

Abstract
Interactions between the enteric nervous system (ENS), immune system, and gut microbiota regulate intestinal homeostasis in adults, but their development and role(s) in early life are relatively underexplored. In early life, these interactions are dynamic, as the mucosal immune system, microbiota, and the ENS are developing and influencing each other. Moreover, disrupting gut microbiota and gut immune system development, and potentially ENS development, by early life antibiotic exposure increases the risk of diseases affecting the gut. Here, we review the development of the ENS and immune/epithelial cells, and identify potential critical periods for their interactions and development. We highlight knowledge gaps that, when addressed, may help promote intestinal homeostasis, including in the settings of early life antibiotic exposure.
Keywords: Enteric nervous system, muscularis macrophages, innate lymphoid cells, goblet cells, neonatal microbiome, early life antibiotics
Early Life Intestinal Immune Interactions
As a baby transitions from intrauterine to extrauterine life, the gut takes on new roles: it begins to extract energy from luminal contents while appropriately identifying pathogens and protecting the body from infection [1]. To adapt to these changes, the neonatal gut undergoes maturation, specifically regarding the enteric nervousosal immune system, and the microbiota [2–6]. Neuroimmune interactions allow for communication between the ENS and intestinal immune cells to coordinate their responses to promote homeostasis or host defense [7,8]. The microbiome plays an important role in facilitating these connections. Disruption of neuroimmune interactions has been implicated in the pathophysiology of intestinal inflammatory disorders [9–13]. Most studies of ENS/immune/gut microbiota interactions have been performed in adults. In recent years, studies have increasingly addressed their role(s) in early life [14–17].
The foregoing considerations may be of particular relevance to premature neonates, who experience the same stresses of the transition to extrauterine life as term infants, but must do so with even more immature physiological systems. Additionally, in many countries, more than 75% of extremely premature neonates are exposed to broad-spectrum antibiotics in the first days of life [18]. This is life-saving for infants with early-onset sepsis (EOS; a perinatally acquired infection manifesting in the first three days of life), but increases the risk of other complications that increase the morbidity and mortality including late-onset sepsis (LOS; sepsis occurring after 72 hours of life) and necrotizing enterocolitis (NEC) [19,20]. While it is known that the neonatal ENS, mucosal immune cells, and microbiota interact in early life and can influence each other’s maturation, the critical timing of these interactions and the mechanisms by which they influence each other’s development are just beginning to be investigated. In this review, we summarize early postnatal murine development of the ENS as a basis for further discussion into ENS/immune/gut microbiota interactions and development. We then discuss how the ENS interacts with select mucosal immune cell types (macrophages, innate lymphocytes, and goblet cells in early life and the relevance of these interactions in neonatal mice.
Enteric Nervous System Postnatal Development Requires Interaction with the Developing Microbiota and Mucosal Immune Cells
ENS development in mice
The enteric nervous system (ENS) is the intrinsic nervous system of the gut and consists of a complex network of neurons and glia that control most gut functions [21]. The ENS is organized into two plexuses (Figure 1) [22]. The myenteric plexus has cell bodies located between the circular and longitudinal muscle layers of the gut. The submucosal plexus has cell bodies closer to the lumen. These plexuses sense luminal stimuli and respond with appropriate effector signals to promote digestion, motility, and host defense [23]. The ENS begins to form early in fetal development when vagal neural crest cells invade the foregut and colonize the intestine in a complex and coordinated spatial and temporal process of migration, proliferation, and differentiation during early gestation in both mice and humans. [22,24,25]. This allows for the blueprint of the ENS to be present at birth, but significant development is still required for the ENS to become the mature network observed in adults. Notably, mice are born with immature intestines, including the ENS, compared to humans. The neonatal murine intestine is thought to be roughly equivalent to the human fetal intestine at weeks 22-24 of gestation [26]. This suggests that much of the maturation that normally occurs in the early postnatal period in mice also occurs postnatally for human neonates born prematurely.
Figure 1: Basic Anatomy of the Enteric Nervous System.
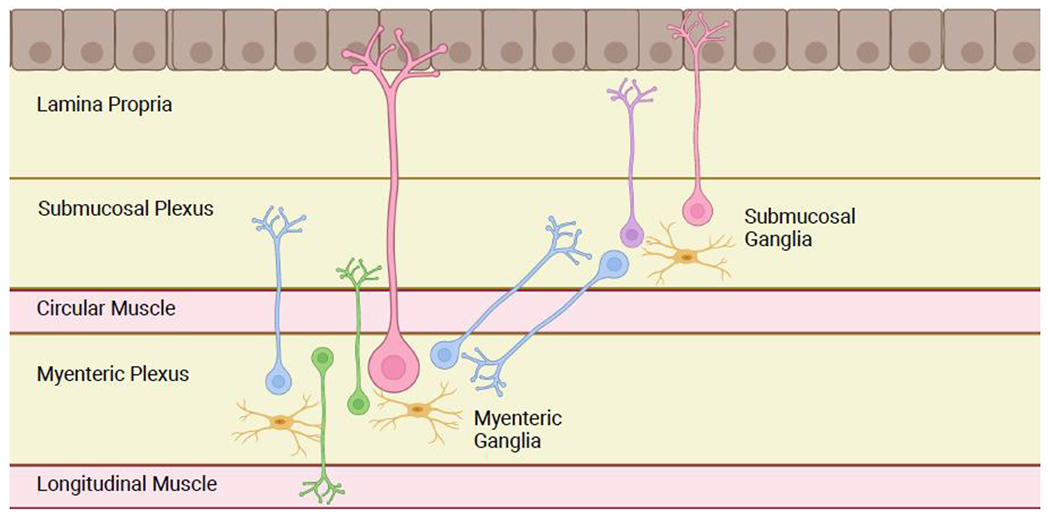
The ENS is organized into two plexuses: the myenteric plexus (located between the circular and longitudinal muscle layers) and the submucosal plexus (located closer to the lumen, deep to the mucosa). Neurons are organized into ganglia. Myenteric motor neurons (green) project to the muscle layers to coordinate the contraction and relaxation required for peristalsis. Sensory neurons (pink) in both plexuses have projections to the luminal surface to detect intestinal contents. Submucosal secretomotor neurons (purple) project to the epithelial layer to coordinate secretion, absorption, and local blood flow. Additional neurons (blue) connect ganglia to each other within and between plexuses. Glia (yellow) are present throughout the bowel wall and are necessary for ENS function. Figure generated using BioRender.com.
In general, ENS development occurs in a pattern progressing rostral to caudal and from the myenteric to submucosal plexus. This suggests that perturbations of gut microbiota/immune/ENS interactions at a given time in development will affect regions and layers of the gut differently. Several excellent reviews have profiled the mechanics of postnatal murine ENS development, and we will highlight the most relevant points for the discussion of neuroimmune interaction development [2,14].
Timing of ENS development in mice
Early postnatal enteric neuronal development has two main focuses: permanent neurochemical identity of the neurons (what neurotransmitters/neuropeptides neurons express) and efficient, directed signaling (neuron morphology, electrochemical properties and synapse formation). ENS neuron “birthdates” (when progenitors leave the cell cycle for terminal differentiation), begin around mid-gestation but extend into the postnatal period, with most neurons being born by the end of the first week of postnatal life [27–30]. Most of these later born neurons will be part of the submucosal plexus, which lags behind the myenteric plexus throughout ENS development and is still undergoing gangliogenesis in the first days of postnatal life [28]. Neurons of a particular neurochemical subtype leave the cell cycle at approximately the same time; therefore, a stereotypical set of neurotransmitters and neuropeptides is not present in the ENS until late fetal or early neonatal development. These neurotransmitters and neuropeptides include neuropeptide Y, vasoactive intestinal peptide (VIP) and calcitonin gene related peptide (all mediators of important neuroimmune connections in the adult intestine) [7,28]. Some enteric neurons will transiently express certain neurotransmitters (e.g. neuronal nitric oxides synthase (nNOS) and tyrosine hydroxylase) in early life, but not as adults [31,32]. The ratio of neurons of different neurochemical subtypes is also in flux during the first weeks of postnatal life. nNOS, classically expressed in inhibitory motor neurons in the myenteric plexus, is also expressed in approximately half of submucosal neurons at birth but decreases to less than 5% by adulthood[2,31,33]. nNOS expression appears to be especially influenced by the microbiota, as germ free mice, antibiotic treated mice and mice with genetic ablation of TLRs have decreased nNOS+ neurons in the myenteric plexus [34–36]. This suggests that the microbiota influences neurochemical subtype expression and that a normal microbiota is required for normal enteric neuron specification in the postnatal period.
Neuronal morphology and circuit formation are also active processes in the neonatal period [27–29]. In mice, neuronal control over migrating motor complexes in the colon does not exist until the first week of life and is not fully mature until after weaning [14]. In the first week of life, enteric neurons have filamentous dendrites that mature into a lamellar morphology over time [37]. These changes in dendrite shape could suggest a change in the sources of input to the neuron given the dendrites’ classical role of receiving signals. Enteric neuron axonal projections also change over the first weeks of life [29,37,38]. At birth, most enteric neurons project their axons caudally. Development of rostral axonal projections is apparent only after birth, meaning the bidirectional communication necessary for coordinated peristalsis is not possible prenatally. Circumferential neuronal connections also do not develop until late fetal development/early postnatal life. These circumferential projections have to form new connections through the preweaning period as the axons do not grow along with the circumference of the gut [37]. Early life myenteric neurons display a relatively immature electrical profile [37]. Fast electrical signals predominate in the first weeks of life with more mature and varied slow depolarization only identifiable in adult mice. The microbiota and its metabolites can modulate enteric neuronal excitability in adulthood, and alterations to the neonatal microbiota has been demonstrated to alter neuron electrical activity [36,39,40]. These data suggest that the microbiota may alter the patterns of electrical activity in the neonatal intestine that are required for mature circuit formation.
Sensing and affecting microbes and immune responses
Enteric neurons express a variety of receptors for microbial products, metabolites, and cytokines (Table 1) [14,41]. This expression renders the ENS competent to be shaped both directly by the microbiota and indirectly through interactions with local immune cells [40,42]. A unique role for the microbiota in influencing early life ENS development has been demonstrated in the reduced neuronal density and altered subtype specification observed in antibiotic treated neonatal mice [36]. While antibiotic treatment also disrupts the ENS in mature mice, the same antibiotic treatment induces different changes pre-weaning versus post weaning mice [43]. Enteric neurons and glia express pattern recognition receptors including toll-like receptors 2, 4, and 9, and genetic ablation of these receptors (or its downstream effector MyD88) disrupts ENS structure and function in adult mice [35,44,45]. While ENS cells express these receptors in embryonic development, their impact in neonatal intestinal homeostasis has not been thoroughly explored. Intriguingly, germ-free mice are noted to have abnormal ENS density and subtype specification at postnatal day 3 (P3) [46]. This may represent altered fetal development as studies of central nervous system (CNS) development have identified a role for metabolites from the maternal microbiota to influence the nervous system prenatally - a hypothesis that remains to be tested in the context of the ENS [47].
Table 1.
Immune mediators and receptors expressed by enteric neurons and their observed roles in intestinal homeostasis.
| Receptor | Phenotype | References |
|---|---|---|
| TLR2 | TLR2 −/− have reduced GDNF levels causing abnormal ENS architecture, neuronal subtype specification, delayed transit time, and abnormal mucosal secretion |
[44] |
| TLR4 | TLR4 −/− mice have prolonged intestinal motility, decreased nNOS neurons | [35] |
| TLR9 | Promotes macrophage chemoattraction and secretion of proinflammatory cytokines. | [45] |
| MyD88 | ENS-specific deletion of MyD88 causes prolonged intestinal motility and decreased nNOS neurons |
[35] |
| TGR5 | TGR5 −/− mice have slower colonic transit | [111] |
| IL-4Rα | Signaling activated by IL-4, IL-13, potentially involved in infection mediated hypermotility | [41,112,113] |
| TNF SRF | TNFα signaling promotes neuronal production of neuropeptide Y and influences intestinal inflammation and motility |
[114] |
| TGFβR | TGFβ signaling induces neurite outgrowth in cultured neurons | [115] |
| Mediator | ||
| IL-6 | Inhibits Rorγ Treg induction in a microbiota dependent manner in adults | [48] |
| IL-18 | Induces goblet cells to produce antimicrobial peptides in adults | [49] |
| CSF-1 | Facilitates crosstalk between ENS and macrophages, increases with age | [54] |
Neurons and glia of the ENS also express receptors for microbial metabolites including short chain fatty acids and bile acids (Table 1). Notably, microbially derived serotonin is important for enteric neurogenesis in adult mice, and previous studies have also identified it as important for regulating the neonatal ENS [36,40,43]. Enteric neurons express immune mediators (both constitutively and in response to stimuli) allowing cells of the ENS to reciprocally alter function of local immune cells (Table 1) [14,41]. Of note, despite the plethora of cells present in the intestine that may secrete a cytokine, studies have identified that ENS specific production is required for particular phenotypes. For example, ENS-specific IL-6 and IL-18 have been appreciated to have direct effects on regulatory T cells and intestinal goblet cells respectively [48,49].
Early life antibiotic exposure is common and early life antibiotic exposure increases the risk of some intestinal diseases which are related, by either cause or effect, to ENS function. The ability of enteric neurons to sense products from the gut microbiota and immune cells, as well as to affect the microbiota and immune system is well documented, and while these pathways are known to be present in early life (and prenatally), their role(s) in early life development of the ENS is largely unexplored. Below we will review what is known about the development and neonatal function of neuroimmune interactions known to be important in adult mice.
Muscularis Macrophages
Muscularis macrophages (MMs) are a subset of intestinal tissue resident macrophages that closely interact with the ENS to regulate intestinal motility and tissue repair [50]. MMs are classically defined as CD45+ CX3CR1+ F4/80+ CD11b+ CD11c-/lo, although gene expression analyses suggest there are further sub-classifications. As their name suggests, MMs are primarily found in the muscularis of the bowel in close proximity to the neurons of the myenteric plexus, but also exist in the lamina propria near the submucosal neurons. In adult mice, MMs have a role in protecting enteric neurons from death during enteric infection, thereby reducing risk of post viral irritable bowel syndrome, but also appear to inhibit normal bowel motility in the setting of post-operative ileus [51]. Premature and antibiotic exposed infants are at risk for intestinal inflammatory disorders, poor motility, feeding intolerance and functional GI disorders, some with similar pathophysiology to adult pathologies involving MMs [13,52]. Intestinal macrophages are initially yolk-sac-derived, but most are replaced by bone-marrow-derived macrophages over the murine preweaning period [6]. However, the macrophages associating with myenteric and submucosal neurons are primarily long-lived yolk-sac-derived macrophages [53]. It is unknown if some yolk-sac macrophages are predestined to identify and associate with neurons or alternatively if this is a serendipitous encounter prolonging macrophage lifespan. Together these observations suggest that macrophage-neuron interactions could be established in early life.
Growth Factor Secretion
In adult mice, MMs interact with neurons in a microbiota-dependent manner to regulate intestinal motility [54]. Adult mice treated with a prolonged course of broad-spectrum antibiotics have significantly reduced numbers of MMs [55]. Consequently, the levels of bone morphogen protein (BMP) 2, an important trophic factor for enteric neurons secreted by MMs, is also decreased in the colon [56]. Enteric neurons reciprocally produce colony stimulating factor (CSF) 1, a growth factor required for MMs [57]. While this symbiotic secretion of growth factors links the ENS and MMs in adults (Figure 2), the mechanism of communication between the ENS and macrophages in early life is less clear.
Figure 2: Enteric Neuron/Macrophage Crosstalk.
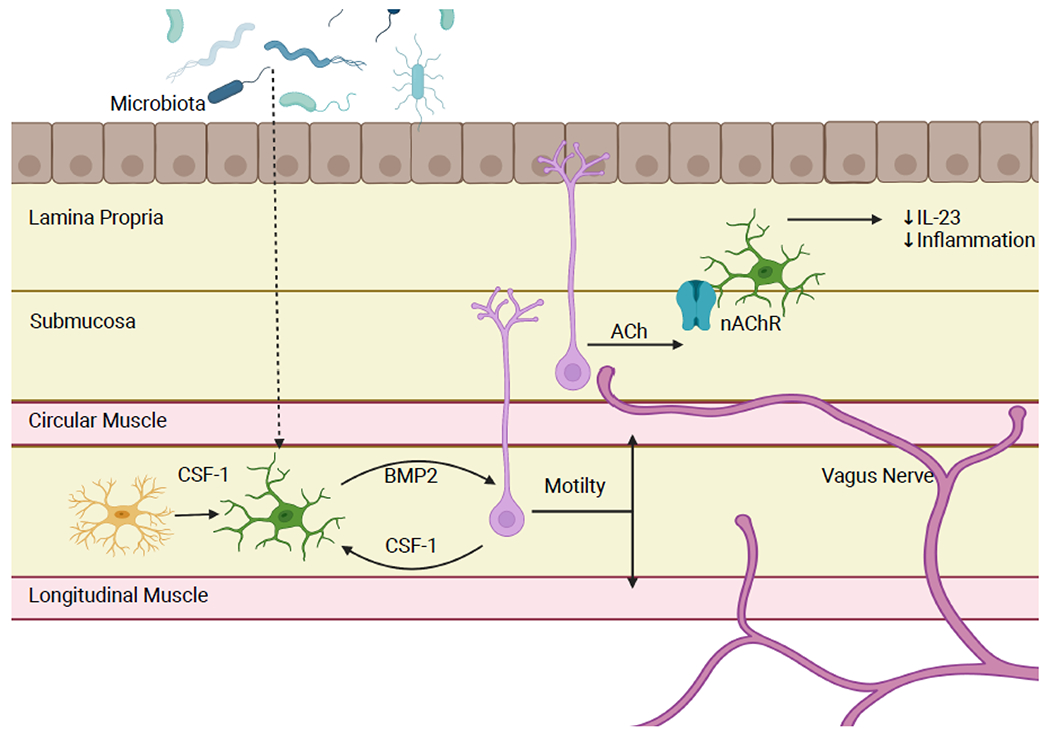
Enteric neuron and tissue resident macrophages have several mechanisms of communication to regulate intestinal homeostasis. One mechanism is symbiotic production of growth factors. Muscularis macrophages produce the enteric neuron growth factor BMP2 while enteric neurons and glia secrete CSF-1, an important growth factor for macrophages. In colons of antibiotic treated mice, there is a reduction in number of macrophages and subsequently in the level of BMP2, demonstrating that this relationship is microbiota dependent. This results in slower motility and abnormal smooth muscle contraction. Additionally, cholinergic enteric neurons signal to neuron associated macrophages in the lamina propria. Acetylcholine binds to the nAChR receptor on macrophages, inducing an anti-inflammatory phenotype and low levels of IL-23 production. Figure generated using BioRender.com.
Fetal MM development does not require the ENS, as mice with total intestinal aganglionosis have normal MM quantity, patterning and function on P1 [25]. Mice with abnormal CSF-1 signaling lack MMs and have increased enteric neuronal density and disorganized ganglia as adults [54]. This suggests that MMs are necessary for normal ENS development, but ENS development in these mice has not been evaluated [54,57]. CSF1-BMP2 cross-communication that is observed in adults is not mature in neonatal mice. The ENS does not become the primary source of intestinal CSF-1 until late in the pre-weaning period (between P14-21), the same point MMs become the primary intestinal source of BMP2 [25]. The trigger for ENS secretion of CSF-1 and MM production of BMP2 is unknown, though likely to be the microbiota or a microbial metabolite given the coinciding time course of microbiome development and the disruption in this crosstalk caused by antibiotics in adult mice.
The recently discovered role for enteric glia utilizing CSF-1 to specifically recruit anti-inflammatory macrophages to an inflamed muscularis is another example of ENS/MM crosstalk manifested by CSF-1 signaling [58,59]. Enteric glia are protective against intestinal inflammation in general, and NEC specifically [60–62]. NEC is associated with decreased glial density in both human patients and animal models [62]. TLR4-mediated glial cell death and dysmotility were identified as early events in animal models of NEC, and enteric glia depletion causes worse intestinal inflammation in NEC models. If glial cells secrete CSF-1 in response to inflammation in neonatal animals as they do in adults, this loss of glia in early NEC could be the trigger for worsening inflammation and allow for the accumulation of the pro-inflammatory macrophages observed in NEC [63].
Because ligand/receptor interactions and mechanisms of communication between enteric neurons, glia, and macrophages appear to contribute to intestinal homeostasis in neonates, further studies will be important to identify these interactions in neonates. Given the developmental delay and likely microbiota-dependence of the BMP2/CSF1 axis, it seems likely that other signaling mechanisms are being utilized in early life instead of or in addition to the BMP2/CSF1 axis.
Acetylcholine Signaling
Acetylcholine-expressing neurons are important mediators of smooth muscle contraction in peristalsis [21]. In addition to this important role in motility, neuronal acetylcholine functions as an important signaling molecule to inhibit intestinal inflammation. Enteric neurons signal to macrophages by releasing acetylcholine that is subsequently bound to the α7-nicotinic acetylcholine receptor (α7nAChR) expressed by macrophages [64,65]. Activation of this pathway in intestinal macrophages induces them to adopt an anti-inflammatory phenotype [66].
Hirschsprung disease (HSCR) is a developmental disorder of the ENS in which a section of the distal bowel is not appropriately colonized by ENS precursors, resulting in aganglionosis. Proinflammatory intestinal macrophages have been implicated in the pathogenesis of neonatal intestinal inflammation, most notably in Hirschsprung-associated enterocolitis (HAEC) [67,68]. HAEC is the major cause of mortality for children with Hirschsprung disease due to severe intestinal inflammation and risk of perforation [69]. The risk of HAEC persists after surgical resection of aganglionic bowel, potentially suggesting that the remaining ENS is dysfunctional such that it disrupts its ability to appropriately regulate barrier function and host defense [67,70]. Recent studies have demonstrated that a relative lack of cholinergic fibers in the ganglionic bowel of HSCR patients is associated with disrupted mucosal immunity and increased risk of HAEC [15,71]. Specifically, the reduced cholinergic signaling is associated with decreased abundance of submucosal neuron associated macrophages in the lamina propria that share phenotype and functionality with the MMs that interact with the myenteric plexus neurons [53]. This results in increased macrophage secretion of IL-23 and promotion of Th17 cell driven inflammatory phenotype in the intestine with decreased innervation and could contribute to the mechanism of increased HAEC risk (Figure 2). As antibiotic treatment impacts neuron excitability and action potential firing, dysbiosis of the gut microbiota could further reduce neuronal acetylcholine release in neonates, allowing for inappropriate proinflammatory polarization of intestinal macrophages.
Innate Lymphoid Cells
Innate lymphoid cells (ILCs) are tissue resident immune cells that have heterogeneous functions including host defense, barrier function, and commensal microbial interactions [72]. Group 3 ILCs (ILC3s) are important intermediaries between the host and commensal microbiota via secretion of mediators, such as the cytokines IL-17A and IL-22. They have well elucidated roles in homeostasis but are also associated with chronic intestinal inflammation. ILC3s have three known subcategories: CCR6+ lymphoid tissue inducer (LTi)-like cells, CCR6− NK-cell receptor (NCR)+ ILC3s and CCR6−NCR− ILCs. All subtypes have roles in fetal and neonatal intestinal physiology and pathophysiology [73,74]. NEC is associated with an increase in NCR− ILC3s and IL-17A producing NCR+ cells. Conversely, LOS is associated with a decrease in NCR+ ILC3s that produce IL-17A. In mice, maternal and neonatal antibiotic exposure causes a decrease in these NCR+ IL-17A ILCs which renders the pup at increased risk of sepsis [75,76]. ILC3s represent an important cell type in potential pathology in the neonatal intestine. These cells requires precise regulation and context in order to promote healthy intestinal homeostasis. Emerging evidence suggests the ENS is one component regulating ILC3s.
Vasoactive Intestinal Peptide
VIP neurons make up a minority but a substantial fraction of submucosal neurons in both juvenile and adult mice [21,33]. VIP+ neurons are activated by feeding to simulate epithelial secretomotor activity and blood vessel dilation. VIP is an important mediator of neuroimmune interactions in the intestine as well as in the lung [7,77]. Mice with genetic ablation of VIP expression have significantly reduced numbers of all ILC3 subtypes in the intestine, identifying VIP as important for ILC3 recruitment [78]. Recent studies demonstrate that activation of VIP receptor 2 (VIPR2) induces ILC3 production of IL-22, but only in the presence of alarmins (e.g. IL-23 and IL-33) (Figure 3) [79]. This augments the immune response against pathogens and protects against injury in DSS colitis in mice (Figure 3) [79]. While IL-22 is important in barrier function, it is also implicated in the pathophysiology of inflammatory bowel disease by promoting inflammation [80]. These results suggest that ILC3s require both neuronal and immunological signals to produce IL-22, and the need for two stimuli may be an important mechanism for regulating ILC3s to maintain homeostasis without inducing chronic inflammation.
Figure 3: Enteric Neuron/ILC3 Crosstalk.
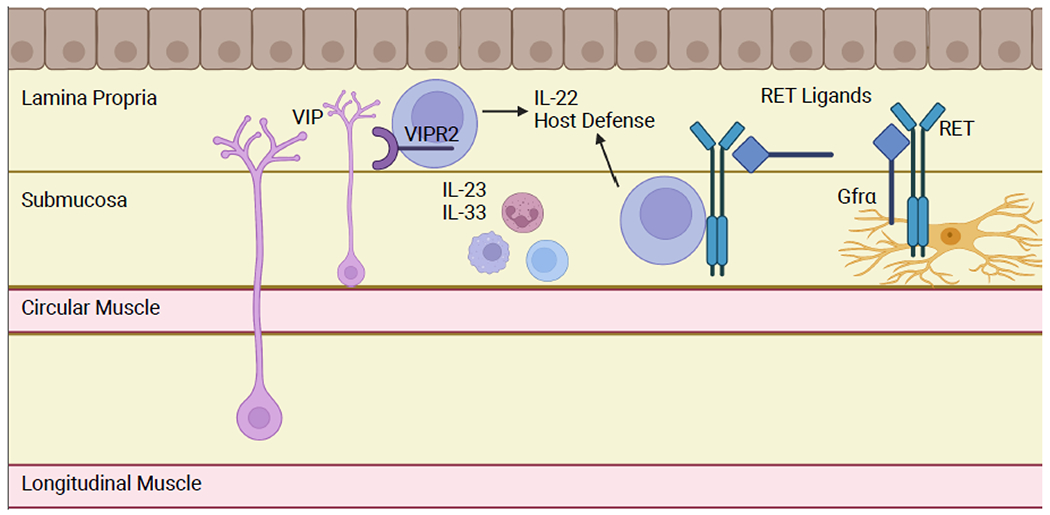
VIP-expressing enteric neurons are activated by feeding and other stimuli. VIP activates ILC3s via VIPR2 activation, inducing IL-22 expression and promoting host defense. This pathway is most effective in the presence of alarmins, i.e. in the setting of an enteric pathogen. Enteric glia activate RET expressing ILC3s by producing RET ligands in a microbiota-dependent manner. Glia and other cells in the intestine also express RET co-receptors to allow trans-activation of RET signaling ILC3s, which do not express the co-receptors autonomously. Figure generated using BioRender.com.
The context in which VIP promotes IL-22 secretion from intestinal ILC3s is not clear, as two concurrently published investigations suggested contradictory effects. In one study, VIP was shown to decrease IL-22 secretion from CCR6+ ILC3s [81]. This facilitated increased lipid absorption from the lumen, but also allowed greater bacterial translocation. These data suggest that VIP is important for the gut’s role in energy extraction but weakens barrier function and host defense. Conversely, in another study, feeding-induced VIP was shown to promote IL-22 secretion by ILC3s [82]. Furthermore, this group found that inhibition of VIP signaling renders mice more sensitive to pathogen-induced inflammation. The seemingly contradictory results of these two studies reinforce the notion that ILC3’s functions are likely to be highly heterogeneous, and underscore the importance of biological and experimental context when examining these functions.
VIP-induced alteration of barrier function via IL-22 in response to feeding could have important consequences in premature neonates. IL-22 expression is relatively low in neonatal intestine in homeostasis but increases under inflammatory conditions [83]. IL-22 has been found to be protective in models of NEC, suggesting that augmentation of IL-22 signaling by VIP could be protective against intestinal inflammation in the neonatal period. The activation of VIP+ neurons by feeding complicates the picture for the extremely premature neonates at highest risk for NEC. In general, extremely low birth weight infants are slowly advanced on feeds over several days to weeks due to concerns for feeding intolerance and NEC [84]. The absence of a feeding stimulus in the presence of a rapidly developing microbiome (and often, the impact of systemic broad-spectrum antibiotics) in these infants may be either protective or harmful, depending on how the human premature intestinal environment influences VIP modulation of IL-22 production by ILC3s. Additionally, if VIP weakens host defense to facilitate nutrient absorption, this could have significant implications for enteral feeding and growth of premature neonates. VIP+ neurons are present in the myenteric plexus of the human fetus by week 18 of gestation, but it is unclear when VIP+ neurons are first present in the human submucosal plexus and when they are first localized to near ILC3s in the intestine. Furthermore, while NEC is associated with decreased numbers of IL-22 expressing ILC3s, LOS is associated with increased numbers [74]. This underlines the importance of understanding how neuroimmune interactions function in neonatal life to best design therapies that mitigate risk of one complication without significantly contributing to the risk of another.
RET Signaling
RET is a receptor tyrosine kinase that is essential for ENS development [24,85]. All ENS precursor cells express RET and a co-receptor (GDNF family receptor, GFR, α1-4) and rely on cis activation by RET ligands to induce proliferation and migration programs required for ENS precursor colonization of the intestine [86,87]. As ENS precursor cells differentiate, neurons maintain RET expression, while glial cells downregulate RET expression. After differentiation, glia maintain expression of GFRα co-receptors, allowing enteric glia to contribute to RET activation in nearby cells via trans activation (RET and the co-receptor are expressed by separate cells). Additionally, other intestinal cells can produce GFRα co-receptors, and they can persist in a soluble form to promote RET transactivation in competent cells (Figure 3). CCR6+ and CCR6− NCR− ILC3 express RET and produce IL-22 to promote host defense in a RET- and ENS-dependent manner [88]. Glial cells in the intestinal lamina propria can adopt a stellate morphology with protrusions that project into cryptopatches to allow for paracrine signaling between the glia and ILC3s. Ablation of Ret in ILC3s rendered affected mice susceptible to DSS-induced inflammation. ILC3s expressing a gain-of-function RET mutant have increased IL-22 expression and confer protection against colonic epithelial damage caused by DSS. Inhibition of microbial sensing in glia cells via cell specific MyD88 knockout reduced ILC3 production of IL-22 and rendered mice more susceptible to colitis, mimicking the phenotype seen in the ILC3 RET knockout [88].
Goblet Cells
Goblet cells are specialized epithelial cells that play an important role in the intestinal mucosal immune system [89]. They are present in fetal development but undergo significant expansion in both number and function in early postnatal murine life [3,90]. Goblet cells contribute to intestinal barrier defense through their production of mucus and antimicrobial peptides. Certain intestinal inflammatory disorders, including HAEC, NEC and ulcerative colitis, are associated with abnormal goblet cell number or function [91–94]. Whether these changes are a cause or an effect of the pathologies is still unknown [91–94].
IL-18
IL-18 is canonically a pro-inflammatory cytokine produced by a host of intestinal cell types, including enteric neurons, and is important in host defense, in part due to its impact on goblet cells [95–99]. IL-18 is an important mediator of the pathophysiology in septic shock in adults. Given that premature neonates have elevated serum IL-18 levels compared to adults at baseline, it was hypothesized that IL-18 could be responsible for severe manifestations of neonatal sepsis as well [100]. In mice, IL-18 appears to mediate its impact on the morbidity and mortality of neonatal sepsis by inducing IL-17A secretion in multiple immune cell types in the small bowel lamina propria [100].
As observed with other cytokines, not all of IL-18’s activities are pathologic. IL-18 is ubiquitously expressed by enteric neurons by the time of weaning [97]. Enteric neurons have been demonstrated to signal to goblet cells to modulate their function and phenotype [101,102]. Neuron-derived IL-18 specifically signals goblet cells to produce antimicrobial peptides [49]. When IL-18 is specifically deleted in enteric nervous system cells (but not hematopoietic or epithelial cells), affected mice were more susceptible to Salmonella infection (Figure 4) [49]. This suggests that enteric neurons have a specific mechanism to signal to goblet cells that is not available to other cell types and could function as a defense mechanism in which neuronal IL-18 induces a protective response from goblet cells in the midst of more global IL-18 induced intestinal inflammation. Additionally, goblet cell dysfunction is noted in HAEC [103,104]. Children with HSCR and mouse models of the disease have altered numbers of goblet cells and disordered mucin secretion/composition [103]. The quantity and quality of AMP in HSCR and HAEC has not been studied, to our knowledge, but given the recent studies discussed above, further investigation is warranted.
Figure 4: Enteric Neuron/Goblet Cell Crosstalk.
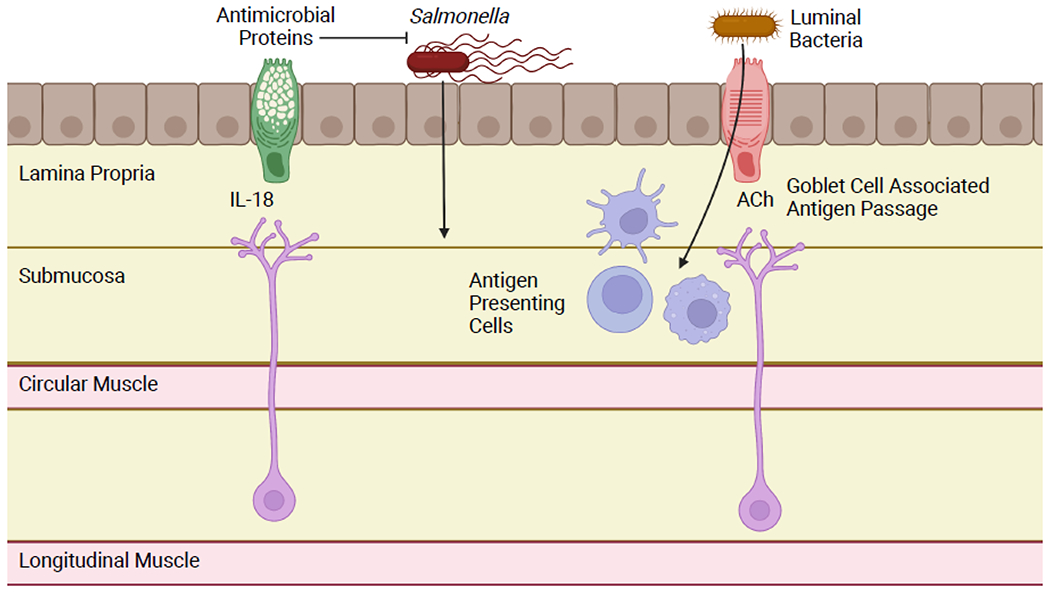
Enteric neurons produce the cytokine IL-18 constitutively. This induces goblet cells to produce antimicrobial proteins under homeostatic conditions and promotes host defense in the context of Salmonella infection. Goblet cells can also form portals, known as goblet cell associated antigen passages (GAPs). GAPs facilitate physiologic and pathologic translocation of luminal bacteria and presentation to antigen presenting cells. Goblet cells are induced to form GAPs by acetylcholine activation of the mAChR4 receptor. Cholinergic enteric neurons have been hypothesized as an important source of acetylcholine for GAP activation. Figure generated using BioRender.com.
Acetylcholine
Additionally, goblet cells contribute to intestinal homeostasis by promoting tolerogenic response to commensal luminal bacteria via goblet cell associated antigen passages (GAPs) [105,106]. In mice, GAPs are dynamic, but tightly regulated in the pre-weaning period. GAPs are induced via activation of muscarinic acetylcholine receptor 4 (mAChR4) on goblet cells and the source of acetylcholine has been hypothesized to be cholinergic enteric neurons [7,107]. Our group has identified that tight temporal regulation of colonic GAPs is required for appropriate immune development in neonatal mice [105]. Specifically, inhibition of colonic GAPs during days of life (DOL) ~10-21 prevents the developing immune system from being exposed to commensal luminal antigens and developing appropriate tolerogenic response [105]. In contrast, open colonic GAPs before DOL10 facilitate pathologic bacterial translocation and clinical sepsis in mouse models [108].
Acetylcholine induces GAP formation by binding to the mAChR4 expressed on goblet cells [107]. GAP inhibition is primarily driven by epidermal growth factor receptor (EGFR) signaling causing decreased sensitivity of the goblet cell to mAChR4 activation. In the absence of mAChR4, and hypothetically acetylcholine, GAPs do not form. While the precise source of the acetylcholine available to goblet cells to induce GAPs has not been identified, the enteric nervous system is known to influence other aspects of goblet cell function including mucus secretion [101,102,104]. While a variety of immune cells also produce acetylcholine, enteric neurons have been hypothesized to provide the acetylcholine necessary to open GAPs [7]. Mouse models of HSCR are associated with abnormal goblet cell function and retained mucus, suggesting the consequences of the absence of the enteric nervous system stimulating goblet cell secretion [104]. The ENS in HSCR is commonly abnormal in the remaining bowel after resection of the distal aganglionic section, resulting in persistent defects in motility and barrier function [109,110]. Quantifying GAPs in the intestine in patients or mouse models of HSCR would be one opportunity to identify the role in enteric neurons regulating GAPs. It will be important to consider the role of extrinsic vagal afferents in identifying the role of intrinsic enteric neuron signaling to goblet cells in intestinal homeostasis and disease.
Concluding Remarks
Neuroimmune interactions are important regulators of intestinal homeostasis in adults, and emerging evidence demonstrates they have a vital role during early development as well. Given the massive cellular turnover and maturation of the microbiota, ENS and mucosal immune cells in the neonatal period, it seems likely that major differences exist between the interactions among these systems during this period than those identified in adults. Macrophages, ILCs, and goblet cells are all important cells of the intestinal mucosal immune system with known microbiota-dependent interactions with the adult ENS. Given that antibiotic exposure and dysbiosis disrupts neuron/immune cell crosstalk, it is conceivable that this disruption contributes to the enhanced risk of NEC and LOS associated with early life antibiotic exposure. Still, but many questions regarding development and malleability of early life neuroimmune interactions remain (see Outstanding Questions). More investigation is necessary to identify how neuroimmune interactions can be fostered in the neonatal period in the setting of perturbations of antibiotics and help mitigate the increased risk of potentially lethal diseases.
Outstanding Questions:
Given that the mammalian ENS develops rostral to caudal and outside to inside (myenteric plexus before submucosal plexus), is development of different regions of the ENS differentially impacted by the microbiota in early postnatal life?
Does early life antibiotic exposure impact CSF-1 production by enteric neurons and BMP2 production by muscularis macrophages?
Do early life antibiotics disrupt neurotransmitter (e.g. acetylcholine and VIP) release? Enteric neurons have altered excitability under germ-free and dysbiotic conditions. This will alter action potential frequency, and therefore could impact acetylcholine and VIP release, which in turn could impact macrophage, ILC3 and goblet cell function.
Is goblet cell AMP production altered in HAEC? Given that enteric neurons constitutively produce IL-18 to promote goblet cell AMP production, the absence or impaired function of enteric neurons in HSCR could weaken barrier defense. This could contribute to the risk of HAEC and therefore represent an avenue for future treatments.
Highlights.
Enteric neurons have dynamic relationships with intestinal macrophages, ILC3s, and goblet cells with mutual effects on each other’s function that shape regulation of intestinal homeostasis.
Neurons and immune cells signal to each other via cytokines, growth factors, and neurotransmitters, and these signaling methods change through the neonatal period into adulthood.
Early life antibiotic exposure alters ENS and mucosal immune system development, but the effect on neuroimmune interactions is not fully understood.
Greater understanding of how neuroimmune interactions develop in the neonatal period will help identify novel strategies to mitigate the deleterious effects of early life antibiotic exposure.
Acknowledgements
We want to thank Shreya Gaddipati, Vini John, and Keely G. McDonald and the members of the Newberry lab for their assistance in preparing this manuscript. Biorender.com was used for generation of figures. This work was supported in part by grants NIH R01DK097317 - RDN, NIH R01AI173220 - RDN, NIH U01AI163073 – RDN, NIH T32DK077653-30 - EMS NIH T32HD43010-20 – EMS, and American Academy of Pediatrics Marshal Klaus Award 2022. The Washington University in Saint Louis School of Medicine Digestive Diseases Research Cores Center is supported by grant NIH P30DK52574.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no conflicts of interest.
References
- 1.Martin RJ et al. (2011) Fanaroff and Martin’s neonatal-perinatal medicine : diseases of the fetus and infant 9th edn), Saunders/Elsevier [Google Scholar]
- 2.Foong JP (2016) Postnatal Development of the Mouse Enteric Nervous System. Adv Exp Med Biol 891, 135–143. 10.1007/978-3-319-27592-5_13 [DOI] [PubMed] [Google Scholar]
- 3.Torow N et al. (2017) Neonatal mucosal immunology. Mucosal Immunol 10, 5–17. 10.1038/mi.2016.81 [DOI] [PubMed] [Google Scholar]
- 4.Koenig JE et al. (2011) Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A 108 Suppl 1, 4578–4585. 10.1073/pnas.1000081107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferretti P et al. (2018) Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe 24, 133–145 e135. 10.1016/j.chom.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bain CC et al. (2014) Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol 15, 929–937. 10.1038/ni.2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobson A et al. (2021) The intestinal neuro-immune axis: crosstalk between neurons, immune cells, and microbes. Mucosal Immunol 14, 555–565. 10.1038/s41385-020-00368-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H et al. (2022) Enteric neuroimmune interactions coordinate intestinal responses in health and disease. Mucosal Immunol 15, 27–39. 10.1038/s41385-021-00443-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kadowaki M et al. (2022) Neuro-immune crosstalk and food allergy: Focus on enteric neurons and mucosal mast cells. Allergol Int. 10.1016/j.alit.2022.03.004 [DOI] [PubMed] [Google Scholar]
- 10.Biskou O et al. (2022) Increased Numbers of Enteric Glial Cells in the Peyer’s Patches and Enhanced Intestinal Permeability by Glial Cell Mediators in Patients with Ileal Crohn’s Disease. Cells 11. 10.3390/cells11030335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holland AM et al. (2021) The enteric nervous system in gastrointestinal disease etiology. Cell Mol Life Sci 78, 4713–4733. 10.1007/s00018-021-03812-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kabata H and Artis D (2019) Neuro-immune crosstalk and allergic inflammation. J Clin Invest 129, 1475–1482. 10.1172/JCI124609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brierley SM and Linden DR (2014) Neuroplasticity and dysfunction after gastrointestinal inflammation. Nat Rev Gastroenterol Hepatol 11, 611–627. 10.1038/nrgastro.2014.103 [DOI] [PubMed] [Google Scholar]
- 14.Foong JPP et al. (2020) Early life interaction between the microbiota and the enteric nervous system. Am J Physiol Gastrointest Liver Physiol 319, G541–G548. 10.1152/ajpgi.00288.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keck S et al. (2021) Lack of Mucosal Cholinergic Innervation Is Associated With Increased Risk of Enterocolitis in Hirschsprung’s Disease. Cell Mol Gastroenterol Hepatol 12, 507–545. 10.1016/j.jcmgh.2021.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Margolis KG et al. (2016) Cellular Organization of Neuroimmune Interactions in the Gastrointestinal Tract. Trends Immunol 37, 487–501. 10.1016/j.it.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McClain JL et al. (2020) Histamine-dependent interactions between mast cells, glia, and neurons are altered following early-life adversity in mice and humans. Am J Physiol Gastrointest Liver Physiol 319, G655–G668. 10.1152/ajpgi.00041.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flannery DD et al. (2018) Temporal Trends and Center Variation in Early Antibiotic Use Among Premature Infants. JAMA Netw Open 1, e180164. 10.1001/jamanetworkopen.2018.0164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cotten CM (2016) Adverse consequences of neonatal antibiotic exposure. Curr Opin Pediatr 28, 141–149. 10.1097/MOP.0000000000000338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stoll BJ et al. (2020) Early-Onset Neonatal Sepsis 2015 to 2017, the Rise of Escherichia coli, and the Need for Novel Prevention Strategies. JAMA Pediatr 174, e200593. 10.1001/jamapediatrics.2020.0593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furness JB (2006) The Enteric Nervous System Blackwell Publishing [Google Scholar]
- 22.Rao M and Gershon MD (2018) Enteric nervous system development: what could possibly go wrong? Nat Rev Neurosci 19, 552–565. 10.1038/s41583-018-0041-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spencer NJ and Hu H (2020) Enteric nervous system: sensory transduction, neural circuits and gastrointestinal motility. Nat Rev Gastroenterol Hepatol 17, 338–351. 10.1038/s41575-020-0271-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lake JI and Heuckeroth RO (2013) Enteric nervous system development: migration, differentiation, and disease. Am J Physiol Gastrointest Liver Physiol 305, G1–24. 10.1152/ajpgi.00452.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Avetisyan M et al. (2018) Muscularis macrophage development in the absence of an enteric nervous system. Proc Natl Acad Sci U S A 115, 4696–4701. 10.1073/pnas.1802490115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stanford AH et al. (2020) A direct comparison of mouse and human intestinal development using epithelial gene expression patterns. Pediatr Res 88, 66–76. 10.1038/s41390-019-0472-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pham TD et al. (1991) Time of origin of neurons in the murine enteric nervous system: sequence in relation to phenotype. J Comp Neurol 314, 789–798. 10.1002/cne.903140411 [DOI] [PubMed] [Google Scholar]
- 28.McKeown SJ et al. (2001) Development of the submucous plexus in the large intestine of the mouse. Cell Tissue Res 303, 301–305 [DOI] [PubMed] [Google Scholar]
- 29.Hao MM and Young HM (2009) Development of enteric neuron diversity. J Cell Mol Med 13, 1193–1210. 10.1111/j.1582-4934.2009.00813.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergner AJ et al. (2014) Birthdating of myenteric neuron subtypes in the small intestine of the mouse. J Comp Neurol 522, 514–527. 10.1002/cne.23423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Young HM and Ciampoli D (1998) Transient expression of neuronal nitric oxide synthase by neurons of the submucous plexus of the mouse small intestine. Cell Tissue Res 291, 395–401. 10.1007/s004410051009 [DOI] [PubMed] [Google Scholar]
- 32.Young HM et al. (1999) Expression of Ret-, p75(NTR)-, Phox2a-, Phox2b-, and tyrosine hydroxylase-immunoreactivity by undifferentiated neural crest-derived cells and different classes of enteric neurons in the embryonic mouse gut. Dev Dyn 216, 137–152. [DOI] [PubMed] [Google Scholar]
- 33.Parathan P et al. (2020) The enteric nervous system undergoes significant chemical and synaptic maturation during adolescence in mice. Dev Biol 458, 75–87. 10.1016/j.ydbio.2019.10.011 [DOI] [PubMed] [Google Scholar]
- 34.Collins J et al. (2014) Intestinal microbiota influence the early postnatal development of the enteric nervous system. Neurogastroenterol Motil 26, 98–107. 10.1111/nmo.12236 [DOI] [PubMed] [Google Scholar]
- 35.Anitha M et al. (2012) Gut microbial products regulate murine gastrointestinal motility via Toll-like receptor 4 signaling. Gastroenterology 143, 1006–1016 e1004. 10.1053/j.gastro.2012.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hung LY et al. (2019) Neonatal Antibiotics Disrupt Motility and Enteric Neural Circuits in Mouse Colon. Cell Mol Gastroenterol Hepatol 8, 298–300 e296. 10.1016/j.jcmgh.2019.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foong JP et al. (2012) Myenteric neurons of the mouse small intestine undergo significant electrophysiological and morphological changes during postnatal development. J Physiol 590, 2375–2390. 10.1113/jphysiol.2011.225938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young HM et al. (2002) The projections of early enteric neurons are influenced by the direction of neural crest cell migration. J Neurosci 22, 6005–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McVey Neufeld KA et al. (2013) The microbiome is essential for normal gut intrinsic primary afferent neuron excitability in the mouse. Neurogastroenterol Motil 25, 183–e188. 10.1111/nmo.12049 [DOI] [PubMed] [Google Scholar]
- 40.De Vadder F et al. (2018) Gut microbiota regulates maturation of the adult enteric nervous system via enteric serotonin networks. Proc Natl Acad Sci U S A 115, 6458–6463. 10.1073/pnas.1720017115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drokhlyansky E et al. (2020) The Human and Mouse Enteric Nervous System at Single-Cell Resolution. Cell 182, 1606–1622 e1623. 10.1016/j.cell.2020.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoo BB and Mazmanian SK (2017) The Enteric Network: Interactions between the Immune and Nervous Systems of the Gut. Immunity 46, 910–926. 10.1016/j.immuni.2017.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hung LY et al. (2020) Antibiotic exposure postweaning disrupts the neurochemistry and function of enteric neurons mediating colonic motor activity. Am J Physiol Gastrointest Liver Physiol 318, G1042–G1053. 10.1152/ajpgi.00088.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brun P et al. (2013) Toll-like receptor 2 regulates intestinal inflammation by controlling integrity of the enteric nervous system. Gastroenterology 145, 1323–1333. 10.1053/j.gastro.2013.08.047 [DOI] [PubMed] [Google Scholar]
- 45.Burgueno JF et al. (2016) TLR2 and TLR9 modulate enteric nervous system inflammatory responses to lipopolysaccharide. J Neuroinflammation 13, 187. 10.1186/s12974-016-0653-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cho SX et al. (2020) Characterization of the pathoimmunology of necrotizing enterocolitis reveals novel therapeutic opportunities. Nat Commun 11, 5794. 10.1038/s41467-020-19400-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vuong HE et al. (2020) The maternal microbiome modulates fetal neurodevelopment in mice. Nature 586, 281–286. 10.1038/s41586-020-2745-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan Y et al. (2021) Interleukin-6 produced by enteric neurons regulates the number and phenotype of microbe-responsive regulatory T cells in the gut. Immunity 54, 499–513 e495. 10.1016/j.immuni.2021.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jarret A et al. (2020) Enteric Nervous System-Derived IL-18 Orchestrates Mucosal Barrier Immunity. Cell 180, 813–814. 10.1016/j.cell.2020.02.004 [DOI] [PubMed] [Google Scholar]
- 50.Viola MF and Boeckxstaens G (2020) Intestinal resident macrophages: Multitaskers of the gut. Neurogastroenterol Motil 32, e13843. 10.1111/nmo.13843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matheis F et al. (2020) Adrenergic Signaling in Muscularis Macrophages Limits Infection-Induced Neuronal Loss. Cell 180, 64–78 e16. 10.1016/j.cell.2019.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salvatore S et al. (2019) Neonatal Antibiotics and Prematurity Are Associated with an Increased Risk of Functional Gastrointestinal Disorders in the First Year of Life. J Pediatr 212, 44–51. 10.1016/j.jpeds.2019.04.061 [DOI] [PubMed] [Google Scholar]
- 53.De Schepper S et al. (2018) Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 175, 400–415 e413. 10.1016/j.cell.2018.07.048 [DOI] [PubMed] [Google Scholar]
- 54.Muller PA et al. (2014) Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell 158, 300–313. 10.1016/j.cell.2014.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Honda M et al. (2020) Perivascular localization of macrophages in the intestinal mucosa is regulated by Nr4a1 and the microbiome. Nat Commun 11, 1329. 10.1038/s41467-020-15068-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chalazonitis A and Kessler JA (2012) Pleiotropic effects of the bone morphogenetic proteins on development of the enteric nervous system. Dev Neurobiol 72, 843–856. 10.1002/dneu.22002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mikkelsen HB and Thuneberg L (1999) Op/op mice defective in production of functional colony-stimulating factor-1 lack macrophages in muscularis externa of the small intestine. Cell Tissue Res 295, 485–493. 10.1007/s004410051254 [DOI] [PubMed] [Google Scholar]
- 58.Grubisic V et al. (2020) Enteric Glia Modulate Macrophage Phenotype and Visceral Sensitivity following Inflammation. Cell Rep 32, 108100. 10.1016/j.celrep.2020.108100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stakenborg M et al. (2022) Enteric glial cells favor accumulation of anti-inflammatory macrophages during the resolution of muscularis inflammation. Mucosal Immunol. 10.1038/s41385-022-00563-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Progatzky F et al. (2021) Regulation of intestinal immunity and tissue repair by enteric glia. Nature 599, 125–130. 10.1038/s41586-021-04006-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langness S et al. (2017) Enteric glia cells are critical to limiting the intestinal inflammatory response after injury. Am J Physiol Gastrointest Liver Physiol 312, G274–G282. 10.1152/ajpgi.00371.2016 [DOI] [PubMed] [Google Scholar]
- 62.Kovler ML et al. (2021) Toll-like receptor 4-mediated enteric glia loss is critical for the development of necrotizing enterocolitis. Sci Transl Med 13, eabg3459. 10.1126/scitranslmed.abg3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Olaloye OO et al. (2021) CD16+CD163+ monocytes traffic to sites of inflammation during necrotizing enterocolitis in premature infants. J Exp Med 218. 10.1084/jem.20200344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matteoli G et al. (2014) A distinct vagal anti-inflammatory pathway modulates intestinal muscularis resident macrophages independent of the spleen. Gut 63, 938–948. 10.1136/gutjnl-2013-304676 [DOI] [PubMed] [Google Scholar]
- 65.Cailotto C et al. (2012) Neuroanatomical evidence demonstrating the existence of the vagal anti-inflammatory reflex in the intestine. Neurogastroenterol Motil 24, 191–200, e193. 10.1111/j.1365-2982.2011.01824.x [DOI] [PubMed] [Google Scholar]
- 66.De Schepper S et al. (2018) Muscularis macrophages: Key players in intestinal homeostasis and disease. Cell Immunol 330, 142–150. 10.1016/j.cellimm.2017.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Imamura A et al. (1992) Mucosal immune defence mechanisms in enterocolitis complicating Hirschsprung’s disease. Gut 33, 801–806. 10.1136/gut.33.6.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen X et al. (2020) Intestinal proinflammatory macrophages induce a phenotypic switch in interstitial cells of Cajal. J Clin Invest 130, 6443–6456. 10.1172/JCI126584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gosain A and Brinkman AS (2015) Hirschsprung’s associated enterocolitis. Curr Opin Pediatr 27, 364–369. 10.1097/MOP.0000000000000210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao L et al. (2010) Murine model of Hirschsprung-associated enterocolitis II: Surgical correction of aganglionosis does not eliminate enterocolitis. J Pediatr Surg 45, 206–211; discussion 211-202. 10.1016/j.jpedsurg.2009.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Muller I et al. (2021) Cholinergic Signaling Attenuates Pro-Inflammatory Interleukin-8 Response in Colonic Epithelial Cells. Front Immunol 12, 781147. 10.3389/fimmu.2021.781147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Artis D and Spits H (2015) The biology of innate lymphoid cells. Nature 517, 293–301. 10.1038/nature14189 [DOI] [PubMed] [Google Scholar]
- 73.Zook EC and Kee BL (2016) Development of innate lymphoid cells. Nat Immunol 17, 775–782. 10.1038/ni.3481 [DOI] [PubMed] [Google Scholar]
- 74.Mirpuri J (2021) The emerging role of group 3 innate lymphoid cells in the neonate: interaction with the maternal and neonatal microbiome. Oxf Open Immunol 2, iqab009. 10.1093/oxfimm/iqab009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Deshmukh HS et al. (2014) The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat Med 20, 524–530. 10.1038/nm.3542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Niu X et al. (2020) Transient neonatal antibiotic exposure increases susceptibility to late-onset sepsis driven by microbiota-dependent suppression of type 3 innate lymphoid cells. Sci Rep 10, 12974. 10.1038/s41598-020-69797-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hewitt RJ and Lloyd CM (2021) Regulation of immune responses by the airway epithelial cell landscape. Nat Rev Immunol 21, 347–362. 10.1038/s41577-020-00477-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu HB et al. (2021) Vasoactive intestinal peptide promotes host defense against enteric pathogens by modulating the recruitment of group 3 innate lymphoid cells. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2106634118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pascal M et al. (2022) The neuropeptide VIP potentiates intestinal innate type 2 and type 3 immunity in response to feeding. Mucosal Immunol. 10.1038/s41385-022-00516-9 [DOI] [PubMed] [Google Scholar]
- 80.Eken A et al. (2014) IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol 7, 143–154. 10.1038/mi.2013.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Talbot J et al. (2020) Feeding-dependent VIP neuron-ILC3 circuit regulates the intestinal barrier. Nature 579, 575–580. 10.1038/s41586-020-2039-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Seillet C et al. (2020) The neuropeptide VIP confers anticipatory mucosal immunity by regulating ILC3 activity. Nat Immunol 21, 168–177. 10.1038/s41590-019-0567-y [DOI] [PubMed] [Google Scholar]
- 83.Mihi B et al. (2021) Interleukin-22 signaling attenuates necrotizing enterocolitis by promoting epithelial cell regeneration. Cell Rep Med 2, 100320. 10.1016/j.xcrm.2021.100320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dutta S et al. (2015) Guidelines for feeding very low birth weight infants. Nutrients 7, 423–442. 10.3390/nu7010423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schuchardt A et al. (1994) Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 367, 380–383. 10.1038/367380a0 [DOI] [PubMed] [Google Scholar]
- 86.Patel A et al. (2012) Differential RET signaling pathways drive development of the enteric lymphoid and nervous systems. Sci Signal 5, ra55. 10.1126/scisignal.2002734 [DOI] [PubMed] [Google Scholar]
- 87.Airaksinen MS and Saarma M (2002) The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 3, 383–394. 10.1038/nrn812 [DOI] [PubMed] [Google Scholar]
- 88.Ibiza S et al. (2016) Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 535, 440–443. 10.1038/nature18644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Knoop KA and Newberry RD (2018) Goblet cells: multifaceted players in immunity at mucosal surfaces. Mucosal Immunol 11, 1551–1557. 10.1038/s41385-018-0039-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pandey U and Aich P (2022) Postnatal intestinal mucosa and gut microbial composition develop hand in hand: A mouse study. Biomed J. 10.1016/j.bj.2022.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Strugala V et al. (2008) Thickness and continuity of the adherent colonic mucus barrier in active and quiescent ulcerative colitis and Crohn’s disease. Int J Clin Pract 62, 762–769. 10.1111/j.1742-1241.2007.01665.x [DOI] [PubMed] [Google Scholar]
- 92.Sodhi CP et al. (2012) Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology 143, 708–718 e705. 10.1053/j.gastro.2012.05.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hodzic Z et al. (2017) The Role of Mucosal Immunity in the Pathogenesis of Necrotizing Enterocolitis. Front Pediatr 5, 40. 10.3389/fped.2017.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nowarski R et al. (2015) Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell 163, 1444–1456. 10.1016/j.cell.2015.10.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dinarello CA (2018) Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev 281, 8–27. 10.1111/imr.12621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chiang HY et al. (2022) IL-22 initiates an IL-18-dependent epithelial response circuit to enforce intestinal host defence. Nat Commun 13, 874. 10.1038/s41467-022-28478-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morarach K et al. (2021) Diversification of molecularly defined myenteric neuron classes revealed by single-cell RNA sequencing. Nat Neurosci 24, 34–46. 10.1038/s41593-020-00736-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Levy M et al. (2015) Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 163, 1428–1443. 10.1016/j.cell.2015.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wlodarska M et al. (2014) NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156, 1045–1059. 10.1016/j.cell.2014.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wynn JL et al. (2016) Targeting IL-17A attenuates neonatal sepsis mortality induced by IL-18. Proc Natl Acad Sci U S A 113, E2627–2635. 10.1073/pnas.1515793113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Herath M et al. (2020) The Role of the Gastrointestinal Mucus System in Intestinal Homeostasis: Implications for Neurological Disorders. Front Cell Infect Microbiol 10, 248. 10.3389/fcimb.2020.00248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schwerdtfeger LA and Tobet SA (2020) Vasoactive intestinal peptide regulates ileal goblet cell production in mice. Physiol Rep 8, e14363. 10.14814/phy2.14363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thiagarajah JR et al. (2014) Altered goblet cell differentiation and surface mucus properties in Hirschsprung disease. PLoS One 9, e99944. 10.1371/journal.pone.0099944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Porokuokka LL et al. (2019) Gfra1 Underexpression Causes Hirschsprung’s Disease and Associated Enterocolitis in Mice. Cell Mol Gastroenterol Hepatol 7, 655–678. 10.1016/j.jcmgh.2018.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Knoop KA et al. (2017) Microbial antigen encounter during a preweaning interval is critical for tolerance to gut bacteria. Sci Immunol 2. 10.1126/sciimmunol.aao1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McDole JR et al. (2012) Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 483, 345–349. 10.1038/nature10863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Knoop KA et al. (2015) Microbial sensing by goblet cells controls immune surveillance of luminal antigens in the colon. Mucosal Immunol 8, 198–210. 10.1038/mi.2014.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Knoop KA et al. (2020) Maternal activation of the EGFR prevents translocation of gut-residing pathogenic Escherichia coli in a model of late-onset neonatal sepsis. Proc Natl Acad Sci U S A 117, 7941–7949. 10.1073/pnas.1912022117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bhave S et al. (2021) Pan-enteric neuropathy and dysmotility are present in a mouse model of short-segment Hirschsprung disease and may contribute to post-pullthrough morbidity. J Pediatr Surg 56, 250–256. 10.1016/j.jpedsurg.2020.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dariel A et al. (2020) Analysis of enteric nervous system and intestinal epithelial barrier to predict complications in Hirschsprung’s disease. Sci Rep 10, 21725. 10.1038/s41598-020-78340-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Alemi F et al. (2013) The receptor TGR5 mediates the prokinetic actions of intestinal bile acids and is required for normal defecation in mice. Gastroenterology 144, 145–154. 10.1053/j.gastro.2012.09.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhao A et al. (2003) Dependence of IL-4, IL-13, and nematode-induced alterations in murine small intestinal smooth muscle contractility on Stat6 and enteric nerves. J Immunol 171, 948–954. 10.4049/jimmunol.171.2.948 [DOI] [PubMed] [Google Scholar]
- 113.Andrews AL et al. (2006) IL-4 receptor alpha is an important modulator of IL-4 and IL-13 receptor binding: implications for the development of therapeutic targets. J Immunol 176, 7456–7461. 10.4049/jimmunol.176.12.7456 [DOI] [PubMed] [Google Scholar]
- 114.Chandrasekharan B et al. (2013) Tumor necrosis factor-neuropeptide Y cross talk regulates inflammation, epithelial barrier functions, and colonic motility. Inflamm BowelDis 19, 2535–2546. 10.1097/01.MIB.0000437042.59208.9f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hagl C et al. (2013) Expression and function of the Transforming Growth Factor-b system in the human and rat enteric nervous system. Neurogastroenterol Motil 25, 601–e464. 10.1111/nmo.12119 [DOI] [PubMed] [Google Scholar]