The ethyl amide group targets a nonconserved His residue on D2 (Asp on D1).16 ABBV-744 is in a phase I clinical trial to study its efficacy and pharmacokinetic profile in acute myeloid leukemia. As a rare example of a BRD4 D1 selective inhibitor, Liu et al. discovered BRD4 D1 selective inhibitor, ZL0516, with 8.5-fold selectivity over BRD4 D2.17 The hydroxyl group formed a water-bridged hydrogen bond with a unique Asn93 on BRD4 D1. ZL0516 can inhibit Toll-like receptor-induced inflammatory gene expression in human small airway epithelial cells.17
Figure 1.

(A) Structures of selective BET bromodomain inhibitors. Key motifs are shown in red. (B) Cocrystal structure of GSK778 (orange) with BRD4 D1 (PDB 6SWN). The pyrrolidyl ring forms a water-bridged hydrogen bond with Asp144 and Asp145. (C) Cocrystal structure of DW34 (green) with BRD4 D1 (PDB 7MLR). The N,N-dimethyl ethylamino group interacts with Asp144 and Asn140 via a water-bridged hydrogen bond. The WPF shelf is shown (residues 81–83). Select structured waters are indicated as red spheres. Hydrogen bonds are shown in yellow dash lines.
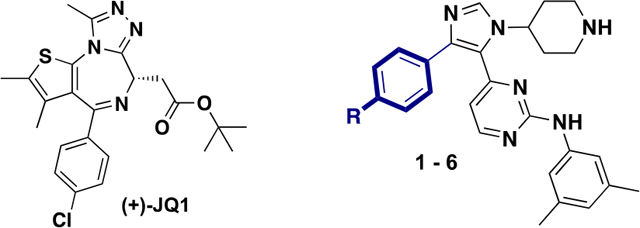
Our lab also reported a 1,4,5-trisubstituted imidazole analogue, derived from a p38α inhibitor scaffold as a BRD4 D1 selective inhibitor, subsequently referred to as UMN627 (Figure 1A).18 The 9–33-fold selectivity for BRD4 D1 over other BET bromodomains was in part obtained by displacement of structured waters in the binding pocket and reorganization of a nonconserved lysine residue in the D1 YNKP motif.18 Targeting conserved water molecules are a potential strategy of bromodomain drug design. However, only a few inhibitors have taken advantage of displacing structured waters for gaining selectivity.19–21 While minimally effective at reducing Myc expression in MM.1S cells, UMN627 reduces chemokine expression from inflammation in liver sinusoidal epithelial cells both in vitro and in vivo.18,22,23 While only an initial set of structure–activity relationship studies were pursued in our report, here we seek to develop more potent and selective inhibitors of BRD4 D1 while also removing residual kinase off-target activity for use as a chemical probe. Using structure-based design, competitive inhibition assays, and direct binding assays, we now report new design rules for increasing potency while improving selectivity for BRD4 D1. These designs have led to two highly potent and selective BRD4 D1 inhibitors. We further demonstrate their effectiveness as anti-inflammatory agents in two model cell lines.
RESULTS AND DISCUSSION
Targeting conserved water molecules within the bromodomain binding site is one mechanism for achieving bromodomain selectivity. For BET bromodomains, this has been supported both experimentally and computationally.18–20,24 One of the reasons for the unique selectivity of UMN627 toward BRD4 D1 is displacement of three conserved structured water molecules associated with in the binding pocket.18 These waters are predicted to be more readily displaced over the conserved water molecules BRD2 and BRD3.24 To further explore the binding pocket, four new analogues (3–6, Table 1) were synthesized using our previously reported protocols with different para-substituents of varying size and electronic properties.18 Competitive fluorescence anisotropy (FA) was used to determine the relative affinities of these analogues relative to two previously described inhibitors containing a para-fluoro group in 1, and a trifluoromethyl group in 2 (Table 1).18,25 (+)-JQ1 was used as a positive control. Halogen atoms (Cl, Br, and I) were first evaluated. The affinity was improved with a Cl atom (3, IC50 = 0.61 μM) relative to a F atom but not as potent as the trifluoromethyl group in 2. Replacing the chloro group with a Br atom improved affinity 3-fold (4, IC50 = 0.20 μM). Further improvement was observed when an iodo group was used (5, IC50 < 0.092 μM), reaching the sensitivity limit of our assay. The trend in this data series was consistent with a halogen-bonding interaction with BRD4 D1. To test steric effects on the affinity with a group lacking a sigma hole, a methoxy group was evaluated. In this case, the affinity was eroded (6, IC50 = 9.4 μM). These same trends were observed by an orthogonal competitive inhibition assay, AlphaScreen (Table 1) with 5 possessing the lowest IC50 of 0.20 μM against BRD4 D1.
Table 1.
IC50 Values of 1 and Analogues 2–6 against BRD4 D1 by Fluorescence Anisotropy and AlphaScreen
| |||
|---|---|---|---|
| compd | R | BRD4 D1 IC50 by FA (μM)a | BRD4 D1 IC50 by AlphaScreen (μM)b |
| (+)-JQ1 | <0.092c | 0.051 | |
| 1 25 | F | 11 ± 0.8 | 3.8 |
| 2 18 | CF3 | 0.31 ± 0.06 | 0.64 |
| 3 | Cl | 0.61 ± 0.09 | 1.3 |
| 4 | Br | 0.20 ± 0.02 | 0.44 |
| 5 | I | <0.092c | 0.20 |
| 6 | OCH3 | 9.4 ± 0.4 | 3.9 |
Data represents the mean and standard deviation of three experimental replicates.
Data represents the mean of two experimental replicates.
IC50 is less than half the concentration of bound protein used.
To better understand the molecular basis of the affinity improvement of 4 and 5, protein cocrystal structures were obtained with BRD4 D1 (Figure 2). Consistent with prior crystal structures, the N-3 atom on the imidazole ring formed a direct hydrogen bonding interaction with Asn140 and did not form a bridging water hydrogen bond to Tyr97 like most BET inhibitors.26 We previously identified an additional mechanism for selectivity targeting the flexible backbone of the YNKP motif for BET D1 domains over D2.18,25 Here, larger backbone movements were observed with 4 and an analogue of 5 that maintains the p-iodophenyl group, 20 (Figure 2B), compared with the previously reported analogue with a p-CF3 group (ΔCαN = 0.94 Å and ΔCαK = 1.7 Å; PDB 6WGX).18 In addition, the p-Br and p-I phenyl ring filled the binding pocket and displaced one more structured water relative to our previous reported inhibitor with a p-CF3 phenyl group.18 The backbone carbonyl of Met105 was predicted to serve as a sigma hole acceptor for halogen bonding. In these two cases, the distances from the oxygen of Met105 to the Br atom (3.25 Å) and the I atom (3.30 Å) were shorter than the sum of their van der Waals radii (Rvdw, Figure 2A).27 The C–Br–O and C–I–O bond angles, 154.4° and 157.6°, respectively, were close to linear and within the acceptable range for observable halogen bonds.27 Thus, we conclude, access to a deeper portion of the histone binding pocket allowed access to halogen-bonding interactions between 4, 20, and BRD4 D1 and supports the origin of the significant affinity improvement. Additional structured water displacement and further perturbation to the YNKP motif may also lead to improved selectivity, vide infra.
Figure 2.

(A) Halogen bonding interaction between 4 and an analogue of 5 (20) and Met105 on BRD4 D1. Conserved water molecules are shown in red (PDB 7RXR, 7RXS). (B) Backbone overlay of BRD4 D1 bound to H4 K5ac,K8ac (PBD 3UVW, violet) and bound to 20 (PDB 7RXS, gray). ΔCαN (the backbone Cα–Cα difference at Asn140 of BRD4 D1 with H4 K5ac,K8ac and with inhibitors) and ΔCαK (the backbone Cα–Cα difference at Lys141 of BRD4 D1 with H4 K5ac,K8ac and with inhibitors) were measured. The side chain of Lys141 was removed for clarity. The structure of 20 can be found in Table 4.
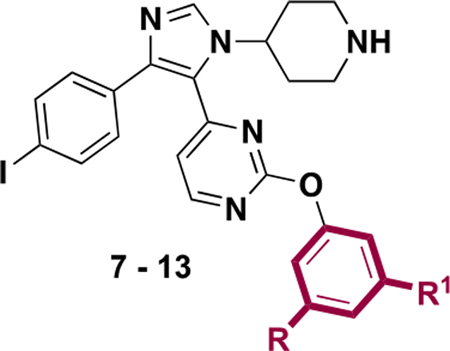
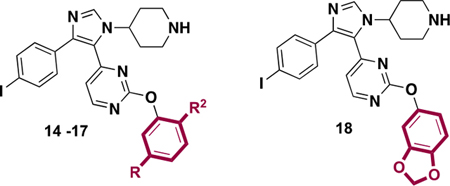
We next evaluated changes to substituents on the pyrimidine ring of our imidazole scaffold, which interacts with a hydrophobic portion of BRD4 D1 called the WPF shelf (Figure 1C). Previously, we reported a strategy to improve BRD4 D1 affinity, and D1 vs D2 selectivity, by using different aryl groups targeting the WPF shelf on a related scaffold.14 Here, several phenyl ethers with alkyl substituents of varying size and hydrophobicity were substituted on the pyrimidine ring (7–18, Tables 2 and 3). Surprisingly, more dramatic changes were observed on the imidazole than on our previously reported triazole scaffold.14 When the exocyclic NH of 5 was replaced by an ether bond (7, IC50 = 0.19 μM), the affinity against BRD4 D1 was reduced by 2.4-fold. Additionally, removing one methyl group led to a slight affinity decrease (9, IC50 = 0.50 μM), and the unsubstituted phenyl group (8) had a 13-fold affinity loss. Alternatively, larger R1 groups, ethyl- (10) and t-butyl- (11), improved affinity up to 0.11 μM. The asymmetric methyl ethyl-substitution had a >3.5-fold affinity increase relative to 7 that was comparable in affinity to our positive control (+)-JQ1 against BRD4 D1. Incorporating electron withdrawing atoms, such as a 3,5-dichloro group, decreased affinity relative to 7 (13, IC50 = 0.43 μM). Changing the substitution pattern of methyl groups (2,6-dimethyl vs 3,5-dimethyl) had modest impact on the affinity (14, IC50 = 0.27 μM). However, with the isopropyl group on the same position, a significant improvement relative to 7, was observed (15, IC50 < 0.092 μM). Affinities reduced sharply when polar ether or hydroxyl groups were installed (16, 17, 18). These results indicated that the aryl groups on the imidazole scaffold form significant hydrophobic interactions with the WPF shelf, which was not observed in our triazole-based inhibitor series.14
Table 2.
IC50 Values of Inhibitors 7–13 against BRD4 D1 by Fluorescence Anisotropy
| |||
|---|---|---|---|
| compd | R | R1 | BRD4 D1 IC50 by FA (μM)a |
| (+)-JQ1 | <0.092b | ||
| 7 | Me | Me | 0.19 ± 0.02 |
| 8 | H | H | 2.4 ± 0.1 |
| 9 | H | Me | 0.50 ± 0.04 |
| 10 | H | Et | 0.20 ± 0.01 |
| 11 | H | t-Bu | 0.11 ± 0.03 |
| 12 | Me | Et | <0.092b |
| 13 | Cl | Cl | 0.43 ± 0.05 |
Data represents the mean and standard deviation of three experimental replicates.
IC50 is less than half the concentration of bound BRD4 D1 used.
Table 3.
IC50 Values of Inhibitors 14–18 against BRD4 D1 by Fluorescence Anisotropy
| |||
|---|---|---|---|
| compd | R | R2 | BRD4 D1 IC50 by FA (μM)a |
| (+)-JQ1 | <0.092b | ||
| 14 | Me | Me | 0.27 ± 0.04 |
| 15 | i-Pr | Me | <0.092b |
| 16 | H | OH | 3.0 ± 0.1 |
| 17 | H | OMe | 2.4 ± 0.1 |
| 18 | 2.7 ± 0.3 | ||
Data represents the mean and standard deviation of three experimental replicates.
IC50 is less than half the concentration of bound BRD4 D1 used.
Having significantly increased the affinity from pyrimidine substitutions, we next turned toward modifications of the N-1 substituted piperidine ring. Previously we installed an ethylamino or N,N-dimethyl-ethylamino group on the solvent exposed piperidyl group of the imidazole scaffold to target a D1-conserved Asp144 on BRD4 D1 for higher affinity and D1/D2 selectivity.18 However, on the basis of the cocrystal structure, these amino groups were unable to engage the conserved water molecule directly to target Asp144 similar to GSK778 and DW34.13,14,18 Analysis of our prior cocrystal structures supported a pyrrolidinyl group for orienting the amino group to target the conserved water molecule binding to Asp144. The two enantiomers, 19 and 21, lacking an ethylamino group, had 5.8- and 10-fold weaker affinity relative to 7 (Table 4). However, when the ethylamino group was installed in 20 and 22, an 11- and 7.3-fold affinity increase was observed with the IC50 values of 0.10 and 0.26 μM, respectively. Similar potency increases were noted with DW34 consistent with forming a new hydrogen bond with the targeted water molecule.14 We again turned to protein crystallography to further support the molecular basis for this improved affinity.
Table 4.
IC50 Values of Pyrrolidinyl Analogues 19–22 against BRD4 D1 by Fluorescence Anisotropy
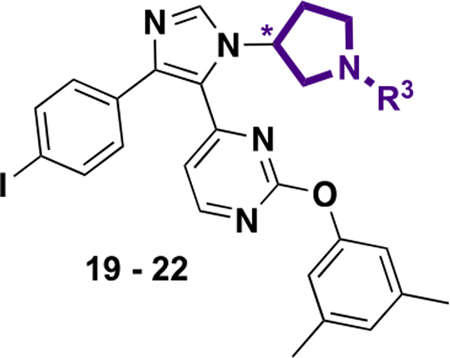
| |||
|---|---|---|---|
| compd | stereocenter | R3 | BRD4 D1 IC50 by FA (μM)a |
| (+)-JQ1 | <0.092b | ||
| 19 | S | H | 1.1 ± 0.1 |
| 20 | S | (CH2)2NH2 | 0.10 ± 0.01 |
| 21 | R | H | 1.9 ± 0.1 |
| 22 | R | (CH2)2NH2 | 0.26 ± 0.01 |
Data represents the mean and standard deviation of three experimental replicates.
IC50 is less than half the concentration of bound BRD4 D1 used.
Cocrystal structures of 20 and 22 with BRD4 D1 were analyzed for new molecular interactions relative to our prior inhibitors to support the affinity gain (Figure 3). In both cases, the p-I phenyl ring remained in the binding pocket and the imidazole ring formed a direct hydrogen bond with Asn140. However, unlike the piperidyl substituted analogue of UMN627, the ethylamino group of 20 could form a water-mediated hydrogen bond with Asp144 (Figure 3A), similar to GSK778 and DW34 (Figure 1B,C).13,14,18 Despite a difference in stereochemistry, the ethylamino group of 22 also engaged Asp 144 but displaced the conserved water molecule (Figure 3B). Together, these interactions support the affinity improvement by targeting the D1-conserved Asp144.
Figure 3.

(A) Cocrystal structure of 20 with BRD4 D1 (PDB 7RXS). The ethylamino group forms a hydrogen bond with Asn140 and Asp144 via a structured water (red sphere). (B) Cocrystal structure of 22 with BRD4 D1 (PDB 7RXT). The ethylamino group forms a direct hydrogen bond with Asn140 and Asp144. Halogen bonds with Met105 are shown in both cases. The two conserved structured waters in the binding pocket are shown as red spheres. Hydrogen bonds are shown in yellow dash lines. Halogen bonds are shown in yellow solid lines.
Our original BRD4 inhibitor, 1, based on the 1,4,5-trisubstituted imidazole was also a p38α inhibitor, with a considerable degree of off-target kinase binding.25 Consistent with the work of Gallagher et al., UMN627 could attenuate p38α binding affinity using a trifluoromethyl group in place of a smaller fluorine atom in a secondary binding pocket.18,31 However, residual kinase activity remained, particularly against CK1.18 To further investigate the selectivity over p38α, a commercial kinase binding assay was used to determine the p38α binding affinity of the p-I molecules (Table 5). Previously, we showed that the exocyclic ether bond had weaker affinity than the N–H bond against p38α, e.g., SB-284851-BT vs 1.25,28 Our new data was also consistent with these findings. Compound 5 with the p-I group had a lower affinity against p38α than 1 but higher than 2, which has a larger CF3 group (Table 5, and Supporting Information, Figure S16). Alternatively, 7, containing the exocyclic ether, was less potent than 5 containing an exocyclic amine. Given the more significant effects with the p-CF3 group, and potential concerns over the stability of the p-I group, we opted to move ahead with the p-CF3 group while maintaining the phenyl ether.
Table 5.
Kinase Affinity against p38α with p-R Substituent
| ||||
|---|---|---|---|---|
| compd | R | size of R (Å3) | X | Kd (nM)a |
| 1 | F | 13.3 | N | 0.4725 |
| SB-284851-BT | F | 13.3 | O | 4128 |
| 2 | CF3 | 39.829 | N | 260 |
| UMN627 | 1900 | |||
| 5 | I | 32.530 | N | 100 |
| 7 | I | 32.530 | O | 1500 |
Kd values were determined by KINOMEscan from Eurofins. Data represents the mean of two experimental replicates.
Compounds 23–32 were thus synthesized according to the findings based on the scaffold containing the p-I group (Table 6). Consistent with our prior designs, our affinity trends held. Notably, 23, 26, and 30 had comparable affinity against BRD4 D1 than (+)-JQ1 by FA. However, given their potency, these inhibitors were approaching the sensitivity limit for the assay. In some cases, Hill slopes were also becoming shallower (Supporting Information, Figure S7).
Table 6.
IC50 Values of Inhibitors 23–32 against BRD4 D1, BRD2 D1, and BRDT D1 by Fluorescence Anisotropy
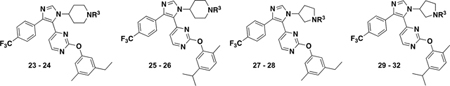
| |||||
|---|---|---|---|---|---|
| compd | R3 | stereocenter | BRD4 D1 IC50 by FA (μM)a | % Inhibition against BRD2 D1 at 50 μMa | % Inhibition against BRDT D1 at 50 μMa |
| (+)-JQ1 | <0.092b | 0.14 ± 0.001c | 0.12 ± 0.04c | ||
| 23 | H | 0.36 ± 0.03 | 79% | 68% | |
| 24 | (CH2)2N(CH3)2 | <0.092b | 88% | 88% | |
| 25 | H | 0.30 ± 0.01 | 84% | 73% | |
| 26 | (CH2)2N(CH3)2 | <0.092b | 89% | 85% | |
| 27 | H | S | 1.1 ± 0.3 | 49% | 49% |
| 28 | (CH2)2N(CH3)2 | S | 0.11 ± 0.02 | 73% | 70% |
| 29 | H | S | 0.88 ± 0.2 | 57% | 54% |
| 30 | (CH2)2N(CH3)2 | S | <0.092b | 87% | 88% |
| 31 | H | R | 3.8 ± 0.5 | 41% | 46% |
| 32 | (CH2)2N(CH3)2 | R | 0.44 ± 0.08 | 59% | 72% |
Data represents the mean and standard deviation of three experimental replicates.
IC50 is less than half the concentration of bound BRD4 D1 used.
IC50 values by fluorescent anisotropy.
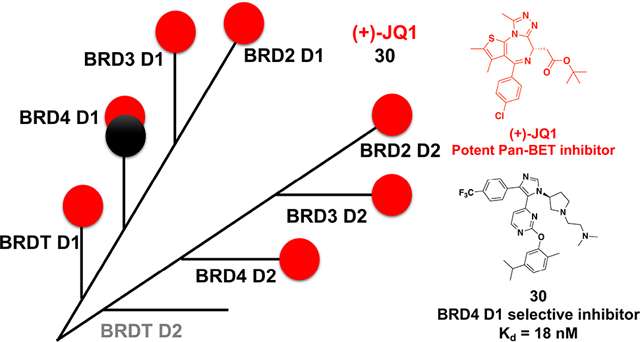
The 1,4,5-trisubsituted imidazole scaffold has been previously found to be BET selective. In the case of 1, BET D1 selectivity was observed via a bromoscan profiling assay; however, modest inhibition to several non-BET bromodomains was observed.25 Affinity to these non-BET bromodomains was subsequently removed through development of UMN627.18 To further quantify the selectivity of our new inhibitors 23–32 within the BET protein family, BRD2 D1 and BRDT D1 were chosen as representative bromodomains. Because of weak binding to these proteins and incomplete inhibition, only the percent inhibition at the highest concentration tested is indicated. The dimethyl-ethylamino group led to an increase in selectivity toward BRD4 D1 (e.g., 23 vs 24 and 25 vs 26). Given the increase in both potency and selectivity over BRD2 D1 and BRDT D1, we sought to evaluate the selectivity against the additional four BET bromodomains of BRD2 and 3 using the FA assay. Molecules 26 and 30 could not fully displace Fl-JQ1 from these bromodomains except for BRD4 D1 (Figure 4). These results were consistent with selectivity trends with thermal shift experiments using molecules 24, 26, 30, and 32 (Supporting Information, Figure S11). Together, these data confirmed the good selectivity against BRD4 D1 over other BET bromodomains.
Figure 4.

Selectivity and affinity of leading molecules 26, 30, and (+)-JQ1 against BET bromodomains by FA and ITC. (A) Competitive binding curves of 30 in red and (+)-JQ1 in gray against D1s. (B) Competitive binding curves of 30 in red and (+)-JQ1 in gray against D2s. (C) IC50 values of (+)-JQ1, 26, and 30 by FA and Kd values by ITC.
Given that several inhibitors had affinity toward BRD4 D1 that approached the sensitivity limit of the FA assay, we turned to isothermal titration calorimetry (ITC) to quantify the binding interactions for 24, 26, 30, and 32. Consistent with our FA assay, 24, 26, and 30 were highly potent, with comparable Kd values of 4.70–18.2 nM and demonstrated stoichiometric binding (Figure 4C, and Supporting Information, Table S2 and Figure S13). Inhibitor 32 bound weaker with a Kd of 37.2 nM. All four compounds exhibited favorable enthalpic and entropic binding (Supporting Information, Figure S12). Finally, preliminary evaluations showed no detectable binding of these compounds to either BRD2 D1 or BRD4 D2, while (+)-JQ1 exhibited potent binding under the same conditions, consistent with the weak binding interactions measured by dose response thermal shift profiling and FA (Figure 4C, and Supporting Information, Figures S14–S15).
While we have yet to cocrystallize 26 or 30, we obtained a cocrystal structure between 32 and BRD4 D1 (Figure 5) to validate our ligand designs. Similar to our previously published crystal structure of an analogue of UMN627 containing the N,N-dimethylamino ethyl group,18 the p-CF3 phenyl ring filled in the binding pocket and the imidazole ring maintained the key hydrogen bond with Asn140. Again, three structured water molecules in the binding pocket were displaced by the p-CF3 phenyl motif. To further evaluate potential roles of water displacement on BRD4 D1 selectivity, we compared D1 and D2 residues surrounding the water channel. Tyr98, located 8 Å from the Kac binding pocket, terminates the water channel in BRD4 D1 and is typically flexible by B-factor measurements (e.g., 36.4 with H4 Kac peptide, Supporting Information, Figure S20A). In comparison, here Tyr98 is engaged at a shorter distance (6 Å, Supporting Information, Figure S20B) and stabilized via two structured waters. A comparison of sequence conservation in BET bromodomains shows Tyr98 is present in BRD4 D1 and both BRDT bromodomains; it is replaced by cysteine or histidine residues in other BET bromodomains (Supporting Information, Figure S20C). Given the distance and hydrogen bonding differences between these residues (Supporting Information, Figure S20D), stabilization of Tyr98 may play a role in determining the observed BRD4 D1 selectivity from a distance through water channel interactions.
Figure 5.

(A) Cocrystal structure of 32 with BRD4 (PDB 7R9C). The structured waters are shown as red spheres. The dimethyl-ethylamino group forms a hydrogen bond with Asn140 and Asp144 via a structured water. Hydrogen bonds are shown in yellow dash. (B) Backbone overlay of BRD4 D1 bound to H4 K5ac,K8ac (PBD 3UVW, violet) and bound to 32 (PDB 7R9C, gray).
Additional favorable interactions were observed with 32 toward the exterior of the Kac binding pocket. Unlike the UMN627 analogue, the dimethyl-ethylamino group oriented toward Asp144, forming a water-mediated hydrogen bond with Asn140 and Asp144 (Figure 5A). Larger lysine backbone movement was also observed (ΔCαK = 1.7 Å, PDB 6WGX vs ΔCαK = 1.9 Å, PDB 7R9C, Figure 5B). The aryl ether group of 32 and the indole of Trp81 were parallel. However, the distance (7.1 Å, Supporting Information, Figure S21) between them was too far away to have the parallel offset π–π stacking interaction. Alternatively, an intramolecular CH–π interaction between the proton on the pyrrolidinyl group and isopropyl methyl phenyl motif may also improve the affinity (Supporting Information, Figure S22).
Given the high affinity and BET selectivity for 26 and 30, p38α kinase inhibitory activity was evaluated with a commercial kinase binding assay. However, both compounds had stronger affinity against p38α compared with UMN627 (Kd = 98 and 780 nM, Supporting Information, Figure S19). CK1 was also tested as an off-target. Encouragingly, the binding affinity of 26 and 30 for CK1 was reduced by 63- and 68-fold relative to UMN627 (Supporting Information, Figure S17). To broadly understand the selectivity over other kinases, 30 was tested at 1 μM with 468 kinases (Supporting Information, Tables S3–S8). 30 had low binding activity against all kinases at this concentration (only three with >50% binding), with the highest affinity toward EGFR (58% bound) supporting 30 as an inhibitor with BRD4 D1 selectivity and only weak kinase off-target binding. Given the high ATP concentration in the cell, kinase inhibition effects are anticipated to be minimal. Together these results support high affinity BRD4 D1 inhibitors, with significant selectivity over kinases for use as chemical probes.
Given our increased potency and selectivity toward BRD4 D1 versus our prior inhibitor, four compounds were selected for evaluation in cells. We previously validated target engagement of BRD4 and downstream cellular effects related to BET inhibition in MM.1S cells.18,25 Therefore, we used a cellular thermal shift assay (CETSA) to measure in-cell target engagement of BET proteins with compounds 23, 24, 26, and 30 in these cells (Figure 6A, and Supporting Information, Figure S23). Surprisingly, compounds 23 and 24 had weak engagement of BRD4, whereas compounds 26 and 30 showed a higher dose-dependent engagement of BRD4. Selectivity toward BET proteins was further confirmed in this assay, as only weak engagement of BRD2 was observed up to 25 μM with 26 and 30 (Figure 6A, and Supporting Information, Figure S23). BRD3 was less amenable to the CETSA experiment (Figure 6A, and Supporting Information, Figure S23).
Figure 6.

Target engagement and cellular effects of select compounds, 23, 24, 26, and 30 in MM.1S cells. (A) Cellular thermal shift assay after 1 h treatment with compounds at indicated concentrations. (B) Western blots for c-Myc after 6 h treatment with compounds at same concentrations as in (A). Data is representative of three replicate experiments, see Supporting Information for uncropped images.
MM.1S cells display a strong dependence of c-Myc on BRD4 inhibition.8 Here, the expression of c-Myc, as measured by Western blot (Figure 6B, and Supporting Information, Figure S23), correlated with BRD4 engagement in these cells. As previously reported with UMN627,18 c-Myc inhibition is not observed at low concentrations of 26 and 30 and may be slightly induced, followed by downregulation at higher concentrations. We have yet to understand the origin of the potential Myc induction, but note that this effect has been seen by us and others using different BRD4 D1 selective inhibitors.18,32 As a second marker for BRD4, we tested effects on inflammation markers, as BET inhibition can downregulate cytokines, such as IL-8.25 We measured IL-8 levels to evaluate the anti-inflammatory effects of our leading inhibitors in nonsmall cell lung cancer A549 cells. At 1 μM concentration, 26 and 30 reduced IL-8 levels to 19%, within 2-fold of positive control (+)-JQ1, without affecting cell viability (Table 7, and Supporting Information, Figure S24–S25).14
Table 7.
Cellular Effects of Inhibitors 26 and 30 on Inflammation and Viability
| (+)-JQ1 | iBET151 | 26 | 30 | |
|---|---|---|---|---|
| EC50 in A549 cells (μM)a | 17 | 3.6 | 14 | |
| % of residual IL-8 at 1 μMa | 12 ± 2 | 19 ± 2 | 19 ± 0.2 | |
| EC50 in LSECs (μM)b | >100 | 7.2 ± 0.7 | 7.0 ± 0.7 | |
| % of residual CCL2 mRNA at 1.3 μMb | 39 ± 26 | 38 ± 15 | 49 ± 2 | |
| % of Residual CXCL1 mRNA at 1.3 μMb | 82 ± 10 | 31 ± 22 | 65 ± 9 |
Viability analysis in A549 cells.
Viability analysis in LSECs.
Previously, we reported that BRD4 inhibition via UMN627 can suppress inflammation from TNFα/NF-κB mediated chemokine expression in human liver sinusoidal endothelial cells and in vivo models of alcoholic hepatitis.22,23 As a final experiment, here, our leading molecules 26 and 30 downregulated CCL2 and CXCL1 mRNA levels at comparable levels to a pan-BET inhibitor, IBET-151, at 1.3 μM (Table 7, and Supporting Information, Figure S27–S28). However, in this case, more significant toxicity was observed for 26 and 30 versus IBET-151, which was well-tolerated up to 100 μM (Supporting Information, Figure S26). Although BRD4 D1 inhibition is not sufficient for reducing c-Myc expression, these results demonstrate strong in vitro selectivity and anti-inflammatory activity in two cell lines comparable to pan-BET inhibitors.
CONCLUSION
In conclusion, we describe the development of BRD4 D1 selective inhibitors with nanomolar affinity through a systematic structure–activity relationship study. The unique selectivity and strong affinity were obtained in part from halogen bonding, flexible backbone conformational effects in BET D1s, displacement of low-energy structured waters to augment BRD4 D1 selectivity, and targeting a BET D1 conserved Asp residue. Our preliminary cellular data demonstrated 26 and 30 can selectively engage BRD4 in MM.1S cells. Similar to UMN627, c-Myc inhibition was not observed at low inhibitor concentrations where BRD4 engagement was demonstrated. Such effects have not been observed with pan-BET or pan-D1 inhibitors in MM.1S cells. Additionally, both inhibitors reduced cytokine IL-8 in a nonsmall lung cancer cell line and showed efficacy in human liver sinusoidal endothelial cells to suppress chemokine expression resulting from inflammation. Together, these studies provide a novel chemical probe for studying the functional significance of inhibiting the BRD4 N-terminal bromodomain in cells over less selective pan-D1 inhibitors. Moreover, the high-resolution structural information presented here for our selective inhibitors offers a unique opportunity for developing BRD4-selective heterobifunctional molecules such as degraders, which would avoid effects from inhibiting BRD2 and BRD3. Preliminary steps have been taken to improve the drug-like properties of DW34 and UMN627, including improving metabolic stability.14 Similarly, future work will focus on optimizing drug metabolism and pharmacokinetic properties of these lead inhibitors to facilitate in vivo studies.
EXPERIMENTAL SECTION
General Procedures.
All chemicals unless otherwise stated were commercially available and used without further purification. Thin-layer chromatography (TLC) was used for monitoring reaction progress and visualized by using UV light. Flash column chromatography was performed on a Teledyne Isco Rf-plus CombiFlash instrument with normal phase precolumn load cartridges and gold high-performance columns. Spectra were collected on a Bruker Avance III HD 400 or a Bruker Avance III HD 500. Chemical shift (δ) are reported in parts per million (ppm) and referenced to residual solvent signal, CDCl3: 1H 7.26 ppm, 13C 77.0 ppm; MeOD: 1H 3.32 ppm, 13C 49.2 ppm; DMSO-d6: 1H 2.50 ppm, 13C 39.5 ppm. Coupling constants (J) are in Hz. Splitting patterns are reported as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and br (broad). High resolution mass spectrometry was used with positive-mode electrospray-ionization methods (ESI-MS) by a Bruker BioTOF II.
Purity Analysis: Purities of 3–32 were checked by reverse-phase high-performance liquid chromatography (RP-HPLC) with a C18 column and 10–60% 0.1% TFA water and acetonitrile over 60 min. HPLC traces were shown in Supporting Information, Figures S1, S2, and S3. All lead compounds used were >95% pure by RP-HPLC.
Synthesis of N-((4-Substituted-phenyl)(tosyl)methyl)-formamide (3a–6a).
A mixture of a para-substituted benzaldehyde (7.1 mmol, 1.5 equiv), p-methylbenzenesulfinic acid (4.8 mmol, 1.0 equiv), camphorsulfonic acid (0.58 mmol, 0.12 equiv), and formamide (17 mmol, 3.5 equiv) were stirred at 60 °C for 18 h. The resulting solid was resuspended in hexane/methanol (4:1, 15 mL) for 30 min. The suspension was filtered and dried to give compound 3a–6a without further purification.
3a (1.2 g, 77%) was isolated as a yellow solid. 1H NMR (400 MHz, DMSO) δ 9.82–9.74 (m, 1H), 7.96 (d, J = 1.3 Hz, 1H), 7.76–7.69 (m, 2H), 7.64–7.56 (m, 2H), 7.54–7.50 (m, 2H), 7.44 (d, J = 8.1 Hz, 2H), 6.47 (d, J = 10.6 Hz, 1H), 2.41 (s, 3H). 13C NMR (100 MHz, DMSO) δ 160.2, 145.0, 134.4, 133.2, 131.2, 129.7, 129.2, 128.4, 69.4, 21.2.
4a (0.97 g, 55%) was isolated as a white solid. 1H NMR (500 MHz, DMSO) δ 9.77 (dd, J = 10.5, 1.5 Hz, 1H), 7.95 (d, J = 1.3 Hz, 1H), 7.74–7.72 (m, 2H), 7.67–7.65 (m, 2H), 7.54–7.51 (m, 2H), 7.45–7.43 (m, 3H), 6.45 (d, J = 10.5 Hz, 1H), 2.41 (s, 3H). 13C NMR (100 MHz, DMSO) δ 160.3, 144.8, 134.4, 130.3, 129.6, 129.48, 129.45, 129.2, 128.3, 70.2, 21.2.
5a (1.4 g, 68%) was isolated as a white solid. 1H NMR (400 MHz, DMSO) δ 9.76 (dd, J = 10.6, 1.5 Hz, 1H), 7.94 (d, J = 1.3 Hz, 1H), 7.85–7.79 (m, 2H), 7.75–7.71 (m, 2H), 7.44 (d, J = 8.0 Hz, 3H), 7.38–7.34 (m, 2H), 6.41 (d, J = 10.5 Hz, 1H), 2.41 (s, 3H). 13C NMR (125 MHz, DMSO) δ160.2, 145.0, 137.1, 133.2 131.5, 130.0, 129.7, 96.6, 69.7, 21.2.
6a (0.74 g, 48%) was isolated as a white yellow solid. 1H NMR (500 MHz, DMSO) δ 9.69 (dd, J = 10.5, 1.6 Hz, 1H), 7.94 (d, J = 1.4 Hz, 1H), 7.70 (d, J = 8.1 Hz, 2H), 7.49–7.45 (m, 2H), 7.42 (d, J = 8.2 Hz, 3H), 7.02–6.95 (m, 2H), 6.31 (d, J = 10.5 Hz, 1H), 3.78 (s, 3H), 2.41 (s, 3H). 13C NMR (100 MHz, DMSO) δ 160.1, 144.7, 133.6, 130.8, 129.6, 129.1, 122.0, 113.7, 69.8, 55.2, 21.1.
Synthesis of 1-Substituted-4-(isocyano(tosyl)methyl)-benzene (3b–6b).
POCl3 (5.2 mmol, 2.0 equiv) was added dropwise to a solution of 3a–6a (2.6 mmol, 1.0 equiv) in anhydrous THF at −10 °C. 2,6-Lutidine (15.8 mmol, 6.0 equiv) was added after 10 min. The reaction was warmed to room temperature and stirred for 16 h. The reaction was quenched by addition of a saturated aqueous solution of NH4Cl. The aqueous phase was extracted with ethyl acetate (3 × 20 mL), dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The crude product was purified by silica gel chromatography using hexanes and ethyl acetate (0–100%) as eluent to give the desired products 3b–6b.
3b (0.55 g, 70%) was isolated as a brown solid. 1H NMR (400 MHz, CDCl3) δ 7.65–7.60 (m, 2H), 7.40–7.33 (m, 4H), 7.31–7.27 (m, 2H), 5.58 (s, 1H), 2.48 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 166.8, 147.0, 137.3, 130.7, 130.1, 129.9, 129.2, 125.3, 75.9, 22.0.
4b (0.48 g, 53%) was isolated as a brown solid. 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 8.1 Hz, 2H), 7.56–7.53 (m, 2H), 7.36 (d, J = 8.1 Hz, 2H), 7.23–7.21 (m, 2H), 5.56 (s, 1H), 2.48 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 166.7, 147.0, 132.1, 130.6, 130.1, 130.0, 129.9, 125.5, 76.0, 22.0.
5b (0.41 g, 43%) was isolated as a brown solid. 1H NMR (500 MHz, CDCl3) δ 7.74 (dq, J = 9.0, 2.4 Hz, 2H), 7.67–7.62 (m, 2H), 7.36 (d, J = 8.1 Hz, 2H), 7.10–7.05 (m, 2H), 5.54 (s, 1H), 2.48 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 166.8, 147.1, 138.1, 130.7, 130.14, 130.08, 126.4, 97.5, 76.2, 22.0.
6b (0.39 g, 50%) was isolated as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.70–7.62 (m, 2H), 7.36 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 2.2 Hz, 1H), 6.96–6.89 (m, 2H), 5.57 (s, 1H), 3.86 (s, 3H), 2.49 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 165.8, 146.6, 130.6, 130. 0, 129.9, 118.4, 114.3, 76.3, 55.6, 21.9.
Synthesis of tert-Butyl 4-(4-(4-Substituted-phenyl)-5-(2-(methylthio)pyrimidin-4-yl)-1H-imidazol-1-yl)piperidine-1-carboxylate (3c–6c).
General procedure I:
A mixture of 3b–6b (0.5 mmol, 1.0 equiv), tert-butyl 4-(((2-(methylthio)pyrimidin-4-yl)methylene)-amino)piperidine-1-carboxylate (0.5 mmol, 1.0 equiv), and potassium carbonate (1.8 mmol, 4.0 equiv) were dissolved in acetonitrile (1.7 mL) and stirred at 40 °C for 16 h. The reaction mixture was quenched by addition of brine and extracted with ethyl acetate (3 × 20 mL). The combined organic phase was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The crude product was purified by silica gel chromatography using hexane and ethyl acetate (0–100%) as eluent to give the desired product.
3c (0.18 g, 73%) was isolated as a brown solid. 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 5.2 Hz, 1H), 7.76 (s, 1H), 7.42–7.37 (m, 2H), 7.33–7.28 (m, 2H), 6.80 (d, J = 5.2 Hz, 1H), 4.81 (tt, J = 12.0, 3.7 Hz, 1H), 4.30 (s, 2H), 2.78 (d, J = 15.4 Hz, 2H), 2.59 (s, 3H), 2.22–2.11 (m, 2H), 1.85 (qd, J = 12.3, 4.3 Hz, 2H), 1.47 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 173.1, 157.8, 157.3, 154.7, 143.3, 136.6, 134.0, 132.8, 129.9, 128.9, 124.4, 117.1, 80.2, 54.5, 43.4 (br), 33.6, 28.5, 14.2.
4c (0.18 g, 68%) was isolated as a brown solid. 1H NMR (400 MHz, CDCl3) δ 8.39 (d, J = 5.2 Hz, 1H), 7.79 (s, 1H), 7.54–7.46 (m, 2H), 7.41–7.32 (m, 2H), 6.84 (d, J = 5.2 Hz, 1H), 4.83 (tt, J = 12.2, 3.8 Hz, 1H), 4.33 (s, 2H), 2.81 (d, J = 13.6 Hz, 2H), 2.62 (s, 3H), 2.19 (d, J = 12.1 Hz, 2H), 1.88 (qd, J = 12.2, 4.3 Hz, 2H), 1.50 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 173.1, 157.7, 157.3, 154.6, 143.2, 136.6, 133.2, 131.8, 130.2, 124.4, 122.2, 117.1, 80.2, 54.4, 43.4 (br), 33.6, 28.5, 14.2.
5c (0.20 g, 70%) was isolated as a brown solid. 1H NMR (500 MHz, CDCl3) δ 8.37 (d, J = 5.2 Hz, 1H), 7.79 (s, 1H), 7.71–7.57 (m, 2H), 7.24–7.16 (m, 2H), 6.82 (d, J = 5.2 Hz, 1H), 4.80 (ddd, J = 12.1, 8.3, 3.7 Hz, 1H), 4.30 (s, 2H), 2.79 (s, 2H), 2.59 (s, 3H), 2.25–2.08 (m, 2H), 1.86 (tt, J = 12.3, 6.1 Hz, 2H), 1.47 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 173.1, 157.6, 157.4, 154.6, 143.1, 137.9, 136.6, 133.5, 130.4, 124.5, 117.14, 94.0, 80.3, 54. 6, 43.4 (br), 33.6, 28.5, 14.2.
6c (0.13 g, 55%) was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.30 (d, J = 5.3 Hz, 1H), 7.74 (s, 1H), 7.40–7.34 (m, 2H), 6.88–6.84 (m, 2H), 6.82 (d, J = 5.2 Hz, 1H), 4.88 (tt, J = 12.1, 3.7 Hz, 1H), 4.29 (s, 2H), 3.81 (s, 3H), 2.78 (d, J = 18.6 Hz, 2H), 2.58 (s, 3H), 2.22–2.11 (m, 2H), 1.84 (qd, J = 12.3, 4.3 Hz, 2H), 1.47 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 172.7, 159.6, 158.2, 156.97, 154. 7, 144.8, 136.5, 130.0, 126.8, 123.6, 117.02, 114.1, 80.2, 55.4, 54.4, 43.4 (br), 33.6, 28.5, 14.2.
Synthesis of tert-Butyl 4-(4-(4-Substituted-phenyl)-5-(2-(methylsulfonyl)pyrimidin-4-yl)-1H-imidazol-1-yl)piperidine-1-carboxylate (3d–6d).
General procedure II:
A solution of oxone (0.69 mmol, 2.4 equiv) in water (5.5 mL) was added dropwise to a solution of 3c–6c (0.29 mmol, 1.0 equiv) in THF (4.0 mL) at −10 °C. The reaction mixture was stirred at room temperature for 18 h and then quenched by the addition of brine and extracted with DCM (3 × 20 mL). The combined organic phase was washed with brine, dried with magnesium sulfate, and concentrated under reduced pressure to give the product without any further purification.
3d (0.13 g, 87%) was a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.59 (d, J = 5.4 Hz, 1H), 7.85 (s, 1H), 7.44–7.39 (m, 2H), 7.39–7.33 (m, 2H), 7.30 (d, J = 5.4 Hz, 1H), 5.03 (tt, J = 12.0, 3.7 Hz, 1H), 4.38–4.18 (m, 2H), 3.38 (s, 3H), 1.47 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 166.3, 159.4, 157.5, 154.7, 146.1, 138.3, 134.9, 132.5, 130.1, 129.4, 123.3, 122.9, 80.1, 55.6, 43.2 (br), 39.1, 33.6 (br), 28.5.
4d (0.15 g, 92%) was a pale-yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.60 (d, J = 5.4 Hz, 1H), 7.86 (s, 1H), 7.57–7.49 (m, 2H), 7.38–7.34 (m, 2H), 7.32 (d, J = 5.4 Hz, 1H), 5.04 (tt, J = 12.0, 3.8 Hz, 1H), 4.37–4.31 (m, 2H), 3.39 (s, 3H), 2.94 (s, 2H), 2.26 (m, 2H), 1.48 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 166.3, 159.4, 157.5, 154.6, 146. 0, 138.3, 132.9, 132.3, 130.4, 123.2, 123.1, 122. 9, 80.1, 55.5, 43.4 (br), 39.1, 33.7 (br), 28.5.
5d (0.14 g, 80%) was isolated as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.60 (d, J = 5.3 Hz, 1H), 7.90 (s, 1H), 7.78–7.66 (m, 2H), 7.32 (d, J = 5.4 Hz, 1H), 7.24–7.16 (m, 2H), 5.01 (tt, J = 12.0, 3.7 Hz, 1H), 4.31–4.18 (m, 3H), 3.38 (s, 3H), 2.36–2.14 (m, 2H), 1.95–1.81 (m, 2H), 1.46 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 166.3, 159.3, 157.6, 154.6, 145.7, 138.3, 133.1, 130.5, 123.3, 123.0, 95.0, 80.1, 55.6, 43.3, 39.1, 33.5, 28.5.
6d (0.14 g, 95%) was isolated as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.54 (d, J = 5.5 Hz, 1H), 7.89 (s, 1H), 7.39–7.37 (m, 2H), 7.33 (d, J = 5.4 Hz, 1H), 6.92–6.90 (m, 2H), 5.08 (tt, J = 12.0, 3.7 Hz, 1H), 4.31 (d, J = 21.6 Hz, 3H), 3.84 (s, 3H), 3.36 (s, 3H), 3.02–2.80 (m, 2H), 2.28–2.19 (m, 2H), 1.86–1.82 (m, 2H), 1.46 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 166.1, 160.3, 159.6, 157.2, 154.7, 147. 1, 138.0, 130.2, 125.9, 122.73, 122.66, 114.6, 80.0, 55.6, 55.5, 43.3, 39.2, 33.7, 28.5.
Synthesis of tert-Butyl 4-(5-(2-((3,5-Dimethylphenyl)-amino)pyrimidin-4-yl)-4-(4-substituted-phenyl)-1H-imidazol-1-yl)piperidine-1-carboxylate (3e–6e).
A mixture of 3d–6d (0.16 mmol, 1.0 equiv) and 3,5-dimethylaniline (0.72 mmol, 4.4 equiv) were heated in a sealed tube at 130 °C for 16 h behind a blast shield. TLC was used to monitor the reaction. The crude product was obtained as a brown oil and purified by silica gel chromatography using hexanes and ethyl acetate (0–100%) as eluent to give the desired products 3e–6e.
3e (36 mg, 40%) was isolated as a brown solid. 1H NMR (500 MHz, CDCl3) δ 8.27 (d, J = 5.1 Hz, 1H), 7.72 (s, 1H), 7.48–7.45 (m, 2H), 7.34 (s, 1H), 7.31–7.28 (m, 2H), 7.19 (d, J = 1.5 Hz, 2H), 6.75 (s, 1H), 6.57 (d, J = 5.1 Hz, 1H), 4.78 (tt, J = 12.1, 3.8 Hz, 1H), 2.43 (t, J = 13.1 Hz, 2H), 2.31 (s, 6H), 2.03 (d, J = 7.0 Hz, 2H), 1.76 (qd, J = 12.3, 4.2 Hz, 2H), 1.45 (s, 10H). 13C NMR (125 MHz, CDCl3) δ 160.6, 158.6, 158.5, 154.6, 142.1, 138.8, 138.7, 136.1, 133.7, 132.9, 129.8, 128. 8, 125.4, 125.2, 118.4, 113.6, 80.1, 53.9, 43.1 (br), 33.5, 28.5, 21.5.
4e (43 mg, 45%) was isolated as a brown solid. 1H NMR (500 MHz, CDCl3) δ 8.27 (d, J = 5.1 Hz, 1H), 7.72 (s, 1H), 7.46–7.43 (m, 2H), 7.42–7.38 (m, 2H), 7.19 (d, J = 1.5 Hz, 2H), 6.75 (s, 1H), 6.57 (d, J = 5.1 Hz, 1H), 4.77 (tt, J = 12.1, 3.8 Hz, 1H), 2.46–2.40 (m, 2H), 1.81–1.72 (m, 3H), 1.45 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 160.6, 158.6, 158.4, 154.6, 142.1, 138.8, 138.7, 136.1, 133.4, 131.7, 130.1, 125.4, 125.2, 121.9, 118.4, 113.6, 80.1, 53.9, 43.1 (br), 33.5, 28.5, 21.6.
5e (42 mg, 40%) was isolated as a brown solid. 1H NMR (500 MHz, CDCl3) δ 8.27 (d, J = 5.1 Hz, 1H), 7.73 (s, 1H), 7.65 (d, J = 8.1 Hz, 2H), 7.36 (s, 1H), 7.28 (s, 1H), 7.18 (s, 2H), 6.74 (s, 1H), 6.58 (d, J = 5.1 Hz, 1H), 4.76 (tq, J = 13.2, 5.4, 4.5 Hz, 1H), 2.47–2.37 (m, 2H), 2.30 (s, 6H), 2.02 (s, 2H), 1.80–1.71 (m, 2H), 1.45 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 160.5, 158.5, 158.4, 154.6, 142.1, 138.8, 138.7, 137.7, 136.1, 133.8, 130.3, 125.5, 125.2, 118.5, 113.6, 93.6, 80.1, 53.9, 43.0 (br), 33.5, 28.5, 21.6.
6e (34 mg, 38%) was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 5.1 Hz, 1H), 7.73 (s, 1H), 7.49–7.45 (m, 2H), 7.26 (s, 1H), 7.22, 6.92–6.86 (m, 2H), 6.76 (s, 1H), 6.62 (d, J = 5.1 Hz, 1H), 4.87 (tt, J = 12.0, 3.8 Hz, 1H), 3.84 (s, 3H), 2.49–2.31 (m, 2H), 2.33 (s, 6H), 1.81–1.74 (m, 2H), 1.45 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 160.3, 159.7, 159.5, 153.0, 145.3, 139.6, 136.6, 130.1, 127.4, 126.8, 123.4, 119.6, 116.3, 114.2, 80.1, 55.4, 54.1, 43.1, 33.4, 28.5, 21.4.
Synthesis of N-(3,5-Dimethylphenyl)-4-(1-(piperidin-4-yl)-4-(4-substituted-phenyl)-1H-imidazol-5-yl)pyrimidin-2-amine (3–6).
General procedure III:
Trifluoroacetic acid (TFA) (0.50 mL) was added to a solution of 3e–6e (64 μmol) in DCM (0.50 mL). After 16 h, the reaction was concentrated under reduced pressure. Cold ether (4.0 mL) was used to precipitate out the desired product 3–6. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt.
3 (27 mg, 98%) was isolated as a yellow solid. 1H NMR (500 MHz, MeOD) δ 8.36 (d, J = 5.1 Hz, 1H), 7.49 (m, 4H), 7.24 (s, 2H), 6.79 (s, 1H), 6.59 (d, J = 5.0 Hz, 1H), 5.02 (ddt, J = 12.2, 7.6, 3.9 Hz, 1H), 3.44–3.38 (m, 2H), 2.81–2.72 (m, 2H), 2.49–2.43 (m, 2H), 2.30 (s, 6H), 2.22–2.14 (m, 2H). 13C NMR (125 MHz, MeOD) δ 162.0, 160.3, 156.8, 140.3, 139.6, 136.8, 131.5, 130.5, 128.9, 127.9, 126.4, 120.5, 114.0, 54.2, 44.3, 30.8, 21.5. 19F NMR (470 MHz, MeOD) δ −77.2. HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H28ClN6+ 459.2059, found 459.2060.
4 (31 mg, 96%) was isolated as a yellow solid. 1H NMR (500 MHz, MeOD) δ 8.96 (s, 1H), 8.37 (d, J = 5.1 Hz, 1H), 7.68–7.60 (m, 2H), 7.45–7.42 (m, 2H), 7.21 (s, 2H), 6.80 (s, 1H), 6.59 (d, J = 5.1 Hz, 1H), 5.02 (tt, J = 12.1, 3.7 Hz, 1H), 3.44–3.35 (m, 2H), 2.79–2.74 (m, 2H), 2.49–2.41 (m, 2H), 2.30 (s, 6H), 2.30–2.19 (m, 2H). 13C NMR (125 MHz, MeOD) δ 162.0, 160.3, 156.8, 140.3, 139.6, 136.8, 131.5, 130.5, 128.9, 127.9, 126.4, 120.5, 114.0, 54.2, 44.3, 30.8, 21.5. 19F NMR (470 MHz, MeOD) δ −77.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H28BrN6+ 503.1554, found 503.1545.
5 (34 mg, 98%) was isolated as a yellow solid. 1H NMR (500 MHz, MeOD) δ 8.45 (s, 1H), 8.38 (d, J = 5.0 Hz, 1H), 7.86–7.83 (m, 2H), 7.30–7.26 (m, 2H), 7.23 (s, 2H), 6.80 (s, 1H), 6.60 (d, J = 5.0 Hz, 1H), 5.01 (ddd, J = 12.2, 8.4, 3.8 Hz, 1H), 3.41 (d, J = 13.0 Hz, 2H), 2.78 (t, J = 13.2 Hz, 2H), 2.46 (d, J = 13.7 Hz, 2H.), 2.31 (s, 6H), 2.20 (qd, J = 13.0, 4.1 Hz, 2H). 13C NMR (125 MHz, MeOD) δ 166.5, 161.9, 159. 5, 154.3, 141.5, 141.1, 139.4, 138.0, 132.2, 131.7, 128.4, 126.3, 120.4, 118.2, 95.9, 53.5, 44.5, 31.0, 21.3. 19F NMR (470 MHz, MeOD) δ −77.2. HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H28IN6+ 551.1415, found 551.1393.
6 (27 mg, 96%) was isolated as a white solid. 1H NMR (400 MHz, MeOD) δ 9.23 (s, 1H), 8.37 (d, J = 5.1 Hz, 1H), 7.47–7.40 (m, 2H), 7.21 (s, 2H), 7.09–7.03 (m, 2H), 6.79 (s, 1H), 6.59 (d, J = 5.1 Hz, 1H), 5.11 (tt, J = 12.0, 3.7 Hz, 1H), 3.85 (s, 3H), 3.41 (d, J = 13.1 Hz, 2H), 2.76 (t, J = 12.6 Hz, 2H), 2.48 (d, J = 13.4 Hz, 2H), 2.30 (s, 6H), 2.22 (td, J = 13.2, 4.2 Hz, 2H). 13C NMR (100 MHz, MeOD) δ 162.9, 162.2, 160.5, 156.2, 140.4, 139.6, 135.9, 131.5, 131.2, 127.2, 126.4, 120.5, 120.2, 115.9, 114.0, 56.0, 54.7, 44.2, 30.7, 21.5. 19F NMR (376 MHz, MeOD) δ −77.1. HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H31N6O+ 455.2554, found 455.2550.
tert-Butyl 4-(5-(2-(3,5-Dimethylphenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)piperidine-1-carboxylate (7f).
General procedure IV:
To a suspension of NaH (60% in mineral oil, 48 mg, 1.2 mmol) in anhydrous THF (3.5 mL) was added 3,5-dimethylphenol (0.15 g, 1.2 mmol) in anhydrous THF (1.7 mL) dropwise at −10 °C. The mixture was stirred for 15 min; this was followed by the addition of 5d (0.17 g, 0.27 mmol) in anhydrous THF (1.7 mL). The reaction mixture was stirred for 16 h, quenched by the addition of water, and extracted with ethyl acetate (3 × 10 mL). The combined organic phase was dried with MgSO4 and concentrated under reduced pressure. Final purification by column chromatography (0–100% hexanes to ethyl acetate) afforded product 7f (7–21, 8–79, 99 mg, 56%). 1H NMR (400 MHz, CDCl3) δ 8.40 (d, J = 5.2 Hz, 1H), 7.70 (d, J = 2.7 Hz, 2H), 7.68 (d, J = 1.9 Hz, 1H), 7.24–7.19 (m, 2H), 6.90 (s, 1H), 6.87 (d, J = 5.2 Hz, 1H), 6.84 (s, 2H), 4.71–4.58 (m, 1H), 2.50–2.44 (m, 2H), 2.34 (s, 6H), 1.91–1.88 (m, 2H), 1.74–1.63 (m, 2H), 1.47 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 165.6, 160.0, 159.9, 154.6, 152.9, 143.9, 139.7, 137.9, 136.8, 133.9, 130.5, 127.5, 124.2, 119.6, 116.5, 94.0, 80.1, 54.2, 43.0 (br), 33.4, 28.5, 21.4.
2-(3,5-Dimethylphenoxy)-4-(4-(4-iodophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)pyrimidine (7).
General procedure III was used to give product 7 (41 mg, 96%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.75 (s, 1H), 8.58 (d, J = 5.1 Hz, 1H), 7.88–7.84 (m, 2H), 7.27–7.22 (m, 2H), 7.03 (d, J = 5.1 Hz, 1H), 6.99 (s, 1H), 6.94 (s, 2H), 4.76 (tt, J = 11.9, 4.0 Hz, 1H), 3.39 (dt, J = 13.0, 2.5 Hz, 2H), 2.75 (td, J = 13.1, 3.0 Hz, 2H), 2.37 (s, 6H), 2.27–2.21 (m, 2H), 2.15 (td, J = 12.8, 4.1 Hz, 2H). 13C NMR (125 MHz, MeOD) δ 166.6, 162.4, 158.4, 154.2, 141.1, 139.7, 138.8, 137.6, 131.7, 129.9, 128.4, 126.8, 120.4, 118.4, 96.9, 54.2, 44.4, 30.8, 21.3. 19F NMR (470 MHz, MeOD) δ −77.4. HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H27IN5O+ 552.1255, found 552.1240.
tert-Butyl 4-(4-(4-Iodophenyl)-5-(2-phenoxypyrimidin-4-yl)-1H-imidazol-1-yl)piperidine-1-carboxylate (8f).
General procedure IV was used to give 8f (M-103, 50 mg, 30%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.41 (d, J = 5.1 Hz, 1H), 7.68 (d, J = 5.9 Hz, 2H), 7.66 (s, 1H), 7.44 (t, J = 7.9 Hz, 2H), 7.28 (d, J = 7.4 Hz, 1H), 7.25–7.22 (m, 2H), 7.19 (d, J = 8.2 Hz, 2H), 6.88 (d, J = 5.1 Hz, 1H), 4.58 (tt, J = 12.0, 3.8 Hz, 1H), 2.45 (s, 2H), 1.89–1.79 (m, 2H), 1.66 (qd, J = 12.4, 4.4 Hz, 2H), 1.46 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 165.4, 160.1, 159.8, 154.5, 153.0, 143.8, 137.9, 136.9, 133.8, 130.5, 129.8, 125.7, 124.1, 122.0, 116.8, 94.0, 80.1, 54.1, 43.1 (br), 33.4, 28.5.
4-(4-(4-Iodophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)-2-phenoxypyrimidine (8).
General procedure III was used to give 8 (40 mg, 96%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.62 (s, 1H), 8.58 (d, J = 5.1 Hz, 1H), 7.83 (d, J = 8.3 Hz, 2H), 7.50 (t, J = 7.9 Hz, 2H), 7.36–7.30 (m, 3H), 7.23–7.18 (m, 2H), 7.01 (d, J = 5.1 Hz, 1H), 4.72 (tt, J = 11.9, 4.1 Hz, 1H), 3.37 (dt, J = 12.9, 2.5 Hz, 2H), 2.75 (td, J = 13.1, 3.1 Hz, 2H), 2.22–2.02 (m, 4H). 13C NMR (125 MHz, MeOD) δ 166.6, 162.6, 158.2, 154.3, 139.7, 138.5, 137.5, 131.7, 131.1, 129.5, 127.0, 126.7, 123.0, 118.6, 97.0, 54.2, 44.4, 30.7. 19F NMR (470 MHz, MeOD) δ −77.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C24H23IN5O+ 524.0942, found 524.0949.
tert-Butyl 4-(4-(4-Iodophenyl)-5-(2-(m-tolyloxy)pyrimidin-4-yl)-1H-imidazol-1-yl)piperidine-1-carboxylate (9f).
General procedure IV was used to give 9f (84 mg, 49%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.40 (dd, J = 5.2, 1.3 Hz, 1H), 7.69 (d, J = 1.1 Hz, 1H), 7.69–7.65 (m, 2H), 7.31 (t, J = 7.8 Hz, 1H), 7.20 (dd, J = 8.2, 1.3 Hz, 2H), 7.07 (d, J = 7.7 Hz, 1H), 7.05–7.00 (m, 2H), 6.87 (dd, J = 5.2, 1.1 Hz, 1H), 4.60 (tt, J = 12.1, 3.9 Hz, 1H), 2.47 (q, J = 13.5, 12.1 Hz, 2H), 2.38 (s, 3H), 1.90–1.81 (m, 2H), 1.67 (qd, J = 12.3, 4.4 Hz, 2H), 1.46 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 165.5, 160.1, 159.8, 154.6, 152.9, 143.8, 140.0, 137.9, 136.8, 133.8, 129.5, 126.5, 124.2, 122.5, 119.0, 116.6, 94.0, 80.1, 54.1, 43.1 (br), 33.4, 28.5, 21.5.
4-(4-(4-Iodophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)-2-(m-tolyloxy)pyrimidine (9).
General procedure III was used to give 9 (83 mg, 99%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.60–8.54 (m, 2H), 7.84 (d, J = 8.1 Hz, 2H), 7.39 (t, J = 7.9 Hz, 1H), 7.23 (d, J = 8.2 Hz, 2H), 7.17 (d, J = 7.1 Hz, 2H), 7.13–7.09 (m, 1H), 7.02 (d, J = 5.1 Hz, 1H), 4.77–4.68 (m, 1H), 3.39 (dt, J = 12.8, 2.5 Hz, 2H), 2.75 (td, J = 13.1, 3.0 Hz, 2H), 2.42 (s, 3H), 2.24–2.17 (m, 2H), 2.12 (td, J = 12.8, 4.0 Hz, 2H). 13C NMR (125 MHz, MeOD) δ 166.6, 162.3, 158.6, 154.3, 141.6, 139.6, 137.7, 131.7, 130.8, 130.4, 127.6, 126.6, 123.4, 119.9, 118.4, 96.7, 54.0, 44.4, 30.8, 21.4. 19F NMR (470 MHz, MeOD) δ −77.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H25IN5O+ 538.1099, found 538.1100.
tert-Butyl 4-(5-(2-(3-Ethylphenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)piperidine-1-carboxylate (10f).
General procedure IV was used to give 10f (95 mg, 54%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.40 (d, J = 5.2 Hz, 1H), 7.71 (s, 1H), 7.69–7.64 (m, 2H), 7.33 (t, J = 7.8 Hz, 1H), 7.23–7.18 (m, 2H), 7.10 (dt, J = 7.9, 1.3 Hz, 1H), 7.08–7.02 (m, 2H), 6.87 (d, J = 5.1 Hz, 1H), 4.62 (tt, J = 12.0, 3.8 Hz, 1H), 4.07 (s, 3H), 2.67 (q, J = 7.6 Hz, 2H), 2.48–2.33 (m, 2H), 1.85 (ddd, J = 12.0, 4.5, 2.2 Hz, 2H), 1.66 (ddt, J = 15.2, 10.9, 5.3 Hz, 2H), 1.45 (s, 10H), 1.24 (t, J = 7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 165.4, 160.1, 159.7, 154.5, 152.9, 146.8, 146.4, 143.7, 137.9, 136.8, 133.7, 130.5, 129.5, 125.3, 124.1, 121.3, 119.2, 116.6, 94.1, 80.1, 54.2, 43.07 (br), 33.4, 28.5, 27.5, 15.4.
2-(3-Ethylphenoxy)-4-(4-(4-iodophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)pyrimidine (10).
General procedure III was used to give 10 (82 mg, 99%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.56 (d, J = 5.1 Hz, 1H), 8.45 (s, 1H), 7.83 (d, J = 8.1 Hz, 2H), 7.41 (t, J = 7.8 Hz, 1H), 7.21 (d, J = 8.1 Hz, 2H), 7.29–7.15 (m, 2H), 7.13 (dd, J = 8.0, 2.3 Hz, 1H), 7.01 (d, J = 5.1 Hz, 1H), 4.78–4.68 (m, 1H), 3.42–3.35 (m, 2H), 2.77–2.68 (m, 4H), 2.21–2.17 (m, 2H), 2.11–2.03 (m, 1H), 1.26 (t, J = 7.6 Hz, 3H). 13C NMR (125 MHz, MeOD) δ 166.5, 161.9, 159.7, 154.5, 148.0, 142.1, 139.3, 138.1, 132.7, 131.7, 130.8, 126.3, 126.1, 122.3, 120.0, 118.3, 95.7, 53.2, 44.6, 31.0, 29.6, 16.0. 19F NMR (470 MHz, MeOD) δ −77.31. HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H27IN5O+ 552.1255, found 552.1267.
tert-Butyl 4-(5-(2-(3-(tert-Butyl)phenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)piperidine-1-carboxylate (11f).
General procedure IV was used to give 11f (0.10 g, 56%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.41 (d, J = 5.2 Hz, 1H), 7.70 (s, 1H), 7.69 (d, J = 8.6 Hz, 3H), 7.36 (t, J = 7.9 Hz, 1H), 7.31–7.28 (m, 1H), 7.22 (d, J = 8.0 Hz, 2H), 7.09–7.05 (m, 1H), 6.88 (d, J = 5.2 Hz, 1H), 4.66 (tt, J = 12.1, 3.8 Hz, 1H), 4.06 (s, 3H), 2.34 (t, J = 13.1 Hz, 2H), 1.89–1.83 (m, 2H), 1.71–1.60 (m, 2H), 1.45 (s, 9H), 1.32 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 165.4, 160.1, 159.7, 154.5, 153.6, 152.7, 144.0, 137.9, 136.8, 133.9, 130.6, 129.1, 124.1, 122.8, 119.1, 118.9, 116.5, 94.1, 80.1, 54.2, 43.3 (br), 35.0, 33.4, 31.4, 28.5.
2-(3-(tert-Butyl)phenoxy)-4-(4-(4-iodophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)pyrimidine (11).
General procedure III was used to give 11 (84 mg, 96%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.57–8.53 (m, 2H), 7.82 (d, J = 7.9 Hz, 2H), 7.42 (t, J = 7.8 Hz, 1H), 7.37 (dd, J = 8.0, 1.5 Hz, 1H), 7.33 (t, J = 1.9 Hz, 1H), 7.22 (d, J = 8.0 Hz, 2H), 7.11 (dd, J = 7.9, 2.1 Hz, 1H), 7.01 (d, J = 5.1 Hz, 1H), 4.77 (tt, J = 12.0, 4.0 Hz, 1H), 3.42–3.36 (m, 2H), 2.79 (td, J = 13.1, 2.8 Hz, 2H), 2.28–2.22 (m, 2H), 2.11 (qd, J = 13.1, 4.2 Hz, 2H), 1.34 (s, 9H). 13C NMR (125 MHz, MeOD) δ 166.6, 162.4, 158.5, 155.0, 154.2, 139.7, 139.0, 137.5, 131.7, 130.5, 130.0, 126.7, 123.9, 120.1, 119.6, 118.5, 96.9, 54.1, 44.4, 35.8, 31.7, 30.9. 19F NMR (470 MHz, MeOD) δ −77.4. HRMS (ESI-TOF) m/z [M + H]+ calcd for C28H31IN5O+ 580.1568, found 580.1573.
tert-Butyl 4-(5-(2-(3-Ethyl-5-methylphenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)piperidine-1-carboxylate (12f).
General procedure IV was used to give 12f (0.13 g, 75%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.39 (dd, J = 5.2, 1.1 Hz, 1H), 7.70 (s, 1H), 7.69–7.65 (m, 2H), 7.21 (d, J = 8.1 Hz, 2H), 6.92 (s, 1H), 6.87 (s, 1H), 6.86 (s, 2H), 4.66 (tt, J = 12.0, 3.8 Hz, 1H), 2.63 (q, J = 7.6 Hz, 2H), 2.43 (q, J = 13.1, 12.4 Hz, 2H), 2.34 (s, 3H), 1.91–1.85 (m, 2H), 1.72–1.63 (m, 2H), 1.46 (s, 9H), 1.22 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 165.5, 160.0, 159.8, 154.6, 152.9, 146.1, 143.9, 139.6, 137.9, 136.8, 133.9, 130.5, 126.2, 124.1, 119.8, 118.4, 116.4, 94.0, 80.1, 54.2, 43.0 (br), 33.4, 28.7, 28.5, 21.5, 15.5.
2-(3-Ethyl-5-methylphenoxy)-4-(4-(4-iodophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)pyrimidine (12).
General procedure III was used to give 12 (42 mg, 99%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.95 (s, 1H), 8.58 (d, J = 5.1 Hz, 1H), 7.88–7.83 (m, 2H), 7.25–7.22 (m, 2H), 7.02 (d, J = 5.1 Hz, 1H), 7.00 (s, 1H), 6.94 (s, 2H), 4.77 (tt, J = 11.8, 4.0 Hz, 1H), 3.38 (dt, J = 13.0, 2.5 Hz, 2H), 2.75 (td, J = 13.1, 3.1 Hz, 2H), 2.65 (q, J = 7.6 Hz, 2H), 2.37 (s, 3H), 2.29–2.22 (m, 2H), 2.18 (td, J = 12.7, 4.0 Hz, 2H), 1.23 (t, J = 7.6 Hz, 3H). 13C NMR (125 MHz, MeOD) δ 165.2, 161.1, 156.8, 152.9, 146.3, 139.8, 138.3, 137.0, 136.1, 130.3, 128.1, 125.8, 125.4, 119.1, 117.9, 117.0, 95.7, 53.0, 43.0, 29.4, 28.2, 20.0, 14.6. 19F NMR (470 MHz, MeOD) δ −77.4. HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H29IN5O+ 566.1412, found 566.1401.
tert-Butyl 4-(5-(2-(3,5-Dichlorophenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)piperidine-1-carboxylate (13f).
General procedure IV was used to give 13f (0.14 g, 75%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.40 (d, J = 5.2 Hz, 1H), 7.74 (s, 1H), 7.70–7.66 (m, 2H), 7.29 (t, J = 1.9 Hz, 1H), 7.20 (d, J = 1.9 Hz, 1H), 7.19 (d, J = 1.9 Hz, 2H), 6.94 (d, J = 5.2 Hz, 1H), 4.63 (tt, J = 12.0, 3.8 Hz, 1H), 4.18 (s, 2H), 2.56 (t, J = 13.0 Hz, 2H), 1.99–1.93 (m, 2H), 1.75 (qd, J = 12.5, 4.3 Hz, 2H), 1.47 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 164.7, 160.3, 160.0, 154.6, 153.7, 144.4, 137.9, 137.1, 135.7, 133.7, 130.5, 126.1, 123.8, 121.2, 117.5, 94.2, 80.2, 54.4, 43.1 (br), 33.4, 28.5.
2-(3,5-Dichlorophenoxy)-4-(4-(4-iodophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)pyrimidine (13).
General procedure III was used to give 13 (0.12 g, 97%) as a yellow solid. 1H NMR (500 MHz, MeOD) δ 8.58 (d, J = 5.2 Hz, 1H), 8.56 (d, J = 2.8 Hz, 1H), 7.80 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 1.8 Hz, 1H), 7.41 (d, J = 1.8 Hz, 2H), 7.24–7.20 (m, 2H), 7.07 (d, J = 5.1 Hz, 1H), 4.75 (tt, J = 11.9, 4.0 Hz, 1H), 3.53–3.49 (m, 2H), 3.48 (d, J = 3.0 Hz, 2H), 2.96 (td, J = 13.1, 3.0 Hz, 2H), 2.34–2.27 (m, 2H), 2.20 (qd, J = 13.0, 4.1 Hz, 2H). 13C NMR (125 MHz, MeOD) δ 166.5, 161.9, 159.8, 154.4, 142.5, 139.3, 138.2, 133.0, 131.7, 131.1, 126.9, 126.0, 123.0, 118.3, 95.5, 53.1, 44.6, 30.9. 19F NMR (470 MHz, MeOD) δ −77.2. HRMS (ESI-TOF) m/z [M + H]+ calcd for C24H21Cl2IN5O+ 592.0163, found 592.0172.
tert-Butyl 4-(5-(2-(2,5-Dimethylphenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)piperidine-1-carboxylate (14f).
General procedure IV was used to give 14f (0.11 g, 60%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.42 (d, J = 5.1 Hz, 1H), 7.75 (s, 1H), 7.70–7.67 (m, 2H), 7.21–7.16 (m, 3H), 7.00 (d, J = 7.9 Hz, 1H), 6.98 (s, 1H), 6.87 (d, J = 5.1 Hz, 1H), 4.62 (tt, J = 12.0, 4.0 Hz, 1H), 2.48 (d, J = 13.9 Hz, 2H), 2.34 (s, 3H), 2.16 (s, 3H), 1.88–1.82 (m, 2H), 1.68 (qd, J = 12.3, 4.3 Hz, 2H), 1.48 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 165.2, 160.1, 160.0, 154.6, 151.3, 143.7, 137.9, 137.3, 136.8, 133.8, 131.3, 130.5, 127.3, 126.8, 124.2, 122.6, 116.4, 94.0, 80.2, 54.1, 43.0 (br), 33.3, 28.5, 21.1, 16.3.
2-(2,5-Dimethylphenoxy)-4-(4-(4-iodophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)pyrimidine (14).
General procedure III was used to give 14 (84 mg, 95%) as a white solid. 1H NMR (400 MHz, MeOD) δ 8.98 (s, 1H), 8.54 (d, J = 5.0 Hz, 1H), 7.83–7.75 (m, 2H), 7.30–7.23 (m, 2H), 7.23–7.14 (m, 2H), 6.99–6.89 (m, 4H), 6.64 (d, J = 1.5 Hz, 2H), 2.34–2.30 (m, 6H). 13C NMR (125 MHz, MeOD) δ 166.6, 162.4, 158.4, 154.2, 141.1, 139.7, 139.0, 137.6, 131.7, 130.0, 128.4, 126.7, 120.4, 118.4, 96.8, 54.2, 44.4, 30.8, 21.3. 19F NMR (376 MHz, MeOD) δ −77.6. HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H27IN5O+ 552.1255, found 552.1250.
tert-Butyl 4-(4-(4-Iodophenyl)-5-(2-(5-isopropyl-2-methylphenoxy)pyrimidin-4-yl)-1H-imidazol-1-yl)piperidine-1-carboxylate (15f).
General procedure IV was used to give 15f (0.10 g, 55%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.39 (d, J = 5.1 Hz, 1H), 7.71 (s, 1H), 7.68 (d, J = 8.1 Hz, 2H), 7.20 (dd, J = 8.2, 3.0 Hz, 3H), 7.07 (dd, J = 7.9, 1.7 Hz, 1H), 7.03 (d, J = 1.7 Hz, 1H), 6.86 (d, J = 5.1 Hz, 1H), 4.65 (tt, J = 12.0, 3.8 Hz, 1H), 2.90 (p, J = 6.9 Hz, 1H), 2.41 (q, J = 10.3, 7.3 Hz, 2H), 2.16 (s, 3H), 1.90–1.82 (m, 2H), 1.67 (qd, J = 12.0, 4.2 Hz, 2H), 1.46 (s, 9H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 165.2, 160.1, 159.9, 154.5, 151.3, 148.5, 143.9, 137.9, 136.8, 133.8, 131.3, 130.5, 127.6, 124.1, 124.0, 120.3, 116.3, 94.1, 80.1, 54.2, 43.0 (br), 33.7, 33.4, 28.5, 24.0, 16.3.
4-(4-(4-Iodophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)-2-(5-isopropyl-2-methylphenoxy)pyrimidine (15).
General procedure III was used to give 15 (84 mg, 98%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.54 (d, J = 5.1 Hz, 1H), 8.43 (s, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 7.8 Hz, 1H), 7.21 (d, J = 8.0 Hz, 2H), 7.15 (dd, J = 7.8, 1.8 Hz, 1H), 7.12 (d, J = 1.7 Hz, 1H), 7.01 (d, J = 5.2 Hz, 1H), 4.75 (tt, J = 12.0, 4.0 Hz, 1H), 3.46–3.40 (m, 2H), 2.94 (p, J = 6.9 Hz, 1H), 2.74 (td, J = 13.1, 2.9 Hz, 2H), 2.21 (s, 6H), 2.11 (qd, J = 13.1, 4.2 Hz, 3H), 1.26 (d, J = 6.9 Hz, 6H). 13C NMR (125 MHz, MeOD) δ 166.3, 162.3, 159.1, 152.5, 150.0, 139.6, 137.7, 132.5, 131.7, 131.1, 128.6, 126.4, 125.1, 121.4, 118.2, 96.6, 54.0, 44.5, 34.8, 30.8, 24.4, 16.1. 19F NMR (470 MHz, MeOD) δ −77.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C28H31IN5O+ 580.1568, found 580.1568.
tert-Butyl 4-(5-(2-(2-Hydroxyphenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)piperidine-1-carboxylate (16f).
General procedure IV was used to give 16f (52 mg, 30%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.36 (d, J = 5.2 Hz, 1H), 7.73 (s, 1H), 7.61 (d, J = 8.1 Hz, 2H), 7.19 (dd, J = 8.0, 1.5 Hz, 1H), 7.12 (dd, J = 8.3, 2.3 Hz, 3H), 7.02 (dd, J = 8.2, 1.5 Hz, 1H), 6.93 (td, J = 7.7, 1.5 Hz, 1H), 6.85 (d, J = 5.0 Hz, 1H), 4.56 (tt, J = 12.0, 4.0 Hz, 1H), 4.06 (s, 3H), 2.49 (s, 2H), 1.78 (dd, J = 12.3, 3.6 Hz, 2H), 1.63 (tt, J = 12.4, 6.2 Hz, 2H), 1.46 (s, 10H). 13C NMR (125 MHz, CDCl3) δ 165.0, 160.0, 159.9, 154.6, 148.5, 143.8, 140.9, 137.9, 137.1, 133.4, 130.5, 127.0, 124.0, 123.0, 120.8, 118.0, 117.0, 94.2, 80.2, 54.1, 43.0 (br), 33.3, 28.5.
2-((4-(4-(4-Iodophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)-pyrimidin-2-yl)oxy)phenol (16).
General procedure III was used to give 16 (43 mg, 98%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.52 (d, J = 5.2 Hz, 1H), 8.30 (s, 1H), 7.78 (d, J = 8.1 Hz, 2H), 7.26–7.20 (m, 1H), 7.17 (dd, J = 8.4, 6.4 Hz, 3H), 7.01–6.98 (m, 1H), 6.95 (dd, J = 11.5, 6.3 Hz, 2H), 4.81–4.70 (m, 1H), 3.45–3.38 (m, 2H), 2.86 (td, J = 13.1, 3.1 Hz, 2H), 2.14 (d, J = 12.8 Hz, 2H), 2.10–1.99 (m, 2H). 13C NMR (125 MHz, MeOD) δ 166.4, 162.0, 159.0, 150.5, 142.3, 139.5, 137.7, 131.6, 131.2, 128.0, 126.4, 124.0, 121.3, 118.4, 118.3, 96.3, 53.6, 44.5, 30.7. 19F NMR (470 MHz, MeOD) δ −77.2. HRMS (ESI-TOF) m/z [M + H]+ calcd for C24H23IN5O2+ 540.0891, found 540.0878.
tert-Butyl 4-(4-(4-Iodophenyl)-5-(2-(2-methoxyphenoxy)-pyrimidin-4-yl)-1H-imidazol-1-yl)piperidine-1-carboxylate (17f).
General procedure IV was used to give 17f (49 mg, 28%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.39 (d, J = 5.1 Hz, 1H), 7.69 (s, 1H), 7.66 (d, J = 8.1 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 7.17 (d, J = 8.1 Hz, 2H), 7.02 (dd, J = 8.3, 6.0 Hz, 2H), 6.86 (d, J = 5.1 Hz, 1H), 4.61 (tt, J = 12.0, 3.9 Hz, 1H), 3.75 (s, 3H), 2.54 (s, 2H), 1.89–1.81 (m, 2H), 1.67 (qd, J = 12.3, 4.4 Hz, 2H), 1.46 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 165.3, 160.0, 159.8, 154.6, 151.7, 143.3, 142.1, 137.8, 136.6, 133.7, 130.4, 126.7, 124.3, 122.9, 121.3, 116.7, 113.3, 93.9, 80.1, 56.2, 54.1, 43.1 (br), 33.3, 28.5.
4-(4-(4-Iodophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)-2-(2-methoxyphenoxy)pyrimidine (17).
General procedure III was used to give 17 (50 mg, 98%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.92 (s, 1H), 8.58 (d, J = 5.1 Hz, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 7.17 (d, J = 8.0 Hz, 3H), 7.06 (t, J = 7.7 Hz, 1H), 7.01 (d, J = 5.0 Hz, 1H), 4.75 (p, J = 7.9 Hz, 1H), 3.76 (s, 3H), 3.48–3.38 (m, 2H), 2.91–2.79 (m, 2H), 2.15 (dt, J = 9.8, 4.8 Hz, 4H). 13C NMR (125 MHz, MeOD) δ 166.4, 162.3, 158.2, 153.0, 143.2, 139.7, 138.4, 137.5, 131.6, 129.6, 128.2, 126.7, 123.6, 122.4, 118.4, 114.6, 97.0, 56.7, 54.2, 44.4, 30.6. 19F NMR (470 MHz, MeOD) δ −77.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H25IN5O2+ 554.1048, found 554.1048.
tert-Butyl 4-(5-(2-(Benzo[d][1,3]dioxol-5-yloxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)piperidine-1-carboxylate (18f).
General procedure IV was used to give 18f (0.11 g, 60%) as a light-purple solid. 1H NMR (500 MHz, CDCl3) δ 8.42 (d, J = 5.1 Hz, 1H), 7.77 (s, 1H), 7.68 (d, J = 8.1 Hz, 2H), 7.20 (d, J = 8.1 Hz, 2H), 6.89 (d, J = 5.1 Hz, 1H), 6.84 (d, J = 8.3 Hz, 1H), 6.75 (d, J = 2.3 Hz, 1H), 6.69 (dd, J = 8.3, 2.4 Hz, 1H), 6.02 (s, 2H), 4.73–4.63 (m, 1H), 4.18 (s, 2H), 2.59 (s, 2H), 1.94 (dt, J = 12.6, 3.0 Hz, 3H), 1.81–1.66 (m, 2H), 1.48 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 165.7, 160.1, 160.0, 154.6, 148.4, 147.3, 145.3, 143.8, 137.9, 136.9, 133.8, 130.5, 124.2, 116.8, 114.3, 108.4, 104.2, 102.0, 94.0, 80.2, 54.2, 43.1 (br), 33.5, 28.5.
2-(Benzo[d][1,3]dioxol-5-yloxy)-4-(4-(4-iodophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)pyrimidine (18).
General procedure III was used to give 18 (88 mg, 96%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.56–8.53 (m, 1H), 7.82–7.77 (m, 2H), 7.19 (d, J = 8.1 Hz, 2H), 7.00 (dd, J = 5.1, 1.3 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 2.3 Hz, 1H), 6.75 (dd, J = 8.5, 2.3 Hz, 1H), 6.03 (s, 2H), 4.74 (td, J = 13.3, 12.7, 5.6 Hz, 1H), 3.49–3.44 (m, 2H), 2.91 (td, J = 13.1, 3.0 Hz, 2H), 2.28–2.21 (m, 2H), 2.14 (qd, J = 13.1, 3.9 Hz, 2H). 13C NMR (125 MHz, MeOD) δ 166.9, 162.3, 158.8, 149.8, 148.7, 146.8, 139.8, 139.5, 137.7, 131.6, 130.8, 126.6, 118.6, 115.2, 109.3, 105.0, 103.4, 96.4, 53.8, 44.5, 30.9. 19F NMR (470 MHz, MeOD) δ −77.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H23IN5O3+ 568.0841, found 568.0850.
Synthesis of tert-Butyl (S,Z)-3-(((2-(Methylthio)pyrimidin-4-yl)methylene)amino)pyrrolidine-1-carboxylate (19a) and tert-Butyl (R,Z)-3-(((2-(Methylthio)pyrimidin-4-yl)methylene)amino)pyrrolidine-1-carboxylate (21a).
To a solution of 2-(methylthio)pyrimidine-4-carbaldehyde (0.15 g, 1.0 mmol) and tert-butyl (S)-3-aminopyrrolidine-1-carboxylate (0.19 g, 1.0 mmol) or tert-butyl (R)-3-aminopyrrolidine-1-carboxylate (0.19 g, 1.0 mmol) in dichloromethane (3.5 mL) was added anhydrous magnesium sulfate (0.12 g, 1.0 mmol), and the reaction was stirred at room temperature for 16 h and monitored by TLC. The reaction mixture was filtered, and the solvent was evaporated under reduced pressure to give the desired product 19a or 21a without further purification.
19a (0.32 g, quant) was obtained as yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.56 (d, J = 5.0 Hz, 1H), 8.23 (s, 1H), 7.58 (dd, J = 5.1, 1.3 Hz, 1H), 4.09 (t, J = 5.7 Hz, 1H), 3.71–3.26 (m, 5H), 2.58 (d, J = 1.3 Hz, 3H), 2.14 (h, J = 6.3 Hz, 1H), 1.99 (dq, J = 12.8, 6.3 Hz, 1H), 1.47 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 161.2, 160.3, 157.9, 154.6, 112.4, 79.5, 69.0, 68.3, 52.4, 52.1, 45.0, 44.6, 33.6, 32.9, 28.7, 14.3.
21a (0.32 g, quant) was obtained as yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.56 (d, J = 5.1 Hz, 1H), 8.23 (s, 1H), 7.58 (d, J = 5.1 Hz, 1H), 4.14–4.06 (m, 1H), 3.70–3.31 (m, 4H), 2.58 (s, 3H), 2.14 (dd, J = 11.4, 6.0 Hz, 1H), 1.99 (dt, J = 12.7, 6.1 Hz, 1H), 1.47 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 161.2, 160.3, 157.9, 154.6, 112.4, 79.5, 69.0, 68.3, 52.4, 52.1, 45.0, 44.6, 33.6, 32.9, 28.7, 14.2.
tert-Butyl (S)-3-(4-(4-Iodophenyl)-5-(2-(methylthio)pyrimidin-4-yl)-1H-imidazol-1-yl)pyrrolidine-1-carboxylate (19b).
General procedure I was used to give 19b (0.22 g, 76%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.37 (d, J = 5.2 Hz, 1H), 7.74–7.69 (m, 1H), 7.68–7.63 (m, 2H), 7.22–7.17 (m, 2H), 6.82 (d, J = 5.2 Hz, 1H), 5.37 (d, J = 22.1 Hz, 1H), 3.88–3.47 (m, 4H), 2.58 (s, 3H), 2.40 (dp, J = 21.1, 7.5, 5.9 Hz, 1H), 2.26 (dtd, J = 13.0, 6.2, 4.6 Hz, 1H), 1.47 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 173.2, 157.6, 157.4, 137.8, 136.7, 133.6, 130.3, 117.2, 94.0, 80.3, 55.8, 55.2, 52.4, 51.8, 44.2, 44.0, 33.2, 32.2, 28.6, 14.3.
tert-Butyl (R)-3-(4-(4-Iodophenyl)-5-(2-(methylthio)pyrimidin-4-yl)-1H-imidazol-1-yl)pyrrolidine-1-carboxylate (21b).
General procedure I was used to give 21b (0.18 g, 63%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.37 (d, J = 5.2 Hz, 1H), 7.71 (d, J = 10.6 Hz, 1H), 7.68–7.63 (m, 2H), 7.22–7.17 (m, 2H), 6.82 (d, J = 5.2 Hz, 1H), 5.37 (d, J = 22.0 Hz, 1H), 3.88–3.45 (m, 4H), 2.58 (s, 3H), 2.40 (dt, J = 14.0, 9.4 Hz, 1H), 2.26 (dtd, J = 13.0, 6.3, 4.7 Hz, 1H), 1.47 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 173.2, 157.6, 157.4, 137.9, 136.7, 133.6, 130.3, 117.2, 94.0, 80.3, 55.8, 55.2, 52.4, 51.8, 44.2, 44.0, 33.2, 32.3, 28.6, 14.3.
tert-Butyl (S)-3-(4-(4-Iodophenyl)-5-(2-(methylsulfonyl)pyrimidin-4-yl)-1H-imidazol-1-yl)pyrrolidine-1-carboxylate (19c).
General procedure II was used to give 19c (98 mg, 57%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.62 (d, J = 5.4 Hz, 1H), 7.79 (d, J = 4.4 Hz, 1H), 7.76–7.69 (m, 2H), 7.34 (d, J = 5.4 Hz, 1H), 7.25–7.17 (m, 2H), 5.52 (s, 1H), 3.94–3.48 (m, 4H), 3.38 (s, 3H), 2.67–2.22 (m, 2H), 1.48 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 159.2, 157.7, 138.33, 138.30, 133.3, 130.5, 123.0, 95.0, 80.4, 56.5, 52.4, 51.7, 44.1, 43.9, 39.2, 33.3, 32.5, 28.6.
tert-Butyl (R)-3-(4-(4-Iodophenyl)-5-(2-(methylsulfonyl)pyrimidin-4-yl)-1H-imidazol-1-yl)pyrrolidine-1-carboxylate (21c).
General procedure II was used to give 21c (91 mg, 53%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.62 (d, J = 5.4 Hz, 1H), 7.79 (s, 1H), 7.75–7.69 (m, 2H), 7.34 (d, J = 5.4 Hz, 1H), 7.23–7.18 (m, 2H), 5.52 (s, 1H), 4.02–3.45 (m, 3H), 3.38 (s, 3H), 2.64–2.24 (m, 2H), 1.48 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 159.2, 157.7, 138.33, 138.30, 133.3, 130.5, 123.0, 95.0, 80.4, 56.5, 51.7, 44.1, 39.2, 33.3, 28.6.
tert-Butyl (S)-3-(5-(2-(3,5-Dimethylphenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)pyrrolidine-1-carboxylate (19d).
General procedure IV was used to give 19d (93 mg, 54%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 5.2 Hz, 1H), 7.71–7.67 (m, 2H), 7.64 (s, 1H), 7.23–7.19 (m, 2H), 6.91 (s, 1H), 6.87 (d, J = 5.2 Hz, 1H), 6.82 (d, J = 1.5 Hz, 2H), 5.29 (p, J = 5.4 Hz, 1H), 3.68–3.28 (m, 4H), 2.34 (s, 6H), 2.00–1.92 (m, 2H), 1.48 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 165.8, 160.0, 159.5, 153.1, 139.7, 137.9, 137.0, 133.9, 130.6, 127.5, 119.6, 116.0, 94.2, 55.9, 55.3, 51.3, 44.0, 32.9, 32.1, 28.6, 21.5.
tert-Butyl (R)-3-(5-(2-(3,5-Dimethylphenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)pyrrolidine-1-carboxylate (21d).
General procedure IV was used to give 21d (0.10 g, 60%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 5.2 Hz, 1H), 7.72–7.67 (m, 2H), 7.64 (s, 1H), 7.24–7.18 (m, 2H), 6.91 (s, 1H), 6.87 (d, J = 5.2 Hz, 1H), 6.82 (s, 2H), 5.34–5.24 (m, 1H), 3.67–3.29 (m, 4H), 2.34 (s, 6H), 2.00–1.93 (m, 2H), 1.48 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 165.8, 160.0, 159.6, 153.1, 139.7, 137.9, 137.1, 130.6, 127.5, 119.6, 116.0, 94.2, 80.3, 55.3, 53.0, 51.9, 51.3, 43.9, 32.9, 28.6, 21.5.
(S)-2-(3,5-Dimethylphenoxy)-4-(4-(4-iodophenyl)-1-(pyrrolidin-3-yl)-1H-imidazol-5-yl)pyrimidine (19).
General procedure III was used to give 19 (75 mg, 96%) as a brown solid. 1H NMR (400 MHz, MeOD) δ 8.57 (s, 1H), 8.50 (d, J = 5.2 Hz, 1H), 7.88–7.78 (m, 2H), 7.26–7.19 (m, 2H), 7.01–6.95 (m, 2H), 6.88 (dt, J = 1.5, 0.7 Hz, 2H), 5.39 (ddd, J = 7.7, 5.1, 2.6 Hz, 1H), 3.64 (dt, J = 12.1, 7.3 Hz, 1H), 3.55 (dd, J = 13.2, 4.7 Hz, 1H), 3.41–3.33 (m, 2H), 2.48–2.38 (m, 1H), 2.35 (s, 6H). 13C NMR (100 MHz, MeOD) δ 166.7, 162.1, 158.7, 154.4, 141.3, 139.6, 138.0, 131.7, 130.8, 128.5, 126.9, 120.4, 117.9, 96.7, 57.9, 51.8, 45.8, 31.9, 21.3. 19F NMR (376 MHz, MeOD) δ −77.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H25IN5O+ 538.1099, found 538.1081. [α]589mm25°C (c 0.074, MeOH) = −0.095.
(R)-2-(3,5-Dimethylphenoxy)-4-(4-(4-iodophenyl)-1-(pyrrolidin-3-yl)-1H-imidazol-5-yl)pyrimidine (21).
General procedure III was used to give 21 (91 mg, quant) as a yellow solid. 1H NMR (400 MHz, MeOD) δ 8.57 (s, 1H), 8.50 (d, J = 5.2 Hz, 1H), 7.85–7.79 (m, 2H), 7.26–7.18 (m, 2H), 7.02–6.95 (m, 2H), 6.90–6.85 (m, 2H), 5.45–5.32 (m, 1H), 3.64 (dt, J = 12.1, 7.4 Hz, 1H), 3.55 (dd, J = 13.2, 4.7 Hz, 1H), 3.43–3.34 (m, 2H), 2.42 (dd, J = 13.5, 6.9 Hz, 1H), 2.35 (s, 6H). 13C NMR (100 MHz, MeOD) δ 165.4, 160.8, 157.2, 153.0, 139.9, 138.2, 136.6, 130.3, 129.2, 127.1, 119.0, 116.5, 95.4, 56.6, 50.4, 44.4, 30.5, 19.9. 19F NMR (376 MHz, MeOD) δ −77.4. HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H25IN5O+ 538.1099, found 538.1097. [α]589mm25°C (c 0.053, MeOH) = 0.25.
tert-Butyl (S)-(2-(3-(5-(2-(3,5-Dimethylphenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)pyrrolidin-1-yl)ethyl)-carbamate (19e).
General procedure V:
A mixture of 19e (0.10 mmol, 1.0 equiv), 2-(Boc-amino)ethyl bromide (0.15 mmol, 1.5 equiv), NaI (0.15 mmol, 1.5 equiv), and K2CO3 (0.70 mmol, 7.0 equiv) was added into methyl ethyl ketone (0.30 mL). The reaction was stirred at room temperature for 48 h then quenched by water and extracted with ethyl acetate (3 × 20 mL). The combined organic phase was dried over anhydrous magnesium sulfate and concentrated under reduced pressure and purified by silica gel chromatography using DCM and methanol (0–20%) as eluent to give the desired product 19e (41 mg, 60%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.37 (d, J = 5.2 Hz, 1H), 8.00 (s, 1H), 7.69–7.65 (m, 2H), 7.22–7.18 (m, 2H), 6.90 (s, 1H), 6.86 (d, J = 5.2 Hz, 1H), 6.82 (d, J = 1.5 Hz, 2H), 5.20–5.14 (m, 1H), 4.87 (s, 1H), 3.25 (q, J = 6.0 Hz, 2H), 3.09 (d, J = 9.2 Hz, 1H), 2.93 (d, J = 10.5 Hz, 1H), 2.57 (t, J = 6.0 Hz, 2H), 2.34 (s, 6H), 2.18 (qd, J = 11.6, 8.3, 4.0 Hz, 1H), 2.11–2.04 (m, 1H), 1.85 (q, J = 9.4, 7.3 Hz, 1H), 1.45 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 165.7, 159.84, 159.81, 153.2, 143.5, 139.7, 138.1, 137.9, 134.1, 130.6, 127.4, 124.4, 119.6, 116.2, 93.9, 61.2, 55.2, 55.1, 39.0, 33.7, 28.6, 21.5.
tert-Butyl (R)-(2-(3-(5-(2-(3,5-Dimethylphenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)pyrrolidin-1-yl)ethyl)-carbamate (21e).
General procedure V was used to give 21e (38 mg, 55%) as a pale-yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.37 (d, J = 5.2 Hz, 1H), 8.00 (s, 1H), 7.69–7.65 (m, 2H), 7.22–7.18 (m, 2H), 6.90 (s, 1H), 6.86 (d, J = 5.2 Hz, 1H), 6.82 (d, J = 1.5 Hz, 2H), 5.17 (dqt, J = 6.7, 3.9, 2.3 Hz, 1H), 4.87 (s, 1H), 3.24 (t, J = 6.0 Hz, 2H), 3.09 (td, J = 8.8, 3.1 Hz, 1H), 2.93 (d, J = 10.5 Hz, 1H), 2.56 (t, J = 6.0 Hz, 2H), 2.34 (s, 6H), 2.32 (s, 1H), 2.17 (q, J = 8.6 Hz, 1H), 2.06 (ddt, J = 17.2, 8.4, 3.3 Hz, 1H), 1.85 (ddd, J = 13.6, 8.3, 4.0 Hz, 1H), 1.45 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 165.7, 159.84, 159.81, 156.1, 153.2, 143.5, 139.7, 138.1, 137.9, 134.1, 130.6, 127.4, 124.4, 119.6, 116.2, 93.9, 61.2, 55.2, 55.1, 53.1, 39.1, 33.8, 28.6, 21.5.
(S)-2-(3-(5-(2-(3,5-Dimethylphenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)pyrrolidin-1-yl)ethan-1-amine (20).
General procedure III was used to give 20 (36 mg, 96%) as a brown solid. 1H NMR (400 MHz, MeOD) δ 8.74 (s, 1H), 8.53 (d, J = 5.1 Hz, 1H), 7.87–7.80 (m, 2H), 7.23–7.17 (m, 2H), 7.00–6.95 (m, 2H), 6.86 (s, 2H), 5.26–5.17 (m, 1H), 3.21–3.06 (m, 3H), 2.92 (ddd, J = 13.4, 8.2, 5.4 Hz, 1H), 2.76 (dt, J = 12.9, 5.3 Hz, 1H), 2.51–2.38 (m, 1H), 2.35 (s, 6H), 2.18 (dtd, J = 14.2, 8.5, 2.9 Hz, 1H), 2.08–1.96 (m, 1H). 13C NMR (100 MHz, MeOD) δ 166.8, 162.1, 158.6, 154.5, 141.2, 139.6, 138.1, 131.7, 128.4, 126.7, 120.5, 117.9, 96.8, 61.4, 58.1, 53.5, 52.9, 38.2, 33.6, 21.4. 19F NMR (376 MHz, MeOD) δ −77.1. HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H30IN6O+ 581.1521, found 581.1527. [α]589mm25°C (c 0.074, MeOH) = −0.041.
(R)-2-(3-(5-(2-(3,5-Dimethylphenoxy)pyrimidin-4-yl)-4-(4-iodophenyl)-1H-imidazol-1-yl)pyrrolidin-1-yl)ethan-1-amine (22).
General procedure III was used to give 22 (37 mg, 98%) as a brown solid. 1H NMR (400 MHz, MeOD) δ 8.71 (s, 1H), 8.52 (d, J = 5.1 Hz, 1H), 7.86–7.77 (m, 2H), 7.25–7.16 (m, 2H), 7.00–6.93 (m, 2H), 6.86 (d, J = 1.6 Hz, 2H), 5.21 (ddq, J = 10.8, 6.4, 3.2, 2.5 Hz, 1H), 3.20–3.07 (m, 3H), 2.91 (ddd, J = 13.4, 8.2, 5.3 Hz, 1H), 2.75 (dt, J = 13.0, 5.3 Hz, 1H), 2.44 (dd, J = 11.1, 6.9 Hz, 1H), 2.35 (s, 6H), 2.32–2.23 (m, 1H), 2.17 (dtd, J = 14.4, 8.7, 2.9 Hz, 1H), 2.00 (dtd, J = 13.5, 8.4, 4.0 Hz, 1H). 13C NMR (100 MHz, MeOD) δ 166.8, 162.1, 158.8, 154.5, 141.2, 139.6, 138.2, 131.7, 128.4, 126.6, 120.5, 117.9, 97.0, 61.5, 58.0, 53.4, 52.9, 38.3, 33.7, 21.4. 19F NMR (376 MHz, MeOD) δ −77.1. HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H30IN6O+ 581.1521, found 581.1496. [α]589mm25°C (c 0.089, MeOH) = 0.16.
tert-Butyl 4-(5-(2-(3-Ethyl-5-methylphenoxy)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)piperidine-1-carboxylate (23b).
General procedure IV was used to give 24b as a white solid (0.11 g, 69%). 1H NMR (400 MHz, CDCl3) δ 8.42 (d, J = 5.1 Hz, 1H), 7.73 (s, 1H), 7.60 (s, 4H), 6.93 (s, 1H), 6.88 (d, J = 1.4 Hz, 2H), 6.85 (d, J = 5.2 Hz, 1H), 4.66 (tt, J = 12.0, 3.8 Hz, 1H), 2.64 (q, J = 7.6 Hz, 2H), 2.46 (q, J = 12.7 Hz, 2H), 2.36 (s, 3H), 1.95–1.85 (m, 2H), 1.73–1.64 (m, 3H), 1.47 (s, 9H), 1.23 (t, J = 7.7 Hz, 4H). 13C NMR (100 MHz, CDCl3) δ 165.6, 160.2, 159.7, 154.6, 152.9, 146.2, 143.3, 139.7, 138.0, 136.9, 130.1 (d, J = 32.5 Hz), 129.0, 126.3, 125.7 (q, J = 3.9 Hz), 124.8, 119.8, 118.4, 116.6, 80.2, 54.3, 43.1, 33.4, 28.8, 28.5, 21.5, 15.5. 19F NMR (376 MHz, CDCl3) δ −62.6.
2-(3-Ethyl-5-methylphenoxy)-4-(1-(piperidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-5-yl)pyrimidine (23).
General procedure III was used to give 23 (31 mg, 96%) as a white solid. 1H NMR (400 MHz, MeOD) δ 8.55 (d, J = 5.1 Hz, 1H), 8.53 (s, 1H), 7.77–7.73 (m, 2H), 7.67–7.63 (m, 2H), 7.01–6.98 (m, 2H), 6.96–6.94 (m, 2H), 4.71 (tt, J = 11.9, 4.0 Hz, 1H), 3.41–3.34 (m, 2H), 2.75 (td, J = 13.1, 3.0 Hz, 2H), 2.66 (q, J = 7.6 Hz, 2H), 2.37 (s, 3H), 2.24 (d, J = 13.3 Hz, 2H), 2.12 (qd, J = 13.1, 4.1 Hz, 2H), 1.24 (t, J = 7.6 Hz, 3H). 13C NMR (125 MHz, MeOD) δ 166.7, 162.4, 158.7, 154.3, 147.6, 141.2, 139.3, 138.0, 135.4, 132.9–131.8 (m), 130.7, 127.1 (q, J = 3.9 Hz), 125.4 (d, J = 271.4 Hz), 120.5, 119.3, 118.5, 53.9, 44.5, 30.9, 29.6, 21.4, 16.0. 19F NMR (376 MHz, MeOD) δ −64.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C28H29F3N5O+ 508.2319, found 508.2314.
2-(4-(5-(2-(3-Ethyl-5-methylphenoxy)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)piperidin-1-yl)-N,N-dimethylethan-1-amine (24).
General procedure VI:
Methyl ethyl ketone (0.30 mL) was added to a mixture of 23 (0.10 mmol, 1.0 equiv), 2-Chloro-N,N-dimethylethylamine hydrochloride (0.15 mmol, 1.5 equiv), NaI (0.15 mmol, 1.5 equiv), and K2CO3 (0.70 mmol, 7.0 equiv). The reaction was stirred at room temperature for 48 h then quenched by water and extracted with ethyl acetate (3 × 20 mL). The combined organic phase was dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give the crude product, which was purified by HPLC using acetonitrile and 0.1% TFA water (40–60%) as eluent to give the desired product 24 (20 mg, 35%). 1H NMR (400 MHz, MeOD) δ 8.55 (s, 1H), 8.53 (d, J = 5.1 Hz, 1H), 7.74 (d, J = 8.3 Hz, 2H), 7.64 (d, J = 8.1 Hz, 2H), 6.98 (d, J = 5.1 Hz, 2H), 6.92 (d, J = 2.1 Hz, 2H), 4.62 (dt, J = 10.7, 5.3 Hz, 1H), 4.00–3.92 (m, 2H), 3.78–3.68 (m, 4H), 3.18 (d, J = 11.2 Hz, 2H), 3.10 (t, J = 6.2 Hz, 2H), 2.99 (s, 6H), 2.65 (q, J = 7.6 Hz, 2H), 2.37 (s, 3H), 2.23–2.12 (m, 2H), 2.11–2.02 (m, 4H), 1.24 (t, J = 7.6 Hz, 3H). 13C NMR (125 MHz, MeOD) δ 166.7, 162.2, 159.0, 154.3, 147.5, 147.5, 141.0, 139.3, 138.1, 135.6, 132.0 (q, J = 32.4 Hz), 130.6, 127.2, 127.1 (q, J = 3.9 Hz), 125.4 (d, J = 271.6 Hz), 120.6, 119.3, 118.3, 61.4, 59.4, 55.1, 53.4, 52.5, 50.7, 44.2, 32.3, 29.7, 21.5, 16.1. 19F NMR (376 MHz, MeOD) δ −64.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C32H38F3N6O+ 579.3054, found 579.3050.
tert-Butyl 4-(5-(2-(5-Isopropyl-2-methylphenoxy)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)piperidine-1-carboxylate (25b).
General procedure IV was used to give 25b as a white solid (82 mg, 51%). 1H NMR (400 MHz, CDCl3) δ 8.44 (d, J = 5.1 Hz, 1H), 7.76 (s, 1H), 7.65–7.59 (m, 4H), 7.24 (d, J = 7.8 Hz, 1H), 7.10 (dd, J = 7.8, 1.8 Hz, 1H), 7.07 (d, J = 1.8 Hz, 1H), 6.87 (d, J = 5.1 Hz, 1H), 4.67 (tq, J = 12.0, 3.8 Hz, 1H), 2.93 (p, J = 6.9 Hz, 1H), 2.47 (t, J = 12.8 Hz, 2H), 2.20 (s, 3H), 1.95–1.86 (m, 2H), 1.78–1.69 (m, 3H), 1.50 (s, 9H), 1.27 (d, J = 6.9 Hz, 7H). 13C NMR (100 MHz, CDCl3) δ 165.3, 160.3, 159.9, 154.6, 151.3, 148. 5, 143.3, 138.0, 136.9, 131.4, 130.1 (d, J = 32.3 Hz), 129.9, 129.0, 127.6, 125.7 (q, J = 3.8 Hz), 124.7, 124.1, 120.3, 116.5, 80.2, 54.2, 43.1 (br), 33.7, 33.4, 28.5, 24.1, 16.3. 19F NMR (470 MHz, MeOD) δ −62.5.
2-(5-Isopropyl-2-methylphenoxy)-4-(1-(piperidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-5-yl)pyrimidine (25).
General procedure III was used to give 25 (33 mg, 99%) as a white solid. 1H NMR (400 MHz, MeOD) δ 8.60–8.54 (m, 2H), 7.80–7.73 (m, 2H), 7.68–7.61 (m, 2H), 7.29 (d, J = 7.7 Hz, 1H), 7.17–7.11 (m, 2H), 7.02 (d, J = 5.1 Hz, 1H), 4.74 (tt, J = 11.4, 4.4 Hz, 1H), 3.48–3.39 (m, 2H), 2.94 (hept, J = 6.9 Hz, 1H), 2.77 (td, J = 12.9, 3.5 Hz, 2H), 2.28–2.09 (m, 7H), 1.26 (d, J = 6.9 Hz, 6H). 13C NMR (100 MHz, MeOD) δ 166.4, 162.3, 159.4, 152.6, 150.0, 140.7, 138.2, 136.5, 132.5, 132.0 (d, J = 32.3 Hz), 130.6, 128.6, 127.0 (q, J = 3.8 Hz), 125.1, 121.4, 118.3, 53.6, 44.6, 34.8, 30.9, 24.4, 16.1. 19F NMR (376 MHz, MeOD) δ −64.2. HRMS (ESI-TOF) m/z [M + H]+ calcd for C29H31F3N5O+ 522.2476, found 522.2474.
2-(4-(5-(2-(5-Isopropyl-2-methylphenoxy)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)piperidin-1-yl)-N,N-dimethylethan-1-amine (26).
General procedure VI was used to give 26 (27 mg,45%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.71 (s, 1H), 8.55 (d, J = 5.1 Hz, 1H), 7.75 (d, J = 8.1 Hz, 2H), 7.63 (d, J = 8.1 Hz, 2H), 7.30–7.25 (m, 1H), 7.12 (dd, J = 7.8, 1.8 Hz, 1H), 7.08 (d, J = 1.8 Hz, 1H), 7.02 (d, J = 5.1 Hz, 1H), 4.73 (tt, J = 12.0, 4.2 Hz, 1H), 3.56 (t, J = 6.6 Hz, 2H), 3.48 (d, J = 12.3 Hz, 2H), 3.36 (t, J = 6.7 Hz, 2H), 2.91 (p, J = 6.9 Hz, 1H), 2.69–2.59 (m, 2H), 2.33 (qd, J = 12.8, 3.8 Hz, 2H), 2.23–2.19 (m, 2H), 2.18 (s, 4H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (125 MHz, MeOD) δ 166.5, 162.3, 159.2, 152.5, 139.7, 138.1, 135.7, 132.5, 132.15 (d, J = 32.3 Hz), 130.6, 128.6, 127.1 (q, J = 3.8 Hz), 125.4 (d, J = 273.5 Hz), 125.1, 121.3, 118.3, 54.5, 53.6, 53.5, 51.8, 44.0, 34.8, 31.7, 24.4, 16.1. 19F NMR (470 MHz, MeOD) δ −64.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C33H40F3N6O+ 593.3211, found 593.3213.
tert-Butyl (S)-3-(5-(2-(Methylthio)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)pyrrolidine-1-carboxylate (27b).
General procedure I was used to give 27b (0.21 g, 85%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.41 (d, J = 5.1 Hz, 1H), 7.82 (s, 1H), 7.59 (s, 4H), 6.81 (d, J = 5.2 Hz, 1H), 5.35 (d, J = 21.2 Hz, 1H), 3.81 (d, J = 27.0 Hz, 1H), 3.62–3.53 (m, 2H), 2.59 (s, 3H), 2.43 (dd, J = 16.5, 8.3 Hz, 1H), 2.34–2.24 (m, 1H), 1.48 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 173.6, 157.7, 157.2, 142.3, 136.6, 130.2 (d, J = 32.8 Hz), 128.8, 125.73 (q, J = 3.7 Hz), 125.71 (q, J = 3.7 Hz), 117.3, 80.5, 56.0, 55.5, 52.4, 51.8, 44.2, 44.0, 33.1, 32.3, 28.6, 14.3. 19F NMR (470 MHz, CDCl3) δ −62.6.
tert-Butyl (R)-3-(5-(2-(Methylthio)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)pyrrolidine-1-carboxylate (27c).
General procedure I was used to give 27c (0.16 g, 65%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.40 (d, J = 5.2 Hz, 1H), 7.79 (s, 1H), 7.58 (s, 4H), 6.81 (d, J = 5.2 Hz, 1H), 5.35 (d, J = 21.3 Hz, 1H), 3.89–3.67 (m, 2H), 3.64–3.53 (m, 2H), 2.59 (s, 3H), 2.43 (d, J = 20.1 Hz, 1H), 2.32–2.19 (m, 1H), 1.48 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 173.5, 157.6, 157.3, 142.5, 137.3, 136.7, 130.1 (d, J = 32.7 Hz), 128.7, 125.7 (d, J = 272.0 Hz), 124.2 (d, J = 272.1 Hz), 117.3, 80.4, 55.9, 55.4, 52.4, 51.8, 44.2, 44.0, 33.1, 32.3, 28.6, 14.3. 19F NMR (470 MHz, CDCl3) δ −62.6.
tert-Butyl (S)-3-(5-(2-(Methylsulfonyl)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)pyrrolidine-1-carboxylate (27d).
General procedure II was used to give 27d (0.13 g, 82%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.66 (d, J = 5.3 Hz, 1H), 7.89 (s, 1H), 7.65 (d, J = 8.2 Hz, 2H), 7.61 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 5.3 Hz, 1H), 5.52 (s, 1H), 3.81 (s, 1H), 3.67–3.50 (m, 2H), 3.39 (s, 3H), 2.58 (d, J = 39.2 Hz, 1H), 2.35 (s, 1H), 1.48 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 166.4, 158.9, 145.2, 138.3, 137.0, 132.0 (q, J = 32.9 Hz), 129.1, 126.1 (q, J = 3.9 Hz), 125.1 (q, J = 274.4 Hz), 123.2, 80.5, 57.3, 56.7, 52.4, 51.8, 44.1, 43.9, 39.2, 33.3, 32.5, 28.6. 19F NMR (470 MHz, CDCl3) δ −62.7.
tert-Butyl (R)-3-(5-(2-(Methylsulfonyl)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)pyrrolidine-1-carboxylate (27e).
General procedure II was used to give 27e (0.13 g, 80%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.65 (d, J = 5.4 Hz, 1H), 7.86 (s, 1H), 7.65 (d, J = 8.3 Hz, 2H), 7.60 (d, J = 8.1 Hz, 2H), 7.32 (d, J = 5.4 Hz, 1H), 5.52 (s, 1H), 3.84–3.79 (m, 1H), 3.66–3.48 (m, 2H), 3.39 (s, 3H), 2.34 (s, 1H), 1.48 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 166.4, 159.0, 158.0, 145.4, 138.3, 137.1, 130.9 (d, J = 32.9 Hz), 129.1, 126.1 (q, J = 3.9 Hz), 125.1, 124.2, 123.2, 122.9 (d, J = 272.2 Hz), 80.5, 57.3, 56.6, 52.4, 51.7, 44.1, 43.9, 39.2, 33.3, 32.5, 30.4, 29.8, 28.6. 19F NMR (470 MHz, CDCl3) δ −62.7.
tert-Butyl (S)-3-(5-(2-(3-Ethyl-5-methylphenoxy)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)-pyrrolidine-1-carboxylate (27f).
General procedure IV was used to give 27f (0.12 g, 77%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.41 (d, J = 5.2 Hz, 1H), 7.71 (s, 1H), 7.61 (d, J = 1.4 Hz, 4H), 6.94 (s, 1H), 6.85 (dd, J = 5.7, 3.7 Hz, 3H), 5.28 (h, J = 5.3, 4.5 Hz, 1H), 3.67–3.32 (m, 4H), 2.65 (q, J = 7.6 Hz, 2H), 2.36 (s, 3H), 1.98 (ddd, J = 10.7, 7.7, 5.4 Hz, 2H), 1.48 (s, 9H), 1.24 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 165.8, 160.2, 159.3, 153.1, 146.3, 139.8, 137.7, 137.0, 130.3 (d, J = 32.4 Hz),129.1, 126.3, 125.8 (q, J = 3.6 Hz), 125.0, 124.2 (d, J = 273.98 Hz), 119.8, 118.4, 116.1, 80.4, 56.1, 55.5, 51.9, 51.4, 44.0, 32.8, 32.1, 28.8, 28.6, 21.5, 15.5. 19F NMR (470 MHz, CDCl3) δ −62.6.
(S)-2-(3-Ethyl-5-methylphenoxy)-4-(1-(pyrrolidin-3-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-5-yl)pyrimidine (27).
General procedure III was used to give 27 (0.10 g, 99%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.81 (s, 1H), 8.53 (d, J = 5.1 Hz, 1H), 7.78 (d, J = 8.2 Hz, 2H), 7.67 (d, J = 8.1 Hz, 2H), 7.01 (s, 1H), 6.99 (d, J = 5.1 Hz, 1H), 6.91 (s, J = 2.0 Hz, 2H), 5.42 (tt, J = 7.6, 5.0 Hz, 1H), 3.69–3.58 (m, 2H), 3.46 (dt, J = 13.2, 6.5 Hz, 1H), 3.36 (ddd, J = 12.1, 8.2, 6.9 Hz, 1H), 2.66 (q, J = 7.6 Hz, 2H), 2.51–2.43 (m, 1H), 2.43–2.38 (m, 1H), 2.37 (s, 3H), 1.24 (t, J = 7.6 Hz, 3H). 13C NMR (125 MHz, MeOD) δ 166.8, 162.2, 158.7, 154.4, 147.8, 141.3, 140.1, 138.2, 135.4, 132.4 (d, J = 32.6 Hz), 130.7, 127.3 (q, J = 3.8 Hz), 125.4 (d, J = 271.6 Hz), 120.7, 119.3, 118.2, 118.1, 57.8, 51.8, 45.9, 32.0, 29.6, 21.4, 16.0. 19F NMR (470 MHz, MeOD) δ −64.2. HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H27F3N5O+ 494.2163, found 494.2160. [α]589mm25°C (c 0.24, MeOH) = −0.14.
(S)-2-(3-(5-(2-(3-Ethyl-5-methylphenoxy)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)pyrrolidin-1-yl)-N,N-dimethylethan-1-amine (28).
General procedure VI was used to give the desired product 28 (17 mg, 30%). 1H NMR (500 MHz, MeOD) δ 8.93 (s, 1H), 8.55 (d, J = 5.1 Hz, 1H), 7.77 (d, J = 8.2 Hz, 2H), 7.65 (d, J = 8.1 Hz, 2H), 7.00 (d, J = 1.8 Hz, 1H), 6.98 (d, J = 5.1 Hz, 1H), 6.89 (dt, J = 3.3, 1.9 Hz, 2H), 5.26 (dddd, J = 9.1, 7.0, 4.4, 2.5 Hz, 1H), 3.45 (ddd, J = 13.9, 8.3, 5.7 Hz, 2H), 3.34 (d, J = 5.5 Hz, 1H), 3.28 (d, J = 2.5 Hz, 1H), 3.15 (ddd, J = 13.8, 8.2, 5.8 Hz, 1H), 2.94 (s, 6H), 2.94–2.88 (m, 1H), 2.65 (q, J = 7.6 Hz, 2H), 2.61 (d, J = 7.1 Hz, 1H), 2.41 (q, J = 8.9 Hz, 1H), 2.36 (s, 3H), 2.23 (dtd, J = 14.4, 8.6, 3.2 Hz, 1H), 2.09 (dtd, J = 13.3, 8.3, 4.3 Hz, 1H), 1.24 (t, J = 7.6 Hz, 3H). 13C NMR (125 MHz, MeOD) δ 166.9, 162.3, 158.6, 154.5, 147.8, 141.2, 138.4, 134.9, 132.4 (q, J = 32.6 Hz), 130.7, 127.4, 127.3, 127.2 (q, J = 3.8 Hz), 125.4 (d, J = 271.4 Hz), 120.8, 119.3, 118.0, 61.5, 58.0, 55.7, 53.6, 50.5, 43.8, 33.6, 29.7, 21.4, 16.1. 19F NMR (471 MHz, MeOD) δ −64.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C31H36F3N6O+ 565.2898, found 565.2895. [α]589mm25°C (c 0.14, MeOH) = −0.089.
tert-Butyl (S)-3-(5-(2-(5-Isopropyl-2-methylphenoxy)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)pyrrolidine-1-carboxylate (29f).
General procedure IV was used to give 29f (0.11 g, 70%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.42 (d, J = 5.2 Hz, 1H), 7.70 (s, 1H), 7.65–7.57 (m, 4H), 7.20 (d, J = 7.8 Hz, 1H), 7.08 (dd, J = 7.8, 1.8 Hz, 1H), 7.01 (d, J = 1.7 Hz, 1H), 6.85 (d, J = 5.2 Hz, 1H), 5.16 (tt, J = 6.3, 3.7 Hz, 1H), 3.63–3.31 (m, 4H), 2.91 (dt, J = 13.7, 6.8 Hz, 1H), 2.15 (s, 3H), 1.98–1.76 (m, 2H), 1.48 (s, 9H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 165.4, 160.4, 159.2, 151.6, 148.7, 137.1, 131.3, 129.1, 125.8 (q, J = 3.7 Hz), 124.1, 120.2, 115.8, 80.4, 55.7, 51.4, 43.8, 33.7, 32.7, 28.6, 24.2, 24.1, 16.1. 19F NMR (376 MHz, CDCl3) δ −62.6.
(S)-2-(5-Isopropyl-2-methylphenoxy)-4-(1-(pyrrolidin-3-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-5-yl)pyrimidine (29).
General procedure III was used to give 29 (0.10 g, 98%) as a yellow solid. 1H NMR (500 MHz, MeOD) δ 8.74 (s, 1H), 8.55 (d, J = 5.1 Hz, 1H), 7.82–7.77 (m, 2H), 7.70–7.64 (m, 2H), 7.30 (dd, J = 7.8, 0.8 Hz, 1H), 7.16 (dd, J = 7.9, 1.8 Hz, 1H), 7.09 (d, J = 1.7 Hz, 1H), 7.00 (d, J = 5.1 Hz, 1H), 5.39 (tt, J = 7.8, 4.7 Hz, 1H), 3.71–3.57 (m, 2H), 3.39 (dd, J = 7.8, 6.1 Hz, 1H), 2.94 (hept, J = 6.9 Hz, 1H), 2.51–2.41 (m, 1H), 2.40–2.29 (m, 1H), 2.18 (s, 3H), 1.27 (d, J = 6.9, 6H). 13C NMR (125 MHz, MeOD) δ 166.4, 162.3, 159.0, 152.7, 150.2, 140.9, 138.3, 135.9, 132.4, 130.7, 129.0, 127.3 (q, J = 3.8 Hz), 125.4 (d, J = 271.3), 125.3, 121.2, 117.9, 57.8, 51.9, 45.9, 34.8, 32.0, 24.4, 16.0. 19F NMR (470 MHz, MeOD) δ −64.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C28H29F3N5O+ 508.2319, found 508.2314. [α]589mm25°C (c 0.080, MeOH) = −0.18.
(S)-2-(3-(5-(2-(5-Isopropyl-2-methylphenoxy)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)pyrrolidin-1-yl)-N,N-dimethylethan-1-amine (30).
General procedure VI was used to give 30 (20 mg, 35%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.92 (s, 1H), 8.57 (d, J = 5.1 Hz, 1H), 7.78 (d, J = 8.1 Hz, 2H), 7.64 (d, J = 8.0 Hz, 2H), 7.27 (dd, J = 7.8, 0.8 Hz, 1H), 7.13 (dd, J = 7.8, 1.8 Hz, 1H), 7.06 (d, J = 1.7 Hz, 1H), 6.98 (d, J = 5.1 Hz, 1H), 5.20 (dddd, J = 9.0, 6.8, 4.1, 2.1 Hz, 1H), 3.42 (dtt, J = 18.0, 8.9, 4.5 Hz, 2H), 3.27–3.21 (m, 1H), 3.13 (ddd, J = 13.9, 8.3, 5.7 Hz, 1H), 2.93–2.83 (m, 1H), 2.55 (dd, J = 11.3, 7.0 Hz, 1H), 2.36 (q, J = 8.9 Hz, 1H), 2.14 (s, 3H), 2.03 (dtt, J = 18.6, 8.8, 4.6 Hz, 1H), 1.26 (d, J = 6.9 Hz, 6H). 13C NMR (125 MHz, MeOD) δ 166.5, 162.5, 158.6, 152.8, 150.2, 138.44, 138.36, 134.83, 132.6 (d, J = 32.1 Hz), 132.4, 130.8, 129.0, 127.2 (q, J = 3.7 Hz), 125.4 (d, J = 271.6 Hz), 125.3, 121.1, 117.9, 61.5, 58.1, 55.9, 53.5, 50.5, 43.8, 34.8, 33.8, 24.5, 16.0. 19F NMR (470 MHz, MeOD) δ −64.3. HRMS (ESI-TOF) m/z [M + H]+ calcd for C32H38F3N6O+ 579.3054, found 579.3059. [α]589mm25°C (c 0.25, MeOH) = −0.098.
tert-Butyl (R)-3-(5-(2-(5-Isopropyl-2-methylphenoxy)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)pyrrolidine-1-carboxylate (31f).
General procedure IV was used to give 31f (0.11 g, 67%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.42 (d, J = 5.2 Hz, 1H), 7.66 (d, J = 4.6 Hz, 1H), 7.65–7.58 (m, 4H), 7.20 (d, J = 7.8 Hz, 1H), 7.08 (dd, J = 7.8, 1.9 Hz, 1H), 7.01 (d, J = 2.0 Hz, 1H), 6.85 (d, J = 5.2 Hz, 1H), 5.16 (tt, J = 6.5, 3.7 Hz, 1H), 3.66–3.31 (m, 4H), 2.91 (hept, J = 6.9 Hz, 1H), 2.15 (s, 3H), 1.97–1.88 (m, 1H), 1.82 (dtd, J = 14.3, 8.5, 6.2 Hz, 1H), 1.48 (s, 9H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 165.4, 160.4, 159.3, 151.6, 148.7, 137.9, 137.1, 131.3, 130.3 (d, J = 32.7 Hz), 129.1, 127.8, 125.8 (q, J = 3.7 Hz), 124.8, 124.2 (d, J = 272.2 Hz), 124.1, 120.2, 115.8, 80.3, 56.2, 51.9, 51.4, 44.9, 43.8, 33.7, 32.7, 31.9, 28.6, 24.2, 24.1, 16.1. 19F NMR (470 MHz, CDCl3) δ −62.6.
(R)-2-(5-Isopropyl-2-methylphenoxy)-4-(1-(pyrrolidin-3-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-5-yl)pyrimidine (31).
General procedure III was used to give 31 (0.10 g, 99%) as a pale-yellow solid. 1H NMR (500 MHz, MeOD) δ 8.69 (s, 1H), 8.53 (d, J = 5.2 Hz, 1H), 7.77 (d, J = 8.2 Hz, 2H), 7.65 (d, J = 8.1 Hz, 2H), 7.28 (d, J = 7.9 Hz, 1H), 7.14 (dd, J = 7.9, 1.9 Hz, 1H), 7.07 (d, J = 1.9 Hz, 1H), 6.98 (d, J = 5.2 Hz, 1H), 5.37 (tt, J = 7.7, 4.8 Hz, 1H), 3.73–3.53 (m, 2H), 3.36 (ddt, J = 11.7, 8.2, 3.1 Hz, 2H), 2.92 (hept, J = 6.9 Hz, 1H), 2.44 (dtd, J = 14.4, 7.3, 5.2 Hz, 1H), 2.32 (dq, J = 15.1, 7.8 Hz, 1H), 2.16 (s, 3H), 1.25 (d, J = 7.0 Hz, 6H). 13C NMR (125 MHz, MeOD) δ 166.4, 162.3, 159.0, 152.7, 150.2, 141.0, 138.3, 136.0, 132.4, 132.3 (d, J = 32.3 Hz) 130.7, 129.0, 127.3, 127.1 (q, J = 4.0 Hz), 125.4 (d, J = 273.7 Hz), 125.3, 121.2, 117.9, 57.7, 51.8, 45.9, 34.8, 32.0, 24.4, 16.0. 19F NMR (470 MHz, MeOD) δ −64.4. HRMS (ESI-TOF) m/z [M + H]+ calcd for C28H29F3N5O+ 508.2319, found 508.2318. [α]589mm25°C (c 2.8, MeOH) = 0.13.
(R)-2-(3-(5-(2-(5-Isopropyl-2-methylphenoxy)pyrimidin-4-yl)-4-(4-(trifluoromethyl)phenyl)-1H-imidazol-1-yl)pyrrolidin-1-yl)-N,N-dimethylethan-1-amine (32).
General procedure VI was used to give 32 (22 mg, 38%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.55 (s, 1H), 8.53 (d, J = 5.2 Hz, 1H), 7.74 (d, J = 8.1 Hz, 2H), 7.62 (d, J = 8.1 Hz, 2H), 7.27 (d, J = 7.8 Hz, 1H), 7.13 (dd, J = 7.8, 1.9 Hz, 1H), 7.06 (d, J = 1.7 Hz, 1H), 6.95 (d, J = 5.2 Hz, 1H), 5.12 (dtd, J = 9.0, 5.2, 4.1, 2.0 Hz, 1H), 3.39 (ddd, J = 13.5, 8.4, 5.2 Hz, 1H), 3.26 (dt, J = 13.5, 5.0 Hz, 1H), 3.13–3.07 (m, 1H), 3.01 (ddd, J = 13.7, 8.4, 5.2 Hz, 1H), 2.93–2.87 (m, 1H), 2.77 (dt, J = 13.7, 5.4 Hz, 1H), 2.39 (dd, J = 11.0, 7.0 Hz, 1H), 2.23 (q, J = 8.9 Hz, 1H), 2.13 (s, 3H), 2.06 (dtd, J = 14.3, 8.7, 3.1 Hz, 1H), 1.95 (dtt, J = 12.9, 8.4, 4.8 Hz, 1H), 1.26 (d, J = 6.9 Hz, 6H). 13C NMR (125 MHz, MeOD) δ 166.3, 162.3, 158.5, 152.6, 150.0, 138.2, 134.7, 132.4 (d, J = 32.0 Hz), 132.3, 130.6, 128.9, 127.1 (q, J = 3.7 Hz), 125.2 (d, J = 271.3 Hz), 125.1, 1201.0, 117.8, 61.4, 58.0, 55.7, 53.3, 50.3, 43.6, 34.7, 33.6, 24.3, 15.8. 19F NMR (470 MHz, MeOD) δ −64.2. HRMS (ESI-TOF) m/z [M + H]+ calcd for C32H38F3N6O+ 579.3054, found 579.3048. [α]589mm25°C (c 0.38, MeOH) = 0.053.
Protein Expression.
BRD3 D1, D2 expression:
The pET-28a(+) plasmid containing the BRD3 D1 and BRD3 D2 were purchased from GenScript. The Escherichia coli strain BL21 star(DE3) was transformed with the BRD3 D1 and BRD3 D2 plasmids and plated onto an agar plates containing kanamycin (100 mg/L) and chloramphenicol (35 mg/L). The plates were incubated overnight at 37 °C. A 5 mL LB culture with kanamycin(100 mg/L) and chloramphenicol (35 mg/L) was inoculated using a single colony from each plate and grown overnight at 37 °C and shaking at 220 rpm. Four primary cultures were used to inoculate 1 L of LB media containing chloramphenicol (35 mg/L) and kanamycin (100 mg/L) until the optical density at 600 nm had reached 0.6–0.8. At this point, an equilibration time of 30 min at 20 °C and 220 rpm was followed by the addition of 1 mM IPTG to induce protein expression. The culture was shaken for 16 h at 20 °C and 220 rpm. Cells were pelleted by centrifugation at 8000g and stored at −20 °C until purification. BRD4 D1, BRD2 D1, and BRDT D1 were expressed and purified as previous reported.33
Fluorescence Anisotropy and AlphaScreen.
Procedures were performed according to our prior report.18
Differential Scanning Fluorimetry (Thermal Stability Profiling).
A real-time PCR instrument (QuantStudio6, Applied Biosystems) was utilized to monitor protein unfolding via SYPRO orange florescent dye (Invitrogen, Carlsbad, CA). Proteins were expressed and purified as described in protein expression section.33 Thermal stability experiments were performed with 10 μM of protein and 5× SYPRO Orange dye in 20 μL volume per well with variable ligand concentration. Final buffer composition for the assay contained 50 mM HEPES, 100 mM NaCl, 1% DMSO, pH 7.4. Proteins were tested from 23 to 90 °C with a ramp rate of 0.05 °C/s. All data analysis was carried out using Protein Thermal Shift Software v1.4 and fitted to a Boltzman Sigmoidal curve to determine the melting temperatures (Tm).
Isothermal Titration Calorimetry (ITC).
Isothermal titration calorimetry (ITC) experiments were performed at 25 °C using a MicroCal iTC200 (GE Healthcare). BRD4D1 was dialyzed at 8 °C overnight into a solution containing 50 mM HEPES and 100 mM NaCl pH 7.4. Then 150 μM BRD4D1 was loaded into the syringe, and experimental compounds were loaded into the cell at 10 μM. Injections were carried out by serial injection of BRD4D1; first, one injection of 0.4 μL followed by 19 incremental injections of 2 μL, at 150 s intervals. Data from the first injection was excluded due to pre-equilibration mixing between contents of cell and syringe at the syringe tip. All experiments were completed in triplicate unless noted otherwise. Data collection, analysis, and plotting were performed using Origin 7 SR4 (version 7.0552). Peak areas were integrated, normalized, and then fitted by nonlinear regression using the one-site model. Binding isotherms provided the equilibrium association or binding constant (Ka), the change in enthalpy (ΔH), and the stoichiometry of binding (N). Binding stoichiometry was 1:1 within experimental error. The change in free energy (ΔG) and change in entropy (ΔS) were determined using the equation:
where R is the universal gas constant, T is the temperature in degrees Kelvin, and other parameters are as defined.
Crystallization Conditions and X-ray Data Collection Methods.
Compounds 4, 20, and 22 were cocrystallized with BRD4 D1 (300 μM, in 10 mM HEPES, 100 mM NaCl, pH 7.4) in 20% (v/v) PEG 3350, and 0.2 M potassium chloride using the hanging drop method. Compound 32 (500 μM) was cocrystallized with BRD4 D1 (300 μM, in 10 mM HEPES, 100 mM NaCl, pH 7.4) in 100 mM Bis-Tris propane, 20% (v/v) PEG 3350, and 20% (v/v) ethylene glycol at pH 8.5 using the hanging drop method. Harvestable crystals grew in 1–2 days at ambient temperature. Crystals were harvested, cryoprotected in ethylene glycol and flash frozen. Data were collected at Advanced Photon Source with the NECAT 24-ID-C and E beamlines. Phaser was used to solve the structure via molecular replacement using PDB 3MXF as a search model.34 Phenix35 and Coot36 were used for structure refinement.
Thermal Stability Profiling.
MM.1S cells (1 × 106) were treated with desired amounts of compound, with DMSO concentrations normalized to 0.1% for all samples. Dosed cells were incubated in DMEM media at 37 °C for 1 h with mild intermittent agitation. Following centrifugation and rinsing with PBS upon completion of the incubation period, cells were resuspended in PBS (100 μL) and thermally denatured for 3 min at the indicated temperatures and equilibrated at room temperature for a further 3 min. Cells were spiked with 10× protease inhibitor cocktail and lysed over three freeze–thaw cycles before soluble protein concentrations of supernatant were determined using the BCA protein assay kit (Pierce). Samples were normalized to the lowest total soluble protein concentration among grouped samples.
NuPAGE 4× LDS sample buffer and NuPAGE 10× sample reducing agent (Invitrogen) were added to normalized protein samples and denatured by heating at 90 °C for 5 min. Protein samples were separated on a gradient 4–12% SDS-PAGE gel (Invitrogen), transferred to a polyvinylidene difluoride membrane (Immobilon), and blocked by incubating in blocking buffer (0.05 g/mL BioRad nonfat milk in PBS) at 4 °C overnight. Proteins were detected by incubation with primary antibodies (BRD4 Rabbit mAb, Cell Signaling Technologies, 13440; BRD4, Cell Signaling Technology, no. 13440, diluted 1:1000 in TBS-T containing 5% nonfat dry milk; BRD3, Abcam, no. AB50818, diluted 1:1000 in TBS-T containing 5% nonfat dry milk; BRD2, Bethyl Laboratories, no. BL167–2A2, diluted 1:1000 in TBS-T containing 5% nonfat dry milk; and Vinculin Mouse mAb, Thermo, 14–9777-82) diluted 1:1000 in blocking buffer for 4 h. at room temperature. The membrane was washed in MQ-water and incubated with HRP-conjugate antimouse (Thermo, A16066) or antirabbit secondary antibodies (Thermo, 65–6120) similarly diluted in blocking buffer, for 1 h, at room temperature. The membrane was again washed in MQ-water before immunocomplexes were visualized upon addition of HRP substrate (Thermo) using the Odyssey FC imaging system (LICOR Biotech.).25,37
Western Blotting.
MM.1S cells were seeded in 12-well plates at a density of 106 cells per well and treated with compounds for 6 h. Cells were harvested by low-speed centrifugation at 500g for 5 min and washed twice with ice-cold PBS. Cells were lysed in 100 μL of RIPA buffer (ThermoFisher Scientific) supplemented with 1× cOmplete Mini Protease (Roche) and Phosphatase Inhibitor Cocktail (ThermoFisher Scientific). After high-speed centrifugation (10 min at 10 000g), protein concentrations were determined by Bradford assay (ThermoFisher Scientific) and normalized by total protein content. Normalized samples were mixed with 4× NuPAGE LDS loading buffer (Invitrogen) and 10× reducing agent (Invitrogen) and heated at 95 °C for 5 min. This was followed by separation on 8–12% SDS-PAGE. Proteins were transferred to PVDF membranes for 7 min on a BioRad Trans-Blot Turbo. Membranes were incubated subsequently with PBS-T containing 5% nonfat dry milk for 16 h at 4 °C. with primary antibodies (c-Myc, Cell Signaling Technology, no. 5605, diluted 1:1000 in PBS-T containing 5% nonfat dry milk; β-actin, Invitrogen no. MA5–11869, diluted 1:1000 in PBS-T containing 5% nonfat dry milk). After the membranes were washed three times with PBS-T, they were incubated with 1000-fold-diluted HRP-conjugated secondary antibodies from Invitrogen (goat antirabbit-IgG, no. G-31460, and goat antimouse-IgG, no. G-21040; in PBS-T containing 5% nonfat dry milk) for 4 h. at room temperature. Membranes were washed three times in PBS-T and treated with SuperSignal West Dura substrates (Cell Signaling Technology) for 1 min and imaged using a LiCor Odessey Fc.25
Viability Assays.
A549 cells were seeded in 96-well plates at approximately 20 000 cells per well and dosed with increasing compound concentrations with 0.5% DMSO with three technical replicates (100 μL final volume). After incubation for 23 h at 37 °C, 10 μL of Alamar Blue reagent was added to each well and the plates were incubated a further 1 h at 37 °C. Fluorescence was determined (Ex, 560 nm; Em, 590 nm), and dose–response data were normalized to untreated and blank wells containing 0.5% DMSO in cell culture media. Data analysis was performed using GraphPad Prism.18,25
IL-8 ELISA.
A549 cells were seeded into 24-well plates at a cell density of 200 000 cells per well. Cells were incubated with the compounds for 24 h and induced with TNF-α (10 ng/mL) 6 h after the initial dosing. Cell media (1 mL) was harvested and centrifuged (10 000g, 5 min) before the protein levels were measured using a human IL-8 ELISA kit (Thermo Scientific, EH2IL8) following the vendor’s instructions. A standard curve was plotted in Microsoft Excel, and unknown samples were plotted against the standard curve to yield IL-8 protein concentrations in picograms per milliliter. Data are reported as means ± SD from three replicate biological experiments with three technical replicates per experiment.25
Drug Toxicity with Human Liver Sinusoidal Endothelial Cells.
Assessment of toxicity was measured with a Cell Signaling XTT Cell Viability Kit (no. 9095). Human liver sinusoidal endothelial cells (LSEC) were seeded on 96-well plates with a density of 12 000 cells/well. LSEC were incubated for 24 h at 37 °C, 5% CO2. A 1.1 mM drug stock was serially diluted (3-fold), and each dilution stock was balanced to 11% DMSO. Old endothelial growth media was removed from LSECs, and 100 μL of fresh media were added to each well. To each respective well, 10 μL of serial drug stocks were added to give final concentrations of 0.1–100 μM drug in 110 μL volume/well. The final DMSO concentration is 1%. LSECs were incubated overnight (20–24 h) with drug at 37 °C, 5% CO2. XTT solution was made as per kit instructions; 50 μL of XTT solution were added to each well plate with a multichannel pipet. Cells were incubated 3 h and then were measured at 462 and 620 nm on a SpectraMax Plus 384 UV–vis spectrometer. Absorbance at 620 nm was used to subtract background signal from 462 nm signal.
In Vitro Drug Efficacy in Human LSEC.
RT-PCR was performed with a RNeasy qPCR Kit (Qiagen). LSECs were seeded on 12-well plates at 100 000 cells/well. LSECs were incubated overnight at 37 °C, 5% CO2. Media was removed from plates and replaced with 1 mL of Endothelial Cell Basal Media (Lonza, CC 3121) for 1.5 h. Afterward, basal media was removed, and 0.5 mL of endothelial growth media were added to each well. Drugs were dissolved in endothelial growth media and 0.5 mL aliquots were added to respective wells. Cells were incubated for 2 h with drug. After 2 h, lipopolysaccharide was added (200 ng/well), and LSECs were incubated for 4 h. Media was aspirated after 4 h, and cells were washed with DPBS. RNA and cDNA were prepared as per kit instructions. qPCR was conducted in duplicate analytical replicates, and SYBR green was used.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by Office of Discovery and Translation Grant at UMN, UL1 TR002494 (W.C.K.P), Center for Regenerative Medicine (V.S.), and the NIH MIRA awards, R35 GM140837–01 (W.C.K.P.) and R35 GM118047 (H.A.) A.D. was supported by the UMN Doctoral Dissertation Fellowship and a NIH chemistry–biology interface training grant (T32-GM008700/T32-GM132029–01). H.Z. was supported by UMN IEM Engineering in Medicine Doctoral Fellowship 2020. D.H. acknowledges funding from the Masonic Cancer Center at the University of Minnesota with resources from Minnesota Masonic Charities. R.L. is supported in part by St. Jude Cancer Center Support grant CA21765 and by the American Lebanese Syrian Associated Charities (ALSAC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health or other funding agencies. X-ray diffraction data were collected at the Northeastern Collaborative Access Team beamlines, which are supported by NIGMS P30 GM124165. The Pilatus 6 M detector on 24-ID-C beamline is funded by a NIH-ORIP HEI grant (S10 RR029205).
ABBREVIATIONS USED
- BET
bromodomain and extra-terminal
- BRD2/3/4/T
bromodomain-containing protein 2/3/4/T
- CETSA
cellular thermal shift assay
- ELISA
enzyme-linked immunosorbent assay
- CCL2
CC chemokine ligand 2
- CXCL1
chemokine ligand 1
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01779.
Corresponding spectra, along with HPLC traces and all relevant biophysical data and additional crystallographic information; full Western blots (PDF)
Molecular-string files for the final compounds (CSV)
Contributor Information
Huarui Cui, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Anand Divakaran, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Zachariah J. Hoell, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
Mikael O. Ellingson, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
Cole R. Scholtz, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
Huda Zahid, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Jorden A. Johnson, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
Elizabeth C. Griffith, Department of Chemical Biology and Therapeutics, St. Jude Children’s Research Hospital, Memphis, Tennessee 38105, United States
Clifford T. Gee, Department of Chemical Biology and Therapeutics, St. Jude Children’s Research Hospital, Memphis, Tennessee 38105, United States
Amani L. Lee, GI Research Unit, Guggenheim 1034 Mayo Clinic, Rochester, Minnesota 55902, United States
Shalil Khanal, GI Research Unit, Guggenheim 1034 Mayo Clinic, Rochester, Minnesota 55902, United States.
Ke Shi, Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Hideki Aihara, Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Vijay H. Shah, GI Research Unit, Guggenheim 1034 Mayo Clinic, Rochester, Minnesota 55902, United States
Richard E. Lee, Department of Chemical Biology and Therapeutics, St. Jude Children’s Research Hospital, Memphis, Tennessee 38105, United States.
Daniel A. Harki, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States; Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
William C. K. Pomerantz, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States; Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
REFERENCES
- (1).Haynes SR; Dollard C; Winston F; Beck S; Trowsdale J; Dawid IB The Bromodomain: A Conserved Sequence Found in Human, Drosophila and Yeast Proteins. Nucleic Acids Res. 1992, 20 (10), 2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Smith SG; Zhou MM The Bromodomain: A New Target in Emerging Epigenetic Medicine. ACS Chem. Biol. 2016, 11 (3), 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Filippakopoulos P; Knapp S The Bromodomain Interaction Module. FEBS Lett. 2012, 586 (17), 2692–2704. [DOI] [PubMed] [Google Scholar]
- (4).Belkina AC; Denis GV BET Domain Co-Regulators in Obesity, Inflammation and Cancer. Nat. Rev. Cancer 2012, 12 (7), 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Anand P; Brown JD; Lin CY; Qi J; Zhang R; Artero C; Alaiti MA; Bullard J; Alazem K; Margulies KB; Cappola TP; Lemieux M; Plutzky J; Bradner JE; Haldar SM BET Bromodomains Mediate Transcriptional Pause Release in Heart Failure. Cell. 2013, 154, 569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Muller S; Filippakopoulos P; Knapp S Bromodomains as Therapeutic Targets. Expert Rev. Mol. Med. 2011, 13, No. e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Huang B; Yang X-D; Zhou M-M; Ozato K; Chen L-F Brd4 Coactivates Transcriptional Activation of NFB via Specific Binding to Acetylated RelA. Mol. Cell. Biol. 2009, 29 (5), 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lovén J; Hoke HA; Lin CY; Lau A; Orlando DA; Vakoc CR; Bradner JE; Lee TI; Young RA Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153 (2), 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Andrieu G; Belkina AC; Denis GV Clinical Trials for BET Inhibitors Run Ahead of the Science. Drug Discovery Today Technol. 2016, 19, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Stonestrom AJ; Hsu SC; Jahn KS; Huang P; Keller CA; Giardine BM; Kadauke S; Campbell AE; Evans P; Hardison RC; Blobel GA Functions of BET Proteins in Erythroid Gene Expression. Blood 2015, 125 (18), 2825–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lamonica JM; Deng W; Kadauke S; Campbell AE; Gamsjaeger R; Wang H; Cheng Y; Billin AN; Hardison RC; Mackay JP; Blobel GA Bromodomain Protein Brd3 Associates with Acetylated GATA1 to Promote Its Chromatin Occupancy at Erythroid Target Genes. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (22), E159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gamsjaeger R; Webb SR; Lamonica JM; Billin A; Blobel GA; Mackay JP Structural Basis and Specificity of Acetylated Transcription Factor GATA1 Recognition by BET Family Bromodomain Protein Brd3. Mol. Cell. Biol. 2011, 31 (13), 2632–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gilan O; Rioja I; Knezevic K; Bell MJ; Yeung MM; Harker NR; Lam EYN; Chung C; Bamborough P; Petretich M; Urh M; Atkinson SJ; Bassil AK; Roberts EJ; Vassiliadis D; Burr ML; Preston AGS; Wellaway C; Werner T; Gray JR; Michon A; Gobbetti T; Kumar V; Soden PE; Haynes A; Vappiani J; Tough DF; Taylor S; Dawson S; Bantscheff M; Lindon M; Drewes G; Demont EH; Daniels DL; Grandi P; Prinjha RK; Dawson MA Selective Targeting of BD1 and BD2 of the BET Proteins in Cancer and Immuno-Inflammation. Science 2020, 368 (6489), 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cui H; Carlson AS; Schleiff MA; Divakaran A; Johnson JA; Buchholz CR; Zahid H; Vail NR; Shi K; Aihara H; Harki DA; Miller GP; Topczewski JJ; Pomerantz WCK 4-Methyl-1,2,3-Triazoles as N-Acetyl-Lysine Mimics Afford Potent BET Bromodomain Inhibitors with Improved Selectivity. J. Med. Chem. 2021, 64, 10497–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Picaud S; Wells C; Felletar I; Brotherton D; Martin S; Savitsky P; Diez-Dacal B; Philpott M; Bountra C; Lingard H; Fedorov O; Müller S; Brennan PE; Knapp S; Filippakopoulos P RVX-208, an Inhibitor of BET Transcriptional Regulators with Selectivity for the Second Bromodomain. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Faivre EJ; McDaniel KF; Albert DH; Mantena SR; Plotnik JP; Wilcox D; Zhang L; Bui MH; Sheppard GS; Wang L; Sehgal V; Lin X; Huang X; Lu X; Uziel T; Hessler P; Lam LT; Bellin RJ; Mehta G; Fidanze S; Pratt JK; Liu D; Hasvold LA; Sun C; Panchal SC; Nicolette JJ; Fossey SL; Park CH; Longenecker K; Bigelow L; Torrent M; Rosenberg SH; Kati WM; Shen Y Selective Inhibition of the BD2 Bromodomain of BET Proteins in Prostate Cancer. Nature 2020, 578 (7794), 306–310. [DOI] [PubMed] [Google Scholar]
- (17).Liu Z; Chen H; Wang P; Li Y; Wold EA; Leonard PG; Joseph S; Brasier AR; Tian B; Zhou J Discovery of Orally Bioavailable Chromone Derivatives as Potent and Selective BRD4 Inhibitors: Scaffold Hopping, Optimization, and Pharmacological Evaluation. J. Med. Chem. 2020, 63 (10), 5242–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cui H; Divakaran A; Pandey AK; Johnson JA; Zahid H; Hoell ZJ; Ellingson MO; Shi K; Aihara H; Harki DA; Pomerantz WCK Selective N-Terminal BET Bromodomain Inhibitors by Targeting Non-Conserved Residues and Structured Water Displacement. Angew. Chemie - Int. Ed 2021, 60, 1220–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Crawford TD; Tsui V; Flynn EM; Wang S; Taylor AM; Côté A; Audia JE; Beresini MH; Burdick DJ; Cummings R; Dakin LA; Duplessis M; Good AC; Hewitt MC; Huang H; Jayaram H; Kiefer JR; Jiang Y; Murray J; Nasveschuk CG; Pardo E; Poy F; Romero AF; Tang Y; Wang J; Xu Z; Zawadzke LE; Zhu X; Albrecht BK; Magnuson SR; Bellon S; Cochran AG Diving into the Water: Inducible Binding Conformations for BRD4, TAF1(2), BRD9, and CECR2 Bromodomains. J. Med. Chem. 2016, 59 (11), 5391–5402. [DOI] [PubMed] [Google Scholar]
- (20).Clegg MA; Bamborough P; Chung CW; Craggs PD; Gordon L; Grandi P; Leveridge M; Lindon M; Liwicki GM; Michon A; Molnar J; Rioja I; Soden PE; Theodoulou NH; Werner T; Tomkinson NCO; Prinjha RK; Humphreys PG Application of Atypical Acetyl-Lysine Methyl Mimetics in the Development of Selective Inhibitors of the Bromodomain-Containing Protein 7 (BRD7)/Bromodomain-Containing Protein 9 (BRD9) Bromodomains. J. Med. Chem. 2020, 63, 5816–5840. [DOI] [PubMed] [Google Scholar]
- (21).Yu Z; Ku AF; Anglin JL; Sharma R; Ucisik MN; Faver JC; Li F; Nyshadham P; Simmons N; Sharma KL; Nagarajan S; Riehle K; Kaur G; Sankaran B; Storl-Desmond M; Palmer SS; Young DW; Kim C; Matzuk MM Discovery and Characterization of Bromodomain 2-Specific Inhibitors of BRDT. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (9), e2021102118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gao J; Wei B; Liu M; Hirsova P; Sehrawat TS; Cao S; Hu X; Xue F; Yaqoob U; Kang N; Cui H; Pomerantz WCK; Kostallari E; Shah VH Endothelial P300 Promotes Portal Hypertension and Hepatic Fibrosis Through C-C Motif Chemokine Ligand 2-Mediated Angiocrine Signaling. Hepatology 2021, 73 (6), 2468–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Liu M; Cao S; He L; Gao J; Arab JP; Cui H; Xuan W; Gao Y; Sehrawat TS; Hamdan FH; Ventura-Cots M; Argemi J; Pomerantz WCK; Johnsen SA; Lee J; Gao F; Ordog T; Mathurin P; Revzin A; Bataller R; Yan H; Shah VH Super Enhancer Regulation of Cytokine-Induced Chemokine Production in Alcoholic Hepatitis. Nat. Commun. 2021, 12, 4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Aldeghi M; Ross GA; Bodkin MJ; Essex JW; Knapp S; Biggin PC Large-Scale Analysis of Water Stability in Bromodomain Binding Pockets with Grand Canonical Monte Carlo. Commun. Chem. 2018, 1, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Divakaran A; Talluri SK; Ayoub AM; Mishra N; Cui H; Widen JC; Berndt N; Zhu J-Y; Carlson AS; Topczewski JJ; Schonbrunn EK; Harki DA; Pomerantz WCK Molecular Basis for the N-Terminal Bromodomain and Extra Terminal (BET) Family Selectivity of a Dual Kinase-Bromodomain Inhibitor. J. Med. Chem. 2018, 61, 9316–9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ember SWJ; Zhu JY; Olesen SH; Martin MP; Becker A; Berndt N; Georg GI; Schonbrunn E Acetyl-Lysine Binding Site of Bromodomain-Containing Protein 4 (BRD4) Interacts with Diverse Kinase Inhibitors. ACS Chem. Biol. 2014, 9 (5), 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Auffinger P; Hays FA; Westhof E; Ho PS Halogen Bonds in Biological Molecules. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (48), 16789–16794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Boehm JC; Bower MJ; Gallagher TF; Kassis S; Johnson SR; Adams JL Phenoxypyrimidine Inhibitors of P38α Kinase: Synthesis and Statistical Evaluation of the P38 Inhibitory Potencies of a Series of 1-(Piperidin-4-Yl)-4-(4-Fluorophenyl)-5-(2-Phenoxypyrimidin-4-Yl) Imidazoles. Bioorg. Med. Chem. Lett. 2001, 11 (9), 1123–1126. [DOI] [PubMed] [Google Scholar]
- (29).Müller K; Faeh C; Diederich F Fluorine in Pharmaceuticals: Looking beyond Intuition. Science (80). 2007, 317 (5846), 1881–1886. [DOI] [PubMed] [Google Scholar]
- (30).Zhao YH; Abraham MH; Zissimos AM Fast Calculation of van Der Waals Volume as a Sum of Atomic and Bond Contributions and Its Application to Drug Compounds. J. Org. Chem. 2003, 68 (19), 7368–7373. [DOI] [PubMed] [Google Scholar]
- (31).Gallagher TF; Seibel GL; Kassis S; Laydon JT; Blumenthal MJ; Lee JC; Lee D; Boehm JC; Fier-Thompson SM; Abt JW; Soreson ME; Smietana JM; Hall RF; Garigipati RS; Bender PE; Erhard KF; Krog AJ; Hofmann GA; Sheldrake PL; McDonnell PC; Kumar S; Young PR; Adams JL Regulation of Stress-Induced Cytokine Production by Pyridinylimidazoles Inhibition of CSBP Kinase. Bioorg. Med. Chem. 1997, 5 (1), 49–64. [DOI] [PubMed] [Google Scholar]
- (32).Raux B; Voitovich Y; Derviaux C; Lugari A; Rebuffet E; Milhas S; Priet S; Roux T; Trinquet E; Guillemot JC; Knapp S; Brunel JM; Fedorov A. Yu; Collette Y; Roche P; Betzi S; Combes S; Morelli X Exploring Selective Inhibition of the First Bromodomain of the Human Bromodomain and Extra-Terminal Domain (BET) Proteins. J. Med. Chem. 2016, 59 (4), 1634–1641. [DOI] [PubMed] [Google Scholar]
- (33).Olson NM; Kroc S; Johnson JA; Zahid H; Ycas PD; Chan A; Kimbrough JR; Kalra P; Schönbrunn E; Pomerantz WCK NMR Analyses of Acetylated H2A.Z Isoforms Identify Differential Binding Interactions with the Bromodomain of the NURF Nucleosome Remodeling Complex. Biochemistry 2020, 59 (20), 1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mccoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Research Papers Phaser Crystallographic Software Research Papers. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Jafari R; Almqvist H; Axelsson H; Ignatushchenko M; Lundbäck T; Nordlund P; Molina DM The Cellular Thermal Shift Assay for Evaluating Drug Target Interactions in Cells. Nat. Protoc. 2014, 9 (9), 2100–2122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.